

Abstract
Objective:
Age-related dementia syndromes are often not related to a single pathophysiological process, leading to multiple neuropathologies found at autopsy. An amnestic dementia syndrome can be associated with Alzheimer’s disease (AD) with comorbid transactive response DNA-binding protein 43 (TDP-43) pathology (AD/TDP). Here, we investigated neuronal integrity and pathological burden of TDP-43 and tau, along the well-charted trisynaptic hippocampal circuit (dentate gyrus [DG], CA3, and CA1) in participants with amnestic dementia due to AD/TDP, amnestic dementia due to AD alone, or non-amnestic dementia due to TDP-43 proteinopathy associated with frontotemporal lobar degeneration (FTLD-TDP).
Methods:
A total of 48 extensively characterized cases (14 AD, 16 AD/TDP, 18 FTLD-TDP) were analyzed using digital HALO software (Indica Labs, Albuquerque, NM, USA) to quantify pathological burden and neuronal loss.
Results:
In AD/TDP and FTLD-TDP, TDP-43 immunoreactivity was greatest in the DG. Tau immunoreactivity was significantly greater in DG and CA3 in AD/TDP compared with pure AD. All clinical groups showed the highest amounts of neurons in DG, followed by CA3, then CA1. The AD and AD/TDP groups showed lower neuronal counts compared with the FTLD-TDP group across all hippocampal subregions consistent with the salience of the amnestic phenotype.
Interpretation:
We conclude that AD/TDP can be distinguished from AD and FTLD-TDP based on differential regional distributions of hippocampal tau and TDP-43. Findings suggest that tau aggregation in AD/TDP might be enhanced by TDP-43.
Introduction
Neurodegeneration is defined as the progressive loss of selectively vulnerable neurons in specific anatomical networks, leading to cognitive impairment over time. Age-related neurodegenerative diseases are therefore characterized by the clinical phenotype (eg, a dementia syndrome), anatomical patterns of atrophy, and pathology at cellular and molecular levels. Dementia syndromes, however, are often not related to a single pathophysiological process and, as a result, multiple pathologies found at autopsy are the rule rather than the exception.
There have been many reports of combined pathologies in the aging brain1–3 associated with the clinical diagnosis of amnestic dementia, most typically due to primary Alzheimer’s disease (AD). Inclusions containing abnormally phosphorylated transactive response DNA-binding protein 43 (TDP-43) are highly common in frontotemporal lobar degeneration (FTLD-TDP) and amyotrophic lateral sclerosis. Such inclusions also co-exist in up to ~60% of AD cases in community-based studies,4 and are also associated with amnestic dementia.3,5 A number of terms have been used to describe the phenomenon in which TDP-43 appears in limbic structures in elderly individuals who present clinically with an amnestic syndrome. Some refer to the phenomenon as “limbic-predominant age-related TDP-43 encephalopathy neuropathological change” (LATE-NC),6 and others as “TDP-43 in non-FTLD”.7 The nomenclature is evolving, in part because biological features remain undefined. Here, we refer to this disease process as “AD/TDP” to reflect the comorbid appearance of AD and TDP-43 pathology at autopsy. Clinicopathological studies examining cases of AD/TDP have found that TDP-43 is associated with loss of episodic and working memory, accelerated cognitive decline, and faster rates of hippocampal atrophy.3–5,8 The abundance of TDP-43 inclusions found in cases alongside hallmarks of AD that include amyloid-β (Aβ) plaques and tau-containing neurofibrillary tangles (NFTs)9 raises central questions about the synergistic relationship between TDP-43 and AD pathology, and their impact on clinical symptoms.
The goal of the present study was to investigate neuronal integrity and pathological burden of TDP-43 in the memory-related hippocampus of participants who were diagnosed in life with: (1) amnestic dementia due to Alzheimer’s disease and TDP-43 (AD/TDP); (2) amnestic dementia due to Alzheimer’s disease alone (AD); and (3) non-amnestic dementia due to TDP-43 associated with FTLD-TDP. The hippocampus was selected for this study because of its serially arranged chain of intrinsic connections that link cytoarchitectonically distinct zones. The pyramidal cells of the cornu ammonis (CA) regions of the hippocampus, particularly the CA1, are vulnerable to the formation of NFTs, whereas dentate gyrus (DG) granule cells of the hippocampal formation are known to be vulnerable to TDP-43 inclusion formation in FTLD.10 Neuronal integrity and pathology were quantified in the DG, CA1, and CA3 to identify the distinct locus of vulnerability in each disease state. Our findings provide evidence that a unique AD/TDP fingerprint exists within the hippocampal complex that distinguishes it from solely tauor TDP-associated neurodegenerative disease processes.
Methods
Participant Characteristics and Demographic Information
Autopsied brains from 48 participants held within the Northwestern University Alzheimer’s Disease Research Center Brain Bank were used. Written informed consent was obtained from all participants, and the study was approved by the Northwestern University Institutional Review Board and in accordance with the Helsinki Declaration. All cases underwent extensive post-mortem neuropathological evaluation. Three groups of cases were included: cases with Alzheimer’s Disease neuropathological change as the primary neuropathologic diagnosis (AD; n = 14), AD with comorbid TDP-43 neuropathology (AD/TDP; n = 16), and frontotemporal lobar degeneration with TDP-43 proteinopathy (FTLD-TDP; n = 18).
AD/TDP and AD cases carried an ante-mortem clinical diagnosis of amnestic mild cognitive impairment or amnestic dementia according to the criteria proposed by the National Institute on Aging–Alzheimer’s Association,11,12 and had a global and Memory Clinical Dementia Rating13 of 0.5–1 at enrollment, with the exception of one case with an initial Global Clinical Dementia Rating of 2. These scores reflected the presence of abnormal memory. Neuropsychological testing utilizing the National Institute on Aging’s Uniform Data Set 2.014 was available for 25 of 30 participants with an amnestic impairment that included a measure of episodic delayed memory (Wechsler Memory Scale-III Logical Memory II15) that was analyzed to provide empirical support for a memory loss. FTLD-TDP cases carried a clinical diagnosis of either primary progressive aphasia (PPA) or behavioral variant frontotemporal dementia (bvFTD), based on published criteria.16,17 Briefly, PPA is an early-onset dementia syndrome characterized by a progressive deterioration of language abilities, and became one of the first syndromes to show that the same clinical phenotype can be caused by heterogeneous pathologies, including FTLD-TDP.18 bvFTD is a dementia syndrome characterized by deterioration of personality, comportment, and at times, executive functioning.17 Both PPA and bvFTD can show underlying FTLD-TDP.19–22 The one common denominator for all neuropathologies associated with PPA and bvFTD is their predilection for the language dominant hemisphere (PPA), or frontal-network anatomical loci (bvFTD)—selectively vulnerable regions to pathology for unknown reasons. FTLD-TDP cases had a global Clinical Dementia Rating range of 0.5 to 1 at initial visit.
All cases were required to be free of medical illness that would interfere with diagnosis. Other exclusion criteria included significant comorbid pathology, such as argyrophilic grain disease, primary age-related tauopathy, FTLD-tau, or Lewy body disease. All amnestic cases met criteria for Braak neurofibrillary tangle staging III-VI, which corresponds to intermediate or high AD neuropathologic change (B2 or B3) according to the National Institute on Aging–Alzheimer’s Association criteria.23,24 AD/TDP cases were positive for at least mild medial temporal TDP-43, corresponding to stages 2 or 3 in the LATE-NC staging criteria.25 One AD/TDP case showed additional diffuse and sparse neocortical Lewy body disease with hippocampal sparing. One case in the AD cohort and 6 cases in the AD/TDP cohort carried a postmortem tertiary diagnosis of hippocampal sclerosis. Four of the FTLD-TDP cases carried a primary GRN mutation and one FTLD-TDP case carried a post-mortem diagnosis of hippocampal sclerosis. APOE genotyping was available for 38 participants. See Table 1 for participant group characteristics and Table S1 for individual case characteristics.
TABLE 1.
Group Characteristics
| Sex (% male) | Age at death (mean yr; SD) | Age at onset (mean yr; SD) | PMI (mean h; SD) | Brain weight (g; SD) | Braak stage V/VI (%) | Aβ plaque score (A3; %) | APOE E4 (%) | |
|---|---|---|---|---|---|---|---|---|
| AD (n = 14) | 57 | 81 (11) | 72 (14) | 19 (9) | 1,197 (131) | 78% | 86% | 33% |
| AD/TDP (n = 16) | 44 | 80 (9) | 69 (12) | 18 (5) | 1,141 (132) | 81% | 100% | 39% |
| FTLD-TDP (n = 18) | 50 | 68 (9) | 59 (9) | 13 (10) | 1,039 (227) | 0% | 0% | 4% |
| TDP type A (n = 5) | 60 | 70 (10) | 64 (12) | 6 (2) | 970 (77) | 0% | 0% | 0% |
| TDP type B (n = 5) | 80 | 67 (14) | 59 (6) | 16 (10) | 1,370 (137) | 0% | 0% | 0% |
| TDP type C (n = 8) | 25 | 68 (4) | 55 (5) | 14 (11) | 1,040 (232) | 0% | 0% | 6% |
Note: APOE genotyping was available for 38 participants and frequencies reflect the proportion of E4 allele present. A3 = severe amyloid-β score criteria according to the NIA-AA; AD = Alzheimer’s disease; AD/TDP = comorbid Alzheimer’s disease and transactive response DNA-binding protein 43; APOE E4 = apolipoprotein allele 4; FTLD-TDP = transactive response DNA-binding protein 43 pathology form of frontotemporal lobar degeneration; PMI = post-mortem interval; TDP = transactive response DNA-binding protein 43; h = hours; yr = years; SD = standard deviation.
Tissue Processing and Histopathology
After autopsy, the cerebral hemispheres were separated in the midsagittal plane, cut into 3- to 4-cm coronal slabs, fixed in 10% formalin for 2 weeks or 4% paraformaldehyde for 36 hours, taken through sucrose gradients (10%–40%) for cryoprotection, and stored in 40% sucrose at 4◦C. Samples of the left hippocampus were embedded in paraffin, and sections were cut at a thickness of 5 μm for immunohistochemical analysis using antibodies against human phosphorylated TDP-43 (pSer409/410; mouse monoclonal, 1:5000; Cosmo Bio, Tokyo, Japan) and AT8 (Ser202, Thr205; mouse monoclonal; Invitrogen MN1020; 1/500) to visualize TDP-43- and tau-positive pathology, respectively. Neuronal loss and gliosis were assessed with histological staining using 1.0% cresyl violet. Pathological diagnosis was rendered by board-certified neuropathologists (E.H.B., Q.M., and M.E.F.) following consensus criteria, and cases in the FTLD-TDP group were classified into type A, B, or C according to published criteria.26 Braak staging27 was surveyed in each case to identify NFT involvement in transentorhinal/entorhinal cortex, other limbic cortical areas, and neocortical regions.
Image Acquisition and Analysis
Digital images of slides at 20x magnification were obtained using an Olympus V200 slide scanner system (Olympus, Tokyo, Japan). Images of hippocampi were analyzed using the Object Colocalization algorithm of the HALO digital image software HALO v3.5.3 (Indica Labs). The CA1, CA3, and dentate gyrus, including the molecular, granule, and polymorphic cell layers, were annotated manually on each slide image using HALO software. Two measures of interest were obtained from the HALO Object Colocalization module: (1) object count per mm2 to determine number of neurons, AT8 positive inclusions, and TDP-43 positive inclusions; and (2) % area occupied (%AO) to determine total percentage of area covered by AT8 and/or TDP-43 immunoreactivity. For each measure (ie, inclusion count, %AO), the optimal color, object blur radius, contrast radius, contrast threshold, minimum optical density, and object size were averaged among a subsection (ie, 20%) of the total slides; these values were used to create a final set of parameters by which every slide was analyzed. See Fig 1 and Table 2 for information on digital quantitation of image analysis. After image analysis, cases with object counts or %AO values outside 1 SD of the mean of their pathological group underwent manual quality control for accuracy and values adjusted accordingly. Collaborators may request digital images and optimized protocols derived from post-mortem histological analysis of cases included in this study on request and with approval (see: https://www.brain.northwestern.edu/scientists-students/collaborative-request.html).
FIGURE 1:
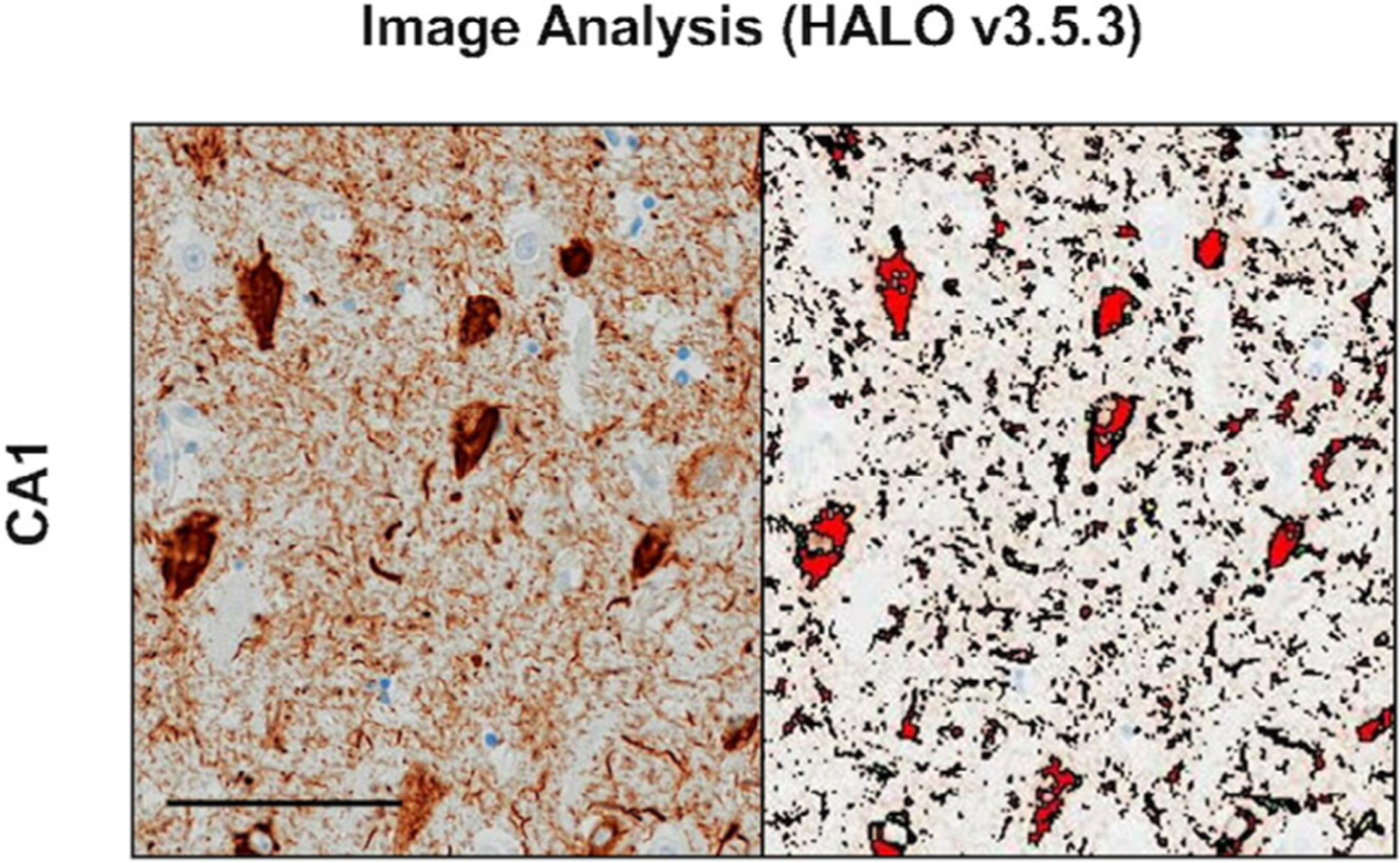
Image analysis (HALO v3.5.3). Hippocampal subregion, CA1, stained with AT8 is shown as an example of image visualization captured with HALO digital overlay. Both neurofibrillary tangles and neurites were included in the percentage of immunoreactivity analyses (see Results section). Photomicrograph shows a 91-year-old woman with a 3-year duration of amnestic dementia due to Alzheimer’s disease with comorbid transactive response DNA-binding protein 43 pathology. Image taken at ×10 magnification. Scale bar represents 100 μm.
TABLE 2.
Parameters for Digital Quantitation of Image Analysis (HALO v3.5.3)
| Object blur radius | Contrast radius | Contrast threshold | Minimum optical density | Object size | |
|---|---|---|---|---|---|
| CV | 2 | 12 | 0.47 | 0.0470 | 64.375 |
| TDP-43 | 1.5 | 11.27 | 0.147 | 0.0565 | 0-max |
| AT8 | 2 | 12 | 0.49 | 0.0550 | 0-max |
Note: By stain, the average parameters amongst a subsection (20%) of the total slides, which was used to generate a set of parametric values by which every slide was analyzed. See “Image Acquisition and Analysis” for more details.
Abbreviations: CV = cresyl violet; TDP-43 = transactive response DNA-binding protein 43.
Statistical Analysis
Data passed tests of normality and were normally distributed. Fisher’s exact tests were used to compare APOE allele frequency, Braak staging, memory performance, and amyloid-β plaque severity between groups. Student t tests and one-way ANOVAs were employed to compare inclusion counts and percentage of immunoreactivity between pathological groups. All statistical analyses were performed using GraphPad Prism 6 (GraphPad Software, La Jolla, CA, USA). Significance level was set to p < 0.05. Ancillary analyses were performed based on Braak staging. When the FTLD-TDP group was included in analyses, subsequent statistical tests were performed to exclude GRN cases and those cases with FTLD-TDP-type C.
Results
As expected, the mean age at death was significantly greater in AD/TDP (79.6 years, SD 9.1) and AD only (M 80.6 years, SD 10.9) groups compared with the FTLD-TDP (68.3 years, SD 8.6) group. There were no significant differences in sex and education between groups. The mean disease duration did not differ between the AD/TDP (10 years, SD 4.7), AD (8.8 years, SD 5.0), and FTLD-TDP (9 years, SD 4.8) groups.
The mean Wechsler Memory Scale-III Logical Memory II memory raw score for the AD group was 4.3 (SD 4.9) versus a mean of 3.27 (SD 5.62) in the AD/TDP group; although lower in the AD/TDP group, differences did not reach statistical significance. Based on WMS-III age-adjusted normative values15 episodic memory performance in these groups fell in the ~5–9% range compared with peers their age, which corresponded to a z-score of approximately −1.3 (scaled score of 6), indicating at least mild amnestic impairment.
There were no significant differences in postmortem interval or brain weight between groups. Analysis of available APOE genotyping showed a significantly lower APOE E4 allele frequency in FTLD-TDP compared with AD and AD/TDP groups (FTLD-TDP: 4%; AD: 33%; AD/TDP: 39%; p < 0.001). There was no difference in APOE E4 allele frequency between AD and AD/TDP groups. In the AD group, 78% showed Braak V/VI staging, which was not significantly different from 81% in the AD/TDP group. Amyloid-β plaque severity was not significantly different between groups either (AD, 86% vs AD/TDP, 100%). All FTLD-TDP cases showed Braak stage and amyloid-β plaque severity score of 0.
Amnestic Syndromes Due to AD or AD/TDP Are Characterized by Distinct Hippocampal Patterns of Tau Pathology
Within both amnestic groups, the CA1 showed significantly higher tau immunoreactivity (%AO of AT8) compared with CA3 (AD: p < 0.0001; AD/TDP: p < 0.05) and DG (both groups, p < 0.0001). Remarkably, the AD/TDP group showed more than double the percentage of area occupied by tau immunoreactivity compared with the AD group in the CA3 (12.0% vs 5.6%; p < 0.05) and DG (7.6% vs 1.8%; p < 0.05), but there were no significant differences in the CA1 subfield (Fig 2). The results remained consistent when all cases with Braak staging of III/IV were excluded.
FIGURE 2:
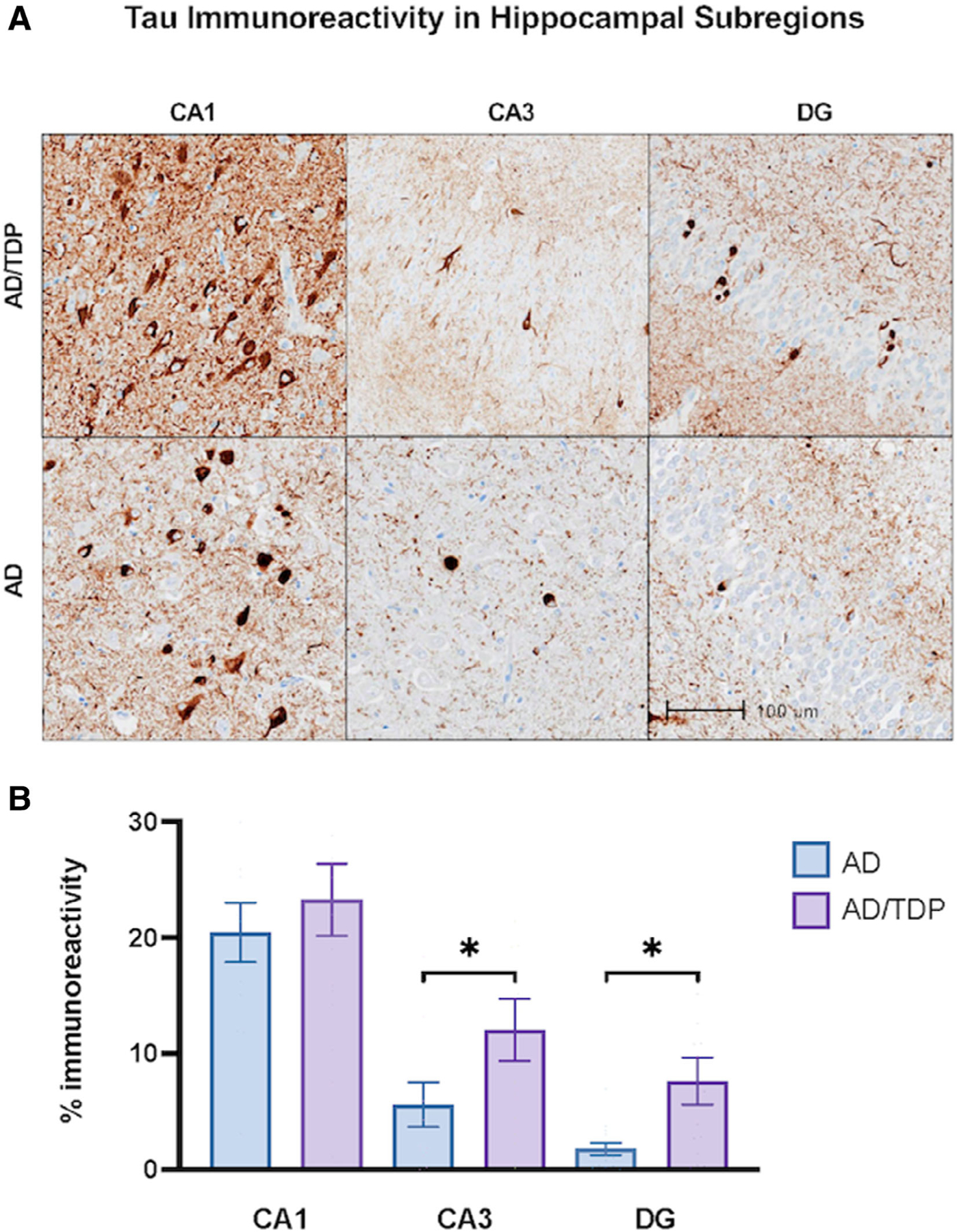
Tau immunoreactivity in amnestic dementias across subregions of the hippocampal complex. (A) Hippocampal subregions immunohistochemically stained with AT8 to visualize phosphorylated tau immunoreactivity in cases with amnestic dementia due to AD alone and due to Alzheimer’s disease (AD) with comorbid transactive response DNA-binding protein 43 (TDP-43) pathology (AD/TDP). The AD/TDP case shown is an 83-year-old woman with an 18-year history of amnestic dementia. The AD case is a 89-year-old woman with a 19-year history of amnestic dementia. Both cases were found to show distribution and extent of neurofibrillary tangle consistent with Braak stage IV/V. Images were taken at 10x magnification. (B) There was significantly more tau immunoreactivity in AD/TDP versus AD cases in both the CA3 and dentate gyrus (DG), but not the CA1. Bars represent mean percentage of area occupied by tau immunoreactivity and standard error is shown. *p < 0.05
Regional Burden of TDP in the Hippocampus Discriminates Between AD/TDP and FTLD-TDP
Within the FTLD-TDP group, the DG was significantly more affected by TDP-43 immunoreactivity (0.94%) compared with CA3 (0.13%, p < 0.005) and CA1 (0.32%, p < 0.05), consistent with prior findings.13 The greatest relative burden of TDP-43 pathology was also observed in the DG subregion in the AD/TDP group despite scarcity across all hippocampal subregions. The percentage of area covered by TDP-43 immunoreactivity was significantly greater in the FTLD-TDP group compared with the AD/TDP group across the CA1 (0.05%; p < 0.05), CA3 (0.02%, p < 0.05), and the DG (0.09%, p < 0.005; see Fig 3). Counts of TDP-43-positive inclusions were consistent with these findings; FTLD-TDP showed significantly greater TDP-43 inclusions per mm2 across the CA1 (14 vs 7 per mm2), CA3 (13 vs 1 per mm2), and DG (88 vs 3 per mm2), compared with the AD/TDP group ( p < 0.005). These dissociations, particularly in the DG, highlight a differential regional affinity of AD and TDP pathology to hippocampal subfields in amnestic versus non-amnestic dementia phenotypes.
FIGURE 3:
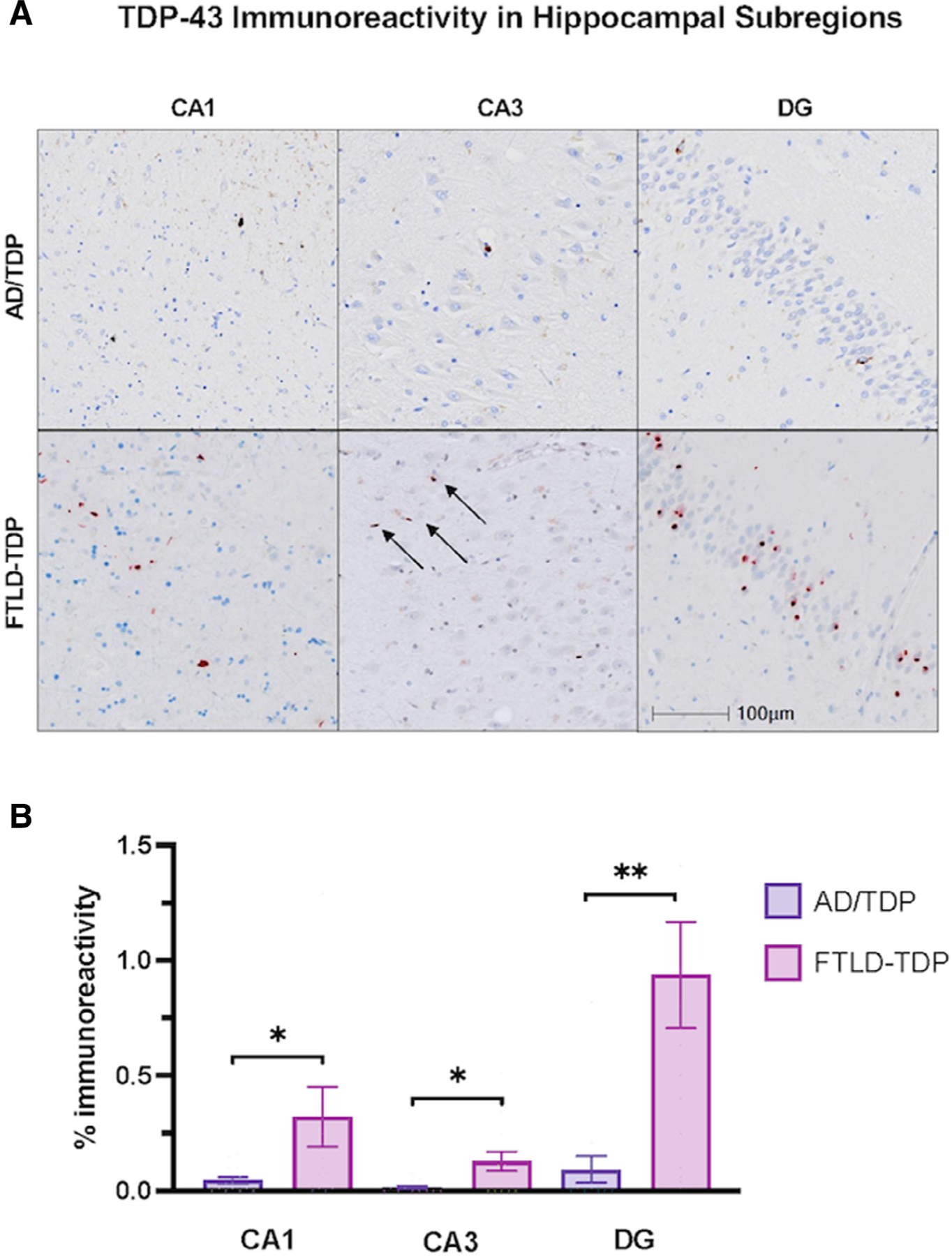
Transactive response DNA-binding protein 43 (TDP-43) immunoreactivity across hippocampal subregions in Alzheimer’s disease (AD) with comorbid transactive response DNA-binding protein 43 (TDP-43) pathology (AD/TDP) versus TDP-43 proteinopathy associated with frontotemporal lobar degeneration (FTLD-TDP). (A) Hippocampal subregions stained with TDP-43 to visualize TDP-43 immunoreactivity in amnestic dementia due to AD/TDP and non-amnestic dementias (PPA and bvFTD) due to FTLD-TDP. The FTLD-TDP case is a 65-year-old woman with a 13-year history of primary progressive aphasia found to have FTLD-TDP type C at autopsy, and the AD/TDP case is 83-year-old woman with an 18-year history of amnestic dementia, diagnosed with Braak stage VI (see Fig 2A). Images were taken at 10x magnification. (B) There was significantly more TDP-43 positivity in FTLD-TDP across all hippocampal subregions compared with AD/TDP. Overall, TDP-43 immunoreactivity within AD/TDP was relatively low across all regions. Bars represent mean percentage of area occupied by TDP-43 immunoreactivity and standard error is shown. *p < 0.05; **p < 0.01
Comparison of Total Pathological Burden in Hippocampal Subregions in AD/TDP Versus AD and FTLD-TDP
Both AD and AD/TDP groups showed significantly higher percentage of area covered by total immunoreactivity (combined tau+TDP-43) in the CA1 compared with the FTLD-TDP group ( p < 0.0001), a finding that was driven by the predominance of tau pathology in this region (Fig 4A). The AD/TDP group showed significantly greater total immunoreactivity in CA3 (12.9%) and DG (7.3%) compared with AD (CA3: 5.7%, p < 0.05; DG: 1.8%, p < 0.01) and FTLD-TDP (CA3: 0.2%, p < 0.0001; DG: 0.9%, p < 0.005) groups (Fig 4B,C). In the AD/TDP group, TDP-43 immunoreactivity comprised of <0.5% of total immunoreactivity in all hippocampal subregions. The results remained consistent when FTLD-TDP type A cases due to a GRN mutation (n = 4) were excluded from analyses.
FIGURE 4:
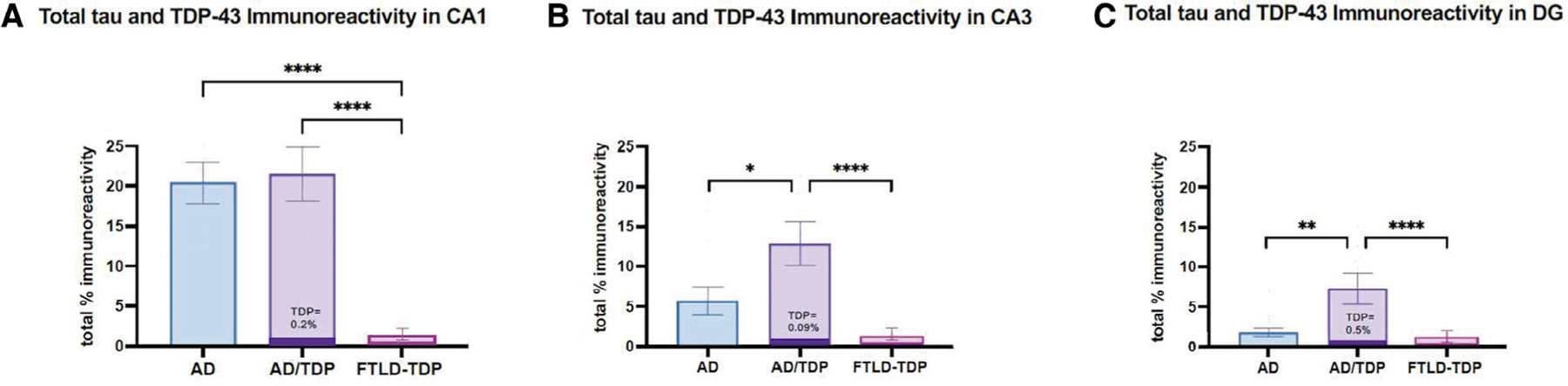
The percentage of area occupied by total tau and transactive response DNA-binding protein 43 (TDP-43) immunoreactivity across cohorts. The mean total percentage of immunoreactivity (sum of AT8 and TDP-43 positivity) is shown across all hippocampal subregions in all groups, where dark purple bars represent the proportion of TDP-43 immunoreactivity relative to total burden in the AD/TDP group. (A) In the CA1, AD/TDP and AD showed nearly equivalent total immunoreactivity, primarily due to an abundance of tau pathology. (B, C) In the CA3 and dentate gyrus (DG), AD/TDP showed significantly greater total immunoreactivity compared with AD and FTLD-TDP groups, also determined by high relative burden of tau pathology. Within the AD/TDP group, TDP-43 immunoreactivity comprised a minimal proportion of total pathology. Bars represent mean percentage of area occupied by total immunoreactivity and standard error is shown. Note, dark purple bar in AD/TDP is not to scale. *p < 0.05; **p < 0.01; data analyses excluding GRN (FTLD-TDP type A) and FTLD-TDP type C are not shown”.
Neuronal Counts across Hippocampal Subfields
Neuronal counts across hippocampal subfields showed a consistent pattern in all groups, whereby the highest counts of granule neurons were found in the DG, followed by pyramidal neurons in CA3, and CA1 (Fig 5). The amnestic groups had significantly lower neuronal counts compared with the FTLD-TDP group in CA1 ( p < 0.0001), DG ( p < 0.0001), and CA3 (FTLD-TDP vs AD only, p < 0.005). This finding of relatively greater hippocampal degeneration in AD and AD/TDP appears concordant with the salience of the memory impairment that characterizes the amnestic syndrome due to AD or AD/TDP. Results remained consistent when cases with hippocampal sclerosis were excluded.
FIGURE 5:
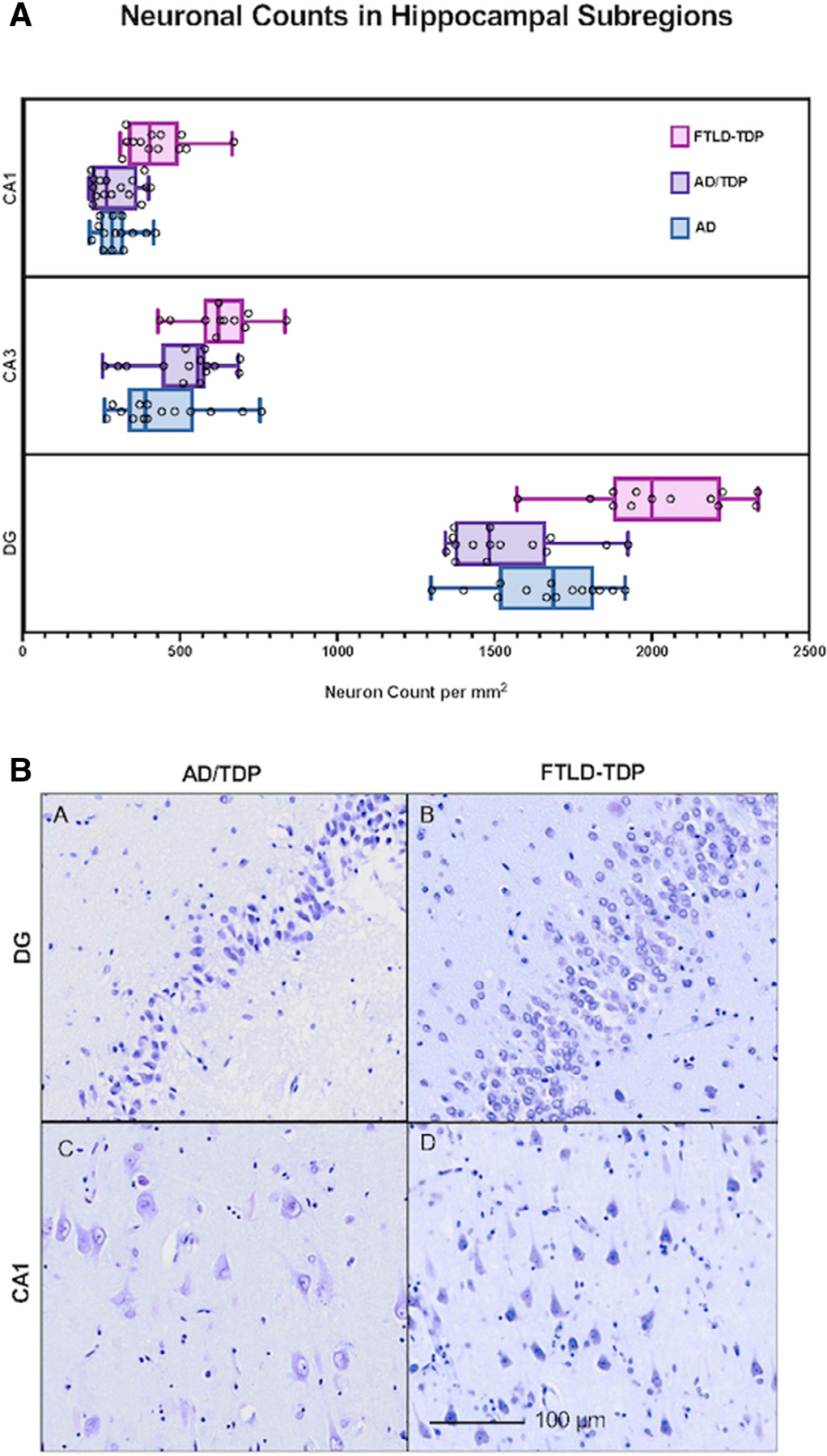
Neuronal counts in hippocampal subregions across amnestic and transactive response DNA-binding protein 43 proteinopathy associated with frontotemporal lobar degeneration (FTLD-TDP) cohorts. (A) Box plots show the distribution of neuronal counts. The box extends from the 25th percentile to the 75th percentile. The median is shown by a line, and “whiskers” extend from minimum to maximum counts. Individual dots represent neuronal counts in a single case. As expected, in all groups the granule cells of the dentate gyrus (DG) were most numerous given their high cell packing density, followed by CA3, then CA1. In accordance with the salience of the amnestic syndrome, the amnestic groups showed significantly lower neuronal counts compared to the FTLD-TDP group in CA1 and DG (p < 0.0001). (B) Photomicrograph of the dentate gyrus and CA1 region of hippocampus stained with cresyl violet to visualize neurons in individuals with AD/TDP versus FTLD-TDP; note significantly fewer neurons in AD/TDP. The FTLD-TDP case is a 65-year-old woman with a 13-year history of PPA found to have FTLD-TDP type C at autopsy, and the AD/TDP case is 83-year-old woman with an 18-year history of amnestic dementia (see Figs 2A and 3A).
Ancillary Analyses Excluding the FTLD-TDP type C
FTLD-TDP has been classified into subtypes based on the morphology and distribution of inclusions and neurites in cortex.28 The Mackenzie harmonized classification scheme (2011) characterizes FTLD-TDP-type A by moderate to numerous cytoplasmic inclusions and short dystrophic neurites in upper cortical layers, and type B by the distribution of cytoplasmic inclusions in all cortical layers.26 The type C species is more easily distinguished from types A and B because of its characteristic of long dystrophic neurites and rare cytoplasmic inclusions in cortex. A study by Josephs et al. described AD/TDP cases that more closely resembled the TDP found along the type A/B spectrum versus type C.29 Therefore, in ancillary analyses, we excluded FTLD-TDP type C cases (n = 8) to ensure that variant-specific TDP would not interfere with findings. Overall, our results remained consistent when excluding TDP type C cases. It is worth noting that when average TDP immunoreactivity was compared between type C cases (1.4% of area covered; SD 0.01) and type A + B cases (0.60% of area covered; SD 0.01), type C cases showed significantly greater TDP immunoreactivity in the DG only (p < 0.05).
Discussion
In recent years, the literature on age-related dementia syndromes has been replete with research that highlights a comorbidity of TDP-43 and AD in patients diagnosed with an ante-mortem amnestic dementia syndrome—up to 40% in population-based studies30 and up to ~60% in community-based studies.4 The neuronal vulnerability profile in AD/TDP remains elusive, and studies have suggested that there is a cumulative effect on neuronal groups susceptible to both NFTs of AD and TDP-43. Some studies have suggested that Aβ42 triggers TDP-43, and that they “cross-seed” each other into a neurotoxic state,31 whereas others suggest that tau may “trigger and aggravate” TDP-43.32 We focused on tau in the present study because of its preference for the hippocampus and clear hierarchical staging. Amyloid-β has been shown to follow a more diffuse distribution in the neocortex.33 Additionally, cognitive decline has been found to correlate with the aggregation of tau.34 Future studies will address amyloid-β distribution in detail. From a pathogenic standpoint, studies on comorbid AD/TDP have shown that both pathologies follow similar, but not identical, progression patterns.25,29 In the present study, we investigated pathological burden and neuronal loss in hippocampal subregions of AD/TDP, and compared with AD alone and to FTLD-TDP. We found a regional susceptibility profile in hippocampal subfields unique to AD/TDP, revealing what appears to be a synergistic relationship between comorbid tau and TDP-43 pathology.
The classical trisynaptic circuit of the hippocampal system is integral to human memory, and its gradual destruction in AD leads to degradation of encoding and consolidation.35 Its major components, the DG, CA3, and CA1, are integrally connected and were chosen for the present study due to their respective susceptibility profiles. In the topographical hierarchy of NFT progression, the granule cells of the dentate gyrus are relatively spared from early involvement.36 It appears that NFTs do not appear in granule cells until later stages of disease.37 With the exception of those with cerebral age-related TDP-43 with significant hippocampal sclerosis (once referred to as “CARTS”),38 dentate granule cells show early vulnerability in FTLD-TDP. It is not known why in early stages of AD, DGcells are resilient compared with neighboring cells in the CA1 subfield of the hippocampal complex.39 These profiles serve as a backdrop to the study of selective vulnerability along the TDP-AD continuum leading to amnestic dementia.
In the current study, the CA1 region of the hippocampus was devastated with tau pathology in AD and AD/TDP groups, aligning with previous studies that found the CA1 selectively vulnerable to NFTs in early stages of AD.39 The granule cells of the DG were relatively spared from tau infiltration.40 Interestingly, the nidus of vulnerability in AD/TDP differed. We found that AD/TDP had significantly greater tau immunoreactivity present in the DG and CA3 compared with AD, rendering AD/TDP neuropathologically distinct. Our results suggest that the presence of TDP-43 is associated with enhanced tau pathology, perhaps providing a putative explanation for why cases with AD/TDP have been associated with greater hippocampal atrophy and more severe clinical deficits.5 Studies on comorbid AD/TDP have shown that both pathologies follow similar, but not exact, progression patterns.25,29 Additionally, TDP-43 and tau have been shown to co-accumulate41 and even co-localize in a single neuron in individuals with AD/TDP.42 One study proposed that TDP-43 may function as a templating agent to trigger tau aggregation.41 The mechanism of TDP-43 propagation is unknown, but evidence supports that pathological aggregates of TDP-43 can be transferred from cell-to-cell in a seed-dependent and self-templating manner.43 We have also found evidence suggesting that TDP-43 inclusions spread throughout the hippocampus in a manner that supports transsynaptic propagation along axonal pathways.10 Another possible mechanism is the propagation of TDP-43 by glial cells, supported by evidence that the overexpression of TDP-43 in astrocytes can induce non-cell autonomous neuronal toxicity.44 Despite its frequent albeit sparse appearance, it seems that TDP-43 is exerting a toxic effect on its surrounding environment via a tau-dependent mechanism that is ripe for further study.
In our cohort, significantly more TDP-43 immunoreactivity was present in FTLD-TDP compared with AD/TDP in each hippocampal subregion in spatial patterns consistent with previous reports.10 We showed previously in FTLD-TDP that mature inclusion deposition occurred in highest abundance in the DG, whereas early or “pre-inclusions” were more commonly found in sequential relay stations (CA3, followed by CA2-CA1) within the hippocampus, providing evidence in support of transsynaptic propagation of TDP-43 aggregates.10 In AD/TDP cases, however, TDP-43 burden made up <0.5% of the total immunoreactivity, the majority of which involved the DG. This suggests that FTLD-TDP and AD/TDP show unique patterns of neuroanatomic vulnerability to TDP-43 in the DG and CA1, respectively, and that the pattern of disease spread throughout corticolimbic circuits are likely distinct. One possibility is that the TDP-43 found alongside AD presents with a distinct molecular signature; a recent study found different TDP-43 aggregate composition based on fluorescence double-labeling experiments with antibodies raised against full-length or ‘fragmented’ TDP-43.45 Additionally, four of the FTLD-TDP cases in our cohort were positive for a GRN mutation, which decreases the anti-inflammatory function of the progranulin protein, and is associated with the pathogenesis of FTLD-TDP and neuronal loss.46 Although findings remained similar when excluding cases with a GRN mutation, additional study is poised to evaluate proliferation of glia, particularly microglia, and ensuing inflammatory changes in cases with TDP-43.
Neurophysiological dysfunction in both AD and FTLD with TDP-43 include gliosis,47 synaptic abnormalities,48 and neuronal loss.49 Both amnestic groups (AD and AD/TDP) in our cohort showed significantly lower counts of neurons in the CA1 and DG of the hippocampal complex compared with cases with non-amnestic phenotype dementia syndromes due to FTLD-TDP; CA1 also showed a significantly higher amount of pathological burden overall. This is the first study to our knowledge that provides empirical support for a root cause of memory loss in AD/TDP, which appears to be driven by tau-dependent neuronal loss. There were no measurable differences between the two amnestic groups across the hippocampal hubs investigated. DG granule cells send a dense, zinc-rich “mossy fiber” bundle that synapses on dendritic spines of neurons in CA3; these neurons then send collaterals of their projecting axons, also known as “Schaffer collaterals,” to CA1 pyramidal cells.50 One worthy avenue of exploration would be a focus on additional connections, such as the mossy fiber bundle, that link distinct zones along limbic circuits responsible for memory function. The present study showed that the type C subtype of FTLD-TDP appears to favor the hippocampal DG granule cells relative to type A or B; a second avenue of exploration would be to investigate more closely the morphological signatures of TDP (eg, nuclear or cytoplasmic vs neuritic) as it coexists with AD in the hippocampus.
We conclude from the present findings that in AD/TDP, tau immunoreactivity is significantly greater in DG and CA3 compared with pure AD, and that the contribution of TDP-43 inclusions, although smaller than that in FTLD-TDP, appears to enhance tau accumulation. Our findings indicate that the AD/TDP profile represents a distinct pathophysiological process different from pure AD and FTLD-TDP. The combined findings of higher tau pathology in CA1 and significant neuronal loss in CA1 and DG in both AD and AD/TDP groups are concordant with the salience of the amnestic syndrome. Hippocampal-dependent episodic memory is especially nuanced, with multiple stages, and dependent on the salience of specific incoming information. One limitation of the study was its reliance on inclusion criteria reflecting only mild amnestic impairment, which precludes the examination of pathology in later stages. Additionally, in patients with neurodegenerative disorders, comorbid pathologies, such as AD/TDP, are the norm; this study carefully removed this confounder to examine the pure impact of TDP-43 versus NFT proteinopathy. Future studies are needed to address concordance between the type and extent of memory impairment in AD/TDP and multiple etiological markers of neurodegeneration at death in this older adult population.
Supplementary Material
Acknowledgments
We are grateful to our research participants for their invaluable contributions to scientific discovery.
This work was supported by grants from the National Institute on Aging (P30AG072977, R01AG062566 (including a Diversity Supplement), R01AG077444, K08AG065463, F31AG07631 and R56AG075600), National Institute of Neurological Disorders and Stroke (R01NS085770, T32NS047987), the National Alzheimer’s Coordinating Center (U01AG016976); and the National Science Foundation (DGE-1842165).
Footnotes
Additional supporting information can be found in the online version of this article.
Potential Conflicts of Interest
Nothing to report.
References
- 1.Rabinovici GD, Carrillo MC, Forman M, et al. Multiple comorbid neuropathologies in the setting of Alzheimer’s disease neuropathology and implications for drug development. Alzheimers Dement 2017;3: 83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rahimi J, Kovacs GG. Prevalence of mixed pathologies in the aging brain. Alzheimers Res Ther 2014;6:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uchino A, Takao M, Hatsuta H, et al. Incidence and extent of TDP-43 accumulation in aging human brain. Acta Neuropathol Commun 2015;20:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.James BD, Wilson RS, Boyle PA, et al. TDP-43 stage, mixed pathologies, and clinical Alzheimer’s-type dementia. Brain 2016;139:2983– 2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Josephs KA, Whitwell JL, Weigand SD, et al. TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Ann Neurol 2014;76:811–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nelson PT, Dickson DW, Trojanowski JQ, et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 2019;142:1503–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Josephs KA, Murray ME, Tosakulwong N, et al. Pathological, imaging and genetic characteristics support the existence of distinct TDP-43 types in non-FTLD brains. Acta Neuropathol 2019;137:227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nag S, Yu L, Wilson RS, et al. TDP-43 pathology and memory impairment in elders without pathologic diagnoses of AD or FTLD. Neurology 2017;88:653–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bejanin A, Schonhaut DR, La Joie R, et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain 2017;140:3286–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jamshidi P, Kim G, Shahidehpour RK, et al. Distribution of TDP-43 pathology in hippocampal synaptic relays suggests transsynaptic propagation in frontotemporal lobar degeneration. J Neuropathol Exp Neurol 2020;79:585–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011;7:270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011;7:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morris JC. The clinical dementia rating (CDR): current version and scoring rules. Neurology 1993;43:2412–2414. [DOI] [PubMed] [Google Scholar]
- 14.Weintraub S, Salmon D, Mercaldo N, et al. The Alzheimer’s disease Centers’ uniform data set (UDS): the neuropsychologic test battery. Alzheimer Dis Assoc Disord 2009;23:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wechsler D Wechsler Memory Scale 3rd ed. San Antonio, TX: The Psychological Corporation, 1997. [Google Scholar]
- 16.Mesulam M Primary progressive aphasia: a dementia of the language network. Dement Neuropsychol 2013;7:2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011;134:2456–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mesulam M, Wicklund A, Johnson N, et al. Alzheimer and frontotemporal pathology in subsets of primary progressive aphasia. Ann Neurol 2008;63:709–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Irwin DJ, Cairns NJ, Grossman M, et al. Frontotemporal lobar degeneration: defining phenotypic diversity through personalized medicine. Acta Neuropathol 2015;129:469–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Irwin DJ, McMillan CT, Xie SX, et al. Asymmetry of post-mortem neuropathology in behavioural-variant frontotemporal dementia. Brain 2018;141:288–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kwong LK, Neumann M, Sampathu DM, et al. TDP-43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathol 2007;114:63–70. [DOI] [PubMed] [Google Scholar]
- 22.Gefen T, Mao Q, Kohler M, et al. Primary progressive aphasia has a unique signature distinct from dementia of the Alzheimer’s type and behavioral variant frontotemporal dementia regardless of pathology. J Neuropathol Exp Neurol 2020;79:1379–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 2012;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 2012;123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Josephs KA, Murray ME, Whitwell JL, et al. Updated TDP-43 in Alzheimer’s disease staging scheme. Acta Neuropathol 2016;131: 571–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mackenzie IR, Neumann M, Baborie A, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 2011;122: 111–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Braak H, Braak E, Bohl J. Staging of Alzheimer-related cortical destruction. Eur Neurol 1993;33:403–408. [DOI] [PubMed] [Google Scholar]
- 28.Lee EB, Porta S, Michael Baer G, et al. Expansion of the classification of FTLD-TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol 2017;134:65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Josephs KA, Murray ME, Whitwell JL, et al. Staging TDP-43 pathology in Alzheimer’s disease. Acta Neuropathol 2014;127:441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nelson PT, Brayne C, Flanagan ME, et al. Frequency of LATE neuropathologic change across the spectrum of Alzheimer’s disease neuropathology: combined data from 13 community-based or population-based autopsy cohorts. Acta Neuropathol 2022;144:27–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fang YS, Tsai KJ, Chang YJ, et al. Full-length TDP-43 forms toxic amyloid oligomers that are present in frontotemporal lobar dementia-TDP patients. Nat Commun 2014;12:4824. [DOI] [PubMed] [Google Scholar]
- 32.McAleese KE, Walker L, Erskine D, et al. TDP-43 pathology in Alzheimer’s disease, dementia with Lewy bodies and ageing. Brain Pathol 2017;27:472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen SD, Lu JY, Li HQ, et al. Staging tau pathology with tau PET in Alzheimer’s disease: a longitudinal study. Transl Psychiatry 2021; 11:483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guillozet AL, Weintraub S, Mash DC, Mesulam MM. Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch Neurol 2003;60:729–736. [DOI] [PubMed] [Google Scholar]
- 35.Anderson P, Lomo T. Mode of activation of hippocampal pyramidal cells by excitatory synapses on dendrites. Exp Brain Res 1966;2: 247–260. [PubMed] [Google Scholar]
- 36.Arnold SE, Hyman BT, Flory J, et al. The topographical and neuroanatomical distribution of neurofibrillary tangles and Neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb Cortex 1991;1:103–116. [DOI] [PubMed] [Google Scholar]
- 37.Stefanits H, Wesseling C, Kovacs GG. Loss of Calbindin immunoreactivity in the dentate gyrus distinguishes Alzheimer’s disease from other neurodegenerative dementias. Neurosci Lett 2014;30: 137–141. [DOI] [PubMed] [Google Scholar]
- 38.Smith VD, Bachstetter AD, Ighodaro E, et al. Overlapping but distinct TDP-43 and tau pathologic patterns in aged hippocampi. Brain Pathol 2018;28:264–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Furcila D, Dominguez-Alvaro M, DeFelipe J, Alonso-Nanclares L. Subregional density of neurons, neurofibrillary tangles and amyloid plaques in the hippocampus of patients with Alzheimer’s disease. Front Neuroanat 2019;13:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen-Plotkin AS, Lee VM, Trojanowski JQ. TAR DNA-binding protein 43 in neurodegenerative disease. Nat Rev Neurol 2010;6:211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Montalbano M, McAllen S, Cascio FL, et al. TDP-43 and tau oligomers in Alzheimer’s disease, amyotrophic lateral sclerosis, and frontotemporal dementia. Neurobiol Dis 2020;146:105130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Higashi S, Watanabe R, Arai T. Fluorescence in-situ hybridization method reveals that carboxyl-terminal fragments of transactive response DNA-binding protein-43 truncated at the amino acid residue 218 reduce poly(A)+ RNA expression. Neuroreport 2018;29:846–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jo M, Lee S, Jeon YM, et al. The role of TDP-43 propagation in neurodegenerative diseases: integrating insights from clinical and experimental studies. Exp Mol Med 2020;52:1652–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tong J, Huang C, Bi F, et al. Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. EMBO J 2013;32:1917–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tome SO, Vandenberghe R, Ospitalieri S, et al. Distinct molecular patterns of TDP-43 pathology in Alzheimer’s disease: relationship with clinical phenotypes. Acta Neuropathol Commun 2020;8:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mao Q, Zheng X, Gefen T, et al. FTLD-TDP with and without GRN mutations cause different patterns of CA1 pathology. J Neuropathol Exp Neurol 2019;78:844–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cairns NJ, Bigio EH, Mackenzie IR, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the consortium for frontotemporal lobar degeneration. Acta Neuropathol 2007;114:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Connelly SJ, Mukaetova-Ladinska EB, Abdul-All Z, et al. Synaptic changes in frontotemporal lobar degeneration: correlation with MAPT haplotype and APOE genotype. Neuropathol Appl Neurobiol 2011;37:366–380. [DOI] [PubMed] [Google Scholar]
- 49.Mackenzie IR, Baborie A, Pickering-Brown S, et al. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol 2006;112: 539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rosene DL, Van Hoesen GW. The hippocampal formation of the primate brain: a review of some comparative aspects of cytoarchitecture and connections. Cereb Cortex 1987;6:345–456. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.