

Abstract
Accurate determination of the clinical significance of genetic variants is critical to the integration of genomics in medicine. To facilitate this process, the NIH-funded Clinical Genome Resource (ClinGen) has assembled Variant Curation Expert Panels (VCEPs), groups of experts and biocurators which provide gene- and disease-specifications to the American College of Medical Genetics & Genomics and Association for Molecular Pathology’s (ACMG/AMP) variation classification guidelines. With the goal of classifying the clinical significance of GAA variants in Pompe disease (Glycogen storage disease, type II), the ClinGen Lysosomal Diseases (LD) VCEP has specified the ACMG/AMP criteria for GAA. Variant classification can play an important role confirming the diagnosis of Pompe disease as well as the identification of carriers. Furthermore, since the inclusion of Pompe disease on the Recommended Uniform Screening Panel (RUSP) for newborns in the USA in 2015, the addition of molecular genetic testing has become an important component in the interpretation of newborn screening results, particularly for asymptomatic individuals. To date, the LD VCEP has submitted classifications and supporting data on 243 GAA variants to public databases, specifically ClinVar and the ClinGen Evidence Repository. Here, we describe the ACMG/AMP criteria specification process for GAA, an update of the GAA-specific variant classification guidelines, and comparison of the ClinGen LD VCEP’s GAA variant classifications with variant classifications submitted to ClinVar. The LD VCEP has added to the publicly available knowledge on the pathogenicity of variants in GAA by increasing the number of expert-curated GAA variants present in ClinVar, and aids in resolving conflicting classifications and variants of uncertain clinical significance.
Keywords: Pompe disease, glycogen storage disease type II, GAA, variant classification, ClinGen Variant Curation Expert Panel, ClinVar
1. Introduction
Accurate determination of the clinical significance of genetic variants is foundational to genomic and personalized medicine. Publication of guidance by the American College of Medical Genetics and Genomics (ACMG) in collaboration with the Association for Molecular Pathology (AMP) in 2015 was an important step towards standardizing variant interpretation1. However, as these guidelines were written to apply to a wide range of Mendelian disorders, the addition of specific gene and disease specifications to the ACMG/AMP criteria is required to facilitate variant classification and to minimize possible discrepancies in classification between different laboratories. To address this issue, the Clinical Genome Resource (ClinGen), an NIH-funded consortium, assembles Variant Curation Expert Panels (VCEPs) which develop specific ACMG/AMP based criteria for different disease genes under guidance from the ClinGen Sequence Variant Interpretation Working Group (SVI). The specified criteria are then used to classify variants in the gene of interest, followed by submission of the classifications and supporting data to public databases, specifically ClinVar and the ClinGen Evidence Repository, following a standardized protocol2,3. ClinGen’s VCEP process has been approved as an FDA-recognized genetic variant database and, as such, variant classifications submitted to ClinVar from ClinGen VCEPs are posted with an “FDA Recognized Database” tag at the three star review level4.
The ClinGen Lysosomal Diseases (LD) VCEP was formed under the umbrella of the ClinGen Inborn Errors of Metabolism Clinical Domain Working Group. This group was formerly known as the Lysosomal Storage Diseases VCEP but recently changed its name in keeping with the recommendations of the WORLDSymposium5. The LD VCEP focused its initial efforts on specification of the ACMG/AMP criteria for interpretation of variants within GAA. GAA encodes the lysosomal enzyme acid alpha-glucosidase (GAA; EC 3.2.1.20). Deficiency of GAA causes Pompe disease (glycogen storage disease type II; acid maltase deficiency; MIM# 232300), an autosomal recessive lysosomal disorder with a broad clinical spectrum. Pompe disease is categorized according to the age of onset and severity of symptoms6,7,8. Patients with classical infantile-onset Pompe disease (IOPD) present in infancy with cardiomegaly and severe hypotonia. Without treatment, infants with IOPD typically die from cardiorespiratory insufficiency in the first year of life. Patients with a non-classical form of IOPD have been reported as presenting in the first year of life but without the severe cardiac features. Late-onset Pompe disease (LOPD) has a wide range of disease onset and rate of progression. Patients can present in childhood or adulthood. LOPD is characterized by progressive limb-girdle weakness and respiratory insufficiency. While severe cardiomyopathy and aortic root dilatation are infrequent in individuals with LOPD, vascular anomalies, such as aneurysms and arterial dolichoectasia have been reported and available data supports screening for cardiac features, particularly arrhythmias9–13. Due to the overlap in clinical features with many other neuromuscular conditions, there is often a lag in the diagnosis of LOPD individuals.
Historically, Pompe disease has been diagnosed by the measurement of GAA enzyme activity in appropriate tissues, such as fibroblasts, muscle, lymphocytes, leukocytes, and dried blood spots, with the identification of GAA variants by DNA sequencing used for confirmation of the diagnosis, and for testing family members14. However, genomic testing is increasingly being used as a first tier approach in diagnostics and carrier testing. As such, GAA variants may be identified when GAA is included on next generation sequencing panels for various clinical indications, as well as when exome or genome sequencing is performed. In addition, GAA sequencing can play a critical role in confirming the results of newborn screening, particularly if the results are ambiguous and the patient is not yet symptomatic. Furthermore, GAA has been added to the secondary findings list, which was published by the ACMG with the goal of providing actionable findings to individuals undergoing genomic analysis for a different indication15.
The currently approved treatment for Pompe disease is enzyme replacement therapy (ERT), which was first approved by the Food and Drug Administration (FDA) in the USA and by the European Medical Agency (EMA) in 2006. Additional therapies, including gene therapy, rescue of normal splicing by allele-specific oligonucleotides, and substrate reduction therapy are under investigation16–20. ERT has been shown to improve the clinical symptoms and longevity of individuals with Pompe disease16,21,22. As the initiation of ERT early in the natural history of disease is essential to the prevention of irreversible damage from occurring, newborn screening for the condition was initiated in various countries, beginning with Taiwan in 200523. In the United States, Pompe disease was added to the Recommended Uniform Screening Panel (RUSP) for newborns in 2015 and screening was implemented by 37 US newborn screening programs by December 202224. While the methods vary, newborn screening programs typically include the measurement of GAA activity in dried blood spots, followed by confirmatory enzyme assay, clinical assessment, and GAA sequencing.
Clearly, there is a critical need to understand the clinical significance of GAA variants in order to provide patients and their families with accurate diagnostic and prognostic information in a timely manner. Herein, we describe the development of the LD VCEP, a group that was initiated to generate ACMG/AMP criteria specifications for GAA variant classification. We describe the ACMG/AMP criteria specification process and the results of the GAA variant classification efforts to date, including submission of 243 variant classifications to ClinVar, a publicly available database, for use by the scientific and medical communities.
2. Methods
2.1. Development of GAA-specific ACMG/AMP criteria, Version 1
The LD VCEP (GAA subgroup) is currently composed of 17 individuals with expertise in clinical/biochemical genetics, clinical laboratory diagnostics, variant interpretation, research, newborn screening, and variant classification, from various institutions across the USA and Canada (https://clinicalgenome.org/affiliation/50009/).
Following the ClinGen VCEP protocol3, the LD VCEP held biweekly conference calls to review the ACMG/AMP variant interpretation criteria1. Some criteria were deemed “not applicable” to GAA or Pompe disease. For other criteria, the group developed gene- or disease-specific specifications, and discussed the appropriate strength of evidence at which the criteria could be applied. Population minor allele frequency (MAF) threshold calculations for BA1, BS1, and PM2 were made using an online calculator (http://cardiodb.org/allelefrequencyapp)25. Additional efforts included identification of key functional domains in GAA, definition of informative functional assays, and characterization of specific phenotypic criteria. Recommendations for using ACMG/AMP criteria from the ClinGen’s Sequence Variant Interpretation working group (SVI) were also incorporated26. The ClinGen SVI is working towards improving the consistency in usage of the ACMG/AMP criteria and transparency in classification rationale and oversees the development of variant classification rules by ClinGen VCEPs.
2.2. Collection and analysis of data
Published literature for each variant was identified by PubMed and GoogleScholar searches, in addition to variant-specific data from the Pompe Variant Database (https://www.pompevariantdatabase.nl/pompe_mutations_list.php). Data was collected and entered in the ClinGen Variant Curation Interface (VCI, https://curation.clinicalgenome.org/)27. under the appropriate ACMP/AMP criteria codes by biocurators following the VCEP Standard Operating Procedure3. The VCI pulls in additional data for curation from public databases such as gnomAD (https://gnomad.broadinstitute.org/)28, and also the results of in silico predictors including REVEL29. Anonymized, unpublished data from two participating clinical laboratories (the Duke Molecular Diagnostics Laboratory and Revvity Omics [formerly Perkin Elmer Genomics]) were included when available. All variants were annotated using NM_000152.5, and all were classified for Pompe disease (MONDO:0009290). Using the GAA-specific ACMG/AMP guidelines, biocurators made a provisional classification for each variant. After three experts reviewed the data and the provisional classification, the classification for each variant was approved. The classification and supporting data were then published in the public ClinGen Evidence Repository and submitted to ClinVar.
2.3. Pilot study
The draft specifications were tested and refined by classifying 50 GAA variants which were selected to represent the spectrum of variant types reported in GAA, including missense, nonsense, frameshift, canonical splice site, intronic, and in-frame indels, as well as a range of classifications in ClinVar. For in-frame insertion and deletions, of which there are few in ClinVar, we classified unpublished variants identified by the Duke Molecular Diagnostic laboratory.
The criteria applied and subsequent variant classifications were reviewed on conference calls to stimulate further discussion, resolve discrepancies, and reach a final consensus on the classification of each variant. Once the specifications were approved by the SVI, the GAA variant classifications were submitted to ClinVar where they are displayed with a 3-star evidence level and the FDA recognition tag.
2.4. Update to Version 2 GAA-specific ACMG/AMP criteria
Concurrent with approval of Version 1 of the specifications, guidance for the use of functional data in variant classification and the recommended use of PM2 at the supporting strength level rather than at the moderate level, were published by the ClinGen Sequence Variant Interpretation Working Group30,31. The specifications were updated to Version 2 by incorporating these recommendations as well as other adjustments based on our experience using Version 1 of the specifications. Variant classifications made using Version 1 of the specifications were updated using Version 2. Differences in the number of times the updated codes were used, the strengths at which these codes were used, and the impact these code changes on final variant classifications was determined.
2.5. ClinVar data
Data from the NCBI’s public database, ClinVar32, were surveyed to determine the number of GAA variants and their classifications, submitted by all submitters (data obtained May 2, 2023). All ClinVar variant records for which the LD VCEP had submitted a classification were analyzed to determine the number of novel variants added to ClinVar by the LD VCEP, and the number of conflicting and VUS classifications that were resolved by the LD VCEP.
3. Results and discussion
3.1. Development of the ClinGen Lysosomal Diseases VCEP
The ClinGen LD VCEP underwent the steps required for a ClinGen-associated group to submit variant classifications to ClinVar at the 3-star review status and with the “FDA Recognized Database” tag.
3.2. Summary of specifications
In Version 1 of the GAA variant classification specifications, 13 of the 28 ACMG/AMP criteria were deemed by the LD VCEP to be inapplicable to GAA variant interpretation (PS2, PS4, PM1, PM6, PP1, PP2, PP5, BS2, BS4, BP1, BP3, BP5, and BP6), 6 were used “as is” with the option to change strength under certain circumstances (PS1, PM3, PM4, PM5, BP2, and BP7) and 11 were modified based on characteristics specific to GAA and Pompe disease (PVS1, PS3, PM2, PP3, PP4, BA1, BS1, BS3, and BP4). Based on further consideration, PM1 and BS2 were specified and included in Version 2 of the specifications. The Version 2 pathogenic code specifications are shown in Table 1, the benign code specifications are shown in Table 2, and a summary of updates made from Version 1 to Version 2 are shown in Table 3. The current specifications are also available in the ClinGen Criteria Specification Registry33.
Table 1.
Summary of the specifications made to Pathogenic ACMG-AMP criteria codes for the classification of variants in GAA (Version 2).
| ACMG/AMP Criterion | ACMG/AMP criterion description | Specification | Strength | Description |
|---|---|---|---|---|
| PVS1 | Null variant in a gene where loss of function is a known mechanism of disease |
Gene-specific, strength | Very Strong | • Any nonsense, frameshift, or splice variant creating a premature stop codon before codon 916a. • In-frame deletions of an entire exon containing critical active site/substrate binding residuesa (exons 8 and 10), or for which another variant removing the exon is known to be pathogenic (exons 2 and 18). |
| Strong | • In-frame loss of an exon which is part of the catalytic barrel domain and contains pathogenic/likely pathogenic non-truncating variants (exons 6 and 9). • Initiator codon variantb. |
|||
| Moderate | • Premature termination codon in the 3’ end of GAA (3’ to codon 916), not predicted to be degraded by nonsense-mediated decay. • Predicted exon-skipping due to canonical splice variant or exon deletion resulting in an in-frame deletion of <10% of the gene product (exons 17, 19, and 20). |
|||
| PS1 | Same amino acid change as a previously established pathogenic variant regardless of nucleotide change. | N/A | Strong | No changes |
| PS2 | De novo (maternity and paternity confirmed) in a patient with the disease and no family history. | Not used | N/A | N/A |
| PS3 | Well-established in vitro or in vivo functional studies supportive of a damaging effect. | Gene-specific, strength | Strong | RT-PCR evidence of mis-splicing for non-canonical intronic variants with no evidence of normal splice products. |
| Moderate | • <5% wild type GAA activity when the variant is expressed in a heterologous cell type and evidence of abnormal GAA synthesis and/or processing. • RT-PCR evidence of mis-splicing for non-canonical intronic variants with evidence of normal splice products. |
|||
| Supporting | • <30% wild type GAA activity when the variant is expressed in a heterologous cell type without additional evidence of abnormal synthesis and/or processing. • RT-PCR evidence of mis-splicing for non-canonical intronic variants with evidence of normal splice products. |
|||
| PS4 | The prevalence of the variant in affected individuals is significantly increased compared to the prevalence in controls. | Not used | N/A | N/A |
| PM1 | Located in a mutational hot spot and/or critical and well-established functional domain without benign variation | Gene-specific | Moderate | Missense substitution or in frame deletion of residues important in the active site architecture and substrate binding of GAA:- Asp282, Trp376, Asp404, Leu405, Ile441, Trp481, Trp516, Asp518, Met519, Arg600, Trp613, Asp616, Trp618, Phe649, Leu650, His674. |
| PM2 | Low frequency in population databases. | Supporting | Minor allele frequency <0.1% (0.001) in all continental populations with >2000 alleles in gnomAD. | |
| PM3 | Detected in trans with a pathogenic variant | Strength | Very Strong | 4 or more points (see SVI recommendations) |
| Strong | >2-4 points (see SVI recommendations) | |||
| Moderate | 2-<4 points (see SVI recommendations) | |||
| Supporting | 1-<2 points (see SVI recommendations) | |||
| PM4 | Protein length changes due to in-frame deletions/insertions in a non-repeat region or stop-loss variants | Strength | Moderate | In-frame deletion/insertions of two or more amino acids but less than one exon. |
| Supporting | In-frame deletion/insertions of one amino acid. | |||
| PM5 | Missense change at an amino acid residue where a different missense change determined to be pathogenic has been seen before. | Strength | Moderate | No changes |
| Supporting | Missense change at an amino acid residue where a different missense change determined to be pathogenic by the LD VCEP has been seen before. |
|||
| PM6 | Confirmed de novo without confirmation of paternity and maternity. | Not used | N/A | N/A |
| PP1 | Co-segregation with disease in multiple affected family members. | Not used | N/A | N/A |
| PP2 | Missense variant in a gene that has a low rate of benign missense variation and where missense variants are a common mechanism of disease. | Not used | N/A | N/A |
| PP3 | Multiple lines of computational evidence support a deleterious effect on the gene or gene product. | Gene-specific | Supporting | • REVEL score >0.7 for missense variants. • In-frame deletion or insertion predicted to be deleterious by 2 out of 3 tools (e.g. PROVEAN, MutationTaster, MutPred-InDel). • Predicted impact on splicing by SpliceAI (score >0.5). |
| PP4 | Phenotype specific for disease with single genetic etiology. | Gene-specific, strength | Moderate Supporting | Points-based system Points-based system |
| PP5 | Reputable source recently reports variant as pathogenic but the evidence is not available to the laboratory to perform an independent evaluation | Not used | N/A | N/A60 |
50 base pairs upstream of the penultimate exon/intron boundary, the point beyond which nonsense-mediated decay is not predicted for a premature termination codon.
Table 2.
Summary of specifications made to Benign ACMG-AMP criteria codes for the classification of variants in GAA (Version 2).
| ACMG/AMP Criterion | ACMG/AMP criterion description | Specification | Strength | Description |
|---|---|---|---|---|
| BA1 | Common in population databases. | Disease-specific | Stand alone | Highest minor allele frequency >0.01 (>1%) in any continental population in gnomAD with >2000 alleles. |
| BS1 | Allele frequency greater than expected for disease. | Disease-specific | Strong | Highest minor allele frequency >0.005 (>0.5%) in any continental population in gnomAD with >2000 alleles. |
| BS3 | Well-established in vitro or in vivo functional studies show no damaging effect on protein function. | Disease-specific, strength | Supporting | >50% activity when the variant is expressed in a heterologous cell type, or >30% activity if there is also evidence of normal synthesis and processing. |
| BS4 | Lack of segregation in affected members of a family. | Not used | N/A | N/A |
| BP1 | Missense variant in gene where only LOF causes disease. | Not used | N/A | N/A |
| BP2 | Observed in cis with a pathogenic variant. | None | Supporting | No changes |
| BP3 | In-frame deletions/insertions in a repetitive region without a known function. | Not used | N/A | N/A |
| BP4 | Multiple lines of computational evidence suggest no impact on gene or gene product. | Gene-specific | Supporting | • REVEL score <0.5 for missense variants. • In-frame deletion or insertion predicted benign by 2 out of 3 tools (e.g. PROVEAN, MutationTaster, and MutPred-InDel). • No predicted impact on splicing by SpliceAI (score <0.2) |
| BP5 | Variant found in a case with an alternate molecular basis for disease. | Not used | N/A | N/A |
| BP6 | Reputable source recently reports the variant as benign but the evidence is not available to the laboratory to perform an independent evaluation. | Not used | N/A | N/A60 |
| BP7 | A synonymous (silent) variant for which splicing prediction algorithms predict no impact to the splice consensus sequence nor the creation of a new splice site AND the nucleotide is not highly conserved. | None | Supporting | No changes |
Table 3.
Summary of the updates made from Version 1 to Version 2 of the ACMG-AMP criteria code specifications for the classification of variants in GAA.
| Version 1 | Version 2 | Main impact | Justification | |
|---|---|---|---|---|
| Pathogenic criteria | ||||
| PS3 | Criterion applied at strong for <5% activity and moderate for 5-30% activity in appropriate functional assays. | Criterion can no longer be applied at strong; applied at moderate for <5% GAA activity AND evidence of abnormal processing; and supporting for <5% GAA activity in appropriate functional assays. | Downgraded | SVI guidance 30 |
| PM1 | Not applicable. | Missense substitution or in frame deletion of residues important in the active site architecture and substrate binding of GAA. | Added | Re-evaluation by VCEP members |
| PM2 | If the threshold is met, apply at moderate strength. | If threshold is met, apply at supporting strength, based on SVI recommendations. | Downgraded | SVI guidance (https://clinicalgenome.org/site/assets/files/5182/pm2_-_svi_recommendation_-_approved_sept2020.pdf) |
| PM3 | Cases must meet PP4 to be counted for PM3 | Cases are no longer required to meet the strict PP4 criterion in order to be counted for PM3. However, some evidence for the diagnosis of Pompe disease must be present. | Allows use of additional data | Re-evaluation by VCEP members |
| PP4 | Specific values for residual activity must be available and meet the criteria. | A point-based system was developed to allow the use of additional data. | Allows use of additional data | Re-evaluation by VCEP members |
| Benign criteria | ||||
| BS2 | Not applicable. | Homozygous individual of any age with normal GAA activity. | Added | Re-evaluation by VCEP members |
| BS3 | Criterion applied at strong for >60% activity, moderate for 40-60% activity in appropriate functional assays, or 30-60% activity if evidence of normal synthesis and processing is also available. | Criterion can no longer be applied at strong; applied at supporting for >50% activity in appropriate functional assays, and >30% activity if evidence of normal synthesis and processing is also available. | Downgraded | SVI guidance 30 |
3.2.1. Computational and predictive data
Null variants (PVS1)
PVS1 is defined as “null variant (nonsense, frameshift, canonical +/−1 or 2 splice sites, initiation codon, single or multi-exon deletion) in a gene where loss of function (LOF) is a known mechanism of disease”1. GAA encodes acid alpha-glucosidase, a monomeric lysosomal enzyme encoded by a single gene with no evidence of alternative active isoforms. As loss of function of GAA is the disease mechanism for Pompe disease, PVS1 may be applied for null variants. PVS1 was applied using additional guidance from the ClinGen SVI34 that included gene-specific considerations (see Supplemental Table 1). For initiator codon variants, after the start methionine, the next in-frame methionine is at position 122 but the likelihood of this start site being used is low and, even if used, the gene product would be missing the signal sequence35, suggesting that GAA protein would be mislocalized and/or degraded. This prompted us to apply PVS1 at strong for initiator codon variants. Indeed, GAA protein has been shown to be absent by Western blot analysis from fibroblasts derived from three patients homozygous for c.1A>G35. For variants involving either the +1 or +2 position of GT donor splice sites, the exon immediately 5’ of the variant is predicted to be skipped, and all variants of either the −1 or −2 position of AG acceptor splice sites, the exon immediately 3’ of the variant is predicted to be skipped, unless there is experimental data to indicate otherwise. The in-frame/out-of-frame consequences for skipping any exon in GAA was determined (Supplemental Table 2) and, for variants predicted to result in in-frame exon skipping, the strength of evidence was based on knowledge functional domains within the relevant exon.
3.2.2. Computational predictors (PP3, BP4)
In silico predictors are useful ancillary tools to support variant interpretation and are incorporated into the ACMG/AMP guidelines as “Multiple lines of computational evidence support a deleterious effect (PP3)/ benign effect (BP4) on the gene or gene product”. Typically, computational predictions are based on amino acid properties, protein folding characteristics, conservation through evolution, region constraint, and allele frequency in population vs disease-based databases. The LD VCEP uses REVEL to predict the impact of missense variants29 and SpliceAI to predict the impact of all variants on splicing36. PP3 was applied to missense variants with a REVEL score ≥0.7, and BP4 was applied for a REVEL score ≤0.5, based on the sensitivity and specificity for detecting Pathogenic/Likely Pathogenic variants and Benign/Likely Benign variants at different thresholds (Supplemental Table 3). All variants were analyzed using SpliceAI to investigate a possible effect on splicing. A SpliceAI score of ≥0.5 met PP3, while a score of <0.2 was taken to indicate no predicted impact on splicing. Since Version 2 of the GAA specifications was approved, additional guidance has been released by the SVI on the use of in silico predictions for missense variants37, and on the incorporation of PP3 for splice variants38. These recommendations will be included in the next iteration of the LD VCEP’s GAA variant classification specifications.
3.2.3. Population data
Allele frequency criteria (BA1, BS1, and PM2)
Consideration of the allele frequency is useful in assessing the pathogenicity of a variant. The allele frequency may be too common to be considered disease causing (BA1), greater than expected for the disorder in question (BS1), or rare enough to be consistent with the disease frequency, penetrance, and inheritance pattern (PM2)1,39. The LD VCEP used the calculator at http://cardiodb.org/allelefrequencyapp25 to determine the minor allele frequency threshold for each of the allele frequency criteria (BA1, BS1, PM2). This online tool requires the user to input the prevalence, maximum allelic contribution, maximum genetic contribution, and penetrance. For all three allele frequency criteria, we set the maximum genetic contribution at 100% under the assumption that all cases of Pompe disease are caused by variants in GAA. We used different values for each of the remaining variables with the goal of setting conservative cutoffs so that no pathogenic variants would meet BS1 or BA1. While the penetrance of Pompe disease is not known, it is assumed to be high and, therefore, we set the penetrance to 100% for all three allele frequency criteria. The prevalence of Pompe disease is variable depending on the population40–42. It is typically quoted to be approximately 1 in 40,000 based on individuals diagnosed after presenting with clinical symptoms. However, with the surge of newborn screening programs for Pompe disease in several countries and different US states, it is now known that this is an underestimate due, at least in part, to underdiagnosis of individuals with LOPD whose symptoms overlap with a myriad of other neuromuscular conditions. Based on newborn screening studies, Pompe disease has an incidence as high as 1 in 4,500 individuals in some populations when combining infantile and late-onset cases43–45, with most studies reporting ~ 1 in 10,000 - 1 in 20,000. Therefore, we used a prevalence value of 1 in 10,000 for the BA1 and BS1, and 1 in 40,000 for PM2.
The maximum allelic contribution, or maximum number of cases attributable to a single variant, varies between populations and clinical cohorts. However, based on overall data from the Pompe Registry46, the most prevalent variant in individuals with onset of symptoms <12 years and cardiomyopathy is c.2560C>T - p.(Arg854Ter), present in 7.5% of the alleles (28/373 alleles), while c.-32-13T>G is the most common variant in late-onset cases, present in 45.1% of the alleles (561/1243 alleles). To calculate the thresholds for the BA1, BS1, and PM2 criteria, we used maximum allelic contribution of 1.0, 0.5, and 0.2.
Based on these parameters, the final minor allele frequency thresholds of BA1, BS1, and PM2 were set to >1% (>0.01), >0.5% (>0.005), and <0.1% (<0.001) respectively, while recognizing the limitations that allele frequencies of pathogenic variants and the prevalence of disease varies considerably between different populations. Of note, some well-known pathogenic variants do not meet our PM2 threshold, but there is sufficient case-level and functional evidence to support their pathogenicity. This includes c.2560C>T - p.(Arg854Ter) (MAF 0.001891 in the African/African-American population), c.1935C>A - p.(Asp645Glu) (MAF 0.001729 in the East Asian population), and c.-32-13T>G (MAF 0.005293 in the European non-Finnish population).
In Version 2 of the GAA-specific ACMG/AMP guidelines, following the guidance of the ClinGen SVI WG31, PM2 was applied at the supporting level to variants meeting the PM2 threshold, rather than at the moderate level used in Version 1.
Observation in controls inconsistent with disease (BS2)
For recessive disorders, BS2 is defined as “observed in a healthy adult individual”1. While this criterion was not included in Version 1 of the specifications, it was specified and included in Version 2. Of note, observation of a GAA variant in homozygosity in a healthy adult does not exclude the variant as being pathogenic because LOPD can present late in life (5th-6th decade), can have mild symptoms, and may remain undiagnosed. It is possible that homozygotes may be present in large population databases such as gnomAD. In fact, a homozygous individual is reported in gnomAD for the most common pathogenic GAA variant in individuals with late-onset Pompe disease (c.−32-13T>G). To apply this criterion, the LD VCEP therefore, requires that there is clear documentation of normal GAA activity in an individual with no clinical features of Pompe disease who is homozygous for the variant, or compound heterozygous for the variant and a known pathogenic variant, with confirmed phase. Because the residual GAA activity is not expected to change throughout the lifetime, this criterion can be applied for an individual of any age, if the requirements are met.
3.2.4. Functional data
Functional assays (PS3 / BS3)
The ACMG/AMP guidelines define functional assays as “well-established in vitro or in vivo” studies that support a deleterious (PS3) or benign (BS3) impact of the variant on the function of the protein, recommending that these codes be applied at the strong level. GAA activity can be measured in an appropriate cell type from patients (fibroblasts, leukocytes, dried blood spots, muscle)47 or in cultured cells expressing GAA cDNA48,49. GAA activity measured in cells from a patient represents a feature of that individual and the interaction of all of the genetic variants that they carry. This data, therefore, is included as evidence for PP4 (“disease-specific phenotype”) rather than PS3 and BS3. Assays that involve transient expression of GAA cDNA containing a variant of interest in COS cells, HEK293 cells, or GAA-deficient fibroblasts, are broadly accepted as in vitro functional assays for PS3 and BS3. Some studies also include analysis of GAA synthesis and processing by Western blot or pulse chase to assess the amount of precursor protein (110 kDa), intermediate (95 kDa) and active enzyme (76, 70 kDa)50. In Version 1 of our specifications, we applied PS3 and BS3 at the strong level for variants with low (<10%) or high (>60%) activity in these in vitro assays. However, subsequent guidance from the ClinGen SVI prompted us to re-specify these criteria based on the recommendation that functional assays include a minimum number of pathogenic and benign controls30.
Unfortunately, the majority of published functional assays for GAA do not include control variants. We therefore attempted to identify variants examined in these historical studies that could retrospectively be considered as pathogenic or benign controls, as these variants could be classified as pathogenic or benign using other evidence, without applying PS3 and BS3. Although this approach identified a number of suitable control variants, the number was insufficient to meet the recommendations of the SVI. Therefore, PS3 may be applied at the supporting level for any variant with <30% activity when transiently expressed in vitro. PS3 may be applied at the moderate level for detailed studies that combine GAA activity with Western blot data48,49. While we recognize that GAA synthesis and processing and GAA activity are closely correlated, analysis of the impact of a variant by two different methods provides some verification of the results.
Critical domain (PM1)
PM1 can be applied for variants that are “located in a mutational hot spot and/or critical and well-established functional domain (e.g. active site of an enzyme) without benign variation”1. This criterion was not included in Version 1 of the GAA variant classification guidelines but, upon further consideration by the members of the LD VCEP, it was included in Version 2. GAA is an exo-hydrolase cleaving both α1-4 and α1-6 glucose linkages in glycogen and releasing α-glucose as a product. The active site region of GAA encompasses amino acids 347-72650. Based on the crystal structures of native GAA and rhGAA, the following residues have been identified as important to the active site architecture, mechanism, and/or substrate binding of GAA: Asp282, Trp376, Asp404, Leu405, Ile441, Trp481, Trp516, Asp518, Met519, Arg600, Trp613, Asp616, Trp618, Phe649, Leu650, His67451,52. No variants at these positions have been classified as benign or likely benign in ClinVar or the Pompe Variant database53, and all variants of these residues were rare or absent in gnomAD v2.1.1. Of note, Asp518 is the catalytic nucleophile and Asp616 is the catalytic acid/base 51,52,54. The other residues are important in active site architecture and substrate binding. In Version 2 of the GAA-specific variant classification guidelines, PM1 was applied to missense substitutions and in-frame deletions of these residues.
3.2.5. Allelic data (PM3)
For recessive disorders, like Pompe disease, observation of the variant of interest in trans with a pathogenic variant supports the pathogenicity of the variant1. The LD VCEP applied PM3 following the guidance of the SVI55 which has developed a points system based on the classification of the second variant and whether or not the phase is known, for compound heterozygotes. The issue of circularity was carefully considered for all variants with case-level data to ensure that the classification of the variant on the other allele did not use evidence from the variant being interrogated, as recommended by the ClinGen SVI WG. The allelic evidence used in evaluating compound heterozygous cases was documented clearly within the VCI and written summary.
3.2.6. Specific phenotype (PP4)
PP4 can be applied if the “patient’s phenotype or family history is highly specific for a disease with a single genetic etiology”1. For Version 1 of the specifications, the LD VCEP considered that deficiency of GAA activity, measured in a relevant sample type such as fibroblasts, leukocytes, dried blood spots or muscle, is highly specific for Pompe disease, in the absence of “pseudodeficiency variants” (c.1726G>A p.(Gly576Ser); c.2065G>A - p(.Glu689Lys); and c.271G>A - p.(Asp91Asn))47 . In Version 1, this was the only specification included for PP4, with the added detail that the data available for a patient must meet PP4 in order for PM3 to be applied. With increased experience in using Version 1 of the guidelines, the LD VCEP found this specification to be overly strict, resulting in the inability to count data for cases that had been diagnosed with Pompe disease but for whom the data required were not provided in publications. It is important to note that a positive newborn screen with reduced GAA enzyme activity is not considered sufficient for the application of PP4. Deficient enzyme activity must be demonstrated by a diagnostic assay, which may be performed on fibroblasts, leukocytes, dried blood spots or muscle47, 56–59. For Version 2 of the specifications, we considered all clinical and laboratory features of Pompe disease and specificity of these features for Pompe disease in comparison to other disorders. A points system was developed to guide curators in weighing the evidence according to the available data; with sufficient evidence PP4 can be applied at supporting or upgraded to the moderate level. In addition, the requirement for cases to meet PP4 in order to count PM3 was removed, as long as there was sufficient evidence to indicate that an individual had the diagnosis of Pompe disease (Table 4).
Table 4.
Points allocated for different types of evidence for PP4.
| Description of evidence | Points |
|---|---|
| Deficient GAA activitya, documented as either 1) <10% of normal mean control level of GAA activity in leukocytes, lymphocytes, or muscleb samples, and/or <30% in cultured fibroblastsc, or 2) Activity in the affected range (which must be provided in the publication) in any appropriate tissue (muscle, cultured skin fibroblasts, leukocytes, lymphocytes, whole blood or dried blood spot diagnostic assay; newborn screening methodologies not appropriate). | 2 |
| Patient reported to have Infantile Onset Pompe disease (IOPD) AND documentation of symptomsd of that condition. At a minimum, cardiomegaly, hypertrophic cardiomyopathy, left ventricular hypertrophy or a related term, and hypotonia, muscle weakness, or a related term, must be reported. | 1 |
| Cross reactive immunological material (CRIM) study of cultured skin fibroblasts or peripheral blood mononuclear cells | 1 |
| reported to show absence of the 76 Da and 70 kDa bands, which represent the mature, active GAA enzymee. This includes patients described as CRIM-negative (with no detectable GAA protein on Western blot), or those who are CRIM-positive but do not make the mature protein (e.g. only 110 kDa and 95 kDa bands are present). | |
| The patient is reported to be on enzyme replacement therapyf for Pompe disease. | 1 |
| GAA activity is reported to be deficienta but the data are not provided (i.e. values for the patient and normal range as determined by the testing laboratory) | 0.5 |
| Individual identified by positive newborn screening results | 0.25 |
| Urinary Glc4g is elevated above the normal range. | 0.25 |
| Muscle MRI shows evidence of Pompe disease. | 0.25 |
| Muscle histologyh is consistent with Pompe disease; there is glycogen storage in the lysosomes of muscle cells appearing as vacuoles that stain positively with periodic acid-Schiff. | 0.25 |
For a total of 2 points or more, PP4_Moderate is applied; for 1 point, PP4 is applied.
Criterion not applied if the variant meets BA1, or otherwise meets criteria for benign or likely benign classification.
If either of the pseudodeficiency variants c.1726G>A - p.(Gly576Ser) or c.2065G>A - p.(Glu689Lys) are present, whether heterozygous or homozygous, deficiency of GAA activity cannot be used to apply PP4. If c.271G>A - p.(Asp91Asn) is present, deficiency of GAA activity cannot be used to apply PP4 if glycogen was the assay substrate, but the data can be used if 4-MU was the substrate36.
<10% activity in muscle is used because activity of GAA in muscle samples can overlap in patients with LOPD and carriers.
To account for higher residual GAA activity in patients with late onset Pompe disease (LOPD)
Symptoms in patients with IOPD are fairly specific with few other conditions mimicking this disorder. Therefore, documentation of symptoms is not considered to be sufficient for application of PP4
For further details on the synthesis and intracellular processing and transport of GAA, and CRIM analysis in patients with Pompe disease, please see refs 35 and 63.
If a patient is receiving Enzyme Replacement Therapy, the assumption is that their diagnosis of Pompe disease is well supported by clinical and laboratory evaluations
Elevated urinary glucose tetrasaccharide (Glc4, also known as hexose tetrasaccharide or Hex4), which is derived from the amylolytic degradation of glycogen, has a high sensitivity and specificity for Pompe disease64, 65.
While histochemical evidence of glycogen storage in muscle is supportive of a glycogen storage disorder it is not specific for Pompe disease.
3.2.7. Criteria not included
Various codes were not included in the specifications because they were found to be not applicable to GAA or Pompe disease. This includes the codes for de novo variants (PS2, PM6) because de novo variants are rarely reported in GAA (PMID: 7981676, 27142047) and any de novo variants would be assessed based on the variant type, functional evidence, and in trans data as described in these guidelines. PS4 was not included because there are no case-control studies for Pompe disease and, as this is a recessive disorder, the number of patients with the variant is, instead, addressed by the PM3 evidence code. The segregation codes (PP1, BS4) are not used because GAA is the only gene involved in Pompe disease and, therefore, any variant in the gene would likely co-segregate with the disease, regardless of whether that variant is pathogenic. Benign and pathogenic missense variation is spread throughout the gene, with no known constrained regions, ruling out the use of PP2 and BP1. BP5 was not used because there is no known alternate molecular basis for deficiency of GAA activity, other than variants in GAA. The reputable source criteria (PP5, and BP6) were removed, based on recommendations from the ClinGen SVI60.
3.4. Variant classification using GAA-specific ACMG/AMP variant classification guidelines
To date the LD VCEP has classified 243 variants using GAA-specific variant classification guidelines and has submitted those classifications to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and the ClinGen Evidence Repository (https://erepo.clinicalgenome.org/evrepo/) (Figure 1). One hundred and thirty seven variants were classified initially using Version 1 of the specifications. As mentioned, the differences between Version 1 and Version 2 of the specifications included less strict requirements for the application of PP4, the application of PP4 is no longer a requirement to count points for PM3, the downgrading of PM2 to PM2_Supporting for all variants meeting the PM2 allele frequency threshold, and the downgrading of functional evidence from PS3 to PS3_Moderate or PS3_Supporting. PP4, at any strength, was applied to 15 variants using the Version 1 specifications and 21 times with Version 2, reflecting the allowance to use additional types of data. For the majority of variants, the strength increased from supporting to moderate, again due to increased data use, but also the increase in evidence strength for data indicating GAA deficiency. Similarly, PM3, at any strength, was applied for 15 variants with Version 1 but 37 times using Version 2, and applied at higher strengths, again reflecting less strict requirements which allowed the use of additional data. Conversely, PS3 was applied at strong for 12 variants using Version 1, but downgraded to moderate strength for 8 of those variants and to supporting strength for 4 variants. A comparison of criterion strength utilization for both Versions of the specifications is shown in Supplemental Table 4.
Figure 1.
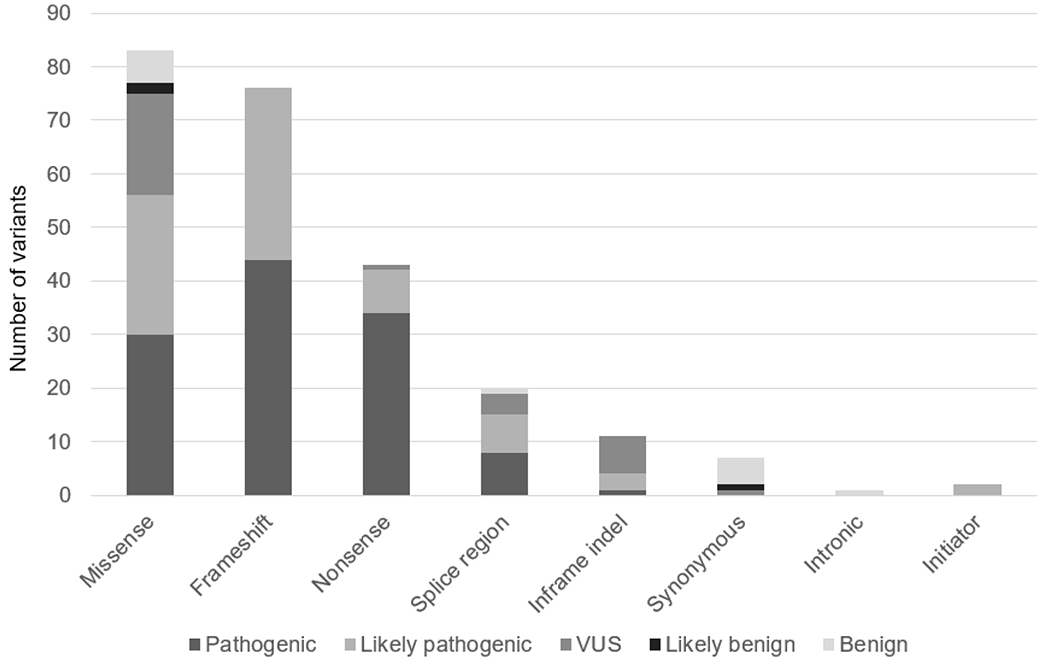
Classification of GAA variants submitted to ClinVar by the ClinGen LD VCEP (n=243)
To assess the overall impact of these criteria changes, the classifications of all 137 variants classified with Version 1 were assessed using Version 2 (Supplemental Table 5). Importantly, no new data was added for this analysis so that any differences in classification were solely due to the criteria specifications. Eight of 34 loss-of-function variants (nonsense and frameshift) for which only PVS1 and PM2 had been applied initially, were upgraded from likely pathogenic to pathogenic based on the ability to use case-level evidence (PM3 and/or PP4) in Version 2. However, the classifications of 2/22 missense variants and 2/10 in frame deletions were downgraded (three downgraded from P to LP and one from LP to VUS) due to the decrease in strength of functional data in Version 2 as recommended by the SVI30. The SVI has concluded that traditional enzyme based functional expression studies require the assessment of multiple known benign variants in addition to known pathogenic variants in order to be used at a moderate strength30. The availability of rigorous functional studies that comply with current recommendations is important for variant classification; such studies are underway for GAA61.
3.5. Use of unpublished data from clinical laboratories
The use of unpublished data can have a powerful impact on variant classification62. Two clinical laboratories participate with the ClinGen LD VCEP by providing anonymized laboratory data on variants of interest. Specifically, for variants that are of uncertain significance (VUS) or likely pathogenic, the application of additional case-level data (e.g. PP4, PM3) could result in a change of classification to likely pathogenic or pathogenic. These laboratories provided the VCEP with a list of all variants identified in GAA. If a variant being classified is on the list, the VCEP then requests the de-identified data from the laboratory and incorporates the data into the curation, while being careful not to include a patient that has already been curated from the literature. To date, laboratory data obtained in this manner has been included in the classifications of 13 variants, 8 of which changed classification based on the laboratory data (6 VUS upgraded to likely pathogenic, and 2 likely pathogenic upgraded to pathogenic) (data not shown).
3.6. ClinVar submissions
Analysis of data from ClinVar (accessed July 7, 2023) showed that, out of 2368 GAA variants submitted, 845 (36%) were variants of uncertain significance (VUS), and 197 (8%) had conflicting interpretations (P/LP vs VUS, n=48; VUS vs LB/B, n= 149). This illustrates the importance of collecting and consistently evaluating data with the goal of reclassifying these variants, to either benign/likely benign or pathogenic/likely pathogenic, in order to facilitate the clinical utility of this information.
To date, the LD VCEP has submitted classifications and supporting data on 243 GAA variants to ClinVar (Figure 1). Of these variants, 32 (13%) had not been previously submitted to ClinVar by any submitter and therefore provided new information; 28 of those 32 were classified by the LD VCEP as either pathogenic or likely pathogenic (Supplemental Table 6). Most of these variants were classified because they were identified in the published literature as being either in compound heterozygosity with another variant of interest (PM3), or occurring at the same amino acid position (PS1, PM5). Thus, in order to accurately apply these criteria to the variant of interest, the second variant was also classified. In addition, two variants, not previously submitted to ClinVar, were in-frame indel variants identified by a clinical diagnostic laboratory. These in-frame variants were included as part of the LD VCEP’s pilot study, recognizing that this variant type was lacking in the ClinVar database.
Of the 211 variants with a previous ClinVar record, 40 had conflicting classifications prior to classification by the ClinGen LD VCEP; of those 40 variants, 17 were classified by the LD VCEP as pathogenic or likely pathogenic, 6 were classified as likely benign or benign, and 17 were classified as a VUS.
Regarding resolution of VUS, 2 out of 32 variants previously classified in ClinVar as VUS were classified by the LD VCEP as likely pathogenic. These variants are c.1194+3G>C, for which all other 9 submitters to ClinVar classified the variant as a VUS but unpublished data from participating clinical laboratories allowed us to classify the variant as likely pathogenic; and c.2314T>C - p.(Trp772Arg), which had one previous submission in ClinVar, as VUS. However, no additional information for comparison was provided from the submitter.
4. Conclusions
The LD VCEP has significantly added to the publicly available knowledge on the pathogenicity of variants in GAA by increasing the number of variants present in public variant resources, such as ClinVar and the ClinGen Evidence Repository, and helping to resolve conflicting classifications and variants of uncertain clinical significance. Future plans include continued work on novel variants, or variants with conflicting classifications and those with uncertain significance, collection and use of unpublished laboratory data to facilitate variant interpretation as well as expansion to other genes involved in lysosomal diseases.
Supplementary Material
Acknowledgements
This work was funded by a grant from the NHGRI, U24 HG009650. We are grateful to the ClinGen Sequence Variant Interpretation Working Group and ClinGenVariant Curation Working Group for advice on specification and application of ACMG/AMP criteria for GAA in addition to their input on this manuscript.
Conflict of Interest Statement
The following authors work or have worked in a fee-for-service diagnostic laboratory that offers GAA analysis or enzymatic testing for Pompe disease: JM, DK, SG, YP, BD, TD, RU, DB, CH, LR, RS, FV, YW, and CR. RS is a medical consultant for the Wisconsin Newborn Screening Program; consults and has equity interest in Acer Therapeutics, and PTC Therapeutics; consults for DNARx, Best Doctors/Teladoc, Leadiant, Travere; has received honoraria from Medscape/WebMD, and research support from Alexion.
Abbreviations
- ACMG
American College of genetics and Genomics
- AMP
Association for Molecular Pathology
- FDA
Food and Drug Administration (USA)
- GAA
acid alpha-glucosidase
- LD
Lysosomal Diseases
- SVI
ClinGen Sequence Variant Interpretation Working Group
- VCEP
Variant Curation Expert Panel
- VCI
ClinGen Variant Curation Interface
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015. May;17(5):405–24. doi: 10.1038/gim.2015.30. Epub 2015 Mar 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rivera-Muñoz EA, Milko LV, Harrison SM, Azzariti DR, Kurtz CL, Lee K, Mester JL, Weaver MA, Currey E, Craigen W, Eng C, Funke B, Hegde M, Hershberger RE, Mao R, Steiner RD, Vincent LM, Martin CL, Plon SE, Ramos E, Rehm HL, Watson M, Berg JS. ClinGen Variant Curation Expert Panel experiences and standardized processes for disease and gene-level specification of the ACMG/AMP guidelines for sequence variant interpretation. Hum Mutat. 2018. Nov;39(11):1614–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.ClinGen Variant Curation Expert Panel protocol: https://clinicalgenome.org/docs/clingen-variant-curation-expert-panel-vcep-protocol/.
- 4.FDA Recognition of ClinGen Assertions in ClinVar: https://clinicalgenome.org/docs/fda-recognizes-clingen-assertions-in-clinvar-frequently-asked-questions/
- 5.WORLDSymposium description of Lysosomal Disease: https://worldsymposia.org/about-worldsymposia-lysosomal-cell-biology/
- 6.Leslie N, Bailey L. 2007 Aug 31 [updated 2017 May 11]. Pompe Disease. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2023. [PubMed] [Google Scholar]
- 7.Hannah WB, Derks TGJ, Drumm ML, Grünert SC, Kishnani PS, Vissing J. Glycogen storage diseases. Nat Rev Dis Primers. 2023. Sep 7;9(1):46. doi: 10.1038/s41572-023-00456-z. [DOI] [PubMed] [Google Scholar]
- 8.Davison JE. Advances in diagnosis and management of Pompe disease. J Mother Child. 2020. Oct 2;24(2):3–8. doi: 10.34763/jmotherandchild.20202402si.2001.000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sacconi S, Wahbi K, Theodore G, et al. Atrio-ventricular block requiring pacemaker in patients with late onset Pompe disease. Neuromuscul Disord. 2014; 24(7): 648–650, doi: 10.1016/j.nmd.2014.04.005, [DOI] [PubMed] [Google Scholar]
- 10.Herbert M, Cope H, Li JS, Kishnani PS. Severe Cardiac Involvement Is Rare in Patients with Late-Onset Pompe Disease and the Common c.−32-13T>G Variant: Implications for Newborn Screening. J Pediatr. 2018. Jul;198:308–312. doi: 10.1016/j.jpeds.2018.02.007. Epub 2018 Apr 4. [DOI] [PubMed] [Google Scholar]
- 11.van Kooten HA, Roelen CHA, Brusse E, van der Beek NAME, Michels M, van der Ploeg AT, Wagenmakers MAEM, van Doom PA. Cardiovascular disease in non-classic Pompe disease: A systematic review. Neuromuscul Disord. 2021. Feb;31(2):79–90. doi: 10.1016/j.nmd.2020.10.009. Epub 2020 Nov 9. [DOI] [PubMed] [Google Scholar]
- 12.Jastrzębska A, Kostera-Pruszczyk A. Multisystem presentation of Late Onset Pompe Disease: what every consulting neurologist should know. Neurol Neurochir Pol. 2023;57(2):143–150. doi: 10.5603/PJNNS.a2022.0075. Epub 2022 Dec 7. [DOI] [PubMed] [Google Scholar]
- 13.Sniderman King L, Pan Y, Nallamilli BRR, Hegde M, Jagannathan L, Ramachander V, Lucas A, Markind J, Colzani R. Pompe disease ascertained through The Lantern Project, 2018-2021: Next-generation sequencing and enzymatic testing to overcome obstacles to diagnosis. Mol Genet Metab. 2023. May;139(1):107565. doi: 10.1016/j.ymgme.2023.107565. Epub 2023 Apr 5. [DOI] [PubMed] [Google Scholar]
- 14.Pompe Disease Diagnostic Working Group; Winchester B, Bali D, Bodamer OA, Caillaud C, Christensen E, Cooper A, Cupler E, Deschauer M, Fumić K, Jackson M, Kishnani P, Lacerda L, Ledvinová J, Lugowska A, Lukacs Z, Maire I, Mandel H, Mengel E, Müller-Felber W, Piraud M, Reuser A, Rupar T, Sinigerska I, Szlago M, Verheijen F, van Diggelen OP, Wuyts B, Zakharova E, Keutzer J. Methods for a prompt and reliable laboratory diagnosis of Pompe disease: report from an international consensus meeting. Mol Genet Metab. 2008. Mar;93(3):275–81. doi: 10.1016/j.ymgme.2007.09.006. Epub 2007 Dec 19. [DOI] [PubMed] [Google Scholar]
- 15.Miller DT, Lee K, Chung WK, Gordon AS, Herman GE, Klein TE, Stewart DR, Amendola LM, Adelman K, Bale SJ, Gollob MH, Harrison SM, Hershberger RE, McKelvey K, Richards CS, Vlangos CN, Watson MS, Martin CL; ACMG Secondary Findings Working Group. Correction to: ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021. Aug;23(8): 1582–1584. doi: 10.1038/s41436-021-01278-8. Erratum for: Genet Med. 2021 Aug;23(8):1381-1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bolano-Diaz C, Diaz-Manera J. Therapeutic Options for the Management of Pompe Disease: Current Challenges and Clinical Evidence in Therapeutics and Clinical Risk Management. Ther Clin Risk Manag. 2022. Dec 13;18:1099–1115. doi: 10.2147/TCRM.S334232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guémy C, Laforêt P. The new horizons for treatment of Late-Onset Pompe Disease (LOPD). Rev Neurol (Paris). 2023. Jan-Feb;179(1-2):81–89. doi: 10.1016/j.neurol.2022.12.004. Epub 2023 Jan 4. [DOI] [PubMed] [Google Scholar]
- 18.Goina E, Peruzzo P, Bembi B, Dardis A, Buratti E. Glycogen Reduction in Myotubes of Late-Onset Pompe Disease Patients Using Antisense Technology. Mol Ther. 2017. Sep 6;25(9):2117–2128. doi: 10.1016/j.ymthe.2017.05.019. Epub 2017 Jun 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van der Wal E, Bergsma AJ, Pijnenburg JM, van der Ploeg AT, Pijnappel WWMP. Antisense Oligonucleotides Promote Exon Inclusion and Correct the Common c.-32-13T>G GAA Splicing Variant in Pompe Disease. Mol Ther Nucleic Acids. 2017. Jun 16;7:90–100. doi: 10.1016/j.omtn.2017.03.001. Epub 2017 Mar 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van der Wal E, Bergsma AJ, van Gestel TJM, In ‘t Groen SLM, Zaehres H, Araúzo-Bravo MJ, Schöler HR, van der Ploeg AT, Pijnappel WWMP. GAA Deficiency in Pompe Disease Is Alleviated by Exon Inclusion in iPSC-Derived Skeletal Muscle Cells. Mol Ther Nucleic Acids. 2017. Jun 16;7:101–115. doi: 10.1016/j.omtn.2017.03.002. Epub 2017 Mar 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khan AA, Case LE, Herbert M, DeArmey S, Jones H, Crisp K, Zimmerman K, ElMallah MK, Young SP, Kishnani PS. Higher dosing of alglucosidase alfa improves outcomes in children with Pompe disease: a clinical study and review of the literature. Genet Med. 2020. May;22(5):898–907. doi: 10.1038/s41436-019-0738-0. Epub 2020 Jan 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dornelles AD, Junges APP, Pereira TV, Krug BC, Gonçalves CBT, Llerena JC Jr, Kishnani PS, de Oliveira HA Jr, Schwartz IVD. A Systematic Review and Meta-Analysis of Enzyme Replacement Therapy in Late Onset Pompe Disease. J Clin Med. 2021. Oct 21;10(21):4828. doi: 10.3390/jcm10214828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chien YH, Hwu WL, Lee NC. Newborn screening: Taiwanese experience. Ann Transl Med. 2019. Jul;7(13):281. doi: 10.21037/atm.2019.05.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh S, Ojodu J, Kemper AR, Lam WKK, Grosse SD. Implementation of Newborn Screening for Conditions in the United States First Recommended during 2010-2018. Int J Neonatal Screen. 2023. Apr 6;9(2):20. doi: 10.3390/ijns9020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whiffin N, Minikel E, Walsh R, O’Donnell-Luria AH, Karczewski K, Ing AY, Barton PJR, Funke B, Cook SA, MacArthur D, Ware JS. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet Med. 2017. Oct;19(10):1151–1158. doi: 10.1038/gim.2017.26. Epub 2017 May 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guidance from the ClinGen SVI: https://clinicalgenome.org/working-groups/sequence-variant-interpretation/
- 27.Preston CG, Wright MW, Madhavrao R, Harrison SM, Goldstein JL, Luo X, et al. , ClinGen Variant Curation Interface: a variant classification platform for the application of evidence criteria from ACMG/AMP guidelines. Genome Med. 2022. Jan 18;14(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA MacArthur DG. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E, Karyadi D, Cannon-Albright LA, Teerlink CC, Stanford JL, Isaacs WB, Xu J, Cooney KA, Lange EM, Schleutker J, Carpten JD, Powell IJ, Cussenot O, Cancel-Tassin G, Giles GG, MacInnis RJ, Maier C, Hsieh CL, Wiklund F, Catalona WJ, Foulkes WD, Mandal D, Eeles RA, Kote-Jarai Z, Bustamante CD, Schaid DJ, Hastie T, Ostrander EA, Bailey-Wilson JE, Radivojac P, Thibodeau SN, Whittemore AS, Sieh W. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am J Hum Genet. 2016. Oct 6;99(4):877–885. doi: 10.1016/j.ajhg.2016.08.016. Epub 2016 Sep 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brnich SE, Abou Tayoun AN, Couch FJ, Cutting GR, Greenblatt MS, Heinen CD, Kanavy DM, Luo X, McNulty SM, Starita LM, Tavtigian SV, Wright MW, Harrison SM, Biesecker LG, Berg JS; Clinical Genome Resource Sequence Variant Interpretation Working Group. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 2019. Dec 31;12(1):3. doi: 10.1186/s13073-019-0690-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.SVI Recommendation for Absence/Rarity (PM2) - https://clinicalgenome.org/site/assets/files/5182/pm2_-_svi_recommendation_-_approved_sept2020.pdf
- 32.Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, Karapetyan K, Katz K, Liu C, Maddipatla Z, Malheiro A, McDaniel K, Ovetsky M, Riley G, Zhou G, Holmes JB, Kattman BL, Maglott DR. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018. Jan 4;46(D1):D1062–D1067. doi: 10.1093/nar/gkx1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Version 2 of the ClinGen Lysosomal Diseases VCEP’s ACMG/AMP specifications for GAA: https://cspec.genome.network/cspec/ui/svi/doc/GN010
- 34.Abou Tayoun AN, Pesaran T, DiStefano MT, Oza A, Rehm HL, Biesecker LG, Harrison SM; ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI). Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. 2018. Nov;39(11):1517–1524. doi: 10.1002/humu.23626. Epub 2018 Sep 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bali DS, Goldstein JL, Banugaria S, Dai J, Mackey J, Rehder C, Kishnani PS. Predicting cross-reactive immunological material (CRIM) status in Pompe disease using GAA mutations: lessons learned from 10 years of clinical laboratory testing experience. Am J Med Genet C Semin Med Genet. 2012. Feb 15;160C(1):40–9. doi: 10.1002/ajmg.c.31319. Epub 2012 Jan 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, Kosmicki JA, Arbelaez J, Cui W, Schwartz GB, Chow ED, Kanterakis E, Gao H, Kia A, Batzoglou S, Sanders SJ, Farh KK. Predicting Splicing from Primary Sequence with Deep Learning. Cell. 2019. Jan 24;176(3):535–548.e24. doi: 10.1016/j.cell.2018.12.015. Epub 2019 Jan 17. [DOI] [PubMed] [Google Scholar]
- 37.Pejaver V, Byrne AB, Feng BJ, Pagel KA, Mooney SD, Karchin R, O’Donnell- Luria A, Harrison SM, Tavtigian SV, Greenblatt MS, Biesecker LG, Radivojac P, Brenner SE; ClinGen Sequence Variant Interpretation Working Group. Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am J Hum Genet. 2022. Dec 1;109(12):2163–2177. doi: 10.1016/j.ajhg.2022.10.013. Epub 2022 Nov 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walker LC, de la Hoya M, Wiggins GA, Lindy A, Vincent LM, Parsons M, Canson DM, Bis-Brewer D, Cass A, Tchourbanov A, Zimmermann H, Byrne AB, Pesaran T, Karam R, Harrison SM; ClinGen Sequence Variant Interpretation Working Group; Spurdle AB. Application of the acmg/amp framework to capture evidence relevant to predicted and observed impact on splicing: recommendations from the ClinGen SVI splicing subgroup. medRxiv [Preprint]. 2023. Feb 26:2023.02.24.23286431. doi: 10.1101/2023.02.24.23286431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ghosh R, Harrison SM, Rehm HL, Plon SE, Biesecker LG; ClinGen Sequence Variant Interpretation Working Group. Updated recommendation for the benign stand-alone ACMG/AMP criterion. Hum Mutat. 2018. Nov;39(11):1525–1530. doi: 10.1002/humu.23642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poorthuis BJ, Wevers RA, Kleijer WJ, Groener JE, de Jong JG, van Weely S, Niezen-Koning KE, van Diggelen OP. The frequency of lysosomal storage diseases in The Netherlands. Hum Genet. 1999. Jul-Aug;105(1-2):151–6. doi: 10.1007/s004399900075. [DOI] [PubMed] [Google Scholar]
- 41.Martiniuk F, Chen A, Mack A, Arvanitopoulos E, Chen Y, Rom WN, Codd WJ, Hanna B, Alcabes P, Raben N, Plotz P. Carrier frequency for glycogen storage disease type II in New York and estimates of affected individuals born with the disease. Am J Med Genet. 1998. Aug 27;79(1):69–72. doi: . [DOI] [PubMed] [Google Scholar]
- 42.Pinto R, Caseiro C, Lemos M, Lopes L, Fontes A, Ribeiro H, Pinto E, Silva E, Rocha S, Marcão A, Ribeiro I, Lacerda L, Ribeiro G, Amaral O, Sá Miranda MC. Prevalence of lysosomal storage diseases in Portugal. Eur J Hum Genet. 2004. Feb;12(2):87–92. doi: 10.1038/sj.ejhg.5201044. [DOI] [PubMed] [Google Scholar]
- 43.Chien YH, Hwu WL, Lee NC. Newborn screening: Taiwanese experience. Ann Transl Med. 2019. Jul;7(13):281. doi: 10.21037/atm.2019.05.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ficicioglu C, Ahrens-Nicklas RC, Barch J, Cuddapah SR, DiBoscio BS, DiPerna JC, Gordon PL, Henderson N, Menello C, Luongo N, Ortiz D, Xiao R. Newborn Screening for Pompe Disease: Pennsylvania Experience. Int J Neonatal Screen. 2020. Nov 13;6(4):89. doi: 10.3390/ijns6040089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sawada T, Kido J, Sugawara K, Momosaki K, Yoshida S, Kojima-Ishii K, Inoue T, Matsumoto S, Endo F, Ohga S, Hirose S, Nakamura K. Current status of newborn screening for Pompe disease in Japan. Orphanet J Rare Dis. 2021. Dec 18;16(1):516. doi: 10.1186/s13023-021-02146-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reuser AJJ, van der Ploeg AT, Chien YH, Llerena J Jr, Abbott MA, Clemens PR, Kimonis VE, Leslie N, Maruti SS, Sanson BJ, Araujo R, Periquet M, Toscano A, Kishnani PS, On Behalf Of The Pompe Registry Sites. GAA variants and phenotypes among 1,079 patients with Pompe disease: Data from the Pompe Registry. Hum Mutat. 2019. Nov;40(11):2146–2164. doi: 10.1002/humu.23878. Epub 2019 Aug 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Niño MY, Wijgerde M, de Faria DOS, Hoogeveen-Westerveld M, Bergsma AJ, Broeders M, van der Beek NAME, van den Hout HJM, van der Ploeg AT, Verheijen FW, Pijnappel WWMP. Enzymatic diagnosis of Pompe disease: lessons from 28 years of experience. Eur J Hum Genet. 2021. Mar;29(3):434–446. doi: 10.1038/s41431-020-00752-2. Epub 2020 Nov 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kroos M, Pomponio RJ, van Vliet L, Palmer RE, Phipps M, Van der Helm R, Halley D, Reuser A; GAA Database Consortium. Update of the Pompe disease mutation database with 107 sequence variants and a format for severity rating. Hum Mutat. 2008. Jun;29(6):E13–26. doi: 10.1002/humu.20745. [DOI] [PubMed] [Google Scholar]
- 49.Kroos M, Hoogeveen-Westerveld M, Michelakakis H, Pomponio R, Van der Ploeg A, Halley D, Reuser A; GAA Database Consortium. Update of the pompe disease mutation database with 60 novel GAA sequence variants and additional studies on the functional effect of 34 previously reported variants. Hum Mutat. 2012. Aug;33(8):1161–5. doi: 10.1002/humu.22108. Epub 2012 May 29. [DOI] [PubMed] [Google Scholar]
- 50.Kroos M, Hoogeveen-Westerveld M, van der Ploeg A, Reuser AJ. The genotype-phenotype correlation in Pompe disease. Am J Med Genet C Semin Med Genet. 2012. Feb 15;160C(1):59–68. doi: 10.1002/ajmg.c.31318. Epub 2012 Jan 17. [DOI] [PubMed] [Google Scholar]
- 51.Deming D, Leeb K, McSherry T, Weib RR, Edmunds T, Garman SC. The molecular basis for Pompe disease revealed by the structure of human acid α-glucosidase. bioRxiv, 10.1101/212837v1.full.pdf [DOI] [Google Scholar]
- 52.Roig-Zamboni V, Cobucci-Ponzano B, Iacono R, Ferrara MC, Germany S, Bourne Y, Parenti G, Moracci M, Sulzenbacher G. Structure of human lysosomal acid α-glucosidase-a guide for the treatment of Pompe disease. Nat Commun. 2017. Oct 24;8(1):1111. doi: 10.1038/s41467-017-01263-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Faria DOS, ’t Groen SLMI, Hoogeveen-Westerveld M, Nino MY, van der Ploeg AT, Bergsma AJ, Pijnappel WWMP. Update of the Pompe variant database for the prediction of clinical phenotypes: Novel disease-associated variants, common sequence variants, and results from newborn screening. Hum Mutat. 2021. Feb;42(2):119–134. doi: 10.1002/humu.24148. Epub 2020 Dec 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hermans MM, Kroos MA, van Beeumen J, Oostra BA, Reuser AJ. Human lysosomal alpha-glucosidase. Characterization of the catalytic site. J Biol Chem. 1991. Jul 25;266(21):13507–12. (de Faria et al, 2021) [PubMed] [Google Scholar]
- 55.SVI Recommendation for in trans Criterion (PM3): https://clinicalgenome.org/site/assets/files/3717/svi_proposal_for_pm3_criterion_-Version_1.pdf
- 56.Zhang H, Kallwass H, Young SP, Carr C, Dai J, Kishnani PS, Millington DS, Keutzer J, Chen YT, Bali D. Comparison of maltose and acarbose as inhibitors of maltase-glucoamylase activity in assaying acid alpha-glucosidase activity in dried blood spots for the diagnosis of infantile Pompe disease. Genet Med. 2006. May;8(5):302–6. doi: 10.1097/01.gim.0000217781.66786.9b. [DOI] [PubMed] [Google Scholar]
- 57.Pompe Disease Diagnostic Working Group; Winchester B, Bali D, Bodamer OA, Caillaud C, Christensen E, Cooper A, Cupler E, Deschauer M, Fumić K, Jackson M, Kishnani P, Lacerda L, Ledvinová J, Lugowska A, Lukacs Z, Maire I, Mandel H, Mengel E, Müller-Felber W, Piraud M, Reuser A, Rupar T, Sinigerska I, Szlago M, Verheijen F, van Diggelen OP, Wuyts B, Zakharova E, Keutzer J. Methods for a prompt and reliable laboratory diagnosis of Pompe disease: report from an international consensus meeting. Mol Genet Metab. 2008. Mar;93(3):275–81. doi: 10.1016/j.ymgme.2007.09.006. Epub 2007 Dec 19. [DOI] [PubMed] [Google Scholar]
- 58.Goldstein JL, Young SP, Changela M, Dickerson GH, Zhang H, Dai J, Peterson D, Millington DS, Kishnani PS, Bali DS. Screening for Pompe disease using a rapid dried blood spot method: experience of a clinical diagnostic laboratory. Muscle Nerve. 2009. Jul;40(1):32–6. doi: 10.1002/mus.21376. [DOI] [PubMed] [Google Scholar]
- 59.Reuser AJ, Verheijen FW, Bali D, van Diggelen OP, Germain DP, Hwu WL, Lukacs Z, Mühl A, Olivova P, Piraud M, Wuyts B, Zhang K, Keutzer J. The use of dried blood spot samples in the diagnosis of lysosomal storage disorders--current status and perspectives. Mol Genet Metab. 2011. Sep-Oct;104(1-2):144–8. doi: 10.1016/j.ymgme.2011.07.014. Epub 2011 Jul 23. [DOI] [PubMed] [Google Scholar]
- 60.Biesecker LG, Harrison SM; ClinGen Sequence Variant Interpretation Working Group. The ACMG/AMP reputable source criteria for the interpretation of sequence variants. Genet Med. 2018. Dec;20(12):1687–1688. doi: 10.1038/gim.2018.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goomber S, Huggins E, Rehder CW, Cohen JL, Bali DS, Kishnani PS. Development of a clinically validated in vitro functional assay to assess pathogenicity of novel GAA variants in patients with Pompe disease identified <i>via</i> newborn screening. Front Genet. 2022. Sep 30;13:1001154. doi: 10.3389/fgene.2022.1001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kelly MA, Caleshu C, Morales A, Buchan J, Wolf Z, Harrison SM, Cook S, Dillon MW, Garcia J, Haverfield E, Jongbloed JDH, Macaya D, Manrai A, Orland K, Richard G, Spoonamore K, Thomas M, Thomson K, Vincent LM, Walsh R, Watkins H, Whiffin N, Ingles J, van Tintelen JP, Semsarian C, Ware JS, Hershberger R, Funke B. Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inherited cardiomyopathies: recommendations by ClinGen’s Inherited Cardiomyopathy Expert Panel. Genet Med. 2018. Mar;20(3):351–359. doi: 10.1038/gim.2017.218. Epub 2018 Jan 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moreland RJ, Jin X, Zhang XK, Decker RW, Albee KL, Lee KL, Cauthron RD, Brewer K, Edmunds T, Canfield WM. Lysosomal acid alpha-glucosidase consists of four different peptides processed from a single chain precursor. J Biol Chem. 2005. Feb 25;280(8):6780–91. doi: 10.1074/jbc.M404008200. Epub 2004 Nov 1. [DOI] [PubMed] [Google Scholar]
- 64.Young SP, Piraud M, Goldstein JL, Zhang H, Rehder C, Laforet P, Kishnani PS, Millington DS, Bashir MR, Bali DS. Assessing disease severity in Pompe disease: the roles of a urinary glucose tetrasaccharide biomarker and imaging techniques. Am J Med Genet C Semin Med Genet. 2012. Feb 15;160C(1):50–8. doi: 10.1002/ajmg.c.31320. Epub 2012 Jan 17. [DOI] [PubMed] [Google Scholar]
- 65.Piraud M, Pettazzoni M, de Antonio M, Vianey-Saban C, Froissart R, Chabrol B, Young S, Laforêt P; French Pompe study group. Urine glucose tetrasaccharide: A good biomarker for glycogenoses type II and III? A study of the French cohort. Mol Genet Metab Rep. 2020. May 1;23:100583. doi: 10.1016/j.ymgmr.2020.100583. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.