

SUMMARY
The bacterial PASTA kinase, IreK, is required for intrinsic cephalosporin resistance in the Gram-positive opportunistic pathogen, Enterococcus faecalis. IreK activity is enhanced in response to cell wall stress, such as cephalosporin exposure. The downstream consequences of IreK activation are not well understood in E. faecalis, but recent work in other low-GC Gram-positive bacteria demonstrated PASTA kinase-dependent regulation of MurAA, an enzyme which performs the first committed step in the peptidoglycan synthesis pathway. Here, we used genetic suppressor selections to identify MurAA as a downstream target of IreK signaling in E. faecalis. Using complementary genetic and biochemical approaches we demonstrated that MurAA abundance is regulated by IreK signaling in response to physiologically relevant cell wall stress to modulate substrate flux through the peptidoglycan synthesis pathway. Specifically, the IreK substrate, IreB, promotes proteolysis of MurAA through a direct physical interaction in a manner responsive to phosphorylation by IreK. MurAB, a homolog of MurAA, also promotes MurAA proteolysis and interacts directly with IreB. Our results therefore establish a connection between the cell wall stress sensor IreK and one critical physiological output to modulate peptidoglycan synthesis and drive cephalosporin resistance.
Keywords: Cephalosporin resistance, PASTA kinase, peptidoglycan synthesis, Enterococcus
Graphical Abstract

The cell wall stress sensing PASTA-kinase, IreK, is required for intrinsic cephalosporin resistance in Enterococcus faecalis. Using unbiased genetic selections we identified the peptidoglycan synthesis enzyme, MurAA, as a downstream effector of IreK. We demonstrate that IreK signaling modulates the cellular abundance of MurAA in response to stress, which has consequences on flux through the peptidoglycan synthesis pathway, and in turn, cephalosporin resistance.
INTRODUCTION
Cephalosporin antibiotics, which target bacterial cell wall synthesis, are a commonly prescribed class of beta-lactams used to treat a broad range of infections. Despite their utility, treatment with cephalosporins has the potential to result in secondary, nosocomial infections caused by enterococcal species (primarily Enterococcus faecalis and Enterococcus faecium) which possess intrinsic resistance to this class of antibiotics (Carmeli et al., 2002; Murray, 1990; Shepard & Gilmore, 2002). Enterococci are ovoid, Gram-positive, commensals of the gastrointestinal tract. Administration of cephalosporins can lead to abnormal proliferation of these resistant organisms and subsequent dissemination (Brandl et al., 2008; Donskey et al., 2000; Ubeda et al., 2010) resulting in urinary tract infections, bacteremia, endocarditis, and other infections (Magill et al., 2018; Weiner et al., 2016). Enterococcal species are the third-most common cause of infective endocarditis (Baddour et al., 2015; Holland et al., 2016) a life-threatening disease in which bacteria grow in a biofilm-like state on the heart valves, with 97% of these cases caused by E. faecalis (Baddour et al., 2015). The emergence of multi-drug resistant strains of enterococci has made treating these infections a unique clinical challenge (Hollenbeck and Rice, 2012; Kristich, Rice and Arias, 2014; Miller et al., 2016) and underscores the importance of understanding the molecular mechanisms that underlie both acquired and intrinsic antimicrobial resistance (such as that to cephalosporins) to aid with the prevention and treatment of these infections.
The mode of action of cephalosporins is to inhibit the penicillin-binding proteins (PBPs) which catalyze crosslinking of the bacterial peptidoglycan (PG) (Bush and Bradford, 2016), a mesh-like structure composed of linear chains of repeating N-acetylmuramic acid and N-acetylglucosamine moieties linked by peptide bridges. Crosslinking of PG is essential for maintenance of cell-shape and osmotic support. Enterococci encode at least two PBPs that exhibit low-reactivity towards cephalosporins, contributing to their resistance to these antibiotics (Signoretto, Boaretti and Canepari, 1994; Arbeloa et al., 2004; Djorić, Little and Kristich, 2020). However, proper flux of PG precursors though the peptidoglycan synthesis pathway upstream of the terminal crosslinking step is also required for cephalosporin resistance (Mascari et al., 2022).
E. faecalis, like most low-GC Gram-positive bacteria (Du et al., 2000), encodes two UDP-N-acetylglucosamine 1-carboxyvinyl transferases capable of catalyzing the first-committed step in PG biosynthesis: MurAA and MurAB. Neither enzyme is individually essential in E. faecalis, but MurAA seems to play a more central role in PG biosynthesis (Mascari et al., 2022) and deletion of murAA, but not murAB, leads to impaired growth and loss of cephalosporin resistance (Vesić and Kristich, 2012). MurAA orthologs tend to be considered the “primary” UDP-N-acetylglucosamine 1-carboxyvinyl transferases in most organisms that encode two copies of the enzyme (Blake et al., 2009; Kedar et al., 2008; Kock et al., 2004; Rismondo et al., 2017) and are essential in both Bacillus subtilis and Listeria monocytogenes (Kock, Gerth and Hecker, 2004a; Rismondo, Bender and Halbedel, 2017). A notable exception is Streptococcus pneumoniae, in which the MurAB ortholog, MurZ was recently demonstrated to be the predominant enzyme (Tsui et al., 2023).
Recent work in L. monocytogenes (a rod-shaped bacterium) demonstrated that the stability of the L. monocytogenes MurAA homolog, LmMurA, is regulated by the PASTA-domain containing serine/threonine kinase, PrkA (Wamp et al., 2020, 2022). The PrkA substrate, ReoM, promotes degradation of LmMurA by the ClpCP proteolytic complex – a pathway that is shut off upon phosphorylation of ReoM by PrkA. Regulation of LmMurA via this mechanism has downstream consequences on PG biosynthesis, virulence and both laboratory and intracellular growth (Kelliher et al., 2021; Wamp et al., 2020, 2022). Further work demonstrated that this pathway is conserved in another rod-shaped bacterium, B. subtilis (Wamp et al., 2020; Sun, Hürlimann and Garner, 2023) as well as in the spherical bacterium, Staphylococcus aureus (Kelliher et al., 2021). In contrast, recent work in the ovoid bacterium S. pneumoniae by Tsui, Joseph, and colleagues provides genetic evidence that although MurZ and/or MurA (the S. pneumoniae homologs of MurAB and MurAA, respectively) are downstream of the S. pneumoniae PASTA kinase, StkP, multiple lines of evidence demonstrated that neither MurZ nor MurA is proteolytically regulated by ClpP (Tsui et al., 2023). Tsui, Joseph, and coworkers propose a model in which the StkP substrate and LmReoM homolog, IreB, regulate the activity instead of the abundance of one or both MurZ and MurA in S. pneumoniae. Hence, while PASTA-kinase dependent regulation of MurAA/AB homologs seems to be conserved amongst low-GC Gram-positive bacteria, the specific mechanisms by which this occurs may vary from species to species. Our previous data suggested that MurAA is downstream of the E. faecalis PASTA kinase (Vesić and Kristich, 2012), but the mechanism of regulation in E. faecalis remained unknown.
In E. faecalis, the PASTA kinase/substrate pair homologous to PrkA/ReoM (L. monocytogenes) and StkP/IreB (S. pneumoniae) is IreK/IreB. The kinase, IreK, is activated at low levels during exponential growth, and its activity is enhanced in response to cell wall stress such as that imposed by cell wall active antibiotics. In contrast, during stationary phase the kinase is nearly completely inactivated (Labbe and Kristich, 2017). Deletion of ireK is associated with marked loss of resistance to cephalosporins (Kristich, Wells and Dunny, 2007) in addition to other cell wall targeting agents such as sodium cholate (a component of bile), nisin and lysozyme (Kristich, Wells and Dunny, 2007; Banla et al., 2018). Deletion of the IreK substrate, ireB, results in cephalosporin hyper-resistance and enhances resistance of the ΔireK mutant to both sodium cholate and nisin (Hall et al., 2013) suggesting that IreB is a negative regulator of downstream pathways that contribute to these resistant phenotypes. However, the specific molecular function of enterococcal IreB was previously unknown. The cognate phosphatase for the system, IreP, dephosphorylates both IreK and downstream substrates. Accordingly, deletion of ireP results in elevated phosphorylation and activity of IreK and enhanced cephalosporin resistance (Kristich et al., 2011).
To determine how IreB impacts cephalosporin resistance in E. faecalis, we used unbiased genetic selections coupled with biochemical approaches to identify MurAA as a downstream regulatory target for IreB. Our results provide direct evidence that IreK regulates a physical interaction between MurAA and IreB through phosphorylation of IreB. We further demonstrate that the MurAA homolog MurAB is also capable of interacting with IreB, and that the presence of MurAB is required for MurAA proteolysis. Overall, our data suggest that control of MurAA cellular abundance by IreB is one critical role of IreK signaling in response to multiple environmental stressors. However, regulation of E. faecalis MurAA was not sufficient to fully explain the role of IreK and IreB in cephalosporin resistance, suggesting that IreB may have additional, yet-undefined downstream targets that are important for providing this resistance.
RESULTS
Point mutations in murAA suppress phenotypes associated with impaired IreK signaling
To elucidate the pathway downstream of IreB, we performed two distinct genetic suppressor selections (outlined in Supplemental figure 1). The first (selection 1, Supplemental figure 1a) used strain BL102 in which IreK is catalytically inactive due to a K41R substitution (Labbe and Kristich, 2017). In the absence of normal IreK kinase activity, IreB remains in an unphosphorylated and constitutively “active” state (negatively regulating cephalosporin resistance), which contributes to the reduced cephalosporin resistance of BL102. We reasoned that it might be possible to identify mutants in which IreB was no longer able to inhibit a downstream target, thereby restoring cephalosporin resistance despite constitutive activation of IreB, and therefore selected for spontaneous suppressor mutants of BL102 exhibiting elevated resistance to ceftriaxone (a representative cephalosporin). As expected (Hall et al., 2013), some resistant suppressor mutants acquired loss-of-function mutations in ireB. However, we also identified other resistant suppressor mutants with wild-type ireB sequence that were subsequently subjected to whole genome sequencing to identify the responsible mutations. In independently isolated mutants, we identified two different substitutions at alanine 115 of MurAA (A115T and A115V) which exhibited increased ceftriaxone resistance relative to the BL102 background strain (Table 1). The MurAA A115 residue is just upstream of a conserved L/MPGGC sequence which contains the catalytic cysteine residue (MurAA C120).
TABLE 1.
Mutations identified from suppressor selections
| Parent strain† | Challenge condition | Suppressor strain | Mutation identified | Ceftriaxone MIC (μg ml−1)‡ |
|---|---|---|---|---|
| OG1 ireK K41R | - | - | - | 1 |
| Ceftriaxone | JL600 | murAA A115T | 8 | |
| JL599 | murAA A115V | 8 | ||
| OG1RF ΔireK | - | - | - | 1 |
| P-ireB§ | JL400 | murAA N188K | 16 | |
| JL436 | murAA D163Y | 4 | ||
| JL439 | murAA D163Y | 4 | ||
| JL402 | murAA D163E | 8 | ||
| JL398 | murAA A212D | 4 | ||
| JL401 | murAA P265L | 8 | ||
| OG1 ΔireK | - | - | - | 1 |
| P-ireB§ | JL350 | clpP A46E | 8 |
Strains used were OG1 ireK K41R, BL102; OG1RF ΔireK, CK119, OG1 ΔireK, JL206
Minimal inhibitory concentration for ceftriaxone were determined as the median MIC from at least 2 independent biological replicates.
plasmid used was pCJK227
Because of its design, the aforementioned selection had the potential to reveal other targets of IreK that were not necessarily downstream of IreB (given that IreK is known to have other substrates that play a role in cephalosporin resistance (Kellogg and Kristich, 2018; Iannetta et al., 2021; Minton et al., 2022)). As an alternative approach to more specifically identify factors downstream of IreB, in a second selection (selection 2, Supplemental figure 1b) we exploited a previous observation that an IreB expression vector could not be successfully introduced into an E. faecalis strain lacking IreK (Hall et al., 2013), suggesting that overexpression of unphosphorylated (i.e. active) IreB leads to unsustainable inhibition of a downstream target that is critical for growth. Reasoning that it might be possible to identify mutations that uncouple IreB from inhibition of its downstream target, we took advantage of this phenotype by selecting for spontaneous suppressor mutations that allowed growth upon overexpression of ireB in the absence of ireK. Through this selection, we identified multiple independently isolated mutants with single amino acid substitutions in murAA (Table 1). Analysis of ceftriaxone resistance revealed that all such mutants also led to increases in the ceftriaxone MIC relative to the background strain (Table 1), despite the fact that we did not select for elevated cephalosporin resistance in selection 2. Under similar conditions a suppressor mutant was identified with a single-amino acid substitution in the caseinolytic protease proteolytic subunit, clpP, locus (A46E) that allowed overexpression of ireB (Table 1).
When mapped to an AlphaFold model (Jumper et al., 2021; Varadi et al., 2022) of the E. faecalis MurAA structure, the A115 residue is located directly adjacent to a flexible loop that has been proposed to serve as a lid over the catalytic pocket located between the two globular domains of the protein (Skarzynski et al., 1996). In contrast, the residues identified in selection 2 are located in what appears to be a surface-exposed cluster distal from the active site (Supplemental figure 2a), suggesting that the murAA mutations from selection 2 might be affecting MurAA function in a distinct way from those of selection 1. We chose one residue identified in each selection to focus on for further analysis (A115T (selection 1) and N188K (selection 2)). The N188K substitution was selected from the selection 2 subset on the basis that it had greatest impact on cephalosporin resistance (Table 1). Both the A115 and N188 residues are well conserved amongst MurAA homologs from the Firmicutes, although S. pneumoniae MurA has a serine residue in place of alanine 115 (Supplemental figure 2b).
To validate their role in the observed suppressor phenotypes, both the A115T and N188K substitutions were re-constructed in the genome of the ΔireK strain of E. faecalis. Unlike the ΔireK strain, the ΔireK murAA N188K strain was able to accept a plasmid expressing ireB (Supplemental figure 3) and MIC analysis showed that both the N188K and A115T mutations in the ΔireK background led to similar increases in ceftriaxone resistance (Table 2) as were seen during the initial suppressor selection (Table 1). When introduced into an otherwise wild-type background, both substitutions led to a ceftriaxone hyper-resistance phenotype (Table 3). Importantly, deletion of ireB also results in ceftriaxone hyper-resistance ((Hall et al., 2013), Table 3). Together with our earlier observation that ectopic overexpression of murAA could rescue the ceftriaxone resistance defect of ΔireK strain (Vesić and Kristich, 2012), the identification of these mutations in murAA suggests that MurAA is likely an important downstream target of IreK signaling in E. faecalis and is specifically downstream of IreB.
TABLE 2.
The impact of mutations on ceftriaxone resistance of the ireK deletion strain.
| Strain† | Ceftriaxone MIC (μg ml−1)‡ |
|---|---|
| Wild-type | 64 |
| ΔireK | 1 (≤1–1) |
| ΔireK murAA A115T | 8 (8–16) |
| ΔireK murAA N188K | 8 (8–16) |
| ΔireK ireB | 32 |
| ΔireK clpP A46E | 4 |
| ΔireK clpC | 8 |
| ΔireK murAB | 4 |
| ΔireK murAB C116S | ≤0.5 |
Strains used were Wild-type, OG1; ΔireK, JL206; ΔireK murAA A115T, CM27; ΔireK murAA N188K, CM28; ΔireK ireB, CM52; ΔireK clpP A46E, CM39; ΔireK clpC, CM38; ΔireK murAB, CM44; ΔireK murAB C116S, CM45.
Minimal inhibitory concentration for ceftriaxone were determined as the median MIC from at least 3 independent biological replicates. In cases where the replicate values were not identical, the range of values is reported in parentheses.
TABLE 3.
Ceftriaxone resistance of mutants in the presence of IreK
| Strain† | Ceftriaxone MIC (μg ml−1)b |
|---|---|
| Wild-type | 64 |
| murAA A115T | 1024 |
| murAA N188K | 512 |
| ΔireB | 1024 (512–1024) |
| ΔireB murAA A115T | 2048 |
| ΔireB murAA N188K | 1024 |
| ΔclpP | 128 |
| ΔclpC | 64 |
| clpP A46E | 128 |
| ΔmurAB | 32 |
Strains used were Wild-type, OG1; murAA A115T, CM2; murAA N188K, CM3; ΔireB, JL367; ΔireB murAA A115T, CM29; ΔireB murAA N188K, CM30; ΔclpP, CM34; ΔclpC, CM34; clpP A46E, CM36; ΔmurAB, CM1.
Minimal inhibitory concentration for ceftriaxone were determined as the median MIC from at least 3 independent biological replicates. In cases where the replicate values were not identical, the range of values is reported in parentheses.
IreK impacts substrate flux through the PG synthesis pathway
We recently demonstrated that control of substrate flux through the peptidoglycan biosynthesis pathway is a key driver of cephalosporin resistance (Mascari et al., 2022), and that the cellular abundance of MurAA is a major factor determining this flux. The observations that MurAA might be downstream of IreK suggest that IreK signaling could regulate flux of substrates through the PG synthesis pathway in response to growth and cell wall stress. To test this, incorporation of [14C] GlcNAc into the SDS-insoluble peptidoglycan was monitored over time in actively growing cells as we and others have described previously (Bisicchia et al., 2007; Mesnage et al., 2008; Djorić, Little and Kristich, 2020; Mascari et al., 2022). The ΔireK mutant exhibited an ~30% reduction in the rate of [14C] GlcNAc incorporation relative to wild-type, indicative of impaired flux of substrates in the peptidoglycan synthesis pathway. Introduction of the murAA N188K or A115T substitutions into the ΔireK mutant restored the [14C] GlcNAc incorporation rate to wild-type (Figure 1a), suggesting that impaired PG precursor flux is a critical consequence of disruption of IreK.
Figure 1. PG substrate flux is impaired in the absence of IreK but rescued by the murAA N188K and A115T mutations, and by deletion of ireB or clpC.

[14C] GlcNAc incorporation into the SDS-insoluble PG following pulse-labeling of exponentially growing E. faecalis strains (A) ΔireK, ΔireK murAA A115T and ΔireK murAA N188K or (B) ΔireK, ΔireK ireB and ΔireK clpC and compared to wild-type. Incorporation rates were determined from data points between 10 and 25 min and are reported in the table. Data represent mean ± standard deviation for two independent replicates. Strains were wild-type, OG1; ΔireK, JL206; murAA A115T ΔireK, CM27; murAA N188K ΔireK, CM28; ΔireK ireB, CM52; ΔireK clpC, CM38.
The cellular abundance of MurAA is impacted by both IreB and the suppressor mutations
Given that overexpression of MurAA overcomes defects associated with loss of normal PASTA kinase signaling in both E. faecalis and other organisms (Vesić and Kristich, 2012; Wamp et al., 2022; Tsui et al., 2023) we asked whether the N188K and/or A115T substitutions might be impacting the cellular abundance of MurAA. In addition to their role in cell wall stress-sensing in actively dividing cells, PASTA kinase/phosphatase pairs in other organisms have also been implicated in regulation of stationary phase processes (Gaidenko, Kim and Price, 2002), and furthermore, MurAA abundance has been demonstrated to be growth-phase dependent in B. subtilis, such that upon the transition to stationary phase (when there is reduced demand for peptidoglycan synthesis) the cells become depleted of MurAA (Kock, Gerth and Hecker, 2004a). We observed similar growth-phase dependent trends in MurAA abundance in wild-type E. faecalis (Supplemental figure 4), leading us to evaluate MurAA levels during both exponential and stationary growth phases in our strains of interest. Using immunoblot analysis we found either statistically significant or reproducibly trending increases in MurAA abundance associated with the murAA N188K and murAA A115T mutations (Figure 2a).
Figure 2. Immunoblot analysis and quantification of MurAA steady-state abundance.

Whole cell lysates were prepared from E. faecalis cells collected during exponential phase (left) or stationary phase (right) and subjected to SDS-PAGE. Membranes with probed with an anti-MurAA anti-serum and MurAA signal was normalized to total protein in each lane (total protein images shown in Supplemental figure 5 A–B). Quantification was done for three biological replicates and error bars show standard deviation. Unless otherwise indicated, stars represent statistical differences relative to wild-type (*, p=0.01 to 0.05; **, p=0.001 to 0.01; ***, p=0.0001 to 0.001) based on One-way ANOVA with Tukey’s multiple comparisons. One representative image is shown for each blot. Strains are wild-type, OG1; murAA N188K, CM3; murAA A115T, CM2; ΔireB, JL367; ΔireB murAA N188K, CM30; ΔireB murAA A115T, CM29; ΔireP, JL455; ΔireK, JL206; ΔireK murAA N188K, CM28; ΔireK murAA A115T, CM27; ΔclpP, CM34; ΔclpC, CM35; clpP A46E, CM36; ΔireB clpC, CM42.
MurAA abundance was also elevated in an ireB deletion strain, with statistically significant increases observed during both exponential and stationary phase (Figure 2a). To determine whether the increased MurAA accumulation seen in the ireB deletion and MurAA point mutant strains was occurring through a shared mechanism we constructed double mutant strains. No further increase in MurAA steady-state abundance was observed in either double mutant relative to ΔireB, and in fact both double mutant strains exhibited trending decreases in MurAA levels relative to ΔireB during exponential phase (Figure 2a). While it is not clear what is driving this dampened effect in the double mutants, the lack of an additive effect on MurAA levels is consistent with a shared mechanism for accumulation of MurAA in the ΔireB and point mutant strains.
We did not observe any reproducible decrease in MurAA abundance in the ireK deletion strain (Figure 2b) despite the fact that IreB is constitutively active when IreK is absent. Given that these experiments used unstressed growth conditions under which IreK is not highly activated (and therefore IreB is largely active), we suspect that differences in MurAA abundance might be subtle and possibly below our limit of detection. Moreover, the IreK/IreP pair has many substrates and deletion of ireK likely exerts pleiotropic effects, which could influence MurAA indirectly. The N188K and A115T mutations in murAA, however, were still associated with trending increases in MurAA levels in the ΔireK background (Figure 2b), suggesting that the effect of these mutations can overcome constitutive activation of IreB.
To determine if changes in MurAA abundance are influenced by the phosphorylation state of IreB, we analyzed a mutant lacking ireP, which encodes the cognate phosphatase for IreK. In the absence of IreP, IreK is hyper-activated, resulting in substantial phosphorylation (and therefore inactivation) of IreB (Labbe and Kristich, 2017) as well as other phosphorylation substrates of IreK. Although the effect is modest, MurAA levels trend upwards in the ΔireP mutant (Figure 2b), which would be consistent with phosphorylation of IreB enabling MurAA accumulation. However, the IreK/IreP pair has many substrates and deletion of ireP likely exerts pleiotropic effects, which might account for the difference in MurAA abundance between the ΔireB and ΔireP mutants via an as-yet-unknown mechanism. In wild-type E. faecalis cells, cell wall stresses such as exposure to cephalosporins triggers enhanced phosphorylation of IreB by IreK (Labbe and Kristich, 2017). We therefore monitored MurAA abundance during exposure of E. faecalis cells to ceftriaxone (at half the MIC, which did not significantly alter the growth kinetics of these strains (Supplemental figure 7)), revealing increased MurAA abundance upon ceftriaxone challenge of wild-type cells (Figure 3). No change in MurAA abundance was observed for the ΔireK or ΔireB strains when grown in the presence of half-MIC concentrations of ceftriaxone (Figure 3), consistent with this effect being mediated by signaling through the IreK-IreB pathway. Phosphorylation of IreB in response to physiologically relevant stresses, therefore, also impacts MurAA levels.
Figure 3. MurAA protein level is increased upon ceftriaxone treatment.
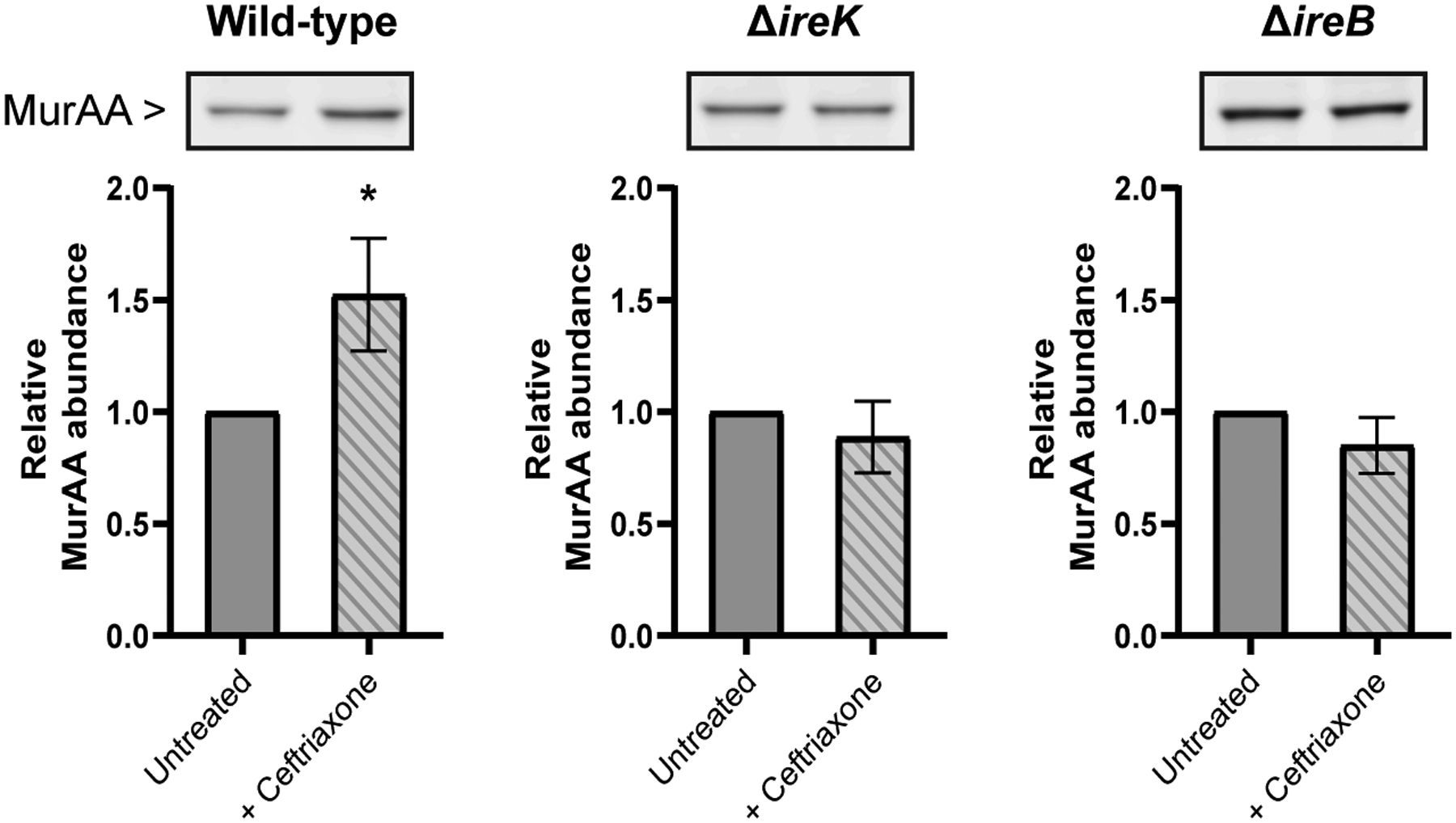
Wild-type, ΔireK and ΔireB E. faecalis cells were grown in the presence (striped bars) or absence (solid bars) of 0.5X MIC ceftriaxone (32, 0.5 and 512 ug mL−1 respectively) in MHB at 37°C. Whole cell lysates were generated from exponentially growing cells (OD600=0.2). Immunoblot was performed with anti-sera to MurAA and MurAA signal was normalized to total protein in each lane (total protein images shown in Supplemental figure 6a). Quantification was done for three biological replicates and error bars show standard deviation. Statistical significance was determined using an unpaired t-test (*, p=0.01 to 0.05). One representative immunoblot image is shown for each strain.
Taken together, these findings suggest that IreB regulates MurAA abundance in E. faecalis, while the MurAA N188K and A115T mutations seem to disconnect control of the MurAA abundance from IreB. Like the murAA N188K and A115T substitutions, deletion of ireB was also able to fully rescue the [14C] GlcNAc incorporation defect of the ΔireK strain and restored ceftriaxone resistance nearly to wild-type (Figure 1b, Table 2).
The N188K substitution stabilizes MurAA from ClpCP-mediated proteolysis
In L. monocytogenes the IreB homolog, ReoM, regulates proteolysis of LmMurA by the ClpCP proteolytic complex in response to changes in the phosphorylation state of ReoM (Wamp et al., 2020). The ClpCP complex is made up of two heptameric rings of the serine peptidase, ClpP, and at least one hexameric ring of the ATPase subunit, ClpC, which are responsible for substrate recognition, unfolding and translocation (reviewed in (Frees et al., 2007)). ClpCP complexes have been implicated in both stress response and growth-phase dependent regulation of biosynthetic enzymes (Krüger et al., 2000; Kock, Gerth and Hecker, 2004b; Gerth et al., 2008; Zheng et al., 2020) including the stationary phase depletion of MurAA in B. subtilis (Kock, Gerth and Hecker, 2004a). Recently, MurAA was identified in a mass spectrometry-based proteomics analysis as a differentially abundant protein in an E. faecalis clpP deletion strain (Zheng et al., 2020), suggesting that MurAA may be proteolytically regulated by ClpCP in E. faecalis as well, potentially in response to changes in demand for peptidoglycan synthesis.
To test this, we constructed mutants lacking either clpC or clpP and analyzed MurAA abundance, revealing that MurAA abundance was increased in both clpC and clpP deletion strains during both exponential and stationary growth phases (Figure 2c). The fact that MurAA accumulates to the same level in both strains (Figure 2c) is consistent with the ClpC/P pair specifically being responsible for this phenotype, as opposed to other ATPases which are known to partner with ClpP such as ClpE and ClpX (Frees et al., 2007). We observed a slight growth defect for ΔclpP strain that was not seen for the ΔclpC strain during unstressed growth (data not shown) suggesting that deletion of clpP has additional negative consequences on growth unrelated to MurAA levels, prompting us to focus on the clpC deletion for our studies. Further, the increase in MurAA abundance seen in both the clpC and clpP deletion strains was comparable to that of the ΔireB mutant. Construction and analysis of a ΔclpC ireB double mutant revealed no further increases in MurAA abundance, suggesting a shared mechanism driving MurAA accumulation. Protein stability experiments demonstrated that following treatment with chloramphenicol to block new protein synthesis, MurAA abundance rapidly decreases in wild-type cells, but not in the absence of clpC or ireB (Figure 4). The MurAA N188K mutant exhibited partial stabilization while no stabilizing effect was seen in the presence of the A115T substitution (Figure 4). No differences in murAA transcript level were detected in the murAA N188K or ΔireB strains (Supplemental figure 9) consistent with the hypothesis that changes in protein stability are responsible for the increased steady state abundance of MurAA in these strains. Taken together, our results indicate that unphosphorylated IreB stimulates proteolysis of MurAA in E. faecalis, while the N188K mutation disconnects MurAA from IreB-mediated proteolysis. Note that the MurAA homolog, MurAB, did not exhibit any difference in steady-state abundance in any strain tested in Figure 2 or in response to ceftriaxone treatment (data not shown) and was stable following chloramphenicol treatment even in the wild-type strain (Supplemental figure 10), demonstrating that MurAB is not regulated by this mechanism.
Figure 4. MurAA is stabilized in the absence of IreB or ClpC and by the N188K substitution.

Immunoblot analysis of whole cell lysates prepared from E. faecalis cultures pre- and post-chloramphenicol treatment to evaluate degradation of MurAA over time. Whole cell lysates were subjected to SDS-PAGE and immunoblotting analysis using a MurAA anti-serum. MurAA signal was normalized to total protein in each lane (total protein images shown in Supplemental Figure 8a) and compared to t0. MurAA half-life (t1/2) was calculated by fitting the normalized signal intensity over time to first order decay. The half-life for the ΔclpC and ΔireB strains was not determined (n.d.) due to nearly complete stabilization of MurAA over the course of the experiment. Quantification was done for three biological replicates and mean ± standard error is reported. Stars represent statistical differences relative to wild-type (*, p=0.01 to 0.05; **, p=0.001 to 0.01) based on Two-way ANOVA with Dunnett’s multiple comparisons. One representative immunoblot is shown for each strain. Strains are wild-type, OG1; ΔclpC, CM35; ΔireB, JL367; murAA N188K, CM3; murAA A115T, CM2).
A point mutation in clpP also emerged from our suppressor selection 2 (Table 1) as noted above. Reconstruction of this mutation in the ireK deletion background enabled transformation of the ireB expression plasmid and modestly improved cephalosporin resistance (Supplemental figure 3, Table 2). To test if the clpP A46E substitution affected MurAA abundance, we introduced the mutation into an otherwise wild-type strain and found that MurAA abundance is increased in the clpP A46E mutant to the same level as that of the clpP deletion strain (Figure 2c), consistent with A46E being a loss of function allele. Additionally, deletion of clpC in the ΔireK background not only led to a similar subtle increase in the abundance of MurAA during exponential phase as the N188K substitution (Supplemental figure 11, Figure 2b), but also a similar increase in the ceftriaxone MIC (Table 2) and fully rescued the [14C] GlcNAc incorporation defect of the ΔireK strain (Figure 1b). Collectively, these results are consistent with the model previously established in L. monocytogenes (Wamp et al., 2020, 2022), in which the ClpCP complex is responsible for proteolytic degradation of MurAA in a manner that depends on unphosphorylated IreB.
The N188K substitution blocks a physical interaction between MurAA and IreB.
Wamp et al. previously reported that the L. monocytogenes homologs of IreB and MurAA physically associate both in vivo (Wamp et al., 2020) and in vitro (Wamp et al., 2022) and that surface-exposed mutations in LmMurA that stabilize the protein from proteolytic degradation impair this interaction in vivo (Wamp et al., 2022). Given that the mutations we identified in suppressor selection 2 were located in a surface-exposed patch on the predicted MurAA structure (Supplemental figure 2a), we hypothesized that a protein-protein interaction at this surface could similarly be required for IreB-mediated proteolysis of MurAA and, by extension, the N188K substitution could be stabilizing MurAA by impairing this protein-protein interaction.
To determine whether MurAA and IreB from E. faecalis physically interact in vitro, we performed thermal shift assays (TSAs) (Layton and Hellinga, 2011; Foulquier and Galinier, 2017). Thermal denaturation of purified recombinant MurAA was monitored in the presence of absence of IreB using SYPRO Orange dye. Changes in the thermal denaturation temperature of a protein can occur upon binding of a ligand or interaction partner, reflecting differences in stability in the bound vs. unbound state (i.e. the thermal shift). TSAs can be used to detect physical interactions between proteins in vitro if the proteins of interest have sufficiently different melt temperatures (Tm) such that the individual unfolding profiles do not overlap (Layton and Hellinga, 2011), as is the case with MurAA and IreB (Figure 5a, Supplemental figure 12b). Using this approach, we initially observed no shift in the Tm for MurAA when IreB was added at 10-fold molar excess (Figure 5a, solid black vs. solid purple). To test if MurAA needed to be in an enzymatically active conformation to bind IreB, we added the MurAA substrate UDP-GlcNAc along with Fosfomycin (a non-hydrolyzable analog of the other MurAA substrate PEP) in order to effectively lock MurAA in a substrate-bound conformation. The addition of UDP-GlcNAc and Fosfomycin (in the absence of IreB) led to a 9.8°C shift in the MurAA Tm indicating stabilization caused by their binding (Figure 5a, solid black vs. dashed black). When IreB was added in the presence of UDP-GlcNAc and Fosfomycin we observed an additional 16.5°C shift in the MurAA Tm (Figure 5a, dashed black vs. dashed purple). A similar IreB-dependent shift was seen in the presence of UDP-GlcNAc and PEP, while addition of UDP-GlcNAc alone led to what appears to be a partial stabilization in the presence of IreB. Addition of PEP or Fosfomycin alone did not result in IreB-dependent stabilization of MurAA (Supplemental figure 12a). Addition of UDP-GlcNAc and Fosfomycin to IreB alone had no impact on its Tm (Supplemental figure 12b). As a further test for specificity, we added a nonspecific control protein with a high Tm (Lysozyme) at 10-fold molar excess and observed no effect on the MurAA Tm in the presence or absence of UDP-GlcNAc and Fosfomycin (Supplemental figure 12c). Taken together, these data demonstrate that IreB binds directly to MurAA and does so specifically when MurAA is bound to both UDP-GlcNAc and Fosfomycin/PEP.
Figure 5. MurAA interacts with unphosphorylated IreB.

Thermal denaturation profiles of purified recombinant (A) wild-type MurAA (B) MurAA N188K and (C) MurAA A115T were plotted as the first derivative of the fluorescence emission as a function of temperature (-d(RFU)/dT) in the presence or absence of a 10-fold molar excess of IreB. Experiments were done in the presence or absence of the MurAA substrate UDP-GlcNAC (UNAG) and the PEP analog Fosfomycin. One representative profile from three technical replicates is shown for each condition and the average Tm (°C) is indicated. (D) Bacterial two-hybrid assay in which IreB and MurAA were cloned as genetic fusions to the T18 and T25 domains of Bordetella pertussis adenylate cyclase and introduced into the DHM1 strain of E. coli. Cells co-expressing the indicated combinations of fusions were spotted onto media containing X-gal. The known IreB-IreB self-interaction was used a positive control. Constructs containing the leucine zipper domains of GCN4 fused to the T18 and T25 domains (Zip) were used as negative controls. (E) Thermal denaturation profiles of MurAA in the presence of unphosphorylated IreB vs. IreB that had been phosphorylated in vitro using recombinant IreK kinase domain. IreB binding to MurAA was assessed in the presence of UNAG and Fosfomycin. One representative profile from three technical replicates is shown for each condition and the average Tm ± standard deviation is indicated.
To determine if the MurAA N188K or A115T substitutions altered the ability of MurAA to interact with IreB, we purified recombinant MurAA variants and analyzed them using TSAs. The MurAA N188K variant still displayed a UDP-GlcNAc/Fosfomycin-dependent shift in Tm (Figure 5b, solid black vs. dashed black), reflecting substrate binding and indicating that the N188K variant was properly folded. However, no IreB-dependent Tm shift occurred for the N188K variant in the presence or absence of substrates (dashed black vs. dashed purple and solid black vs. solid purple lines respectively), indicating that the N188K substitution impairs binding to IreB in vitro. In contrast, the MurAA A115T variant exhibited shifts in Tm similar to wild-type MurAA (Figure 5c), indicating that the A115T substitution did not affect IreB binding. In agreement with these in vitro findings, we were also able to detect the MurAA-IreB interaction using a bacterial two-hybrid approach, and found that the N188K, but not the A115T mutation in MurAA prevented the interaction (Figure 5d). Note that while the bacterial two-hybrid assay spot for the IreB-MurAA A115T interaction was not completely blue in both orientations, color development was still obvious relative to non-interacting controls (Figure 5d), consistent with retention of the IreB-MurAA interaction. Similar variability in the extent of blue color when testing interactions between the L. monocytogenes homologs was reported by Wamp et al., 2022. Together these results are consistent with the hypothesis that the MurAA N188K variant escapes IreB-dependent proteolysis in vivo by interfering with the IreB-MurAA protein-protein interaction.
Given that stabilization of MurAA in E. faecalis cells appears to be dependent on the phosphorylation state of IreB, we asked whether phosphorylation of IreB would alter its interaction with MurAA. To do this, we phosphorylated IreB in vitro using purified IreK kinase domain (Supplemental figure 13) and used the resulting phosphorylated IreB for TSAs. While this preparation of phosphorylated IreB does contain a small amount of IreK kinase domain, the equivalent molar amount of the IreK kinase domain on its own did not impact the MurAA thermal denaturation profile (data not shown). While MurAA showed the expected thermal shift in the presence of unphosphorylated IreB (Figure 5e, dashed black vs. dashed purple), no shift was detected in the presence of phosphorylated IreB (Figure 5e, dashed black vs. dashed orange). These data suggest that IreB facilitates MurAA proteolysis via a physical interaction between these two proteins, and that disruption of this interaction upon phosphorylation of IreB is what leads to the accumulation of MurAA. The MurAA homolog, MurAB, contributes to the proteolytic regulation of MurAA.
Wamp et al. demonstrated that in L. monocytogenes, deletion of LmMurZ (the homolog of E. faecalis MurAB) was associated with stabilization of LmMurA (Wamp et al., 2020). We similarly found that the steady-state abundance of MurAA is increased in the ΔmurAB mutant of E. faecalis (Figure 6a) due to stabilization of MurAA (Figure 6b). While loss of murAB is not associated with ceftriaxone hyper-resistance in otherwise wild-type E. faecalis (Vesić and Kristich, 2012; Table 3), it does increase the ceftriaxone resistance of the ireK deletion strain (Table 2).
Figure 6. MurAB promotes proteolysis of MurAA and interacts with IreB.
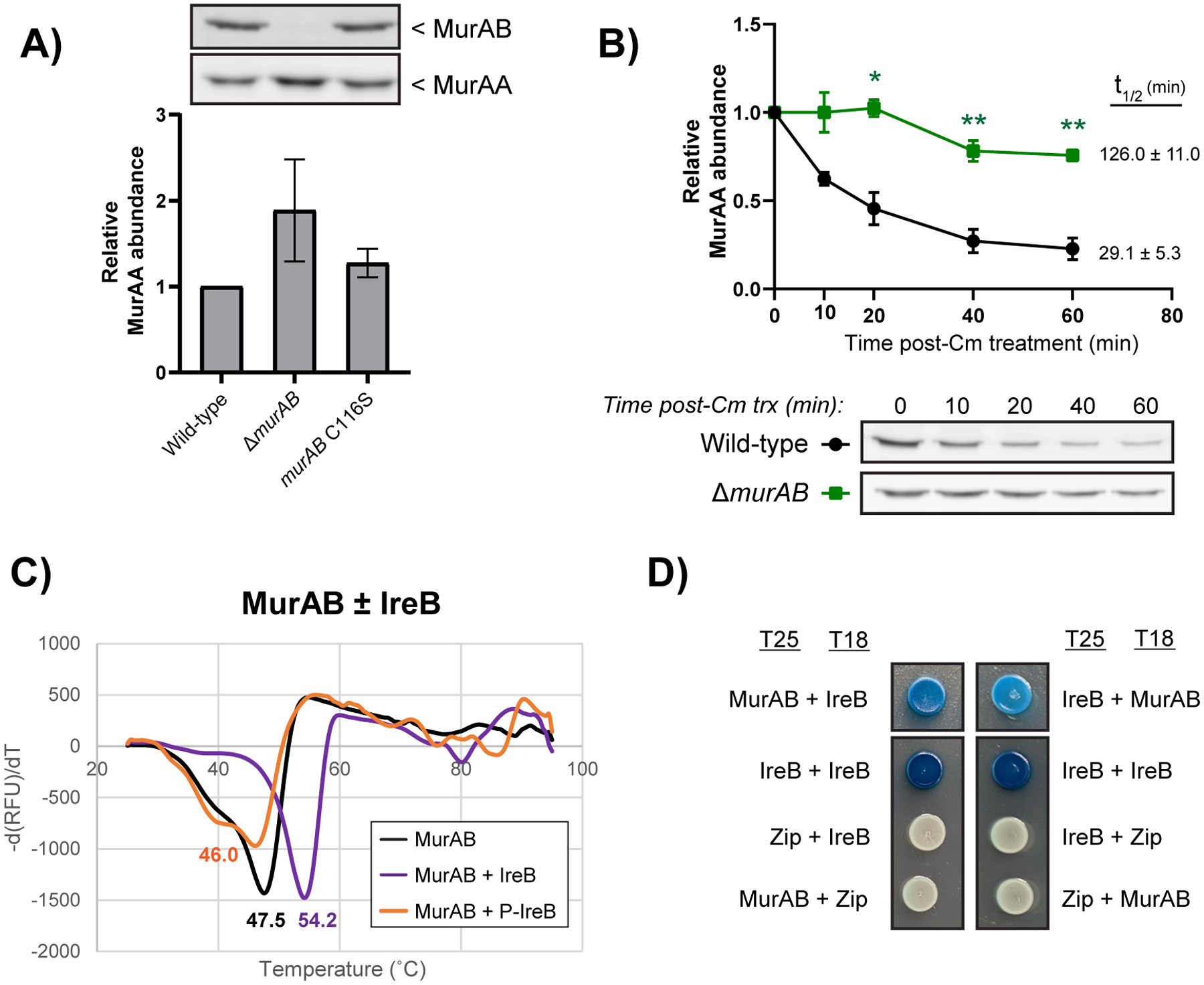
(A) Immunoblots of E. faecalis lysates from the indicated strains collected during exponential phase and subjected to SDS-PAGE. Membranes were probed with either anti-MurAB or anti-MurAA anti-serum. MurAA signal was normalized to total protein in each lane (Supplemental figure 6b and c) and quantified for three replicates, with error bars indicating standard deviation. One representative image is shown for each blot. (B) Protein stability assay in which the indicated strains were treated during exponential phase with chloramphenicol to block protein synthesis and MurAA signal was monitored over time. MurAA signal was normalized to total protein in each lane (Supplemental figure 8b) and compared to t0. MurAA half-life (t1/2) was extrapolated from normalized signal intensity over time and reported. Quantification was done for three biological replicates and error bars show standard error. Stars represent statistical differences relative to wild-type (*, p=0.01 to 0.05; **, p=0.001 to 0.01) based on Two-way ANOVA with Dunnett’s multiple comparisons. (C) Thermal denaturation profiles of purified recombinant MurAB in the presence or absence of phosphorylated or unphosphorylated IreB added in 10-fold molar excess. One representative profile from three technical replicates is shown for each condition and the average Tm (°C) is indicated. (D) Bacterial two-hybrid assay in which IreB and MurAB were cloned as genetic fusions to the T18 and T25 domains of Bordetella pertussis adenylate cyclase, introduced into the DHM1 strain of E. coli and spotted on media containing X-gal. The IreB-IreB self-interaction is used as a positive control while leucine zipper domains (Zip) were used for negative controls. Strains are wild-type, OG1; ΔmurAB, CM1; murAB C116S, CM40.
To determine if these effects were due to loss of MurAB-mediated UDP-N-acetylglucosamine 1-carboxyvinyl transferase activity or loss of the MurAB protein itself, we constructed a mutant in which MurAB is genetically inactivated via an amino acid substitution at the catalytic cysteine residue (C116S). The MurAB C116S substitution mutant did not appreciably alter the abundance of either MurAB or MurAA in cells (Figure 6a) or increase the ceftriaxone resistance level of the ireK deletion strain (Table 2). In fact, the ΔireK murAB C116S was more susceptible to cephalosporins than the ΔireK strain and readily acquired suppressor mutations (data not shown). Thus, it appears that the MurAB protein physically promotes degradation of MurAA independently of MurAB catalytic activity, possibly through direct interaction.
Given (i) the impact of MurAB on MurAA abundance; (ii) the similarity between MurAA and MurAB; and (iii) the interaction between MurAA and IreB, we hypothesized that MurAB might also interact with IreB. Indeed, we were able to detect an interaction between purified MurAB and IreB using TSAs (Figure 6c) and bacterial two-hybrid approaches (Figure 6d). Unlike the MurAA-IreB interaction, addition of substrates was not necessary for detection of the MurAB-IreB interaction in vitro. However, similar to MurAA, MurAB interacted only with the unphosphorylated form of IreB (Figure 6c), consistent with unphosphorylated IreB complexing with MurAB to promote proteolysis of MurAA in E. faecalis.
In L. monocytogenes, a protein called ReoY has also been demonstrated to be important for proteolytic regulation of MurAA (Wamp et al. 2020). Both E. faecalis and B. subtilis encode homologs of ReoY, but no ReoY homolog is encoded S. pneumoniae. The E. faecalis ReoY homolog, OG1RF_11272, was previously identified as a suppressor of a gastrointestinal tract colonization defect of the ΔireK strain (Banla et al, 2018). Here, we found that deletion of OG1RF_11272 leads to accumulation of MurAA to comparable levels as deletion of ireB (Supplemental figure 14) and increases the ceftriaxone MIC of the ΔireK strain (Table S1) in agreement with a role for OG1RF_11272 in IreK-dependent proteolytic regulation of MurAA in E. faecalis.
Accumulation of MurAA restores growth of the ΔireK strain during stress but is not sufficient for cephalosporin resistance.
The ΔireK mutant exhibits a growth defect in the presence of bile (Kristich, Wells and Dunny, 2007) and at elevated temperature (Minton et al., 2022). Here, we also observed a growth defect for the ireK deletion mutant relative to wild-type in the presence of 2 mM hydrogen peroxide (Figure 7). Deletion of either ireB or clpC, or introduction of the murAA N188K allele, in the ΔireK background provided either partial or complete rescue of the growth defects observed for the ΔireK mutant in each of these conditions (Figure 7), suggesting that increasing MurAA levels improves (and in some cases restores) the ability of the ΔireK strain to tolerate stress. Given this observation, and the ceftriaxone hyper-resistance phenotype seen for the ΔireB and murAA N188K mutants, we wondered whether MurAA accumulation (in the presence of IreK) might similarly improve growth in the presence of other stresses relative to a wild-type strain. In the presence of IreK, accumulation of MurAA largely did not impact growth under these conditions, however both the ΔireB and murAA N188K strains grew slightly worse than wild-type during hydrogen peroxide stress (Supplemental figure 15a) suggesting, unsurprisingly, that dysregulation of this pathway is detrimental under some conditions. Supplementation of the growth media with 20 mM MgCl2 also partially rescued the growth defect of the ΔireK strain during heat-stress (Supplemental figure 15b). Mg2+ has been proposed to have stabilizing effects on peptidoglycan through unknown mechanisms (Schulz et al., 2022; Tesson et al., 2022). Taken together, these results are consistent with a hypothesis that in the absence of IreK, cells cannot properly adapt to cell envelope stress, at least in part, due to their inability to modulate MurAA levels. However, we cannot rule out the possibility that other pleiotropic effects of ireK deletion contribute to the observed phenotypes.
Figure 7: Mutations that stabilize MurAA improve the growth defect of the ireK deletion strain during diverse stress conditions.

E. faecalis strains were grown in Mueller-Hinton broth under the indicated conditions. Strains were evaluated in biological triplicate and one representative growth curves is shown for each. Strains are wild-type, OG1; ΔireK, JL206; ΔireK ireB, CM52; ΔireK clpC, CM38, ΔireK murAA N188K, CM28; ΔireK murAA A115T, CM27.
We were surprised to find that mutants which accumulated MurAA to similar extents (Figure 2) exhibited different degrees of cephalosporin resistance (Table 2, Table 3). While all mutations that result in MurAA stabilization increase the ceftriaxone MIC of the ireK deletion strain to some extent (consistent with the finding that MurAA levels are linked to cephalosporin resistance), only deletion of ireB restores ceftriaxone resistance nearly back to that of the wild-type (Table 2). Similar differences in the magnitude of the ceftriaxone resistance phenotype were observed in otherwise wild-type cells: while the murAA N188K and ΔireB strains exhibit substantial hyper-resistant to cephalosporins, the ΔclpC, ΔclpP, ΔmurAB, and ΔOG1RF_11272 strains do not (Table 3, Supplemental Table 1). In fact, the murAB deletion strain is marginally less resistant to cephalosporins than wild-type. Given that the ClpCP complex likely regulates proteolysis of other targets in addition to MurAA, we considered that deletion of clpC or clpP might have other consequences that could indirectly block the ceftriaxone hyper-resistance phenotype. To test this, we introduced a murAA expression plasmid (which drives ceftriaxone hyper-resistance in wild-type cells) into the ΔclpC and ΔclpP strains and found that the plasmid-bearing strains were hyper-resistant (Table S2), indicating that there is no inherent block to ceftriaxone resistance in the absence of functional ClpCP complexes. Along the same lines, ectopic overexpression of MurAA was able to drive ceftriaxone hyper-resistance in the ΔmurAB strain, demonstrating that MurAB itself is also not required for hyper-resistance (Table S2). We conclude that stabilization of MurAA is necessary but not fully sufficient to explain the cephalosporin hyper-resistance phenotype observed in some strains.
The effect of the MurAA A115T mutation is independent from regulation by IreB
Like the N188K substitution, the other murAA substitution we chose to investigate from our suppressor selections, A115T, led to increased steady-state abundance of MurAA (Figure 2) and improved the PG synthesis defect (Figure 1a) and growth of the ireK deletion strain under diverse stress conditions (Figure 7). However, the A115T substitution was not associated with stabilization of MurAA to proteolysis (Figure 4) and did not block the interaction with IreB. (Figure 5c, Figure 5d). Further, unlike the ΔireB murAA N188K double mutant strain which phenocopied the ΔireB strain in terms of ceftriaxone resistance, the ΔireB murAA A115T mutant exhibited greater ceftriaxone resistance than either of the single mutants (Table 3), suggesting that deletion of ireB and the murAA A115T substitution act independently to impact resistance. No differences in murAA transcript level were observed for this mutant (Supplemental figure 9), leaving open the possibility that the A115T substitution has an effect on MurAA translation. Further investigation is required to explore this possibility.
Based on the proximity of the A115 residue to the active site of MurAA and its positioning at the base of the catalytic lid (Supplemental figure 2a) we also considered whether this mutation impacted the catalytic activity of the enzyme (independent of its effects on MurAA abundance). To test this, we purified both wild-type and mutant MurAA variants and evaluated enzymatic activity in vitro, revealing no differences in the reaction rates for MurAA A115T (nor MurAA N188K) relative to the wild-type protein (Supplemental figure 16). It remains possible, however, that the A115T substitution could have a more indirect consequence on activity such as altering the binding of a yet-unknown protein or small molecule inhibitor.
DISCUSSION
Regulation of MurA family proteins by PASTA kinases in low-GC Gram-positive bacteria has been revealed in recent years to be critical for proper cell physiology, antibiotic resistance, and virulence. Through their work in the rod-shaped L. monocytogenes, Wamp and colleagues demonstrated PASTA-kinase dependent control of MurA proteolysis by ClpCP (Wamp et al., 2020), which built upon earlier findings by Rismondo et al. that established LmMurA as a ClpCP substrate (Rismondo, Bender and Halbedel, 2017). Kelliher et al. (2021) and Wamp et al (2022) subsequently demonstrated that modulation of LmMurA abundance via this pathway is a point of regulation of PG synthesis and is critical for adaptation during intracellular growth of L. monocytogenes. Regulation of MurA family proteins by PASTA kinases has now been shown to occur in another rod-shaped bacterium, B. subtilis (Wamp et al., 2020; Sun, Hürlimann and Garner, 2023), and in the spherical S. aureus (Kelliher et al., 2021). However, in the ovoid bacterium S. pneumoniae, Tsui, Joseph, and colleagues recently demonstrated that while SpMurA and SpMurZ appear to be regulated by PASTA kinase signaling, they are not subject to ClpCP-dependent proteolysis, consistent with a distinct regulatory mechanism in this organism (Tsui et al., 2023). Our results reveal that E. faecalis, another ovoid bacterium, does not share the mechanism used by S. pneumoniae and instead employs a PASTA-kinase regulated ClpCP-dependent proteolysis mechanism to regulate its primary MurA family member (MurAA).
We used unbiased genetic approaches to reveal mutations in murAA that restored cephalosporin resistance to strains lacking IreK activity, or enabled overexpression of constitutively active IreB (IreK substrate) in the absence of IreK. Thus, MurAA acts downstream of IreK/IreB in E. faecalis consistent with our previous observations (Vesić and Kristich, 2012). Further studies revealed that IreK signaling regulates ClpCP-dependent proteolysis of MurAA through IreB (Figure 8), analogous to the mechanism in rod-shaped L. monocytogenes (Wamp et al., 2020, 2022; Kelliher et al., 2021) and in contrast to the non-ClpCP dependent mechanism proposed in the ovoid S. pneumoniae (Tsui et al., 2023). E. faecalis, but not S. pneumoniae (Tsui et al. 2023), also encodes a homolog of LmReoY (OG1RF_11272), which was demonstrated to be important for regulation of LmMurA proteolysis by ClpCP (Wamp et al. 2020) and impacts MurAA levels in E. faecalis (Supplemental Figure 14), further supporting a shared mechanism for MurAA regulation in E. faecalis and L. monocytogenes, but not S. pneumoniae. While E. faecalis and S. pneumoniae share the same ovoid cell morphology, they are evolutionarily distant and possess distinct metabolisms. Our results reveal that cell shape does not appear to be a useful predictor for the regulatory strategy used to control the activity of MurA family enzymes. Importantly, we demonstrated activity of the IreK-IreB-ClpCP regulatory pathway in wild-type cells exposed to physiologically relevant stimuli that trigger enhanced IreK activity. Namely, we demonstrate that MurAA abundance is impacted by exposure of E. faecalis cells to ceftriaxone and provide direct evidence that phosphorylation of IreB disrupts the MurAA-IreB interaction. We also provide additional insight into the role of MurAB in regulation of MurAA proteolysis, a major gap in the current understanding of this pathway. Further, and in contrast to what has been suggested in L. monocytogenes (Wamp et al., 2022), our results suggest that regulation of MurAA abundance in E. faecalis may not be the sole critical regulatory point for IreK-signaling downstream of IreB (see discussion below).
Figure 8. Model of MurAA regulation by IreK signaling in E. faecalis.

(A) When IreK is absent or not activated (such as during stationary phase), IreB is fully active and interacts with both MurAA and MurAB. These interactions promote degradation of MurAA by ClpCP which results in downregulation of peptidoglycan substrate flux. (B) During cell wall stress, IreK, and subsequently IreB, become phosphorylated, blocking the interaction with MurAA and leading to an accumulation of MurAA and release of MurAB. Accumulation of MurAA increases substrate flux into the PG synthesis pathway. Proteolysis of MurAA can also be blocked by genetic disruption of ClpCP, loss of MurAB and mutation in murAA (N188K) that blocks the MurAA-IreB interaction. During unstressed exponential-phase growth IreK and IreB are partially phosphorylated. MurAA is retained at sufficient levels to drive flux into the PG synthesis pathway to support cell growth but is not fully released from inhibition potentially to prevent unintended consequences of excessive PG synthesis.
Upon challenge of a ΔireK strain with ectopic overexpression of ireB (which is normally not tolerated due to constitutive activation of IreB in the absence of IreK) we identified an asparagine to lysine substitution at residue 188 of MurAA that stabilized the MurAA protein by blocking a physical interaction with IreB. A direct protein-protein interaction between the L. monocytogenes homologs of MurAA and IreB has previously been demonstrated. Mutations in LmMurA that stabilize the protein in vivo disrupted this interaction when tested via bacterial two-hybrid assay but did not block the interaction in vitro under the conditions tested (Wamp et al., 2022). Here, we confirmed that stabilization of MurAA is associated specifically with disruption of a direct physical interaction between MurAA and IreB, because the MurAA N188K mutant is stabilized in vivo and no longer interacts with IreB in vitro when tested via thermal shift assay using purified proteins. We also found that phosphorylation of IreB by IreK similarly blocks the physical interaction of IreB with MurAA, which has not been previously demonstrated. These data confirm that a direct physical interaction between IreB and MurAA is required for proteolysis of the PG synthesis enzyme and importantly that activation of IreK and subsequent phosphorylation of IreB regulates this pathway by toggling the physical interaction between IreB and MurAA “on” and “off”.
We also found that addition of the MurAA substrate UDP-GlcNAc and either PEP (the other MurAA substrate) or Fosfomycin (a non-hydrolyzable PEP-analog) was required to detect the interaction between MurAA and IreB in vitro by thermal shift assay (Figure 5, Supplemental Figure 12a), suggesting that substrate loading is a prerequisite for this interaction to occur. It is possible that substrate availability may provide an additional layer of regulation for IreB-dependent proteolysis of MurAA. Detection of the LmMurA-ReoM interaction in vitro by Wamp et al. did not require addition of substrates, although the impact of substrate loading was not explicitly tested (Wamp et al., 2022). Possibly, addition of substrates would help to uncover differences in ReoM-binding that were expected to be seen for wild-type versus the LmMurA stabilizing mutants.
We previously demonstrated that IreB forms domain-swapped dimers in vitro and in vivo (Hall et al., 2017). Mutations that disrupted dimer formation led to loss of IreB function and reduced steady-state abundance of IreB, suggesting that monomeric IreB is not stable in cells. Based on this, we hypothesize that IreB binds MurAA and MurAB as a dimer and depict it as such in our model (Figure 8). We attempted to evaluate whether dimerization was a requirement for these interactions by performing bacterial two-hybrid assays with non-dimerizing mutants of IreB (M73R and L81R; Hall et al., 2017). While these non-dimerizing IreB mutants did not interact with MurAB (data not shown), we could not confirm that the IreB mutants were actually expressed and stable in this assay. It remains possible that an interaction between IreB and another protein could stabilize the monomeric form of IreB, but this requires further investigation. It remains unclear exactly how IreB binding to MurAA helps facilitate its proteolysis, but it has been proposed that IreB may act as an adaptor for targeting to ClpCP (Wamp et al., 2020). Further, while MurAB homologs have been previously demonstrated to be important for this process (Rismondo, Bender and Halbedel, 2017; Wamp et al., 2020, 2022), the specific role of MurAB has been even less clear. Here, we provide the first evidence that like MurAA, MurAB can also directly interact with IreB, and this interaction specifically occurs when IreB is unphosphorylated. Moreover, MurAB-dependent regulation of MurAA proteolysis does not require catalytic activity of MurAB. Instead, we hypothesize that unphosphorylated IreB must physically interact with MurAB and that the IreB-MurAB complex is required to promote MurAA proteolysis. Such a requirement that MurAB is physically present in the complex and presumably properly folded (which we assume is required for interaction with IreB) could serve as a safeguard to prevent the cell from destroying all of its capacity to initiate PG synthesis by ensuring that some basal level of peptidoglycan synthesis can still occur (via MurAB) to sustain the cell during MurAA downregulation (i.e. MurAA cannot be degraded if MurAB levels are insufficient).
The changes we observed in MurAA steady-state abundance during exponential growth upon disruption of the proteolytic cascade were modest, especially relative what has been reported in L. monocytogenes (Rismondo, Bender and Halbedel, 2017; Wamp et al., 2020). Although notably, Sun and colleagues recently reported similarly modest increases in MurAA levels upon deletion of clpC and ireB homologs in B. subtilis (Sun, Hürlimann and Garner, 2023) and in S. pneumoniae, chromosomal gene duplications of MurZ or MurA were sufficient to overcome phenotypes associated with loss of the PASTA-kinase, StkP (Tsui et al., 2023). More substantial differences were observed in these strains in stationary phase, which seems consistent with the fact that in wild-type cells, IreK (and thus IreB) is largely unphosphorylated during stationary phase, in contrast to the partially phosphorylated state even in the absence of stress during exponential phase (Labbe and Kristich, 2017). MurAA, therefore, is already partially stabilized during normal conditions in exponential-phase cells (leading to more modest effects of stabilizing mutations), in contrast to stationary phase. However, upon exposure of exponentially growing cells to cell wall stress (such as cell wall acting antibiotics), IreK becomes more highly activated (Labbe and Kristich, 2017) resulting in increased stabilization of MurAA. Hence it appears that even during otherwise unstressed exponential growth the cells possess ‘excess’ capacity to ramp up cell wall synthesis in times of need. Sun and coworkers recently described a similar phenomenon in Bacillus subtilis mediated by the B. subtilis PASTA kinase pathway (Sun, Hürlimann and Garner, 2023). In any case, our data suggest that modulating MurAA abundance within a narrow window is sufficient to drive measurable changes in the phenotype of the cells. Mutations that lead to similar minor increases in MurAA steady-state abundance (murAA N188K, ΔclpC/P, ΔireB) provide similar improvement to ΔireK strain in terms of PG substrate flux, growth, and antibiotic resistance. Overall, this seems to suggest that E. faecalis (and perhaps B. subtilis) is highly sensitive to the cellular abundance of MurAA.
Furthermore, we found that stabilization of MurAA not only increased ceftriaxone resistance of the ireK deletion strain, but also restored growth under various stress conditions (Figure 7, Table 2). Additionally, loss of OG1RF_11272 (the E. faecalis ReoY homolog), which was previously demonstrated to restore the ability of the ireK deletion strain to colonize the gastrointestinal tract (Banla et al., 2018), increased MurAA levels (Supplemental Figure 14). Together, these findings may suggest that IreK-dependent regulation of MurAA levels may be one mechanism by which IreK supports adaptation to stress more broadly; however loss of IreK likely has pleiotropic effects whose contributions to the observed phenotypes remain unknown. Our observation that IreK is important for growth during H2O2-mediated stress was not previously known. Generation of reactive oxygen species following Fenton chemistry is known to cause damage to DNA, lipids and proteins (reviewed in Ezraty et al. 2017) which could have both direct and indirect consequences on cell envelope integrity such as via damage to membrane lipids or to proteins required for ongoing cell wall synthesis or maintenance during growth. In that way, the requirement for IreK during H2O2-mediated stress would be consistent with prior observations which suggest that the PASTA kinase is broadly important for sensing disruption of the cell envelope (Banla et al., 2018; Hall et al., 2013, Labbe and Kristich, 2017).
In any case, the fact that the murAA N188K, murAA A115T and ΔireB strains all exhibit H2O2-dependent growth defects relative to wild-type in the presence of IreK (Supplemental figure 15) is consistent with an evolutionary disadvantage associated with MurAA being fully uninhibited under some stress conditions. Similarly, L. monocytogenes strains that accumulate MurAA grow slower in the presence of high salt (Rouquette et al., 1996; Wamp et al., 2020, 2022), and overexpression of SpMurZ (the primary UDP-N-acetylglucosamine enolpyruvyl transferase in S. pneumoniae) is detrimental for growth even under non-stress conditions (Tsui et al., 2023). Together, this suggests that tight regulation of MurAA abundance is critical for both normal growth and the cellular response to stress, and therefore regulation by the IreK PASTA kinase is likely important for fitness. Consistent with this, co-culture competition experiments previously demonstrated that a strain with dysregulated IreK signaling exhibits a fitness defect (Kristich et al., 2011).
It has been suggested that regulation of LmMurA and SpMurZ/MurA is the critical role of signaling via the IreK and IreB homologs in L. monocytogenes (Wamp et al., 2022) and S. pneumoniae (Tsui et al., 2023) respectively. Our data demonstrating that stabilization of MurAA is sufficient to completely rescue growth of the ΔireK strain during heat and bile acid stress aligns with this. However, we found that while deletion of ireB restored the cephalosporin resistance level of the ΔireK strain nearly to wild-type, the clpC deletion had more modest effects on resistance (Table 2). In an otherwise wild-type background, deletion of ireB, but not clpC supports ceftriaxone hyper-resistance (Table 3). This is in contrast to what has been reported in L. monocytogenes where deletion of both clpC and reoM (the IreB homolog) leads to increased ceftriaxone resistance relative to wild-type, with a more pronounced hyper-resistance phenotype in the ΔclpC strain (Wamp et al., 2020). Neither the murAB nor OG1RF_11272 deletion strains of E. faecalis were found to have increased ceftriaxone resistance either, (Table 3, Table S1) despite accumulation of MurAA (Figure 6, Supplemental figure 14), suggesting that this observation is not directly related to other consequences of loss of ClpC. One potential explanation is that IreB may regulate additional targets important for mitigation of ceftriaxone stress through ClpC-independent mechanisms. Given the interaction between MurAB and IreB, it seems possible that IreB may also provide some regulation of MurAB, albeit not via regulation of proteolysis. Further, it remains unclear why the murAB deletion strain appears to be slightly less resistant to cephalosporins than the wild-type strain despite stabilization of MurAA, especially given that ectopic overexpression of murAA supports hyper-resistance in this strain (Table S2). Possibly, MurAB plays a larger role in cephalosporin resistance than we have previously appreciated that is masked by non-physiologic levels of murAA. Additional studies are warranted to explore the relationship between MurAB, IreB and cephalosporin resistance.
Finally, the impact of the murAA A115T mutation remains elusive. While steady-state levels of MurAA were increased in the A115T strain (Figure 2a), MurAA was not found to be protected from proteolysis (Figure 4) nor were transcript levels increased (Supplemental figure 9). Tsui, Joseph and colleagues recently reported that loss of the RNA-binding proteins KhpA and KhpB increased SpMurZ protein levels without changing transcript levels for SpMurZ (Tsui et al., 2023), although the specific mechanism is unknown. Possibly, similar post-transcriptional regulation of MurAA occurs in E. faecalis as well. Another possibility is that the A115T mutation has an indirect effect on MurAA activity that was not captured by our in vitro enzymatic assay. The E. coli MurAA homolog has been demonstrated to be feedback regulated by UDP-MurNAc, the product of the subsequent step in the PG synthesis pathway that is catalyzed by MurB (Mizyed et al., 2005). Enterococcus faecium MurAA has recently been suggested to purify in complex with UDP-MurNAc (Zhou et al., 2023), lending credence to the idea that this mode of regulation may also occur in enterococci, but additional studies are necessary to evaluate this possibility and the potential impact of the A115T mutation on this mechanism of regulation.
EXPERIMENTAL PROCEDURES
Bacterial strains and growth conditions.
Bacterial strains and plasmids used in this study are listed in Table S3. E. coli strains were grown in Lysogeny broth (LB) or agar. Both TOP10 and DH5α were used as routine cloning hosts. E. faecalis strains were propagated on Mueller-Hinton broth (MHB) or agar (prepared according to the manufacturer’s instructions; Difco) at 30 ℃ unless otherwise indicated. Antibiotics were included when necessary for maintenance of plasmids at the following concentrations: chloramphenicol, 20 μg ml−1 (E. coli) or 10 μg ml−1 (E. faecalis); kanamycin, 50 μg ml−1; ampicillin, 100 μg ml−1; erythromycin, 100 μg ml−1 (E. coli) or 10 μg ml−1 (E. faecalis).
Growth during stress was evaluated in microbroth cultures using a Bioscreen C instrument with OD600 measurements taken every 15 min following brief shaking over a course of 24 hr. Stationary-phase cultures were diluted to a normalized density of OD600 = 4×10−5 (~1×105 CFU) and grown in MHB at either 37 or 45 °C and in the presence of the indicated additives.
Genetic manipulation of enterococci.
Mutant strains of E. faecalis were generated as previously described (Kellogg et al., 2017), using pJH086 a temperature-sensitive, counter-selectable allelic exchange plasmid. Counterselection plates contained 20% sucrose and p-Cl-Phe. Deletion alleles were generated in-frame. Fragments of genomic DNA upstream and downstream of the gene of interest which included a small number of codons at the beginning and end of each gene were amplified and introduced into pJH086 by isothermal assembly (Gibson et al., 2009). Specific details about deletion alleles are included in Supplemental table S3. The murAA N188K, murAA A115T and clpP A46E alleles were amplified from suppressor strains JL400, JL600 and JL350 respectively for introduction into pJH086. The murAB C116S allele was generated by amplifying the murAB gene in two fragments (including upstream and downstream sequence) with the mutated residues included the primers used to generate the junction point of the two fragments. All mutant or complemented strains were constructed independently at least twice and analyzed to ensure that their phenotypes were concordant.
Generation of plasmids.
Unless otherwise indicated, recombinant plasmids were generated using isothermal assembly (Gibson et al., 2009) and inserts were sequenced in their entirety to ensure the absence of any undesired mutations. Expression of wild-type and mutant MurAA, MurAB and IreK-n for protein purification was accomplished using the IPTG (isopropyl β-D-1-thiogalactopyranoside)-inducible expression vector pET28b. IreB was expressed from the IPTG-inducible vector, pET28a-His-smt3, which encodes a cleavable His6-SUMO fusion. Plasmids used for expression of murAA A115T (pCAM24) and murAA N188K (pCAM28) were generated via Q5 site-directed mutagenesis (New England BioLabs) of pDV20. Plasmids used for bacterial two-hybrid assays were pUT18c and pKT25 or pKNT25-based and included fusions to the c-terminus of cya T18 and T25 domains or n-terminus of the T25 domain respectively. Wild-type and mutant alleles were amplified from genomic DNA for the generation of these plasmids.
Isolation of suppressor mutants.
(A). Selection 1.
Independent stationary-phase cultures of strain BL102, which encodes a catalytically inactive variant (K41R) of IreK and is therefore susceptible to ceftriaxone, were spread onto MHB agar supplemented with inhibitory concentrations of ceftriaxone and incubated at 37°C. Ceftriaxone-resistant colonies were retested to confirm that they retained increased ceftriaxone resistance after passage in the absence of drug. After verifying that the ireB, ireP and ireK genes did not contain any mutations (other than ireK K41R) by Sanger sequencing of PCR amplicons, genomic DNA (gDNA) was isolated from suppressor mutants using Qiagen genomic tip 100/G columns according to the manufacturer’s instructions following an initial incubation with lysozyme (10 mg ml−1) and mutanolysin (500 U mL−1) at 37°C for 10 min. Next-generation Illumina sequencing of the gDNA was performed at Elim Biopharm. Sequencing reads were mapped to the E. faecalis OG1RF genome and SNVs found in the suppressor mutant genomes that were not present in BL102 were verified by Sanger sequencing of PCR amplicons from the corresponding genomic loci.
(B). Selection 2.
Conjugation was used to introduce an ireB-expressing plasmid into the ireK-deficient CK119 strain of E. faecalis OG1RF. IreB-strep was expressed under the control of a constitutively active promoter from pCJK227 which contains an oriT that allows mobilization by conjugation from E. faecalis donors carrying pCF10–101 (non-mobilizable derivative of pCF10; (Kristich, Wells and Dunny, 2007)). pCJK227 was mobilized from a rifampicin-susceptible donor strain CK111/pCF10–101 into the rifampicin-resistant CK119 on BHI agar. Cells were then grown on Brain-Heart Infusion (BHI) agar supplemented with 200 μg ml−1 rifampicin and 10 μg ml−1 erythromycin to select for recipient cells (CK119) carrying pCJK227. After confirming the absence of mutations in IreB by Sanger sequencing of PCR amplicons, genomic sequencing was performed as described for selection 1.
Antimicrobial susceptibility assays.
MICs for ceftriaxone were determined using a microbroth dilution method. Briefly, bacteria from stationary-phase cultures were inoculated into wells containing 2-fold serial dilutions of antibiotic in MHB at a normalized density of OD600 = 4×10−5 (~1×105 CFU). Plates were incubated in a Bioscreen C plate reader at 37 ℃ for 24 h with brief shaking before each measurement. The optical density at 600 nm (OD600) was determined every 15 min, and the lowest concentration of antibiotic that prevented growth was recorded as the MIC.
Peptidoglycan synthesis assays.
Uptake of [14C] GlcNAc into SDS-insoluble peptidoglycan was monitored as described previously (Bisicchia et al., 2007; Mesnage et al., 2008; Djorić, Little and Kristich, 2020). Briefly, cells were grown to exponential phase in MHB then diluted 6-fold into prewarmed media containing 0.33 μCi ml−1 [14C] GlcNAc (Perkin Elmer) and incubated at 37°C. At intervals, aliquots were mixed with equal volume of 0.2% SDS, pelleted and radioactivity in the SDS-insoluble pellets was measured using scintillation counting and normalized to optical density measurements taken in triplicate for parallel unlabeled cultures.
Immunoblot analysis.
Stationary-phase cultures were diluted to OD600 = 0.01 in MHB (supplemented with 0.5X MIC ceftriaxone when indicated) and incubated at 37 ℃ with shaking. For standard immunoblots, cultures were grown until OD600 ~0.2. At this point, an aliquot was removed for exponential phase analysis and harvested by mixing with an equal volume of cold EtOH/acetone (1:1). When stationary phase samples were collected, the remaining culture was allowed to grow overnight and harvested the next day in the same manner. For protein stability assays cultures were grown until OD600 ~0.15, an aliquot was removed and harvested with EtOH/acetone for t0, and the remaining culture was treated with 150 μg ml−1 chloramphenicol. Aliquots were then harvested at 10, 20, 40 and 60 min post-chloramphenicol treatment. After harvesting, cells were pelleted by centrifugation and washed once with water. To prepare total cell lysates, bacteria were suspended in lysozyme buffer (10 mM Tris, 50 mM NaCl, 20% sucrose [pH 8]), normalized to equivalent OD600, treated with lysozyme (5 mg ml−1) for 30 min at 37°C, mixed with Laemmli SDS sample buffer containing 100 mM DTT and boiled for 5 min. After SDS-PAGE, proteins were transferred to polyvinylidene difluoride (PVDF; Bio-Rad) using a Trans-blot Turbo system (Bio-Rad) and incubated with No-Stain Protein Labeling reagent (Invitrogen) for visualization of total protein prior to blocking with 5% skim milk in Tris-buffered saline. Membranes were then probed with rabbit anti-MurAA custom polyclonal antiserum followed by goat anti-rabbit, Alexa Fluor 647-conjugated secondary antibody (Invitrogen). Imaging of membranes was performed using an Amersham Typhoon 5 (Cytiva, Marlborough, MA).
Quantification of immunoblots was performed using AzureSpot software with minimum profile background subtraction. The MurAA signal was normalized to total protein in each respective lane. For standard immunoblots, the average x-fold change relative to wild-type for each strain is reported for an n of 3 individual replicates. Error bars reflect standard deviation. For protein stability assays, the normalized MurAA signal at each timepoint was reported relative to the normalized signal for each respective strain at t0. MurAA half-life was calculated by fitting the normalized signal intensity over time to first order decay as described in Belle et al. 2006. Mean +/− SEM is reported for stability assay data. Statistical analysis was performed using GraphPad Prism.
Protein purification.
Recombinant MurAA-His6 (wild-type, N188K and A115T) was purified from E. coli BL21(DE3) cells. Stationary phase cultures were diluted to OD600 = 0.01 in LB supplemented with 50 μg mL−1 kanamycin (for plasmid maintenance) and incubated at 37°C with shaking. At OD600 = 0.6 cultures were treated with 0.4 mM IPTG and incubated for 3hr at 30°C. Cells were pelleted by centrifugation, resuspended in binding buffer (50 mM Na2HPO4, 300 mM NaCl, 5 mM imidazole [pH=7.3]) supplemented with 1x Halt protease inhibitor cocktail (ThermoFisher) and lysed using a French press. Whole cell lysates were clarified using centrifugation and filtered with a 0.2 μm syringe filter. The filtered supernatant was then added to a 3 mL bed volume of Cobalt-charged TALON resin (Takara) pre-loaded into a column and equilibrated with binding buffer. The resin was washed 3x with 8-column volumes of wash buffer (50 mM Na2HPO4, 300 mM NaCl, 10 mM imidazole [pH=7.3]) and the bound protein was eluted with 5-column volumes of elution buffer (50 mM Tris, 300 mM NaCl, 100 mM EDTA [pH=8]). The eluted sample was concentrated using an Amicon Ultra 15 mL, 10 kDa cut-off centrifugal concentrator and subjected to size-exclusion chromatography using a HiLoad 16/600 Superdex column (Cytiva, Marlborough, MA) with a 50 mM Tris, 300 mM NaCl, 20% glycerol [pH=8] buffer at a 0.9 mL min−1 flow rate. MurAA-His6 containing fractions were confirmed via SDS-PAGE analysis and pooled. The pooled sample was concentrated, aliquoted, flash frozen in a EtOH/dry ice slurry and stored at −80°C.
Recombinant MurAB-His6 was expressed in E. coli C43(DE3) cells. Transformations were achieved through heat-shock following the protocol provided by Lucigen. Cultures were grown to stationary phase in the presence of 0.2% glucose to prevent target protein expression prior to induction, then diluted 1:100 in LB supplemented with 50 μg mL−1 kanamycin (no glucose), grown to an OD600 ~0.8 and induced with 0.1 mM IPTG for 3 h at 30°C. Purification was as described above for MurAA.
His6-SUMO-IreB was expressed and purified from E. coli BL21(DE3) cells as described previously (Hall 2017) with modifications. Stationary phase cultures were diluted 1:100 in LB supplemented with 50 μg mL−1 kanamycin and grown at 37°C with shaking until OD600 ~0.7. IPTG was added at a final concentration of 0.4 mM and cultures were induced overnight at 30°C. Cultures were pelleted with centrifugation and resuspended in 50 mM Na2HPO4, 300 mM NaCl, 5 mM Imidazole [pH=7.4] buffer supplemented with 1x Halt protease inhibitor cocktail (ThermoFisher) and lysed using a French press. After lysis, purification steps proceeded at ambient temperature. Soluble, clarified lysates were batch-bound to a Ni-charged resin (GoldBio) which was then washed with 50 mM Na2HPO4, 300 mM NaCl, 20 mM Imidazole [pH=7.4] and eluted with 50 mM Na2HPO4, 300 mM NaCl, 500 mM Imidazole [pH=7.4]. His-GB1-Ulp1 protease and 1 mM DTT were added to the eluate for SUMO cleavage and the mixture was dialyzed overnight at ambient temperature against 2L 50 mM Na2HPO4, 350 mM NaCl [pH=6.6]. After dialysis the cleaved protein was loaded onto Ni-charged resin and the flow through was collected and concentrated using an Amicon Ultra 15 mL, 3 kDa cut-off centrifugal concentrator. Following removal of the His-tag, the concentrated protein was subjected to size-exclusion chromatography using a HiLoad 16/600 Superdex column (Cytiva, Marlborough, MA) with a 50 mM Na2HPO4, 350 mM NaCl [pH=6.6] buffer at 4°C. Fractions containing IreB were pooled, concentrated, and stored at room temperature.
His6-IreK-n (intracellular domain only) used for in vitro phosphorylation of recombinant IreB was purified as described previously (Minton et al., 2022).
Thermal shift assays.
Protocol was adapted from Foulquier and Galinier (2017). Purified, recombinant proteins (2.5 μM wild-type or mutant MurAA or MurAB) were combined with either IreB (2.5 or 25 μM as indicated) or equivalent volume IreB buffer (50 mM Na2HPO4, 350 mM NaCl [pH=6.6]) and 10X SYPRO Orange (Invitrogen), in a 50 mM Tris, 25 mM NaCl buffer [pH=7.5]. When indicated, UDP-N-acetylglucosamine (UNAG) and Fosfomycin were added at a final concentration of 1 mM. Reactions were heated at a rate 0.5°C/10sec to 95°C and using a Bio-Rad CFX Connect Real-Time PCR instrument and fluorescence measurements were taken at each 0.5°C increment. The first derivate of the fluorescence emission was plotted as a function of temperature and the Tm was determined as the lowest point of the curve. All thermal shift measurements were conducted in triplicate.
In vitro phosphorylation of IreB was performed as previously described (Minton 2022). Briefly, purified IreB was incubated in 100-fold molar excess with purified recombinant IreK-n (intracellular domain) in the presence of 5 mM MgCl2 in 50 mM Tris, 25 mM NaCl [pH=7.5]. 2 mM ATP was added to start the reaction which proceeded for 40 min at 37°C. Additional reactions were set up under the same conditions but in the absence of IreK (for non-phosphorylated IreB) or in the absence of both IreB and IreK (for no IreB reactions). SDS-PAGE followed by staining with Pro-Q Diamond Phosphoprotein Gel Stain (Invitrogen) and SYPRO Ruby Protein Gel Stain (Invitrogen) was used to confirm phosphorylation of IreB under these conditions when IreK was present and lack of phosphorylation in reactions that did not include IreK. The kinase and control reaction products were then added to 5 μM MurAA or MurAB such that IreB was at 10-fold molar excess in the presence of 10X SYPRO Orange and analysis proceeded as described above.
Bacterial two-hybrid.
Pairs of plasmids expressing a gene of interested fused to either the T18 or T25 fragments of Bordetella pertussis adenylate cyclase were co-electroporated into E. coli DHM1 cells which are adenylate cyclase-deficient. Three individual transformants for each combination were isolated then grown to stationary phase in LB supplemented with 50 μg mL−1 kanamycin, 100 μg mL−1 ampicillin, and 0.1 mM IPTG. Stationary-phase cultures were spotted onto LB agar supplemented with 50 μg mL−1 kanamycin, 100 μg mL−1 ampicillin, 0.1 mM IPTG and 150 μg mL−1 X-gal. Plates were incubated at ambient temperature for 5 days then photographed.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Nicole Minton and Matthew Bluma for use of purified IreK-n and IreB respectively as well as Dr. Dušanka Djorić and Dr. Stephanie Kellogg for the generation of plasmids used in this study. The pathway model was generated using BioRender.com under a license funded by the Medical College of Wisconsin Department of Microbiology and Immunology.
This study was supported in part by grants AI134660 and F31AI156980 from the National Institutes of Health (NIH). The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- Arbeloa A, Segal H, Hugonnet JE, Josseaume N, Dubost L, Brouard JP, Gutmann L, Mengin-Lecreulx D and Arthur M (2004) ‘Role of Class A Penicillin-Binding Proteins in PBP5-Mediated β-Lactam Resistance in Enterococcus faecalis’, J Bacteriol, 186(5), pp. 1221–1228. 10.1128/JB.186.5.1221-1228.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baddour LM, Wilson WR, Bayer AS, Fowler VG, Tleyjeh IM, Rybak MJ, Barsic B, Lockhart PB, Gewitz MH, Levison ME, Bolger AF, Steckelberg JM, Baltimore RS, Fink AM, O’Gara P and Taubert KA (2015) ‘Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications’, Circulation, 132, pp. 1435–1486. 10.1161/CIR.0000000000000296. [DOI] [PubMed] [Google Scholar]
- Banla IL, Kommineni S, Hayward M, Rodrigues M, Palmer KL, Salzman NH and Kristich CJ (2018) ‘Modulators of Enterococcus faecalis cell envelope integrity and antimicrobial resistance influence stable colonization of the mammalian gastrointestinal tract’, Infect Immun, 86(1), pp. 1–15. 10.1128/IAI.00381-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belle A, Tanay A, Bitincka L, Shamir R and O’Shea EK (2006) ‘Quantification of protein half-lives in the budding yeast proteome’, Proc Natl Acad Sci U S A, 103(35), pp. 13004–13009. 10.1073/pnas.0605420103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisicchia P, Noone D, Lioliou E, Howell A, Quigley S, Jensen T, Jarmer H and Devine KM (2007) ‘The essential YycFG two-component system controls cell wall metabolism in Bacillus subtilis ’, Mol Microbiol, 65(1), pp. 180–200. 10.1111/j.1365-2958.2007.05782.x. [DOI] [PubMed] [Google Scholar]
- Blake KL, O’Neill AJ, Mengin-Lecreulx D, Henderson PJF, Bostock JM, Dunsmore CJ, Simmons KJ, Fishwick CWG, Leeds JA and Chopra I (2009) ‘The nature of Staphylococcus aureus MurA and MurZ and approaches for detection of peptidoglycan biosynthesis inhibitors’, Mol Microbiol, 72(2), pp. 335–343. 10.1111/j.1365-2958.2009.06648.x. [DOI] [PubMed] [Google Scholar]
- Brandl K, Plitas G, Mihu CN, Ubeda C, Jia T, Fleisher M, Schnabl B, Dematteo RP and Pamer EG (2008) ‘Vancomycin-resistant enterococci exploit antibiotic-induced innate immune deficits’, Nature, 455(7214), pp. 804–807. 10.1038/nature07250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush K and Bradford PA (2016) ‘B-Lactams and B-lactamase inhibitors: an overview’, Cold Spring Harb Perspect Med, 6:a025247. 10.1101/cshperspect.a025247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeli Y, Eliopoulos GM and Samore MH (2002) ‘Antecedent treatment with different antibiotic agents as a risk factor for vancomycin-resistant Enterococcus’, Emerg Infect Dis, 8(8), pp. 802–807. 10.3201/eid0808.010418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djorić D, Little JL and Kristich CJ (2020) ‘Multiple low-reactivity class B penicillin-binding proteins are required for cephalosporin resistance in enterococci’, Antimicrob Agents Chemother, 64(4), pp. 1–13. 10.1128/AAC.02273-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donskey CJ, Chowdhry TK, Hecker MT, Hoyen CK, Hanrahan JA, Hujer AM, Hutton-Thomas RA, Whalen CC, Bonomo RA and Rice LB (2000) ‘Effect of antibiotic therapy on the density of vancomycin-resistance Enterococci in the stool of colonized patients’, N Engl J Med, 343(26), pp. 1925–1932. 10.1056/NEJM200012283432604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W, Brown JR, Sylvester DR, Huang J, Chalker AF, So CY, Holmes DJ, Payne DJ and Wallis NG (2000) ‘Two active forms of UDP-N-acetylglucosamine enolpyruvyl transferase in Gram-positive bacteria’, J Bacteriol, 182(15), pp. 4146–4152. 10.1128/JB.182.15.4146-4152.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezraty B, Gennaris A, Barras F and Collet J-F (2017) ‘Oxidative stress, protein damage and repair in bacteria’, Nature Rev Microbiol, 15, pp. 385–396. 10.1038/nrmicro.2017.26 [DOI] [PubMed] [Google Scholar]
- Foulquier E and Galinier A (2017) ‘YvcK, a protein required for cell wall integrity and optimal carbon source utilization, binds uridine diphosphate-sugars’, Sci Rep, 7(1), pp. 1–11. 10.1038/s41598-017-04064-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frees D, Savijoki K, Varmanen P and Ingmer H (2007) ‘Clp ATPases and ClpP proteolytic complexes regulate vital biological processes in low GC, Gram-positive bacteria’, Mol Microbiol, 63(5), pp. 1285–1295. 10.1111/j.1365-2958.2007.05598.x. [DOI] [PubMed] [Google Scholar]
- Gaidenko TA, Kim T-J and Price CW (2002) ‘The PrpC Serine-Threonine Phosphatase and PrkC Kinase Have Opposing Physiological Roles in Stationary-Phase Bacillus subtilis Cells’, J Bacteriol, 184(22), pp. 6109–6114. 10.1128/JB.184.22.6109-6114.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerth U, Kock H, Kusters I, Michalik S, Switzer RL and Hecker M (2008) ‘Clp-dependent proteolysis down-regulates central metabolic pathways in glucose-starved Bacillus subtilis’, J Bacteriol, 190(1), pp. 321–331. 10.1128/JB.01233-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA and Smith HO (2009) ‘Enzymatic assembly of DNA molecules up to several hundred kilobases’, Nat Methods, 6(5), pp. 343–345. 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Hall CL, Tschannen M, Worthey EA and Kristich CJ (2013) ‘IreB, a Ser/Thr kinase substrate, influences antimicrobial resistance in Enterococcus faecalis’, Antimicrob Agents Chemother, 57(12), pp. 6179–6186. 10.1128/AAC.01472-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall CL, Lytle BL, Jensen D, Hoff JS, Peterson FC, Volkman BF and Kristich CJ (2017) ‘Structure and Dimerization of IreB, a Negative Regulator of Cephalosporin Resistance in Enterococcus faecalis’, J Mol Biol, 429(15), pp. 2324–2336. 10.1016/j.jmb.2017.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland TL, Baddour LM, Bayer AS, Hoen B, Miro JM and Fowler VG (2016) ‘Infective endocarditis’, Nat Rev Dis Primers, 2(16059). 10.1038/nrdp.2016.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck BL and Rice LB (2012) ‘Intrinsic and acquired resistance mechanisms in Enterococcus’, Virulence, 3(5), pp. 421–433. 10.4161/viru.21282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannetta AA, Minton NE, Uitenbroek AA, Little JL, Stanton CR, Kristich CJ and Hicks LM (2021) ‘IreK-Mediated, Cell Wall-Protective Phosphorylation in Enterococcus faecalis’, J Proteome Res, 20(11), pp. 5131–5144. 10.1021/acs.jproteome.1c00635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P and Hassabis D (2021) ‘Highly accurate protein structure prediction with AlphaFold’, Nature, 596(7873), pp. 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedar GC, Brown-Driver V, Reyes DR, Hilgers MT, Stidham MA, Shaw KJ, Finn J and Haselbeck RJ (2008) ‘Comparison of the essential cellular functions of the two murA genes of Bacillus anthracis’, Antimicrob Agents Chemother, 52(6), pp. 2009–2013. 10.1128/AAC.01594-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelliher JL, Grunenwald CM, Abrahams RR, Daanen ME, Lew CI, Rose WE and Sauer J-D (2021) ‘PASTA kinase-dependent control of peptidoglycan synthesis via ReoM is required for cell wall stress responses, cytosolic survival, and virulence in Listeria monocytogenes’, PLoS Pathog, 17(10), p. e1009881. 10.1371/journal.ppat.1009881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg SL and Kristich CJ (2018) ‘Convergence of PASTA kinase and two-component signaling in response to cell wall stress in Enterococcus faecalis’, J Bacteriol, 200(12), pp. 1–14. 10.1128/JB.00086-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg SL, Little JL, Hoff JS and Kristich CJ (2017) ‘Requirement of the CroRS two-component system for resistance to cell wall-targeting antimicrobials in Enterococcus faecium’, Antimicrobial Agents and Chemotherapy. 61(5), e02461–16. 10.1128/AAC.02461-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kock H, Gerth U and Hecker M (2004a) ‘MurAA, catalysing the first committed step in peptidoglycan biosynthesis, is a target of Clp-dependent proteolysis in Bacillus subtilis’, Mol Microbiol, 51(4), pp. 1087–1102. 10.1046/j.1365-2958.2003.03875.x. [DOI] [PubMed] [Google Scholar]
- Kock H, Gerth U and Hecker M (2004b) ‘The ClpP peptidase is the major determinant of bulk protein turnover in Bacillus subtilis’, J Bacteriol, 186(17), pp. 5856–5864. 10.1128/JB.186.17.5856-5864.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristich CJ, Little JL, Hall CL and Hoff JS (2011) ‘Reciprocal regulation of cephalosporin resistance in Enterococcus faecalis’, mBio, 2(6), e00199–11. 10.1128/mBio.00199-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristich CJ, Rice LB and Arias CA (2014) ‘Enterococcal Infection - Treatment and Antibiotic Resistance’, in Enterococci: From Commensals to Leading Causes of Drug Resistant Infection. [Google Scholar]
- Kristich CJ, Wells CL and Dunny GM (2007) ‘A eukaryotic-type Ser/Thr kinase in Enterococcus faecalis mediates antimicrobial resistance and intestinal persistence’, Proc Natl Acad Sci U S A, 104(9), pp. 3508–3513. 10.1073/pnas.0608742104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger E, Witt E, Ohlmeier S, Hanschke R and Hecker M (2000) ‘The Clp proteases of Bacillus subtilis are directly involved in degradation of misfolded proteins’, J Bacteriol, 182(11), pp. 3259–3265. 10.1128/JB.182.11.3259-3265.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbe BD and Kristich CJ (2017) ‘Growth- and stress-induced PASTA kinase phosphorylation in Enterococcus faecalis’, J Bacteriol, 199(21), e00363–17. 10.1128/JB.00363-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layton CJ and Hellinga HW (2011) ‘Quantitation of protein-protein interactions by thermal stability shift analysis’, Protein Sci, 20(8), pp. 1439–1450. 10.1002/pro.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magill SS, O’Leary E, Janelle SJ, Thompson DL, Dumyati G, Nadle J, Wilson LE, Kainer MA, Lynfield R, Greissman S, Ray SM, Beldavs Z, Gross C, Bamberg W, Sievers M, Concannon C, Buhr N, Warnke L, Maloney M, Ocampo V, Brooks J, Oyewumi T, Sharmin S, Richards K, Rainbow J, Samper M, Hancock EB, Leaptrot D, Scalise E, Badrun F, Phelps R and Edwards JR (2018) ‘Changes in Prevalence of Health Care–Associated Infections in U.S. Hospitals’, N Engl J Med, 379(18), pp. 1732–1744. 10.1056/nejmoa1801550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascari CA, Djoric D, Little JL and Kristich CJ (2022) ‘Use of an Interspecies Chimeric Receptor for Inducible Gene Expression Reveals that Metabolic Flux through the Peptidoglycan Biosynthesis Pathway is an Important Driver of Cephalosporin Resistance in Enterococcus faecalis’, J Bacteriol, 204(4). 10.1128/jb.00602-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesnage S, Chau F, Dubost L and Arthur M (2008) ‘Role of N-acetylglucosaminidase and N-acetylmuramidase activities in Enterococcus faecalis peptidoglycan metabolism’, J Biol Chem, 283(28), pp. 19845–19853. 10.1074/jbc.M802323200. [DOI] [PubMed] [Google Scholar]
- Miller WR, Murray BE, Rice LB and Arias CA (2016) ‘Vancomycin-Resistant Enterococci: Therapeutic Challenges in the 21st Century’, Infectious Disease Clinics of North America, 30(2), pp. 415–439. 10.1016/j.idc.2016.02.006. [DOI] [PubMed] [Google Scholar]
- Minton NE, Djorić D, Little J and Kristich CJ (2022) ‘GpsB Promotes PASTA Kinase Signaling and Cephalosporin Resistance in Enterococcus faecalis’, J Bacteriol, 204(10). 10.1128/jb.00304-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizyed S, Oddone A, Byczynski B, Hughes DW and Berti PJ (2005) ‘UDP-N-acetylmuramic Acid (UDP-MurNAc) Is a Potent Inhibitor of MurA (Enolpyruvyl-UDP-GlcNAc Synthase)’, Biochemistry, 15;44(10), pp. 4011–7. 10.1021/bi047704w. [DOI] [PubMed] [Google Scholar]
- Murray BE (1990) ‘The life and times of the Enterococcus’, Clin Microbiol Rev, 3(1), pp. 46–65. 10.1080/00472331003798640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rismondo J, Bender JK and Halbedel S (2017) ‘Suppressor mutations linking gpsB with the first committed step of peptidoglycan biosynthesis in Listeria monocytogenes’, J Bacteriol, 199(1), pp. 1–16. 10.1128/JB.00393-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouquette C, Ripio MT, Pellegrini E, Bolla JM, Tascon RI, Vázquez-Boland JA and Berche P (1996) ‘Identification of a ClpC ATPase required for stress tolerance and in vivo survival of Listeria monocytogenes’, Mol Microbiol, 21(5), pp. 977–987. 10.1046/j.1365-2958.1996.641432.x. [DOI] [PubMed] [Google Scholar]
- Schulz LM, Rothe P, Halbedel S, Gründling A and Rismondo J (2022) ‘Imbalance of peptidoglycan biosynthesis alters the cell surface charge of Listeria monocytogenes’, The Cell Surface, 8 (2022) 100085. 10.1016/j.tcsw.2022.100085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard BD and Gilmore MS (2002) ‘Antibiotic-resistant enterococci: the mechanisms and dynamics of drug introduction and resistance’, Microbes Infect, 4, pp. 215–224. 10.1016/s1286-4579(01)01530-1. [DOI] [PubMed] [Google Scholar]
- Signoretto C, Boaretti M and Canepari P (1994) ‘Cloning, sequencing, and expression in Escherichia coli of the low-affinity penicillin binding protein of Enterococcus faecalis’, FEMS Microbiol Lett, 123, pp. 99–106. 10.1111/j.1574-6968.1994.tb07207.x [DOI] [PubMed] [Google Scholar]
- Skarzynski T, Mistry A, Wonacott A, Hutchinson SE, Kelly VA and Duncan K (1996) ‘Structure of UDP-N-acetylglucosamine enolpyruvyl transferase, an enzyme essential for the synthesis of bacterial peptidoglycan, complexed with substrate UDP-N-acetylglucosamine and the drug fosfomycin’, Structure, 4, pp. 1465–1474. 10.1016/S0969-2126(96)00153-0. [DOI] [PubMed] [Google Scholar]
- Sun Y, Hürlimann S and Garner E (2023) ‘Growth rate is modulated by monitoring cell wall precursors in Bacillus subtilis’, Nature Microbiol, 8(3), pp. 469–480. 10.1038/s41564-023-01329-7. [DOI] [PubMed] [Google Scholar]
- Tesson B, Dajkovic A, Keary R, Marlière C, Dupont-Gillain CC and Carballido-López R (2022) ‘Magnesium rescues the morphology of Bacillus subtilis mreB mutants through its inhibitory effect on peptidoglycan hydrolases’, Scientific Reports, 12(1). 10.1038/s41598-021-04294-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui H-CT, Joseph M, Zheng JJ, Perez AJ, Manzoor I, Rued BE, Richardson JD, Branny P, Doubravová L, Massidda O and Winkler ME (2023) ‘Negative regulation of MurZ and MurA underlies the essentiality of GpsB- and StkP-mediated protein phosphorylation in Streptococcus pneumoniae D39’, Mol Microbiol. 10.1111/mmi.15122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubeda C, Taur Y, Jenq RR, Equinda MJ, Son T, Samstein M, Viale A, Socci ND, Van Den Brink MRM, Kamboj M and Pamer EG (2010) ‘Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans’, J Clin Invest, 120(12), pp. 4332–4341. 10.1172/JCI43918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, Yuan D, Stroe O, Wood G, Laydon A, Zídek A, Green T, Tunyasuvunakool K, Petersen S, Jumper J, Clancy E, Green R, Vora A, Lutfi M, Figurnov M, Cowie A, Hobbs N, Kohli P, Kleywegt G, Birney E, Hassabis D and Velankar S (2022) ‘AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models’, Nucleic Acids Res, 50(D1), pp. D439–D444. 10.1093/nar/gkab1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesić D and Kristich CJ (2012) ‘MurAA is required for intrinsic cephalosporin resistance of Enterococcus faecalis’, Antimicrob Agents Chemother, 56(5), pp. 2443–2451. 10.1128/AAC.05984-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamp S, Rothe P, Stern D, Holland G, Dö Hling J and Halbedel S (2022) ‘MurA escape mutations uncouple peptidoglycan biosynthesis from PrkA signaling’, PLoS Pathog, 18(3), pp. 1–30. 10.1371/journal.ppat.1010406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamp S, Rutter ZJ, Rismondo J, Jennings CE, Lewis RJ and Halbedel S (2020) ‘PrkA controls peptidoglycan biosynthesis through the essential phosphorylation of ReoM’, eLife, 9, p. e56048. 10.7554/eLife.56048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner LM, Webb AK, Limbago B, Dudeck MA, Patel J, Kallen AJ, Edwards JR and Sievert DM (2016) ‘Antimicrobial-Resistant Pathogens Associated with Healthcare-Associated Infections: Summary of Data Reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2011–2014’, Infection Control and Hospital Epidemiology, 37(11), pp. 1288–1301. 10.1017/ice.2016.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Wu Y, Lin Z, Wang G, Jiang S, Sun X, Tu H, Yu Z and Qu D (2020) ‘ClpP participates in stress tolerance, biofilm formation, antimicrobial tolerance, and virulence of Enterococcus faecalis’, BMC Microbiol, 20(1), pp. 1–18. 10.1186/s12866-020-1719-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Utama B, Pratap S, Supandy A, Song X, Tran TT, Mehta HH, Arias CA and Shamoo Y (2023) ‘Enolpyruvate transferase MurAAA149E, identified during adaptation of Enterococcus faecium to daptomycin, increases stability of MurAA-MurG interaction’, J Biol Chem, 299(3), 102912. 10.1016/j.jbc.2023.102912. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.