

Abstract
Objectives
Endothelial dysfunction is known to be a key characteristic of preeclampsia (PE) and can contribute to progression of symptoms and injury to multiple organ systems. Delivery is the only treatment for progression of PE, but development of an endothelial-based therapy for PE presents a promising strategy. Growth factors and cytokines are dysregulated in PE and can impact endothelial function, manifesting changes in Ca2+ signaling and interruptions in monolayer barrier function that contribute to symptoms of hypertension, proteinuria, and edema. In this study, we highlight Src kinase as a partial mediator of growth factor and cytokine mediated endothelial dysfunction.
Study Design
Fura-2 Ca2+ imaging and Electrical Cell Impedance Sensing (ECIS) assays are performed on growth factor or cytokine exposed human umbilical vein endothelial cells (HUVECs). Inhibitors to MEK/ERK (U0126) or Src (PP2) are used to determine the contribution of kinase signaling pathways.
Main Outcome Measures
Decreases in HUVEC Ca2+ signaling or monolayer resistance measure endothelial dysfunction. Reversal of endothelial dysfunction by kinase inhibitors reveals the respective contibutions of MEK/ERK and Src kinase.
Results
We show that Src inhibition protects Ca2+ signaling responses against insults induced by VEGF165, bFGF, PlGF, TNFα, and IL-1β. Additionally, we show that Src inhibition protects the endothelial monolayer from the full impact of TNFα insult. Further, we find that MEK/ERK inhibition does not offer protection from growth factor-mediated endothelial dysfunction.
Conclusions
The results of this study suggest cytokine and growth factor-stimulated Src kinase plays a partial role on promoting endothelial dysfunction in HUVECs.
Keywords: Preeclampsia, cytokine, growth factor, Src kinase, HUVEC, endothelial, endothelial dysfunction
INTRODUCTION
Preeclampsia (PE) is a gestational hypertensive disorder in which elevated blood pressure occurs after 20 weeks of gestation, accompanied with other symptoms such as proteinuria. PE affects 5-10% of all pregnancies and there is no treatment to prevent disease progression except for delivery, a high-risk scenario for both mother and neonate [1]. Fetal vascular abnormalities are also observed in PE, including abnormal umbilical doppler ultrasound readings [2; 3], impaired in vitro umbilical vein endothelial Ca2+ signaling [4; 5], and increased in vitro vascular leakiness in response to PE serum [6]. Children born from PE pregnancies also have increased rates of cardiovascular disease later in life [7].
An underlying phenomenon behind these symptoms is wide-spread maternal endothelial dysfunction likely caused, at least in part, by circulating factors released from the maternal-fetal interface [1]. We have previously shown that growth factors (VEGF165, EGF, and bFGF) and cytokines (TNFα, IL-1β, IL-6, and IL-8) contribute to endothelial dysfunction through disruption of Ca2+ signaling and/or monolayer integrity [8], indicating the importance of these factors in the endothelial dysfunction characteristic of PE. Many of these factors are dysregulated in PE and likely are capable of contributing to endothelial dysfunction. Endothelial dysfunction is linked to symptoms of hypertension, proteinuria, and edema, as is often observed in PE [9; 10; 11]. In vitro measures of Ca2+ signaling and monolayer integrity are used as surrogate measures of in vivo hypertension and edema/proteinuria.
In uterine artery endothelial cells isolated from pregnant sheep, insults to Ca2+ signaling caused by VEGF165 and TNFα were found to be primarily due to Src actions, with MEK/ERK’s involvement to a lesser degree [12; 13]. The mechanism by which Src and MEK/ERK disrupted Ca2+ signaling in these studies was through phosphorylation of Cx43 gap junctions, inhibiting intercellular communications supportive of pregnancy-adapted Ca2+ signaling [12; 13]. It has also been shown that the importance of the phosphorylation status of Cx43 gap junctions to Ca2+ signaling capacity extends to HUVEC [14; 15]. We previously demonstrated in HUVEC that protective effects of the nutraceutical conjugated linoleic acid on growth factor and cytokine-mediated Ca2+ signaling dysfunction was likely due to the Src inhibiting properties of its c10,t12 isomer, via selective reversal of the Src-specific Y265 phosphorylation on Cx43 [16; 17]. This insult to Ca2+ signaling contributes to endothelial dysfunction by weakening nitric oxide-mediated vasodilation, encouraging development of hypertension [11]. Impaired endothelial Ca2+ signaling has been linked to preeclampsia in multiple studies and is observed in maternal and fetal tissues [5; 18; 19].
Src kinase activity has also been implicated in the signaling environment that maintains endothelial monolayer permeability. Low levels of Src activity are required in the non-pathologic state for maintenance of endothelial monolayer barrier function [20]. However, increased Src activity, like what would result from growth factor or cytokine activation in the inflammatory microenvironment, results in increased endothelial monolayer permeability [20]. It has been reported that Src inhibition is able to prevent TNFα-mediated phosphorylation of VE-cadherin, a component of adherens junctional complexes, strengthening the monolayer’s ability to withstand TNFα insult [20]. A review by Yuan details evidence of ERK 1/2 action in monolayer permeability, reporting that while the use of an ERK 1/2 inhibitor can prevent VEGF165-mediated insult to the monolayer, the contributions of ERK 1/2 signaling can vary between vascular beds and between different experimental techniques (e.g. Type of insult, duration of experiment) [21].
In this study, we aimed to evaluate the mechanistic contribution of Src and MEK/ERK signaling in the growth factor- and cytokine-mediated insults to Ca2+ signaling and monolayer integrity. We accomplished this by utilizing the putative Src inhibitor PP2 and the MEK/ERK inhibitor U0126 as pretreatments to our growth factor and cytokine panel consisting of VEGF165, bFGF, EGF, PlGF, TNFα, IL-1β, IL-6, or IL-8. We hypothesize that inhibiting Src will protect Ca2+ signaling capacity and monolayer integrity from growth factor or cytokine insult, whereas MEK/ERK inhibition would be less protective. With successful rescue of endothelial function, inhibitors of Src and/or MEK/ERK may present intriguing therapeutic targets for PE, worthy of future study.
METHODS
Materials
ATP (disodium salt), Heparin sodium salt, PP2, and all other chemicals were purchased from Sigma- Aldrich (St Louis, MO, USA) unless otherwise stated. Growth factors and cytokines (bFGF, EGF, VEGF165, PIGF, TNFα, IL-1β, IL-6, IL-8) were purchased from R & D systems (Minneapolis, MN, USA). U0126 was purchased from Promega (Madison, WI, USA). Glass bottom microwell dishes for Ca2+ imaging studies were from MatTek Corporation (Ashland, MA, USA). Minimum Essential Medium (MEM), Fura2-AM, and pluronic acid were purchased from Invitrogen (Life Technologies Inc., Grand Island, NY). Serum used in culture medium was fetal bovine serum (FBS) from Invitrogen and Endothelial Cell Growth Supplement ECGS was from Millipore (Temecula, CA). Experimental buffer for the Ca2+ signaling assay was Krebs buffer (125mM NaCl, 5 mM KCl, 1 mM MgSO4, 1 mM KH2PO4, 6 mM glucose, 25 mM HEPES, 2 mM CaCl2, pH 7.4). The growth medium used for the Ca2+ signaling and western blot assays is referred to as HEH medium (Human, ECGS, Heparin) and is as described in [5]. The growth medium used for the monolayer integrity assay is Endothelial Cell Medium (ECM) from ScienCell (Carlsbad, CA) and the assay media for the monolayer integrity is the basal Endothelial Cell Medium also from ScienCell. Cx43 antibodies were from Santa Cruz Biotechnology (Dallas, TX, USA).
Cell culture
HUVEC were isolated from umbilical cords in accordance with approval from the institution review board of UnityPoint Health-Meriter Hospital (Madison, WI) and the University of Wisconsin-Madison (Madison, WI). Isolation methods for the HUVEC used in this study followed the methods previously described [5], in which HUVEC were pooled from 5 individuals and used at passage 3.
Ca2+ signaling assay
HUVEC were plated onto 35mm glass bottom dishes and grown to confluence in HEH media. Cells were loaded with Fura2-AM (10uM with 0.05% Pluronic acid) and incubated for one hour at 37 °C before Krebs buffer washes and incubation at room temperature for 30 minutes. Imaging was then completed using a Nikon inverted microscope (Diaphot 150; Nikon, Melville, NY). Up to 99 cells are hand circled within the field of vision, identifying the regions where measurements were obtained. After a 5-minute baseline period, HUVEC were treated with 100uM ATP and measurements of excitation at 340nm and 380nm at 1 second intervals were collected for 30 minutes. The number of cells responding to ATP stimulation with at least 3 bursts was recorded and a 30-minute Krebs wash was done before moving to the next experiment stage. HUVEC were then pretreated with 10uM PP2 or U0126 for 20 minutes before a 30-minute treatment with 10ng/mL of growth factor or cytokine. Following treatment, HUVEC underwent a second 100uM ATP stimulation while maintaining focus on the same field of cells, thus the first stimulation serves as an internal control. This strategy is illustrated in Figure 1. Results from the PP2/U0126 pretreatment experiments were compared to both treatments of the growth factors and cytokines alone (referred to as “Treatment” in Figures 2 and 3), and to an ATP-only control where cells underwent subsequent ATP additions with only vehicle treatments in between (referred to as “Control” in Figures 2 and 3).
Figure 1: Representative tracings of Ca2+ bursting responses in individual HUVEC.
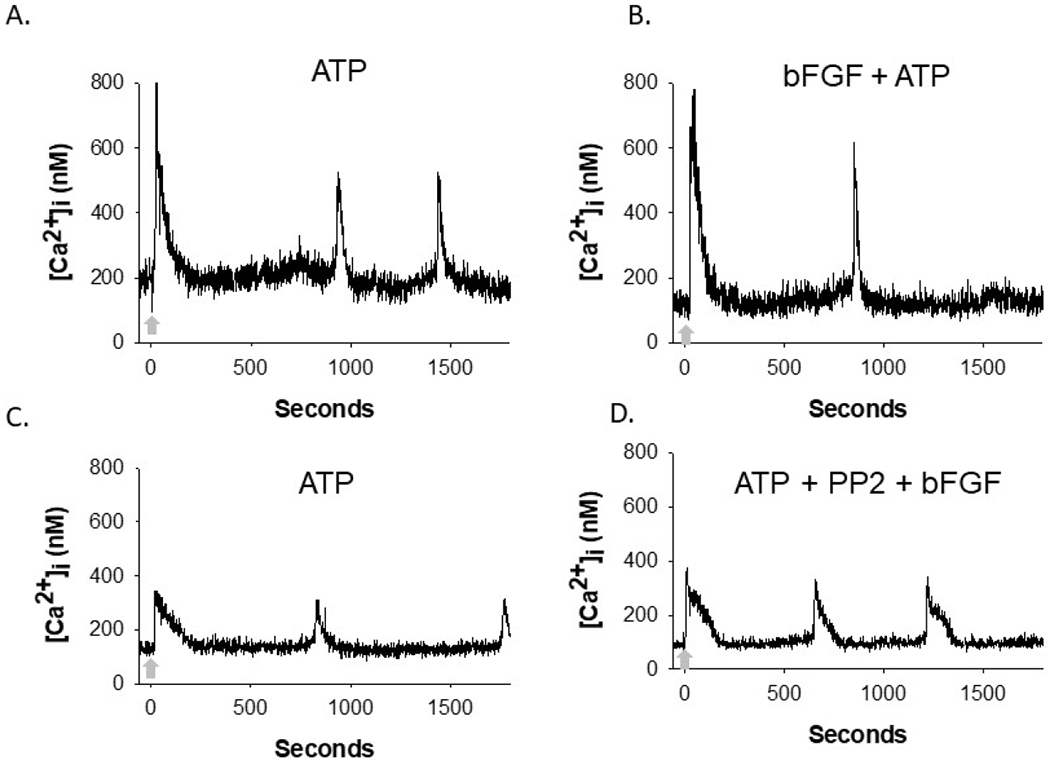
Confluent HUVEC were exposed to an initial 100 uM ATP stimulation prior to subsequent ATP stimulation (indicated by arrow) after inhibitor and growth factor or cytokine treatment, all while focus is retained on each individual cell. Here, bFGF treatment and bFGF with PP2 pretreatment representative tracings are shown as an example. Panels A and B display the same cell, with panel A showing its initial ATP stimulation, and panel B showing its subsequent ATP stimulation after it has been treated with bFGF. Panels C and D display the same cell, with panel C showing its initial ATP stimulation, and panel D showing its subsequent ATP stimulation after treatment with PP2 and bFGF. For quantitative analysis in Figures 2 and 3, the number of bursts counted in the second ATP stimulation (B or D) are compared to the initial, internal control, ATP stimulation (A or C) for each experimental condition.
Figure 2: Src inhibition protects Ca2+ signaling capacity against insult from VEGF165, bFGF, PlGF, TNFα, and IL-1β.
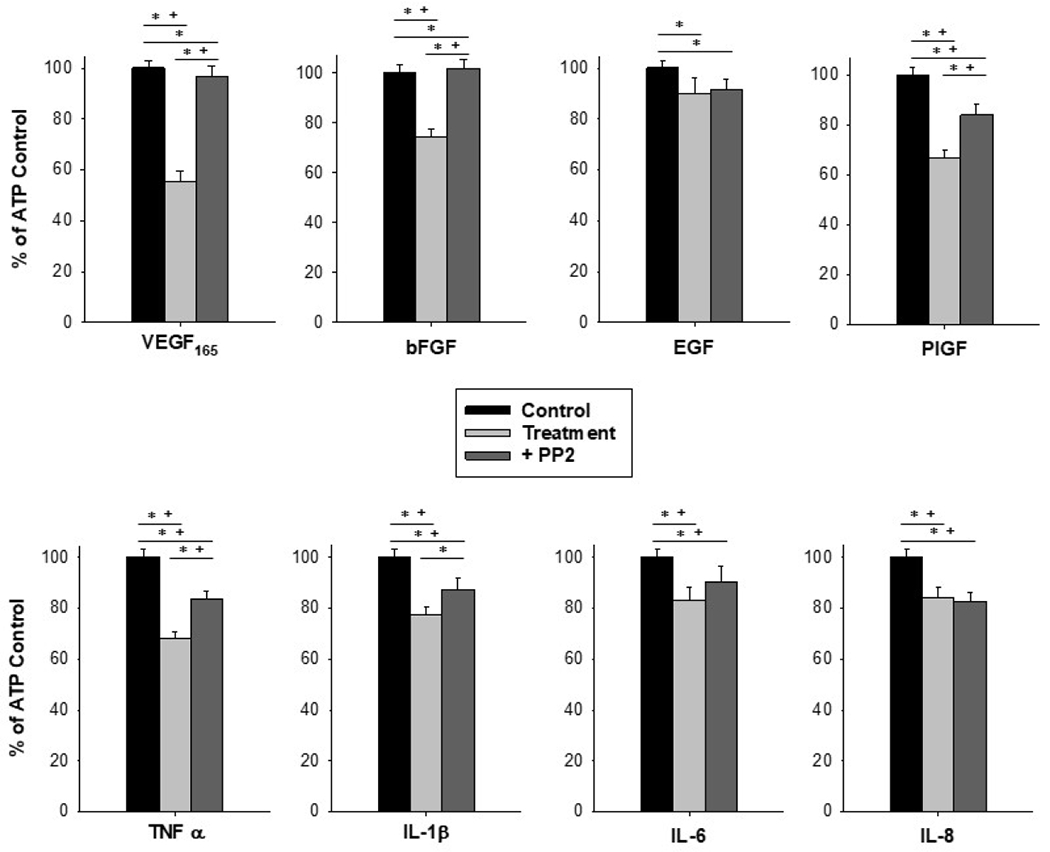
Pretreatment with the Src inhibitor PP2 at 10uM for 20 minutes before treatment with a panel of growth factors and cytokines at 10ng/mL for 30 minutes offered protection of ATP-stimulated Ca2+ bursting responses in HUVEC. Stimulation with 100uM ATP only is depicted as Control in black bars, stimulation with ATP after growth factor or cytokine treatment is depicted in light gray bars, and ATP after PP2 pretreatment prior to growth factor or cytokine treatment is in dark gray bars. Data is shown as mean count of Ca2+ bursts in cells that responded with at least 3 Ca2+ bursts upon initial ATP stimulation, ± SEM for at least 80 cells from at least 4 separate dishes of pooled cells. Statistical analysis was done by Rank-sum test, with * indicating p<0.05 and Kruskal-Wallis with + indicating p<0.05.
Figure 3: MEK/ERK inhibition worsens Ca2+ signaling capacity in the case of TNFα and IL-8.
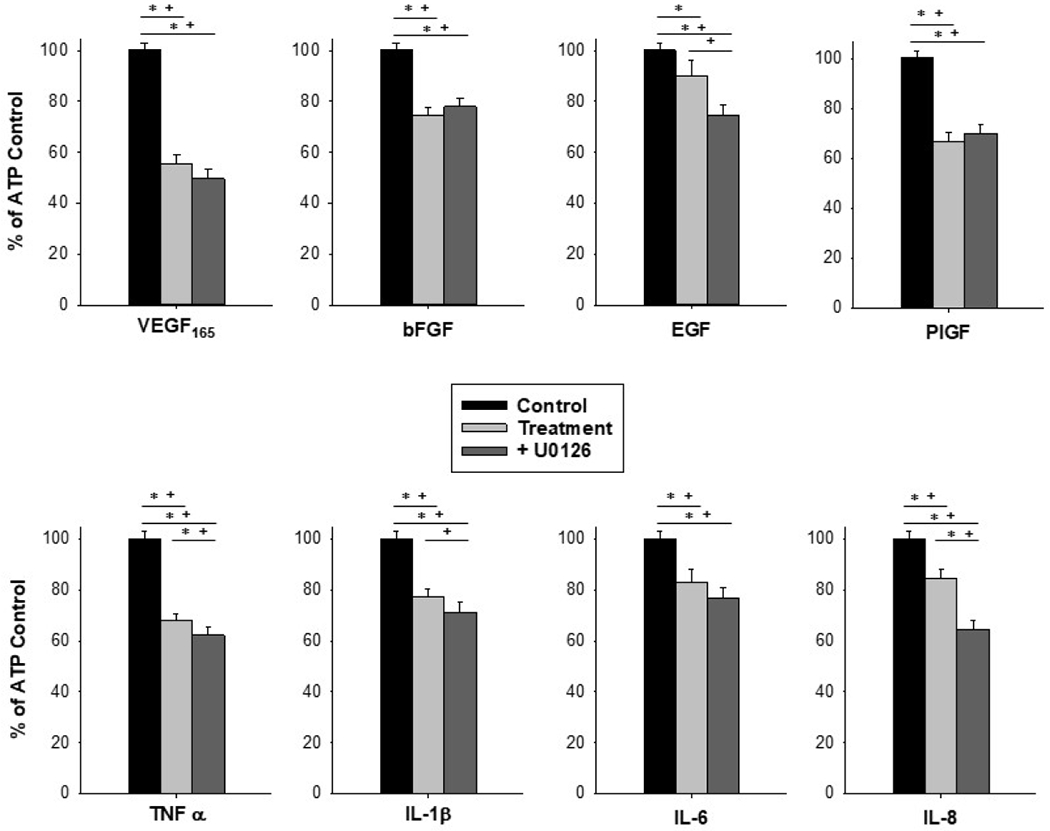
Pretreatment with the MEK/ERK inhibitor U0126 at 10uM for 20 minutes before treatment with a panel of growth factors and cytokines at 10ng/mL for 30 minutes did not protect ATP-stimulated Ca2+ bursting responses in HUVEC. Stimulation with 100uM ATP only is depicted as Control in black bars, stimulation with ATP after growth factor or cytokine treatment is depicted in light gray bars, and ATP after U0126 pretreatment prior to growth factor or cytokine treatment is in dark gray bars. Data is shown as mean count of Ca2+ bursts in cells that responded with at least 3 Ca2+ bursts upon initial ATP stimulation, ± SEM for at least 80 cells from at least 4 separate dishes of pooled cells. Statistical analysis was done by Rank-sum test, with * indicating p<0.05 and Kruskal-Wallis with + indicating p<0.05.
Monolayer integrity assay
HUVEC were plated onto 96 well plates featuring gold electrodes for measurement of electrical resistance using the electric cell-substrate impedance sensing (ECIS) system (Applied BioPhysics, Troy, New York). HUVEC were grown to confluence (48 hours with a media change at 24 hours) in ECM before undergoing serum withdrawal with ECMb (basal), which occurs for 3 hours allowing for resistance measurements to return to preserum withdrawal levels. Pretreatments of 10uM PP2 or U0126 were then administered, with 10ng/mL growth factor and cytokines treatments administered 30 minutes later. Data was collected for 24 hours after growth factor/cytokine treatment with alternate excitation wavelengths of 340 nm and 380 nm at 1 s intervals.
Statistical analysis
For the Ca2+ signaling assay each treatment condition was administered to 4-6 dishes with at least 90 cells circled for each dish. The difference in burst numbers from treatment and internal ATP control was used as the data points for this assay, with statistical analysis completed by paired t-test for each cell against internal control and Mann-Whitney Rank-Sum test against vehicle control. Additional analysis by Kruskal-Wallis was done to confirm multiple comparisons. Data are presented as means ± S.E.M. and p ≤ 0.05 was considered statistically significant. Data for the monolayer integrity assay are presented as resistance normalized to control with statistical analysis using student’s t test.
RESULTS
Impact of Src inhibition on Ca2+ signaling capacity.
Analysis of Ca2+ bursting responses was done by comparing the initial ATP stimulation burst numbers to the post-treatment ATP stimulation burst numbers (Figure 1). Although growth factors are known to stimulate Ca2+, the 30-minute stimulation time was enough for cells to return to baseline. We therefore only present ATP-stimulated Ca2+ responses and not growth factor-stimulated Ca2+ responses. Pretreatment with the Src inhibitor, PP2, shielded HUVEC from growth factor and cytokine-mediated insults to ATP-stimulated Ca2+ signaling capacity, as measured by number of Ca2+ bursts relative to ATP treated control (Figure 2). PP2 treatment alone results in a reduction in Ca2+ signaling, as represented by % of ATP control, to 75.6% (Data not shown). PP2 pretreatment increased Ca2+ signaling from 55.6% ± 3.7 for VEGF165 alone to 96.5% ± 4.4 for VEGF165 + PP2 (p<0.01 vs VEGF165 alone), and 74.6% ± 3.0 for bFGF alone to 101.7% ± 3.8 for bFGF+PP2 (p<0.001 vs bFGF alone), indicating full protection of Ca2+ signaling. PlGF treatment alone resulted in a Ca2+ response of 66.49% ± 3.7 of ATP control, which increased to 84.08% ± 4.5 with PP2 pretreatment (p<0.001 vs PlGF alone). This indicates partial protection from PlGF insult. PP2 did not alter the Ca2+ response caused by EGF (EGF alone = 90.2% ±5.9, EGF + PP2 = 91.6% ± 3.9, p=0.8 vs EGF alone). PP2 treatment improved the Ca2+ response for TNFα from 67.9% ± 2.7 to 83.8% ± 2.9 (p<0.05 vs TNFα alone) and improved (by Rank-sum test but not Kruskal-Wallis) the Ca2+ response for IL-1β from 77.4% ± 3.0 to 87.5% ± 4.6 (p<0.001 vs IL-1β alone). The Ca2+ response to IL-6 and IL-8 was unchanged with PP2 treatment (IL-6 = 83.2% ± 4.9 IL-6+PP2 = 90.3% ± 6.4, p=0.35 vs IL-6 alone; IL-8=84.3% ± 4.1 IL-8+PP2= 82.6% ± 3.5, p=0.92 vs IL-8 alone.
Impact of MEK/ERK inhibition on Ca2+ signaling capacity.
Pretreatment with the MEK/ERK inhibitor U0126 failed to protect HUVEC from growth factor and cytokine-mediated insult to Ca2+ signaling capacity (Figure 3). U0126 treatment decreased Ca2+ signaling as represented by % of ATP control from 67.93% for TNFα alone to 62.17% for TNFα + U0126 (p<0.05 vs TNFα alone), and 82.26% for IL-8 alone to 64.57% for IL-8+U0126 (p<0.01 vs IL-8 alone) by Rank-sum test, indicating further inhibition of Ca2+ signaling. Additional decreases after U0126 treatment for EGF and IL-1β are seen by Kruskal-Wallis test. For all other factors assessed, U0126 did not offer any changes to the Ca2+ signaling responses from the action of the factor alone.
Impact of Src and MEK/ERK inhibition on endothelial monolayer integrity.
Broadly speaking, Src inhibition via PP2 had the capacity to offer improvements in monolayer integrity whereas MEK/ERK inhibition via U0126 was injurious to the monolayer over the measurement period, and this is displayed for growth factors in Figure 4 and for cytokines in Figure 5. For VEGF165, EGF, IL-1β, IL-6 and IL-8, pretreatment with PP2 did not result in statistically significant changes in monolayer resistance compared to each factor alone. For bFGF, PP2 pretreatment resulted in about a 20% reduction in resistance measurements compared to bFGF alone for the entirety of the experiment (p<0.05), but resistance measurements were elevated compared to control beginning at hour 18 (p<0.05). For PlGF, PP2 pretreatment resulted in a slight increase in monolayer resistance compared to PlGF alone from hours 16-22 (p<0.05) and from control for hours 8-13 (p<0.05). For TNFα, PP2 pretreatment results in monolayer resistance measurements returning to control levels beginning at hour 16 (p>0.05 hours 16-24), offering protection from the full severity of TNFα insult. For all factors assessed, with the exception of bFGF, U0126 pretreatment resulted in a severe and sustained reduction in monolayer resistance measurements to a 50% reduction from control levels. For bFGF, U0126 pretreatment resulted in a decrease in monolayer resistance of about 30% compared to bFGF treatment alone from hours 10-25 (p<0.05), but was not different from control.
Figure 4: MEK/ERK inhibition is broadly deleterious to the monolayer, whereas Src inhibition offers benefits with PlGF and bFGF treatment.
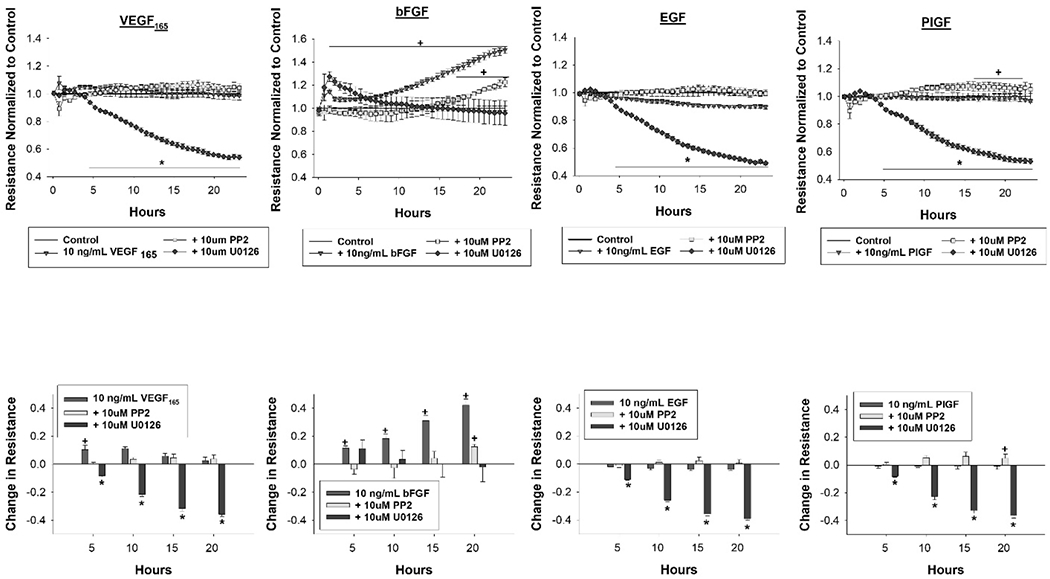
HUVEC were pretreated with the MEK/ERK inhibitor U0126 or Src inhibitor PP2 at 10um 30 minutes before treatment with a panel of growth factors (VEGF165, bFGF, EGF and PlGF) at 10ng/mL. Data is shown as mean ± SEM, with resistance normalized to control in the top row and the quantitative change in resistance relative to control at hours 5, 10 ,15 and 20 in the bottom row. Data was collected for 24 hours after growth factor and cytokine treatments. Treatments were run in triplicate with n=6 96 well plates. Statistical analysis was done by student’s t test with * indicating p<0.05 reduction in resistance measurements and + indicating p<0.05 increase in resistance measurements.
Figure 5: MEK/ERK inhibition is broadly deleterious to the monolayer, whereas Src inhibition offers protection from the worst of TNFα insult.
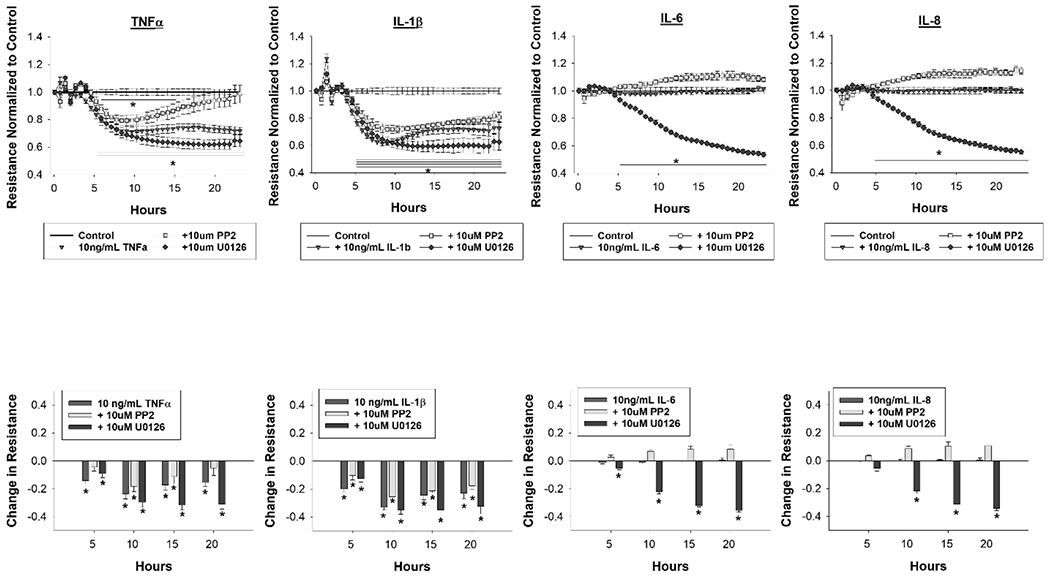
HUVEC were pretreated with the MEK/ERK inhibitor U0126 or Src inhibitor PP2 at 10um 30 minutes before treatment with a panel of cytokines (TNFα, IL-1β, IL-6 and IL-8) at 10ng/mL. Data is shown as mean ± SEM, with resistance normalized to control in the top row and the quantitative change in resistance relative to control at hours 5, 10 ,15 and 20 in the bottom row. Data was collected for 24 hours after growth factor and cytokine treatments. Treatments were run in triplicate with n=6 96 well plates. Statistical analysis was done by student’s t test with * indicating p<0.05 reduction in resistance measurements.
DISCUSSION
Previous work has indicated that Src inhibition could serve as a protective strategy against growth factor- and cytokine-mediated insult to Ca2+ signaling capacity, but in those instances either a nutraceutical compound with potentially promiscuous activity or ovine endothelial cell type were utilized [12; 16]. In the present study we show that Src inhibition in HUVEC offers complete protection of Ca2+ signaling capacity from VEGF165 and bFGF insult and partial protection against PlGF, TNFα and IL-1β insult. Growth factor and cytokine signaling is known to promote closure and increased protein turnover of gap junctions. We have previously shown a crucial link between sustained Ca2+ responses and functional gap junctions in HUVEC [14; 16]. To date, numerous studies have demonstrated that Y265 on Cx43 is a specific site for Src kinase phosphorylation [12; 16; 22; 23]. further supported by the PP2 specificity reported herein. Of note, it is particularly promising that Src inhibition offers full protection from VEGF165 insult to Ca2+ signaling and some protection from PlGF insult as these two factors are commonly dysregulated in PE [1; 11; 24; 25]. The observation that PlGF itself caused a marked decrease in Ca2+ signaling capacity was unexpected. Previous work in ovine uterine artery cells from pregnant ewes did not show a strong inhibitory role for PlGF on Ca2+ signaling, reporting that PlGF treatment only reduced the Ca2+ response to 90.7% of ATP control [12]. Additionally, in the ovine model, it had been established that signaling via VEGFR-2 but not VEGFR-1, which PlGF is primarily the agonist for, was responsible for the observed inhibition in Ca2+ signaling by VEGF165 [12; 26]. A stronger PlGF-mediated insult could suggest a more robust role for VEGFR1 signaling upon ATP-stimulated Ca2+ responses than was previously appreciated. However, further experiments will be needed to interrogate this hypothesis.
At least one study demonstrates coupling of bFGF to Src and MEK/ERK activation [27]. In this study, MEK/ERK activation appears to be involved in the bFGF-mediated rise in monolayer integrity, as U0126 prevented the consistent increase in monolayer resistance typically caused by bFGF treatment. Furthermore, our results herein suggest that there is a stronger role for Src activation in the bFGF-mediated insult to Ca2+ signaling, as Src inhibition fully rescued the ATP-stimulated Ca2+ response while MEK/ERK inhibition offered no protection. In a study utilizing an anterior pituitary cell line it was found that bFGF treatment resulted in inhibitory phosphorylation of Cx43 at S368, and that PKCα was involved in the mechanism behind this insult, but not ERK 1/2 [28]. There are reports of inter-connected action of PKC and Src, with some studies reporting PKC action upstream of Src activation and other implicating Src action in PKC activation [29; 30]. This observation may be due to unique characteristics of different cell types. However, both scenarios could be true and result in these mediators co-amplifying the same signal in our model system. This suggests that inhibiting Src with PP2, as we have done here, could be sufficient to dampen both Src and PKC action. Notably, Src activation can coactivate other kinases, including PKC, and collectively phosphorylate multiple residues of Cx43 [31].
Focusing our attention on the most potent cytokines, TNFα and IL-1β, Src inhibition was only partially successful in protecting against TNFα insult to Ca2+ signaling. However, Src inhibition proved an effective strategy to protect the monolayer from the full impact of TNFα insult. Others have shown that protecting HUVEC from TNFα mediated disruption of VE-cadherin resulted in an endothelium that was more resistant to leukocyte extravasation, thus displaying a more anti-inflammatory phenotype [32].
However, the failure of Src inhibition to protect against IL-1β insult to the monolayer highlights that there are other signaling pathways mediating this insult. Since MEK/ERK inhibition was almost universally deleterious to the monolayer in this study we can conclude that inhibiting the MEK/ERK pathway, especially with compounds functionally similar to U0126 may not be a useful strategy for improving cytokine-related endothelial dysfunction, at least in HUVEC. A further confounding factor is that there will be a large degree of heterogeneity in circulating growth factor and cytokine levels between PE patients, complicating the signaling microenvironment.
CONCLUSION
Inhibition of Src kinase activity is partially effective in reducing cytokine-mediated endothelial dysfunction in HUVECs. Other kinase signaling cascades are likely involved in the mechanics of Ca2+ signaling and monolayer insult and will need to be identified in further studies. Future studies will be needed to determine whether inhibiting Src kinase activity may be a beneficial therapeutic strategy for combating cytokine-mediated endothelial dysfunction in PE. However, this study provides important insights into the partial role of Src kinase in growth factor and cytokine-mediated Ca2+ signaling and monolayer integrity dysfunction in HUVECs.
Highlights.
Endothelial dysfunction related to Ca2+ signaling loss in VEGF165, bFGF, PlGF, TNFα, or IL-1β treated HUVEC is improved by the Src kinase inhibor PP2
Src kinase inhibitor PP2 also improves TNFα-mediated losses in HUVEC monolayer integrity
The MEK/ERK signaling inhibitor U0126 offers no improvement to growth factor or cytokine-mediated endothelial dysfunction in HUVEC
ACKNOWLEDGEMENTS
Thank you to the University of Wisconsin-Madison School of Medicine and Public Health, Wisconsin Alumni Research Foundation, and the University of Wisconsin-Madison Endocrinology and Reproductive Physiology Program for supporting the research and the training of AKM. The current institution of N Khurshid is Promedica Toledo Hospital, Toledo, Ohio.
FUNDING
This work was funded by the National Institutes of Health grant award HD079865. This work represent work towards degree requirements of AKM towards PhD completion in the Endocrinology and Reproductive Physiology Training Program at the University of Wisconsin-Madison. Further, AKM was supported by NIH T32 predoctoral training award HD041921. LC was supported by the NIH T32 postdoctoral training award HD101384.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTEREST
There are no conflicts of interest to disclose.
References
- [1].Tomimatsu T, Mimura K, Matsuzaki S, Endo M, Kumasawa K, Kimura T, Preeclampsia: Maternal Systemic Vascular Disorder Caused by Generalized Endothelial Dysfunction Due to Placental Antiangiogenic Factors, Int J Mol Sci 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Harrington K, Thompson MO, Carpenter RG, Nguyen M, Campbell S, Doppler fetal circulation in pregnancies complicated by pre-eclampsia or delivery of a small for gestational age baby: 2. Longitudinal analysis, Br J Obstet Gynaecol 106 (1999) 453–66. [DOI] [PubMed] [Google Scholar]
- [3].Bodelsson G, Marsal K, Stjernquist M, Reduced contractile effect of endothelin-1 and noradrenalin in human umbilical artery from pregnancies with abnormal umbilical artery flow velocity waveforms, Early Hum Dev 42 (1995) 15–28. [DOI] [PubMed] [Google Scholar]
- [4].Steinert JR, Wyatt AW, Poston L, Jacob R, Mann GE, Preeclampsia is associated with altered Ca2+ regulation and NO production in human fetal venous endothelial cells, The FASEB journal : official publication of the Federation of American Societies for Experimental Biology 16 (2002) 721. [DOI] [PubMed] [Google Scholar]
- [5].Krupp J, Boeldt DS, Yi FX, Grummer MA, Bankowski Anaya HA, Shah DM, Bird IM, The loss of sustained Ca(2+) signaling underlies suppressed endothelial nitric oxide production in preeclamptic pregnancies: implications for new therapy, Am J Physiol Heart Circ Physiol 305 (2013) H969–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhang Y, Gu Y, Li H, Lucas MJ, Wang Y, Increased endothelial monolayer permeability is induced by serum from women with preeclampsia but not by serum from women with normal pregnancy or that are not pregnant, Hypertension in pregnancy : official journal of the International Society for the Study of Hypertension in Pregnancy 22 (2003) 99. [DOI] [PubMed] [Google Scholar]
- [7].A.C.o.O.a. Gynecologists, Task Force on Hypertension in Pregnancy, Task Force on Hypertension in Pregnancy2013.
- [8].Mauro AK, Khurshid N, Berdahl DM, Ampey AC, Adu D, Shah DM, Boeldt DS, Cytokine concentrations direct endothelial function in pregnancy and preeclampsia, J Endocrinol 248 (2021) 107–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Guan C, Zhao F, Yang Z, Tang Q, Wang L, Li X, Zhang L, Deng Z, Hou H, Wang J, Xu Y, Zhang R, Lin Y, Tan P, Zhang Y, Liu S, Zhang L, A review of key cytokines based on gene polymorphism in the pathogenesis of pre-eclampsia, Am J Reprod Immunol 87 (2022) e13503. [DOI] [PubMed] [Google Scholar]
- [10].Umapathy A, Chamley LW, James JL, Reconciling the distinct roles of angiogenic/antiangiogenic factors in the placenta and maternal circulation of normal and pathological pregnancies, Angiogenesis 23 (2020) 105–117. [DOI] [PubMed] [Google Scholar]
- [11].Boeldt DS, Bird IM, Vascular adaptation in pregnancy and endothelial dysfunction in preeclampsia, J Endocrinol 232 (2017) R27–R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Boeldt DS, Grummer MA, Yi F, Magness RR, Bird IM, Phosphorylation of Ser-279/282 and Tyr-265 positions on Cx43 as possible mediators of VEGF-165 inhibition of pregnancy-adapted Ca2+ burst function in ovine uterine artery endothelial cells, Mol Cell Endocrinol 412 (2015) 73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ampey AC, Boeldt DS, Clemente L, Grummer MA, Yi F, Magness RR, Bird IM, TNF-alpha inhibits pregnancy-adapted Ca(2+) signaling in uterine artery endothelial cells, Mol Cell Endocrinol 488 (2019) 14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Boeldt DS, Krupp J, Yi FX, Khurshid N, Shah DM, Bird IM, Positive versus negative effects of VEGF165 on Ca2+ signaling and NO production in human endothelial cells, Am J Physiol Heart Circ Physiol 312 (2017) H173–H181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Van Rijen H, van Kempen MJ, Analbers LJ, Rook MB, van Ginneken AC, Gros D, Jongsma HJ, Gap junctions in human umbilical cord endothelial cells contain multiple connexins, Am J Physiol 272 (1997) C117–30. [DOI] [PubMed] [Google Scholar]
- [16].Mauro AK, Berdahl DM, Khurshid N, Clemente L, Ampey AC, Shah DM, Bird IM, Boeldt DS, Conjugated linoleic acid improves endothelial Ca2+ signaling by blocking growth factor and cytokine-mediated Cx43 phosphorylation, Mol Cell Endocrinol 510 (2020) 110814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Shahzad MMK, Felder M, Ludwig K, Van Galder HR, Anderson ML, Kim J, Cook ME, Kapur AK, Patankar MS, Trans10,cis12 conjugated linoleic acid inhibits proliferation and migration of ovarian cancer cells by inducing ER stress, autophagy, and modulation of Src, PLoS One 13 (2018)e0189524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Steinert JR, Wyatt AW, Poston L, Jacob R, Mann GE, Preeclampsia is associated with altered Ca2+ regulation and NO production in human fetal venous endothelial cells, FASEB J 16 (2002) 721–3. [DOI] [PubMed] [Google Scholar]
- [19].Mahdy Z, Otun HA, Dunlop W, Gillespie JI, The responsiveness of isolated human hand vein endothelial cells in normal pregnancy and in pre-eclampsia, J Physiol 508 ( Pt 2) (1998) 609–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hu G, Place AT, Minshall RD, Regulation of endothelial permeability by Src kinase signaling: vascular leakage versus transcellular transport of drugs and macromolecules, Chem Biol Interact 171 (2008) 177–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yuan SY, Protein kinase signaling in the modulation of microvascular permeability, Vascular Pharmacology 39 (2002) 213–223. [DOI] [PubMed] [Google Scholar]
- [22].Giepmans BN, Hengeveld T, Postma FR, Moolenaar WH, Interaction of c-Src with gap junction protein connexin-43. Role in the regulation of cell-cell communication, J Biol Chem 276 (2001) 8544–9. [DOI] [PubMed] [Google Scholar]
- [23].Solan JL, Lampe PD, Specific Cx43 phosphorylation events regulate gap junction turnover in vivo, FEBS Lett 588 (2014) 1423–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Maynard SE, Min J-Y, Merchan J, Lim K-H, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP, Karumanchi SA, Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia, Journal of Clinical Investigation 111 (2003) 649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF, Schisterman EF, Thadhani R, Sachs BP, Epstein FH, Sibai BM, Sukhatme VP, Karumanchi SA, Circulating angiogenic factors and the risk of preeclampsia, N Engl J Med 350 (2004) 672–83. [DOI] [PubMed] [Google Scholar]
- [26].Boeldt DS, Grummer MA, Magness RR, Bird IM, Altered VEGF-stimulated Ca2+ signaling in part underlies pregnancy-adapted eNOS activity in UAEC, J Endocrinol 223 (2014) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Deo DD, Axelrad TW, Robert EG, Marcheselli V, Bazan NG, Hunt JD, Phosphorylation of STAT-3 in response to basic fibroblast growth factor occurs through a mechanism involving platelet-activating factor, JAK-2, and Src in human umbilical vein endothelial cells. Evidence for a dual kinase mechanism, J Biol Chem 277 (2002) 21237–45. [DOI] [PubMed] [Google Scholar]
- [28].Vitale ML, Barry A, Biphasic Effect of Basic Fibroblast Growth Factor on Anterior Pituitary Folliculostellate TtT/GF Cell Coupling, and Connexin 43 Expression and Phosphorylation, J Neuroendocrinol 27 (2015) 787–801. [DOI] [PubMed] [Google Scholar]
- [29].Katz S, Boland R, Santillan G, Modulation of ERK 1/2 and p38 MAPK signaling pathways by ATP in osteoblasts: involvement of mechanical stress-activated calcium influx, PKC and Src activation, Int J Biochem Cell Biol 38 (2006) 2082–91. [DOI] [PubMed] [Google Scholar]
- [30].Tan M, Li P, Sun M, Yin G, Yu D, Upregulation and activation of PKC alpha by ErbB2 through Src promotes breast cancer cell invasion that can be blocked by combined treatment with PKC alpha and Src inhibitors, Oncogene 25 (2006) 3286–95. [DOI] [PubMed] [Google Scholar]
- [31].Solan JL, Lampe PD, Connexin 43 in LA-25 cells with active v-src is phosphorylated on Y247, Y265, S262, S279/282, and S368 via multiple signaling pathways, Cell Commun Adhes 15 (2008) 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Herrera-Garcia AM, Dominguez-Luis MJ, Arce-Franco M, Armas-Gonzalez E, Alvarez de La Rosa D, Machado JD, Pec MK, Feria M, Barreiro O, Sanchez-Madrid F, Diaz-Gonzalez F, Prevention of neutrophil extravasation by alpha2-adrenoceptor-mediated endothelial stabilization, J Immunol 193 (2014) 3023–35. [DOI] [PubMed] [Google Scholar]