

Abstract
Biallelic pathogenic variants in PGAP3 cause a rare glycosylphosphatidyl-inositol biogenesis disorder, PGAP3-CDG. This multisystem condition presents with a predominantly neurological phenotype, including developmental delay, intellectual disability, seizures, and hyperphosphatemia. Here, we summarized the phenotype of sixty-five individuals including six unreported individuals from our CDG natural history study with a confirmed PGAP3-CDG diagnosis. Common additional features found in this disorder included brain malformations, behavioral abnormalities, cleft palate, and characteristic facial features. This report aims to review the genetic and metabolic findings and characterize the disease’s phenotype while highlighting the necessary clinical approach to improve the management of this rare CDG.
Keywords: PGAP3, glycophosphatidylinositol anchor biogenesis defects, GPI-anchor, alkaline phosphatase, psychomotor developmental delay
INTRODUCTION
PGAP3 gene (Post-GPI attachment to proteins phospholipase 3), is located on chromosome 17q12 and encodes the glycosylphosphatidylinositol (GPI)-specific phospholipase in the Golgi apparatus. Post-GPI attachment to proteins phospholipase 3 deficiency was previously named hyperphosphatasia with mental retardation syndrome 4 (HPMRS4), (recently changed to hyperphosphatasia with impaired intellectual development syndrome 4 (OMIM 615716)) results from biallelic pathogenic variants in PGAP3. This rare subtype of glycosylphosphatidyl-inositol biogenesis disorders (GPIBD) is classified under congenital disorders of glycosylation (PGAP3-CDG) [1].
Most patients with PGAP3-CDG manifest signs and symptoms at birth or in the first years of life. The typical symptoms are often neurological and include hypotonia, ataxia, severe developmental delay, intellectual disability (ID), distinctive facial features, and seizures with onset between the first days of life to a few years of age. In some patients, seizures are treatable with anti-epileptic drugs (AED), but in many, epilepsy is difficult to treat. Autistic behavior and sleep disturbances are very common in PGAP3-CDG. Brain imaging may show structural abnormalities of the brain. And typically, patients show an elevation of alkaline phosphatase (ALP) in the blood [2].
To date, fifty-nine individuals with a confirmed PGAP3-CDG diagnosis have been reported. In this study, we describe an additional six individuals with PGAP3-CDG, in addition to a detailed review of previously reported individuals aiming to characterize the phenotypic spectrum of PGAP3-CDG and direct the clinical management of this rare CDG.
MATERIALS AND METHOD
We evaluated six patients with confirmed pathogenic PGAP3 variants from the 208 enrolled CDG patients followed by the Frontiers of Congenital Disorders of Glycosylation Consortium (FCDGC) natural history study (institutional review board [IRB]: 19–005187; NCT04199000). Prospective data, as well as retrospective data were available for frequent patients’ follow-ups are part of standard care at each site.
We also reviewed all publications on patients diagnosed with PGAP3-CDG with confirmed pathogenic variants, focusing on their clinical, genetic, and metabolic characteristics by using PubMed database search from the date of the first clinical description until May 2023 using the following terms: hyperphosphatasia with mental retardation syndrome 4 (HPMRS4), hyperphosphatasia with impaired intellectual development syndrome 4, Mabry syndrome, PGAP3, and PGAP3-CDG.
RESULTS
This review included sixty-five molecularly confirmed PGAP3-CDG patients (26 males and 39 females). Data from 59 patients were collected from previously published articles (n=13) including case cohorts (n=6) [3,4,5,6,7,8], and case reports (n=7) [9,10,11,12,13,14,15]. Data for the so far unreported, additional six patients were collected from the shared natural history database.
The median age was 7 years whereas the youngest and oldest patients were 2 and 25 years old, respectively. All patients shared the common phenotype of severe psychomotor developmental delay, with a characteristic high serum alkaline phosphatase (ALP) level, in addition to multi-system involvement and severity as detailed below.
Systems involvement in PGAP3-CDG
Development
All reported patients including our cohort had a global developmental delay with two or more affected developmental domain milestones. The three major delayed developmental domains are: cognition, reported in 95% (n=62); motor function, in 96% (n=63); and speech, in 93% of patients (n=61), 53 patients were reported as nonverbal while only eight patients used limited verbal communication. No data was available for 4 patients.
Neurological manifestation
Neurological involvement was a consistent finding in all patients. Hypotonia was present in 90% of patients (n=59). Ataxic and unsteady gait was described in 27% of patients (n=18) including our cohort. Microcephaly was reported in 38% (n=25) while macrocephaly was noted in five patients. Nearly half of the patients had seizures (n=32). Different seizure subtypes included generalized tonic-clonic, myoclonic, absence, partial, and atypical febrile seizures. Seizures were medically controlled in eight patients and only two patients failed to respond to multiple AEDs including one newly reported 16-year-old patient. Different AEDs have been used to control the seizure either as monotherapy or as multiple therapies including sodium valproate, levetiracetam, diazepam, pregabalin, clonazepam, lamotrigine, and phenobarbital. Ketogenic diet was trialed on one patient who had refractory seizures. (Supplementary material 1)
Brain magnetic resonance imaging (MRI) abnormalities
Different structural brain malformations were detected by brain MRIs performed on 31 patients. The most common finding was corpus callosum hypoplasia, detected in 54% of patients (n=17), followed by cerebellar vermis hypoplasia or atrophy in five patients, brain atrophy in four, and hypoplastic cerebellum in three patients. Other rare brain MRI findings included delayed myelination, cerebroventricular hemorrhage, Dandy-Walker malformation, cortical dysplasia, small internal capsule, Rathke cyst, and ventriculomegaly with frontoparietal atrophy.
Craniofacial manifestation
Almost all patients showed distinctive facial features such as arched eyebrows, hypertelorism, broad nasal bridge and tip, short and bulbous nose, long palpebral fissures, long philtrum, thin and wide upper lip, large fleshy earlobes, low set ears, and abnormal dentation. Cleft palate was stated in 61% of patients (n=40). Figure 1 illustrates the commonly reported facial features of PGAP3-CDG.
Fig.1.
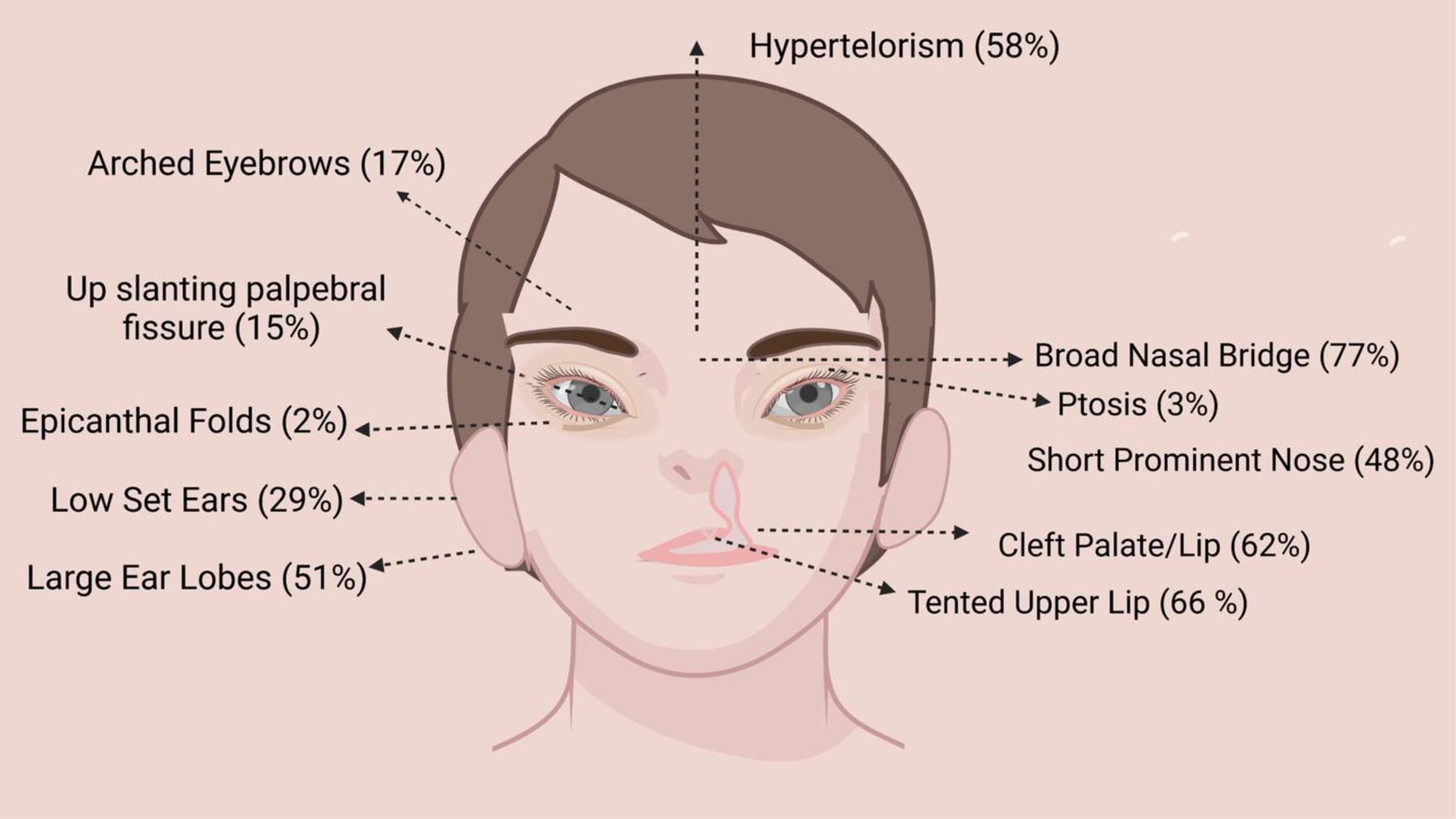
Commonly reported facial features in PGAP3-CDG
Behavioral abnormalities
About 44% of patients manifested with one or a combination of two behavioral symptoms. Autism was the most commonly stated abnormality, reported in 29% of patients (n=19), followed by hyperactivity, in 18% (n=12), and sleep disturbance in 12% (n=8). Less reported behavioral issues included bruxism, hypersomnolence, aggression, obsessive-compulsive behavior, and involuntary midline hand movement.
Other less common involved systems include cardiovascular, gastrointestinal, ophthalmological, skeleton, and hearing
Cardiovascular and gastrointestinal involvement are not commonly reported in patients with PGAP3-CDG with a rate of 12% for each (n=8).
Structural cardiac defects were found only in eight patients including atrial septal defect, ventricular septal defect, or concentric ventricular hypertrophy. The main reported gastrointestinal manifestations were failure to thrive, reported in 35% (n=23), followed by dysphagia, gastroesophageal reflux, esophagitis, constipation, or intestinal malrotation.
The ophthalmological manifestation was documented in only 20% of patients, (n=13) where the most common abnormality was megalocornea, stated in seven patients. Other less reported eye phenotypes include nystagmus, microphthalmia, poor vision, and optic disc abnormality.
Short stature was stated in 40% of patients (n=26). Skeletal deformities are rarely noted including plagiocephaly, scoliosis, pectus excavatum, clinodactyly camptodactyly of 5th finger, hip dysplasia, pes equinovarus, and coxa valga. Sensorineural hearing loss was documented in 20% of patients (n=13). There was no reported brachytelephalangy in PGAP3-CDG patients.
Pyridoxine (vitamin B6) therapy and ketogenic diet
Pyridoxine therapy was initiated in only four patients including three from our cohort. The ketogenic diet was trialed on two of our patients. Supplementary material 1.
PGAP3 variants
Twenty-four different pathogenic variants were reported in PGAP3. Homozygous variants were detected in 78% of reported patients who are born to consanguineous families. The most commonly observed variants more than once were His284Tyr, followed by Met135fs. Figure 2 lists all previously reported PGAP3 variants. There was no genotype/phenotype found in this review.
Fig.2.
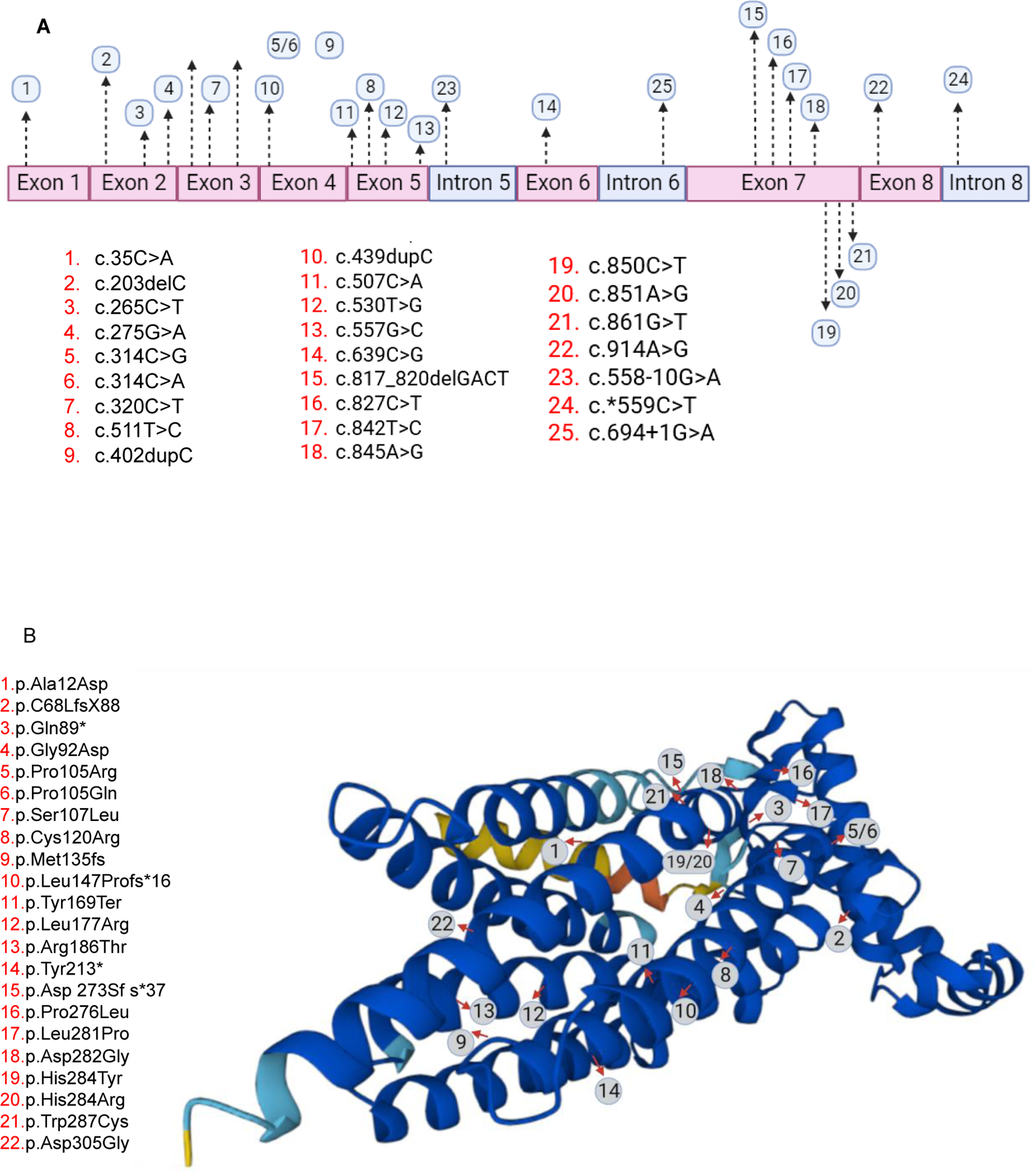
Reported pathogenic PGAP3 variants
Disease course and outcome
A total of 7 patients were above 14 years of age (three were >18 years of age). All had static conditions. Their main phenotype included ID, speech delay, behavioral abnormalities, and seizures [3,4,6]. Based on our data we have knowledge of one patient who succumbed to aspiration pneumonia at 18 months of age.
DISCUSSION:
The PGAP3 gene encodes a glycophosphatidylinositol (GPI)-specific phospholipase that is expressed in the Golgi apparatus. The enzyme is involved in fatty acid GPI remodeling which is critical for a proper association between GPI-anchored proteins and lipid rafts [16]. Mutations in PGAP3 results in a rare autosomal recessive CDG subtype; PGAP3-CDG or previously called Mabry syndrome/ HPMRS4. In this review, we describe the genetic, biochemical and phenotypic spectrum of 65 patients with PGAP3-CDG.
PGAP3 has a critical role in brain morphogenesis and oligodendrocytes formation and function which explains the predominant and significant neurological involvement in PGAP3 deficiency [13]. All patients had major neurological involvement with high ALP (65/65).
PGAP3 is involved in the later step of GPI-anchor synthesis and defects in PGAP3 leads to GPI anchored proteins instability including Alkaline phosphatase (ALP). As result, hyperphosphatemia is observed in PGAP3-CDG and some other GPIBDs [1]. Thus, it is considered a useful marker for biochemical diagnosis of some GPIBDs.
Other GPI-anchored proteins (GPI-APs) like CD59, CD55 and urokinase plasminogen activator receptor can be measured in individuals affected with PGAP3 deficiency. Reduced cell surface levels of GPI-APs in patients’ blood granulocytes was noted in PGAP3-CDG [3]
The most common features observed across PGAP3-CDG patients are developmental delay, (ID), hypotonia, seizures, and distinctive dysmorphic features. The frequency of phenotypic features in all patients with PGAP3-CDG is shown in Figure 3. The main presenting phenotype is often noticed either at birth with congenital craniofacial abnormalities or during the first or second year of life with failure to acquire major developmental milestones mainly the motor and language skills, with or without seizures. Behavioral abnormalities, autism spectrum disorders, and sleep disturbance are recognizable phenotypes of GPIBDs including PGAP3-CDG [17]. Significant behavioral involvement was found in our review, autism and sleeping difficulties were the main reported behavioral phenotype.
Fig.3.
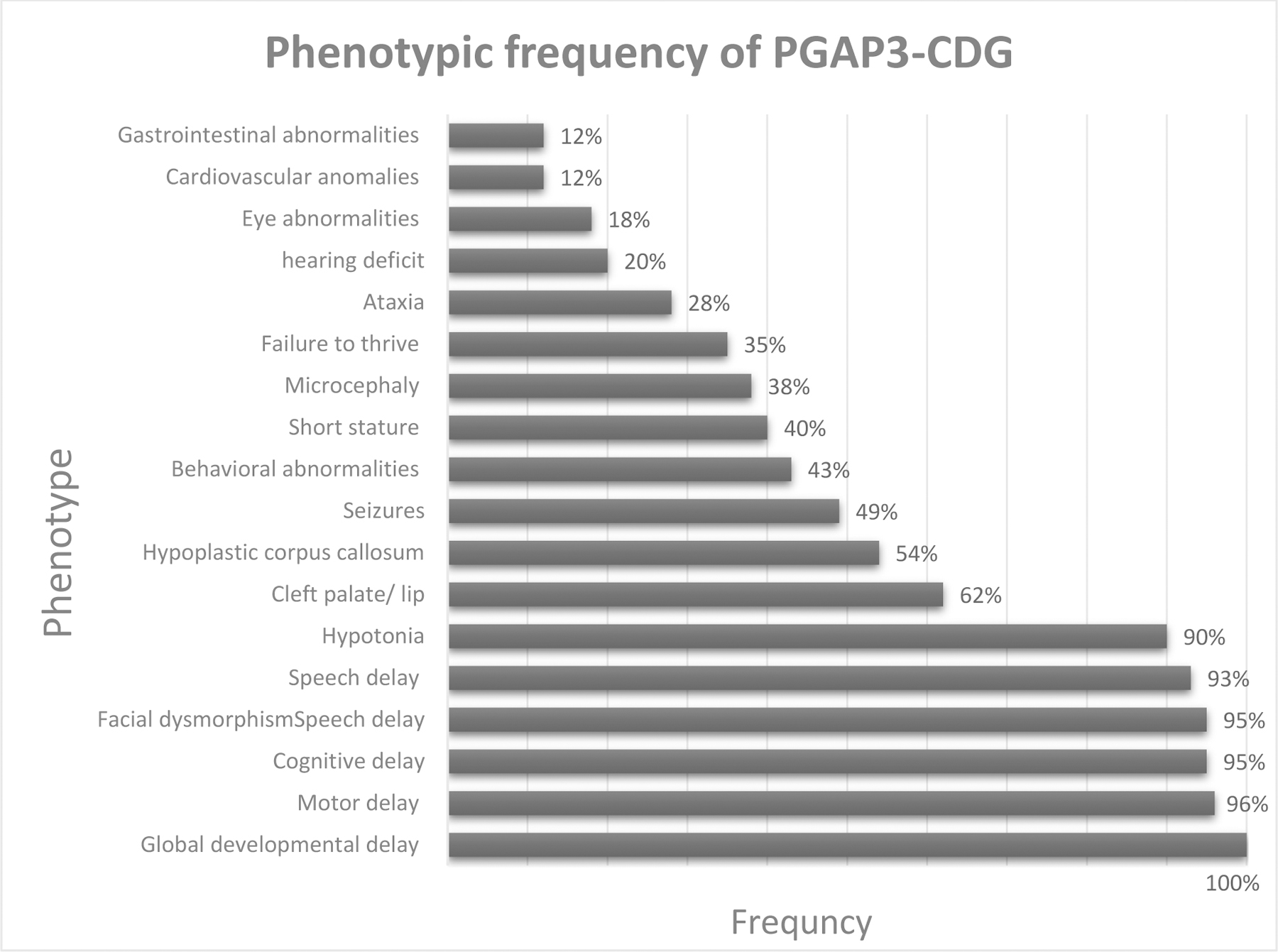
Phenotypic frequency of PGAP3-CDG
The most common neuroimaging abnormalities reported are hypoplastic corpus callosum, and cerebellar and, or cerebral atrophy. Neuroimaging data were missing in 50% of previous reports. The pathomechanism of the neurostructural abnormalities has been shown in a knockdown Pgap3 zebrafish which also may contribute to the craniofacial dysmorphism as a sequence of early fetal brain dysmorphogenesis [13].
In comparison to other CDGs, multisystem involvement was not well illustrated in GPIBD. Data on the multisystem involvement in PGAP3-CDG may be underestimated due to the scarce data available from previous reports. PGAP3-CDG shares the other GPIBDs’ main phenotype including global developmental delay, hypotonia, seizures, and facial dysmorphism. Brachytelephalangy which is a notable skeletal anomaly in GPIBDs was not reported in PGAP3-CDG patients.
From reviewing previous reports including our cohort, PGAP3-CDG was found to present with at least one or more extra-neurological involvement including cardiac anomalies, gastrointestinal abnormalities, eye dysmorphology, hearing loss, or skeletal defects and short stature. Deafness can contribute to the failure of acquiring language skills in PGAP3-CDG which is significantly affected in PGAP3-CDG.
Although the cause of the seizure in GPIBD is not well understood, patients with hyperphosphatemia secondary to defects in GPI-anchored tissue nonspecific alkaline phosphatase manifest with seizures that might be refractory to conventional AED therapy [18]. ALP is a key enzyme for dephosphorylation of pyridoxal 5- phosphate to pyridoxal (the active form of pyridoxine). Pyridoxine (B6) can cross the blood-brain barrier and transported to the brain in the absence of effective ALP in GPIBDs [19]. Furthermore, oral B6 supplements are hypothesized to enhance the g-aminobutyric acid (GABA) synthesis which is depleted in GPIBDs [20,21]. There are few reports in the literature about the benefits of pyridoxine (B6) supplementation in managing untreatable seizures in GPIBDs [18, 20, 21, 22]. Treatment with a 200 mg daily B6 supplement in a child with PGAP3-CDG improved his seizures frequency and sleep pattern [10]. In our cohort, three patients were supplemented with daily 50 mg of B6, and two of them did not manifest seizures yet.
Ketogenic diet has been shown improvement in some patients with GPIBDs [23]. The exact mechanism of ketogenic diet effectiveness in controlling seizures is debated. Many theoretical hypotheses have been proposed [24]. In GPIBDs, restoring the depleted GABA in the brain by the high ketone bodies enhances the inhibitory neurotransmission [25]. Few patients with PIGA-CDG have been managed with ketogenic diet which showed either termination or a significant reduction in seizures frequency [26]. In our cohort, two patients were trialed on ketogenic diet, it was indicated for refractory seizures in a 16-year-old male. However, the diet did not affect his clinical seizures, but his ALP levels showed a significant decline while he was on the ketogenic diet. In another 2-year-old female who did not manifest seizures, a trial of ketogenic diet showed subjective improvement in her motor function and ataxic movement with a reduction of her ALP levels during the therapy (Figure 3). This observatory result suggests that dietary alterations might improve the GPI-anchoring of ALP and could be considered in PGAP3-CDG and GPIBDs. Further studies on the ketogenic diet effect on GPIBDs are required.
Unfortunately, there are gaps in the published literature about details of clinical management, including seizure treatment and, or the use of ketogenic diet. One of the major limitations of our study is the source of retrieved data was based on the high-level clinical descriptions from published case cohorts and case series. The ongoing natural history study on CDG patients: ClinicalTrials.gov (NCT03173300) will provide a richer data set for this group of metabolic disorders.
In conclusion, PGAP3-CDG is considered a multisystem CDG with major neurological involvement and high ALP levels. Early recognition of the disease in individuals with ID and/or seizures by screening ALP levels or use of broad genetic testing as a first-tier diagnostic evaluation can help in directing those patients’ symptomatic management. Care can include seizure reduction by supplementing B6 and/or trying a ketogenic diet which has shown positive effects in the ALP levels, improving motor function, and controlling the seizures in GPIBDs. In addition, early enrollment in rehabilitation centers enhances patients’ development and independency. Management of this rare disease is still only supportive and symptomatic but targeted therapies for several GPIBDs are on the horizon [27].
Supplementary Material
Fig.4.
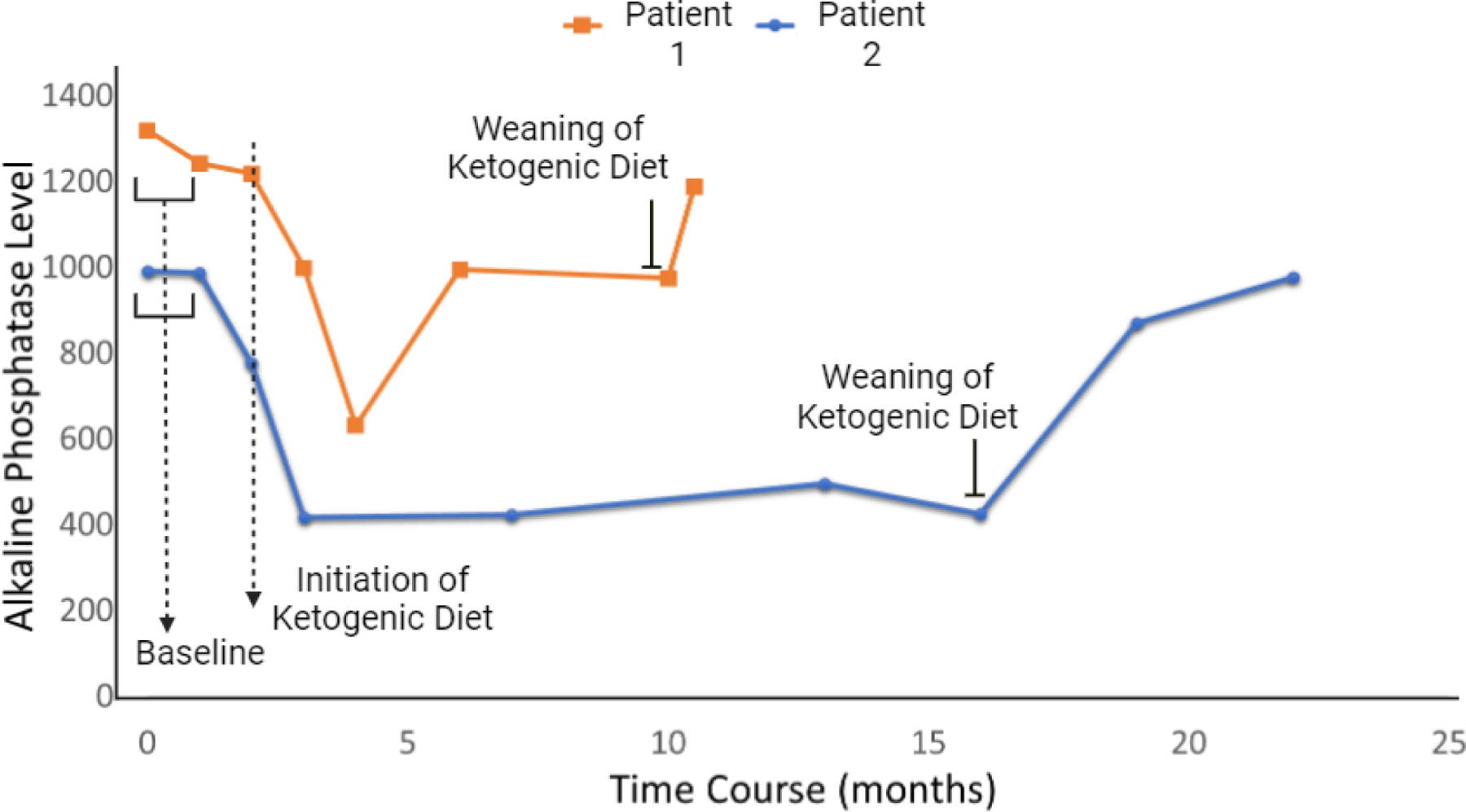
ALP levels during ketogenic diet therapy in two PGAP3-CDG patients
Table 1.
Assessment and management guidance for PGAP3-CDG
| Detailed developmental assessment Hypotonia severity Motor function Abnormal movement Speech assessment Hearing assessment Eye exam Seizure evaluation (EEG, neuroimaging) Behavioral assessment Echocardiogram Swallowing assessment and upper GI study Growth evaluation Alkaline phosphatase level Other glycoproteins (liver function test, coagulation profiles, and hormonal assays) Vitamin B6 supplements Ketogenic diet for seizures management ALP monitoring during ketogenic diet |
ACKNOWLEDGMENTS
This work was funded by the grant titled Frontiers in Congenital Disorders of Glycosylation (1U54NS115198–01) from the National Institute of Neurological Diseases and Stroke (NINDS), the National Center for Advancing Translational Sciences (NCATS), National Institute of Child Health and Human Development (NICHD) and the Rare Disorders Consortium Disease Network (RDCRN), at the National Institute of Health. DDG was supported by a Fellowship of the Belgian American Educational Foundation.
Abbreviation
- PGAP3
Post-GPI attachment to proteins 3
- GPI
glycophosphatidylinositol
- GPIBD
GPI biogenesis disorders
- GPI-APs
glycophosphatidylinositol anchored proteins
- CDG
Congenital disorder of glycosylation
- HPMRS4
hyperphosphatasia with mental retardation syndrome 4
- ALP
alkaline phosphatase
- ID
intellectual disability
- AED
anti-epileptic drugs
- GABA
g-aminobutyric acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Jaeken J Congenital disorders of glycosylation (CDG): it’s (nearly) all in it! J Inherit Metab Dis 2011. Aug;34(4):853–8. [DOI] [PubMed] [Google Scholar]
- 2.Rare disease network. https://fcdgc.rarediseasesnetwork.org/diseases-studied/pgap3-cdg.
- 3.Howard MF, Murakami Y, Pagnamenta AT, Daumer-Haas C, Fischer B, Hecht J, Keays DA, Knight SJ, Kölsch U, Krüger U, Leiz S, et al. Mutations in PGAP3 impair GPI-anchor maturation, causing a subtype of hyperphosphatasia with mental retardation. Am J Hum Genet 2014. Feb 6;94(2):278–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knaus A, Awaya T, Helbig I, Afawi Z, Pendziwiat M, Abu-Rachma J, Thompson MD, Cole DE, Skinner S, Annese F, et al. Rare Noncoding Mutations Extend the Mutational Spectrum in the PGAP3 Subtype of Hyperphosphatasia with Mental Retardation Syndrome. Hum Mutat 2016. Aug;37(8):737–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abdel-Hamid MS, Issa MY, Otaify GA, Abdel-Ghafar SF, Elbendary HM, Zaki MS. PGAP3-related hyperphosphatasia with mental retardation syndrome: Report of 10 new patients and a homozygous founder mutation. Clin Genet 2018. Jan;93(1):84–91 [DOI] [PubMed] [Google Scholar]
- 6.Balobaid A, Ben-Omran T, Ramzan K, Altassan R, Almureikhi M, Musa S, Al-Hashmi N, Al-Owain M, Al-Zaidan H, Al-Hassnan Z, et al. Delineating the phenotypic spectrum of hyperphosphatasia with mental retardation syndrome 4 in 14 patients of Middle-Eastern origin. Am J Med Genet A 2018. Dec;176(12):2850–2857 [DOI] [PubMed] [Google Scholar]
- 7.Maddirevula S, Alsahli S, Alhabeeb L, Patel N, Alzahrani F, Shamseldin HE, Anazi S, Ewida N, Alsaif HS, Mohamed JY, et al. Expanding the phenome and variome of skeletal dysplasia. Genet Med 2018. Dec;20(12):1609–1616 [DOI] [PubMed] [Google Scholar]
- 8.Pagnamenta AT, Murakami Y, Taylor JM, Anzilotti C, Howard MF, Miller V, Johnson DS, Tadros S, Mansour S, Temple IK, et al. Analysis of exome data for 4293 trios suggests GPI-anchor biogenesis defects are a rare cause of developmental disorders. Eur J Hum Genet 2017. Jun;25(6):669–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nampoothiri S, Hebbar M, Roy AG, Kochumon SP, Bielas S, Shukla A, Girisha KM. Hyperphosphatasia with Mental Retardation Syndrome Due to a Novel Mutation in PGAP3. J Pediatr Genet 2017. Sep;6(3):191–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakaguchi T, Žigman T, Petković Ramadža D, Omerza L, Pušeljić S, Ereš Hrvaćanin Z, Miyake N, Matsumoto N, Barić I. A novel PGAP3 mutation in a Croatian boy with brachytelephalangy and a thin corpus callosum. Hum Genome Var 2018. Mar 8;5:18005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akgün Doğan Ö, Demir GÜ, Kosukcu C, Taskiran EZ, Simsek-Kiper PÖ, Utine GE, Alikaşifoğlu M, Boduroğlu K. Hyperphosphatasia with mental retardation syndrome type 4 In two siblings-expanding the phenotypic and mutational spectrum. Eur J Med Genet 2019. Jun;62(6):103535. [DOI] [PubMed] [Google Scholar]
- 12.Abi Farraj L, Khatoun WD, Abou Chebel N, Wakim V, Dawali K, Ghassibe-Sabbagh M. Clinical, genetic, and molecular characterization of hyperphosphatasia with mental retardation: a case report and literature review. Diagn Pathol 2019. Nov 4;14(1):123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Da’as SI, Aamer W, Hasan W, Al-Maraghi A, Al-Kurbi A, Kilani H, AlRayahi J, Zamel K, Stotland MA, Fakhro KA. PGAP3 Associated with Hyperphosphatasia with Mental Retardation Plays a Novel Role in Brain Morphogenesis and Neuronal Wiring at Early Development. Cells 2020. Jul 27;9(8):1782. doi: 10.3390/cells9081782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bezuidenhout H, Bayley S, Smit L, Kinnear C, Möller M, Uren C, Urban MF. Hyperphosphatasia with mental retardation syndrome type 4 in three unrelated South African patients. Am J Med Genet A 2020. Oct;182(10):2230–2235 [DOI] [PubMed] [Google Scholar]
- 15.Alhaidari AI, Albakri AS, Alhumaidi SS. A Novel PGAP3 Gene Mutation-Related Megalocornea Can Be Misdiagnosed as Primary Congenital Glaucoma. Cureus 2022. Sep 21;14(9):e29387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kinoshita T, Fujita M. Biosynthesis of GPI-anchored proteins: special emphasis on GPI lipid remodeling. J Lipid Res 2016. Jan;57(1):6–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carmody LC, Blau H, Danis D et al. Significantly different clinical phenotypes associated with mutations in synthesis and transamidase+remodeling glycosylphosphatidylinositol (GPI)-anchor biosynthesis genes. Orphanet J Rare Dis 15, 40 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Messina M, Manea E, Cullup T, Tuschl K, Batzios S. Hyperphosphatasia with mental retardation syndrome 3: Cerebrospinal fluid abnormalities and correction with pyridoxine and Folinic acid. JIMD Rep 2022. Nov 22;64(1):42–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thompson MD, Killoran A, Percy ME, Nezarati M, Cole DE, Hwang PA. Hyperphosphatasia with neurologic deficit: a pyridoxine-responsive seizure disorder? Pediatr Neurol 2006. Apr;34(4):303–7 [DOI] [PubMed] [Google Scholar]
- 20.Kuki Ichiro, Takahashi Yukitoshi, Okazaki Shin, Kawawaki Hisashi, Ehara Eiji, Inoue Norimitsu, Kinoshita Taroh, Murakami Yoshiko. Vitamin B6–responsive epilepsy due to inherited GPI deficiency. Neurology Oct 2013, 81 (16) 14671469; DOI: 10.1212/WNL.0b013e3182a8411a [DOI] [PubMed] [Google Scholar]
- 21.Wilson MP, Plecko B, Mills PB, Clayton PT. Disorders affecting vitamin B6 metabolism. J Inherit Metab Dis 2019. Jul;42(4):629–646 [DOI] [PubMed] [Google Scholar]
- 22.Bayat A, Aledo-Serrano A, Gil-Nagel A, Korff CM, Thomas A, Boßelmann C, Weber Y, Gardella E, Lund AM, de Sain-van der Velden MGM, Møller RS. Pyridoxine or pyridoxal-5-phosphate treatment for seizures in glycosylphosphatidylinositol deficiency: A cohort study. Dev Med Child Neurol 2022. Jun;64(6):789–798 [DOI] [PubMed] [Google Scholar]
- 23.Boyer SW, Johnsen C, Morava E. Nutrition interventions in congenital disorders of glycosylation. Trends Mol Med 2022. Jun;28(6):463–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rho JM. How does the ketogenic diet induce anti-seizure effects? Neurosci Lett 2017. Jan 10;637:4–10. doi: 10.1016/j.neulet.2015.07.034. Epub 2015 Jul 26. [DOI] [PubMed] [Google Scholar]
- 25.Bayat A, Knaus A, Pendziwiat M, Afenjar A, Barakat TS, Bosch F, Callewaert B, Calvas P, Ceulemans B, Chassaing N, et al. Lessons learned from 40 novel PIGA patients and a review of the literature. Epilepsia 2020. Jun;61(6):1142–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joshi C, Kolbe DL, Mansilla MA, Mason S, Smith RJ, Campbell CA. Ketogenic diet - A novel treatment for early epileptic encephalopathy due to PIGA deficiency. Brain Dev 2016. Oct;38(9):848–51 [DOI] [PubMed] [Google Scholar]
- 27.Brasil S, Allocca M, Magrinho SCM, Santos I, Raposo M, Francisco R, Pascoal C, Martins T, Videira PA, Pereira F, et al. Systematic Review: Drug Repositioning for Congenital Disorders of Glycosylation (CDG). Int J Mol Sci 2022. Aug 5;23(15):8725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jumper J et al. Highly accurate protein structure prediction with AlphaFold. Nature (2021) [DOI] [PMC free article] [PubMed]
- 29.Varadi M et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Research (2021) [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.