

Abstract
Previous work has associated polymorphisms in the dopamine transporter gene (rs6347 in DAT1/SLC6A3) and brain derived neurotrophic factor gene (Val66Met in BDNF) with atrophy and memory decline. However, it is unclear whether these polymorphisms relate to atrophy and cognition through associations with Alzheimer’s disease pathology. We tested for effects of DAT1 and BDNF polymorphisms on cross-sectional and longitudinal β-amyloid (Aβ) and tau pathology (measured with positron emission tomography (PET)), hippocampal volume, and cognition. We analyzed a sample of cognitively normal older adults (cross-sectional n=321) from the Alzheimer’s Disease Neuroimaging Initiative (ADNI). DAT1 and BDNF interacted to predict Aβ-PET, tau-PET, and hippocampal atrophy. Carriers of both “non-optimal” DAT1 C and BDNF Met alleles demonstrated greater pathology and atrophy. Our findings provide novel links between dopamine and neurotrophic factor genes and AD pathology, consistent with previous research implicating these variants in greater risk for developing AD.
Keywords: Dopamine transporter gene, Brain derived neurotrophic factor gene, Tau PET, Amyloid PET, Hippocampal atrophy, ADNI
1. INTRODUCTION
β-amyloid (Aβ) pathology is a central component of Alzheimer’s disease (AD) and has been associated with a cascade of events including hyperphosphorylated tau proteins, neurodegeneration, and cognitive deficits (Hardy and Higgins, 1992). Identifying factors that influence pathological disease progression will be critical for understanding individual differences in AD risk and identifying candidate targets for intervention (Karran et al., 2011). Dysfunctional neurotransmitter activity is strongly implicated in AD, with most research focused on acetylcholine and norepinephrine (Berry and Harrison, 2023; Ciampa et al., 2022; Hampel et al., 2018; Jacobs et al., 2021). However, emerging lines of evidence identify the suboptimal dopamine function observed in AD (Pan et al., 2019) as a potential exacerbator of AD pathophysiology. In AD models, declines in dopamine signaling are associated with impaired memory and hippocampal plasticity (Nobili et al., 2017). Notably, dopamine agonists have been shown to reverse Aβ-mediated reductions in hippocampal plasticity (Yuan Xiang et al., 2016). Dopamine may also exert protective effects against Aβ-induced neurotoxicity as in vivo studies demonstrate dopamine and its metabolites can disassemble Aβ fibrils and inhibit Aβ aggregation (Li et al., 2004).
The dopamine transporter (DAT) protein is a key regulator of dopamine signaling (Jaber et al., 1997), as it controls extracellular dopamine levels through reuptake into the presynaptic neuron (Vaughan and Foster, 2013). A single nucleotide polymorphism (SNP; rs6347) in the DAT gene (DAT1/SLC6A3; T>C substitution, minor allele frequency ~ .27; Phan et al., 2020) is located on chromosome 5 in exon 9. The minor C allele of rs6347 occurs at a higher frequency in AD patients compared with healthy controls and is linked to faster ventricular expansion and lower scores on the Mini Mental State Exam (MMSE) in both healthy controls and clinically-diagnosed AD patients (Roussotte et al., 2015). Further, the C allele is associated with more severe dementia compared with the T allele in patients with clinical diagnoses of AD (Lin et al., 2012). However, cognitive decline and atrophy may be present in aging absent of pathology or in other forms of dementia. It is unclear whether rs6347 is related to cognition and atrophy through links with AD pathology or through other pathways that are not associated with AD.
Brain-derived neurotrophic factor (BDNF) is a growth factor critical for the development and maintenance of neurons (Gorski et al., 2003). BDNF is most highly concentrated in the hippocampus (Yan et al., 1997) and is richly expressed in the dopaminergic midbrain and striatal regions (Fenner et al., 2014; Seroogy et al., 1994). In AD, Aβ impairs BDNF signaling (Jerónimo-Santos et al., 2015), and lower BDNF levels are related to greater tau burden in autopsy studies (Ginsberg et al., 2019). One polymorphism in the BDNF gene (Val66Met, rs6265) has been repeatedly associated with AD (Franzmeier et al., 2021; Lim et al., 2013). Val66Met is a SNP located on chromosome 11 (minor allele frequency = .19; Phan et al., 2020) that results in a methionine (Met) amino acid substitution for valine (Val) at codon 66. Met carriers who are also Aβ positive exhibit more severe declines in hippocampal volume and episodic memory compared with Val/Val carriers who are Aβ positive and individuals who are Aβ negative with any BDNF genotype (Lim et al., 2013). This suggests that sub-optimal BDNF function may exacerbate the negative effects of AD pathology. It is worth noting that there are substantial inconsistencies in reported BDNF Val66Met results, including null effects (Ji et al., 2015) and findings suggesting the Val/Val allele is implicated in AD (Voineskos et al., 2011). We expand upon these inconsistencies in the Discussion. The mechanisms underlying BDNF’s impact on AD trajectories is an area of active research, though evidence suggests BDNF may support resistance to neurodegeneration, in part, through modification of dopamine signaling (Meisner et al., 2008; Zhu et al., 2015).
While BDNF Val66Met is a contributor to AD polygenic risk scores (Porter et al., 2018), dopamine polymorphisms are rarely considered despite links between dementia and DAT1 rs6347 (Roussotte et al., 2015). Neither BDNF nor DAT1 polymorphisms have been identified in genome-wide association studies of AD, and likely have small effects independently. There is considerable functional interaction between BDNF and the dopamine system as most mesencephalic dopamine-producing neurons express the high affinity BDNF receptor tyrosine kinase receptor B (TrkB; Numan and Seroogy, 1999). Relevant to cognition, studies in rodent models have defined conjoint effects of BDNF and dopamine signaling on memory (Rossato et al., 2009). In humans, effects of dopamine genetic polymorphisms on episodic memory depend on BDNF genotype in aging (Papenberg et al., 2019), and DAT1*BDNF interactions predict trait neuroticism, a well-known risk-factor for dementia (Hünnerkopf et al., 2007). Together, these studies motivate further examination of the interactive effects of BDNF and DAT1 on AD.
The goal of the current study is to establish relationships among AD pathology, atrophy, cognition, and genetic polymorphisms that have previously been implicated in risk for AD but lack direct links to Aβ and tau pathology. We focused our analyses on a large sample of cognitively normal older adults, as studying healthy individuals is key to understanding the relative risk for developing AD and many interventions target the pre-clinical stage of AD (van Bokhoven et al., 2021). We first hypothesized that DAT1 rs6347 and BDNF Val66Met would relate to Aβ and tau pathology such that individuals carrying one or more “non-optimal” variants (i.e., DAT1 CC and BDNF Met) would exhibit higher PET measures of pathology. Second, we hypothesized that carriers of non-optimal variants would display lower hippocampal volume and worse cognition.
2. METHODS
2.1. Participants
Our cross-sectional sample consisted of 321 cognitively normal adults over the age of 60 (mean age=73.8 years, SD=7.0, range=61.2–94.4; 56% female) from the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Inclusion criteria for cognitively normal participants included scoring above the education cutoffs for the Logical Memory component of the Wechsler Memory Scale (≥ 3 for 0–7 years of education, ≥ 5 for 8–15 years of education, and ≥ 9 for 16 or more years of education), Mini Mental State Exam = 24–30, Clinical Dementia Rating = 0, Geriatric Depression Scale ≤ 5, no significant impairments in cognitive function or daily activities, and no history of depression within the last year. Participants had at least 12 years of education and had no major medical illnesses or MR contraindications. For cross-sectional analyses we used each participant’s first tau-PET scan and the Aβ-PET scan closest to the first tau-PET. Participants were required to have known rs6347 and rs6265 genotypes, [18F]Flortaucipir tau PET and Aβ PET ([18F]Florbetapir or [18F]Florbetaben PET). A subset of these participants with at least one follow-up scan or session were included in longitudinal analyses (Aβ: n=235; tau: n=135; MRI: n=215, cognition: n=236). Due to smaller longitudinal sample sizes, we consider longitudinal analyses to be exploratory and the results should be interpreted with caution. For longitudinal Aβ analyses, mean age=74.4 years, SD=7.0, range=61.2 – 91.5. For longitudinal tau analyses, mean age=74.1, SD=6.5, range=62.4 – 90.5. For longitudinal MR analyses, mean age=74.5, SD=6.9, range=61.2 – 91.5. For longitudinal cognitive analyses, mean age=73.8, SD=7.0, range=61.2 – 94.4. All participants provided informed consent.
2.2. Genetic data
DNA from peripheral blood samples was genotyped using either the Ilumina Omni 2.5M BeadChip or the Ilumina Global Screening Array v2. Genotype data was in Hardy-Weinberg equilibrium for DAT1 rs6347 and BDNF Val66Met (rs6347: 178 T homozygotes (55%), 111 heterozygotes (35%), and 32 C homozygotes (10%); Val66Met: 208 Val homozygotes (65%), 100 heterozygotes (31%), and 13 Met homozygotes (4%)). Due to a low number of BDNF Met/Met carriers, we grouped together any individuals carrying a Met allele (Met/Met and Val/Met), as done previously (Lim et al., 2013).
2.3. Aβ and tau PET acquisition and processing
Documentation on PET data acquisition and processing is available on the ADNI website (https://adni.loni.usc.edu/). PET imaging was performed at multiple sites using one of several different scanners: GE Healthcare PET/CT or PET only, Philips Medical Systems PET/CT or PET only, or Siemens Medical Solutions PET/CT or PET only. There were no differences in radiotracer yield, acquisition time, or number of frames across different scanners or sites. There were also no differences in imaging protocols across different sites.
To measure tau pathology, participants were given a 10 mCi ± 10% bolus injection into an antecubital vein 75–105 minutes before scanning. Dynamic acquisition frames were obtained over 30 minutes (6 × 5 min frames). [18F]Flortaucipir standardized uptake ratios (SUVRs) were calculated by coregistering each participant’s PET scan to the MRI scan closest to the PET scan. MRI scans were reconstructed and segmented using FreeSurfer (v.7.1.1). [18F]Flortaucipir scans were partial volume corrected using the Geometric Transfer Matrix (Rousset et al., 1998) and an inferior cerebellar reference region. Our analyses included tau ROIs measured in the entorhinal cortex, which is one of the earliest sites of cortical tau accumulation (Braak and Braak, 1985; Kaufman et al., 2018), and a meta-temporal lobe region consisting of the entorhinal cortex, amygdala, fusiform, parahippocampal gyrus, inferior temporal gyrus, and middle temporal gyrus (Jack et al., 2020).
Aβ was measured with two different PET tracers ([18F]Florbetapir and [18F]Florbetaben) depending on when participants joined ADNI. Participants received a bolus injection of either 10 mCi ± 10% (Florbetapir) or 8.1 mCi ± 10% (Florbetaben) and dynamic acquisition frames were obtained over 20 minutes of continuous scanning (4 × 5 minutes frames) either 50 minutes (Florbetapir) or 90 minutes (Florbetaben) post-injection. Using the Aβ-PET scan closest in time to baseline [18F]Flortaucipir, PET images were coregistered to the MRI scan closest to the Aβ-PET scan and a cortical summary region was created (including frontal, anterior/posterior cingulate, lateral parietal, and lateral temporal regions). SUVRs were calculated by dividing the cortical summary region by the whole cerebellum, which was used as the reference region for both cross-sectional and longitudinal analyses. SUVRs were normalized to the amyloid centiloid scale to enable comparison of scans obtained using the two different tracers (Royse et al., 2021).
For longitudinal analyses, participants had an average of 3.19 follow up Aβ-PET scans (SD=1.41, range=2–6) and there was an average follow up time (time between first and last scan) of 4.99 years (SD=3.00, range=1.52–11.01) for Aβ-PET. There was an average of 2.60 follow up tau-PET scans (SD=.77, range=2–5), with an average follow up time of 2.83 years (SD=1.37, range=.80–5.86).
2.4. Hippocampal volume
T1-weighted MRIs are available in the ADNI database. Analyses relied on FreeSurfer software (version 7.0.0). Automatic segmentation of subcortical regions is based upon an atlas of probabilistic information on the location of structures, as previously described (Fischl et al., 2002). Right and left hippocampal volumes were segmented separately and added together to create a bilateral volume. Estimated total intracranial volume was used as a covariate in analyses involving hippocampal volume to adjust for differences in head size. Longitudinal change in hippocampal volume was assessed in a subset of n=215 participants with a mean follow-up time of 4.04 years (SD=2.69, range=.98–10.51) and a mean of 3.87 follow-up scans (SD=2.40, range=2 – 9).
2.5. Cognitive assessments
Cognitive measures included the ADNI University of Washington (UW) Memory (Crane et al., 2012) and Executive Function (EF) (Gibbons et al., 2012) composites and the Preclinical Alzheimer’s Cognitive Composite (PACC) (Donohue et al., 2014) Cross-sectional cognition was measured at the cognitive testing session closest to the baseline [18F]Flortaucipir PET scan. Longitudinal cognition was measured using all sessions after the baseline [18F]Flortaucipir scan (n=236 participants). Mean follow-up time was 2.47 years (SD=.98, range=.96–5.56). Mean number of follow up sessions was 2.98 (SD=1.28, range=2–8).
2.6. Statistical analyses
We first investigated whether carrying both “non-optimal” genotypes was associated with higher Aβ and tau pathology by testing DAT1*BDNF interactions on Aβ and tau PET, as well as main effects of DAT1 and BDNF. We used multiple regression models with cross-sectional and longitudinal measures of pathology as dependent variables. Longitudinal change in pathology over time was analyzed using linear mixed effects modeling with both random slope and random intercept in the lme4 R package. Individual slopes for each participant were extracted from the model and used as dependent variables in linear regression analyses testing for DAT1*BDNF interactions on longitudinal change in pathology (Model 1 in the SPSS PROCESS Macro). We next conducted an exploratory moderated mediation analysis to test whether Aβ mediates the effects of the polymorphisms on tau pathology. This analysis involved a bias-corrected and accelerated (BCa) 95% confidence intervals bootstrap estimation (10,000 samples). We used the moderated mediation model to test whether DAT1 rs6347 (independent variable, X) affects tau-PET (dependent variable, Y) both directly and indirectly through effects of rs6347 on Aβ-PET (mediator, M), and whether this mediation is moderated by BDNF Val66Met (moderator, W). The moderated mediation was run using Model 8 in the PROCESS Macro (version 4.0; Hayes, 2013) in SPSS (version 28.0.1.1). Finally, we used multiple regression models to test for direct and interactive effects of the polymorphisms on cross-sectional and longitudinal hippocampal volume and cognition. All regression analyses and the moderated mediation included age, sex, and years of education as covariates. Longitudinal models also adjusted for number of follow-up scans/sessions, and mean follow-up time. Effect sizes were calculated using Cohen’s f2.
Based on our cross-sectional sample size (n=321), a sensitivity analysis using G*Power (version 3.1, Faul et al., 2007) revealed a sensitivity to detect small effect sizes (f2=.041, 80% power, alpha .05, Fixed Model R2 increase).
3. RESULTS
3.1. Sample characteristics
Participant demographics, genotype information, baseline tau-PET SUVRs, and Aβ-PET centiloids closest to baseline tau-PET are presented in Table 1. Linear regression models with each genotype as the predictor demonstrated no genotype differences in age, years of education, MMSE score, GDS score, or PET measures (see Table 1 for p-values). Additionally, logistic regression models revealed no associations between genotype and sex, no associations between genotype and Aβ status (rs6347: p=.31; Val66Met: p=.35), and no associations between rs6347 and Val66Met genotypes (p=.44).
Table 1.
Participant characteristics at first tau-PET scan.
| Variable | Total (N=321), 35% Aβ+ | rs6347 CC (N=32), 44% Aβ+ | rs6347 TC (N=111), 36% Aβ+ | rs6347 TT (N=178), 34% Aβ+ | p-value (comparing rs6347 genotypes) 39% Aβ+ | rs6265 Met carriers (N=113), | rs6265 Val/Val (N=208), 34% Aβ+ | p-value (comparing rs6265 genotypes) |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| N female (%) | 179 (56) | 17 (53) | 58 (52) | 104 (58) | .35 | 48 (42) | 94 (45) | .64 |
| Age, years | 73.8 | 72.4 | 73.9 | 73.9 | .38 | 73.0 | 74.2 | .14 |
| (SD) | (7.0) | (7.1) | (7.1) | (7.1) | (7.2) | (6.8) | ||
| Education, | 16.9 | 16.6 | 16.6 | 17.2 | .06 | 17.2 | 16.7 | .16 |
| years (SD) | (2.3) | (2.4) | (2.4) | (2.2) | (2.3) | (2.2) | ||
| MMSE (SD) | 29.1 (1.2) | 29.1 (1.1) | 29.0 (1.4) | 29.1 (1.2) | .50 | 29.2 (1.2) | 29.0 (1.2) | .23 |
| GDS (SD) | 0.7 (1.2) | 0.8 (1.6) | 0.7 (1.0) | 0.8 (1.3) | 0.71 | 0.7 (1.1) | 0.8 (1.3) | 0.34 |
| FBP/FBB in | 21.8 | 25.4 | 23.7 | 19.9 | .23 | 24.5 | 20.3 | .25 |
| centiloids (SD) | (30.7) | (29.5) | (33.1) | (29.4) | (33.0) | (29.4) | ||
| Entorhinal FTP SUVR (SD) | 1.1 (.1) | 1.2 (.2) | 1.1 (.1) | 1.1 (.1) | .13 | 1.2 (.1) | 1.1 (.1) | .07 |
Abbreviations: MMSE, Mini Mental State Exam; GDS, Geriatric Depression Scale; FBP/FBB, Florbetapir/Florbetaben Aβ-PET normalized to the centiloid scale; FTP SUVR, Flortaucipir standardized uptake value ratio (tau-PET measured in the entorhinal cortex).
3.2. Carriers of both DAT1 CC and BDNF Met genotypes exhibit higher Aβ and tau pathology
We first investigated DAT1*BDNF interactions predicting cross-sectional and longitudinal Aβ-PET. DAT1 and BDNF interacted to predict cross-sectional Aβ-PET (t(314)=2.35, p=.019, f2=.02; Figure 1A, left; Table 2A) such that individuals carrying both “non-optimal” genotypes (DAT1 CC and BDNF Met) exhibited higher Aβ centiloids. Carriers of both DAT1 CC and BDNF Met showed numerically larger rates of increase in longitudinal Aβ, but this was not statistically significant (t(226)=1.69, p = 0.09, f2=.01; Figure 1A, right; Table 2B). Direct effects of DAT1 rs6347 and BDNF Val66Met on cross-sectional and longitudinal Aβ-PET were null (cross-sectional: rs6347 p=.16, Val66Met p=.13; longitudinal: rs6347 p=.26, Val66Met p=.91; adjusting for age, sex, and years of education).
Figure 1.
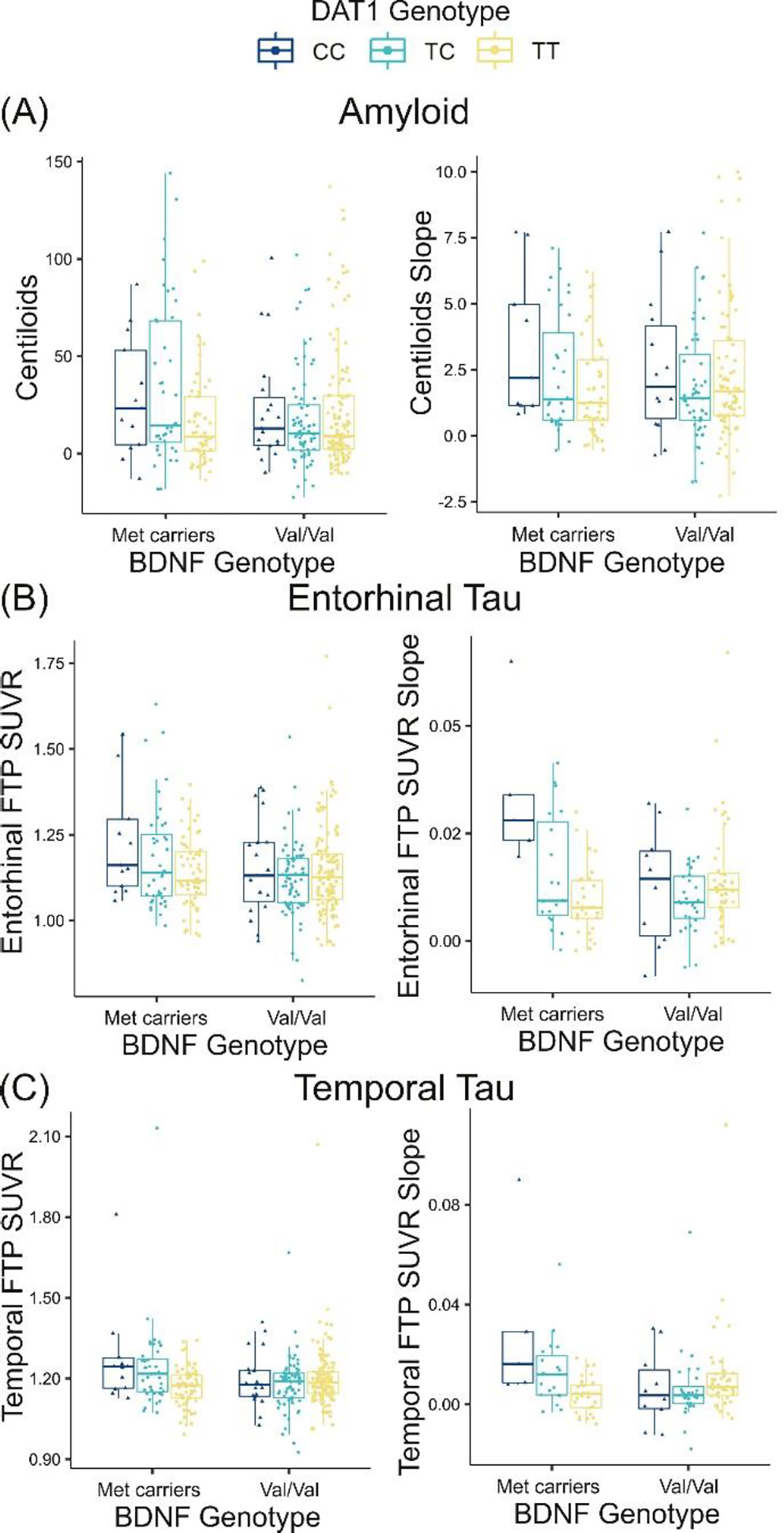
DAT1 rs6347 interacts with BDNF Val66Met to predict cross-sectional and longitudinal Aβ and tau pathology such that individuals carrying both “non-optimal” alleles (DAT1 CC and BDNF Met) exhibit higher Aβ Centiloids and tau SUVR compared with other genotypes. (A) DAT1 and BDNF interact to predict cross-sectional Aβ-PET measured in centiloids (p=.019; left). The DAT1*BDNF interaction predicting longitudinal change in Aβ-PET did not reach significance (p=.09; right). (B) Significant DAT1*BDNF interactions predicting entorhinal tau-PET SUVR (cross-sectional: p=.013, left; longitudinal: p=.0008, right). (C) Significant DAT1*BDNF interactions predicting temporal lobe tau-PET (cross-sectional: p=.001, left; longitudinal: p=.001, right). The interaction effect on temporal lobe tau-PET remained significant after removing four outliers (p=.005).
Table 2.
DAT1 rs6347*BDNF Val66Met interactions on Aβ-PET (A), (B), entorhinal tau-PET (C), (D), and meta-temporal lobe tau-PET (E), (F).
| Variable | Unstandardized Coef. | SE | t | p | 95% CI |
|---|---|---|---|---|---|
|
| |||||
| (A) Aβ-PET (cross-sectional) | R2 = .070, F(6, 314) = 3.914, p = .0009 | ||||
|
| |||||
| rs6347*Val66Met | 12.273 | 5.219 | 2.352 | .019 | 2.004, 22.542 |
| rs6347 | −3.121 | 2.528 | −1.235 | .218 | −8.095, 1.852 |
| Val66Met | −5.012 | 3.527 | −1.421 | .156 | −11.951, 1.927 |
| Sex | 4.155 | 3.471 | 1.197 | .232 | −2.675, 10.985 |
| Age | .943 | .245 | 3.845 | .0001 | .460, 1.425 |
| Years Ed | −.586 | .749 | −.783 | .434 | −2.059, .887 |
| (B) Aβ-PET (longitudinal) | R2 = .048, F(8, 226) = 3.914, p = .188 | ||||
| rs6347*Val66Met | .861 | .473 | 1.691 | .092 | −.133, 1.737 |
| rs6347 | −.257 | .229 | −1.119 | .265 | −.710, .196 |
| Val66Met | −.023 | .315 | −.074 | .942 | −.645, .598 |
| Sex | .358 | .316 | 1.131 | .259 | −.266, .981 |
| Age | .060 | .024 | 2.470 | .041 | .012, .108 |
| Years Ed | −.033 | .067 | −.493 | .622 | −165, .108 |
| Follow-up Time | −.106 | .154 | −.689 | .492 | −.410, .198 |
| Number of scans | .060 | .322 | .185 | .853 | −.575, .695 |
| (C) Entorhinal tau-PET (cross-sectional) | R2 = .058, F(6, 314) = 3.247, p = .004 | ||||
| rs6347*Val66Met | .055 | .022 | 2.493 | .013 | .012, .098 |
| rs6347 | −.018 | .011 | −1.669 | .096 | −.039, .003 |
| Val66Met | −.025 | .015 | −1.703 | .090 | −.055, .004 |
| Sex | .005 | .015 | .328 | .743 | −.024, .034 |
| Age | .002 | .001 | 1.759 | .080 | −.0002, .004 |
| Years Ed | .007 | .003 | 2.115 | .035 | .0005, .013 |
| (D) Entorhinal tau-PET slope | R2 = .201, F(8, 126) = 3.956, p = .0003 | ||||
| rs6347*Val66Met | .012 | .003 | 3.437 | .0008 | .005, .017 |
| rs6347 | −.003 | .001 | −1.988 | .049 | −.006, −.001 |
| Val66Met | −.002 | .002 | −.737 | .463 | −.006, .003 |
| Sex | .003 | .002 | 1.232 | .220 | −.002, .007 |
| Age | .0003 | .0002 | 1.928 | .056 | .00002 .001 |
| Years Ed | .001 | .0004 | 1.129 | .199 | −.0003, .001 |
| Follow-up Time | −.001 | .001 | −1.663 | .099 | −.003, .0003 |
| Number of scans | .003 | .001 | 2.063 | .041 | .0001, .006 |
| (E) Meta-temporal tau-PET (cross-sectional) | R2 = .056, F(6, 314) = 3.108, p = .006 | ||||
| rs6347*Val66Met | .067 | .020 | 3.344 | .0009 | .028, .106 |
| rs6347 | −.017 | .010 | −1.722 | .086 | −.036, .002 |
| Val66Met | −.010 | .014 | −.756 | .450 | −.037, .016 |
| Sex | −.0006 | .013 | −.046 | .964 | −.027, .026 |
| Age | .002 | .0009 | 1.455 | .147 | −.0005, .003 |
| Years Ed | .004 | .003 | 1.355 | .176 | −.002, .010 |
| (F) Meta-temporal tau-PET slope | R2 = .122, F(8, 126) = 2.181, p = .033 | ||||
| rs6347*Val66Met | .014 | .004 | 3.265 | .001 | .006, .023 |
| rs6347 | −.003 | .002 | −1.274 | .205 | −.007, .001 |
| Val66Met | −.001 | .003 | −.232 | .817 | −.006, .005 |
| Sex | .0001 | .003 | .281 | .794 | −.001, .001 |
| Age | .0001 | .0002 | .505 | .614 | −.0003, .001 |
| Years Ed | .0001 | .001 | .228 | .820 | −.001, .001 |
| Follow-up Time | −.001 | .001 | −.811 | .419 | −.003, .001 |
| Number of scans | .003 | .002 | 1.476 | .143 | −.001, .007 |
We next tested whether DAT1 and BDNF would interact to predict cross-sectional and longitudinal tau-PET (entorhinal and meta-temporal ROIs). Similar to the Aβ analyses, participants carrying both DAT1 CC and BDNF Met exhibited the highest tau SUVR and the greatest rates of longitudinal increase. DAT1*BDNF significantly predicted cross-sectional and longitudinal entorhinal tau (cross-sectional: t(314)=2.49, p=.013, f2=.02; longitudinal: t(126)=3.44, p=.0008, f2=.07; Figure 1B; Table 2C, D) and meta-temporal tau (cross-sectional: t(314)=3.34, p=.0009, f2=.03; longitudinal: t(126)=3.26, p=.001, f2=.09; Figure 1C; Table 2E, F). Paralleling Aβ-PET analyses, direct effects of DAT1 rs6347 and BDNF Val66Met on cross-sectional and longitudinal tau-PET were null for entorhinal ROIs (cross-sectional entorhinal: rs6347 p=.06, Val66Met p=.07; longitudinal entorhinal: rs6347 p=.14, Val66Met p=.50) and meta-tau ROIs (cross-sectional meta-ROI: rs6347 p=.06, Val66Met p=.40; longitudinal meta-ROI: rs6347 p=.17, Val66Met p=.98).
Given the interactive effects of DAT1 and BDNF on both Aβ and tau pathology, we conducted a moderated mediation analysis to test whether Aβ mediates the relationship between the polymorphisms and tau. This analysis is in line with prominent models describing the role of Aβ in AD, which suggest that Aβ contributes to tau spread (Hardy and Higgins, 1992; Karran et al., 2011). The moderated mediation was performed on cross-sectional data only, as there were few participants with both longitudinal Aβ and longitudinal tau. The moderated mediation model (Figure 2) demonstrated that BDNF Val66Met significantly moderated the mediation among DAT1 rs6347, Aβ-PET, and tau-PET (moderated mediation index = .02, 95% CI [.002, .036]), adjusting for age, sex, and years of education as covariates. As demonstrated by our multiple regression analyses, BDNF Val66Met significantly moderates relationships between DAT1 and Aβ-PET, as determined by a significant conditional indirect effect of DAT1 and BDNF on tau-PET through Aβ-PET as a mediator. The conditional indirect effect is demonstrated by a significant effect of DAT1 on Aβ-PET for BDNF Met carriers (b=−.015, 95% CI [−.03, −.003]) but not for BDNF Val/Val homozygotes (b=.002, 95% CI [−.006, .020]), and a significant relationship between Aβ and tau PET measures (b=.002, 95% CI [.001, .002]). These results indicate that DAT1 rs6347 and BDNF Val66Met together relate to both amyloid and tau pathology and may have a synergistic effect in which carrying both rs6347 C and Val66Met Met relates to higher pathology.
Figure 2.
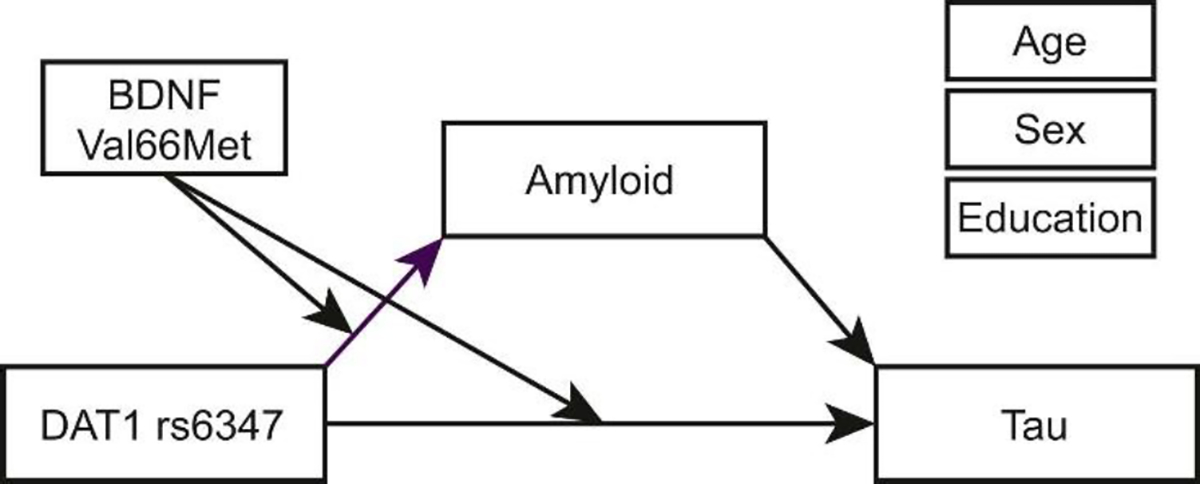
Diagram illustrating exploratory moderated mediation model. Val66Met moderates the mediation between rs6347, Aβ, and tau pathology, with age, sex, and years of education added to the model as covariates.
We replicated this moderated mediation using tau-PET measured within the temporal lobe tau meta-ROI (Jack et al., 2020), rather than entorhinal tau-PET. This analysis yielded similar significant results (moderated mediation index=.012, 95% CI [.002, .025]), indicating a significant moderating effect of BDNF Val66Met on the DAT1→Aβ→tau mediation such that individuals carrying both DAT1 CC and BDNF Met demonstrate higher Aβ and temporal lobe tau pathology. As there were four outliers in the tau meta-ROI data (greater than three SD above the mean), we also re-ran this analysis after removing these potentially influential datapoints (n=318 participants included in analysis) and found that there was still a significant moderated mediation (moderated mediation index=.010, 95% CI [.002, .021]), suggesting that the effect is not driven by individuals with highest tau-PET values.
3.3. DAT1 and BDNF interact to predict change in hippocampal volume
After determining that DAT1 CC and BDNF Met variants are related to higher pathology, we investigated whether these same variants would relate to lower hippocampal volume. Bilateral hippocampal volume was measured using FreeSurfer-derived ROIs (Figure 3A). All regression analyses included age, sex, years of education, and estimated total intracranial volume as covariates. There was no DAT1*BDNF interaction on cross-sectional hippocampal volume (p=.68). However, there was a significant DAT1*BDNF interaction on longitudinal change in hippocampal volume (t(205)=−2.19, p=.03, f2=.02; Figure 3B). Individuals carrying both non-optimal genotypes exhibited greater decline in hippocampal volume over time. Similar to analyses of PET data, there were no main effects of DAT1 rs6347 and BDNF Val66Met on hippocampal volume (cross-sectional: rs6347 p=.53, Val66Met p=.60; longitudinal: rs6347 p=.97, Val66Met p=.17).
Figure 3.
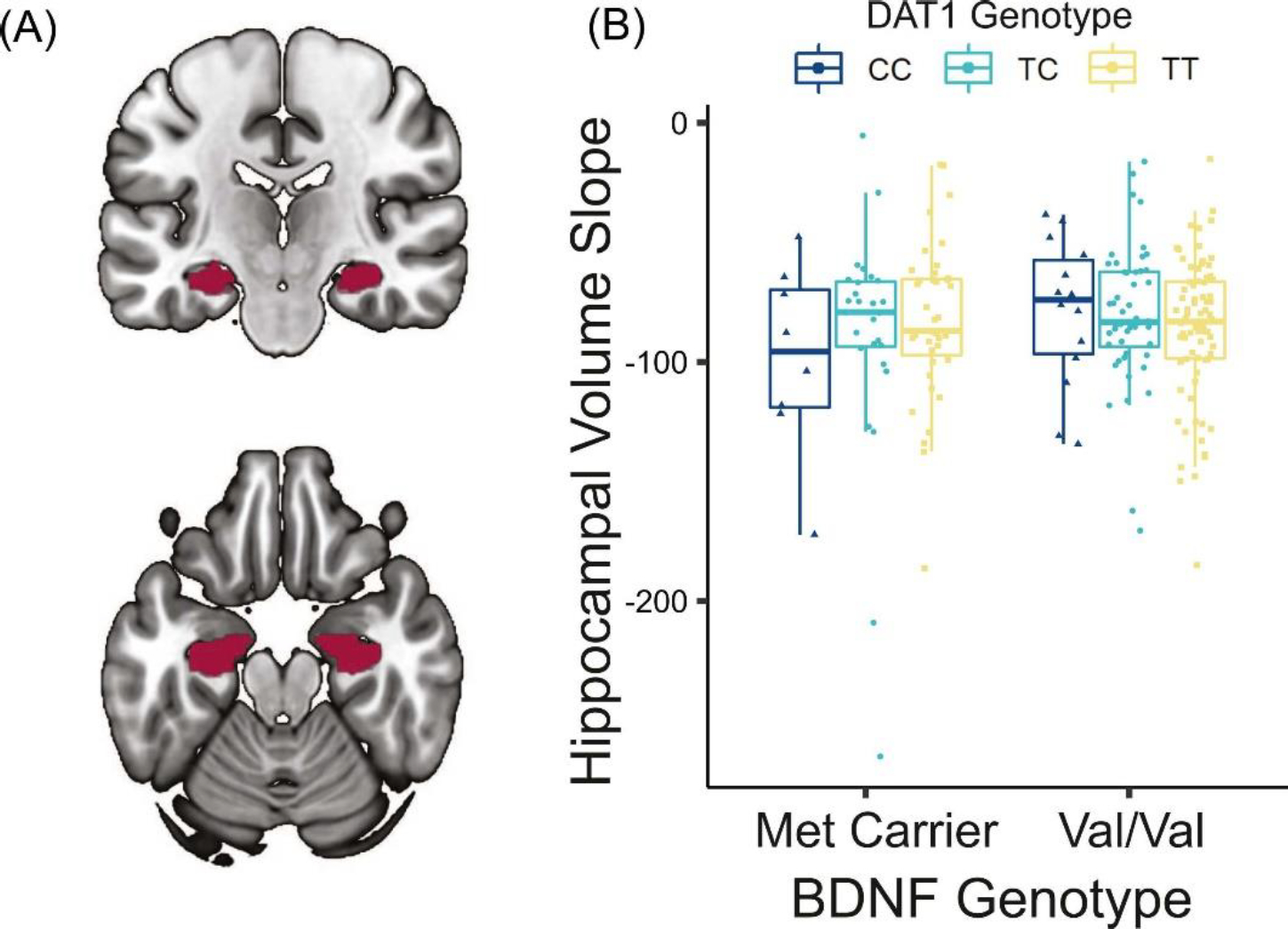
Relationship between polymorphisms and change in hippocampal volume. (A) FreeSurfer-derived ROI of bilateral hippocampus overlaid on MNI152 template. (B) Significant DAT1*BDNF interaction predicting longitudinal change in hippocampal volume (p=.03, adjusting for age, sex, years of education, and total intracranial volume) such that individuals carrying both DAT1 CC and BDNF Met demonstrate greater decline in hippocampal volume. Hippocampal slopes for individual participants were extracted from a linear mixed effects model.
3.4. DAT1 and BDNF genotypes do not relate to cognition
DAT1 and BDNF did not interact to predict any cross-sectional cognitive measures (UW Memory: p=.65, UW EF: p=.51, PACC: p=.87). Similarly, DAT1*BDNF did not predict longitudinal change in cognition (UW Memory: p=.77, UW EF: p=.22, PACC: p=.87). There were no main effects of DAT1 rs6347 or BDNF Val66Met on UW Memory (cross-sectional: rs6347 p=.82, Val66Met p=.80; longitudinal: rs6347 p=.39, Val66Met p=.95), UW Executive Function (cross-sectional: rs6347 p=.71, Val66Met p=.23; longitudinal: rs6347 p=.20, Val66Met p=.76), or PACC (cross-sectional: rs6347 p=.85, Val66Met p=.58; longitudinal: rs6347 p=.55, Val66Met p=.97).
4. DISCUSSION
We investigated relationships among AD-related pathology and two genetic polymorphisms that have previously been associated with increased risk for dementia but have not been directly related to pathology. Our analyses demonstrate that interactions between DAT1 rs6347 and BDNF Val66Met predict PET measures of Aβ and tau pathology and change in hippocampal volume in cognitively normal older adults. Carriers of both rs6347 CC and Val66Met Met demonstrated higher cross-sectional Aβ and tau pathology and greater longitudinal tau and hippocampal atrophy. Our findings extend previous research implicating these variants in AD vulnerability (Lin et al., 2012; Roussotte et al., 2015) by demonstrating associations with greater AD pathology. All of these analyses focused on cognitively normal older adults and demonstrated small effect sizes (Cohen’s f2=.01-.09).
Our moderated mediation analysis suggests that together the DAT1 and BDNF polymorphisms are related to higher Aβ pathology, which then contributes to higher tau pathology. While our analyses do not demonstrate causality, these findings are consistent with models of AD by which Aβ drives increases in tau pathology (Hardy and Higgins, 1992; Karran et al., 2011). Our results are also in line with work linking sub-optimal dopamine function to AD (Nobili et al., 2017; Pan et al., 2019), and research defining protective roles of BDNF (Buchman et al., 2016; Lim et al., 2013). Our study suggests that individuals carrying both “optimal” alleles (DAT1 T and BDNF Val) may show greater resistance to AD-related pathology. While it is difficult to determine whether entorhinal tau in a cognitively unimpaired sample relates to AD or to primary age-related tauopathy (PART), we demonstrate consistent findings in a tau meta-ROI (Jack et al., 2020) consisting of temporal lobe regions, suggesting our results may be relevant to AD-related processes.
While we found no evidence that “optimal” alleles were associated with greater hippocampal volume cross-sectionally, our exploratory longitudinal analyses suggest that these alleles relate to less hippocampal atrophy. Similar discrepancies between cross-sectional versus longitudinal effects have been reported for aging studies evaluating hippocampal volume, which has suggested that longitudinal measures of hippocampal volume may, in some cases, be more sensitive than cross-sectional (Pfefferbaum and Sullivan, 2015). Previous analyses of the DAT1 rs6347 polymorphism have found associations with longitudinal measures of ventricular volume, which were absent for cross-sectional analyses (Roussotte et al., 2015). While BDNF Val66Met has been linked to both cross-sectional and longitudinal hippocampal volume, some research suggests that this polymorphism relates best to longitudinal volume changes (Lim et al., 2017).
It is unclear why we observed genetic effects on pathology and hippocampal atrophy in the absence of effects on cognition. BDNF and DAT1 polymorphisms have been previously linked with individual differences in cognitive function in aging (Baeuchl et al., 2019; van den Bosch et al., 2021) though genetic effects on cognitive function are often small and mediated by diverse factors (Dang et al., 2013). It is also possible that indirect effects of these polymorphisms on cognition (via effects on Aβ, tau and hippocampal atrophy) are only evident with greater disease progression, which would be in general agreement with observations that cognitive dysfunction temporally trails neurodegeneration and accumulation of Aβ and tau (Karran et al., 2011). As mentioned in the introduction, there are mixed findings regarding the role of BDNF in AD, which has often been studied in the context of BDNF interactions with APOE. Multiple studies suggest that carrying both BDNF Met and APOE e4 alleles relates to greater pathology (Adamczuk et al., 2013; Stonnington et al., 2020), while findings are more mixed for analyses focused on cognition (Stonnington et al., 2020; Ward et al., 2014). This is in line with evidence that BDNF may be more sensitive to early changes in pathology and neurodegeneration than to cognitive function (Lim et al., 2016; Stonnington et al., 2020).
DAT1 and BDNF interacted to predict cross-sectional Aβ and tau and longitudinal tau, but, unexpectedly, there was no significant interaction predicting longitudinal Aβ (p=.09, f2=.01). While our exploratory moderated mediation suggested a path by which genetic effects on tau are mediated by Aβ, this was complicated by the lack of polymorphism effects on longitudinal Aβ. Additional research is needed to establish DAT1*BDNF effects on Aβ-independent, age-related tau accumulation. Important limitations of this work are the relatively small number of participants with both non-optimal genotypes and the small effect sizes observed throughout. Due to the small sample and small effect sizes, we do not want to strongly interpret this null result for longitudinal Aβ in the absence of replication in another PET dataset, or exploration within a larger fluid biomarker dataset.
Additional research is needed to more clearly define the mechanisms by which BDNF and the dopamine system interact to influence AD pathology and hippocampal atrophy. BDNF maintains the health of dopamine-producing neurons via TrkB receptors (Numan and Seroogy, 1999) and regulates dopamine receptor expression (Guillin et al., 2001). Dopamine neurons can, in turn, increase BDNF expression via dopaminergic signaling (Okazawa et al., 1992). Thus, non-optimal function of dopamine and BDNF can create a “vicious cycle”, magnifying deficits in each system. Broadly, non-optimal dopamine and BDNF signaling may create a more vulnerable environment in which Aβ and tau are more likely to accumulate. The positive impact of enhanced BDNF/TrkB signaling on dopamine system health has been studied in the context of excitotoxity in AD and Parkinson’s disease models (Meisner et al., 2008; Zhu et al., 2015), which provides initial groundwork for establishing effects of these systems on hippocampal atrophy. Relevant to pathology, there is some evidence suggesting dopamine can disaggregate Aβ fibrils in vivo (Li et al., 2004), and that Aβ oligomers decrease BDNF expression (Garzon and Fahnestock, 2007). Optimal dopaminergic tone also plays a key role in stabilizing neural activity, with direct effects on hippocampal synaptic plasticity (Rossato et al., 2009; Yuan Xiang et al., 2016), and GABAergic inhibition (Seamans et al., 2001). Thus, dysregulation of dopamine and BDNF signaling may enhance pathology development and spread by increasing neural hyperactivity, a known promoter of Aβ and tau aggregation (Bero et al., 2011; Wu et al., 2016). Other indirect pathways may arise via associations with other neuromodulator systems. Increased dopamine transmission can prevent Aβ-induced internalization of acetylcholine receptors (Jürgensen et al., 2011), while the BDNF receptor TrkB supports neuroprotective effects of norepinephrine against Aβ toxicity (Liu et al., 2015). It will be critical to replicate our findings and extend them with in vitro research defining the cellular mechanisms that might drive associations between reduced BDNF, non-optimal dopamine function, and elevated AD pathology.
5. CONCLUSION
Understanding mechanisms that contribute to individual differences in Aβ and tau pathology in cognitively normal older adults will be critical for advancing our understanding of variation in AD risk. A recent review of AD drug trials highlighted the potential of genetic pathways as diagnostic indicators and targets of preventative drugs (van Bokhoven et al., 2021). Here, we demonstrate novel associations among polymorphisms in the dopamine transporter and BDNF genes, AD pathology, and hippocampal atrophy. Our results provide a direct link between AD pathology and variants of these genes previously associated with worse disease trajectories.
HIGHLIGHTS.
DAT1 and BDNF polymorphisms relate to Aβ and tau pathology in healthy older adults.
Carriers of both DAT1 CC and BDNF Met exhibit higher Aβ and tau.
DAT1 CC and BDNF Met are also linked with greater hippocampal atrophy.
ACKNOWLEDGEMENTS
This study was funded by National Institutes of Health Grant R21 AG081759-01A1. Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Footnotes
CONFLICTS OF INTEREST
SM Landau is on the DSMB for Keife RX and the NIH IPAT study and has received speaking honoraria from Eisai. Authors CJ Ciampa, TM Morin, A Murphy, R La Joie, and AS Berry have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain
REFERENCES
- Adamczuk K, De Weer A-S, Nelissen N, Chen K, Sleegers K, Bettens K, Van Broeckhoven C, Vandenbulcke M, Thiyyagura P, Dupont P, Van Laere K, Reiman EM, Vandenberghe R, 2013. Polymorphism of brain derived neurotrophic factor influences β amyloid load in cognitively intact apolipoprotein E ε4 carriers. NeuroImage: Clinical 2, 512–520. 10.1016/j.nicl.2013.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeuchl C, Chen H-Y, Su Y-S, Hämmerer D, Klados MA, Li S-C, 2019. Interactive effects of dopamine transporter genotype and aging on resting-state functional networks. PLOS ONE 14, e0215849. 10.1371/journal.pone.0215849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee J-M, Holtzman DM, 2011. Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat Neurosci 14, 750–756. 10.1038/nn.2801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry AS, Harrison TM, 2023. New perspectives on the basal forebrain cholinergic system in Alzheimer’s disease. Neuroscience & Biobehavioral Reviews 150, 105192. 10.1016/j.neubiorev.2023.105192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E, 1985. On areas of transition between entorhinal allocortex and temporal isocortex in the human brain. Normal morphology and lamina-specific pathology in Alzheimer’s disease. Acta Neuropathol 68, 325–332. 10.1007/BF00690836 [DOI] [PubMed] [Google Scholar]
- Buchman AS, Yu L, Boyle PA, Schneider JA, Jager PLD, Bennett DA, 2016. Higher brain BDNF gene expression is associated with slower cognitive decline in older adults. Neurology 86, 735–741. 10.1212/WNL.0000000000002387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciampa CJ, Parent JH, Harrison TM, Fain RM, Betts MJ, Maass A, Winer JR, Baker SL, Janabi M, Furman DJ, D’Esposito M, Jagust WJ, Berry AS, 2022. Associations among locus coeruleus catecholamines, tau pathology, and memory in aging. Neuropsychopharmacol. 47, 1106–1113. 10.1038/s41386-022-01269-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane PK, Carle A, Gibbons LE, Insel P, Mackin RS, Gross A, Jones RN, Mukherjee S, Curtis SM, Harvey D, Weiner M, Mungas D, for the Alzheimer’s Disease Neuroimaging Initiative, 2012. Development and assessment of a composite score for memory in the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Brain Imaging and Behavior 6, 502–516. 10.1007/s11682-012-9186-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang LC, O’Neil JP, Jagust WJ, 2013. Genetic effects on behavior are mediated by neurotransmitters and large-scale neural networks. NeuroImage 66, 203–214. 10.1016/j.neuroimage.2012.10.090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohue MC, Sperling RA, Salmon DP, Rentz DM, Raman R, Thomas RG, Weiner M, Aisen PS, for the Australian Imaging, B., and Lifestyle Flagship Study of Ageing; the Alzheimer’s Disease Neuroimaging Initiative; and the Alzheimer’s Disease Cooperative Study, 2014. The Preclinical Alzheimer Cognitive Composite: Measuring Amyloid-Related Decline. JAMA Neurology 71, 961–970. 10.1001/jamaneurol.2014.803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faul F, Erdfelder E, Lang A-G, Buchner A, 2007. G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behavior Research Methods 39, 175–191. 10.3758/BF03193146 [DOI] [PubMed] [Google Scholar]
- Fenner ME, Achim CL, Fenner BM, 2014. Expression of full-length and truncated trkB in human striatum and substantia nigra neurons: implications for Parkinson’s disease. J Mol Hist 45, 349–361. 10.1007/s10735-013-9562-z [DOI] [PubMed] [Google Scholar]
- Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, van der Kouwe A, Killiany R, Kennedy D, Klaveness S, Montillo A, Makris N, Rosen B, Dale AM, 2002. Whole Brain Segmentation: Automated Labeling of Neuroanatomical Structures in the Human Brain. Neuron 33, 341–355. 10.1016/S0896-6273(02)00569-X [DOI] [PubMed] [Google Scholar]
- Franzmeier N, Ren J, Damm A, Monté-Rubio G, Boada M, Ruiz A, Ramirez A, Jessen F, Düzel E, Rodríguez Gómez O, Benzinger T, Goate A, Karch CM, Fagan AM, McDade E, Buerger K, Levin J, Duering M, Dichgans M, Suárez-Calvet M, Haass C, Gordon BA, Lim YY, Masters CL, Janowitz D, Catak C, Wolfsgruber S, Wagner M, Milz E, Moreno-Grau S, Teipel S, Grothe MJ, Kilimann I, Rossor M, Fox N, Laske C, Chhatwal J, Falkai P, Perneczky R, Lee J-H, Spottke A, Boecker H, Brosseron F, Fliessbach K, Heneka MT, Nestor P, Peters O, Fuentes M, Menne F, Priller J, Spruth EJ, Franke C, Schneider A, Westerteicher C, Speck O, Wiltfang J, Bartels C, Araque Caballero MÁ, Metzger C, Bittner D, Salloway S, Danek A, Hassenstab J, Yakushev I, Schofield PR, Morris JC, Bateman RJ, Ewers M, 2021. The BDNFVal66Met SNP modulates the association between beta-amyloid and hippocampal disconnection in Alzheimer’s disease. Mol Psychiatry 26, 614–628. 10.1038/s41380-019-0404-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzon DJ, Fahnestock M, 2007. Oligomeric Amyloid Decreases Basal Levels of Brain-Derived Neurotrophic factor (BDNF) mRNA via Specific Downregulation of BDNF Transcripts IV and V in Differentiated Human Neuroblastoma Cells. J. Neurosci. 27, 2628–2635. 10.1523/JNEUROSCI.5053-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons LE, Carle AC, Mackin RS, Harvey D, Mukherjee S, Insel P, Curtis SM, Mungas D, Crane PK, for the Alzheimer’s Disease Neuroimaging Initiative, 2012. A composite score for executive functioning, validated in Alzheimer’s Disease Neuroimaging Initiative (ADNI) participants with baseline mild cognitive impairment. Brain Imaging and Behavior 6, 517–527. 10.1007/s11682-012-9176-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, Malek-Ahmadi MH, Alldred MJ, Chen Y, Chen K, Chao MV, Counts SE, Mufson EJ, 2019. Brain-derived neurotrophic factor (BDNF) and TrkB hippocampal gene expression are putative predictors of neuritic plaque and neurofibrillary tangle pathology. Neurobiology of Disease 132, 104540. 10.1016/j.nbd.2019.104540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, Zeiler SR, Tamowski S, Jones KR, 2003. Brain-Derived Neurotrophic Factor Is Required for the Maintenance of Cortical Dendrites. J. Neurosci. 23, 6856–6865. 10.1523/JNEUROSCI.23-17-06856.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillin O, Diaz J, Carroll P, Griffon N, Schwartz J-C, Sokoloff P, 2001. BDNF controls dopamine D3 receptor expression and triggers behavioural sensitization. Nature 411, 86–89. 10.1038/35075076 [DOI] [PubMed] [Google Scholar]
- Hampel H, Mesulam M-M, Cuello AC, Farlow MR, Giacobini E, Grossberg GT, Khachaturian AS, Vergallo A, Cavedo E, Snyder PJ, Khachaturian ZS, 2018. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 141, 1917–1933. 10.1093/brain/awy132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA, 1992. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 256, 184–185. 10.1126/science.1566067 [DOI] [PubMed] [Google Scholar]
- Hayes AF, 2013. Introduction to mediation, moderation, and conditional process analysis: A regression-based approach, Introduction to mediation, moderation, and conditional process analysis: A regression-based approach. Guilford Press, New York, NY, US. [Google Scholar]
- Hünnerkopf R, Strobel A, Gutknecht L, Brocke B, Lesch KP, 2007. Interaction between BDNF Val66Met and Dopamine Transporter Gene Variation Influences Anxiety-Related Traits. Neuropsychopharmacol 32, 2552–2560. 10.1038/sj.npp.1301383 [DOI] [PubMed] [Google Scholar]
- Jaber M, Jones S, Giros B, Caron MG, 1997. The dopamine transporter: a crucial component regulating dopamine transmission. Mov Disord 12, 629–633. 10.1002/mds.870120502 [DOI] [PubMed] [Google Scholar]
- Jack CR Jr, Wiste HJ., Weigand SD., Therneau TM., Lowe VJ., Knopman DS., Botha H., Graff-Radford J., Jones DT., Ferman TJ., Boeve BF., Kantarci K., Vemuri P., Mielke MM., Whitwell J., Josephs K., Schwarz CG., Senjem ML., Gunter JL., Petersen RC., 2020. Predicting future rates of tau accumulation on PET. Brain 143, 3136–3150. 10.1093/brain/awaa248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs HIL, Riphagen JM, Ramakers IHGB, Verhey FRJ, 2021. Alzheimer’s disease pathology: pathways between central norepinephrine activity, memory, and neuropsychiatric symptoms. Molecular Psychiatry 26, 897–906. 10.1038/s41380-019-0437-x [DOI] [PubMed] [Google Scholar]
- Jerónimo-Santos A, Vaz SH, Parreira S, Rapaz-Lérias S, Caetano AP, Buée-Scherrer V, Castrén E, Valente CA, Blum D, Sebastião AM, Diógenes MJ, 2015. Dysregulation of TrkB Receptors and BDNF Function by Amyloid-β Peptide is Mediated by Calpain. Cerebral Cortex 25, 3107–3121. 10.1093/cercor/bhu105 [DOI] [PubMed] [Google Scholar]
- Ji H, Dai D, Wang Y, Jiang D, Zhou X, Lin P, Ji X, Li J, Zhang Y, Yin H, Chen R, Zhang L, Xu M, Duan S, Wang Q, 2015. Association of BDNF and BCHE with Alzheimer’s disease: Meta‑analysis based on 56 genetic case‑control studies of 12,563 cases and 12,622 controls. Experimental and Therapeutic Medicine 9, 1831–1840. 10.3892/etm.2015.2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jürgensen S, Antonio LL, Mussi GEA, Brito-Moreira J, Bomfim TR, De Felice FG, Garrido-Sanabria ER, Cavalheiro ÉA, Ferreira ST, 2011. Activation of D1/D5 Dopamine Receptors Protects Neurons from Synapse Dysfunction Induced by Amyloid-β Oligomers*. Journal of Biological Chemistry 286, 3270–3276. 10.1074/jbc.M110.177790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karran E, Mercken M, Strooper BD, 2011. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov 10, 698–712. 10.1038/nrd3505 [DOI] [PubMed] [Google Scholar]
- Kaufman SK, Del Tredici K, Thomas TL, Braak H, Diamond MI, 2018. Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in Alzheimer’s disease and PART. Acta Neuropathol 136, 57–67. 10.1007/s00401-018-1855-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhu M, Manning-Bog AB, Di Monte DA, Fink AL, 2004. Dopamine and L-dopa disaggregate amyloid fibrils: implications for Parkinson’s and Alzheimer’s disease. The FASEB Journal 18, 962–964. 10.1096/fj.03-0770fje [DOI] [PubMed] [Google Scholar]
- Lim YY, Hassenstab J, Cruchaga C, Goate A, Fagan AM, Benzinger TLS, Maruff P, Snyder PJ, Masters CL, Allegri R, Chhatwal J, Farlow MR, Graff-Radford NR, Laske C, Levin J, McDade E, Ringman JM, Rossor M, Salloway S, Schofield PR, Holtzman DM, Morris JC, Bateman RJ, on behalf of the Dominantly Inherited Alzheimer Network, 2016. BDNF Val66Met moderates memory impairment, hippocampal function and tau in preclinical autosomal dominant Alzheimer’s disease. Brain 139, 2766–2777. 10.1093/brain/aww200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim YY, Rainey-Smith S, Lim Y, Laws SM, Gupta V, Porter T, Bourgeat P, Ames D, Fowler C, Salvado O, Villemagne VL, Rowe CC, Masters CL, Zhou XF, Martins RN, Maruff P, 2017. BDNF Val66Met in preclinical Alzheimer’s disease is associated with short-term changes in episodic memory and hippocampal volume but not serum mBDNF. International Psychogeriatrics 29, 1825–1834. 10.1017/S1041610217001284 [DOI] [PubMed] [Google Scholar]
- Lim YY, Villemagne VL, Laws SM, Ames D, Pietrzak RH, Ellis KA, Harrington KD, Bourgeat P, Salvado O, Darby D, Snyder PJ, Bush AI, Martins RN, Masters CL, Rowe CC, Nathan PJ, Maruff P, 2013. BDNF Val66Met, Aβ amyloid, and cognitive decline in preclinical Alzheimer’s disease. Neurobiology of Aging 34, 2457–2464. 10.1016/j.neurobiolaging.2013.05.006 [DOI] [PubMed] [Google Scholar]
- Lin WY, Wu BT, Lee CC, Sheu JJ, Liu SH, Wang WF, Tsai CH, Liu HP, Tsai FJ, 2012. Association analysis of dopaminergic gene variants (Comt, Drd4 And Dat1) with Alzheimer s disease. J Biol Regul Homeost Agents 26, 401–410. [PubMed] [Google Scholar]
- Liu X, Ye K, Weinshenker D, 2015. Norepinephrine Protects against Amyloid-β Toxicity via TrkB. J Alzheimers Dis 44, 251–260. 10.3233/JAD-141062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisner F, Scheller C, Kneitz S, Sopper S, Neuen-Jacob E, Riederer P, Meulen V. ter, Koutsilieri E., 2008. Memantine Upregulates BDNF and Prevents Dopamine Deficits in SIV-Infected Macaques: A Novel Pharmacological Action of Memantine. Neuropsychopharmacol 33, 2228–2236. 10.1038/sj.npp.1301615 [DOI] [PubMed] [Google Scholar]
- Nobili A, Latagliata EC, Viscomi MT, Cavallucci V, Cutuli D, Giacovazzo G, Krashia P, Rizzo FR, Marino R, Federici M, De Bartolo P, Aversa D, Dell’Acqua MC, Cordella A, Sancandi M, Keller F, Petrosini L, Puglisi-Allegra S, Mercuri NB, Coccurello R, Berretta N, D’Amelio M, 2017. Dopamine neuronal loss contributes to memory and reward dysfunction in a model of Alzheimer’s disease. Nat Commun 8, 14727. 10.1038/ncomms14727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numan S, Seroogy KB, 1999. Expression of trkB and trkC mRNAs by adult midbrain dopamine neurons: A double-label in situ hybridization study. Journal of Comparative Neurology 403, 295–308. [DOI] [PubMed] [Google Scholar]
- Okazawa H, Murata M, Watanabe M, Kamei M, Kanazawa I, 1992. Dopaminergic stimulation up-regulates the in vivo expression of brain-derived neurotrophic factor (BDNF) in the striatum. FEBS Letters 313, 138–142. 10.1016/0014-5793(92)81430-T [DOI] [PubMed] [Google Scholar]
- Pan X, Kaminga AC, Wen SW, Wu X, Acheampong K, Liu A, 2019. Dopamine and Dopamine Receptors in Alzheimer’s Disease: A Systematic Review and Network Meta-Analysis. Frontiers in Aging Neuroscience 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papenberg G, Karalija N, Salami A, Andersson M, Axelsson J, Riklund K, Lindenberger U, Nyberg L, Bäckman L, 2019. The Influence of Hippocampal Dopamine D2 Receptors on Episodic Memory Is Modulated by BDNF and KIBRA Polymorphisms. Journal of Cognitive Neuroscience 31, 1422–1429. 10.1162/jocn_a_01429 [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Sullivan EV, 2015. Cross-sectional versus longitudinal estimates of age-related changes in the adult brain: overlaps and discrepancies. Neurobiol Aging 36, 2563–2567. 10.1016/j.neurobiolaging.2015.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan L, Jin Y, Zhang H, Qiang W, Shekhtman E, Shao D, Revoe D, Villamarin R, Ivanchenko E, Kimura M, Wang Z, Hao L, Sharopova N, Bihan M, Sturcke A, Lee M, Popova N, Wu W, Bastiani C, Ward M, Holmes J, Lyoshin V, Kaur K, Moyer E, Feolo M, Kattman B, 2020. ALFA: Allele Frequency Aggregator [WWW Document]. National Center for Biotechnology Information, U.S. National Library of Medicine. URL https://www.ncbi.nlm.nih.gov/snp/docs/gsr/alfa/ (accessed 7.28.23). [Google Scholar]
- Porter T, Villemagne VL, Savage G, Milicic L, Ying Lim Y, Maruff P, Masters CL, Ames D, Bush AI, Martins RN, Rainey-Smith S, Rowe CC, Taddei K, Groth D, Verdile G, Burnham SC, Laws SM, 2018. Cognitive gene risk profile for the prediction of cognitive decline in presymptomatic Alzheimer’s disease. Personalized Medicine in Psychiatry 7–8, 14–20. 10.1016/j.pmip.2018.03.001 [DOI] [Google Scholar]
- Rossato JI, Bevilaqua LRM, Izquierdo I, Medina JH, Cammarota M, 2009. Dopamine Controls Persistence of Long-Term Memory Storage. Science 325, 1017–1020. 10.1126/science.1172545 [DOI] [PubMed] [Google Scholar]
- Rousset OG, Ma Y, Evans AC, 1998. Correction for partial volume effects in PET: principle and validation. Journal of Nuclear Medicine 39, 904–911. [PubMed] [Google Scholar]
- Roussotte FF, Gutman BA, Hibar DP, Madsen SK, Narr KL, Thompson PM, 2015. Carriers of a common variant in the dopamine transporter gene have greater dementia risk, cognitive decline, and faster ventricular expansion. Alzheimer’s & Dementia 11, 1153–1162. 10.1016/j.jalz.2014.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royse SK, Minhas DS, Lopresti BJ, Murphy A, Ward T, Koeppe RA, Bullich S, DeSanti S, Jagust WJ, Landau SM, for the Alzheimer’s Disease Neuroimaging Initiative, 2021. Validation of amyloid PET positivity thresholds in centiloids: a multisite PET study approach. Alzheimer’s Research & Therapy 13, 99. 10.1186/s13195-021-00836-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamans JK, Gorelova N, Durstewitz D, Yang CR, 2001. Bidirectional Dopamine Modulation of GABAergic Inhibition in Prefrontal Cortical Pyramidal Neurons. J. Neurosci. 21, 3628–3638. 10.1523/JNEUROSCI.21-10-03628.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seroogy KB, Lundgren KH, Tran TMD, Guthrie KM, Isackson PJ, Gall CM, 1994. Dopaminergic neurons in rat ventral midbrain express brain-derived neurotrophic factor and neurotrophin-3 mRNAs. Journal of Comparative Neurology 342, 321–334. 10.1002/cne.903420302 [DOI] [PubMed] [Google Scholar]
- Stonnington CM, Velgos SN, Chen Y, Syed S, Huentelman M, Thiyyagura P, Lee W, Richholt R, Caselli RJ, Locke DEC, Lu B, Reiman EM, Su Y, Chen K, 2020. Interaction between BDNF Val66Met and APOE4 on biomarkers of Alzheimer’s disease and cognitive decline. J Alzheimers Dis 78, 721–734. 10.3233/JAD-200132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bokhoven P, de Wilde A, Vermunt L, Leferink PS, Heetveld S, Cummings J, Scheltens P, Vijverberg EGB, 2021. The Alzheimer’s disease drug development landscape. Alzheimer’s Research & Therapy 13, 186. 10.1186/s13195-021-00927-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bosch KA, Verberk IMW, Ebenau JL, van der Lee SJ, Jansen IE, Prins ND, Scheltens P, Teunissen CE, Van der Flier WM, 2021. BDNF-Met polymorphism and amyloid-beta in relation to cognitive decline in cognitively normal elderly: the SCIENCe project. Neurobiology of Aging 108, 146–154. 10.1016/j.neurobiolaging.2021.08.018 [DOI] [PubMed] [Google Scholar]
- Vaughan RA, Foster JD, 2013. Mechanisms of dopamine transporter regulation in normal and disease states. Trends in Pharmacological Sciences 34, 489–496. 10.1016/j.tips.2013.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineskos AN, Lerch JP, Felsky D, Shaikh S, Rajji TK, Miranda D, Lobaugh NJ, Mulsant BH, Pollock BG, Kennedy JL, 2011. The Brain-Derived Neurotrophic Factor Val66Met Polymorphism and Prediction of Neural Risk for Alzheimer Disease. Archives of General Psychiatry 68, 198–206. 10.1001/archgenpsychiatry.2010.194 [DOI] [PubMed] [Google Scholar]
- Ward DD, Summers MJ, Saunders NL, Janssen P, Stuart KE, Vickers JC, 2014. APOE and BDNF Val66Met polymorphisms combine to influence episodic memory function in older adults. Behavioural Brain Research 271, 309–315. 10.1016/j.bbr.2014.06.022 [DOI] [PubMed] [Google Scholar]
- Wu JW, Hussaini SA, Bastille IM, Rodriguez GA, Mrejeru A, Rilett K, Sanders DW, Cook C, Fu H, Boonen RACM, Herman M, Nahmani E, Emrani S, Figueroa YH, Diamond MI, Clelland CL, Wray S, Duff KE, 2016. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci 19, 1085–1092. 10.1038/nn.4328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Radeke MJ, Matheson CR, Talvenheimo J, Welcher AA, Felnstein SC, 1997. Immunocytochemical localization of TrkB in the central nervous system of the adult rat. Journal of Comparative Neurology 378, 135–157. [DOI] [PubMed] [Google Scholar]
- Yuan Xiang P, Janc O, Grochowska KM, Kreutz MR, Reymann KG, 2016. Dopamine agonists rescue Aβ–induced LTP impairment by Src-family tyrosine kinases. Neurobiology of Aging 40, 98–102. 10.1016/j.neurobiolaging.2016.01.008 [DOI] [PubMed] [Google Scholar]
- Zhu G, Li J, He L, Wang X, Hong X, 2015. MPTP-induced changes in hippocampal synaptic plasticity and memory are prevented by memantine through the BDNF-TrkB pathway. British Journal of Pharmacology 172, 2354–2368. 10.1111/bph.13061 [DOI] [PMC free article] [PubMed] [Google Scholar]