

Abstract
As a core chromatin-regulatory scaffolding protein, WDR5 mediates numerous protein–protein interactions (PPIs) with other partner oncoproteins. However, small-molecule inhibitors that block these PPIs exert limited cell-killing effects. Here, we report structure–activity relationship studies in pancreatic ductal adenocarcinoma (PDAC) cells that led to the discovery of several WDR5 proteolysis-targeting chimer (PROTAC) degraders, including 11 (MS132), a highly potent and selective von Hippel–Lindau (VHL)-recruiting WDR5 degrader, which displayed positive binding cooperativity between WDR5 and VHL, effectively inhibited proliferation in PDAC cells, and was bioavailable in mice and 25, a cereblon (CRBN)-recruiting WDR5 degrader, which selectively degraded WDR5 over the CRBN neo-substrate IKZF1. Furthermore, by conducting site-directed mutagenesis studies, we determined that WDR5 K296, but not K32, was involved in the PROTAC-induced WDR5 degradation. Collectively, these studies resulted in a highly effective WDR5 degrader, which could be a potential therapeutic for pancreatic cancer and several potentially useful tool compounds.
Graphical Abstract
INTRODUCTION
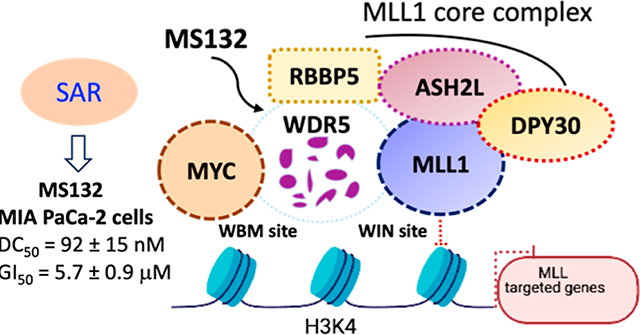
WD repeat domain 5 (WDR5), a highly conserved member of the WD40 repeat-containing protein family among multicellular organisms, functions as a core scaffolding component in many multiprotein complexes that are associated with epigenetic machinery and chromatin regulation.1–3 WDR5 has a characteristic donut-shaped seven-bladed β-propeller structure and contains two major binding sites: WDR5 interaction (WIN) site and WDR5 binding motif (WBM) site, both of which mediate the essential biological functions of WDR5.4–8 At the WIN site, WDR5 binds with the mixed lineage leukemia 1 (MLL1)/SET family proteins (MLL1–4, SETd1A, and SETd1B) with a set of core partner proteins, such as retinoblastoma-binding protein 5 (RBBP5), absent small homeotic-2-like protein (ASH2L), and DPY30, to form MLL1/SET core complexes that deposit histone H3 lysine 4 di- and trimethylation (H3K4me2 and H3K4me3).5,9 In addition, WDR5 also engages in the recruitment and localization of the oncogenic transcription factor MYC to chromatin, which promotes tumorigenesis at the opposite WBM cleft that also interacts with RBBP5.8,10 Aberrant WDR5 expression is associated with a wide range of cancers, including mixed-lineage leukemias,11–13 pancreatic,8 breast,14,15 prostate,16 gastric,17 colorectal,18 bladder cancers,19 and neuroblastoma.20,21 Therefore, WDR5 protein has emerged as a potential pharmacological target for treating both hematological and solid tumors. Blockage of the protein–protein interactions (PPIs) between the WDR5 WIN site and MLL1 or WBM site and MYC has been pursued to inhibit the histone methylation and suppress the transcription function of MYC, respectively.22,23
Over the past decade, substantial progress has been made toward the development of WDR5 WIN-site inhibitors, including macrocyclic peptidomimetics24–26 and small-molecule inhibitors.27–35 However, most of the initial small-molecule PPI inhibitors, such as OICR-9429, showed limited effect on inhibiting cancer cell growth in in vitro and in vivo models.28,29 In 2021, Guo’s group reported a potent and orally bioavailable WDR5/MLL1 PPI inhibitor DDO-2213 that moderately suppressed the growth of MV4;11 xenograft tumors in mice.36 Recently, Fesik’s group and co-workers optimized their dihydroisoquinolinone-containing WDR5 WIN-site inhibitors by incorporating bicyclic heteroaryl P7 units, resulting in potent and orally bioavailable WDR5 inhibitors that are effective in the MV4;11 xenograft tumor models.37 To date, only a few WBM site-targeting chemical probes have been discovered to disrupt the PPIs between WDR5 and MYC. However, no anticancer activity has been reported.38–40 Although none of the WDR5 small-molecule inhibitors are under clinical investigation, the preclinical studies with these WDR5 PPI inhibitors have demonstrated the therapeutic potential of blocking WDR5’s PPIs with either MLL1 or MYC. However, because WDR5 has multifaceted scaffolding functions, these PPI inhibitors that target only one binding site are unlikely to be able to diminish all of the oncogenic functions of WDR5.
Proteolysis targeting chimeras (PROTACs) are heterobifunctional small molecules that consist of ligands to bind proteins of interest (POIs), ligands to recruit E3 ligases, and chemical linkers to connect both ligands. PROTACs bring E3 ligases into the close proximity of the POIs, leading to selective polyubiquitination and the subsequent degradation of the POIs via the 26S proteasome.41–45 Compared with occupancy-driven small-molecule inhibitors that specifically target the enzymatic functions of the POIs, PROTACs pharmacologically induce the degradation of the POIs, resulting in the suppression of both the enzymatic and nonenzymatic functions of the POIs.46,47 Recently, based on WDR5 WIN-site inhibitor OICR-9429, our group and Knapp group independently developed von Hippel–Lindau (VHL)-recruiting WDR5 PROTACs, such as MS33, MS67, and 8g (Figure 1).48,49 By conducting a limited structure–activity relationship (SAR) study in mixed lineage leukemia rearranged (MLL-r) leukemia cells, we discovered MS67, a potent and selective VHL-recruiting WDR5 degrader, which effectively suppressed in vivo tumor growth in both MLL-r cell line xenograft and patient-derived xenograft (PDX) mouse models.48 In addition, our group also developed a cereblon (CRBN)-recruiting WDR5 PROTAC, MS40, which potently downregulated the protein levels of WDR5, IKZF1/3, and LIMD1 (Figure 1).50 Here, we report our SAR studies in pancreatic ductal adenocarcinoma (PDAC) cells that resulted in the discovery of multiple VHL- and CRBN-recruiting WDR5 degraders, including 11 (MS132), which is a potent and selective VHL-recruiting WDR5 degrader that effectively suppressed proliferation in several PDAC cell lines and 25, which is a CRBN-recruiting WDR5 degrader that selectively degraded WDR5 over the CRBN neo-substrate IKZF1. While it has been understudied in the PROTAC field, by conducting site-directed mutagenesis studies, we also determined that WDR5 lysine 296 (K296), but not lysine 32 (K32), was involved in PROTAC-induced degradation of WDR5.
Figure 1.

Chemical structures of representative WDR5 PROTAC degraders.
RESULTS AND DISCUSSION
Design and Biological Evaluation of VHL-Recruiting WDR5 Degraders in Pancreatic Cancer Cells.
Based on our previously reported crystal structures of ternary complexes, WDR5–MS33–VCB (PDB ID: 7JTO) and WDR5–MS67–VCB (PDB ID: 7JTP), the morpholine ring in OICR-9429 (1) was not required for WDR5 degraders.48 Therefore, a carboxylic group was anchored at the meta-position of the upper phenyl ring to bridge the WDR5 binder moiety to the linker moieties. In addition, the introduction of 2,4-dimethyl groups to the bottom methylpiperazine moiety and a fluoro group to the ortho-position of the upper phenyl ring significantly enhanced the binding affinity to WDR5 (Figure 2A).48,51 Based on these structural insights, we designed and synthesized a set of putative WDR5 degraders 3–20 by conjugating precursor 2 to the VHL-recruiting ligand VHL-152,53 through a variety of PEG and alkyl linkers with different compositions and lengths (Figure 2B).
Figure 2.

Schematic design of WDR5 binder precursors (A) and putative degraders (B).
The WDR5 degradation effect of these putative VHL-recruiting WDR5 degraders was assessed using Western blotting (WB) analysis in a PDAC cell line, MIA PaCa-2. The cells were treated with degraders or OICR-9429 at 0.1 and 0.5 μM compound concentrations for 18 h (Figure 3). As expected, the inhibitor OICR-9429 had no effect on the protein levels of WDR5 at the tested concentrations. Compounds 3–10 bearing polyethylene glycol (PEG) linkers were unable to induce effective degradation of WDR5. On the other hand, compound 11 (MS132) with the shortest alkyl linker effectively decreased the WDR5 protein levels at both 0.1 and 0.5 μM. Compounds bearing longer alkyl linkers, 12–20, did not degrade WDR5 as effectively as compound 11. These SAR results demonstrated that a short alkyl linker is preferred for VHL-recruiting WDR5 degraders.
Figure 3.

Effects of compounds 3–20 on inducing WDR5 degradation in MIA PaCa-2 cells. MIA PaCa-2 cells were treated with DMSO or the indicated compounds at concentrations of 0.1 and 0.5 μM, respectively, for 18 h. The cell lysates were analyzed by WB analysis to measure the WDR5 protein levels. Tubulin was used as the loading control. The results shown are representative of at least two independent experiments.
Design and Biological Evaluation of Novel CRBN-Recruiting WDR5 Degraders.
Previously, we discovered MS40, a CRBN-recruiting WDR5 and IKZF1 dual degrader, which also had an off-target effect on inducing LIMD1 protein degradation in MV4;11 cells.50 To improve the selectivity of CRBN-recruiting WDR5 degraders, we conducted a brief SAR study, which focused on modifying the bridge moieties that connect the linker moiety to either the E3 ligase ligand or the WDR5 ligand (Figure 4). First, we replaced the amino group of the pomalidomide moiety on MS40 with a glycolamide bridge group to provide compound 21.54 We also designed the corresponding negative control compound 22, which contains a methylated glutarimide ring that is incapable of engaging CRBN. We next designed and synthesized another glycolamide-containing compound 23 and its corresponding negative control 24 by maintaining the linker length of MS40. We found that 21 and 23 to a less extent effectively downregulated WRD5 protein levels, but both compounds were completely ineffective in degrading neo-substrate IKZF1, and protein LIMD1 in MV4;11 cells (Figure 5A). Notably, negative controls 22 and 24 did not have an effect on the protein levels of WDR5, IKZF1, and LIMD1, supporting the CRBN-dependent mechanism for 21 and 23-mediated WDR5 degradation. Moreover, as illustrated in Figure 5A, compound 21 was slightly more effective than MS40 and 23 in downregulating WDR5 protein levels. Last, we replaced the piperazinyl-containing bridge on the WRD5 binder moiety of 21 to the acetamide bridge on the intermediate 2, to provide compound 25 (Figure 4). We also designed the corresponding negative control compound, 26. As illustrated in Figures 5B and S1, compound 25 exhibited similar potency as 21 in degrading WDR5 protein and had no effect on IKZF1 protein levels in MV4;11 cells. As expected, the negative control 26 did not affect the protein levels of WDR5 and IKZF1. We also evaluated the effect of MS40, 21, 25, and 26 on degrading WDR5 in the MIA PaCa-2 pancreatic cancer cell line, which does not express IKZF1. We found that these compounds, but not the negative control compound 26, effectively reduced the cellular WDR5 protein levels (Figure 5C). Taken together, these SAR results indicate that the α-oxyacetamide bridge added to the CRBN binder moiety significantly improved the selectivity of CRBN-recruiting WDR5 degraders, resulting in several potentially useful tool compounds such as 25 that effectively degraded WDR5 but not the CRBN neo-substrate IKZF1.
Figure 4.

Chemical structures of designed CRBN-recruiting WDR5 degraders and their corresponding negative control compounds.
Figure 5.

Effects of compounds 21–26 on reducing WDR5, IKZF1, and LIMD1 protein levels in MV4;11 and MIA PaCa-2 cells. (A) MV4;11 cells were treated with DMSO or the indicated compounds at concentrations of 0.1 and 0.5 μM, respectively, for 18 h. The cell lysates were analyzed by WB to measure the WDR5, IKZF1, and LIMD1 protein levels. Tubulin was used as the loading control. (B,C) MV4;11 (B) and MIA PaCa-2 (C) cells were treated with DMSO or the indicated compounds at the indicated concentrations for 18 h. The cell lysates were analyzed by WB to examine the WDR5 (B,C) and IKZF1 (B) protein levels. Tubulin was used as the loading control. The results shown are representative of at least two independent experiments.
Because these CRBN-recruiting WDR5 degraders were less effective than the VHL-recruiting compound 11 in degrading WDR5 in MIA PaCa-2 cells, we selected compound 11 as a featured WDR5 degrader for further characterization in a set of biochemical and cellular assays. To study the mechanism of action (MOA) of 11-mediated WDR5 degradation, we also designed and synthesized a structurally similar analogue of compound 11, 27 (MS132N), by incorporating a diastereomer of VHL-1 to block the VHL engagement (Figure 6).
Figure 6.

Chemical structure of compound 27 (MS132N), a negative control of compound 11.
Compound 11 Induces Positive Binding Cooperativity between WDR5 and von Hippel–Lindau Elongin C-Elongin B (VCB).
We first sought to determine the binding affinities of degrader 11 to the VCB E3 ligase complex and target protein WDR5 using isothermal titration calorimetry (ITC). As illustrated in Figures 7A and S2A,B, compound 11 retained high binding affinities to both VCB (KD = 534 ± 48 nM) and WDR5 (KD = 122 ± 56 nM). We next assessed binding cooperativity between WDR5 and VCB induced by compound 11 by titrating WDR5 into the saturated WDR5-11 binary complex, with the titration of VCB into 11 as the reference. Notably, we observed a significant enhancement in VCB binding to the WDR5-11 binary complex (α = KD (binary)/KD (ternary) = 2.30), demonstrating that compound 11 favorably induced the formation of the WDR5-11–VCB ternary complex. As expected, the negative control compound 27 did not display any noticeable binding to VCB but maintained similar binding affinity to WDR5 (KD = 106 ± 43 nM) as degrader 11 (Figures 7B and S3A,B).
Figure 7.

Compound 11 induces positive binding cooperativity between WDR5 and VCB. (A) Representative inverse ITC titrations of VCB into compound 11 (left) and 11-WDR5 complex (middle) and of WDR5 into compound 11 (right). Three replicates of inverse ITC were performed for VCB into compound 11 (left), VCB into 11-WDR5 complex (middle), and WDR5 into compound 11 (right) to calculate the binding kinetic and determine the cooperativity (α) for compound 11. The calculated values represent the mean ± SD from three independent experiments. The first injection has been removed from the fitting. (B) Inverse ITC titrations of VCB (left) and WDR5 (right) into compound 27. Three replicates of inverse ITC titrations were performed for VCB into compound 27 (left) and WDR5 into compound 27 (right) and for measuring binding kinetics. The calculated values represent the mean ± SD from three independent experiments. The first injection has been removed from the fitting.
Compound 11 Effectively Induces WDR5 Degradation in a Concentration-, Time-, and Ubiquitin/Proteasome-Dependent Manner.
We assessed the effect of compound 11 on inducing WDR5 degradation in several PDAC cell lines, including MIA PaCa-2, BxPC-3, HPAF-II, Panc10.05, and PANC-1. As shown in Figures 8A and S4, compound 11, but not the negative control 27, induced WDR5 degradation in a concentration-dependent manner in these PDAC cells with no “hook effect” observed at concentrations up to 50 μM. The half maximal degradation concentration (DC50) of 11-induced WDR5 degradation was determined to be approximately 92 ± 35 nM, with the maximal level of degradation (Dmax) of 73 ± 12% in MIA PaCa-2 cells (Figures 8B and S4). We next evaluated the kinetics of the WDR5 degradation induced by 11 in MIA PaCa-2 cells. As illustrated in Figure 8C, compound 11 effectively induced WDR5 degradation in a time-dependent manner with a significant reduction observed at 2 h and the maximal degradation achieved at 24 h. We also conducted a series of rescue experiments in MIA PaCa-2 cells to validate the MOA of 11-mediated WDR5 degradation (Figure 8D,E). Pretreatment of MIA PaCa-2 cells with 0.4 μM of the proteasome inhibitor carfilzomib for 2 h completely blocked 11-induced WDR5 degradation, suggesting that the observed WDR5 degradation induced by compound 11 is mediated through the proteasome. Additionally, pretreatment with the WDR5 inhibitor OICR-9429 concentration-dependently reduced the WDR5 degradation induced by compound 11 (Figure 8E), demonstrating that WDR5 binding is required for the 11-induced WDR5 degradation. Furthermore, the negative control compound 27 was unable to degrade WDR5 in all tested pancreatic cancer cells, indicating that the VHL E3 ligase is essential for 11-mediated WDR5 degradation (Figure 8A, right panel). Collectively, these mechanistic studies demonstrate that the WDR5 degradation induced by 11 is mediated by the ubiquitin-proteasome system (UPS) and requires the engagement of both the target protein WDR5 and the E3 ligase VHL.
Figure 8.

Compound 11 induces WDR5 degradation in a concentration-, time-, and UPS-dependent manner in PDAC cells. (A) MIA PaCa-2, BxPC-3, HPAF-II, Panc10.05, and PANC-1 cells were treated with DMSO, 11, or 27 at the indicated concentrations for 18 h. The cell lysates were analyzed by WB to examine the WDR5 protein levels. (B) WDR5 DC50 curve of compound 11 in MIA PaCa-2 cells, generated using the data shown in Figure S4 (from 4 independent experiments). (C) MIA PaCa-2 cells were treated with DMSO or 0.1 μM of compound 11, and the cells were collected at the indicated time for WB. (D,E) MIA PaCa-2 cells were pretreated with 0.4 μM of carfilzomib (D) or OICR-9429 at the indicated concentrations (E) for 2 h and then treated with DMSO or 0.5 μM of compound 11 for an additional 4 h. Tubulin was used as the loading control in (A,C–E). The results shown in (A,C–E) are representative of at least two independent experiments.
Compound 11 Is a Highly Selective WDR5 Degrader.
To assess the proteome-wide degradation selectivity of compound 11, we performed an unbiased tandem mass tagged (TMT) mass spectrometry (MS)-based global proteomic study in MIA PaCa-2 cells (Figure 9). To minimize the potential complications from secondary effects caused by WDR5 degradation, we performed proteomics analysis with a 2 h treatment rather than a longer treatment (e.g., 18 h). As shown in Figure S5, compared to DMSO treatment, compound 11 significantly reduced WDR5 protein levels after the cells were treated with 11 at 1.5 μM for 2 h. As illustrated in Figure 9, out of 4040 quantified proteins, WDR5 is the only protein whose abundance was significantly decreased by compound 11, with a cutoff of a P value less than 0.01 and a Log2 fold change greater than 0.5. Overall, these quantitative global proteomic study results demonstrate that compound 11 is highly selective for inducing WDR5 degradation at an early time point (such as 2 h).
Figure 9.

Compound 11 selectively induces WDR5 degradation in MIA PaCa-2 cells in an unbiased TMT MS-based global proteomic study. MIA PaCa-2 cells were treated with DMSO or 11 at 1.5 μM for 2 h before the cell lysates were harvested for MS analysis. The dashed lines indicate a cutoff of a P value less than 0.01 (y axis) and a Log2 fold change greater than 0.5 (x axis) in three independent biological replicates.
Compound 11 Effectively Suppresses the Growth of PDAC Cells.
We evaluated the antiproliferative activity of compound 11 and the negative control 27 in multiple PDAC cell lines including MIA PaCa-2, BxPC-3, HPAF-II, Panc10.05, and PANC-1. As shown in Figure 10A–F, compound 11, but not the control 27, inhibited the growth of these cells with GI50 values ranging from 4.0 to 32 μM. Of particular note, the negative control 27 did not induce any significant WDR5 degradation in all tested PDAC cell lines (Figure 8A). 27’s lack of antiproliferative activity in PDAC cells is consistent with the lack of antiproliferative activity previously reported for the WDR5 PPI inhibitor OICR-9429.48 Taken together, these results suggest that the WDR5 degradation induced by compound 11 results in its antiproliferative activity in PDAC cells.
Figure 10.

Compound 11 inhibits the proliferation of PDAC cells. (A–E) Growth inhibition curves of compounds 11 and 27 in pancreatic cancer cell lines, MIA PaCa-2, BxPC-3, HPAF-II, Panc10.05, and PANC-1. Y-axis, presented in the mean ± SEM of data from three independent experiments, shows the relative cell growth post-treatment with compounds 11 and 27 at the indicated concentrations (x-axis) for 6 days, normalized to DMSO-treated. (F) Summary of GI50 values of compounds 11 and 27 in the five pancreatic cancer cell lines after a 6 day treatment.
Compound 11 Is Bioavailable in Mice.
We next assessed the in vivo mouse pharmacokinetic (PK) properties of compound 11 (Figure 11). After a single intraperitoneal (IP) injection of compound 11 at a dose of 75 mg/kg, the maximum plasma concentration (Cmax = 5 μM) was achieved at 0.5 h post-treatment, and the concentrations of 11 in plasma were maintained around 1 μM for the initial 4 h and above 100 nM over 12 h with a sufficient systemic exposure AUC0–last (area under the plasma concentration–time curve) value of 7300 h*ng/mL. These results indicate that 11 has the potential to be used in in vivo animal studies. Importantly, no clinical signs were observed in the tested mice during the PK study.
Figure 11.

Plasma concentrations of compound 11 over a 12 h period in mice after a single IP injection of compound 11 at 75 mg/kg. The plasma concentrations represent the means ± SEM of data from three mice per time point.
WDR5 K296, but Not K32, Is Involved in PROTAC-Mediated WDR5 Degradation.
In our previously conducted structural and biophysical studies, we investigated the PPIs between WDR5 and VHL induced by our WDR5 degrader MS67 and the cross protein–ligand (MS67) interactions.48 However, elucidation of which lysine residues of WDR5 are polyubiquitinated and are involved in PROTAC-induced WDR5 degradation has not been performed. It is worth noting that while many PROTAC degraders have been reported, very few studies have attempted to determine the lysine ubiquitination sites of the target protein in the PROTAC field. To determine which lysine residues of WDR5 are involved in PROTAC-induced WDR5 degradation, we analyzed the crystal structure of the WDR5–MS67–VCB ternary complex (PDB ID: 7JTP) we solved previously and selected K32 and K296 for examination because they are solvent-exposed residues and could be potential ubiquitination sites (Figure 12A). Next, we performed lysine-to-arginine mutagenesis to get WDR5 K32R and K296R mutants and evaluated the effect of MS67 on degrading WDR5 wild-type (WT) and K32R and K296R mutants. As expected, MS67 induced robust degradation of WDR5 WT in a concentration-dependent manner (Figure 12A). On the other hand, while the K32R mutant displayed similar sensitivity as that of the MS67 treatment, the K296R mutant was resistant to the MS67 treatment (Figure 12B). These results suggest that K296 is likely a polyubiquitination site and is involved in PROTAC-induced WDR5 degradation.
Figure 12.
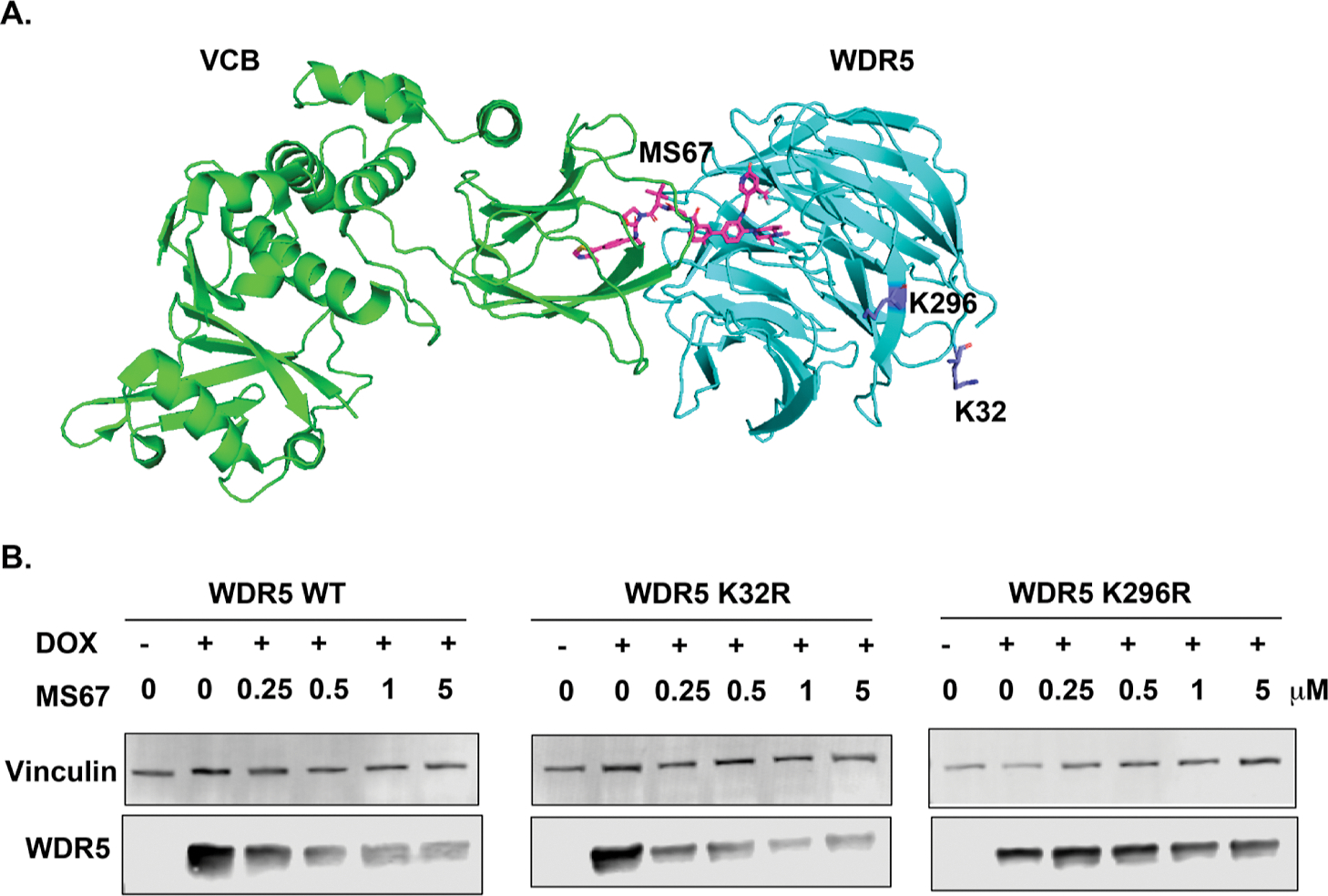
WDR5 K296, but not K32, is involved in PROTAC-mediated WDR5 degradation. (A) Crystal structure of the WDR5–MS67–VCB ternary complex (PDB ID: 7JTP) with WDR5 K296 and K32 residues colored in purple. (B) Immunoblot analysis of WDR5 WT (left), K32R (middle), and K296R (right) mutants after MS67 treatment. HEK293T cells, which are ectopically overexpressed with WDR5 WT, K32R, and K296R mutants, respectively, upon treatment with doxycycline (Dox), were treated with DMSO or MS67 at the indicated concentrations for 3 days. Vinculin was used as the loading control. The results shown are representative of at least two independent experiments.
Chemical Synthesis.
The key intermediate 2 and linkers 28a–h and 29a–j were synthesized following our previously reported procedures.48,52,55 Compounds 3–20 were synthesized through amide-coupling of intermediate 2 and linkers 28a–h and 29a–j (Scheme 1).
Scheme 1. Syntheses of Compounds 3–20a.

aReagents and conditions: (a) 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDCI), 1-hydroxy-7-azabenzo-triazole (HOAt), N-methylmorpholine (NMM), DMSO, rt, and 12 h.
The synthetic route for the preparation of compounds 21–26 is outlined in Scheme 2. Intermediate 30 and the N-methylated analogue 31 were prepared according to our previously reported procedures.56 Amide coupling reactions between intermediates 30–31 and commercially available linkers 32–33, followed by tert-butyl ester hydrolysis, afforded carboxylic acid intermediates 34–37, which were further converted into compounds 21–24 by coupling with intermediate 38.48,50 Similarly, amide coupling between intermediates 30–31 and commercially available tert-butyl (7-aminoheptyl)carbamate (39) and subsequent removal of the Boc-group provided intermediates 40–41. Amide coupling between 40–41 and 2 yielded compounds 25 and 26.
Scheme 2. Syntheses of Compounds 21–26a.

aReagents and conditions: (a) EDCI, HOAt, NMM, DMSO, rt, and 12 h and (b) TFA/DCM, rt, and 30 min.
The synthetic route for the negative control compound 27 is outlined in Scheme 3. The diastereomer of VHL-1, 42, was prepared following our previously reported procedures.57,58 Amide coupling between 42 and commercially available (tert-butoxycarbonyl)glycine (43) and subsequent Boc-deprotection afforded intermediate 44.52 Amide coupling between intermediates 2 and 44 provided compound 27.
Scheme 3. Synthesis of the Negative Control 27a.

aReagents and conditions: (a) EDCI, HOAt, NMM, DMSO, rt, and 12 h and (b) TFA/DCM, rt, and 30 min.
CONCLUSIONS
In summary, based on the structural insights revealed by our previously reported crystal structures of the WDR5–MS33–VCB and WDR5–MS67–VCB ternary complexes,48 we designed and synthesized a set of WDR5 PROTAC degraders, utilizing intermediate 2 with high WDR5 binding affinity. We evaluated the effect of these compounds on degrading WDR5 in pancreatic cancer cells. From these studies, we discovered compound 11, a highly potent and selective VHL-recruiting WDR5 degrader. Compound 11 induced positive binding cooperativity between WDR5 and VCB, which led to highly effective degradation of WDR5 in five PDAC cell lines. The WDR5 degradation induced by compound 11 was dependent on the E3 ligase VHL and proteasome and occurred very rapidly (as early as 2 h). Notably, out of more than 4000 proteins detected, WDR5 is the only protein that was significantly degraded by compound 11 in an unbiased MS-based global proteomic study at an early time point (2 h). We also developed a negative control of compound 11, compound 27, which binds WDR5 but not VHL and did not degrade WDR5 in PDAC cell lines. Importantly, compound 11, but not the negative control 27, effectively suppressed the growth in five PDAC cell lines, indicating that WDR5 degradation is the main contributor to the observed antiproliferative activity of compound 11 in PDAC cells. Moreover, compound 11 was bioavailable in a mouse PK study via IP injection, making it suitable for in vivo efficacy studies in PDAC xenograft mouse models.
In addition, we optimized our previously reported CRBN-recruiting WDR5 degrader MS40,50 which degraded WDR5 and the CRBN neo-substrate IKZF1. By replacing the amino group of the CRBN ligand with a glycolamide bridge, we discovered novel and selective CRBN-recruiting WDR5 degraders, 21 and 25, which effectively degraded WDR5 but did not degrade IKZF1.
Lastly, while a large number of PROTAC degraders have been developed for various target proteins, the studies that were intended to determine the lysine ubiquitination sites of the target protein have been scarce in the PROTAC field. To elucidate which WDR5 lysine residues are likely polyubiquitinated and are implicated in the PROTAC-induced WDR5 degradation, we conducted site-directed mutagenesis studies and determined that WDR5 K296, but not K32, was involved in the PROTAC-induced WDR5 degradation.
Overall, we discovered a highly potent and selective VHL-recruiting WDR5 PROTAC degrader, which could be a potential therapeutic for pancreatic cancer and several selective CRBN-recruiting WDR5 PROTAC degraders, which did not degrade CRBN neosubstrates. These compounds and their corresponding negative controls are potentially useful tool compounds for further investigating the oncogenic functions of WDR5 in pancreatic cancer and other WDR5-dependent cancers.
EXPERIMENTAL SECTION
Chemistry General Procedures.
All commercially available solvents and reagents were used without further purification. Normal and reverse phase flash chromatography were performed using a Teledyne ISCO CombiFlash instrument and HP C18 RediSep Rf reverse phase silica columns, respectively. Final compounds for biological evaluation were purified with preparative high-performance liquid chromatography (HPLC) on an Agilent Prep 1200 series with the UV detector set to 254 nm with solvent A (0.1% of TFA in water) and solvent B (acetonitrile) as eluents with a flow rate of 40 mL/min at room temperature. Purities of the final compounds were assessed at >95% by HPLC under the following conditions: Agilent 1200 series system, 2.1 mm × 150 mm Zorbax 300SB-C18 5 μm column, 1–99% gradient of 0.1% TFA in water, and 0.1% TFA in acetonitrile; flow rate, 0.4 mL/min; high-resolution mass spectra (HRMS) data were acquired on an Agilent G1969A API-TOF with an electrospray ionization (ESI) source. Proton (1H NMR) and carbon (13C NMR) nuclear magnetic resonance spectra were recorded on either an AVANCE NEO 600 or 400 MHz NMR spectrometer and reported in parts per million (ppm) on the δ scale in the following format: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quarter, and m = multiplet), coupling constant (J, Hz), and integration. Intermediates 2, 28a–h, 29a–j, 30–31, 38, and 42 were synthesized following our previously reported procedures.52,55,56
N-(2′-Fluoro-5′-((2-(2-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-2-oxoethoxy)ethyl)carbamoyl)-4-((3S,5R)-3,4,5-trimethylpiperazin-1-yl)-[1,1′-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide (3).
To a solution of intermediate 2 (11.9 mg, 0.022 mmol) in DMSO (1 mL) were added linker 28a (12.4 mg, 0.022 mmol, 1.0 equiv), EDCI (6.3 mg, 0.033 mmol, 1.5 equiv), HOAt (4.5 mg, 0.033 mmol, 1.5 equiv), and NMM (6.7 mg, 0.066 mmol, 3.0 equiv). After stirring overnight at room temperature, the resulting mixture was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford compound 3 as a white solid (15.1 mg, yield 65%). 1H NMR (600 MHz, CD3OD): δ 8.93 (s, 1H), 8.14 (s, 1H), 8.05–7.99 (m, 2H), 7.94–7.87 (m, 1H), 7.49–7.34 (m, 6H), 7.28–7.18 (m, 1H), 6.92 (d, J = 3.2 Hz, 1H), 4.68 (s, 1H), 4.59–4.47 (m, 3H), 4.36–4.30 (m, 1H), 4.13–4.00 (m, 2H), 3.88–3.82 (m, 1H), 3.79–3.67 (m, 5H), 3.64–3.57 (m, 2H), 3.55–3.46 (m, 2H), 3.38–3.30 (m, 2H), 2.98 (d, J = 3.4 Hz, 3H), 2.44 (s, 3H), 2.24–2.18 (m, 1H), 2.10–2.05 (m, 1H), 1.47–1.39 (m, 6H), 0.97 (s, 9H). HRMS (m/z): for C53H62F4N9O8S+ [M + H]+ calcd, 1060.4373; found, 1060.4389.
N-(2′-Fluoro-5′-((2-(3-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-3-oxopropoxy)ethyl)carbamoyl)-4-((3S,5R)-3,4,5-trimethylpiperazin-1-yl)-[1,1′-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide (4).
Compound 4 was synthesized following the standard procedures for preparing compound 3 from intermediates 2 (11.9 mg, 0.022 mmol) and 28b (17 mg, 0.022 mmol, 1.0 equiv). Compound 4 was obtained as a white solid (17.7 mg, yield 75%). 1H NMR (600 MHz, CD3OD): δ 8.94 (d, J = 1.2 Hz, 1H), 8.15 (s, 1H), 8.00 (d, J = 7.3 Hz, 2H), 7.91–7.82 (m, 1H), 7.52–7.33 (m, 6H), 7.26 (dd, J = 10.3, 8.6 Hz, 1H), 6.92 (s, 1H), 4.62 (s, 1H), 4.56 (dd, J = 9.4, 7.3 Hz, 1H), 4.50–4.41 (m, 2H), 4.32 (d, J = 15.5 Hz, 1H), 3.86 (d, J = 10.9 Hz, 1H), 3.81–3.74 (m, 2H), 3.74–3.69 (m, 1H), 3.69–3.59 (m, 3H), 3.58–3.49 (m, 3H), 3.37–3.32 (m, 2H), 2.98 (s, 5H), 2.56–2.49 (m, 2H), 2.44 (d, J = 1.2 Hz, 3H), 2.21 (dd, J = 13.1, 7.7 Hz, 1H), 2.11–2.03 (m, 1H), 1.44 (d, J = 6.5 Hz, 6H), 0.97 (s, 9H). HRMS (m/z): for C54H64F4N9O8S+ [M + H]+ calcd, 1074.4529; found, 1074.4576.
N-(2′-Fluoro-5′-((2-(2-(2-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-2-oxoethoxy)ethoxy)ethyl)carbamoyl)-4-((3S,5R)-3,4,5-trimethylpiperazin-1-yl)-[1,1′-biphenyl]-3-yl)-6-oxo4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide (5).
Compound 5 was synthesized following the standard procedures for preparing compound 3 from intermediates 2 (11.9 mg, 0.022 mmol) and 28c (13.5 mg, 0.022 mmol, 1.0 equiv). Compound 5 was obtained as a white solid (21.8 mg, yield 90%). 1H NMR (600 MHz, CD3OD): δ 8.95 (s, 1H), 8.14 (s, 1H), 8.01 (s, 1H), 7.94 (dd, J = 7.5, 2.4 Hz, 1H), 7.88–7.79 (m, 1H), 7.54–7.33 (m, 6H), 7.32–7.19 (m, 1H), 6.92 (s, 1H), 4.72 (s, 1H), 4.54–4.45 (m, 3H), 4.28 (d, J = 15.5 Hz, 1H), 4.05–3.88 (m, 2H), 3.84–3.77 (m, 1H), 3.73–3.63 (m, 6H), 3.61–3.50 (m, 5H), 3.36–3.32 (m, 4H), 2.98 (s, 3H), 2.46 (s, 3H), 2.26–2.15 (m, 1H), 2.13–1.99 (m, 1H), 1.44 (d, J = 6.4 Hz, 6H), 1.01 (s, 9H). HRMS (m/z): for C55H66F4N9O9S+ [M + H]+ calcd, 1104.4635; found, 1104.4599.
N-(2′-Fluoro-5′-((2-(2-(3-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-3-oxopropoxy)ethoxy)ethyl)carbamoyl)-4-((3S,5R)-3,4,5-trimethylpiperazin-1-yl)-[1,1′-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide (6).
Compound 6 was synthesized following the standard procedures for preparing compound 3 from intermediates 2 (11.9 mg, 0.022 mmol) and 28d (17.9 mg, 0.022 mmol, 1.0 equiv). Compound 6 was obtained as a white solid (22.8 mg, yield 92%). 1H NMR (600 MHz, CD3OD): δ 8.97 (s, 1H), 8.15 (d, J = 2.0 Hz, 1H), 8.06–7.90 (m, 2H), 7.86–7.82 (m, 1H), 7.51–7.40 (m, 3H), 7.40–7.32 (m, 3H), 7.27 (dd, J = 10.3, 8.6 Hz, 1H), 6.92 (s, 1H), 4.63 (s, 1H), 4.59–4.45 (m, 3H), 4.33 (d, J = 15.5 Hz, 1H), 3.91–3.83 (m, 1H), 3.78 (dd, J = 10.9, 3.9 Hz, 1H), 3.74–3.47 (m, 12H), 3.36–3.30 (m, 2H), 3.02–2.92 (m, 5H), 2.53–2.38 (m, 5H), 2.23–2.17 (m, 1H), 2.10–2.01 (m, 1H), 1.44 (d, J = 6.5 Hz, 6H), 1.00 (s, 9H). HRMS (m/z): for C56H68F4N9O9S+ [M + H]+ calcd, 1118.4791; found, 1118.4823.
N-(2′-Fluoro-5′-(((S)-13-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidine-1-carbonyl)-14,14-dimethyl-11-oxo-3,6,9-trioxa-12-azapentadecyl)carbamoyl)-4-((3S,5R)-3,4,5-trimethylpiperazin-1-yl)-[1,1′-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide (7).
Compound 7 was synthesized following the standard procedures for preparing compound 3 from intermediates 2 (13 mg, 0.024 mmol) and 28e (20.3 mg, 0.024 mmol, 1.0 equiv). Compound 7 was obtained as a white solid (17.4 mg, yield 63%). 1H NMR (600 MHz, CD3OD): δ 8.95 (s, 1H), 8.15 (d, J = 2.1 Hz, 1H), 8.06–7.93 (m, 2H), 7.89–7.76 (m, 1H), 7.52–7.32 (m, 6H), 7.31–7.23 (m, 1H), 6.92 (s, 1H), 4.67 (s, 1H), 4.61–4.45 (m, 3H), 4.34 (d, J = 15.5 Hz, 1H), 4.01–3.89 (m, 2H), 3.88–3.74 (m, 2H), 3.69–3.47 (m, 14H), 3.36–3.34 (m, 1H), 3.33–3.32 (m, 1H), 3.09–2.92 (m, 5H), 2.46 (s, 3H), 2.28–2.16 (m, 1H), 2.13–2.02 (m, 1H), 1.44 (d, J = 6.5 Hz, 6H), 1.01 (s, 9H). HRMS (m/z): for C57H70F4N9O10S+ [M + H]+ calcd, 1148.4897; found, 1148.4917.
N-(2′-Fluoro-5′-(((S)-14-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidine-1-carbonyl)-15,15-dimethyl-12-oxo-3,6,9-trioxa-13-azahexadecyl)carbamoyl)-4-((3S,5R)-3,4,5-trimethylpiperazin-1-yl)-[1,1′-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide (8).
Compound 8 was synthesized following the standard procedures for preparing compound 3 from intermediates 2 (13 mg, 0.024 mmol) and 28f (20.7 mg, 0.024 mmol, 1.0 equiv). Compound 8 was obtained as a white solid (11.7 mg, yield 42%). 1H NMR (600 MHz, CD3OD): δ 8.97 (s, 1H), 8.15 (s, 1H), 8.07–7.94 (m, 2H), 7.85 (ddd, J = 8.5, 4.5, 2.4 Hz, 1H), 7.53–7.32 (m, 6H), 7.28 (dd, J = 10.3, 8.6 Hz, 1H), 6.92 (s, 1H), 4.62 (s, 1H), 4.58–4.45 (m, 3H), 4.34 (d, J = 15.5 Hz, 1H), 3.86 (d, J = 11.0 Hz, 1H), 3.77 (dd, J = 11.0, 3.9 Hz, 1H), 3.71–3.49 (m, 16H), 3.37–3.31 (m, 2H), 2.98 (s, 5H), 2.54–2.38 (m, 5H), 2.24–2.17 (m, 1H), 2.09–2.03 (m, 1H), 1.43 (d, J = 6.5 Hz, 6H), 1.00 (s, 9H). HRMS (m/z): for C58H72F4N9O10S+ [M + H]+ calcd, 1162.5053; found, 1162.5043.
N-(2′-Fluoro-5′-(((S)-17-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidine-1-carbonyl)-18,18-dimethyl-15-oxo-3,6,9,12-tetraoxa-16-azanonadecyl)carbamoyl)-4-((3S,5R)-3,4,5-trimethylpiperazin-1-yl)-[1,1′-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide (9).
Compound 9 was synthesized following the standard procedures for preparing compound 3 from intermediates 2 (13 mg, 0.024 mmol) and 28g (17.1 mg, 0.024 mmol, 1.0 equiv). Compound 9 was obtained as a white solid (22.6 mg, yield 78%). 1H NMR (600 MHz, CD3OD): δ 9.03 (s, 1H), 8.16 (d, J = 1.9 Hz, 1H), 8.05–7.94 (m, 2H), 7.93–7.80 (m, 1H), 7.54–7.35 (m, 6H), 7.29 (dd, J = 10.3, 8.6 Hz, 1H), 6.92 (s, 1H), 4.63 (s, 1H), 4.60–4.46 (m, 3H), 4.35 (d, J = 15.5 Hz, 1H), 3.92–3.84 (m, 1H), 3.78 (dd, J = 11.0, 3.9 Hz, 1H), 3.74–3.47 (m, 20H), 3.38–3.30 (m, 2H), 3.05–2.91 (m, 5H), 2.58–2.50 (m, 1H), 2.50–2.39 (m, 4H), 2.24–2.18 (m, 1H), 2.10–2.04 (m, 1H), 1.44 (d, J = 6.5 Hz, 6H), 1.01 (s, 9H). HRMS (m/z): for C60H76F4N9O11S+ [M + H]+ calcd, 1206.5316; found, 1206.5287.
N-(2′-Fluoro-5′-(((S)-20-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidine-1-carbonyl)-21,21-dimethyl-18-oxo-3,6,9,12,15-pentaoxa-19-azadocosyl)carbamoyl)-4-((3S,5R)-3,4,5-trimethylpiperazin-1-yl)-[1,1′-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide (10).
Compound 10 was synthesized following the standard procedures for preparing compound 3 from intermediates 2 (13 mg, 0.022 mmol) and 28h (22.8 mg, 0.024 mmol, 1.0 equiv). Compound 10 was obtained as a white solid (15.3 mg, yield 51%). 1H NMR (600 MHz, CD3OD): δ 9.02 (s, 1H), 8.16 (d, J = 1.9 Hz, 1H), 8.06–7.97 (m, 2H), 7.93–7.82 (m, 1H), 7.54–7.36 (m, 6H), 7.29 (dd, J = 10.3, 8.6 Hz, 1H), 6.93 (s, 1H), 4.63 (s, 1H), 4.60–4.45 (m, 3H), 4.35 (d, J = 15.5 Hz, 1H), 3.87 (d, J = 11.2 Hz, 1H), 3.78 (dd, J = 11.0, 3.9 Hz, 1H), 3.74–3.48 (m, 24H), 3.38–3.32 (m, 2H), 2.98 (d, J = 3.0 Hz, 5H), 2.59–2.51 (m, 1H), 2.51–2.41 (m, 4H), 2.24–2.16 (m, 1H), 2.11–2.03 (m, 1H), 1.44 (d, J = 6.5 Hz, 6H), 1.02 (s, 9H). HRMS (m/z): for C62H80F4N9O12S+ [M + H]+ calcd, 1250.5578; found, 1250.5534.
N-(2′-Fluoro-5′-((2-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-2-oxoethyl)carbamoyl)-4-((3S,5R)-3,4,5-trimethylpiperazin-1-yl)-[1,1′-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide (11).
Compound 11 was synthesized following the standard procedures for preparing compound 3 from intermediates 2 (13 mg, 0.024 mmol) and 29a (17.2 mg, 0.024 mmol, 1.0 equiv). Compound 11 was obtained as a white solid (21.2 mg, yield 89%). 1H NMR (400 MHz, CD3OD): δ 8.99 (s, 1H), 8.15 (s, 1H), 8.04 (dd, J = 7.4, 2.4 Hz, 1H), 8.00 (s, 1H), 7.93–7.87 (m, 1H), 7.46 (dd, J = 10.0, 7.3 Hz, 3H), 7.41–7.34 (m, 3H), 7.29 (dd, J = 10.3, 8.6 Hz, 1H), 6.91 (s, 1H), 4.66 (s, 1H), 4.60–4.47 (m, 3H), 4.34 (d, J = 15.5 Hz, 1H), 4.10 (d, J = 11.7 Hz, 2H), 3.89 (d, J = 11.2 Hz, 1H), 3.79 (dd, J = 11.0, 3.8 Hz, 1H), 3.56–3.48 (m, 2H), 3.32–3.27 (m, 4H), 2.98 (s, 3H), 2.46 (s, 3H), 2.25–2.17 (m, 1H), 2.10–2.01 (m, 1H), 1.44 (d, J = 6.5 Hz, 6H), 1.04 (s, 9H). 13C NMR (101 MHz, CD3OD): δ 174.44, 172.03, 171.33, 169.27, 165.27, 164.36, 162.90 (d, 1JC–F = 208.1 Hz, 1C, −*C–F), 161.15, 153.63, 147.65, 143.76, (q, 2JC–F = 46.67 Hz, 1C), 141.57, 140.78, 133.62, 133.60, 131.84 (d, 3JC–F = 3.56 Hz, 1C), 131.50 (d, 3JC–F = 3.03 Hz, 1C), 130.75, 130.37 (2C), 130.04, 129.95, 129.62, 129.53, 129.47, 129.06 (2C), 128.15, 128.12, 123.39 (q, 1JC–F = 282.4 Hz, 1C, −*CF3), 117.60, 117.36, 71.11, 62.45, 60.88, 59.07, 58.03, 57.61, 57.48, 57.27, 44.08, 43.67, 38.91, 37.40, 36.94, 26.98 (3C), 15.21, 15.15 (2C). tR = 4.16 min, HRMS (m/z): for C51H58F4N9O7S+ [M + H]+ calcd, 1016.4111; found, 1016.4124.
N-(2′-Fluoro-5′-((3-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-3-oxopropyl)carbamoyl)-4-((3S,5R)-3,4,5-trimethylpiperazin-1-yl)-[1,1′-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide (12).
Compound 12 was synthesized following the standard procedures for preparing compound 3 from intermediates 2 (13 mg, 0.024 mmol) and 29b (17.5 mg, 0.024 mmol, 1.0 equiv). Compound 12 was obtained as a white solid (19 mg, yield 77%). 1H NMR (600 MHz, CD3OD): δ 8.96 (s, 1H), 8.14 (s, 1H), 8.07–7.93 (m, 2H), 7.89–7.77 (m, 1H), 7.50–7.24 (m, 7H), 6.92 (s, 1H), 4.61 (s, 1H), 4.58–4.46 (m, 3H), 4.33 (d, J = 15.5 Hz, 1H), 3.92 (d, J = 11.2 Hz, 1H), 3.78 (dd, J = 10.9, 3.9 Hz, 1H), 3.71–3.67 (m, 1H), 3.65–3.58 (m, 1H), 3.53 (q, J = 7.6, 5.4 Hz, 2H), 3.37–3.32 (m, 2H), 2.98 (d, J = 5.2 Hz, 5H), 2.67–2.54 (m, 2H), 2.46 (s, 3H), 2.23–2.16 (m, 1H), 2.09–2.02 (m, 1H), 1.44 (d, J = 6.5 Hz, 6H), 1.00 (s, 9H). HRMS (m/z): for C52H60F4N9O7S+ [M + H]+ calcd, 1030.4267; found, 1030.4276.
N-(2′-Fluoro-5′-((4-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-4-oxobutyl)carbamoyl)-4-((3S,5R)-3,4,5-trimethylpiperazin-1-yl)-[1,1′-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide (13).
Compound 13 was synthesized following the standard procedures for preparing compound 3 from intermediates 2 (13 mg, 0.024 mmol) and 29c (17.8 mg, 0.024 mmol, 1.0 equiv). Compound 13 was obtained as a white solid (18.7 mg, yield 75%). 1H NMR (600 MHz, CD3OD): δ 9.02 (s, 1H), 8.25–8.09 (m, 1H), 8.09–7.92 (m, 2H), 7.86 (ddd, J = 8.5, 4.5, 2.4 Hz, 1H), 7.55–7.34 (m, 6H), 7.29 (dd, J = 10.2, 8.6 Hz, 1H), 6.92 (s, 1H), 4.64–4.43 (m, 4H), 4.35 (d, J = 15.5 Hz, 1H), 3.90 (d, J = 11.0 Hz, 1H), 3.78 (dd, J = 11.0, 4.0 Hz, 1H), 3.60–3.50 (m, 2H), 3.41 (h, J = 6.7 Hz, 2H), 3.35–3.32 (m, 2H), 2.98 (s, 5H), 2.47 (s, 3H), 2.43–2.31 (m, 2H), 2.23–2.16 (m, 1H), 2.12–2.03 (m, 1H), 1.99–1.85 (m, 2H), 1.44 (d, J = 6.5 Hz, 6H), 1.03 (s, 9H). HRMS (m/z): for C53H62F4N9O7S+ [M + H]+ calcd, 1044.4424; found, 1044.4445.
N-(2′-Fluoro-5′-((5-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-5-oxopentyl)carbamoyl)-4-((3S,5R)-3,4,5-trimethylpiperazin-1-yl)-[1,1′-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide (14).
Compound 14 was synthesized following the standard procedures for preparing compound 3 from intermediates 2 (13 mg, 0.024 mmol) and 29d (13.6 mg, 0.024 mmol, 1.0 equiv). Compound 14 was obtained as a white solid (8.1 mg, yield 32%). 1H NMR (600 MHz, CD3OD): δ 8.96 (s, 1H), 8.15 (s, 1H), 8.05–7.93 (m, 2H), 7.88–7.82 (m, 1H), 7.55–7.22 (m, 7H), 6.93 (s, 1H), 4.63–4.41 (m, 4H), 4.34 (d, J = 15.5 Hz, 1H), 3.89 (d, J = 11.0 Hz, 1H), 3.78 (dd, J = 10.9, 3.9 Hz, 1H), 3.52 (d, J = 6.9 Hz, 2H), 3.40 (t, J = 6.7 Hz, 2H), 3.33 (t, J = 10.9 Hz, 2H), 2.98 (d, J = 7.6 Hz, 5H), 2.47 (s, 3H), 2.39–2.27 (m, 2H), 2.20 (dd, J = 13.2, 7.5 Hz, 1H), 2.13–2.03 (m, 1H), 1.75–1.60 (m, 4H), 1.44 (d, J = 6.5 Hz, 6H), 1.02 (s, 9H). HRMS (m/z): for C54H64F4N9O7S+ [M + H]+ calcd, 1058.4580; found, 1058.4565.
N-(2′-Fluoro-5′-((7-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-7-oxoheptyl)carbamoyl)-4-((3S,5R)-3,4,5-trimethylpiperazin-1-yl)-[1,1′-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide (15).
Compound 15 was synthesized following the standard procedures for preparing compound 3 from intermediates 2 (13 mg, 0.024 mmol) and 29e (13.9 mg, 0.024 mmol, 1.0 equiv). Compound 15 was obtained as a white solid (15.9 mg, yield 63%). 1H NMR (600 MHz, CD3OD): δ 8.99 (s, 1H), 8.15 (s, 1H), 8.03–7.95 (m, 2H), 7.88–7.70 (m, 1H), 7.64–7.35 (m, 6H), 7.28 (dd, J = 10.2, 8.6 Hz, 1H), 6.92 (s, 1H), 4.63–4.44 (m, 4H), 4.34 (d, J = 15.5 Hz, 1H), 3.89 (dd, J = 11.1, 2.1 Hz, 1H), 3.78 (dd, J = 11.0, 3.9 Hz, 1H), 3.60–3.48 (m, 2H), 3.42–3.37 (m, 2H), 3.34–3.30 (m, 2H), 3.06–2.93 (m, 5H), 2.47 (s, 3H), 2.36–2.25 (m, 2H), 2.22–2.17 (m, 1H), 2.13–2.02 (m, 1H), 1.72–1.56 (m, 4H), 1.48–1.34 (m, 8H), 1.00 (s, 9H). HRMS (m/z): for C55H66F4N9O7S+ [M + H]+ calcd, 1072.4737; found, 1072.4713.
N-(2′-Fluoro-5′-((8-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-8-oxooctyl)carbamoyl)-4-((3S,5R)-3,4,5-trimethylpiperazin-1-yl)-[1,1′-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide (16).
Compound 16 was synthesized following the standard procedures for preparing compound 3 from intermediates 2 (13 mg, 0.024 mmol) and 29f (14.3 mg, 0.024 mmol, 1.0 equiv). Compound 16 was obtained as a white solid (12.6 mg, yield 48%). 1H NMR (600 MHz, CD3OD): δ 8.97 (s, 1H), 8.14 (s, 1H), 8.01 (s, 1H), 7.96 (dd, J = 7.4, 2.4 Hz, 1H), 7.86–7.80 (m, 1H), 7.49–7.41 (m, 3H), 7.43–7.35 (m, 3H), 7.27 (dd, J = 10.3, 8.5 Hz, 1H), 6.93 (s, 1H), 4.62 (s, 1H), 4.60–4.46 (m, 3H), 4.34 (d, J = 15.5 Hz, 1H), 3.90 (d, J = 11.0 Hz, 1H), 3.79 (dd, J = 11.0, 3.9 Hz, 1H), 3.53–3.50 (m, 2H), 3.42–3.30 (m, 4H), 3.04–2.93 (m, 5H), 2.47 (s, 3H), 2.33–2.17 (m, 3H), 2.11–2.04 (m, 1H), 1.66–1.60 (m, 4H), 1.55–1.30 (m, 10H), 1.00 (s, 9H). HRMS (m/z): for C56H68F4N9O7S+ [M + H]+ calcd, 1086.4893; found, 1086.4910.
N-(2′-Fluoro-5′-((10-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-10-oxodecyl)carbamoyl)-4-((3S,5R)-3,4,5-trimethylpiperazin-1-yl)-[1,1′-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide (17).
Compound 17 was synthesized following the standard procedures for preparing compound 7 from intermediates 2 (13 mg, 0.024 mmol) and 29g (19.2 mg, 0.024 mmol, 1.0 equiv). Compound 17 was obtained as a white solid (16.7 mg, yield 63%). 1H NMR (600 MHz, CD3OD): δ 8.96 (s, 1H), 8.15 (s, 1H), 8.02–7.93 (m, 2H), 7.86–7.80 (m, 1H), 7.50–7.34 (m, 6H), 7.27 (dd, J = 10.3, 8.6 Hz, 1H), 6.92 (s, 1H), 4.62 (s, 1H), 4.59–4.46 (m, 3H), 4.35 (d, J = 15.4 Hz, 1H), 3.89 (d, J = 11.0 Hz, 1H), 3.79 (dd, J = 10.9, 3.9 Hz, 1H), 3.58–3.54 (m, 2H), 3.40–3.33 (m, 4H), 2.98 (d, J = 5.5 Hz, 5H), 2.47 (s, 3H), 2.33–2.16 (m, 3H), 2.10–2.02 (m, 1H), 1.70–1.52 (m, 4H), 1.44 (d, J = 6.5 Hz, 6H), 1.42–1.30 (m, 6H), 1.01 (s, 9H). HRMS (m/z): for C57H70F4N9O7S+ [M + H]+ calcd, 1100.5050; found, 1100.5076.
N-(2′-Fluoro-5′-((11-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-11-oxoundecyl)carbamoyl)-4-((3S,5R)-3,4,5-trimethylpiperazin-1-yl)-[1,1′-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide (18).
Compound 18 was synthesized following the standard procedures for preparing compound 3 from intermediates 2 (13 mg, 0.024 mmol) and 29h (14.9 mg, 0.024 mmol, 1.0 equiv). Compound 18 was obtained as a white solid (19.1 mg, yield 71%). 1H NMR (600 MHz, CD3OD): δ 8.99 (s, 1H), 8.16–8.13 (m, 1H), 8.01 (s, 1H), 7.97 (dd, J = 7.4, 2.4 Hz, 1H), 7.86–7.80 (m, 1H), 7.52–7.34 (m, 6H), 7.31–7.24 (m, 1H), 6.93 (s, 1H), 4.62 (s, 1H), 4.60–4.46 (m, 3H), 4.35 (d, J = 15.5 Hz, 1H), 3.92–3.87 (m, 1H), 3.79 (dd, J = 11.0, 3.9 Hz, 1H), 3.57–3.47 (m, 2H), 3.40–3.31 (m, 4H), 2.98 (d, J = 3.9 Hz, 5H), 2.47 (s, 3H), 2.33–2.17 (m, 3H), 2.11–2.03 (m, 1H), 1.66–1.55 (m, 4H), 1.44 (d, J = 6.5 Hz, 6H), 1.39–1.32 (m, 8H), 1.02 (s, 9H). HRMS (m/z): for C58H72F4N9O7S+ [M + H]+ calcd, 1114.5206; found, 1114.5243.
N-(2′-Fluoro-5′-((10-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-10-oxodecyl)carbamoyl)-4-((3S,5R)-3,4,5-trimethylpiperazin-1-yl)-[1,1′-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide (19).
Compound 19 was synthesized following the standard procedures for preparing compound 3 from intermediates 2 (13 mg, 0.024 mmol) and 29i (19.8 mg, 0.024 mmol, 1.0 equiv). Compound 19 was obtained as a white solid (12.6 mg, yield 33%). 1H NMR (600 MHz, CD3OD): δ 8.99 (s, 1H), 8.15 (s, 1H), 8.03–7.94 (m, 2H), 7.83 (ddd, J = 8.7, 4.6, 2.4 Hz, 1H), 7.51–7.45 (m, 2H), 7.48–7.35 (m, 4H), 7.31–7.25 (m, 1H), 6.93 (s, 1H), 4.62 (s, 1H), 4.60–4.47 (m, 3H), 4.35 (d, J = 15.4 Hz, 1H), 3.89 (d, J = 11.0 Hz, 1H), 3.79 (dd, J = 10.9, 3.9 Hz, 1H), 3.54–3.51 (m, 2H), 3.40–3.30 (m, 4H), 3.06–2.90 (m, 5H), 2.48 (s, 3H), 2.32–2.17 (m, 3H), 2.11–2.03 (m, 1H), 1.61 (dd, J = 16.1, 8.9 Hz, 4H), 1.44 (d, J = 6.5 Hz, 6H), 1.41–1.22 (m, 10H), 1.02 (s, 9H). HRMS (m/z): for C59H74F4N9O7S+ [M + H]+ calcd, 1128.5363; found, 1128.5341.
N-(2′-Fluoro-5′-((11-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-11-oxoundecyl)carbamoyl)-4-((3S,5R)-3,4,5-trimethylpiperazin-1-yl)-[1,1′-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide (20).
Compound 20 was synthesized following the standard procedures for preparing compound 3 from intermediates 2 (13 mg, 0.024 mmol) and 29j (15.6 mg, 0.024 mmol, 1.0 equiv). Compound 20 was obtained as a white solid (16 mg, yield 58%). 1H NMR (600 MHz, CD3OD): δ 8.94 (s, 1H), 8.17–8.13 (m, 1H), 8.03–7.94 (m, 2H), 7.84 (ddd, J = 8.5, 4.6, 2.4 Hz, 1H), 7.51–7.35 (m, 6H), 7.28 (dd, J = 10.3, 8.6 Hz, 1H), 6.93 (s, 1H), 4.62 (s, 1H), 4.59–4.47 (m, 3H), 4.35 (d, J = 15.5 Hz, 1H), 3.89 (d, J = 11.0 Hz, 1H), 3.79 (dd, J = 11.0, 3.9 Hz, 1H), 3.52 (s, 2H), 3.40–3.30 (m, 4H), 2.98 (s, 5H), 2.47 (s, 3H), 2.32–2.17 (m, 3H), 2.09–2.05 (m, 1H), 1.65–1.56 (m, 4H), 1.44 (d, J = 6.5 Hz, 6H), 1.43–1.20 (m, 12H), 1.02 (s, 9H). HRMS (m/z): for C60H76F4N9O7S+ [M + H]+ calcd, 1142.5519; found, 1142.5487.
N-(3′-((4-(2-(8-(2-((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)octanamido)ethyl)piperazin-1-yl)methyl)4-(4-methylpiperazin-1-yl)-[1,1′-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide (21).
Compound 21 was synthesized following the standard procedures for preparing compound 3 from intermediates 38 (63.4 mg, 0.11 mmol) and 34 (50.2 mg, 0.11 mmol, 1.0 equiv). Compound 21 was obtained as a white solid (79.5 mg, 69%). 1H NMR (600 MHz, CD3OD): δ 8.27 (d, J = 2.1 Hz, 1H), 8.06 (s, 1H), 7.83–7.62 (m, 3H), 7.57–7.44 (m, 4H), 7.39 (dd, J = 8.4, 6.9 Hz, 2H), 6.93 (s, 1H), 5.13 (dd, J = 12.9, 5.5 Hz, 1H), 4.73 (s, 2H), 4.34 (s, 2H), 3.63 (d, J = 11.7 Hz, 2H), 3.55–3.40 (m, 10H), 3.37 (s, 2H), 3.35–3.30 (m, 3H), 3.24–3.14 (m, 4H), 2.98 (s, 3H), 2.95–2.83 (m, 1H), 2.79–2.69 (m, 2H), 2.67 (s, 1H), 2.21 (t, J = 7.6 Hz, 2H), 2.18–2.10 (m, 1H), 1.63–1.51 (m, 4H), 1.41–1.27 (m, 6H). 13C NMR (151 MHz, CD3OD): δ 176.09, 173.20, 170.09, 168.34, 166.91, 166.34, 163.87, 162.50, 154.81, 142.33, 140.99, 140.08 (q, 2JC–F = 33.0 Hz, 1C, −*C–F), 138.37, 137.32, 136.86, 133.43, 132.64, 130.91, 129.59, 129.48, 128.99, 127.86, 124.43, 122.07, 121.02 (q, 1JC–F = 273.0 Hz, 1C, −*C–F), 120.99, 120.20, 119.22 (d, 3JC–F = 3.0 Hz, 1C, −*C–F), 117.74, 116.54, 113.02, 67.85, 63.73, 60.13, 56.32, 53.77, 53.59, 49.64, 49.18, 49.10, 48.91, 48.48, 42.31, 39.05, 38.71, 35.40, 34.39, 30.80, 28.71 (2C), 28.54, 26.24, 25.11, 22.22. HRMS (m/z): for C54H64F3N10O9+ [M + H]+ calcd, 1053.4804; found, 1053.4811.
N-(3′-((4-(2-(8-(2-((2-(1-Methyl-2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)octanamido)ethyl)piperazin-1-yl)methyl)-4-(4-methylpiperazin-1-yl)-[1,1′-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide (22).
Compound 22 was synthesized following the standard procedures for preparing compound 3 from intermediates 38 (29.6 mg, 0.05 mmol) and 35 (24.2 mg, 0.05 mmol, 1.0 equiv). Compound 22 was obtained as a white solid (40.2 mg, 75%). 1H NMR (600 MHz, CD3OD): δ 8.28 (d, J = 2.1 Hz, 1H), 8.05 (s, 1H), 7.84–7.77 (m, 2H), 7.77–7.70 (m, 1H), 7.59–7.51 (m, 3H), 7.48 (d, J = 7.6 Hz, 1H), 7.40 (dd, J = 16.0, 8.4 Hz, 2H), 6.94 (s, 1H), 5.16 (dd, J = 13.0, 5.4 Hz, 1H), 4.76 (s, 2H), 4.32 (s, 2H), 3.63 (d, J = 11.8 Hz, 2H), 3.51 (t, J = 5.9 Hz, 2H), 3.47–3.35 (m, 14H), 3.22–3.07 (m, 7H), 2.98 (s, 3H), 2.95–2.86 (m, 2H), 2.76–2.57 (m, 1H), 2.21 (t, J = 7.6 Hz, 2H), 2.16–2.08 (m, 1H), 1.69–1.51 (m, 4H), 1.41–1.25 (m, 6H). 13C NMR (151 MHz, CD3OD): δ 176.01, 172.17, 169.82, 168.38, 166.90, 166.37, 163.87, 162.50, 154.88, 142.31, 140.99, 140.08 (q, 2JC–F = 33.0 Hz, 1C, −*C–F), 138.36, 137.41, 136.86, 133.47, 132.64, 131.15, 129.54, 129.45, 128.94, 127.80, 124.42, 122.02 (q, 1JC–F = 273.0 Hz, 1C, −*C–F), 122.11, 120.97, 120.27, 119.24 (d, 3JC–F = 3.0 Hz, 1C, −*C–F), 117.80, 116.56, 113.03, 67.92, 60.17, 56.33, 53.75, 53.60, 49.82, 49.77, 49.28, 48.92, 48.46, 42.30, 38.69, 35.38, 34.48, 31.07, 28.73, 28.69, 28.50, 26.23 (2C), 26.01 (2C), 25.11, 21.45. HRMS (m/z): for C55H66F3N10O9+ [M + H]+ calcd, 1067.4961; found, 1067.4968.
N-(3′-((4-(2-(5-(2-((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)pentanamido)ethyl)piperazin-1-yl)methyl)-4-(4-methylpiperazin-1-yl)-[1,1′-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide (23).
Compound 23 was synthesized following the standard procedures for preparing compound 3 from intermediates 38 (62.7 mg, 0.105 mmol) and 36 (45.3 mg, 0.105 mmol, 1.0 equiv). Compound 23 was obtained as a white solid (72 mg, 68%). 1H NMR (600 MHz, CD3OD): δ 8.26 (d, J = 2.2 Hz, 1H), 8.05 (s, 1H), 7.87–7.69 (m, 3H), 7.58–7.52 (m, 2H), 7.49 (dd, J = 7.6, 4.7 Hz, 2H), 7.37 (dd, J = 8.4, 4.8 Hz, 2H), 6.93 (s, 1H), 5.12 (dd, J = 12.9, 5.5 Hz, 1H), 4.73 (s, 2H), 4.39 (s, 2H), 3.62 (d, J = 11.8 Hz, 2H), 3.59–3.40 (m, 10H), 3.38–3.31 (m, 5H), 3.29–3.26 (m, 1H), 3.23–3.15 (m, 4H), 2.98 (s, 3H), 2.92–2.80 (m, 1H), 2.80–2.63 (m, 2H), 2.26 (t, J = 7.3 Hz, 2H), 2.19–2.07 (m, 1H), 1.71–1.51 (m, 4H). 13C NMR (151 MHz, CD3OD): δ 175.63, 173.15, 170.15, 168.46, 166.88, 166.39, 163.88, 162.51, 154.92, 142.38, 140.08 (q, 2JC–F = 31.5 Hz, 1C, −*C–F), 140.13, 138.38, 137.24, 136.86, 133.43, 132.60, 130.39, 129.70, 129.52, 129.11, 128.00, 124.45, 122.02 (q, 1JC–F = 276.0 Hz, 1C, −*C–F), 122.14, 120.97, 120.32, 119.24 (d, 3JC–F = 3.0 Hz, 1C, −*C–F), 117.73, 116.56, 113.00, 67.95, 60.03, 56.31, 53.76, 53.58 (2C), 49.43, 49.16, 48.90, 48.47, 42.31, 38.38, 34.84, 34.30, 30.77 (2C), 28.36 (2C), 22.31, 22.20. HRMS (m/z): for C51H58F3N10O9+ [M + H]+ calcd, 1011.4335; found, 1011.4329.
N-(3′-((4-(2-(5-(2-((2-(1-Methyl-2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)pentanamido)ethyl)piperazin-1-yl)methyl)-4-(4-methylpiperazin-1-yl)-[1,1′-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide (24).
Compound 24 was synthesized following the standard procedures for preparing compound 3 from intermediates 38 (71.6 mg, 0.12 mmol) and 37 (51.4 mg, 0.12 mmol, 1.0 equiv). Compound 24 was obtained as a white solid (97.8 mg, 80%). 1H NMR (600 MHz, CD3OD): δ 8.26 (d, J = 2.3 Hz, 1H), 8.06 (s, 1H), 7.86–7.65 (m, 3H), 7.59–7.52 (m, 2H), 7.52–7.44 (m, 2H), 7.38 (dd, J = 8.4, 5.6 Hz, 2H), 6.92 (s, 1H), 5.14 (dd, J = 13.0, 5.4 Hz, 1H), 4.75 (s, 2H), 4.37 (s, 2H), 3.63 (d, J = 11.7 Hz, 2H), 3.56–3.38 (m, 8H), 3.37 (s, 8H), 3.26–3.15 (m, 4H), 3.11 (s, 3H), 2.98 (s, 3H), 2.93–2.81 (m, 2H), 2.76–2.58 (m, 1H), 2.25 (t, J = 7.3 Hz, 2H), 2.16–2.06 (m, 1H), 1.72–1.50 (m, 4H). 13C NMR (151 MHz, CD3OD): δ 175.50, 172.15, 169.86, 168.45, 166.90, 166.35, 163.86, 162.49, 154.94, 142.39, 140.99, 140.08 (q, 2JC–F = 31.5 Hz, 1C, −*C–F), 138.40, 137.26, 136.86, 133.45, 132.63, 130.64, 129.64, 129.49, 129.07, 127.93, 124.44, 122.02 (q, 1JC–F = 276.0 Hz, 1C, −*C–F), 122.15, 120.97, 120.27, 119.23 (d, 3JC–F = 3.0 Hz, 1C, −*C–F), 117.70, 116.54, 113.00, 67.91, 60.06, 56.27, 53.78, 53.58, 49.81, 49.52, 49.07, 48.89, 48.48 (2C), 42.31, 38.38, 34.84, 34.33, 31.07, 28.39, 26.03 (2C), 22.33, 21.43. HRMS (m/z): for C52H60F3N10O9+ [M + H]+ calcd, 1025.4481; found, 1025.4486.
N-(5′-((7-(2-((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)heptyl)carbamoyl)-2′-fluoro-4-((3S,5R)-3,4,5-trimethylpiperazin-1-yl)-[1,1′-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide (25).
Compound 25 was synthesized following the standard procedures for preparing compound 3 from intermediates 2 (27.3 mg, 0.05 mmol) and 40 (27.2 mg, 0.05 mmol, 1.0 equiv). Compound 25 was obtained as a white solid (34.6 mg, 71%). 1H NMR (400 MHz, CD3OD): δ 8.16 (s, 1H), 8.04 (s, 1H), 7.97 (dd, J = 7.6, 2.1 Hz, 1H), 7.87–7.80 (m, 1H), 7.80–7.72 (m, 1H), 7.54–7.44 (m, 2H), 7.42–7.34 (m, 2H), 7.33–7.20 (m, 1H), 6.93 (s, 1H), 5.21–5.08 (m, 1H), 4.73 (d, J = 1.8 Hz, 2H), 3.64–3.48 (m, 2H), 3.34–3.27 (m, 4H), 3.01 (d, J = 1.8 Hz, 3H), 2.92–2.66 (m, 3H), 2.18–2.10 (m, 1H), 2.06 (s, 6H), 1.68–1.52 (m, 4H), 1.51–1.35 (m, 10H). HRMS (m/z): for C49H53F4N8O9+ [M + H]+ calcd, 973.3866; found, 973.3869.
N-(2′-Fluoro-5′-((7-(2-((2-(1-methyl-2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)heptyl)carbamoyl)-4-((3S,5R)-3,4,5-trimethylpiperazin-1-yl)-[1,1′-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide (26).
Compound 26 was synthesized following the standard procedures for preparing compound 3 from intermediates 2 (54.7 mg, 0.1 mmol) and 41 (45.8 mg, 0.1 mmol, 1.0 equiv). Compound 26 was obtained as a white solid (64.8 mg, 66%). 1H NMR (400 MHz, CD3OD): δ 8.16 (s, 1H), 8.04 (s, 1H), 7.97 (dd, J = 7.7, 2.3 Hz, 1H), 7.86–7.80 (m, 1H), 7.79–7.72 (m, 1H), 7.47 (q, J = 4.1 Hz, 2H), 7.38 (dd, J = 8.4, 6.3 Hz, 2H), 7.27 (t, J = 9.5 Hz, 1H), 6.92 (s, 1H), 5.15 (dd, J = 13.1, 5.3 Hz, 1H), 4.73 (s, 2H), 3.57 (t, J = 8.2 Hz, 2H), 3.34–3.24 (m, 4H), 3.12 (d, J = 1.8 Hz, 3H), 3.00 (s, 3H), 2.98–2.75 (m, 2H), 2.73–2.60 (m, 1H), 2.18–2.09 (m, 1H), 2.08–1.98 (m, 6H), 1.67–1.52 (m, 4H), 1.52–1.42 (m, 4H), 1.42–1.29 (m, 6H). HRMS (m/z): for C50H55F4N8O9+ [M + H]+ calcd, 987.4023; found, 987.4034.
N-(2′-Fluoro-5′-((2-(((S)-1-((2R,4S)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-2-oxoethyl)carbamoyl)-4-((3S,5R)-3,4,5-trimethylpiperazin-1-yl)-[1,1′-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide (27).
Compound 27 was synthesized following the standard procedures for preparing compound 3 from intermediates 2 (18.3 mg, 0.033 mmol) and 4452 (16.3 mg, 0.02 mmol, 1.0 equiv). Compound 20 was obtained as a white solid (29.2 mg, 89%). 1H NMR (400 MHz, CD3OD): δ 9.13 (s, 1H), 8.12 (s, 1H), 8.01 (d, J = 8.1 Hz, 1H), 7.94 (dd, J = 7.4, 2.4 Hz, 1H), 7.83–7.79 (m, 1H), 7.41–7.37 (m, 1H), 7.33 (d, J = 8.3 Hz, 1H), 7.30 (s, 4H), 7.23 (dd, J = 10.3, 8.5 Hz, 1H), 6.92 (s, 1H), 4.54 (t, J = 7.5 Hz, 1H), 4.52–4.48 (m, 1H), 4.47–4.39 (m, 2H), 4.29 (d, J = 15.5 Hz, 1H), 4.08 (s, 2H), 3.97 (q, J = 5.5, 5.1 Hz, 2H), 3.72 (dd, J = 10.9, 3.3 Hz, 1H), 3.56–3.46 (m, 2H), 3.02–2.92 (m, 6H), 2.43 (s, 3H), 2.27–2.21 (m, 1H), 2.13–2.09 (m, 1H), 1.44 (dd, J = 6.2, 4.1 Hz, 6H), 1.09 (s, 9H). 13C NMR (101 MHz, CD3OD): δ 174.25, 172.27, 168.93, 165.19, 162.84 (d, 1JC–F = 216.14 Hz, 1C, −*C–F), 152.85, 148.84, 143.61, 141.55, 141.22, 140.02 (q, 2JC–F = 47.47 Hz, 1C), 133.64, 131.70 (d, 3JC–F = 3.0 Hz, 1C), 131.42 (d, 3JC–F = 3.0 Hz, 1C), 131.34, 130.57, 130.29 (2C), 129.95, 129.86, 129.65, 129.50, 129.36, 128.79 (2C), 128.07 (d, 2JC–F = 16.16 Hz, 1C), 125.75, 123.36 (q, 1JC–F = 288.9 Hz, 1C), 120.62, 117.57, 117.33, 114.39, 70.52, 62.43, 60.96, 60.10, 60.00, 57.60, 56.91, 43.81, 43.43, 39.09, 37.43, 35.69, 27.00 (3C), 15.80, 15.14 (2C). tR = 4.31 min, HRMS (m/z): for C51H57F4N9O7S+ [M + H]+ calcd, 1016.4111; found, 1016.4111.
8-(2-((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)octanoic Acid (34).
To a solution of intermediate 30 (99.6 mg, 0.3 mmol) in DMSO (1 mL) were added commercially available linker 32 (64.5 mg, 0.3 mmol, 1.0 equiv), EDCI (86.4 mg, 0.45 mmol, 1.5 equiv), HOAt (61.2 mg, 0.45 mmol, 1.5 equiv), and NMM (91 mg, 0.9 mmol, 3.0 equiv). After stirring overnight at rt, the resulting mixture was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford the Boc-protected intermediate. This intermediate was dissolved in TFA/DCM (1 mL/1 mL), and the mixture was stirred at rt for 30 min. The solvent was removed under reduced pressure, and the resulting mixture was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford compound 34 as a white solid (50.2 mg, yield 36%). 1H NMR (600 MHz, CD3OD): δ 7.80 (dd, J = 8.4, 7.3 Hz, 1H), 7.53 (d, J = 7.3 Hz, 1H), 7.43 (d, J = 8.5 Hz, 1H), 5.15 (dd, J = 12.8, 5.4 Hz, 1H), 4.76 (s, 2H), 3.35 (d, J = 21.9 Hz, 2H), 2.98–2.88 (m, 1H), 2.83–2.65 (m, 2H), 2.28 (t, J = 7.4 Hz, 2H), 2.21–2.09 (m, 1H), 1.66–1.50 (m, 4H), 1.44–1.26 (m, 6H). ESI (m/z): [M + H]+, 474.5.
8-(2-((2-(1-Methyl-2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)octanoic Acid (35).
Compound 35 was synthesized following the standard procedures for preparing compound 34 from intermediates 31 (50 mg, 0.029 mmol) and 32 (31 mg, 0.029 mmol, 1.0 equiv). Compound 35 was obtained as a white solid (24.2 mg, 29%). 1H NMR (600 MHz, CD3OD): δ 7.88–7.77 (m, 1H), 7.54 (d, J = 7.3 Hz, 1H), 7.44 (d, J = 8.4 Hz, 1H), 5.18 (dd, J = 13.0, 5.4 Hz, 1H), 4.77 (s, 2H), 3.35–3.28 (m, 2H), 3.17 (s, 3H), 2.99–2.86 (m, 2H), 2.74–2.71 (m, 1H), 2.28 (t, J = 7.4 Hz, 2H), 2.18–2.10 (m, 1H), 1.62–1.58 (m, 4H), 1.39–1.31 (m, 6H). ESI (m/z): [M + H]+, 488.8.
5-(2-((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)pentanoic Acid (36).
Compound 36 was synthesized following the standard procedures for preparing compound 34 from intermediates 30 (22.9 mg, 0.07 mmol) and 33 (14.5 mg, 0.07 mmol, 1.0 equiv). Compound 36 was obtained as a white solid (23.9 mg, 79%). 1H NMR (600 MHz, DMSO-d6): δ 12.00 (s, 1H), 11.12 (s, 1H), 7.98 (t, J = 5.8 Hz, 1H), 7.81 (dd, J = 8.5, 7.3 Hz, 1H), 7.50 (d, J = 7.3 Hz, 1H), 7.39 (d, J = 8.5 Hz, 1H), 5.12 (dd, J = 12.9, 5.4 Hz, 1H), 3.17 (s, 2H), 3.15 (q, J = 6.6 Hz, 2H), 2.90 (ddd, J = 17.1, 13.9, 5.5 Hz, 1H), 2.70–2.53 (m, 2H), 2.22 (t, J = 7.1 Hz, 2H), 2.08–2.01 (m, 1H), 1.58–1.34 (m, 4H). ESI (m/z): [M + H]+, 432.6.
5-(2-((2-(1-Methyl-2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)pentanoic Acid (37).
Compound 37 was synthesized following the standard procedures for preparing compound 34 from intermediates 31 (50 mg, 0.14 mmol) and 33 (30 mg, 0.14 mmol, 1.0 equiv). Compound 37 was obtained as a white solid (51.4 mg, 83%). 1H NMR (600 MHz, CD3OD): δ 7.70 (dd, J = 8.5, 7.3 Hz, 1H), 7.42 (d, J = 7.3 Hz, 1H), 7.33 (d, J = 8.4 Hz, 1H), 5.07 (dd, J = 13.0, 5.4 Hz, 1H), 4.67 (s, 2H), 3.26–3.21 (m, 2H), 3.06 (s, 3H), 2.86–2.73 (m, 2H), 2.65–2.58 (m, 1H), 2.23 (t, J = 7.1 Hz, 2H), 2.07–2.01 (m, 1H), 1.62–1.42 (m, 4H). ESI (m/z): [M + H]+, 446.8.
N-(7-Aminoheptyl)-2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamide (40).
Compound 40 was synthesized following the standard procedures for preparing compound 34 from intermediates 30 (66.4 mg, 0.2 mmol) and 39 (92.2 mg, 0.4 mmol, 2.0 equiv). Compound 40 was obtained as a white solid (55.4 mg, 64%). 1H NMR (400 MHz, CD3OD): δ 7.82–7.80 (m, 1H), 7.56 (dd, J = 7.5, 1.7 Hz, 1H), 7.46 (dd, J = 8.5, 1.7 Hz, 1H), 5.19–5.16 (m, 1H), 4.78 (d, J = 1.8 Hz, 2H), 3.37 (d, J = 1.7 Hz, 2H), 2.95 (dd, J = 11.1, 5.4 Hz, 4H), 2.75–2.71 (m, 1H), 2.24v–2.11 (m, 1H), 1.67–1.63 (m, 4H), 1.42–1.38 (m, 6H). ESI (m/z): [M + H]+, 445.6.
N-(7-Aminoheptyl)-2-((2-(1-methyl-2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamide (41).
Compound 41 was synthesized following the standard procedures for preparing compound 34 from intermediates 31 (80 mg, 0.23 mmol) and 39 (106 mg, 0.46 mmol, 2.0 equiv). Compound 41 was obtained as a white solid (45.8 mg, 43%). 1H NMR (400 MHz, CD3OD): δ 7.83–7.81 (m, 1H), 7.55 (dd, J = 7.5, 1.7 Hz, 1H), 7.45 (dd, J = 8.5, 1.7 Hz, 1H), 5.18 (ddd, J = 12.8, 5.4, 1.8 Hz, 1H), 4.78 (d, J = 1.8 Hz, 2H), 3.37 (d, J = 1.7 Hz, 2H), 3.17 (d, J = 1.7 Hz, 3H), 2.93 (dd, J = 11.1, 5.4 Hz, 4H), 2.76–2.72 (m, 1H), 2.22–2.10 (m, 1H), 1.66–1.61 (m, 4H), 1.42–1.40 (m, 6H). ESI (m/z): [M + H]+, 459.5.
(2R,4S)-1-((S)-2-(2-Aminoacetamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (44).
Compound 44 was synthesized following the standard procedures for preparing compound 34 from intermediates 42 (215 mg, 0.5 mmol) and 43 (175 mg, 1.0 mmol, 2.0 equiv). Compound 44 was obtained as a white solid (136 mg, 56%). 1H NMR (600 MHz, CD3OD): δ 8.97 (s, 1H), 7.54–7.38 (m, 4H), 4.67 (s, 1H), 4.63–4.52 (m, 3H), 4.38 (d, J = 15.4 Hz, 1H), 3.99–3.93 (m, 1H), 3.87–3.70 (m, 3H), 2.51 (s, 3H), 2.29–2.24 (m, 1H), 2.14–2.09 (m, 1H), 1.09 (s, 9H). HRMS m/z: for C24H34N5O4S+ [M + H]+ calcd, 488.2326; found, 488.2322.
Cell Lines and Tissue Culture.
Human leukemia MV4;11 cells (American Tissue Culture Collection [ATCC], CRL-9591) were cultured in the RPMI 1640 base medium supplemented with 10% FBS and 1% penicillin/streptomycin. Human PDAC cells used in the work include BxPC-3 (ATCC, CRL-1687), HPAF-II (ATCC, CRL-1997), PANC-1 (ATCC, CRL-1469), and Panc 10.05 (ATCC, CRL-2547) cells, which were cultured in RPMI 1640 supplemented with 10% FBS and 1% penicillin/streptomycin. The MIA PaCa-2 (ATCC, CRL-1420) PDAC cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin.
Authentication of cell line identities, including those of parental and derived lines, was ensured by the tissue culture facility affiliated to the UNC Lineberger Comprehensive Cancer Center with genetic signature profiling and fingerprinting analysis. Every 1–2 months, a routine examination of cell lines in culture for any possible mycoplasma contamination was performed using commercially available detection kits (Lonza).
Preparation of Proteins for ITC Studies.
For the expression of the full-length N-terminal His-tagged WDR5 (Uniprot accession number P61964), the pET28-LIC-WDR5 vector was obtained from Addgene (plasmid number 25489). The plasmid was transformed into Escherichia coli BL21 (DE3) cells and grown at 37 °C until the culture reached an OD600 of ~2.0. The temperature was then reduced to 18 °C, and the expression was induced by the addition of 0.5 mM IPTG, followed by incubation for 16 h. The cells were then resuspended in a lysis buffer (50 mM Tris pH 7.5, 500 mM NaCl, 5% glycerol, 0.01% IGEPAL, 25 mM imidazole, and 5 mM 2-mercaptoethanol) in the presence of Pierce protease inhibitor tablets, EDTA-free (Thermo Fisher), and 1 mM PMSF. The cells were lysed by sonication, clarified by centrifugation, the filtered supernatant was loaded onto a HisTrap HP affinity column (GE Healthcare), and the protein was eluted using an imidazole gradient ranging from 25 to 250 mM. The fractions containing WDR5 protein were concentrated and subjected to size exclusion chromatography by HiLoad 26/600 Superdex 200 (GE Healthcare), preequilibrated with 20 mM HEPES pH 7.5, 150 mM NaCl, and 2 mM TCEP.
For the expression of the His-tagged VCB complex, a plasmid containing pVHL54–213 (P40337) with an N-terminal His tag and a TEV protease cleavable site was cotransformed with a pCDF duet plasmid containing EloB1–104 (Q15370) and EloC1–112 (Q15369) into E. coli BL21 (DE3) cells. The cells were grown at 37 °C until OD600 reached 2.0, and then the temperature was reduced to 18 °C and IPTG was added to a final concentration of 0.4 mM. The cells were harvested at 16 h post induction and resuspended in a lysis buffer (50 mM Tris pH 7.5, 500 mM NaCl, 5% glycerol, 0.01% IGEPAL, 25 mM imidazole, and 5 mM 2-mercaptoethanol) in the presence of Pierce protease inhibitor tablets, EDTA-free (Thermo Fisher), and 1 mM PMSF. The clarified lysate after centrifugation was loaded onto a 5 mL HisTrap HP affinity column and purified with an imidazole gradient. The protein was further purified by gel filtration using HiLoad 26/600 Superdex 200. The final purified protein was flash frozen in liquid nitrogen and stored at −80 °C in 50 mM Tris pH 7.5, 150 mM NaCl, and 2 mM TCEP.
Antibodies and Immunoblotting.
Total cell lysate was used for Western blots as previously described.59 The following primary antibodies were used in the study: WDR5 (Santa Cruz, sc-393080), tubulin (Cell Signaling Technology, 2144), and vinculin (Cell Signaling, 13901S). Blots were imaged using fluorescence-labeled secondary antibodies on ChemiDoc Imaging Systems.
Cell Proliferation Assay by Counting.
1–5 × 105/mL of cells were seeded in triplicate in the 24-well plates. Compounds were added at a range of concentrations indicated in the study. Mediums with fresh compounds were changed every 2 days. Cells were passaged with dilution to keep cell density under 1 × 106/mL at all times. Cells were counted by an automated cell counter (Biorad, TC10) every 2 days. 50% of maximal growth inhibition (GI50) values were calculated using GraphPad Prism software using a nonlinear regression analysis, and the mean ± SEM was calculated from triplicated treatment data.
MTS (3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) Assay.
Pancreatic cancer cells were seeded at a density of 500 or 1000 cells per well in 96-well plates and incubated with the indicated compounds at different time points. Fresh medium with compound was changed every 2 days. At each time point, MTS reagent (Promega) was added to the cell culture medium and incubated for 0.5–2 h under standard culture conditions. Then, the plates were briefly shaken and subjected to measure absorbance at 490 nm using a CYTATION-5 imaging reader (BioTek).
Proteomics Profiling.
Four million MIA PaCa-2 cells were seeded in each of the 10 cm plates, and after the cells became adhered to the dish bottom, they were treated with DMSO or 1.5 μM of compound 11 for 2.5 h. Cells were then harvested and washed three times in 1 × PBS, followed by lysis in the lysis buffer (8 M urea, 50 mM Tris–HCl, and pH 8.0) with the use of the tip probe sonicator (Fisher) to help the lysis process. The resulting lysis samples were centrifuged at maximum speed for 15 min in a refrigerated microfuge to completely remove cell debris. The protein concentration was measured by a Bradford assay, and the samples were then frozen at −80 °C until further analysis. For MS analysis, 200 μg of each sample was reduced with 5 mM DTT at 56 °C for 30 min, alkylated with 15 mM iodoacetamide at room temperature in the dark for 45 min, then diluted to 1 M urea with 50 mM ammonium bicarbonate (pH 7.8). The samples were digested with LysC (Wako) and trypsin (Promega) at a 1:50 ratio overnight at 37 °C. After desalting using Waters SepPak C18 cartridges, a Pierce colorimetric peptide quantitation assay was performed to determine the digested peptide concentration. Then, 50 μg of each sample was labeled with an individual tandem mass tag (TMT) 10plex reagent at room temperature with shaking for 2 h. The reactions were quenched and combined into a single multiplexed sample. A 100 μg aliquot of the mixed sample was fractionated into 8 fractions using the Pierce high pH reversed phase fractionation spin columns. Peptide fractions were analyzed on the Easy nLC 1200-QExactive HF (carried out in the UNC Proteomics Core). Raw data were searched against a reviewed Uniprot human database (containing 20,245 sequences) using Andromeda within MaxQuant software (version 1.6.2.10). Proteins were quantified using TMT intensities, and the data were further analyzed in Perseus, Excel, and GraphPad.
Mouse PK Study.
Compound 11 (in its HCl salt form) was dissolved in a solution formulation of 5% NMP, 5% solutol HS-15, and 90% normal saline. Six male Swiss albino mice were administered intraperitoneally with a solution formulation of compound 11 at 75 mg/kg. Blood samples (approximately 60 μL) were collected under light isoflurane anesthesia from a set of three mice at 0.5, 1, 2, 4, 8, and 12 h. Plasma was harvested by centrifugation of blood and stored at −70 ± 10 °C until analysis. The plasma concentration–time of compound 11 was used for the PK analysis. Plasma samples were quantified by the fit-for-purpose LC–MS/MS method (LLOQ: 5.03 ng/mL). PK analysis was performed using GraphPad Prism software in the form of nonlinear regression analysis. The compound concentrations in plasma at each time point are the average values from three test mice. Error bars represent ± SEM.
Mutagenesis on Human WDR5 and Generation of Stable Cell Lines with WDR5 Mutants.
The WDR5 WT and mutant nucleotide sequences were modified from p3x Flag-CMV-14-WDR5 (Addgene plasmid #59974). Point mutations were introduced by PCR using the Q5 site-directed mutagenesis kit (New England E0554S). The mutagenic primers used followed the same procedures described in previous reports.48 PCRs for single lysine to arginine mutations were run for 25 cycles of 30 s at 98 °C and 1 min at 68 to 74 °C, 2 min 30 s at 72 °C, followed by 10 min at 72 °C. The resulting mutant plasmids were verified by Sanger sequencing. The WDR5 WT and mutant PCR fragments with 3X flag-tag were cloned into lentiviral TetO-Puro vectors. The lentiviral TetO-WDR5-Puro vectors were utilized to transduce HEK293T cells to generate inducible/stable cell lines that overexpress WDR5 WT, K32R, and K296R mutants. For lentivirus production, HEK293T cells were seeded in DMEM medium supplemented with 10% fetal bovine serum. Plasmid DNA vectors containing human WDR5 WT and its mutants were transfected using lipofectamine (3000) according to the manufacturer’s protocol. After 48 and 72 h of transfection, lentivirus packaging cell supernatant was collected and concentrated using the Lenti-X concentrator (Takara, 631232). Concentrated lentivirus was quantified using Lenti-GoStix Plus (Takara 631280) and applied to HEK293T cells for 24 h in the presence of 5 μg/mL Polybrene (Sigma). WDR5 overexpression was induced by the addition of 1 μg/mL doxycycline to the medium. After puromycin selection for 4 days, the stable cell lines were utilized for further experiments. Cell lysates were collected at 72 h after the stable cell lines were treated with the indicated concentrations of MS67. Supernatants were run on 4–12% gradient SDS-PAGE gels and transferred to a nitrocellulose membrane (BioRad) using the Trans-Blot Turbo system (BioRad). Membranes were probed with WDR5 (SantaCruz, G-9) and vinculin (Cell Signaling, 13901S) antibodies. Western blot images were detected using the LI-COR machine (LI-COR Biosciences). The images were cropped at specific protein bands of interest using Image Studio (LI-COR Biosciences) to improve the clarity of the data presentation.
Statistics and Reproducibility.
Experimental data are presented as the mean ± SD or SEM of three independent experiments unless otherwise noted. Statistical analysis was performed using an unpaired two-sided Student’s t-test for comparing two sets of data with an assumed normal distribution. The results for immunoblotting are representative of at least two biologically independent experiments unless otherwise noted. All statistical analyses and visualizations were performed using GraphPad (Prism v8.4.2) or Excel.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the grants R01CA268384 (to J.J. and G.G.W.) and R35GM131780 (to A.K.A.) from the U.S. National Institutes of Health (NIH) and an endowed professorship from the Icahn School of Medicine at Mount Sinai (to A.K.A.). This work utilized the NMR spectrometer systems at Mount Sinai acquired with funding from the NIH’s SIG grants 1S10OD025132 and 1S10OD028504.
ABBREVIATIONS USED
- ASH2L
absent small homeotic-2-like protein
- CRBN
cereblon
- EDCI
1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide
- H3K4me2 and H3K4me3
histone H3 lysine 4 di- and trimethylation
- HOAt
1-hydroxy-7-azabenzo-triazole
- IKZF1
ikaros
- IP
intraperitoneal
- ITC
isothermal titration calorimetry
- MLL1
mixed lineage leukemia 1
- MLL-r
mixed lineage leukemia rearranged
- MOA
mechanism of action
- MS
mass spectrometry
- PDAC
pancreatic ductal adenocarcinoma
- PEG
polyethylene glycol
- PK
pharmacokinetic
- POI
protein of interest
- PPIs
protein–protein interactions
- PROTAC
proteolysis targeting chimera
- RBBP5
retinoblastoma-binding protein 5
- SAR
structure–activity relationship
- TMT
tandem mass tags
- TPD
targeting protein degradation
- UPS
ubiquitin proteasome system
- VHL
von Hippel-Lindau
- WB
Western blotting
- WBM
WDR5 binding motif
- WDR5
WD repeat domain 5
- WIN
WDR5 interaction
- WT
wild-type
Footnotes
The authors declare the following competing financial interest(s): J.J., G.G.W., J.L., D.L., and X.Y. are inventors of a patent application filed by the Icahn School of Medicine at Mount Sinai. J.J. is a cofounder and equity shareholder in Cullgen, Inc., a scientific cofounder and scientific advisory board member of Onsero Therapeutics, Inc., and a consultant for Cullgen, Inc., EpiCypher, Inc., Accent Therapeutics, Inc., and Tavotek Biotherapeutics, Inc. The Jin laboratory received research funds from the Celgene Corporation, Levo Therapeutics, Inc., Cullgen, Inc., and Cullinan Oncology, Inc.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c01521.
Molecular formula strings for all compounds (CSV) Effects of compounds 21 and 25 on inducing WDR5 degradation in MV4;11 cells; inverse ITC titrations of VCB into 11 and WDR5-11 complex and of WDR5 in to 11; inverse ITC titrations of VCB and WDR5 into 27; effects of compound 11 on inducing WDR5 degradation in MIA PaCa-2 cells; Western blotting verification of the WDR5 degradation induced by compound 11 in MIA PaCa-2 cell lysates for proteomic studies; and 1H NMR, 13C NMR, and HPLC spectra of compounds 11 and 27 (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.3c01521
Contributor Information
Xufen Yu, Mount Sinai Center for Therapeutics Discovery, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States; Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Dongxu Li, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, North Carolina 27599, United States; Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Jithesh Kottur, Mount Sinai Center for Therapeutics Discovery, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States; Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Huen Suk Kim, Mount Sinai Center for Therapeutics Discovery, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States; Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Laura E. Herring, Department of Pharmacology, University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, North Carolina 27599, United States
Yao Yu, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, North Carolina 27599, United States; Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States; Duke Cancer Institute, Department of Pathology, and Department of Pharmacology and Cancer Biology, Duke University School of Medicine, Durham, North Carolina 27710, United States.
Ling Xie, Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Xiaoping Hu, Mount Sinai Center for Therapeutics Discovery, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States; Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Xian Chen, Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Ling Cai, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, North Carolina 27599, United States; Duke Cancer Institute and Department of Pathology, Duke University School of Medicine, Durham, North Carolina 27710, United States.
Jing Liu, Mount Sinai Center for Therapeutics Discovery, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States; Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Aneel K. Aggarwal, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States
Gang Greg Wang, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, North Carolina 27599, United States; Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States; Department of Pharmacology, University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, North Carolina 27599, United States; Duke Cancer Institute, Department of Pathology, and Department of Pharmacology and Cancer Biology, Duke University School of Medicine, Durham, North Carolina 27710, United States.
Jian Jin, Mount Sinai Center for Therapeutics Discovery, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States; Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
REFERENCES
- (1).Guarnaccia AD; Tansey WP Moonlighting with WDR5: A Cellular Multitasker. J. Clin. Med. 2018, 7 (2), 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Trievel RC; Shilatifard A WDR5, a complexed protein. Nat. Struct. Mol. Biol. 2009, 16 (7), 678–680. [DOI] [PubMed] [Google Scholar]
- (3).Schapira M; Tyers M; Torrent M; Arrowsmith CH WD40 repeat domain proteins: a novel target class? Nat. Rev. Drug Discovery 2017, 16 (11), 773–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Jain BP; Pandey S WD40 Repeat Proteins: Signalling Scaffold with Diverse Functions. Protein J. 2018, 37 (5), 391–406. [DOI] [PubMed] [Google Scholar]
- (5).Han Z; Guo L; Wang H; Shen Y; Deng XW; Chai J Structural basis for the specific recognition of methylated histone H3 lysine 4 by the WD-40 protein WDR5. Mol. Cell 2006, 22 (1), 137–144. [DOI] [PubMed] [Google Scholar]
- (6).Couture JF; Collazo E; Trievel RC Molecular recognition of histone H3 by the WD40 protein WDR5. Nat. Struct. Mol. Biol. 2006, 13 (8), 698–703. [DOI] [PubMed] [Google Scholar]
- (7).Dharmarajan V; Lee JH; Patel A; Skalnik DG; Cosgrove MS Structural basis for WDR5 interaction (Win) motif recognition in human SET1 family histone methyltransferases. J. Biol. Chem. 2012, 287 (33), 27275–27289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Carugo A; Genovese G; Seth S; Nezi L; Rose JL; Bossi D; Cicalese A; Shah PK; Viale A; Pettazzoni PF; et al. In Vivo Functional Platform Targeting Patient-Derived Xenografts Identifies WDR5-Myc Association as a Critical Determinant of Pancreatic Cancer. Cell Rep. 2016, 16 (1), 133–147. [DOI] [PubMed] [Google Scholar]
- (9).Alicea-Velazquez NL; Shinsky SA; Loh DM; Lee JH; Skalnik DG; Cosgrove MS Targeted Disruption of the Interaction between WD-40 Repeat Protein 5 (WDR5) and Mixed Lineage Leukemia (MLL)/SET1 Family Proteins Specifically Inhibits MLL1 and SETd1A Methyltransferase Complexes. J. Biol. Chem. 2016, 291 (43), 22357–22372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Thomas LR; Adams CM; Wang J; Weissmiller AM; Creighton J; Lorey SL; Liu Q; Fesik SW; Eischen CM; Tansey WP Interaction of the oncoprotein transcription factor MYC with its chromatin cofactor WDR5 is essential for tumor maintenance. Proc. Natl. Acad. Sci. U.S.A. 2019, 116 (50), 25260–25268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ge Z; Song EJ; Kawasawa YI; Li J; Dovat S; Song C WDR5 high expression and its effect on tumorigenesis in leukemia. Oncotarget 2016, 7 (25), 37740–37754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ge Z; Song E; Li J; Dovat S; Song C Clinical Significance of WDR5 High Expression and Its Effect on Tumorigenesis in Adult Leukemia. Blood 2015, 126 (23), 3657. [Google Scholar]
- (13).Wu M; Shu HB MLL1/WDR5 complex in leukemogenesis and epigenetic regulation. Chin. J. Cancer 2011, 30 (4), 240–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Dai X; Guo W; Zhan C; Liu X; Bai Z; Yang Y WDR5 Expression Is Prognostic of Breast Cancer Outcome. PLoS One 2015, 10 (9), No. e0124964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Cai WL; Chen JF; Chen H; Wingrove E; Kurley SJ; Chan LH; Zhang M; Arnal-Estape A; Zhao M; Balabaki A; et al. Human WDR5 promotes breast cancer growth and metastasis via KMT2-independent translation regulation. Elife 2022, 11, No. e78163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Malek R; Gajula RP; Williams RD; Nghiem B; Simons BW; Nugent K; Wang H; Taparra K; Lemtiri-Chlieh G; Yoon AR; et al. TWIST1-WDR5-Hottip Regulates Hoxa9 Chromatin to Facilitate Prostate Cancer Metastasis. Cancer Res. 2017, 77 (12), 3181–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sun W; Guo F; Liu M Up-regulated WDR5 promotes gastric cancer formation by induced cyclin D1 expression. J. Cell. Biochem. 2018, 119 (4), 3304–3316. [DOI] [PubMed] [Google Scholar]
- (18).Tan X; Chen S; Wu J; Lin J; Pan C; Ying X; Pan Z; Qiu L; Liu R; Geng R; et al. PI3K/AKT-mediated upregulation of WDR5 promotes colorectal cancer metastasis by directly targeting ZNF407. Cell Death Dis. 2017, 8 (3), No. e2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Chen X; Xie W; Gu P; Cai Q; Wang B; Xie Y; Dong W; He W; Zhong G; Lin T; et al. Upregulated WDR5 promotes proliferation, self-renewal and chemoresistance in bladder cancer via mediating H3K4 trimethylation. Sci. Rep. 2015, 5, 8293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Sun Y; Bell JL; Carter D; Gherardi S; Poulos RC; Milazzo G; Wong JW; Al-Awar R; Tee AE; Liu PY; et al. WDR5 Supports an N-Myc Transcriptional Complex That Drives a Protumorigenic Gene Expression Signature in Neuroblastoma. Cancer Res. 2015, 75 (23), 5143–5154. [DOI] [PubMed] [Google Scholar]
- (21).Han QL; Zhang XL; Ren PX; Mei LH; Lin WH; Wang L; Cao Y; Li K; Bai F Discovery, evaluation and mechanism study of WDR5-targeted small molecular inhibitors for neuroblastoma. Acta Pharmacol. Sin. 2023, 44 (4), 877–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chen X; Xu J; Wang X; Long G; You Q; Guo X Targeting WD Repeat-Containing Protein 5 (WDR5): A Medicinal Chemistry Perspective. J. Med. Chem. 2021, 64 (15), 10537–10556. [DOI] [PubMed] [Google Scholar]
- (23).Thomas LR; Adams CM; Fesik SW; Eischen CM; Tansey WP Targeting MYC through WDR5. Mol. Cell Oncol. 2020, 7 (2), 1709388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Karatas H; Townsend EC; Cao F; Chen Y; Bernard D; Liu L; Lei M; Dou Y; Wang S High-affinity, small-molecule peptidomimetic inhibitors of MLL1/WDR5 protein-protein interaction. J. Am. Chem. Soc. 2013, 135 (2), 669–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Cao F; Townsend EC; Karatas H; Xu J; Li L; Lee S; Liu L; Chen Y; Ouillette P; Zhu J; et al. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol. Cell 2014, 53 (2), 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Karatas H; Li Y; Liu L; Ji J; Lee S; Chen Y; Yang J; Huang L; Bernard D; Xu J; et al. Discovery of a Highly Potent, Cell-Permeable Macrocyclic Peptidomimetic (MM-589) Targeting the WD Repeat Domain 5 Protein (WDR5)-Mixed Lineage Leukemia (MLL) Protein-Protein Interaction. J. Med. Chem. 2017, 60 (12), 4818–4839. [DOI] [PubMed] [Google Scholar]
- (27).Senisterra G; Wu H; Allali-Hassani A; Wasney GA; Barsyte-Lovejoy D; Dombrovski L; Dong A; Nguyen KT; Smil D; Bolshan Y; et al. Small-molecule inhibition of MLL activity by disruption of its interaction with WDR5. Biochem. J. 2013, 449 (1), 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Grebien F; Vedadi M; Getlik M; Giambruno R; Grover A; Avellino R; Skucha A; Vittori S; Kuznetsova E; Smil D; et al. Pharmacological targeting of the Wdr5-MLL interaction in C/EBPα N-terminal leukemia. Nat. Chem. Biol. 2015, 11 (8), 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zhu J; Sammons MA; Donahue G; Dou Z; Vedadi M; Getlik M; Barsyte-Lovejoy D; Al-awar R; Katona BW; Shilatifard A; et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 2015, 525 (7568), 206–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Getlik M; Smil D; Zepeda-Velazquez C; Bolshan Y; Poda G; Wu H; Dong A; Kuznetsova E; Marcellus R; Senisterra G; et al. Structure-Based Optimization of a Small Molecule Antagonist of the Interaction Between WD Repeat-Containing Protein 5 (WDR5) and Mixed-Lineage Leukemia 1 (MLL1). J. Med. Chem. 2016, 59 (6), 2478–2496. [DOI] [PubMed] [Google Scholar]
- (31).Li DD; Chen WL; Wang ZH; Xie YY; Xu XL; Jiang ZY; Zhang XJ; You QD; Guo XK High-affinity small molecular blockers of mixed lineage leukemia 1 (MLL1)-WDR5 interaction inhibit MLL1 complex H3K4 methyltransferase activity. Eur. J. Med. Chem. 2016, 124, 480–489. [DOI] [PubMed] [Google Scholar]
- (32).Wang F; Jeon KO; Salovich JM; Macdonald JD; Alvarado J; Gogliotti RD; Phan J; Olejniczak ET; Sun Q; Wang S; et al. Discovery of Potent 2-Aryl-6,7-dihydro-5 H-pyrrolo[1,2- a]imidazoles as WDR5-WIN-Site Inhibitors Using Fragment-Based Methods and Structure-Based Design. J. Med. Chem. 2018, 61 (13), 5623–5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Aho ER; Wang J; Gogliotti RD; Howard GC; Phan J; Acharya P; Macdonald JD; Cheng K; Lorey SL; Lu B; et al. Displacement of WDR5 from Chromatin by a WIN Site Inhibitor with Picomolar Affinity. Cell Rep. 2019, 26 (11), 2916–2928.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Tian J; Teuscher KB; Aho ER; Alvarado JR; Mills JJ; Meyers KM; Gogliotti RD; Han C; Macdonald JD; Sai J; et al. Discovery and Structure-Based Optimization of Potent and Selective WD Repeat Domain 5 (WDR5) Inhibitors Containing a Dihydroisoquinolinone Bicyclic Core. J. Med. Chem. 2020, 63 (2), 656–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Teuscher KB; Meyers KM; Wei Q; Mills JJ; Tian J; Alvarado J; Sai J; Van Meveren M; South TM; Rietz TA; et al. Discovery of Potent Orally Bioavailable WD Repeat Domain 5 (WDR5) Inhibitors Using a Pharmacophore-Based Optimization. J. Med. Chem. 2022, 65 (8), 6287–6312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Chen W; Chen X; Li D; Zhou J; Jiang Z; You Q; Guo X Discovery of DDO-2213 as a Potent and Orally Bioavailable Inhibitor of the WDR5-Mixed Lineage Leukemia 1 Protein-Protein Interaction for the Treatment of MLL Fusion Leukemia. J. Med. Chem. 2021, 64 (12), 8221–8245. [DOI] [PubMed] [Google Scholar]
- (37).Teuscher KB; Chowdhury S; Meyers KM; Tian J; Sai J; Van Meveren M; South TM; Sensintaffar JL; Rietz TA; Goswami S; et al. Structure-based discovery of potent WD repeat domain 5 inhibitors that demonstrate efficacy and safety in preclinical animal models. Proc. Natl. Acad. Sci. U.S.A. 2023, 120 (1), No. e2211297120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Macdonald JD; Chacon Simon S; Han C; Wang F; Shaw JG; Howes JE; Sai J; Yuh JP; Camper D; Alicie BM; et al. Discovery and Optimization of Salicylic Acid-Derived Sulfonamide Inhibitors of the WD Repeat-Containing Protein 5-MYC Protein-Protein Interaction. J. Med. Chem. 2019, 62 (24), 11232–11259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Chacon Simon S; Wang F; Thomas LR; Phan J; Zhao B; Olejniczak ET; Macdonald JD; Shaw JG; Schlund C; Payne W; et al. Discovery of WD Repeat-Containing Protein 5 (WDR5)-MYC Inhibitors Using Fragment-Based Methods and Structure-Based Design. J. Med. Chem. 2020, 63 (8), 4315–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ding J; Li G; Liu H; Liu L; Lin Y; Gao J; Zhou G; Shen L; Zhao M; Yu Y; et al. Discovery of Potent Small-Molecule Inhibitors of WDR5-MYC Interaction. ACS Chem. Biol. 2023, 18 (1), 34–40. [DOI] [PubMed] [Google Scholar]
- (41).Dale B; Cheng M; Park KS; Kaniskan HU; Xiong Y; Jin J Advancing targeted protein degradation for cancer therapy. Nat. Rev. Cancer 2021, 21 (10), 638–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Li K; Crews CM PROTACs: past, present and future. Chem. Soc. Rev. 2022, 51 (12), 5214–5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Bekes M; Langley DR; Crews CM PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discovery 2022, 21 (3), 181–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).He M; Cao C; Ni Z; Liu Y; Song P; Hao S; He Y; Sun X; Rao Y PROTACs: great opportunities for academia and industry (an update from 2020 to 2021). Signal Transduction Targeted Ther. 2022, 7 (1), 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Kabir M; Yu X; Kaniskan HU; Jin J Chemically induced degradation of epigenetic targets. Chem. Soc. Rev. 2023, 52 (13), 4313–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Gao H; Sun X; Rao Y PROTAC Technology: Opportunities and Challenges. ACS Med. Chem. Lett. 2020, 11 (3), 237–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Sun D; Zhang J; Dong G; He S; Sheng C Blocking Non-enzymatic Functions by PROTAC-Mediated Targeted Protein Degradation. J. Med. Chem. 2022, 65 (21), 14276–14288. [DOI] [PubMed] [Google Scholar]
- (48).Yu X; Li D; Kottur J; Shen Y; Kim HS; Park KS; Tsai YH; Gong W; Wang J; Suzuki K; et al. A selective WDR5 degrader inhibits acute myeloid leukemia in patient-derived mouse models. Sci. Transl. Med. 2021, 13 (613), No. eabj1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Dolle A; Adhikari B; Kramer A; Weckesser J; Berner N; Berger LM; Diebold M; Szewczyk MM; Barsyte-Lovejoy D; Arrowsmith CH; et al. Design, Synthesis, and Evaluation of WD-Repeat-Containing Protein 5 (WDR5) Degraders. J. Med. Chem. 2021, 64 (15), 10682–10710. [DOI] [PubMed] [Google Scholar]
- (50).Li D; Yu X; Kottur J; Gong W; Zhang Z; Storey AJ; Tsai YH; Uryu H; Shen Y; Byrum SD; et al. Discovery of a dual WDR5 and Ikaros PROTAC degrader as an anti-cancer therapeutic. Oncogene 2022, 41 (24), 3328–3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Al-Awar RZ-V; Zepeda-Velazquez CA; Poda G; Isaac M; Uehling D; Wilson B; Joseph B; Liu Y; Subramanlan P; Mamai A; Prakesch M; Stille JK Inhibitors of WDR5 protein-protein binding cross-reference to related applications. WO 2017147700 A1, 2017. [Google Scholar]
- (52).Yu X; Xu J; Xie L; Wang L; Shen Y; Cahuzac KM; Chen X; Liu J; Parsons RE; Jin J Design, Synthesis, and Evaluation of Potent, Selective, and Bioavailable AKT Kinase Degraders. J. Med. Chem. 2021, 64 (24), 18054–18081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Yu X; Xu J; Cahuzac KM; Xie L; Shen Y; Chen X; Liu J; Parsons RE; Jin J Novel Allosteric Inhibitor-Derived AKT Proteolysis Targeting Chimeras (PROTACs) Enable Potent and Selective AKT Degradation in KRAS/BRAF Mutant Cells. J. Med. Chem. 2022, 65 (20), 14237–14260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Jiang B; Wang ES; Donovan KA; Liang Y; Fischer ES; Zhang T; Gray NS Development of Dual and Selective Degraders of Cyclin-Dependent Kinases 4 and 6. Angew. Chem., Int. Ed. Engl. 2019, 58 (19), 6321–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Yu X; Xu J; Shen Y; Cahuzac KM; Park KS; Dale B; Liu J; Parsons RE; Jin J Discovery of Potent, Selective, and In Vivo Efficacious AKT Kinase Protein Degraders via Structure-Activity Relationship Studies. J. Med. Chem. 2022, 65 (4), 3644–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Meng F; Xu C; Park KS; Kaniskan HU; Wang GG; Jin J Discovery of a First-in-Class Degrader for Nuclear Receptor Binding SET Domain Protein 2 (NSD2) and Ikaros/Aiolos. J. Med. Chem. 2022, 65 (15), 10611–10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Cheng M; Yu X; Lu K; Xie L; Wang L; Meng F; Han X; Chen X; Liu J; Xiong Y; et al. Discovery of Potent and Selective Epidermal Growth Factor Receptor (EGFR) Bifunctional Small-Molecule Degraders. J. Med. Chem. 2020, 63 (3), 1216–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Yu X; Cheng M; Lu K; Shen Y; Zhong Y; Liu J; Xiong Y; Jin J Exploring Degradation of Mutant and Wild-Type Epidermal Growth Factor Receptors Induced by Proteolysis-Targeting Chimeras. J. Med. Chem. 2022, 65 (12), 8416–8443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Cai L; Rothbart SB; Lu R; Xu B; Chen WY; Tripathy A; Rockowitz S; Zheng D; Patel DJ; Allis CD; et al. An H3K36 methylation-engaging Tudor motif of polycomb-like proteins mediates PRC2 complex targeting. Mol. Cell 2013, 49 (3), 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.