

Abstract
A 6-year-old female with a history of Aicardi-Goutières syndrome (AGS) presented to dermatology clinic with hypopigmented and hyperpigmented macules and patches consistent with dyschromatosis symmetrica hereditaria (DSH). Previous genetic workup demonstrated a de novo, heterozygous mutation in the adenosine deaminase acting on RNA 1 (ADAR) gene. While the co-occurrence of AGS and DSH has previously been described in mutations of the ADAR gene, our case highlights the potential association between these disorders that may aid in earlier future diagnosis of AGS.
Keywords: Aicardi-Goutieres syndrome, dyschromatosis symmetrica hereditaria, ADAR1 gene, genodermatosis, pigmentary disorder
Case Report:
A 6-year-old African-American female with a history of Aicardi-Goutières syndrome (AGS) presented to dermatology clinic for evaluation of xerosis with hyperpigmented and hypopigmented macules and patches. Birth history was notable for term induction due to idiopathic intrauterine growth restriction. At 6 months of age, she was noted to have developmental delays that progressed to developmental regression including loss of head control and nystagmus. CT head was notable for scattered brain calcifications, and MRI brain similarly showed calcifications and white mater degenerative disease. TORCH workup for infectious etiology was negative. By 2 years of age, prior to genetic testing, patient had developed patchy areas of hypopigmentation on cheeks, hands, and feet. The patient was found to have a de novo, heterozygous mutation in adenosine deaminase acting on RNA 1 (ADAR) gene (c.3247C>T p.Arg1083Cys) on whole exome sequencing, confirming diagnosis of AGS at the age of 2 years. She was subsequently started on 2mg baricitinib via a clinical trial protocol (NCT03921554) with no further developmental regression. At time of evaluation in dermatology clinic, there were hypopigmented and hyperpigmented macules and patches concentrated on the dorsal hands and feet most consistent with dyschromatosis symmetrica hereditaria (DSH) (Figures 1A&B). There was no other family history of developmental delays, seizures, systemic lupus erythematosus, chilblains, or similar skin dyspigmentation.
Figure 1:

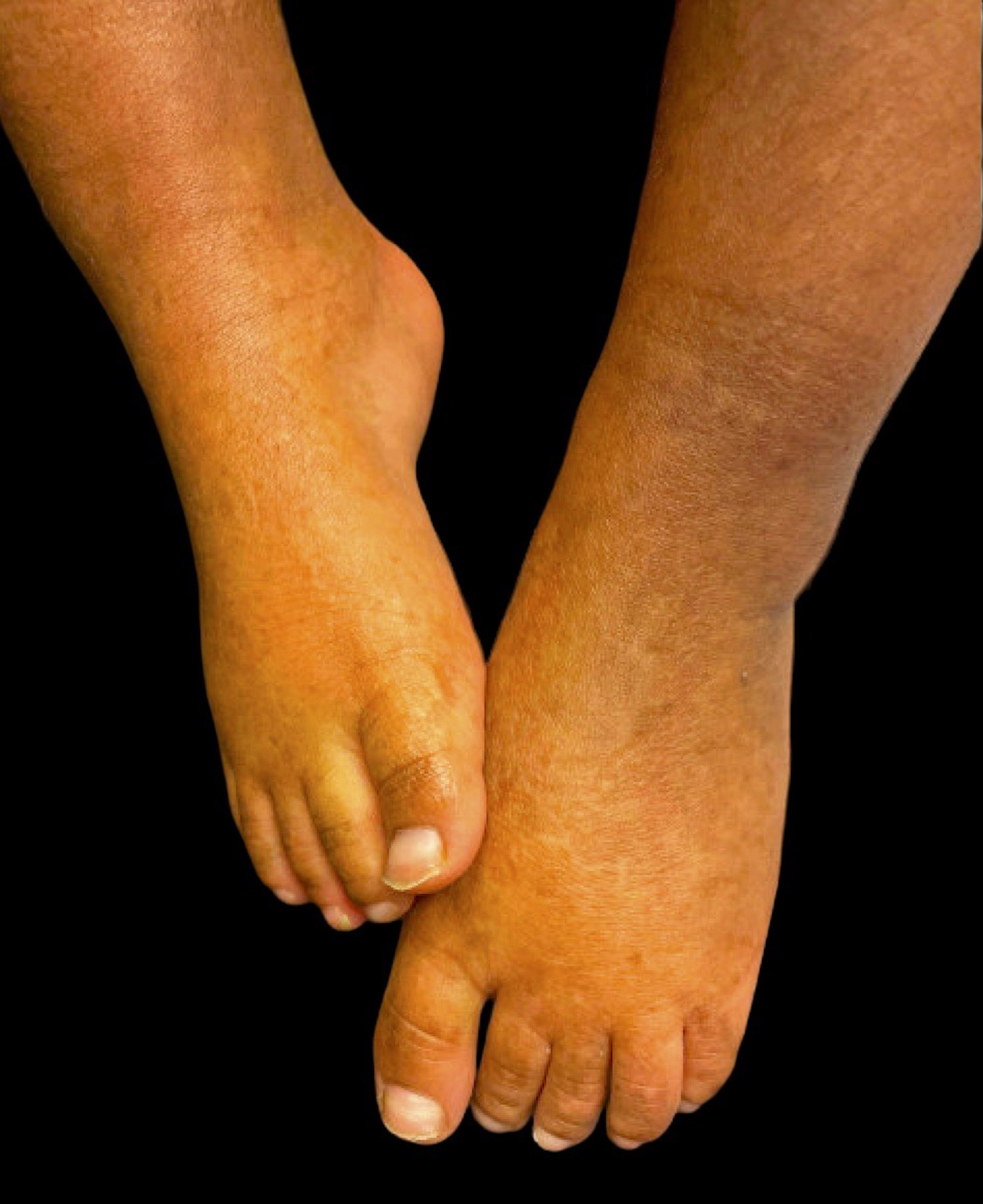
Clinical photos from dermatology examination at 6 years of age demonstrating hypo- and hyper-pigmented macules and patches concentrated on (A) dorsal hand and (B) dorsal feet consistent with DSH.
Discussion:
AGS represents a family of genetic conditions with related clinical presentations, most commonly reflecting failure to properly degrade host RNA or DNA. It is characterized by basal ganglia calcification, white matter degeneration, and elevated IFN-alpha levels in CSF. Multiple genetic causes have been recognized, including variants in ADAR gene. Pigmentary changes with ADAR mutations are hypothesized to be due to alterations in melanocyte survival.
ADAR encodes a double stranded RNA-specific adenosine deaminase necessary for RNA modification involved in innate immunity. Deficits with ADAR are known to result in inflammation secondary to interferon induction. ADAR variants are also known to be the underlying genetic mutation seen in DSH, characterized by hyper- and hypopigmented macules on the dorsal surfaces of distal extremities, sparing the palms, soles, and mucosal membrane. Lesions arise in infancy or early childhood and tend to increase in number until adolescence. Concurrent presentation of DSH and AGS 6 has been previously described with compound heterozygous and homozygous ADAR mutations1–3. Our case reports on a patient with de novo, heterozygous ADAR variants associated with DSH and AGS 6. However, we cannot exclude the possibility of another unknown mutated allele in the non-coding region of ADAR is responsible for the present findings. Given the association between AGS and interferon-1 induction, JAK inhibition is one target of clinical trials; initial data from one such trial suggests that JAK inhibition may contribute to improvement with achievement of new developmental milestones even within 3 months of therapy5. Thus, early diagnosis is essential to prevent long-term developmental deficits from the disease. As the diagnosis of AGS is often delayed, our case highlights a rare association between DSH and AGS that could potentially aid in an earlier diagnosis of AGS.
Funding:
This study was supported by the National Institute of Neurological Disorders and Stroke (NNDS) via Award ID: U01NS106845.
Footnotes
Conflicts of interest: The authors declare no conflicts of interest with respect to the research, authorship, and/or publication of this article.
References:
- 1.Liu L, Zhang L, Huang, P, et al. Case Report: Aicardi-Goutières Syndrome Type 6 and Dyschromatosis Symmetrica Hereditaria With Congenital Heart Disease and Mitral Valve Calcification - Phenotypic Variants Caused by Adenosine Deaminase Acting on the RNA 1 Gene Homozygous Mutations. Frontiers in pediatrics 2022;10,852903. doi: 10.3389/fped.2022.852903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sathishkumar D, Muthusamy K, Gupta A, et al. Co-occurrence of Aicardi-Goutières syndrome type 6 and dyschromatosis symmetrica hereditaria due to compound heterozygous pathogenic variants in ADAR1: a case series from India. Clin Exp Dermatol 2021;46(4):704–709. doi: 10.1111/ced.14531. [DOI] [PubMed] [Google Scholar]
- 3.Linares-Navarro R, Delgado-Vicente S, Jiménez-González A, et al. Aicardi-Goutières syndrome and dyschromatosis symmetrica hereditaria due to compound heterozygous mutation of ADAR1, presentation of two cases. Int J Dermatol 2023; Epub ahead of print. doi: 10.1111/ijd.16692. [DOI] [PubMed] [Google Scholar]
- 4.Suganuma M, Kono M, Yamanaka M, et al. Pathogenesis of a variant in the 5’ untranslated region of ADAR1 in dyschromatosis symmetrica hereditaria. Pigment Cell Melanoma Res 2020;33(4):591–600. doi: 10.1111/pcmr.12863. [DOI] [PubMed] [Google Scholar]
- 5.Vanderver A, Adang L, Gavazzi F, et al. Janus Kinase Inhibition in the Aicardi-Goutières Syndrome. N Engl J Med 2020;383(10):986–989. doi: 10.1056/NEJMc2001362. [DOI] [PMC free article] [PubMed] [Google Scholar]