

Abstract
Background
Even though prostate cancer (PCa) patients initially respond to androgen deprivation therapy, some will eventually develop castration resistant prostate cancer (CRPC). Androgen receptor (AR) mediated cell signaling is a major driver in the progression of CRPC while only a fraction of PCa becomes AR negative. This study aimed to understand the regulation of AR levels by N- myristoyltransferase in PCa cells.
Methods
Two enantiomers, (1S,2S)-D-NMAPPD and (1R,2R)-D-NMAPPD (LCL4), were characterized by various methods (1H and 13C NMR, UHPLC, high-resolution mass spectra, circular dichroism) and evaluated for the ability to bind to N-myristoyltransferase 1 (NMT1) using computational docking analysis. Structure-activity relationship analysis of these compounds led to the synthesis of (1R,2R)-LCL204 and evaluation as a potential NMT1 inhibitor utilizing the purified full length NMT1 enzyme. The NMT inhibitory activity wase determined by Click chemistry and immunoblotting. Regulation of NMT1 on tumor growth was evaluated in a xenograft tumor model.
Results
(1R,2R)-D-NMAPPD, but not its enantiomer (1S,2S)-D-NMAPPD, inhibited NMT1 activity and reduced AR protein levels. (1R,2R)-LCL204, a derivative of (1R,2R)-D-NMAPPD, inhibited global protein myristoylation. It also suppressed protein levels, nuclear translocation, and transcriptional activity of AR full-length or variants in PCa cells. This was due to enhanced ubiquitin and proteasome-mediated degradation of AR. Knockdown of NMT1 levels inhibited tumor growth and proliferation of cancer cells.
Conclusion
Inhibitory efficacy on N-myristoyltransferase activity by D-NMAPPD is stereospecific. (1R,2R)-LCL204 reduced global N-myristoylation and androgen receptor protein levels at low micromolar concentrations in prostate cancer cells. pharmacological inhibition of NMT1 enhances ubiquitin-mediated proteasome degradation of AR. This study illustrates a novel function of N-myristoyltransferase and provides a potential strategy for treatment of CRPC.
Keywords: NMT1, Myristoylation, AR, Ubiquitination, prostate cancer
Introduction
Prostate cancer (PCa) is the second leading cause of cancer-related deaths in men in developed countries (1). Androgen receptor (AR) signaling is crucial in prostate development and cancer progression (2, 3). Medical or surgical androgen deprivation therapy (ADT) is usually a standard treatment for PCa patients (4). Tremendous effort has been made in developing effective treatments by targeting androgen synthesis and/or AR signaling, however patients usually develop resistance to ADT with progression to castration resistance prostate cancer (CRPC) (5–7). Multiple molecular mechanisms are responsible for CRPC including an increase of AR expression, AR mutations, intratumoral androgen synthesis, AR splice variants, and others (8, 9). As a result, despite that a portion of prostate tumors becomes AR negative, AR signaling remains active in a majority of prostate tumors (3, 10), and therefore targeting AR signaling still remains as an effective treatment approach for CRPC (11, 12).
While current treatment strategies mainly rely on inhibition of circulated androgen and blockade of androgen/AR interactions, direct degradation of AR levels has been considered another effective strategy to inhibit AR-pathway mediated cancer progression (13). Post-translational modifications including phosphorylation, methylation, acylation, SUMOylation, and ubiquitination, regulate AR stability and activity (14). Some E3 ligases have been reported to associate with the AR ubiquitination process (15–20). AR ubiquitination regulates AR protein turnover and transcriptional activity (14, 15, 21). Therefore, facilitation of ubiquitination-mediated AR degradation becomes an attractive approach for treatment of PCa.
N-Myristoylation is a co/post-translational lipid modification process where myristic acid, a 14 carbon saturated fatty acid, is attached to an N-terminal glycine (22). In humans, there are two N-myristoyltransferase (NMT) enzymes that facilitate N-myristoylation, NMT1 and NMT2, which share 77% sequence similarity (23). A variety of proteins undergo this modification in mammalian cells including the oncogenic proteins Src, PKA, and AMPK that require myristoylation for their function (24–28). N-myristoylation regulates the stability of these proteins and their association with the cytoplasmic membrane, and facilitates protein-protein interactions (26, 29, 30). Therefore, NMT1 controls cancer cell proliferation (31, 32) by regulating the functions of these proteins through myristoylation (33–36).
Inhibition of NMT to slow cancer progression makes it an attractive therapeutic target for anticancer drug development (37). Several small molecule compounds have been discovered that inhibit NMT activity. We have previously reported that genetic ablation of NMT1 or the small molecule compound D-NMAPPD (B13) inhibited PCa proliferation through suppression of Src kinase activity (38). In this study, we demonstrated that the absolute stereochemical configuration of D-NMAPPD is important in inhibition of NMT1 activity. Inhibition of NMT1 altered AR protein levels, nuclear translocation, and AR transcriptional activity in PCa cells. Furthermore, pharmacological inhibition of NMT1 promoted AR degradation through the ubiquitin proteasome pathway. Given the important role of NMT1 in mediating PCa progression, our study demonstrates a novel function of NMT1 and provides a potential strategy for treatment of CRPC through AR degradation.
Materials and Methods:
Cell culture and reagents
The human prostate cancer cell lines LNCaP, 22RV1, C4–2B, and VCaP were purchased from the American Type Culture Collection (ATCC). All the cell lines were cultured using the ATCC recommended medium of RPMI-1640 with 10% fetal bovine serum (FBS) (Corning) and 1% streptomycin/penicillin (Thermo Fischer Scientific). Cells were maintained in a 37 °C incubator with 5% carbon dioxide (CO2). All the cell lines purchased from the ATCC had a certificate indicating they are mycoplasma-free.
Synthesis of (1R, 2R)-D-NMAPPD (LCL4) and (1R,2R)-LCL204
For (1R,2R)-D-NMAPPD (LCL4) synthesis, (2R,3R)-2-amino-1-(4-nitrophenyl)-1,3-propanediol (0.1 mmol, 212 mg) was dissolved in dry THF (50 mL) and cooled to 4 °C followed by addition of EDCI (156 mg, 1 mol), HOBt (135 mg, 1 mmol), and tetradecanoic acid (228 mg, 1 mmol). The mixture was kept under N2 at room temperature overnight. The reaction mixture was then taken to dryness, and the product was extracted with ethyl acetate. The extracts were washed with aqueous sodium carbonate and then 3 M hydrochloric acid, dried and evaporated to give crude (1R,2R)- D-NMAPPD. Product (1R,2R)-D-NMAPPD (39) was then purified by column chromatography over silica gel using ethyl acetate:hexane 1:10 as eluent.
For (1R,2R)-LCL204 synthesis, (2R,3R)-2-amino-1-(4-nitrophenyl)-1,3-propanediol (1 mmol, 212 mg) was reacted with tetradecanal (255 mg, 1.2 mmol) in methanol/0.05 N acetic acid, 9:1 for 15 min and sodium cyanoborohydride (NaCNBH3, 130 mg, 2 mmol) was added in portions over 1 h. The mixture was stirred overnight at room temperature, evaporated to dryness and the residue dissolved in DCM/MeOH, 2:1. The product was purified by recrystallization from DCM/pentane 1:3 to give (1R,2R)-LCL204 (40) as white solid in 72 % yield.
Characterization of synthetic and commercial D-NMAPPD by NMR spectroscopy and polarimetry
(1R, 2R)-D-NMAPPD was synthesized in-house as described above, and D-NMAPPD [also called (1R, 2R)-B13] was purchased from Cayman Chemical (Item No. 10006305; CAS No. 35922–06-6). Due to different biological efficacies in inhibition of global protein myristoylation reported by two different labs (38, 41), we examined the chemical differences of these two compounds. After various measurements, it was established that the D-NMAPPD from Cayman Chemical is the enantiomer of the in-house synthesized (1R,2R)-D-NMAPPD. Identical NMR spectra were obtained for the two samples, indicating that they have the same relative configuration (additional measurements also in the Supplemental Materials and Methods):
1H-NMR spectra were recorded in methanol-d4 using a 500 MHz instrument. The (1S,2S)-D-NMAPPD (from Cayman Chemical) was characterized as follows: 1H NMR (500 MHz, CD3OD) δ 8.15 (d, J = 8.8 Hz, 2H, 2Ar), 7.61 (d, J = 8.7 Hz, 2H, 2Ar), 5.11 (d, J = 2.8 Hz, 1H, benzylic), 4.16 (ddd, J = 7.5, 5.8, 2.8 Hz, 1H), 3.74 (dd, J = 10.8, 7.5 Hz, 1H, CH2׳), 3.54 (dd, J = 10.7, 5.9 Hz, 1H, CH2׳׳), 2.06 (t, J = 7.3 Hz, 2H, 2CH2), 1.43 – 0.98 (m, 22H, 11CH2), 0.88 (t, J = 6.9 Hz, 3H, CH3). 13C NMR spectra were recorded in CD3OD using a 500 MHz: δ 174.9, 150.9, 147.1, 127.0, 122.7, 70.2, 61.3, 56.2, 35.6, 31.7, 29.4, 29.4, 29.3, 29.2, 29.1, 28.7, 25.6, 22.4, 13.1.
The (1R, 2R)-D-NMAPPD was characterized as follows: 1H NMR (500 MHz, CD3OD) δ 8.15 (d, J = 8.8 Hz, 2H, 2Ar), 7.61 (d, J = 8.7 Hz, 2H, 2Ar), 5.11 (d, J = 2.8 Hz, 1H, benzylic), 4.16 (ddd, J = 7.5, 5.8, 2.8 Hz, 1H), 3.74 (dd, J = 10.8, 7.5 Hz, 1H, CH2׳), 3.54 (dd, J = 10.7, 5.9 Hz, 1H, CH2׳׳), 2.06 (t, J = 7.3 Hz, 2H, 2CH2), 1.43 – 0.98 (m, 22H, 11CH2), 0.88 (t, J = 6.9 Hz, 3H, CH3).
Similarly, it was characterized with 13C-NMR (500 MHz, CD3OD) δ 174.9, 150.9, 147.1, 127.0, 122.7, 70.2, 61.3, 56.2, 35.6, 31.7, 29.4, 29.4, 29.3, 29.2, 29.1, 28.7, 25.6, 22.4, 13.1.
The melting point range for (1S,2S)-D-NMAPPD and (1R,2R)-D-NMAPPD was 79 – 80 °C. Specific rotations for the two samples were measured in methanol on an automatic polarimeter with a path length of 10 cm, and were found to be equal within experimental error but opposite in sign pointing to their enantiomeric nature: (1S,2S)-D-NMAPPD had [α]20D + 4.2 (c = 0.19, CH3CH2OH), while (1R, 2R)-D-NMAPPD had [α]20D −3.6 (c = 0.21, CH3CH2OH).
Molecular docking of two D-NMAPPD stereoisomers
The crystal structure of NMT1 used in this study was retrieved from the RCSB PDB database (PDB file ID: 5UUT) (38). Models of the enantiomers of (1R,2R)-D-NMAPPD and (1S,2S)-D-NMAPPD were prepared by the Ligprep module of Schrodinger Maestro, and the process includes converting 2D structures of ligands to 3D ones, adding hydrogens, calculating partial charges, and optimizing the structures. The protein structure preparation process which includes adding all hydrogens and missing side chains/loops, deleting some water molecules, and correcting bond information, were applied to NMT1 structure by Protein Preparation Wizard. Water molecules beyond 5.00 Å from het groups (i.e., ligand atoms) were deleted and generated het status’s pH = 7.0 ± 2.0. An optimization of H-bond assignment was performed. The PROPKA was kept at 7.0. The system was minimized to cover heavy atoms to RMSD 0.30 Å using force field OPLS3e. For ligand preparation, the same features mentioned above were applied to keep pH = 7.0 ± 2.0. Enantiomers of D-NMAPPD were generated. Finally, XP (extra-precision) docking was applied for identification of the binding interactions. As for Van der Waals radii scaling, the scaling factor was set to 0.80 and partial charge cutoff was set to 0.15 to soften the potential for non-polar regions of the ligands.
Characterization of two different sources of D-NMAPPD by high resolution mass spectrometry (HRMS) and UHPLC
All reactions were carried out under argon. Solvents used for column chromatography were analytical grade. Thin-layer chromatography was carried out with 250 μm glass backed silica (XHL) plates. Plates were visualized under UV light (254 nm) and stained with ninhydrin in n-butanol/AcOH. Purification of crude residues was performed over silica gel chromatography using 230–400 mesh grade 60 silica unless otherwise stated.
High-resolution mass spectrometry (HRMS) data were recorded in the electrospray mode using an orbitrap mass analyzer (ESI-Orbtrap). The m/z for D-NMAPPD (from Cayman Chemical) was calculated for C23H39N2O5 [M + H] to be 423.2859 and found to be 423.2845. Similarly, the m/z of (1R,2R)-D-NMAPPD (LCL4) was calculated for C23H39N2O5 [M + H] to be 423.2859 and found to be 423.2847.
UHPLC analysis was performed with an Acclaim™ 120 C18 5μm (4.6 × 50) column equipped with mass and a UV detector. D-NMAPPD (from Cayman Chemical) and (1R,2R)-D-NMAPPD (LCL4) were characterized with a UHPLC retention time of 5.55 min (flow rate: 0.4 mL/min; initial: H2O 30/CH3CN 70: 2 min; H2O 5/CH3CN 95: 4 min).
Formation of (1R,2R)-2-Amino-1-(4-nitrophenyl)-1,3-propanediol (Chloramphenicol D base)
(1R,2R)-D-NMAPPD (LCL4) (12 mg) was heated with 10 % HCl in CH3OH (0. 95 ml) at 80 °C for 18 h. The reaction mixture was concentrated in vacuo. The residue was dissolved in saturated NaHCO3 (1 mL) and extracted 5 times with ethyl acetate (2 mL). The combined organic layer was dried over Na2SO4, filtered and concentrated. The crude product was purified by column chromatography (CH2Cl2/ CH3OH/ NH3OH 90 : 9 : 1 to 60 : 30 : 10) to yield chloramphenicol D base (3.67 mg, 61%) as white solid.
Characterization of (1R,2R)-2-Amino-1-(4-nitrophenyl)-1,3-propanediol (Chloramphenicol D base) by NMR, high resolution mass spectrometry (HRMS), and UHPLC
The melting point range for (1R,2R)-2-amino-1-(4-nitrophenyl)-1,3-propanediol was 160–161 °C (literature m.p. 162–163 °C) (42). Specific rotations were measured in methanol on an automatic polarimeter with a path length of 10 cm. (1R,2R)-2-amino-1-(4-nitrophenyl)-1,3-propanediol rotation was [α]21D −22.9 (c = 0.07 , CH3OH) (literature [α]27D −23.1 (c = 1.58 , CH3OH)) (42).
NMR spectra were recorded in methanol-d4 using a 500 MHz instrument. The (1R,2R)-2-Amino-1-(4-nitrophenyl)-1,3-propanediol was characterized as follows: 1H NMR (500 MHz, CD3OD) δ 8.20 (d, J = 8.8 Hz, 2H, 2Ar), 7.61 (d, J = 8.6 Hz, 2H, 2Ar), 4.77 (d, J = 5.7 Hz, 1H), 3.52 (dd, J = 10.9, 5.1 Hz, 1H, CH2׳), 3.37 (dd, J = 10.9, 5.9 Hz, 1H, CH2׳׳), 2.91 (q, J = 5.6 Hz, 1H); 13C NMR (500 MHz, CD3OD) δ 150.8, 127.3, 123.0, 72.5, 62.4, 58.4.
High-resolution mass spectra (HRMS) were recorded in the electrospray mode using an orbitrap mass analyzer (ESI-Orbtrap). The (1R,2R)-2-Amino-1-(4-nitrophenyl)-1,3-propanediol was characterized as follows: ESI-HRMS m/z calculated for C9H13O4N2 [M + H] 213.0870 found 213.0867.
Plasmid construction and lentiviral production
To knockdown human NMT1, shRNA target sequences were inserted into psiRNA-W [H1.4] vector at the BbsI site. The H1 promoter together with shRNA was further sub-cloned into FUCRW or FUCGW at the PacI site. The primers used for cloning are listed in Table S1. Lentivirus was generated in HEK293T cells by co-transfecting the lentiviral packaging vectors including MDL, REV, and VSV and a lentiviral vector expressing the gene of interest such as NMT1, AR, or shRNA-NMT1. Virus infection was performed as described previously (43). All lentivirus procedures followed the guidelines and regulations of the University of Georgia.
Western blot and antibodies
Cells were lysed by incubation in RIPA buffer [20 mM Tris-HCl (pH 7.4), 137 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 10% glycerol, 0.1% SDS, and EDTA-free protease inhibitor cocktail set V (Millipore, 539137)] on ice for 20 minutes. After short sonication, cell lysates were centrifuged at 12,000 rpm for 20 minutes, and supernatants were collected for downstream analysis. Twenty micrograms of proteins were subjected to SDS-polyacrylamide gels before being transferred onto nitrocellulose membranes (Bio-Rad). Membranes were blocked with 5% milk powder (Lab1 Scientific) in 1X TBS containing 1% Tween-20 (TBST) for 1 h, washed with TBST, and incubated with the specific antibodies overnight at 4 °C. After washing the membranes 3 times with TBST, they were incubated with the appropriate secondary antibody for 1 h at room temperature.
AR antibody (1:1000) was purchased from Santa Cruz Biotechnology (Dallas, TX) (#441) or Millipore Sigma (Burlington, MA) (#06–680). Antibodies against ubiquitin (#3933), HDAC-1 (#5356) (1:1000), and GAPDH (#2118) (1:2500) were purchased from Cell Signaling Technology (Beverly, MA). The anti-NMT1 (#HPA022949) and γ-tubulin (#T6557) antibody were purchased from Sigma Chemical Co. (St. Louis, MO) and Calbiochem (San Diego, CA).
NMT1 gene expression and protein purification
A modified full length NMT1 genetic sequence containing a His6-tag at the N-terminus was synthesized by GenScript. The gene was cloned in the pET-11a vector and transformed into Rosetta 2 competent cells by heat shock. Cells were grown at 37 °C in LB broth, supplemented with 100 μg/ml ampicillin and 35 μg/ml chloramphenicol, to OD600 0.6–0.8. Cultures were induced with 1 mM IPTG and grown overnight at 18 °C. The cells were harvested by centrifugation at 5000 x g for 10 min. The cells were suspended in a buffer containing 20 mM Tris (pH 7.5), 500 mM NaCl, 10 mM imidazole, 1 mM MgCl2, and 0.1% (v/v) Triton X-100 and lysed by four 1 min rounds of sonication, 5 sec on 5 sec off, at 50% amplitude, on ice. The lysate was cleared by centrifugation at 48,000 x g for 20 min at 4 °C. Cleared lysate was applied to high-density nickel agarose beads (Gold Biotechnology, Olivette, MO) equilibrated with 20 mM Tris (pH 7.5), 500 mM NaCl and 10 mM imidazole. Protein was eluted with the equilibration buffer containing 250 mM imidazole. This elution was diluted 20-fold in Buffer A (20 mM Tris pH 8.9 and 1 mM DTT) and loaded onto a Mono Q anion-exchange column (GE Healthcare, Pittsburgh, PA). NMT1 was eluted in a 0–50% NaCl gradient over 20 column volumes of Buffer A and Buffer B (20 mM Tris pH 8.9, 1 mM DTT, and 1 M NaCl). The His6-tag was cleaved using HRV 3C protease. The remaining free His-tag, His-tagged NMT1, and HRV 3C protease were separated from cleaved NMT1 by nickel affinity chromatography.
Quantitative RT-PCR analysis
Total RNA was extracted from cultured cells using TRIzol reagent according to the manufacturer’s instructions. One μg of total RNA was reverse transcribed using the High-Capacity cDNA Kit (Applied Biosystem). The cDNA samples were used for quantitative RT-PCR reactions. Each PCR reaction contained the synthesized cDNA, SYBR Green (PerfeCTa SYBR Green FastMix, Quantabio), and primers at concentrations of 10 pM each. The primers are listed in Table S1. StepOne Software v2.1 (Applied Biosystem) was used to quantify PCR reactions and expression levels of a target gene were normalized to GAPDH levels in the corresponding samples. Each PCR reaction was done in triplicate.
Isolation of the cytoplasmic and nuclear protein fractions
Isolation of nuclear and cytoplasmic fractions were prepared using NE-PER Nuclear and Cytoplasmic extraction kit (Cat# 78833, ThermoFisher) according to manufacturer’s protocol. Briefly, cell pellets were washed with cold PBS and centrifuged at 600 g for 5 min. The cell pellet was resuspended with 200 μL cytoplasmic extraction reagent containing protease inhibitor. The cell lysates were vortexed for 15 sec followed by incubation on ice for 10 min. The cytoplasmic extraction reagent II (11 μL) was added to the sample and vortexed for 5 sec. After incubation on ice for 5 min, the lysates were centrifuged at 15,000 g for 5 min. The cytoplasmic fraction (supernatant) was transferred to a new tube. Nuclear extraction reagent (100 μL) was added to the insoluble pellet and incubated on ice for 40 min with vortexing for 15 sec at 10 min intervals. After samples were centrifuged at 15,000 g for 10 min, the supernatant (nuclear fraction) was collected and transferred to a new tube.
Quantitative analysis of AR fluorescence
Subcellular localization of androgen receptor (AR) protein and its fluorescence intensity were determined by fluorescence microscopy using a Zeiss Axio observer A1 microscopy with a 40x objective lens (Carl Zeiss Microimaging Inc., Germany). LNCaP cells were treated with 5 μM LCL204 for 48 h. Cells were fixed for 10 min with 4% paraformaldehyde/PBS (Affymetrix, USA), washed with PBS and permeabilized with 0.5% Triton-X100 in PBS for 5 min. Cells were blocked with 5% goat serum and incubated with AR antibody overnight. Cells were washed with PBS and incubated with Alexa Fluor 488 Goat Anti-Rabbit IgG (Invitrogen, USA) secondary antibody (1:700). Cells were mounted with PBS/DAPI. To evaluate AR intensity, samples were imaged and analyzed using ZEN software. In brief, channels of images were split using ZEN image channels split tool and the threshold values of fluorescence signals by the ZEN threshold tool were used to determine the fluorescence signal intensity values.
Protein stability and degradation
To assess the effect of shRNA-NMT1 on AR protein stability, LNCaP and 22Rv1 cells were treated with LCL204 for 24 h followed by addition of 10 μM cycloheximide (Cayman Chemicals; 14126) for 2, 4, 6 h. Cell lysates were prepared and protein levels of AR, NMT1, and GAPDH were determined by Western blotting. AR protein degradation was also examined in LNCaP and 22Rv1 cells treated with 10 μM bortezomib for 9 h (Cayman Chemical; 10008822) after 24 h treatment with LCL204. Protein levels of AR, NMT1, and GAPDH were determined by Western blotting.
Immunoprecipitation for the analysis of AR ubiquitination
For the detection of AR ubiquitination, LNCaP cells were transduced with shRNA-NMT1 by lentiviral infection, or LNCaP and 22Rv1 cells were treated with 5 μM LCL204 for 48 h. The transduced or LCL204-treated cells were grown in 10 cm dishes, and cell lysates were extracted with the immunoprecipitation (IP) lysis buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM EGTA, 10% glycerol, 1% Triton X-100, 1.5 mM MgCl2) containing protease inhibitor and phosphatase inhibitor. One mg of protein was incubated with androgen receptor (AR) (#441) antibody for 16 h at 4 °C. Protein A agarose beads (30 μL) were added to the lysate, and the mixtures were incubated for 2 h at 4 °C. After the incubation, the beads were washed three times with IP lysis buffer followed by addition of 2X SDS sample buffer and heated at 98 °C for 10 min. Ubiquitinated AR was detected by the ubiquitin antibody and AR antibody (Santa Cruz Biotechnology, Inc., SC-7305).
MTT assay for measuring cell proliferation
293T or C4–2B cells were initially seeded in 96-well plates at a density of 8000 cells per well. Once the cells adhered to the plate, the culture medium was aspirated and replaced with 100 μl of fresh medium containing 0 μM, 1.25 μM, 2.5 μM, or 5 μM of LCL204. The cells were cultured in the above medium for 0, 24, 48, and 72 hours, respectively. To measure cell proliferation, 10 μL of a 12 mM MTT solution was introduced into each well and incubated at 37 °C for 4 hours. To halt the reaction, 100 μL of DMSO was added to each well and incubated at room temperature for 15 minutes with agitation. Finally, the absorbance was measured at a wavelength of 595 nm.
Xenograft tumors
To examine the effect of NMT1 knockdown on AR protein expression and tumor growth in vivo, 22Rv1 (5 × 105) and C4–2B (2 × 106) cells transduced with shRNA-NMT1 or control by lentiviral infection were subcutaneously inoculated in the flank side of SCID male mice. Body weight and tumor size were measured (length × width × width/2) weekly. 22Rv1 and C4–2B xenograft tumors were harvested after 8 weeks or when the humane endpoint was reached. Grafts were subjected for hematoxylin and eosin (H&E) and immunohistochemistry (IHC) analysis. All animals were maintained according to the surgical and experimental procedures of the protocol A2013 03–008 approved by the institutional animal use and care committee at the University of Georgia.
Immunohistochemistry and quantification
Formalin-fixed/paraffin-embedded grafts were sectioned at 5 μm thickness and mounted to positively charged microscopic slides. H&E and immunohistochemistry (IHC) staining analysis were performed as described previously (44). The following primary antibodies and dilutions were used for IHC analysis: AR (Santa Cruz Biotechnology, Inc., SC-7305; 1:400), NMT1 (Sigma Aldrich, HPA022963; 1:200), Ki67 (Novus Biologicals, NB500–170; 1:400), and cleaved caspase-3 (Cell Signaling Technology, 9661; 1:200). To evaluate NMT1, AR, and Ki67 intensity, images for samples were taken using 40X magnification under the same light intensity and exposure time. The images were processed by ImageJ software. To quantify the staining intensity, images were uploaded into the ImageJ software and split into three color channels (by selection of Image> Color> Split channels functions). The blue channel was further processed (by selection of Image> Adjust> Threshold functions). The value of the threshold was obtained where all the positive pixels were selected, then the intensity of the positive pixels was measured (by selection of Analyze> Measure functions). The procedures were applied to the other images to obtain intensity of AR staining. The intensity of AR staining is defined as Area, which represents the area where an image was positively stained for AR. A representative image of each sample was displayed.
Statistical analysis
Statistical analysis was performed using GraphPad prism (GraphPad Inc., La Jolla, CA). The data are presented as mean ± S.E. and analyzed using Student’s t-test for experiments containing two groups and one-way analysis of variance (ANOVA) followed by Tukey post-hoc test for experiments containing more than two groups. All t-tests were performed at the two-sided 0.05 level for significance. *, p < 0.05; **, p < 0.01.
RESULTS:
Characterization of (1S,2S)-D-NMAPPD and its enantiomer (1R,2R)-D-NMAPPD (LCL4).
N-myristoyltransferases (NMTs) are involved in numerous essential cellular functions (22). Loss of NMT1 is embryonic lethal (45) and inhibits the proliferation of cancer cells (31, 38, 46). We have previously reported that an NMT1 inhibitor, (1R,2R)-D-NMAPPD (LCL4), which was synthesized in-house, inhibits NMT1 enzymatic activity with IC50 = 77.6 μM (38). However, a recent study showed that commercial D-NMAPPD (from Cayman Chemical, Cat#10006305) has a limited inhibitory effect on protein myristoylation (41). Seeking to resolve this discrepancy, we have determined that the two compounds from different sources are enantiomers. Both compounds have identical 1H (Fig. 1A) and 13C NMR spectra (Fig. 1B), molecular weight, elution times in UHPLC (Fig. 1C), and melting point ranges [(1S,2S)-D-NMAPPD: 79 – 80 °C; (1R,2R)-D-NMAPPD (LCL4): 78 – 80 °C]. However, the circular dichroism spectra of both compounds showed an enantiomeric relationship, with approximately equal but opposite traces (Fig. 1D). The enantiomeric relationship was further confirmed by polarimetry: (1S,2S)-D-NMAPPD, [α]20D +4.2; (1R,2R)-D-NMAPPD, [α]20D −3.6) (Fig. 1E).
Figure 1. Characterization of two D-NMAPPD stereoisomers, (1R,2R)-D-NMAPPD and (1S,2S)-D-NMAPPD.

A) Characterization of (1S,2S)-D-NMAPPD (top panel, from Cayman Chemical, Cat #10006305) and (1R,2R)-D-NMAPPD (lower panel, synthesized in-house) by 1H NMR. NMR spectra of both stereoisomers were recorded in methanol-d4 using a 500 MHz instrument. The 1H NMR spectrum of (1S,2S)-D-NMAPPD (top panel) and (1R,2R)-D-NMAPPD (bottom panel) were identical. B) Characterization of (1S,2S)-D-NMAPPD (top panel) and (1R,2R)-D-NMAPPD (lower panel) by 13C NMR. NMR spectra were recorded in methanol-d4 using a 500 MHz instrument. Signals were observed at δ 174.9, 150.9, 147.1, 127.0, 122.7, 70.2, 61.3, 56.2, 35.6, 31.7, 29.4, 29.4, 29.3, 29.2, 29.1, 28.7, 25.6, 22.4, 13.1. C) (1S,2S)-D-NMAPPD and (1R,2R)-D-NMAPPD have identical UHPLC chromatograms and high-resolution mass spectra (HRMS). Both isomers of D-NMAPPD were subjected to UHPLC analysis (top panel) and mass spectrometry (lower panel). UHPLC analysis was performed with an Acclaim™ 120 C18 5 μm (4.6 × 50) equipped with mass and UV detectors. High-resolution mass spectra (HRMS) were recorded in the electrospray mode using an orbitrap mass analyzer (ESI-Orbtrap). D) Characterization of (1R,2R)-D-NMAPPD and (1S,2S)-D-NMAPPD by circular dichroism. Both (1S,2S)-D-NMAPPD and (1R,2R)-D-NMAPPD were dissolved in methanol and circular dichroism spectra recorded from 180 nm to 320 nm for 200 scans (0.2 sec integration time). E) Specific rotation ([α]20D) of (1S,2S)-D-NMAPPD and (1R,2R)-D-NMAPPD and their docking energy with NMT1. The docking scores and MM-GBSA values for two stereoisomers were generated by using the XP (extra-precision) docking.
The absolute configuration of (1R,2R)-D-NMAPPD was confirmed by hydrolysis of the parent amide to the corresponding amine. Thus, treatment of (1R,2R)-D-NMAPPD with 10% HCl in methanol generated a (−)-base with identical properties to those previously reported by Rebstock et al. (47) (Fig. S1). The (−)-base is a precursor of the antibiotic chloramphenicol, a known inhibitor of Shigella paradysenteriae (42, 47), whose configuration has been confirmed by optical rotation data (47), optical rotatory dispersion and circular dichroism measurements (48–50).
(1R,2R)-D-NMAPPD had lower docking energy to the myristoyl-CoA binding site of NMT1 protein.
To examine if the two enantiomers function differently in inhibition of NMT1, both compounds (1R,2R)-D-NMAPPD and (1S,2S)-D-NMAPPD were docked with the crystal structure of NMT1 (38). (1R,2R)-D-NMAPPD exhibited lower binding free energy in comparison to (1S,2S)-D-NMAPPD (Fig. 1E). Several favorable interactions between (1R,2R)-D-NMAPPD and the myristoyl-CoA binding pocket of NMT1 were revealed. These interactions include 1) the hydroxy group of (1R,2R)- D-NMAPPD with Tyr180 or Leu248 of NMT1; 2) the hydroxymethyl of (1R,2R)- D-NMAPPD with Thr282 or Ile245; and 3) the amide group with Thr282 (Fig. 2A–B). In contrast, only two favorable interactions were observed between (1S,2S)-D-NMAPPD and the NMT1 binding pocket (Fig. 2C–D).
Figure 2. Docking analysis of two stereoisomers, (1R,2R)-D-NMAPPD and (1S,2S)-D-NMAPPD, to the myristoyl-CoA binding site of NMT1 protein.

The crystal structure of NMT1 (truncated form) has been resolved in previous studies (38) . The structure was retrieved from the RCSB PDB database (PDB ID: 5UUT). The structures of (1R,2R)-D-NMAPPD and (1S,2S)-D-NMAPPD were prepared by Ligprep. The interaction of the compounds with the surrounding functional groups of amino acids in NMT1 was revealed for (1R,2R)-D-NMAPPD (A) or (1S,2S)-D-NMAPPD (C). The binding site of myristoyl-CoA in the NMT1 structure was replaced with (1R,2R)-D-NMAPPD (B) or (1S,2S)-D-NMAPPD (D) in the docking analysis.
In the docking of (1R,2R)-D-NMAPPD with NMT1, multiple interactions of NMT1 with the compound include 1) the hydroxy group of (1R,2R)- D-NMAPPD with the Tyr180 or Leu248; 2) the hydroxymethyl of (1R,2R)- D-NMAPPD with Thr282 or Ile245; and 3) the amide group with Thr282. In the docking of (1S,2S)-D-NMAPPD with NMT1, two favorable interactions were revealed including 1) the hydroxy group of (1S,2S)- D-NMAPPD with the Tyr180 or Leu248; and 2) the hydroxymethyl of (1S,2S)- D-NMAPPD with Thr282 or Ile245 in the myristoyl-CoA binding pocket of NMT1.
The (1R,2R)-LCL204, a derivative of (1R,2R)-D-NMAPPD, had a higher inhibitory potential in targeting NMT1 activity.
(1R,2R)-LCL204 is a derivative of (1R,2R)-D-NMAPPD (Fig. 3A), which we previously reported inhibits NMT1 activity with IC50 = 8.7 μM using purified recombinant NMT1 without the N-terminal inhibitory motif (38, 51). To more accurately examine the inhibitory potential of (1R,2R)-LCL204 on NMT1, we further purified the full-length NMT1 protein (Fig. 3B). (1R,2R)-LCL204 inhibited full-length NMT1 activity with IC50 of 2.3 μM (Fig. 3C), confirming that loss of the N-terminal motif indeed enhances NMT1 activity (51). The inhibition of three different compounds on global protein myristoylation was further investigated using click chemistry. (1R,2R)-D-NMAPPD and (1R,2R)-LCL204, but not (1S,2S)-D-NMAPPD inhibited global protein myristoylation at 20–30 μM and 5 μM, respectively. More importantly, (1R,2R)-D-NMAPPD and (1R,2R)-LCL204 also significantly inhibited AR protein levels (Fig. 3D). For simplicity, (1R,2R)-LCL204 will be called LCL204 from this point forward.
Figure 3. Synthesis and inhibitory activity of 1R,2R-LCL204.

A) Synthesis of 1R,2R-LCL204. The reaction of (2R,3R)-2-amino-1-(4-nitrophenyl)-1,3-propanediol with tetradecanal in methanol/0.05 N acetic acid and sodium cyanoborohydride. The final product was purified by recrystallization using DCM/pentane. B) NMT1 sequence, expression and purification. The human full length NMT1 DNA sequence was optimized for bacterial expression and synthesized by GenScript. The NMT1 protein was His6-tagged and expressed in E. coli. The protein was purified by Ni-NTA affinity chromatography, anion exchange chromatography, and the His6-tag was removed by digestion with HRV 3C protease. The purified NMT1 protein was detected by Coomassie blue staining (i) and Western blot analysis using anti-human NMT1 antibody (ii) (Lane 1 contains the purified full length NMT1 protein; Lane 2: PBS buffer). C) Determination of IC50 of (1R,2R)-LCL204 on the full length NMT1 enzyme. NMT1 catalyzes the incorporation of myristoyl group to the N-terminus of glycine of the Gly-Ser-Asn-Lys-Ser-Lys-Pro-Lys peptide and releases CoA. The amount of the released CoA could be measured by its reaction with 7-diethylamino-3-(4’-maleimidylphenyl)-4-methylcoumarin (76). The IC50 of (1R,2R)-LCL204 on NMT1 enzymatic activity was 2.3 μM. Each data point represents three repeats. D) 22Rv1 cells were grown with DMSO or various concentrations of (1S,2S)-D-NMAPPD, (1R,2R)-D-NMAPPD, or (1R,2R)-LCL204 for 3 h, and cells were further treated with/without 20 μM myristic acid-azide for 24 h. Expression levels of AR, GAPDH, and myristoylated proteins were measured by Western blot or in combination with click chemistry.
LCL204 reduced AR protein levels in the nucleus and its transcriptional activity.
AR activity requires its translocation to the nucleus to activate its downstream genes (3). Therefore, we investigated if LCL204 affects AR levels in the nucleus and its transcriptional activity in prostate cancer cells. LCL204 inhibited proliferation of C4–2B cells, but was less toxic in 293T cells (Fig. S2). LCL204 treatment reduced both total and nuclear AR protein levels in 22Rv1 cells (Fig. 4A). Of note, LCL204 decreased levels of both AR full length (FL) (110 kDa) and an AR variant (V7, an AR variant without AR ligand binding domain, which leads to ligand independent AR signaling and nuclear localization) in 22Rv1 cells (Fig. 4A). Additionally, LCL204 inhibited nuclear translocation of the AR-FL protein (110 kDa) in the presence or absence of AR agonist (R1881) and AR-V7 variant in 22Rv1 cells in the absence of R1881 (Fig. 4A). This result was further confirmed by immuno-fluorescence analysis. As expected, AR nuclear levels (green fluorescence) were significantly elevated in the presence of R1881 but were inhibited by LCL204 in the presence or absence of R1881 stimulation (Fig. 4B). In addition, as expected, R1881 increased mRNA levels of PSA and KLK2 in C4–2B cells (Fig. 4C). LCL204 did not change AR mRNA levels in C4–2B and 22Rv1 cells (Fig. 4C), but inhibited PSA and KLK2 gene expression with or without R1881 (Fig. 4C). Taken together, the data suggest that LCL204 reduces AR protein levels and transcription of its regulated downstream genes, but reduced protein levels are not due to reduced AR transcription.
Figure 4. (1R,2R)-LCL204 reduces total AR protein levels, its nuclear translocation and transcriptional activity.

A) 22Rv1 cells were grown with charcoal stripped FBS for 2 days and then treated with/without 10 nM R1881 and 5 μM LCL204 for 48 h. AR expression levels were measured in total cell lysate, the cytoplasmic and nuclear protein fractions. GAPDH and HDAC1 were used as the cytoplasmic and nuclear protein loading controls, respectively. B) LNCaP cells were grown with charcoal stripped FBS for 1 day, then treated with/without 10 nM R1881 and 5 μM LCL204 for 24 h. Immunofluorescence staining for AR (green) was followed by counterstaining with DAPI (blue). The merged images are also shown. Fluorescence intensity was measured by ImageJ. C) 22Rv1 and C4–2B cells were grown with DMSO, 1, or 5 μM LCL204 in the presence or absence of 10 nM R1881 for 48 h. Expression levels of AR and its downstream genes, PSA and KLK2 in C4–2B and 22Rv1 cells were measured by qRT-PCR. Expression levels of AR, PSA, or KLK2 in cells grown without LCL 204 and R1881 were normalized to 1. Data are shown as mean ± SEM. N.S.: not significance; *p < 0.05; **p < 0.01.
LCL204 promoted AR ubiquitination and degradation.
AR can be poly-ubiquitinated and subsequently degraded (19). We examined if LCL204 regulation of AR protein levels was associated with the ubiquitination-proteasome degradation pathway. Down-regulation of AR protein levels by LCL204 was rescued by treatment with bortezomib, a proteasome inhibitor, in LNCaP and 22Rv1 cells (Fig. 5A–B). Additionally, de novo protein synthesis was blocked by cycloheximide (CHX), and AR degradation was observed over time with and without LCL204 (Fig. 5C–D). Next, treatment with LCL204 increased the levels of ubiquitinated AR in both LNCaP and 22Rv1 cells (Fig. 5E). The data indicate that a decrease of AR protein levels by inhibition of NMT1 activity is mediated through the ubiquitination-proteasome degradation pathway.
Figure 5. (1R,2R)-LCL204 promotes AR degradation through protein ubiquitination.
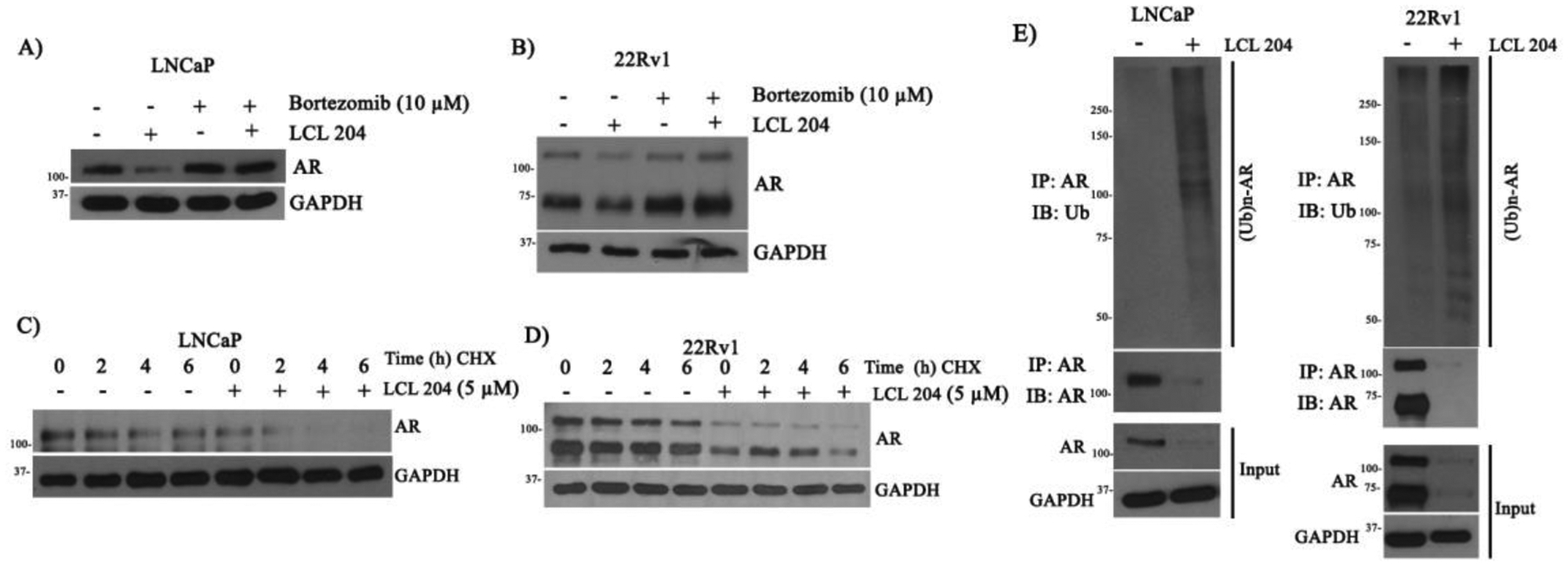
A-D) LNCaP and 22Rv1 cells were grown with/without 5 μM LCL204 and 10 μM Bortezomib, a small molecule proteasome inhibitor, for 24 h (A-B). Additionally, LNCaP and 22Rv1 cells were grown in charcoal-stripped medium with/without 5 μM LCL 204 for 18 h and further treated with 50 μM cycloheximide for 0, 2, 4, 6 h (C-D). Expression levels of AR and GAPDH protein was measured by Western blot. E) LNCaP and 22Rv1 cells were treated with DMSO or 5 μM LCL204 for 24 h. Protein levels of AR, NMT1, and GAPDH were measured in total cell lysates. Additionally, cell lysates were immunoprecipitated with AR antibody and AR and AR-ubiquitin levels were determined by Western blotting with anti-AR or anti-ubiquitin antibodies.
Knockdown of NMT1 inhibits AR levels and growth of prostate xenograft tumors.
We further investigated if knockdown of NMT1 led to inhibition of AR levels and suppressed growth of prostate xenograft tumors. C4–2B cells were transduced with shRNA-NMT1 by lentiviral infection. Knockdown of NMT1 and a decrease of AR levels were determined (Fig. 6A). Additionally, the size and weight of xenograft tumors were significantly inhibited in cells expressing shRNA-NMT1 in comparison with the control (Fig. 6B–E). As expected, expression levels of NMT1 were suppressed in the xenograft tumors expressing shRNA-NMT1 along with levels of AR and Ki67 (Fig. 6F–G). The data suggest that down-regulation of NMT1 inhibits growth of prostate xenograft tumors and is associated with a decrease of AR levels.
Figure 6. Knockdown of NMT1 inhibits growth of prostate cancer xenograft tumors.

C4–2B cells were transduced with shRNA-NMT1 or control by lentiviral infection and inoculated subcutaneously in SCID mice. A) To confirm the knockdown of NMT1, total lysate was harvested from the transduced cells and subjected to Western blotting. B-E) The size of xenograft tumors were examined weekly, and the weights were measured after tumors were harvested (mean ± SEM). The experiment represents 3 grafts per group (one graft per mouse). (*, p < 0.05). F) Expression levels of NMT1, AR, and Ki-67 in the xenograft tumors were analyzed by IHC staining. Scale bar, 100 μm.
DISCUSSION:
Stereochemistry plays an important role in determining selectivity, affinity, and efficacy of inhibitors (52). Our study demonstrates that inhibitor absolute configuration dictates the efficacy of inhibiting NMT1 enzymatic activity. Recently, numerous small molecule compounds targeting NMT activity have been developed for tumor treatment including COPP24, IMP-366, and (1R,2R)-D-NMAPPD (LCL4) (32, 38, 53). By comparing our previously reported compound (1R,2R)-D-NMAPPD (LCL4) (29) with the commercial similar compound (1S,2S)-D-NMAPPD (41), we clearly showed that (1S,2S)-D-NMAPPD was significantly less effective at inhibiting global protein myristoylation in prostate cancer cells. In contrast to (1R,2R)-D-NMAPPD (LCL4), (1S,2S)-D-NMAPPD was also predicted to bind poorly within the myristoyl-CoA binding site of NMT1 by docking analysis. Our study provides an example that stereochemistry regulates the efficacy of an inhibitor in drug design. We further demonstrated that LCL204, a derivative of (1R,2R)-D-NMAPPD, improved inhibition of global protein myristoylation and induced AR degradation in prostate cancer cells (Fig. 7). Similar to LCL4, LCL204 is an analogy of myristic acid that is highly insoluble. Therefore, a delivery vehicle such as nanoparticles or liposomes to encapsulate LCL204 might be necessary to maintain its inhibitory efficiency to target prostate tumors. Therefore, further research should provide an effective approach to encapsulate LCL204 for delivery and reveal its inhibitory function on growth of PCa tumors and AR levels in vivo.
Figure 7. NMT1 regulates AR protein levels and its transcriptional activity through ubiquitination-proteasome pathway.

NMT1 catalyzes myristoylation of a panel of proteins and regulates their molecular functions. Inhibition of NMT activity by LCL204 suppress global proteins myristoylation and enhance AR-ubiquitination and the proteasome degradation pathway. We hypothesize that the protein(s) whose activity are required by protein myristoylation regulate AR stability.
More importantly, this study has revealed a potential novel molecular function of NMT1 in regulating AR protein levels in prostate cancer cells. Knockdown of NMT1 or pharmacological inhibition of NMT1 activity led to a decrease of AR protein levels and its transcriptional activity (Fig. 7). Interestingly, LCL204 decreased both AR full-length and variants by an increase of AR ubiquitination-proteasome degradation. AR ubiquitination plays a role in the regulation of AR stability and its activity (14). There are three major AR ubiquitination sites: K311, K845, and K847 (15, 21). Of these three sites, K845 and K847 are located within the AR ligand binding domain. Since the AR-variant, which lacks the AR ligand binding domain at the C-terminus, is also regulated by NMT1, we reasoned that NMT1 regulation of AR-ubiquitination likely takes place at the K311 site in the AR N-terminus domain (21, 54) or at other unknown lysine sites. Identification of AR ubiquitination site(s) regulated by NMT1 will further provide mechanistic understanding of this degradation pathway.
NMTs regulate global protein myristoylation. This study has not yet revealed which E3 ligase might be potentially involved in NMT1-mediated AR ubiquitination and degradation (Fig. 7). Different E3 ligases have been reported to be involved in AR ubiquitination, which could either promote AR transcriptional activity or lead to degradation of AR (21). For instance, RNF6 is reported to be an E3 ligase for AR ubiquitination leading to an increase of AR transcriptional activity (15). On the other hand, Mdm2 is reported to bind with AR in a phosphorylation-dependent manner to promote its ubiquitination and degradation (16). Other E3 ligases including CHIP (c-terminus of Hsp-70-interacting protein), SKP2, and Siah2 could also potentially be involved in ubiquitination and degradation of AR (17, 19, 20). Future studies should focus on identification of the E3 ligase whose activity is sensitive to protein myristoylation. This knowledge will be very valuable for development of a novel AR degradation strategy.
Directly targeting AR for degradation remains an effective therapeutic strategy for treatment of CRPC. Numerous selective androgen receptor degraders (SARDs) such as UT-69 and SARD033 or Proteolysis Targeting Chimeric molecules such as ARCC-4 and ARV-330 have been developed for targeting full length of AR (55–61). However, the efficacy of these compounds relies on interaction with the ligand binding domain of AR. AR splice variants such as AR-V1 and AR-V7 which lack the ligand binding domain could frequently occur in tumors after androgen deprivation therapy (62). As a result, ARV-330 and ARCC-4 usually lose their efficacy in targeting AR-V7 for degradation. Currently other AR degraders such as ASC-J9 (a curcumin derivative) and 17-allylamino-17-demethoxygeldanamycin (17-AAG) are reported to enhance AR degradation and inhibit growth of CRPC cells (63, 64). However, the biological mechanisms of ASC-J9 are still unknown, and 17-AAG induced AR degradation via inhibition of Hsp90 function might be non-specific for AR degradation since it could also down-regulate a subset of cellular proteins such as HER2, HER3, and Akt (65). In this study, LCL204 pharmacologically inhibits NMT1 activity which leads to the degradation of full-length AR as well as its variant at low micromolar concentrations. In comparison with other AR degraders, our study provides an alternative approach for treatment of androgen sensitive and CRPC by inhibiting both AR-FL and AR-V7 levels in prostate cancer cells.
Our data show that expression levels of AR-full length (AR-FL) and AR-V7 were inhibited by LCL204, but the inhibition was compromised by the co-treatment with R1881 (Fig. 4A), suggesting that the binding of androgen protected AR ubiquitination of both AR-FL and AR-V7. It has been reported that AR-FL may form heterodimers with AR-V7 (66, 67). This interaction could be mediated between the N-terminal domain of AR-V7 and the C-terminal domain of AR-FL, or between DNA binding domains of AR-FL and AR-V7 (68). Our study suggests that the stabilization of AR-FL in the presence of the agonist may indirectly stabilize AR-V7 through the interaction. Therefore, NMT inhibitors might have a synergistic effect with inhibitors targeting AR signaling for treatment of prostate cancer.
Our study shows the functional role of NMT1 in prostate tumor progression. Knockdown of NMT1 inhibited growth of prostate xenograft tumors. This is consistent with other studies regarding NMT1 in cancer progression of various cancer types (29, 31, 37, 38, 46). NMT1 promotes myristoylation of a set of its downstream proteins such as AMPKA, Src kinase, and cAMP-dependent protein kinase (PKA) (26, 38, 69, 70). These proteins require myristoylation modification to carry out their molecular functions, and further synergize with AR in tumor progression. For example, Src kinase requires myristoylation for its kinase activity and oncogenic potential to synergize with AR in prostate tumorigenesis (38). The catalytic subunit of PKA is another protein that requires myristoylation for its molecular function (70–72). PKA regulates AR protein levels, nuclear translocation, and its transcriptional activity (27, 73). Elevation of PKA expression is associated with the progression of CRPC (74, 75). Further studies will be required to illustrate how loss of the myristoylation of these proteins will mediate inhibitory growth of PCa cells.
Supplementary Material
Acknowledgements:
This work was supported by NIH (U01CA225784), NIH (R21AI157831) and AICR (AICR850724) to HC. Mr. Omar Alsaidan is supported by a scholarship from Jouf University, Skaka, Saudi Arabia.
Footnotes
The authors disclose no potential conflicts of interest.
References
- 1.Bray F et al. , Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68, 394–424 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Huang H, Tindall DJ, The role of the androgen receptor in prostate cancer. Crit Rev Eukaryot Gene Expr 12, 193–207 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Tan MH, Li J, Xu HE, Melcher K, Yong EL, Androgen receptor: structure, role in prostate cancer and drug discovery. Acta Pharmacol Sin 36, 3–23 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dell’oglio P et al. , Treatment trends and Medicare reimbursements for localized prostate cancer in elderly patients. Can Urol Assoc J 12, E338–E344 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holzbeierlein J et al. , Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am J Pathol 164, 217–227 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harris WP, Mostaghel EA, Nelson PS, Montgomery B, Androgen deprivation therapy: progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat Clin Pract Urol 6, 76–85 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rice MA, Malhotra SV, Stoyanova T, Second-Generation Antiandrogens: From Discovery to Standard of Care in Castration Resistant Prostate Cancer. Front Oncol 9, 801 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chandrasekar T, Yang JC, Gao AC, Evans CP, Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl Androl Urol 4, 365–380 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakazawa M, Paller C, Kyprianou N, Mechanisms of Therapeutic Resistance in Prostate Cancer. Curr Oncol Rep 19, 13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abate-Shen C, Shen MM, Molecular genetics of prostate cancer. Genes Dev 14, 2410–2434 (2000). [DOI] [PubMed] [Google Scholar]
- 11.Scher HI, Buchanan G, Gerald W, Butler LM, Tilley WD, Targeting the androgen receptor: improving outcomes for castration-resistant prostate cancer. Endocr Relat Cancer 11, 459–476 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Schalken J, Fitzpatrick JM, Enzalutamide: targeting the androgen signalling pathway in metastatic castration-resistant prostate cancer. BJU Int 117, 215–225 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ge R et al. , Degradation of Androgen Receptor through Small Molecules for Prostate Cancer. Curr Cancer Drug Targets 18, 652–667 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Gioeli D, Paschal BM, Post-translational modification of the androgen receptor. Mol Cell Endocrinol 352, 70–78 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Xu K et al. , Regulation of androgen receptor transcriptional activity and specificity by RNF6-induced ubiquitination. Cancer Cell 15, 270–282 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin HK, Wang L, Hu YC, Altuwaijri S, Chang C, Phosphorylation-dependent ubiquitylation and degradation of androgen receptor by Akt require Mdm2 E3 ligase. EMBO J 21, 4037–4048 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sarkar S, Brautigan DL, Parsons SJ, Larner JM, Androgen receptor degradation by the E3 ligase CHIP modulates mitotic arrest in prostate cancer cells. Oncogene 33, 26–33 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.An J, Wang C, Deng Y, Yu L, Huang H, Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell Rep 6, 657–669 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qi J et al. , The E3 ubiquitin ligase Siah2 contributes to castration-resistant prostate cancer by regulation of androgen receptor transcriptional activity. Cancer Cell 23, 332–346 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li B et al. , Skp2 regulates androgen receptor through ubiquitin-mediated degradation independent of Akt/mTOR pathways in prostate cancer. Prostate 74, 421–432 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McClurg UL et al. , Identification of a novel K311 ubiquitination site critical for androgen receptor transcriptional activity. Nucleic Acids Res 45, 1793–1804 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boutin JA, Myristoylation. Cell Signal 9, 15–35 (1997). [DOI] [PubMed] [Google Scholar]
- 23.Farazi TA, Waksman G, Gordon JI, The biology and enzymology of protein N-myristoylation. J Biol Chem 276, 39501–39504 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Thinon E et al. , Global profiling of co- and post-translationally N-myristoylated proteomes in human cells. Nat Commun 5, 4919 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Varkaris A, Katsiampoura AD, Araujo JC, Gallick GE, Corn PG, Src signaling pathways in prostate cancer. Cancer Metastasis Rev 33, 595–606 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patwardhan P, Resh MD, Myristoylation and membrane binding regulate c-Src stability and kinase activity. Mol Cell Biol 30, 4094–4107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sarwar M, Sandberg S, Abrahamsson PA, Persson JL, Protein kinase A (PKA) pathway is functionally linked to androgen receptor (AR) in the progression of prostate cancer. Urol Oncol 32, 25 e21–12 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Khan AS, Frigo DE, A spatiotemporal hypothesis for the regulation, role, and targeting of AMPK in prostate cancer. Nat Rev Urol 14, 164–180 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Q et al. , Pharmacologically targeting the myristoylation of the scaffold protein FRS2alpha inhibits FGF/FGFR-mediated oncogenic signaling and tumor progression. J Biol Chem 293, 6434–6448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim S et al. , Myristoylation of Src kinase mediates Src-induced and high-fat diet-accelerated prostate tumor progression in mice. J Biol Chem 292, 18422–18433 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ducker CE, Upson JJ, French KJ, Smith CD, Two N-myristoyltransferase isozymes play unique roles in protein myristoylation, proliferation, and apoptosis. Mol Cancer Res 3, 463–476 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thinon E, Morales-Sanfrutos J, Mann DJ, Tate EW, N-Myristoyltransferase Inhibition Induces ER-Stress, Cell Cycle Arrest, and Apoptosis in Cancer Cells. ACS Chem Biol 11, 2165–2176 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gordon JI, Duronio RJ, Rudnick DA, Adams SP, Gokel GW, Protein N-myristoylation. J Biol Chem 266, 8647–8650 (1991). [PubMed] [Google Scholar]
- 34.Resh MD, Trafficking and signaling by fatty-acylated and prenylated proteins. Nat Chem Biol 2, 584–590 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Zha J, Weiler S, Oh KJ, Wei MC, Korsmeyer SJ, Posttranslational N-myristoylation of BID as a molecular switch for targeting mitochondria and apoptosis. Science 290, 1761–1765 (2000). [DOI] [PubMed] [Google Scholar]
- 36.Li H, Dou J, Ding L, Spearman P, Myristoylation is required for human immunodeficiency virus type 1 Gag-Gag multimerization in mammalian cells. J Virol 81, 12899–12910 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Das U, Kumar S, Dimmock JR, Sharma RK, Inhibition of protein N-myristoylation: a therapeutic protocol in developing anticancer agents. Curr Cancer Drug Targets 12, 667–692 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Kim S et al. , Blocking Myristoylation of Src Inhibits Its Kinase Activity and Suppresses Prostate Cancer Progression. Cancer Res 77, 6950–6962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhabak KP, Kleuser B, Huwiler A, Arenz C, Effective inhibition of acid and neutral ceramidases by novel B-13 and LCL-464 analogues. Bioorg Med Chem 21, 874–882 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Dagan A, Wang CB, Fibach E, Gatt S, Synthetic, non-natural sphingolipid analogs inhibit the biosynthesis of cellular sphingolipids, elevate ceramide and induce apoptotic cell death. Bba-Mol Cell Biol L 1633, 161–169 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Kallemeijn WW et al. , Validation and Invalidation of Chemical Probes for the Human N-myristoyltransferases. Cell Chem Biol 26, 892–900 e894 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Controulis J, Rebstock MC, Crooks HM, Chloramphenicol (Chloromycetin) .5. Synthesis. J Am Chem Soc 71, 2463–2468 (1949). [Google Scholar]
- 43.Xin L, Ide H, Kim Y, Dubey P, Witte ON, In vivo regeneration of murine prostate from dissociated cell populations of postnatal epithelia and urogenital sinus mesenchyme. Proc Natl Acad Sci U S A 100 Suppl 1, 11896–11903 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cai H et al. , Differential transformation capacity of Src family kinases during the initiation of prostate cancer. Proc Natl Acad Sci U S A 108, 6579–6584 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang SH et al. , N-myristoyltransferase 1 is essential in early mouse development. J Biol Chem 280, 18990–18995 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Deng L et al. , NMT1 inhibition modulates breast cancer progression through stress-triggered JNK pathway. Cell Death Dis 9, 1143 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rebstock MC, Crooks HM, Controulis J, Bartz QR, Chloramphenicol (Chloromycetin) .4. Chemical Studies. J Am Chem Soc 71, 2458–2462 (1949). [Google Scholar]
- 48.Mitscher LA, Kautz F, Lapidus J, Optical Rotatory Dispersion and Circular Dichroism of Diastereoisomers .I. Ephedrines and Chloramphenicols. Can J Chemistry 47, 1957-& (1969). [Google Scholar]
- 49.Dillon J, Nakanishi K, Absolute Configurational Studies of Vicinal Glycols and Amino Alcohols .2. With Pr(Dpm)3. J Am Chem Soc 97, 5417–5422 (1975). [DOI] [PubMed] [Google Scholar]
- 50.Dillon J, Nakanishi K, Absolute Configurational Studies of Vicinal Glycols and Amino Alcohols .1. With Ni(Acac)2. J Am Chem Soc 97, 5409–5417 (1975). [DOI] [PubMed] [Google Scholar]
- 51.Kumar S, Sharma RK, N-terminal region of the catalytic domain of human N-myristoyltransferase 1 acts as an inhibitory module. PLoS One 10, e0127661 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McConathy J, Owens MJ, Stereochemistry in Drug Action. Prim Care Companion J Clin Psychiatry 5, 70–73 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.French KJ et al. , Cyclohexyl-octahydro-pyrrolo[1,2-a]pyrazine-based inhibitors of human N-myristoyltransferase-1. J Pharmacol Exp Ther 309, 340–347 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Gao W, Bohl CE, Dalton JT, Chemistry and structural biology of androgen receptor. Chem Rev 105, 3352–3370 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ponnusamy S et al. , Novel Selective Agents for the Degradation of Androgen Receptor Variants to Treat Castration-Resistant Prostate Cancer. Cancer Res 77, 6282–6298 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gustafson JL et al. , Small-Molecule-Mediated Degradation of the Androgen Receptor through Hydrophobic Tagging. Angew Chem Int Ed Engl 54, 9659–9662 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Han X et al. , Discovery of ARD-69 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Androgen Receptor (AR) for the Treatment of Prostate Cancer. J Med Chem 62, 941–964 (2019). [DOI] [PubMed] [Google Scholar]
- 58.Salami J et al. , Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun Biol 1, 100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Teply BA, Antonarakis ES, Novel mechanism-based therapeutics for androgen axis blockade in castration-resistant prostate cancer. Curr Opin Endocrinol Diabetes Obes 23, 279–290 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Neklesa TK et al. , ARV-330: Androgen receptor PROTAC degrader for prostate cancer. Journal of Clinical Oncology 34, 267–267 (2016). [Google Scholar]
- 61.Neklesa T et al. , ARV-110: An oral androgen receptor PROTAC degrader for prostate cancer. Journal of Clinical Oncology 37, 259–259 (2019). [Google Scholar]
- 62.Sharp A et al. , Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J Clin Invest 129, 192–208 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yamashita S et al. , ASC-J9 suppresses castration-resistant prostate cancer growth through degradation of full-length and splice variant androgen receptors. Neoplasia 14, 74–83 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang R et al. , ASC-J9((R)) suppresses castration resistant prostate cancer progression via degrading the enzalutamide-induced androgen receptor mutant AR-F876L. Cancer Lett 379, 154–160 (2016). [DOI] [PubMed] [Google Scholar]
- 65.Solit DB et al. , 17-Allylamino-17-demethoxygeldanamycin induces the degradation of androgen receptor and HER-2/neu and inhibits the growth of prostate cancer xenografts. Clin Cancer Res 8, 986–993 (2002). [PubMed] [Google Scholar]
- 66.Sun S et al. , Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest 120, 2715–2730 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Antonarakis ES, Armstrong AJ, Dehm SM, Luo J, Androgen receptor variant-driven prostate cancer: clinical implications and therapeutic targeting. Prostate Cancer Prostatic Dis 19, 231–241 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu D et al. , Androgen Receptor Splice Variants Dimerize to Transactivate Target Genes. Cancer Res 75, 3663–3671 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mitchelhill KI et al. , Posttranslational modifications of the 5’-AMP-activated protein kinase beta1 subunit. J Biol Chem 272, 24475–24479 (1997). [DOI] [PubMed] [Google Scholar]
- 70.Tillo SE et al. , Liberated PKA Catalytic Subunits Associate with the Membrane via Myristoylation to Preferentially Phosphorylate Membrane Substrates. Cell Rep 19, 617–629 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bastidas AC et al. , Role of N-terminal myristylation in the structure and regulation of cAMP-dependent protein kinase. J Mol Biol 422, 215–229 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gaffarogullari EC et al. , A myristoyl/phosphoserine switch controls cAMP-dependent protein kinase association to membranes. J Mol Biol 411, 823–836 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tien AH, Sadar MD, Keys to unlock androgen receptor translocation. J Biol Chem 294, 8711–8712 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pollack A et al. , The importance of protein kinase A in prostate cancer: relationship to patient outcome in Radiation Therapy Oncology Group trial 92–02. Clin Cancer Res 15, 5478–5484 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dagar M, Singh JP, Dagar G, Tyagi RK, Bagchi G, Phosphorylation of HSP90 by protein kinase A is essential for the nuclear translocation of androgen receptor. J Biol Chem 294, 8699–8710 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gao T, Yang C, Zheng YG, Comparative studies of thiol-sensitive fluorogenic probes for HAT assays. Analytical and bioanalytical chemistry 405, 1361–1371 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.