

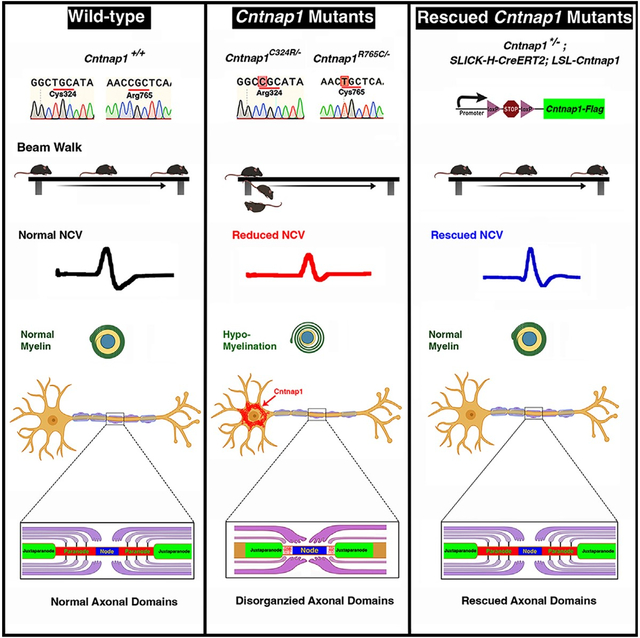
SUMMARY
The Contactin-associated protein 1 (Cntnap1) mouse mutants fail to establish proper axonal domains in myelinated axons. Human CNTNAP1 mutations are linked to hypomyelinating neuropathy-3, which causes severe neurological deficits. To understand the human neuropathology and to model human CNTNAP1C323R and CNTNAP1R764C mutations, we generated Cntnap1C324R and Cntnap1R765C mouse mutants, respectively. Both Cntnap1 mutants show weight loss, reduced nerve conduction, and progressive motor dysfunction. The paranodal ultrastructure shows everted myelin loops and the absence of axo-glial junctions. Biochemical analysis reveals that these Cntnap1 mutant proteins are nearly undetectable in the paranodes, have reduced surface expression and stability, and are retained in the neuronal soma. Postnatal transgenic expression of Cntnap1 in the mutant backgrounds rescues the phenotypes and restores the organization of axonal domains with improved motor function. This study uncovers the mechanistic impact of two human CNTNAP1 mutations in a mouse model and provides proof of concept for gene therapy for CNTNAP1 patients.
In brief
Chang and Sell et al. report the generation and phenotypic characterization of mouse models of human CNTNAP1 mutations. The mouse mutants show hypomyelination and disrupted paranodal axo-glial junctions, reduced nerve conduction, and motor dysfunction. These phenotypes are rescued by postnatal expression of Cntnap1 providing a proof of concept for CNTNAP1 gene therapy.
Graphical Abstract
INTRODUCTION
Bidirectional cellular and molecular interactions between neuronal axons and myelin-producing glial cells form the underlying basis for myelination to promote saltatory nerve conduction. A major feature of myelinated axons is their organization into highly specialized anatomical and molecular domains, named the nodes of Ranvier, the paranodes, the juxtaparanodes, and the internodes.1–15 The paranodes flanking the nodes of Ranvier are the main interaction regions between the myelinating glial membrane and the axons to form the paranodal axo-glial septate junctions.4–7 The paranodes also serve as a molecular barrier between the nodal domain and the juxtaparanodal domain as this barrier function is compromised in the Contactin-associated protein 1 (Cntnap1), Contactin, and Neurofascin (NfascNF155) mutants.4,5,7,16–19 Disorganization of axon-glial interactions at the node/paranodal domains results in severe pathological conditions,4,5,7,17,18,20 and abnormal paranodal domains have also been reported in human myelination disorders such as Charcot-Marie-Tooth disease21–24 and multiple sclerosis.25,26
Cntnap1 is a transmembrane cell-adhesion protein, which forms a cis interacting complex with axonal paranodal Contactin, a neural cell-adhesion molecule, and the 155-kDa isoform of glial paranodal Neurofascin (NF155).27–30 Mutant mice that lack the paranodal junction structure display ataxia, motor dysfunction, and severely reduced nerve conduction properties.4,5,7 Homozygous Cntnap1-null mice display progressive neurologic defects starting in the second week of life, including lack of mobility, tremors, wide-based gait, and generalized motor paresis. Most Cntnap1-null mice die around 3 weeks of their postnatal life with severely compromised motor functions.4 Structure-function analyses of Cntnap1, Contactin, and NF155 have revealed distinct domains that are involved in the protein-protein interactions between Cntnap1, Contactin, and NF155 both in in vitro and in vivo.10,16,27,28 These earlier studies highlighted specific domains that are involved in interactions between these proteins, their association into biochemical complexes, as well as their role in paranodal domain organization.
Mutations in human CNTNAP1 are implicated in human neurological diseases that are characterized by polyhydramnios, severe neonatal hypotonia, arthrogryposis, and severe motor paralysis.31 In subsequent years, a large number of CNTNAP1 mutations have been identified in over 40 families with subjects carrying frameshift, nonsense, and missense mutations.32,33 The mutations resulted in a wide range of phenotypes and variable survival rates, ranging from infancy to early childhood with support.33–36 The most severe phenotypes resulted in acute respiratory distress and muscle atrophy.32 Despite the mounting evidence of CNTNAP1 mutations in human disease, so far there are no mouse models available for studying the impact of Cntnap1 mutations and the potential mechanisms through which the Cntnap1 mutant protein functions are altered leading to neurological deficits.
The current study has recapitulated two human CNTNAP1 mutations in a mouse model by generating two single nucleotide substitutions in the Cntnap1 gene (Cntnap1C324R and Cntnap1R765C). Compared to control, the Cntnap1C324R/− and Cntnap1R765C/− mutant mice suffered severe weight loss from birth and showed progressive motor and neurological deficits, disorganization of the axo-glial paranodal domains, and diminished nerve conduction properties. The Cntnap1C324R and Cntnap1R765C proteins were nearly undetectable in the paranodal domains and severely disrupted the paranodal organization and function. Using in vivo and in vitro biochemical studies, we show that the Cntnap1C324R and Cntnap1R765C mutant proteins are less stable than the wild-type Cntnap1 and are retained in the cell soma with dramatically reduced cell surface expression as well as reduced binding with Contactin. An inducible Cntnap1 mouse strain was generated that expresses the wild-type Cntnap1 protein in a spatiotemporal manner. When this wild-type Cntnap1 protein was expressed in Cntnap1C324R/− and Cntnap1R765C/− mice, it allowed progressive restoration of the paranodal domains, nerve conduction electrophysiological properties, and motor functions. Together, our studies demonstrate that Cntnap1C324R and Cntnap1R765C mutations are loss-of-function mutations that are rescued by the wild-type Cntnap1 protein and that strategies could be developed to express human CNTNAP1 using the gene therapy methodologies to restore neurological and motor functions in human subjects carrying CNTNAP1 mutations.
RESULTS
Human CNTNAP1 mutations in mice severely affect postnatal growth
Recent whole-exome sequencing of children displaying generalized motor deficits with hypomyelination-associated neuropathologies has led to the identification of mutations in the CNTNAP1 gene. These mutations include single nucleotide changes in the CNTNAP1 coding sequence and reading frame shifts leading to terminations across the length of the CNTNAP1 protein.31–33 One of these mutations occurs at the coding nucleotide number 967, changing a TGC codon (cysteine [Cys] 323) to CGC (arginine [Arg] 323). Another mutation occurs at the coding nucleotide number 2290, changing a CGC codon (Arg 764) to TGC (Cys 764). The Cys at 323 is located in the first laminin G-like domain, and the Arg at 764 is located in the fibrinogen CT domain (Figure 1A).33,36 The amino acid sequence comparison between human CNTNAP1 and mouse Cntnap1 shows 93% identity and 96% similarity (Figure 1B).37
Figure 1. Generation of Cntnap1 mouse mutants carrying human CNTNAP1 mutations and their phenotypic analysis.
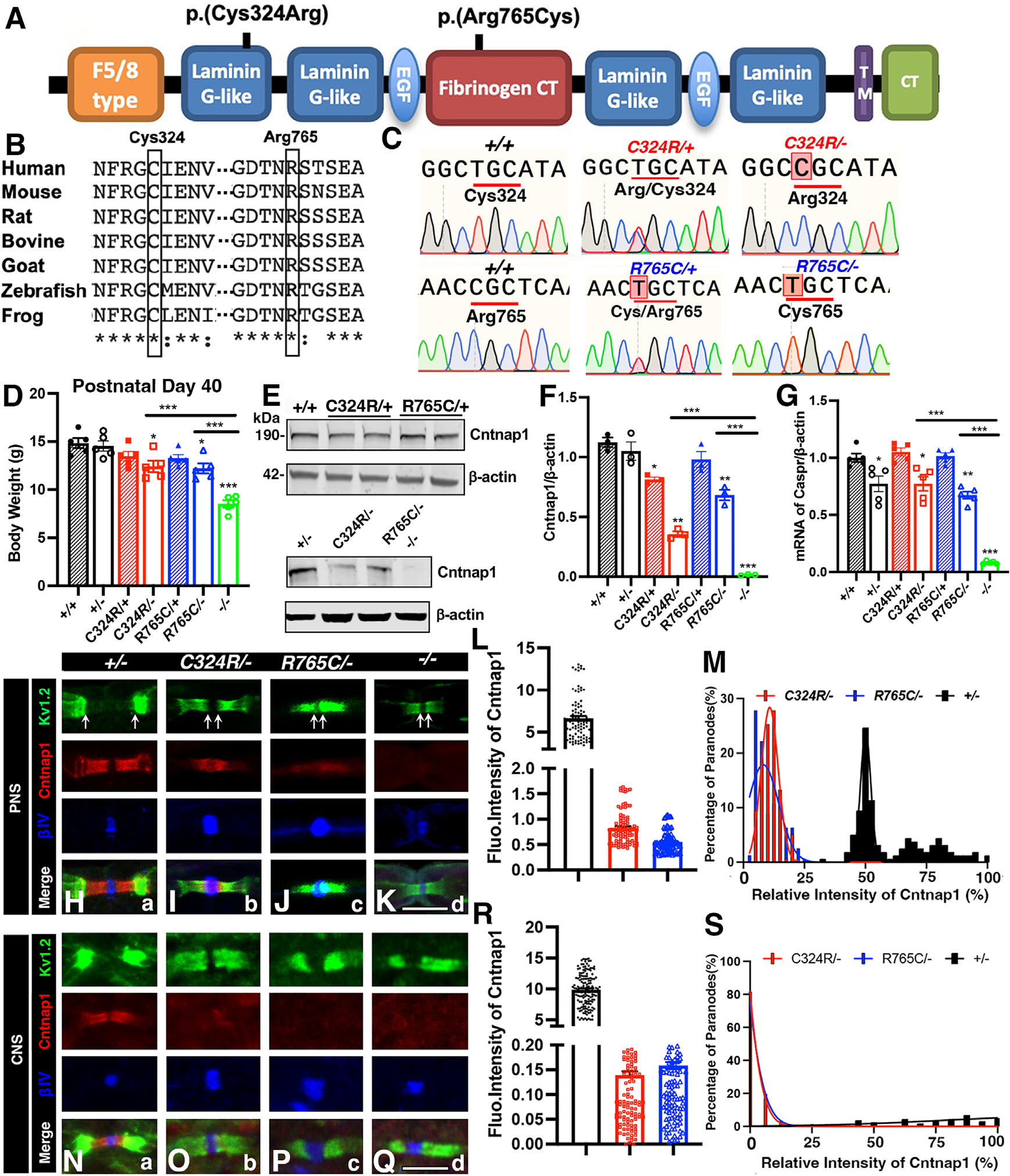
(A) Schematic of Cntnap1 protein and the approximate locations of amino acid changes.
(B) Sequence alignment of the Cntnap1 protein showing high conservation of Cys324 and Arg765 across multiple species.
(C) Genomic DNA sequences from wild-type controls and Cntnap1 mutants.
(D) Body weights at postnatal day 40.
(E) Immunoblots showing expression of Cntnap1 in the CNS (spinal cords). β-actin was used as loading control.
(F) Quantification of relative protein band intensities for Cntnap1 in the CNS.
(G) Quantification of relative mRNA expression for Cntnap1 in the CNS.
(H–K) Immunostaining of sciatic nerves. Scale bar: 5 μm.
(L and M) Measurement of relative fluorescence intensity and distribution of Cntnap1 at the paranodes in the sciatic nerves.
(N–Q) Immunostaining of spinal cords. Scale bar: 5 μm.
(R and S) Measurement of relative fluorescence intensity and distribution of Cntnap1 at the paranodes in the spinal cords. Data are represented as the mean ± SEM of three to six biological replicates.
To examine the consequences of human CNTNAP1 mutations and associated neuropathies, we used CRISPR-Cas9 methodology to create single nucleotide changes that matched the codon substitution changes in human CNTNAP1 for Cys324Arg and Arg765Cys (note that Cys323 corresponds to Cys324 and Arg764 corresponds to Arg765 in mouse Cntnap1 and will be referred to with mouse numbers throughout the text). Upon pronuclear injections, all mouse progeny born were first sequenced at the location of the mutations CGC and TGC codons encoding Arg324 and Cys765, respectively. The mouse lines that positively confirmed the presence of these nucleotide changes were further sequenced for the entire Cntnap1 gene to ensure that no additional mutations were introduced into the Cntnap1 coding sequences during the generation of these lines. Segments of sequences from the homozygous wild-type Cntnap1, heterozygous Cntnap1C324R/+ and Cntnap1R765C/+, and mutant Cntnap1C324R/− and Cntnap1R765C/− genotypes are shown in Figure 1C. The sequence analyses confirmed the successful generation of the mouse models of the two human CNTNAP1C323R and CNTNAP1R764C mutations.
In human patients with CNTNAP1 mutations that cause Cys323Arg and Arg765Cys changes, the CNTNAP1 locus was found to be compound heterozygous, with a paternal stop-codon mutation and a maternal missense mutation.31,33–36,38–40 Thus, to mimic the human disease condition, we crossed Cntnap1C324R/+ and Cntnap1R765C/+ mice with Cntnap1+/− (referred to as +/−) mice, generated previously,4 to obtain the Cntnap1C324R/− and Cntnap1R765C/− (referred as C324R/− or R765C/−) mutants. Starting around P15, we observed a significant reduction in motility, motor paresis, tremors, and general muscle weakness that were easily distinguishable from wild-type or heterozygous littermates. By P40, the weight difference between heterozygous control and Cntnap1 mutants was notable (Figure 1D). Interestingly, the heterozygous mice including +/−, C324R/+, and R765C/+ do not display any phenotype or weight loss in comparison to the wild-type control mice (+/+). These observations suggest that Cntnap1C324R and Cntnap1R765C mutations do not cause any harmful gain-of-function effects.
Single amino acid changes in mouse Cntnap1 affect its stability and cause paranodal axonal domain disorganization
Immunoblotting of spinal cord lysates shows a robust single band of expected molecular weight of ~190 kDa for the full-length Cntnap1 protein (Figure 1E). In C324R/− and R765C/− mutants, the levels of the Cntnap1 protein were significantly reduced in the spinal cord tissues. The C324R/− mutants showed ~64% reduction, while the R765C/− mutants showed ~32% reduction compared to controls (Figures 1E and 1F). This indicates that both C324R and R765C amino acid substitutions reduce the stability of the Cntnap1 protein. Further analysis of the mRNA levels in all mutants revealed no significant changes compared to their respective wild-type and heterozygous controls (Figure 1G). These results suggest that the effects of the mutations are primarily due to post-translational alterations rather than the changes in the levels of mRNA expression.
Next, we examined the expression of Cntnap1 proteins in the paranodal regions of the sciatic nerves (PNS) and the white matter of the spinal cord (CNS). As expected, the intensity and distribution of Cntnap1 proteins in the paranodal area were found to be comparable in both heterozygous mutants (C324R/+ and R765C/+) when compared to wild-type controls, both in PNS and CNS (Figure S1). However, in the case of C324R/− and R765C/− mutants, the mutated Cntnap1 proteins were either present at very low levels or were completely absent at the paranodes in CNS and PNS (Figures 1H–1S). We next examined the localization and distribution of juxtaparanodal K+ channels (Kv1.2) and nodal β-IV spectrin.4,11 In control +/− mice, both β-IV spectrin and K+ channels were separated at the distinct compartments by the paranodal region; in C324R/− and R765C/− mutants, β-IV spectrin was still restricted to the nodal regions but became widened; the juxtaparanodal K+ channels were completely mislocalized into the paranodal region and were in direct proximity to the nodal area, indicating a loss of paranodal function leading to the mislocalization of K+ channels in these Cntnap1 mutants in both the PNS and CNS (Figures 1H–1K and 1N–1Q). The immunostaining of the mutant myelinated fibers reveals a loss of the paranodal domain function in separating the juxtaparanodal proteins from the nodal complex. Together these data show that Cys324Arg and Arg765Cys amino acid substitutions affect the stability of Cntnap1 at the paranodes and also affect the barrier function of the paranodal domains in myelinated axons.
Cntnap1C324R and Cntnap1R765C mutants display mislocalization of key paranodal proteins but not their protein stability
To determine whether the Cntnap1C324R and Cntnap1R765C mutant proteins affected the localization of the other paranodal components, we assessed the expression of NF-155 and Contactin (Figure S2). Immunostaining of PNS and CNS showed the near absence of staining for both NF155 and Contactin in the paranodal regions, similar to what has been observed for Cntanp1-null mutants.4 Quantitatively, the staining intensities of both NF155 and Contactin were proportional to the staining levels of Cntnap1, suggesting that in the absence of proper functional Cntnap1, both NF155 and Contactin are unable to localize properly at the paranodal regions. Surprisingly, immunoblot analysis of the protein expression levels of C324R/− and R765C/− mutants showed that their protein expression levels remained unchanged, both in the spinal cords and sciatic nerves (data not shown). These data indicate that the C324R/− or R765C/− mutant proteins affect the paranodal localization of Contactin and NF155 but not their overall protein expression and stability in the tissues.
Cntnap1C324R/− and Cntnap1R765C/− mutants show severe motor disability and a significant decline in the peripheral nerve conduction properties
To quantitively measure the deficiency in fine motor coordination and balance, we performed beam walking trials (Figure 2A). The C324R/−, R765C/−, and knockout (KO) mice were all unable to hold on to the beam, whereas the control wild-type (+/+), control heterozygous (+/−), and mutant heterozygous C324R/+ and R765C/+ mice were all able to hold steadily and crossed the beam on the first trial, suggesting a severe motor dysfunction in C324R/−, R765C/−, and KO mutant mice (Figure 2B). Next, the rotarod test, which is used to assess fine motor coordination, showed all control (+/+ and +/−) and heterozygous (C324R/+ and R765C/+) mice stayed on the rotarod for the entire experimental time (2 min with up to 25–35 rpm). In contrast, the C324R/−, R765C/−, and KO mutant mice started to fall off when the rotarod started to accelerate (Figures 2C and 2D). We did not observe any significant difference between C324R/−, R765C/−, and KO mutant mice indicating that the single amino acid and null mutants all show severe fine motor coordination deficits.
Figure 2. Cntnap1C324R/− and Cntnap1R765C/− mutants display severe motor disability and decline in nerve conduction properties.
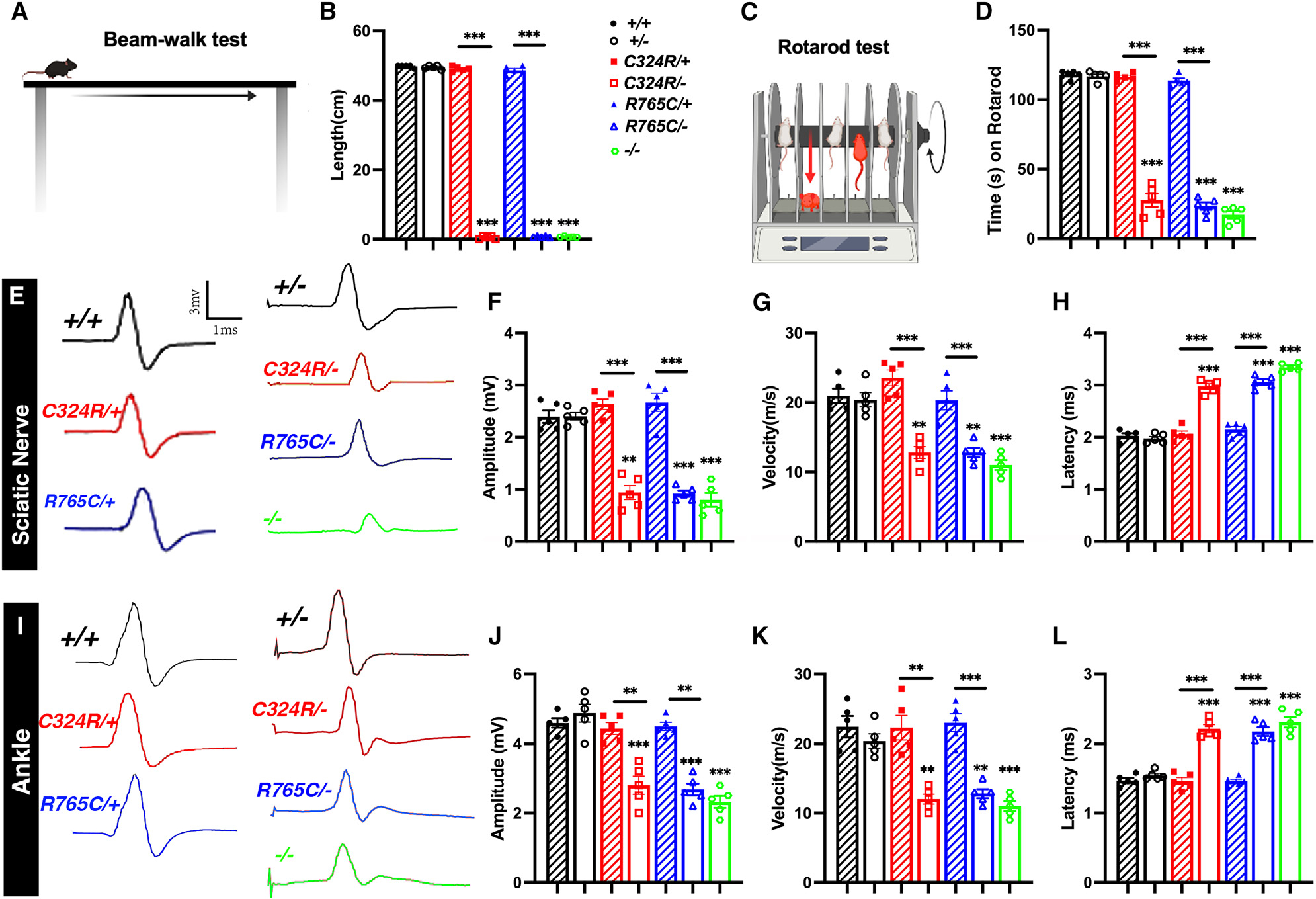
(A and B) Beam walking motor coordination performance.
(C and D) Motor learning ability as measured by rotarod test.
(E–L) Representative electrical impulse traces from P20 sciatic nerves (E) showing amplitude, velocity, and latency (F–H, respectively) and ankle (I) showing amplitude, velocity, and latency (J–L, respectively). Data are represented as the mean ± SEM of five biological replicates.
Next, we analyzed the nerve conduction properties by performing in vivo recording of the sciatic nerves at P30. There was no difference in controls (+/+, +/−, C324R/+, R765C/+), but the nerve conduction amplitudes and velocities for the sciatic nerve and the ankle region were significantly reduced in C324R/−, R765C/−, and KO mutant mice (Figures 2E–2G, and 2I–2K). Meanwhile, the latency measuring the time taken for the fastest nerve fibers to conduct between two stimulation points was significantly increased in C324R/−, R765C/−, and KO mutants (Figures 2H and 2L). Together, the in vivo nerve conduction electrophysiological measurements reveal a significant impact of Cntnap1 mutations on both the strength and velocity of the nerve impulses as is observed in KO mutants, further highlighting the critical role of Cntnap1 at the paranodal domain in myelinated axons.
Cntnap1C324R and Cntnap1R765C mutants display hypomyelination and loss of the paranodal axo-glial junctions
Upon the discovery of mutations in human CNTNAP1, it was observed that these mutations caused hypomyelination in both the central and peripheral nervous system, which has become the hallmark pathology for CNTNAP1-related congenital hypomyelinating neuropathies (CHNs, OMIM: 605253).34–36,41–44 In addition, delayed myelination of white matter was observed in the brain MRIs of human patients.43 To address whether loss of Cntnap1 does impact myelination, we performed detailed electron microscopic (EM) analyses of the white matter axons concerning the extent of myelination and also how these mutations affected the ultrastructural organization of the paranodal region axo-glial junctions. We carried out a comprehensive EM analysis of myelinated axons from the CNS and PNS from 2-month-old control +/− and C324R/−, R765C/−, and KO (−/−) mutants. While the visual inspection of the mutant myelinated fibers suggested hypomyelination in both the CNS and the PNS (Figures 3A–3H), a more quantitative analysis was performed that estimated the g-ratios of the myelinated axons across all genotypes using a semi-automated software.45 The g-ratio was significantly increased in C324R/−, R765C/− and in KO mutants when compared to control +/− mice in both the CNS and PNS (Figures 3I and 3J). In the CNS, the average g-ratio in C324R/− mice was 33% higher than that in control +/− mice, while the average g-ratio in R765C/− mice was 21.6% higher than that in control mice. Interestingly, in the small axons (diameter smaller than 0.5 μm), the difference was even higher compared to control mice. Similarly, the KO mutants also revealed hypomyelination and increased g-ratio in both the CNS and PNS, suggesting that loss of Cntnap1 function results in hypomyelination, and that these Cntnap1 mutations behave as loss-of-function mutations. Further analysis revealed that axons with a higher g-ratio were significantly more frequent in the Cntnap1 mutant mice compared to the heterozygous controls. Specifically, the mutants exhibited markedly higher percentages of axons with a g-ratio of 0.75 or above: C324R/− at 72.04%, R765C/− at 42.1%, and KO at 85.9%, in contrast to 0% in heterozygous controls. Given the software limitations that prevented the capture of completely unmyelinated axons, we conducted a manual analysis of EM images from all genotypes for the presence of unmyelinated axons. In the spinal cord tissues from all genotypes, Cntnap1-KO mice displayed a notable increase in the proportion of unmyelinated axons (5.5%) compared to heterozygous controls (0.9%). Additionally, the C324R/− and R765C/− also presented with unmyelinated axons at 3.8% and 2.4%, respectively (Figure S8). These findings suggest that Cntnap1 C324R/− and R765C/− mutants show variations in the severity of their myelin phenotypes compared to Cntnap1−/− mutants, which was also reflected in the g-ratios observed in Cntnap1 mutants.
Figure 3. Cntnap1C324R/− and Cntnap1R765C/− mutants display hypomyelination and loss of the paranodal axo-glial junctions.
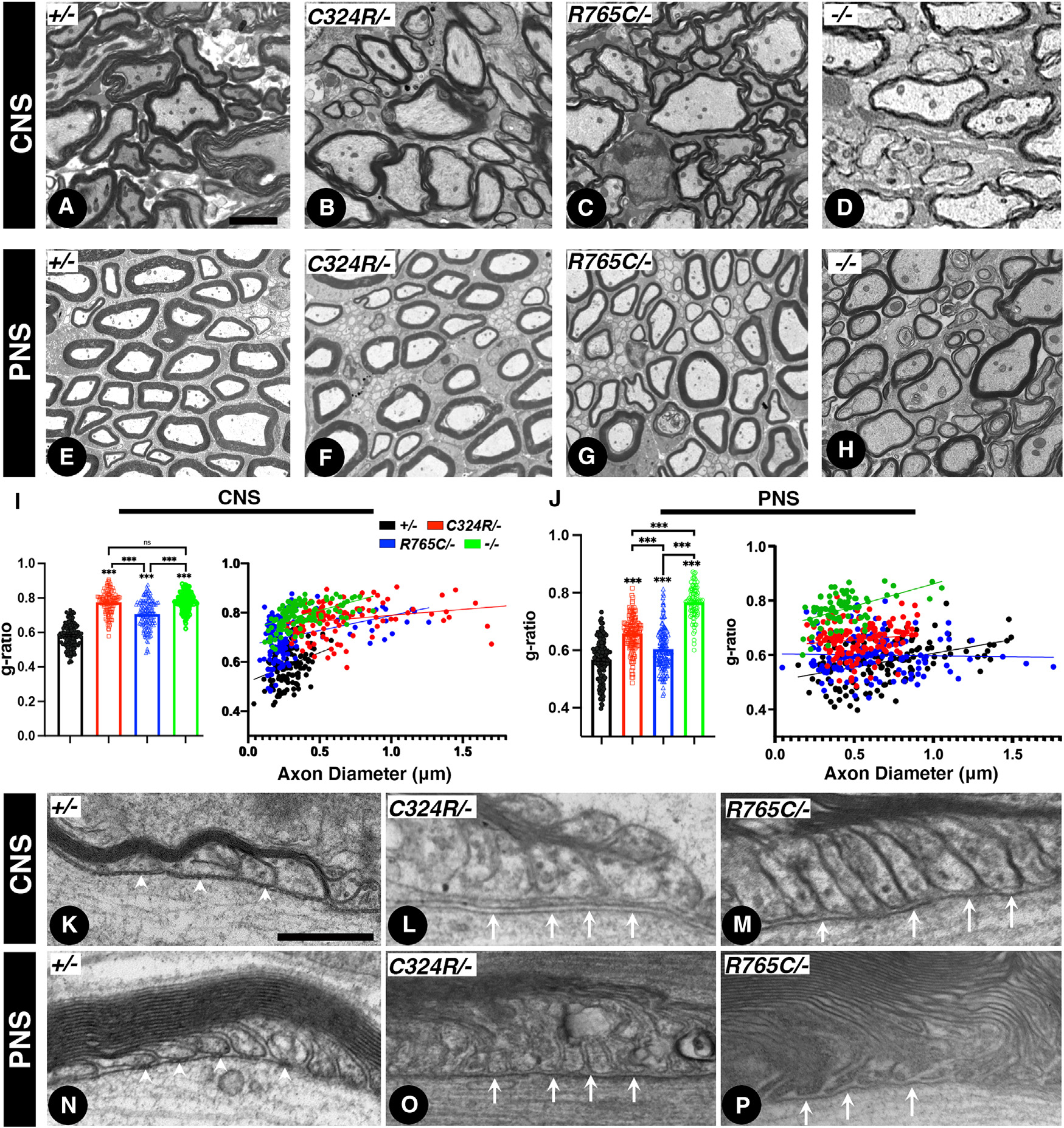
(A–D) Transmission electron microscopy (TEM) images of cross-sections from P21 spinal cords.
(E–H) TEM of cross-sections from P21 sciatic nerves.
(I and J) Morphometric analysis measuring the g-ratios of myelinated axons (I, spinal cords; J, sciatic nerves).
(K–M) TEM images of longitudinal sections of P21 spinal cords at the level of the paranodal region showing axo-glial junctions.
(N–P) TEM images of longitudinal sections of P21 sciatic nerves. White arrowheads in (K) and (N) indicate distinct ladder-like septate junctions between myelin loops. White arrows in (L), (M), (O), and (P) indicate lack of axo-glial septate junctions in the CNS and PNS myelinated axons. Scale bars: 2 μm (A–H) and 0.2 μm (K–P). Data are represented as the mean ± SEM of three biological replicates.
We next examined the ultrastructure of the paranodal axo-glial junctions to determine whether the C324R/− and R765C/− mutant proteins were able to establish axo-glial septate junctions. As shown in Figures 3K–3P, the paranodal region in the CNS and PNS of control +/− mice shows distinct ladder-like septa (white arrowheads) between the myelin loops and the axonal axolemma in the paranodal area. Whereas, in C324R/− and R765C/− mutants, the ladder-like paranodal axo-glial septate is absent (white arrows). No septa-like structures were observed in C324R/− and R765C/− mutants similar to Cntnap1−/−-null mutants,4 showing these mutations cause hypomyelination and loss of the paranodal axo-glial septate junctions.
Neuronal expression of the wild-type Cntnap1 causes gradual restoration of axonal domain organization in Cntnap1C324R/− and Cntnap1R765C/− myelinated axons
Next, we wanted to determine whether timely controlled neuronal expression of the wild-type Cntnap1 gene in C324R/− and R765C/− mutants will be able to restore Cntnap1 expression as well as the functional paranodal domains. To achieve that, we utilized the LoxP-Stop-LoxP (LSL) system,46 to generate a mouse line with the LSL-Cntnap1FLAG sequence integrated into the Rosa26 locus. The presence of the upstream poly(A) (multi-polyadenylation) sequences terminates transcription and prevents the expression of the downstream Cntnap1FLAG cDNA (Figure 4A). Upon expression of tissue-specific Cre recombinase, sequences between the LoxP sites are removed allowing transcription of the Cntnap1FLAG cDNA and expression of Cntnap1FLAG only in selected Cre-expressing cells.
Figure 4. Neuronal expression of the wild-type Cntnap1 gene progressively restores axonal domain organization in Cntnap1C324R/− and Cntnap1R765C/− mutant myelinated axons.
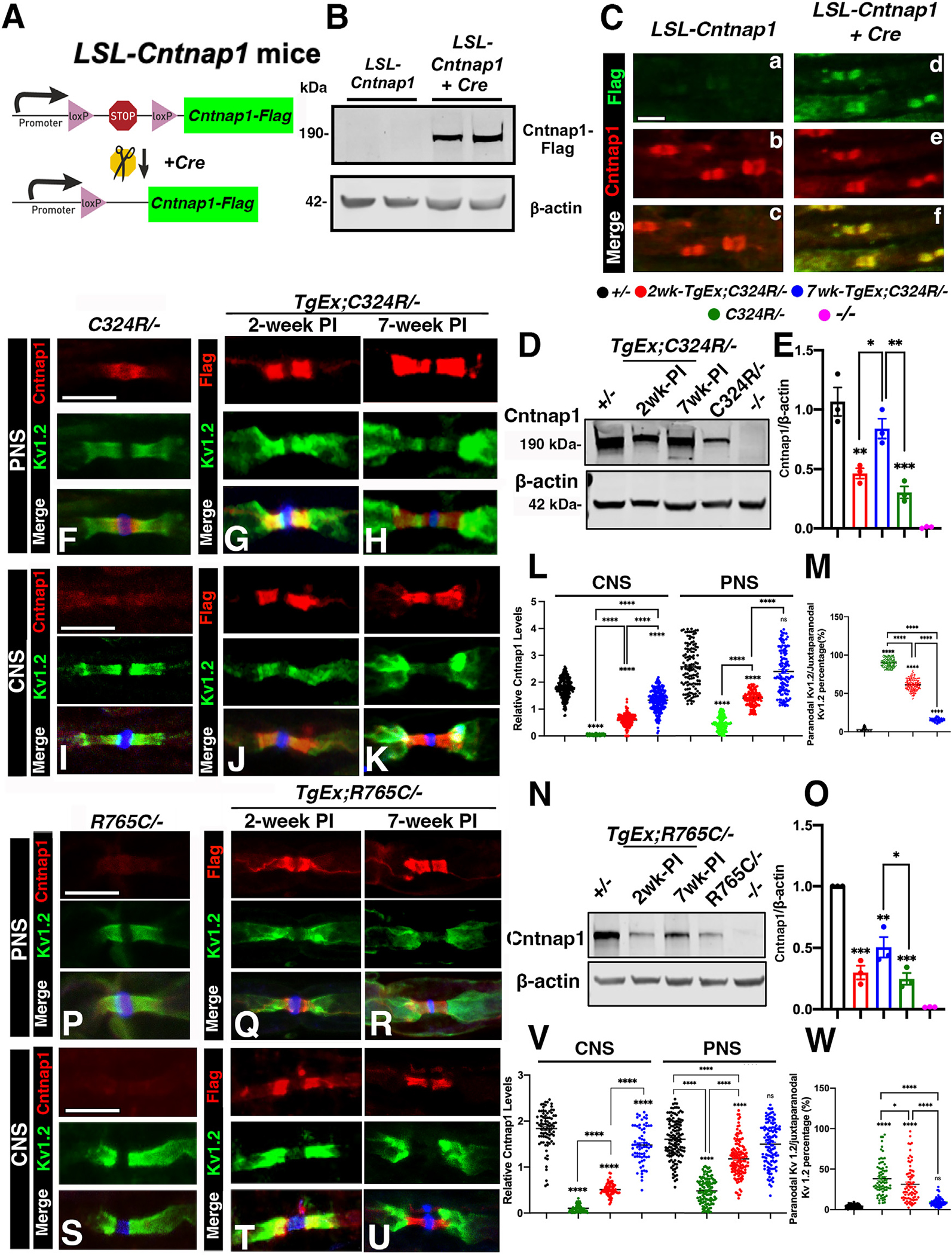
(A) Schematic representation of the generation of LoxP-Stop-LoxP (LSL) Cntnap1FLAG transgenic mice.
(B) Representative immunoblots showing expression of FLAG-tagged Cntnap1 (Cntnap1FLAG) in the spinal cords. β-actin was used as a loading control.
(C) Immunostaining of sciatic nerves. Scale bar: 10 μm.
(D and E) Representative immunoblots showing expression of Cntnap1FLAG in spinal cord tissues at 2 or 7 weeks after tamoxifen injection. β-actin was used as loading control. The relative protein intensities are quantified in (E).
(F–K) Immunostaining of sciatic nerves (F–H) or spinal cords (I–K) from TgEx;C324R/− mice at 2 weeks (G, J) or 7 weeks (H, K) after tamoxifen injection. Scale bar: 5 μm.
(L and M) The relative fluorescence quantification of Cntnap1 in both the CNS and PNS.
(N and O) Representative immunoblots showing the expression of Cntnap1FLAG in the spinal cords from TgEx;R765C/− mice at 2 or 7 weeks after tamoxifen injection. The relative protein intensities of Cntnap1 are quantified in (O).
(P–U) Immunostaining of sciatic nerves (P–R) or spinal cords (S–U) from TgEx;R765C/− mice at 2 weeks (Q, T) or 7 weeks (R, U) after tamoxifen injection. Scale bar: 5 μm.
(V and W) The relative fluorescence quantification of Cntnap1 in both the CNS and PNS. Data are represented as the mean ± SEM of three to six biological replicates.
To confirm the expression of the Cntnap1 protein in LSL-Cntnap1 mice, we crossed this line with the ubiquitous β-Actin-Cre line and examined the expression of the Cntnap1FLAG protein. As shown in Figure 4B, immunoblotting of the spinal cord lysates with an anti-FLAG antibody detected the Cntnap1FLAG protein at ~190 kDa only in Actin-Cre;LSL-Cntnap1FLAG (Act-Ex). The protein was undetectable in the LSL-Cntnap1FLAG spinal cord lysates, which did not express the Cre recombinase. Immunostaining of the sciatic nerve myelinated axons showed that the Cntnap1FLAG protein was properly localized at the paranodes as it colocalized with the endogenous Cntnap1. Without the Cre recombinase expression, the Cntnap1FLAG protein was undetectable in the paranodal domain in LSL-Cntnap1FLAG myelinated axons, which were positively labeled by anti-Cntnap1 antibodies (Figure 4C). In addition, we also crossed this line with Cntnap1 mutant lines, C324R/− and R765C/−, and the relevant genotypes Act-Ex;C324R/− and Act-Ex;R765C/− were further processed for phenotypic analyses and compared to wild-type controls and their mutant background, C324R/− and R765C/−. We did not observe any significant difference between these two lines compared to controls (+/+) during the experimental period (3 months). These Act-Ex;C324R/− and Act-Ex;R765C/− mice were indistinguishable from their control counterparts (Figure S3), regarding their body weight, Cntnap1 protein expression levels or cellular localization, motor function, and nerve conduction properties. Together, these data show that transgenic Cntnap1FLAG protein expression is tightly controlled with no leakiness and when expressed in a Cre-dependent manner can localize at the paranodal region in myelinated axons.
Next, we wanted to determine whether induced expression of Cntnap1 in C324R/− and R765C/− mutant background will allow restoration of the paranodal domain as well as re-localization of the juxtaparanodal proteins. We crossed LSL-Cntnap1 mice with the tamoxifen-inducible SLICK-H-CreERT2 mice, which express the Cre recombinase only in neurons.11,47,48 The final mice of the genotype SLICK-H-CreER;LSL-Cntnap1FLAG;C324R/− (referred to here as TgEx;C324R/−) or SLICK-H-CreER;LSL-Cntnap1FLAG;R765C/− (referred to here as TgEx;R765C/−) received tamoxifen at P21 for 5 consecutive days (at 1 mg/kg body weight). The control animals LSL-Cntnap1FLAG;C324R/− (C324R/−) and LSL-Cntnap1FLAG;R765C/− (R765C/−) without Cre also received the same regimen of tamoxifen. The animals were sacrificed 2 weeks and 7 weeks post tamoxifen injection and processed for immunoblot and immunostaining analysis.
As shown in Figure 4D, transgenic Cntnap1 expression showed a steady increase from 2 to 7 weeks in TgEx;C324R/− mice compared to C324R/− mice. The Cntnap1 protein levels were quantified showing a significant increase in Cntnap1 levels at 7 weeks post tamoxifen treatment (Figure 4E). Immunostaining of the sciatic nerves and spinal cords from TgEx;C324R/− showed expression of Cntnap1FLAG at the paranodes in fibers at 2 weeks post tamoxifen injection and much more prominently in the 7 weeks post tamoxifen. In both the PNS and CNS at 2 weeks, the juxtaparanodal Kv1.2 channels were still observed in the paranodal area, but by 7 weeks post injection, most of the Kv1.2 channels were separated from the paranodal region (Figures 4F–4L). In control C324R/− fibers, the juxtaparanodal Kv1.2 channels are present in the paranodal area adjacent to nodal βIV spectrin. The reorganization of the paranodes and separation of the juxtaparanodal Kv1.2 from the paranodal area were also quantified, which showed that at 7 weeks after expression of Cntnap1FLAG, the paranodal domain reorganization was similar to that of control heterozygous myelinated fibers (Figure 4M).
Similarly, immunoblotting of spinal cords from 2 to 7 weeks after tamoxifen injections in TgEx;R765C/− mice was compared to R765C/− (Figure 4N). The Cntnap1 protein levels were quantified showing a significant increase in Cntnap1 levels at 7 weeks post tamoxifen treatment in TgEx;R765C/− mice (Figure 4O). Immunostaining of the sciatic nerves and spinal cords from R765C/− mutant and TgEx;R765C/− also showed expression of Cntnap1FLAG at the paranodes in fibers at 2 weeks post tamoxifen injection and much more prominently in the 7-week-post-tamoxifen-injection fibers (Figures 4P–4U). In both the PNS and CNS at 2 weeks, the juxtaparanodal Kv1.2 channels were observed in the paranodal area as in TgEx;C324R/− fibers; but by 7 weeks post injection, most of the Kv1.2 channels were separated from the paranodal region. The relative expression of Cntnap1FLAG at the paranodes in TgEx;R765C/− fibers was quantified (Figure 4V), and the reorganization of the paranodes and separation of the juxtaparanodal Kv1.2 from the paranodal area were also quantified (Figure 4W), showing that in 7 weeks after expression of Cntnap1FLAG, the paranodal domain formation is indistinguishable from the control heterozygous myelinated fibers. Additional immunostaining using antibodies against two other well-characterized paranodal proteins (NF155 and Contactin) also revealed their restoration at the paranodes and proper organization of the axonal domains in C324R/− and R765C/− mutants upon Cntnap1FLAG induction (Figure S4). These data demonstrate that the transgenic Cntnap1FLAG protein is able to rescue and restore the paranodal domain and accordingly the organization of the axonal domain in mutant myelinated axons.
Neuronal expression of the wild-type Cntnap1 in Cntnap1C324R/− and Cntnap1R765C/− mutants restores motor coordination
We next sought to determine whether TgEx;C324R/− and TgEx;R765C/− mice will also show improvements in the body weight and motor coordination after tamoxifen injection. The body weight and motor coordination using beam walking and rotarod were recorded at 2, 4, and 8 weeks after tamoxifen injection (Figures S5A–S5H). In the rotarod test, the TgEx;C324R/− and TgEx;R765C/− mutant mice started to perform better from 8 weeks post tamoxifen injection. For beam walking motor performance, within 2 weeks after tamoxifen injection, we observed a significant improvement. The TgEx;C324R/− and TgEx;R765C/− mutant mice were able to walk a short distance on the beam, which improved progressively such that the rescued TgEx;C324R/− and TgEx;R765C/− mutants completed the entire beam distance (50 cm) mirroring Cntnap1+/− control animals (Figures S5C and S5G). The speed on the beam walking also improved progressively and was close to 80% for rescued TgEx;C324R/− mutants or close to 100% for TgEx;R765C/− rescued mutants compared to control animals (Figures S5D and S5H). Furthermore, the nerve conduction velocity (NCV) values were also significantly improved in TgEx;C324R/− and TgEx;R765C/− (Figure S6), compared to control +/− mice in a time-dependent manner. Together, these data demonstrate that the wild-type Cntnap1 can progressively restore motor coordination and function in Cntnap1C324R and Cntnap1R765C mutants.
Expression of Cntnap1 rescues hypomyelination and restores the ultrastructure of paranodal axo-glial junctions in Cntnap1C324R/− and Cntnap1R765C/− mutants
Human subjects carrying CNTNAP1 mutations have shown hypomyelination phenotypes indicative of the role of CNTNAP1 in axonal myelination.35,44 In addition, both C324R/− and R765C/− mutants revealed hypomyelination and reduced g-ratios in myelinated axons (Figure 3). We wanted to determine whether the hypomyelination phenotype in C324R/− and R765C/− mutants could be rescued by neuronal expression of Cntnap1 in both the CNS and PNS myelinated axons. We processed spinal cords and sciatic nerves from +/−, TgEx;C324R/− and TgEx;R765C/− animals that all received tamoxifen at P21 for 5 consecutive days and were analyzed at 8 weeks post tamoxifen injections. Quantification of the g-ratios for the CNS and PNS myelinated axons revealed that the hypomyelination phenotypes in the Cntnap1 mutants were significantly rescued by neuronal expression of Cntnap1, indicating that Cntnap1 is required for proper axonal myelination (Figures 5A–5H). Moreover, the proportion of unmyelinated axons was considerably reduced in the rescued mice compared to the parent Cntnap1 mutations (Figure S8). We also analyzed the longitudinal sections of the myelinated axons to determine whether Cntnap1 expression restored the ultrastructural organization of the paranodal axo-glial junctions. High-resolution transmission electron microscopy (TEM) images from the CNS and PNS of TgEx;C324R/− and TgEx;R765C/− animals showed restoration of the paranodal axo-glial junctions (Figures 5I–5L, black arrowheads). Most notably, the stereotypical septate-like junctions were visible in the rescued animal fibers as observed in control (Cntnap1+/−) myelinated fibers. However, we still noticed the everted as well as inverted myelin loops presented in both TgEx;C324R/− (Figures 5M and 5O) and TgEx;R765C/− (Figures 5N and 5P) mice, suggesting that despite the restoration of paranodal junctions, some myelin loops may remain everted. Together, these data demonstrate that neuronal expression of Cntnap1 can restore proper myelination and paranodal axo-glial junction formation in C324R/− and R765C/− mutants.
Figure 5. Neuronal expression of the wild-type Cntnap1 restores myelination and paranodal axo-glial junctions in C324R/− and R765C/− mutants.
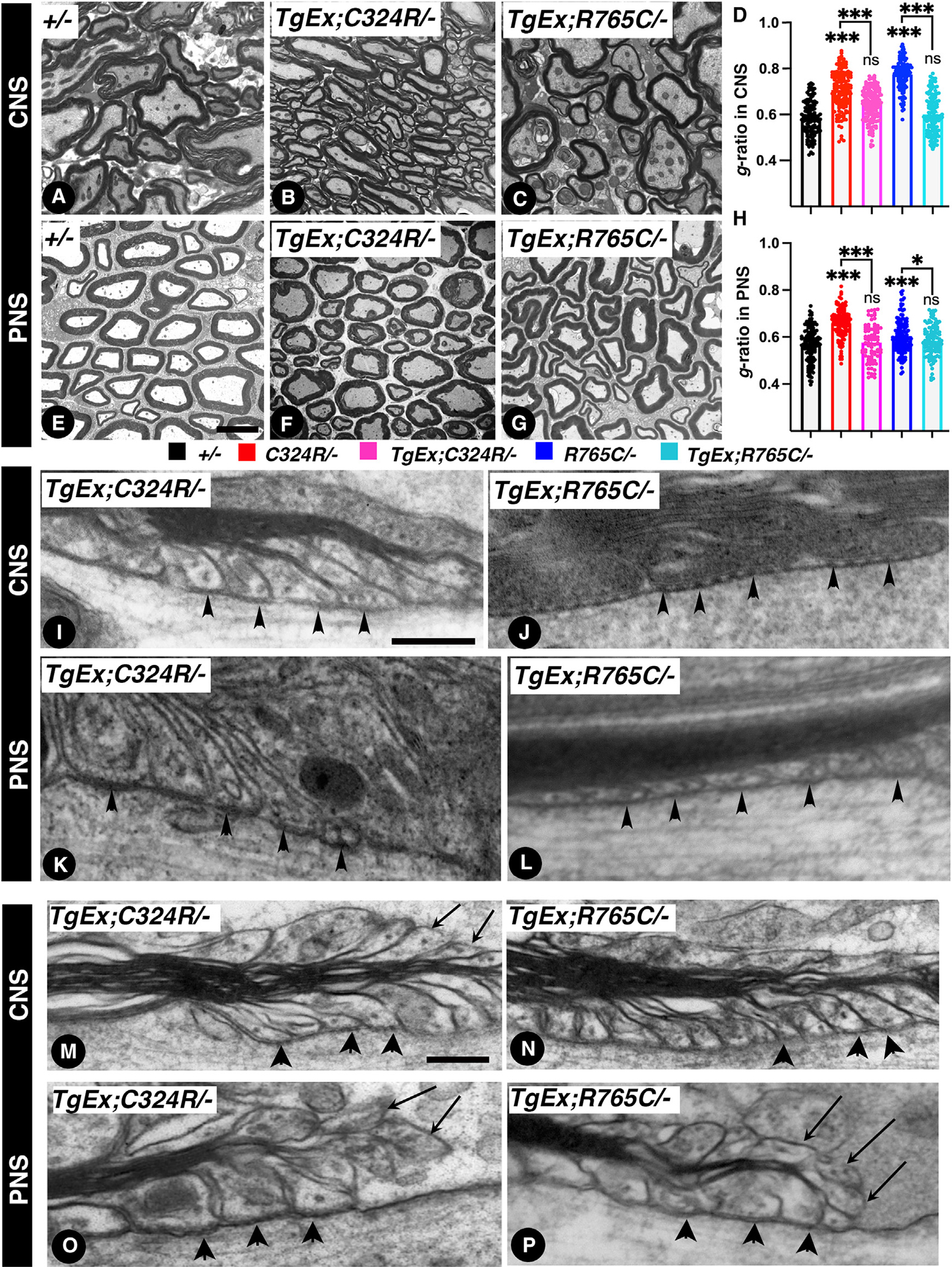
(A–C) 7 weeks after tamoxifen injection, TEM of cross-sections from the spinal cords.
(D) Morphometric analysis showing g-ratios of spinal cord myelinated axons.
(E–G) 7 weeks after tamoxifen injection, TEM of cross-sections from the sciatic nerves.
(H) Morphometric analysis showing g-ratios of sciatic nerve myelinated axons.
(I and J) TEM images of longitudinal sections of spinal cords at the level of the paranodes from 7-week-post-tamoxifen-injection TgEx;C324R/− (I) and TgEx;R765C/− (J). Black arrowheads point to paranodal septa.
(K and L) TEM images of longitudinal sections of sciatic nerves at the level of the paranodes from 7 weeks post tamoxifen injection from TgEx;C324R/− (K) and TgEx;R765C/− (L). Black arrowheads point to paranodal septa.
(M–P) TEM images of longitudinal sections of spinal cords and sciatic nerves. Black arrowheads point to paranodal septa, and black arrows point to everted loops. Scale bars: (A–G) 2 μm; (I–L) 0.2 μm; (M–P) 0.4 μm. Data are represented as the mean ± SEM of three biological replicates.
Early neuronal induction of the wild-type Cntnap1 completely rescues biometric parameters and paranodal axo-glial junctions in C324R/− and R765C/− mutants
Our targeted rescue strategy showed that expression of the wild-type copy of Cntnap1 at P21 restores Cntnap1 expression in the paranodal area and the structure of axonal domains. Moreover, motor functions, such as the ability to cross the beam and improvement in motor coordination on the rotarod test, were restored in R765C/− mice but to a lesser extent in C324R/− mice, even after 8 weeks of Cntnap1 expression. Notably, neither TgEx;C324R/− nor TgEx;R765C/− mice reached body weight levels comparable to controls (+/−), suggesting that the timing of wild-type Cntnap1 induction is critical, and that P21 may already be a late stage for full phenotypic restoration. Considering the severity of CNTNAP1 mutation-related neurological disorders, it is more plausible to induce the expression of wild-type Cntnap1 earlier, offering an intervention to establish the paranodal region and restoration of physiological or motor deficits. To address whether early Cntnap1 expression will restore these deficits, we initiated tamoxifen injections as early as P5–P7 and assessed the mice 2 weeks or 7 weeks after injection (schematic in Figure 6A). At P5 and after 2 weeks of age, all mice showed no significant difference in body weight. However, after 7 weeks of injection, both mutant mice (C324R/− and R765C/−) exhibited considerably lower body weights compared to controls (+/−). In contrast, TgEx;C324R/− and TgEx;R765C/− mice with the induced wild-type Cntnap1 displayed body weights nearly identical to the controls (Figure 6B), indicating that early induction of wild-type Cntnap1 fully rescued the weight loss phenotype in both mutants. Similarly, after 7 weeks of injection, both TgEx;C324R/− and TgEx;R765C/− mice were able to cross the beam without any difficulties and could stay on the rotarod as long as the control mice (Figures 6C and 6D).
Figure 6. Early neuronal expression of Cntnap1 completely rescues deficits in C324R/− and R765C/− mutants.
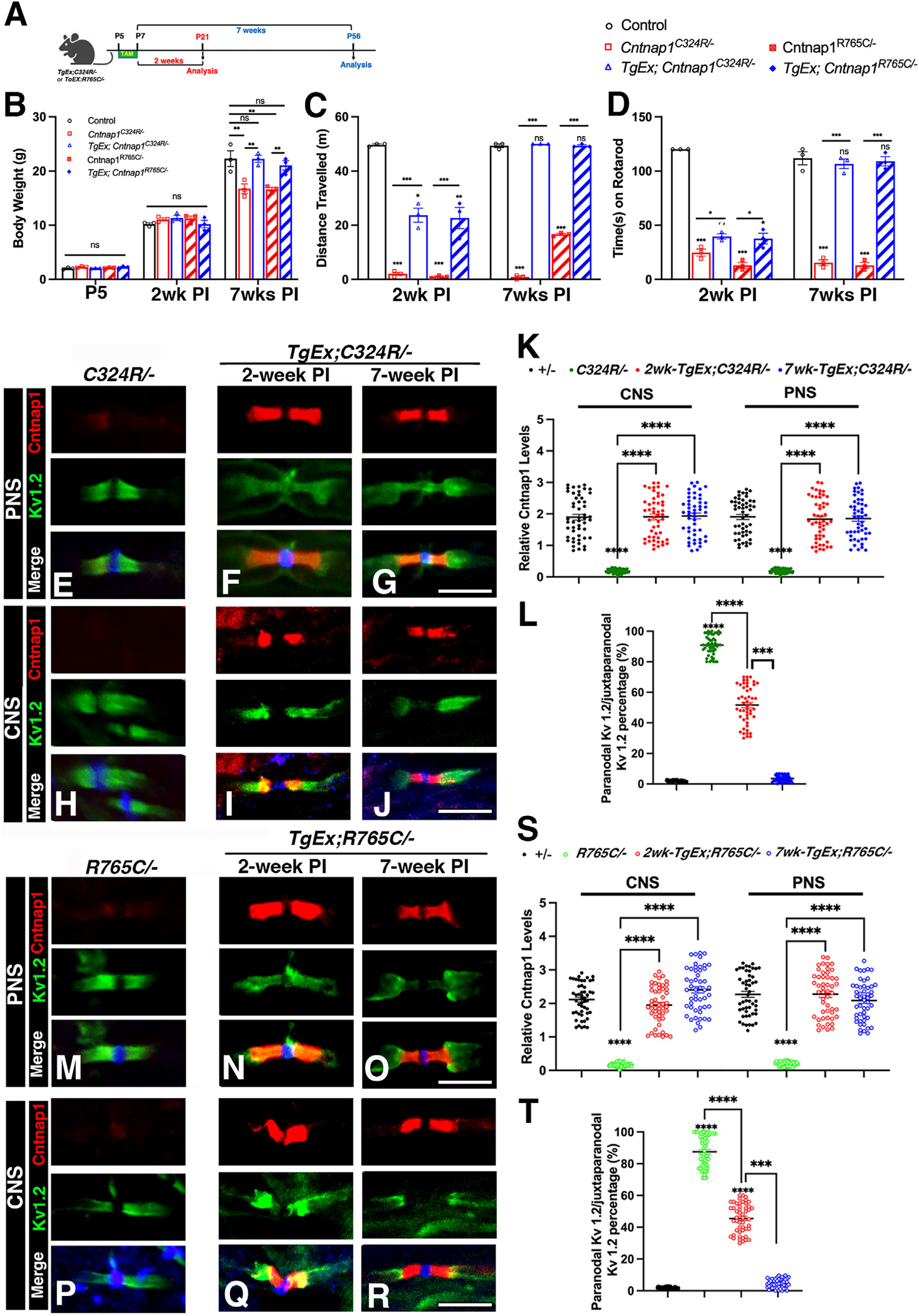
(A) Schematic outline of the experimental strategy.
(B) Body weight at 2 and 7 weeks after P5 tamoxifen injection.
(C) Distance traveled on the beam at 2 or 7 weeks after P5 tamoxifen injection.
(D) Rotarod assay at 2 or 7 weeks after P5 tamoxifen injection.
(E–J) Immunostaining of sciatic nerves (E–G) or spinal cords (H–J) from TgEx;C324R/− mice at 2 weeks (G, J) or 7 weeks (H, K) after P5 tamoxifen injection. Scale bar: 5 μm.
(K and L) The relative fluorescence quantification of Cntnap1 in both the CNS and PNS.
(M–R) Immunostaining of sciatic nerves (M–O) or spinal cords (P–R) from TgEx;R765C/− mice at 2 weeks (N, Q) or 7 weeks (O, R) after P5 tamoxifen injection. Scale bar: 5 μm.
(S and T) The relative fluorescence quantification of Cntnap1 in both the CNS and PNS. Data are represented as the mean ± SEM of three to six biological replicates.
Immunostaining of the sciatic nerves and spinal cords from TgEx;C324R/− mice showed similar expression of Cntnap1 at the paranodes both 2 and 7 weeks after tamoxifen induction (Figures 6E–6L). At 2 weeks, the juxtaparanodal Kv1.2 channels (green) were still partially overlapping with the paranodal area, but by 7 weeks post injection, most of the Kv1.2 channels were separated from the paranodal region (Figures 6G and 6J). The reorganization of the paranodes and separation of the juxtaparanodal Kv1.2 from the paranodal area were quantified, showing that 7 weeks after expressing Cntnap1, the paranodal domain formation was indistinguishable from control heterozygous myelinated fibers (Figure 6L). Similarly, immunostaining at 2 and 7 weeks after tamoxifen induction was compared to R765C/− (Figures 6M–6R). The Cntnap1 protein levels in PNS and CNS from TgEx;R765C/− mice revealed a significant increase in Cntnap1 levels at 2 or 7 weeks post tamoxifen treatment, which is comparable to the control (Figure 6S). The reorganization of the paranodes and separation of the juxtaparanodal Kv1.2 from the paranodal area follow the same trend as the C324R/− rescue (Figure 6T).
Moreover, the NCV values were indistinguishable from controls in mice with earlier induction of TgEx;C324R/− and TgEx;R765C/− (Figures S7A–7F) at 2 and 7 weeks after tamoxifen induction. These results strongly support the idea that early induction of wild-type Cntnap1 leads to a near complete rescue of the phenotypes observed in Cntnap1 mutants.
C324R/− and R765C/− mutant proteins remain in the neuronal soma and reveal reduced binding with Contactin
Mutations in protein-coding genes that alter amino acids often lead to conformational changes in protein structure resulting in altered and potentially misfolded proteins. Mutant proteins within the cell either get retained in the endoplasmic reticulum (ER)49–51 or get further processed through autophagy and degraded through the ubiquitin/proteasome pathway.52 In both Cntnap1C324R and Cntnap1R765C mutant proteins, potential alterations in disulfide bonds or conformational changes are predicted to significantly change the native state of the Cntnap1 protein. Immunostaining of spinal cords revealed that both Cntnap1C324R and Cntnap1R765C mutant proteins were present in the neuronal soma (white arrowheads), which was not observed in the control +/− neurons (Figures 7A–7D). Interestingly, the Contactin protein was also retained in the neuronal somas of C324R/− and R765C/− mutants (white arrows), which was not observed in control somas or the KO mutant neuronal somas. The paranodal staining of Cntnap1 and Contactin is robustly visible in Figure 7Aa, b, overlapped in Figure 7Ad, and is absent in KO, C324R/−, and R765C/− mutant neurons (Figures 7B–7D). These data indicate that Cntnap1C324R and Cntnap1R765C mutant proteins are retained in the neuronal soma, most likely in the ER, and are unable to be properly transported out of the neuron, unlike the normal Cntnap1 protein, whose levels are very low within the neuronal soma but is localized at the paranodal domains of myelinated axons.
Figure 7. Cntnap1C324R and Cntnap1R765C mutant proteins remain in the neuronal soma and show reduced binding with contactin.
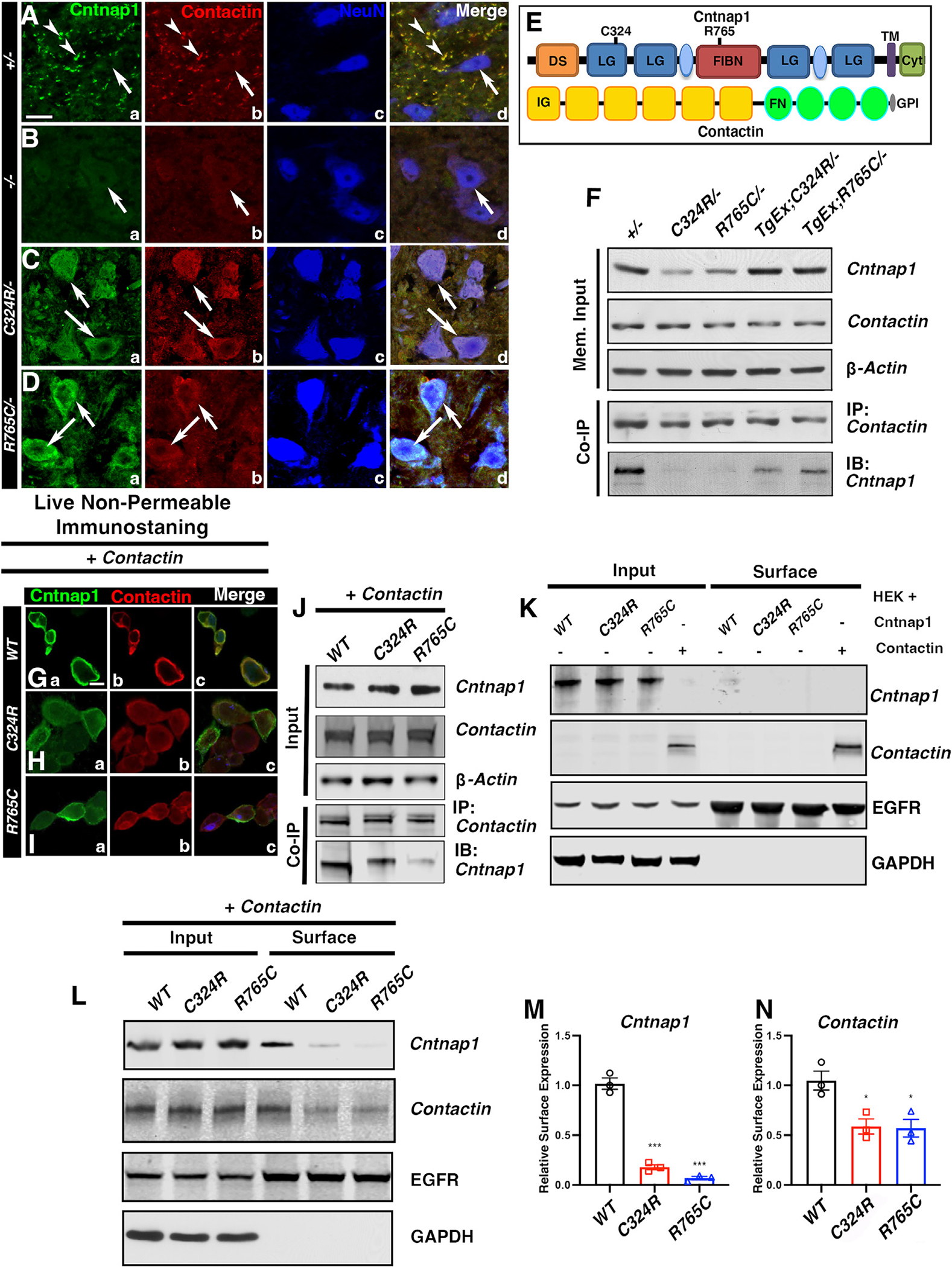
(A–D) Immunostaining of spinal cord cross-sections. Scale bar: 5 μm.
(E) Schematic showing protein domains in Cntnap1 and Contactin.
(F) Co-immunoprecipitation from spinal cord lysates.
(G–I) Representative immunostaining images of Cntnap1 and Contactin co-staining in live non-permeable co-transfected HEK cells. Scale bar: 5 μm.
(J) Co-immunoprecipitation of Cntnap1 and Contactin from HEK cells.
(K) Cell surface/membrane expression of Cntnap1 and Contactin in HEK cells.
(L–N) Cell surface/membrane expression of Cntnap1 (WT) or mutant C324R or R765C in HEK cells co-transfected with cDNAs for Cntnap1 (WT) or Cntnap1 mutants and Contactin. EGFR was used as the cell surface protein marker, and GAPDH was used as negative control for cell surface expression. Data are represented as the mean ± SEM of three independent experiments.
The Cntnap1 protein at the paranodal axo-glial junctions on the axonal membrane side is believed to exist in a cis complex with the paranodal Contactin (schematic in Figure 7E) forming the critical axo-glial junctional complex between the axon and the myelin paranodal loops.4,5,7 To determine if Cntnap1C324R and Cntnap1R765C mutations had any deleterious effects on inter-protein interactions and protein-protein complex formation between Cntnap1 and Contactin, we carried out immunoprecipitations (IPs) using membrane-enriched protein preparations from spinal cords (Figure 7F). This showed the relative levels of Cntnap1 were low in C324R/− and R765C/− lysates compared to other genotypes. The Contactin protein levels were unaffected in C324R/− and R765C/− mutants, and the relative Contactin levels were similar to control +/− mice. The solubilized membrane preparations were immunoprecipitated with anti-Contactin antibodies followed by immunoblotting against Cntnap1. The levels of immunoprecipitated Cntnap1 were significantly low in C324R/− and R765C/− preparations indicating that the binding between mutant Cntnap1C324R and Cntnap1R765C proteins and Contactin was severely affected (Figure 7F). These IP data suggest that C324R and R765C amino acid changes affected protein-protein interactions between Cntnap1 and Contactin.
Next, we obtained human Cntnap1 expression construct (pCMV-Cntnap1FLAG [MR223061]) from OriGene and generated independent constructs of Cntnap1C324R and Cntnap1R765C mutations using site-directed mutagenesis. After confirming the mutations, the wild-type Cntnap1, Cntnap1C324R or Cntnap1R765C cDNAs, and Contactin cDNA (MG50933-CF, SinoBiological) were co-transfected into HEK cells. We first examined Contactin surface expression by immunostaining of live, non-permeabilized HEK cells followed by fixation and permeabilization and immunostaining with an anti-Cntnap1 antibody to detect the expression of Cntnap1. The wild-type Cntnap1 protein was readily localized at the cell surface of the HEK cells; however, the surface localization of Cntnap1C324R and Cntnap1R765C mutant proteins was strongly affected (Figures 7G–7I). Similarly, the Contactin protein surface expression was robust in the presence of wild-type Cntnap1 as visualized by anti-Contactin-FC immunostaining53; in HEK cells co-transfected with either Cntnap1C324R or Cntnap1R765C constructs, the surface expression of Contactin was dramatically reduced. The Cntnap1 protein expression was mostly cytosolic in both Cntnap1C324R or Cntnap1R765C transfected HEK cells as was the Contactin localization. These data strongly indicate that Cntnap1C324R or Cntnap1R765C mutant proteins are unable to co-transport with Contactin to the cell surface and are mostly retained in the cytoplasm possibly ER, as is also observed in vivo in the spinal cord neurons (Figures 7C and 7D). We also carried out IP of cell lysates from HEK cells that co-expressed the wild-type Cntnap1, Cntnap1C324R and Cntnap1R765C proteins, and Contactin. The expression levels of these proteins were similar in HEK cells transfected with respective expression constructs. As shown in Figure 7J, coIP using anti-Contactin antibodies showed reduced levels of Cntnap1 in Cntnap1C324R (60% compared to Cntnap1) or Cntnap1R765C (10% compared to Cntnap1) co-transfected HEK cell lysates, further demonstrating that Cntnap1C324R and Cntnap1R765C mutant proteins fail to bind efficiently with Contactin compared to the wild-type Cntnap1.
Surface biotinylation assay showed surface expression of wild-type Cntnap1 and Cntnap1C324R or Cntnap1R765C mutant proteins was severely impaired when expressed without Contactin; however, Contactin was expressed at the cell surface in the absence of Cntnap1 (Figure 7K). These data demonstrate that in the absence of Contactin, the Cntnap1 protein is unable to get transported to the cell surface indicating that Contactin is necessary for cell surface expression of Cntnap1, which is consistent with previous in vivo studies in the Contactin-null mutants.5 Next, we examined the surface expression of Cntnap1 or Contactin when the wild-type Cntnap1, Cntnap1C324R, or Cntnap1R765C were transfected into HEK cells along with Contactin (Figure 7L). There is ~80% reduction in Cntnap1C324R surface protein levels and ~90% reduction of surface protein expression for Cntnap1R765C protein when compared to surface expression of the wild-type Cntnap1 protein (Figures 7L and 7M). Interestingly, the surface expression of Contactin was also reduced (~50%) in Cntnap1C324R and Cntnap1R765C mutant co-expressed cells compared to wild-type Cntnap1/Contactin co-expression (Figures 7L and 7N). Together, these data demonstrate that Cntnap1C324R and Cntnap1R765C mutations disrupt the protein-protein interactions between Cntnap1 and Contactin, which also leads to defective co-transport and reduced cell surface expression of Cntnap1C324R and Cntnap1R765C mutant proteins.
DISCUSSION
Since the late 1800s, a large spectrum of hereditary neuropathies have been identified, some of which display brain disorders with progressive muscle weakening,21,22,54–56 and others display childhood hypomyelinating neuropathies (CHNs), which have been linked to mutations in several genes associated with myelination.54,57,58 CNTNAP1 mutations in human diseases are associated with CHN. Now, with the rapid development of next-generation sequencing, over 40 cases in individuals from diverse backgrounds (American, Chinese, English, French, Irish, and Palestinian) around the globe have been identified. All presented with hypomyelination and hypotonia, consistent with phenotypes observed in Cntnap1-null mutant mice.4 These rare disorders demonstrate an autosomal recessive pattern of inheritance, whereby an individual must inherit two mutated alleles of CNTNAP1 to manifest disease. Homozygosity or compound heterozygosity for null mutations, such as nonsense, frameshift, or splice site variants in CNTNAP1, are implicated in pathogenesis. Parents who are carriers of a single mutant allele are typically unaffected.32,59 Most cases reported have occurred in the offspring of consanguineous parents, which confers an increased risk for autosomal recessive conditions due to a higher probability of inheriting identical mutant alleles from a common ancestor.32 Delineation of CNTNAP1 mutation status provides insight into diagnosis, prognosis, and recurrence risk for this spectrum of autosomal recessive hypomyelinating disorders.
Here we report the generation and characterization and rescue of two mouse models each harboring a single nucleotide mutation in the mouse Cntnap1 gene, C324R/− and R765C/− representing two human CNTNAP1 mutations. Our studies provide proof of concept of utilizing the wild-type copy of Cntnap1 to provide potential treatment options for human CNTNAP1 genetic deficiency-associated neurological disorders.
Protein stability and transport defects in C324R/− and R765C/− mutants
Phenotypic analysis of C324R/− and R765C/− mutants revealed major defects in the paranodal domain organization even though the mutant proteins are expressed. Our biochemical analyses revealed a marked reduction in the amount of C324R/− and R765C/− mutant proteins at the paranodes in the peripheral and central myelinated axons; and in the CNS, these proteins were essentially undetectable at the paranodes. These observations suggest that the functional deficits and the transport mechanisms may be distinct between the PNS and CNS myelinated axons. Both mutant proteins were retained in the soma of motor neurons as well as the DRG neurons, unlike the wild-type Cntnap1, indicating that these mutant proteins fail to either get properly sorted through the protein transport system and/or are primarily retailed in the ER due to potential misfolding as a result of these amino acid substitutions.
Further in vitro biochemical analysis with co-expression of Cntnap1, Cntnap1C324R, and Cntnap1R765C with Contactin in human HEK cells revealed that the surface expression of the mutant proteins is compromised. Biotin-labeling and pull-down assays revealed that the surface expression of Contactin protein was barely detectable in HEK cells expressing Cntnap1R765C and Contactin, while Cntnap1C324R still maintained detectable surface expression levels of Contactin. Together, our findings on the Cntnap1C324R and Cntnap1R765C mutant proteins indicate that distinct amino acid substitutions in Cntnap1 lead to unique deleterious effects on protein folding, stability, and functionality, and that these changed properties have an impact on proper protein transport to the cell surface. What causes retention of the mutant Cntnap1 proteins and Contactin in these mutants will be addressed as part of our future investigations.
Relevance to human disease and potential therapies
Several mouse and human studies related to axonal domain disorganization have been linked to various neurological diseases with auditory, motor, and nerve conduction impairments. Our study has designed mouse models carrying the human CNTNAP1 mutations, which can be utilized for further clinical studies. These humanized Cntnap1 mouse models showed similar detrimental changes in the structure and function of the axonal domains, affecting the electrical and physiological properties of myelinated axons in both the CNS and PNS, and resulting in severe ataxia and paralysis. Our studies demonstrate that restoring the functional axonal domain organization in Cntnap1 mouse models is possible by re-expressing the wild-type Cntnap1 protein. These rescue strategies could be used to determine the time course of reversing pathologies in human patients carrying CNTNAP1 mutations and provide plausibility to further human studies using viral vector-based strategies with gene therapy. Taken together, our strategies serve as a great starting point for examining the therapeutic potential to alleviate the consequences of human CNTNAP1 mutations-related neurological diseases, and they open avenues to help slow down or cure the genetic diseases related to myelinated axon pathologies.
Limitations of the study
While the current study provides critical insights into the mechanistic impact of human CNTNAP1 mutations, we do find some discrepancies and limitations in the study. Our previous study published more than 2 decades ago reported that Cntnap1−/− mutant mice did not display any significant myelination deficits based on manual estimations of both the myelin thickness and g-ratios,4 which is contradictory to our current results that showed more hypomyelination in the Cntnap1−/− mutants. However, in the current study, we used the latest semi-automated software, MyelTracer.45 This technological progression not only reduces human errors but also permits the assessment of axonal myelination at a significantly larger and more detailed level, which has gained widespread recognition in recent studies.60–66 Using this method, our analyses consistently revealed hypomyelination in Cntnap1C324R, Cntnap1R765C, and Cntnap1−/− mutant mice to varying degrees with Cntnap1−/− mutants displaying the most severe phenotype. Manual analyses also revealed an increase in unmyelinated axons in Cntnap1C324R, Cntnap1R765C, and Cntnap1−/− mutant mice, and these phenotypes were rescued by neuronal expression of the wild-type Cntnap1 transgene. The increase in unmyelinated axons in Cntnap1−/− mutant mice reported in this study is consistent with recently reported findings.67 Furthermore, ultrastructural analysis of sural nerves from human subjects carrying CNTNAP1 mutations also revealed hypomyelination.33,36,43 Collectively, these observations indicate that Cntnap1 does indeed play a role in ensuring proper myelination. Future studies should focus on establishing the role of Cntnap1 and its interacting proteins in developmental myelination as well as in adult myelin formation and/or maintenance, as the outcomes of these studies will be highly relevant to human CNTNAP1-associated hypomyelinating neuropathies.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Manzoor Bhat (bhatm@uthscsa.edu).
Materials availability
All mouse lines and DNA constructs generated in this study are listed in Key resource table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit anti-Cntnap1 | Bhat et al.4 | N/A |
| Guinea Pig anti-Cntnap1 | Bhat et al.4 | N/A |
| Rat anti- NFCT | Pillai et al.7 | N/A |
| Guinea Pig anti- NF186 | Thaxton et al.10 | N/A |
| Rabbit anti- βIV Spectrin | Saifetiarova et al.11 | N/A |
| Rabbit anti-AnkG | Saifetiarova et al.12 | N/A |
| Rabbit anti-Neurofascin155 | Cell Signaling Tech. | Cat# 15035, RRID:AB_2798693 |
| Mouse anti- pan NaV Channels | Sigma-Aldrich | Cat# S8809, RRID:AB_477552 |
| Mouse anti- KV1.2 | NeuroMab | Cat# K14/16, RRID:AB_2877295 |
| Mouse anti-NeuN | Sigma-Aldrich | Cat# MAB377, RRID:AB_2298772 |
| Mouse anti-β-actin | Sigma-Aldrich | Cat# A2228, RRID:AB_476697 |
| Rabbit anti-Flag | Cell Signaling Tech. | Cat# 14793, RRID:AB_2572291 |
| Mouse anti-Flag | Sigma-Aldrich | Cat# F-1804, RRID:AB_262044 |
| Rabbit anti-Contactin-Fc | Generously provided by the laboratory of Dr. James L. Salzer | N/A |
|
| ||
| DNA constructs | ||
|
| ||
| Mouse Cntnap1 ORF clone | OriGene, USA | MR223061 |
| Mouse Cntnap1 C324R clone | This manuscript | N/A |
| Mouse Cntn1 ORF clone | SinoBiological, China | MG50933-CF |
| Mouse Cntnap1 R765C clone | This manuscript | N/A |
| LSL-GAG-Cntnap1Flag | This manuscript | N/A |
|
| ||
| Mouse lines | ||
|
| ||
| Cntnapl KO mice | Bhat et al.4 | N/A |
| Cntnap1C324R mice | This manuscript | N/A |
| Cntnap1R765C mice | This manuscript | N/A |
| LSL-Cntnap1Flag | This manuscript | N/A |
| Slick-H-CreERT2 | Jackson Laboratory | RRID:IMSR_JAX:012708 |
|
| ||
| Software | ||
|
| ||
| ImageJ | NIH | |
| Graphpad Prism 9.0 | Dotmatics | |
| MyelTracer | Kaiser et al.45 | N/A |
|
| ||
| Commercial kits | ||
|
| ||
| Cell Surface Protein Isolation Kit | Pierce™ at ThermoFisher | Cat# 89881 |
Data and code availability
All data, including mouse lines, DNA constructs and original western blot and confocal images, reported in this paper can be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cntnap1 mouse mutants
All transgenic mice described here, except the ones purchased from Jackson laboratory, were generated in the Mouse Genome Engineering and Transgenic Facility at UT Health Science Center in San Antonio. Cntnap1 knockout mice have been reported previously.4 To introduce the Cysteine 324 to Arginine (C324R) and Arginine 765 to Cysteine (R765C) mutations in the mouse Cntnap1 single guide (sg) RNAs 5′-GCCTACCGCCATAACTTCCG-3’ (for C324R) or 5′-CTCAAATTCTGAAGCTCAGT-3’ (for R765C) were selected with minimum off-target effects (designed by CRISPR). 198 base pairs (bp) ssDNA donors with sequences changing TGC>CGC (C324R) or CGC>TGC (R765C) were used along with guide RNAs and Cas9 protein complex for pronuclear microinjection (All gRNA and donor DNA sequences were listed in Table S1). The pups were genotyped and screened by Sanger sequencing (Eurofins, Inc). The entire Cntnap1 cDNAs from each mouse mutant were fully sequenced to ensure no additional mutation/s were created in the Cntnap1 gene.
LoxP-stop-LoxP-Cntnap1 transgenic mice
To generate an inducible tissue-specific overexpression of wild-type Cntnap1, we first generated LoxP-Stop-LoxP-Cntnap1Flag. This construct was linearized with the NheI enzyme and micro-injected into pronuclei. The offspring were screened by using the primers in the Cntnap1 locus that differentiated the endogenous Cntnap1 from the transgenic Cntnap1. This mouse strain is referred to as LSL-Cntnap1. LSL-Cntnap1 was then crossed with SLICK-H-CreERT2 (JAX:012708) to generate SLICK-H-CreERT2;LSL-Cntnap1 mice. To enable the expression of the Cntnap1 transgene in neurons, tamoxifen (MP Biomedicals) at 1 mg/μL was delivered by intraperitoneal (i.p.) injections for 5 consecutive days at postnatal (P)-21 at a dose of 1 mg/12.5 g body weight. At the time of experiments, age-matched control, SLICK-H-CreERT2;LSL-Cntnap1, C324R/− and R765C/− and a combination of appropriate genotype littermates were evaluated by electrophysiological, immunohistochemical, biochemical, and ultrastructural techniques. All animal experiments were performed according to NIH, and Institutional Animal Care and Use Committee approved guidelines for the ethical treatment of laboratory animals at the University of Texas Health Science Center, San Antonio.
METHOD DETAILS
DNA constructs and cell transfections
Wild-type mouse Cntnap1 cDNA plasmid was purchased from OriGene (MR223061); and Contactin cDNA cloning plasmid was obtained from SinoBiological (China) (MG50933-CF). Cntnap1 C324R or R765C mutant forms were generated by using a QuikChange site-directed mutagenesis kit (Stratagene, CA). All DNA primers used are listed in Table S2. The presence of TGC>CGC (for C324R) or CGC>TGC (for R765C) mutations in Cntnap1 and the full Cntnap1 cDNA were verified by full coding sequence sequencing (Eurofins Genomics LLC). HEK cells for Cntnap1 expression were obtained from ATCC and maintained in a humid 5% CO2, 37°C incubator with high glucose DMEM containing 10% fetal bovine serum (FBS) and antibiotics. DNA constructs were transiently transfected into HEK cells using Lipofectamine 2000 (Invitrogen) 24 h after plating of the cells on 60-mm dishes (collecting cells for western blotting) or 4-chamber slides (for immunostaining). Then, the cells were ready for downstream experiments 48–72 h after transfection.68
Antibodies and other reagents
All chemicals and reagents were purchased from Sigma-Aldrich unless otherwise specified. All antibodies used in this study are listed in the Key resources table. Conjugated-secondary antibodies for immunostaining (Alexa Fluor 488, 568, 647) were purchased from Invitrogen (USA).
Rotarod test
Motor function performance was assessed using the Rotarod apparatus (Ugo Basile) (Steele et al., 1998). At least 6 mice per genotype and both male and female mice were included for motor performance studies. Mice were trained on the Rotarod apparatus at 5 RPM for 5 min for three consecutive days. For testing, the speed was gradually accelerated to 25 RPM or 35 RPM over 2 min. Latency to fall was recorded for each trial. Each mouse went through three trials. Results are shown as a mean of the three trials ±SEM.
Beam-walking test
This test was used for the assessment of fine motor coordination, particularly of the hindlimb. Firstly, animals were placed on a round beam (50 cm long, 1.5 cm diameter) that is secured over a tray and allowed to walk across the beam from one end to the other for at least three times. This training step can be useful to achieve a stable baseline measurement (see the videos attached). The time taken to cross the beam was recorded for each trial.
Tissue preparation and immunostaining
Animals were anesthetized with i.p. injection of Avertin (400 mg/kg mouse body weight) and transcardially perfused with phosphate-buffered saline (PBS) (pH 7.2–7.4). The cervical region of the spinal cord (SC) was dissected and postfixed in 4% paraformaldehyde (PFA) overnight at 4°C and then immersed in 30% sucrose in 0.1M phosphate buffer until they settled to the bottom. The tissue was rinsed several times in PBS and frozen in Tissue-Tek O.C.T. Compound (Sakura Finetek USA, Inc). Longitudinal 20 μm sections for SCs were cut with a cryostat (Leica), mounted on slides, and processed for immunostaining. For the peripheral nerves, proximal sciatic nerves (SNs) were dissected out from anesthetized animals, fixed in 4% PFA for 30 min, washed in PBS and teased into individual nerve fibers, dried overnight at room temperature, and stored at −80°C. Samples received primary antibodies at room temp overnight, followed by washing and secondary antibodies for 1 h at room temperature.4,11,68,69
Chemical labeling of surface proteins in HEK cells
The expression of plasma membrane proteins on HEK cells was analyzed with cell surface protein extraction kit. Briefly, Sulfo-NHS-SS-Biotin was first dissolved in 0.1% DMSO in PBS. Forty-eight hours after transfection, HEK cells were washed three times with PBS (with Ca2+ and Mg2+) and then treated with 2 mM Sulfo-NHS-SS-Biotin/PBS for 30 min at 4°C. The cells were then collected in chilled lysis buffer containing protease inhibitors and kept on ice for 1 h. The total protein concentration was determined by MicroBCA kit (Pierce), and equal amounts of total protein were mixed with Streptavidin beads for 1 h at room temperature. Biotinylated proteins were eluted from Streptavidin beads using 100 μL of eluate buffer after 3x washes with washing buffer. The eluted sample was immunoblotted by anti-Cntnap1, and anti-Contactin antibodies. Anti-EGFP or GAPDH were served as a surface protein marker or loading control.
Image analysis
Confocal images were acquired with a Zeiss LSM 710 Microscope.11,70 Briefly, identical settings were used to capture images from the control and all mutant samples, and the images shown are maximal-intensity projections from 5 to 8 Z-stacks with 0.25-μm intervals. For the quantification of nodal intensities, z stack images were used to quantify intensities for each genotype, and a minimum of 50 nodes for each nodal marker per sample were quantified.
Immunoblotting
SCs and SNs were harvested and stored at −80°C until processed. Tissues were homogenized using glass mortar and pestle in ice-cold lysis buffer with protease inhibitors (#A32953, Thermo Scientific). The lysates were incubated for 30 min on ice and then centrifuged at 10,000 × g for 30 min at 4°C. Supernatants were collected, and protein concentrations were estimated using a Pierce BCA protein assay Kit (Thermo Scientific). Equal amounts of protein were resolved by 4–12% SDS-PAGE and transferred onto nitrocellulose membranes. The membranes were blocked in 10% non-fat dry milk for 1 h followed by incubation with appropriate primary antibodies in TBST overnight at 4°C. After three-time washes in TBST, membranes were incubated with IR-conjugated secondary antibodies for 1 h and scanned using Li-COR Odyssey infrared imager. The intensities of immunoblot bands were quantified using ImageJ software (RRID:SCR_003070; NIH) and normalized to β-Actin as the loading control.
In vivo nerve conduction recordings
Animals were anesthetized by continuous isoflurane (5% aerosolized) for in vivo Nerve Conduction Velocity (NCV) recordings using a Nicolet Teca Synergy system (Natus Neurology Inc., Middleton, WI). In order to maintain body temperature to 33°C–34°C during recordings, a warming lamp was kept above mice. NCVs and amplitudes were recorded from the tail and SN of anesthetized mice.70,71 For SN recording, two separate recordings were made in the dorsum of the foot with a stimulus first given at the ankle (0.02ms, 2mA) and secondly at the sciatic notch (0.02ms, 8mA). Once traces were acquired, NCV was calculated by the distance divided by the latency and amplitude was measured as the height of the peak. For sciatic NCV, the distance between the notch and ankle was divided by the latency between the notch and ankle.
Transmission electron microscopy
For transmission electron microscopy (TEM), all tissue were processed using freshly prepared solutions on the day of perfusion.71,72 In brief, animals were anesthetized and transcardially perfused with normal saline followed by 2.5%glutaraldehyde/4% paraformaldehyde EM fixative (dissolved in 0.16 M NaH2PO4/0.11M NaOH buffer, pH 7.2–7.4) for 30 min. After perfusion, entire mouse carcasses were post-fixed for at least 1 week in the same EM fixative. SNs, SCs were dissected out and incubated overnight in 0.1M sodium cacodylate buffer followed by incubation in 2% OsO4 solution and gradient ethanol dehydration. Samples were incubated in propylene oxide, left in 100% PolyBed resin for 36 h and embedded in flat molds at 55°C for 36 h. After embedding, the molds were processed and imaged on a JEOL 1230 electron microscope at the UTHSCSA Electron Microscopy Lab. TEM Images were analyzed by ImageJ software (NIH).
g-ratios of myelinated axons
The g-ratios of SNs and SCs myelinated fibers from TEM images were measured by MyelTracer software.45 At least 20 images per animal taken at 5600× magnification were used for the g-ratio measurement using at least 150 axons in 3 independent mice for each genotype. The g-ratio was measured as the ratio of the inner axonal radius to the outer fiber radius with the myelin sheath.
Co-immunoprecipitation assay
Mouse brains were homogenized in IP lysis buffer (50 mM Tris−HCl pH 7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 5% glycerol with proteinase inhibitors).18 Nuclei and cell debris were removed by low-speed centrifugation (3,000 × g for 10 min at 4°C). The supernatants were centrifuged at 400,000 × g for 60 min at 4°C to obtain the microsomal membrane protein fraction. The membrane pellets were solubilized in Triton X-100 lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton x-100, 5 mM EDTA). After clearing at 10,000 × g for 15min, the membrane fraction supernatants were incubated with desired antibody and Protein A/G beads (Santa Cruz, sc-2003) overnight mixing at 4°C. The beads were washed three times with Trion X-100 lysis buffer the next day, and all binding proteins were eluted with 2x SDS PAGE sample buffer. The membrane input and eluted fractions were subjected to immunoblotting assays.
For in vitro cell experiments, total cell lysates were prepared in IP lysis buffer as described above and only cleaned at 13,000 × g for 30 min at 4°C. Then, the supernatant was incubated with desired antibody and Protein A/G beads overnight and processed as described for brain tissues.
QUANTIFICATION AND STATISTICAL ANALYSIS
For immunostaining, tissues were processed from 3 mice per group per time point, then 50–70 nodes from PNS and CNS were quantified per animal. For immunoblotting, tissues were processed and quantified from 3 mice per group at the terminal time point. For ultrastructure analyses, tissues were processed from 3 mice per group, and for each mouse, a minimum of 200 axons for SN and SC were imaged. Data measurements and analysis were performed by one examiner in a non-blinded manner. All data are presented as mean ± S.E.M. n represents the number of animals (unless otherwise stated statistically significant differences between control and mutant groups were determined by 1- or two-way ANOVA with Tukey’s post hoc test using GraphPad Prism software and are represented by *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001. All p values are provided in the Table S3.
Supplementary Material
Highlights.
Mouse models of human CNTNAP1 mutations display deficits in nerve conduction motor function
Cntnap1 C324R and R765C mutants show hypomyelination and paranodal ultrastructural defects
Cntnap1 C324R and R765C proteins have diminished stability and are retained in neuronal soma
Neuronal Cntnap1 expression rescues neurological deficits, opening avenues for gene therapy
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health/National Institute of General Medical Sciences grant GM063074, the National Multiple Sclerosis Society grant RG-2001–36023, the Doran Family Foundation, the Hereditary Neuropathy Foundation, and the Zachry Foundation Endowment for advancing neuroscience research. We thank all families who willingly shared the CNTNAP1 genetic information for research purposes. We also thank members of the Bhat lab for input and helpful discussions. We also thank J. Salzer for anti-Contactin antibody. All mouse lines carrying single nucleotide changes in Cntnap1 and Lox-stop-Lox-Cntnap1 transgene were generated by the Mouse Genome Engineering and Transgenic Facility in the Department of Cellular and Integrative Phsyiology at UT Health San Antonio. All electron microscopy was performed at the University of Texas Health Science Center at San Antonio. The graphical abstract was created using BioRender.com.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing financial interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113274.
REFERENCES
- 1.Einheber S, Zanazzi G, Ching W, Scherer S, Milner TA, Peles E, and Salzer JL (1997). The axonal membrane protein Caspr, a homologue of neurexin IV, is a component of the septate-like paranodal junctions that assemble during myelination. J. Cell Biol. 139, 1495–1506. 10.1083/jcb.139.6.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhat MA (2003). Molecular organization of axo-glial junctions. Curr. Opin. Neurobiol. 13, 552–559. 10.1016/j.conb.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 3.Buttermore ED, Thaxton CL, and Bhat MA (2013). Organization and maintenance of molecular domains in myelinated axons. J. Neurosci. Res. 91, 603–622. 10.1002/jnr.23197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhat MA, Rios JC, Lu Y, Garcia-Fresco GP, Ching W, St Martin M, Li J, Einheber S, Chesler M, Rosenbluth J, et al. (2001). Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron 30, 369–383. 10.1016/s0896-6273(01)00294-x. [DOI] [PubMed] [Google Scholar]
- 5.Boyle ME, Berglund EO, Murai KK, Weber L, Peles E, and Ranscht B (2001). Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron 30, 385–397. 10.1016/s0896-6273(01)00296-3. [DOI] [PubMed] [Google Scholar]
- 6.Sherman DL, Tait S, Melrose S, Johnson R, Zonta B, Court FA, Macklin WB, Meek S, Smith AJH, Cottrell DF, and Brophy PJ (2005). Neurofascins are required to establish axonal domains for saltatory conduction. Neuron 48, 737–742. 10.1016/j.neuron.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 7.Pillai AM, Thaxton C, Pribisko AL, Cheng JG, Dupree JL, and Bhat MA (2009). Spatiotemporal ablation of myelinating glia-specific neurofascin (Nfasc NF155) in mice reveals gradual loss of paranodal axo-glial junctions and concomitant disorganization of axonal domains. J. Neurosci. Res. 87, 1773–1793. 10.1002/jnr.22015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thaxton C, and Bhat MA (2009). Myelination and regional domain differentiation of the axon. Results Probl. Cell Differ. 48, 1–28. 10.1007/400_2009_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thaxton C, Pillai AM, Pribisko AL, Dupree JL, and Bhat MA (2011). Nodes of Ranvier act as barriers to restrict invasion of flanking paranodal domains in myelinated axons. Neuron 69, 244–257. 10.1016/j.neuron.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thaxton C, Pillai AM, Pribisko AL, Labasque M, Dupree JL, Faivre-Sarrailh C, and Bhat MA (2010). In vivo deletion of immunoglobulin domains 5 and 6 in neurofascin (Nfasc) reveals domain-specific requirements in myelinated axons. J. Neurosci. 30, 4868–4876. 10.1523/JNEUROSCI.5951-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saifetiarova J, Shi Q, Paukert M, Komada M, and Bhat MA (2018). Reorganization of Destabilized Nodes of Ranvier in betaIV Spectrin Mutants Uncovers Critical Timelines for Nodal Restoration and Prevention of Motor Paresis. J. Neurosci. 38, 6267–6282. 10.1523/JNEUROSCI.0515-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saifetiarova J, Taylor AM, and Bhat MA (2017). Early and Late Loss of the Cytoskeletal Scaffolding Protein, Ankyrin G Reveals Its Role in Maturation and Maintenance of Nodes of Ranvier in Myelinated Axons. J. Neurosci. 37, 2524–2538. 10.1523/JNEUROSCI.2661-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dupree JL, Coetzee T, Blight A, Suzuki K, and Popko B (1998). Myelin galactolipids are essential for proper node of Ranvier formation in the CNS. J. Neurosci. 18, 1642–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dupree JL, Coetzee T, Suzuki K, and Popko B (1998). Myelin abnormalities in mice deficient in galactocerebroside and sulfatide. J. Neurocytol. 27, 649–659. 10.1023/a:1006908013972. [DOI] [PubMed] [Google Scholar]
- 15.Dupree JL, Suzuki K, and Popko B (1998). Galactolipids in the formation and function of the myelin sheath. Microsc. Res. Tech. 41, 431–440. . [DOI] [PubMed] [Google Scholar]
- 16.Gollan L, Salomon D, Salzer JL, and Peles E (2003). Caspr regulates the processing of contactin and inhibits its binding to neurofascin. J. Cell Biol. 163, 1213–1218. 10.1083/jcb.200309147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Einheber S, Bhat MA, and Salzer JL (2006). Disrupted axo-glial junctions result in accumulation of abnormal mitochondria at nodes of ranvier. Neuron Glia Biol. 2, 165–174. 10.1017/S1740925X06000275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcia-Fresco GP, Sousa AD, Pillai AM, Moy SS, Crawley JN, Tessarollo L, Dupree JL, and Bhat MA (2006). Disruption of axo-glial junctions causes cytoskeletal disorganization and degeneration of Purkinje neuron axons. Proc. Natl. Acad. Sci. USA. 103, 5137–5142. 10.1073/pnas.0601082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun XY, Takagishi Y, Okabe E, Chishima Y, Kanou Y, Murase S, Mizumura K, Inaba M, Komatsu Y, Hayashi Y, et al. (2009). A novel Caspr mutation causes the shambling mouse phenotype by disrupting axoglial interactions of myelinated nerves. J. Neuropathol. Exp. Neurol. 68, 1207–1218. 10.1097/NEN.0b013e3181be2e96. [DOI] [PubMed] [Google Scholar]
- 20.Takagishi Y, Katanosaka K, Mizoguchi H, and Murata Y (2016). Disrupted axon-glia interactions at the paranode in myelinated nerves cause axonal degeneration and neuronal cell death in the aged Caspr mutant mouse shambling. Neurobiol. Aging 43, 34–46. 10.1016/j.neurobiolaging.2016.03.020. [DOI] [PubMed] [Google Scholar]
- 21.Tooth HH (1886). The Peroneal Type of Progressive Muscular Atrophy (University of Cambridge; ). [Google Scholar]
- 22.Charcot JM (1886). Sur une forme paticuliere d’atrophie musuculaire progressive souvent familial, debutante par les pieds et les jambes et atteignant plus tard les mains. Rev. Med Fr 6, 97–138. [Google Scholar]
- 23.Sahenk Z (1999). Abnormal Schwann cell-axon interactions in CMT neuropathies. The effects of mutant Schwann cells on the axonal cytoskeleton and regeneration-associated myelination. Ann. N. Y. Acad. Sci. 883, 415–426. [PubMed] [Google Scholar]
- 24.Sahenk Z, and Mendell JR (1999). Alterations in nodes of Ranvier and Schmidt-Lanterman incisures in Charcot-Marie-Tooth neuropathies. Ann. N. Y. Acad. Sci. 883, 508–512. [PubMed] [Google Scholar]
- 25.Wolswijk G, and Balesar R (2003). Changes in the expression and localization of the paranodal protein Caspr on axons in chronic multiple sclerosis. Brain 126, 1638–1649. 10.1093/brain/awg151. [DOI] [PubMed] [Google Scholar]
- 26.Coman I, Aigrot MS, Seilhean D, Reynolds R, Girault JA, Zalc B, and Lubetzki C (2006). Nodal, paranodal and juxtaparanodal axonal proteins during demyelination and remyelination in multiple sclerosis. Brain 129, 3186–3195. 10.1093/brain/awl144. [DOI] [PubMed] [Google Scholar]
- 27.Bonnon C, Goutebroze L, Denisenko-Nehrbass N, Girault JA, and Faivre-Sarrailh C (2003). The paranodal complex of F3/contactin and caspr/paranodin traffics to the cell surface via a non-conventional pathway. J. Biol. Chem. 278, 48339–48347. 10.1074/jbc.M309120200. [DOI] [PubMed] [Google Scholar]
- 28.Bonnon C, Bel C, Goutebroze L, Maigret B, Girault JA, and Faivre-Sarrailh C (2007). PGY repeats and N-glycans govern the trafficking of paranodin and its selective association with contactin and neurofascin-155. Mol. Biol. Cell 18, 229–241. 10.1091/mbc.e06-06-0570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Charles P, Tait S, Faivre-Sarrailh C, Barbin G, Gunn-Moore F, Denisenko-Nehrbass N, Guennoc AM, Girault JA, Brophy PJ, and Lubetzki C (2002). Neurofascin is a glial receptor for the paranodin/Caspr-contactin axonal complex at the axoglial junction. Curr. Biol. 12, 217–220. 10.1016/s0960-9822(01)00680-7. [DOI] [PubMed] [Google Scholar]
- 30.Peles E, Nativ M, Lustig M, Grumet M, Schilling J, Martinez R, Plowman GD, and Schlessinger J (1997). Identification of a novel contactin-associated transmembrane receptor with multiple domains implicated in protein-protein interactions. EMBO J. 16, 978–988. 10.1093/emboj/16.5.978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laquérriere A, Maluenda J, Camus A, Fontenas L, Dieterich K, Nolent F, Zhou J, Monnier N, Latour P, Gentil D, et al. (2014). Mutations in CNTNAP1 and ADCY6 are responsible for severe arthrogryposis multiplex congenita with axoglial defects. Hum. Mol. Genet. 23, 2279–2289. 10.1093/hmg/ddt618. [DOI] [PubMed] [Google Scholar]
- 32.Sabbagh S, Antoun S, and Mégarbané A (2020). CNTNAP1 Mutations and Their Clinical Presentations: New Case Report and Systematic Review. Case Rep. Med. 2020, 8795607. 10.1155/2020/8795607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vallat JM, Nizon M, Magee A, Isidor B, Magy L, Péréon Y, Richard L, Ouvrier R, Cogné B, Devaux J, et al. (2016). Contactin-Associated Protein 1 (CNTNAP1) Mutations Induce Characteristic Lesions of the Paranodal Region. J. Neuropathol. Exp. Neurol. 75, 1155–1159. 10.1093/jnen/nlw093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mehta P, Küspert M, Bale T, Brownstein CA, Towne MC, De Girolami U, Shi J, Beggs AH, Darras BT, Wegner M, et al. (2017). Novel mutation in CNTNAP1 results in congenital hypomyelinating neuropathy. Muscle Nerve 55, 761–765. 10.1002/mus.25416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Conant A, Curiel J, Pizzino A, Sabetrasekh P, Murphy J, Bloom M, Evans SH, Helman G, Taft RJ, Simons C, et al. (2018). Absence of Axoglial Paranodal Junctions in a Child With CNTNAP1 Mutations, Hypomyelination, and Arthrogryposis. J. Child Neurol. 33, 642–650. 10.1177/0883073818776157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nizon M, Cogne B, Vallat JM, Joubert M, Liet JM, Simon L, Vincent M, Küry S, Boisseau P, Schmitt S, et al. (2017). Two novel variants in CNTNAP1 in two siblings presenting with congenital hypotonia and hypomyelinating neuropathy. Eur. J. Hum. Genet. 25, 150–152. 10.1038/ejhg.2016.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bellen HJ, Lu Y, Beckstead R, and Bhat MA (1998). Neurexin IV, caspr and paranodin–novel members of the neurexin family: encounters of axons and glia. Trends Neurosci. 21, 444–449. 10.1016/s0166-2236(98)01267-3. [DOI] [PubMed] [Google Scholar]
- 38.Li W, Yang L, Tang C, Liu K, Lu Y, Wang H, Yan K, Qiu Z, and Zhou W (2020). Mutations of CNTNAP1 led to defects in neuronal development. JCI Insight 5, e135697. 10.1172/jci.insight.135697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Letko A, Minor KM, Friedenberg SG, Shelton GD, Salvador JP, Mandigers PJJ, Leegwater PAJ, Winkler PA, Petersen-Jones SM, Stanley BJ, et al. (2020). A CNTNAP1 Missense Variant Is Associated with Canine Laryngeal Paralysis and Polyneuropathy. Genes 11, 1426. 10.3390/genes11121426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lakhani S, Doan R, Almureikhi M, Partlow JN, Al Saffar M, Elsaid MF, Alaaraj N, James Barkovich A, Walsh CA, and Ben-Omran T (2017). Identification of a novel CNTNAP1 mutation causing arthrogryposis multiplex congenita with cerebral and cerebellar atrophy. Eur. J. Med. Genet. 60, 245–249. 10.1016/j.ejmg.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu R, Fu J, Meng L, Lv H, Wang Z, Zhirong J, and Yuan Y (2019). Homozygous splice-site mutation c.78 + 5G>A in PMP22 causes congenital hypomyelinating neuropathy. Neuropathology 39, 441–446. 10.1111/neup.12604. [DOI] [PubMed] [Google Scholar]
- 42.Lesmana H, Vawter Lee M, Hosseini SA, Burrow TA, Hallinan B, Bove K, Schapiro M, and Hopkin RJ (2019). CNTNAP1-Related Congenital Hypomyelinating Neuropathy. Pediatr. Neurol. 93, 43–49. 10.1016/j.pediatrneurol.2018.12.014. [DOI] [PubMed] [Google Scholar]
- 43.Low KJ, Stals K, Caswell R, Wakeling M, Clayton-Smith J, Donaldson A, Foulds N, Norman A, Splitt M, Urankar K, et al. (2018). Phenotype of CNTNAP1: a study of patients demonstrating a specific severe congenital hypomyelinating neuropathy with survival beyond infancy. Eur. J. Hum. Genet. 26, 796–807. 10.1038/s41431-018-0110-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hengel H, Magee A, Mahanjah M, Vallat JM, Ouvrier R, Abu-Rashid M, Mahamid J, Schüle R, Schulze M, Krägeloh-Mann I, et al. (2017). CNTNAP1 mutations cause CNS hypomyelination and neuropathy with or without arthrogryposis. Neurol. Genet. 3, e144. 10.1212/NXG.0000000000000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaiser T, Allen HM, Kwon O, Barak B, Wang J, He Z, Jiang M, and Feng G (2021). MyelTracer: A Semi-Automated Software for Myelin g-Ratio Quantification. eNeuro 8. 10.1523/ENEURO.0558-20.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carofino BL, Ayanga B, and Justice MJ (2013). A mouse model for inducible overexpression of Prdm14 results in rapid-onset and highly penetrant T-cell acute lymphoblastic leukemia (T-ALL). Dis. Model. Mech. 6, 1494–1506. 10.1242/dmm.012575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heimer-McGinn V, and Young P (2011). Efficient inducible Pan-neuronal cre-mediated recombination in SLICK-H transgenic mice. Genesis 49, 942–949. 10.1002/dvg.20777. [DOI] [PubMed] [Google Scholar]
- 48.Taylor AM, Shi Q, and Bhat MA (2018). Simultaneous Ablation of Neuronal Neurofascin and Ankyrin G in Young and Adult Mice Reveals Age-Dependent Increase in Nodal Stability in Myelinated Axons and Differential Effects on the Lifespan. eNeuro 5, ENEURO.0138, 18.2018. 10.1523/ENEURO.0138-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Volpi VG, Touvier T, and D’Antonio M (2016). Endoplasmic Reticulum Protein Quality Control Failure in Myelin Disorders. Front. Mol. Neurosci. 9, 162. 10.3389/fnmol.2016.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ron D, and Walter P (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529. 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 51.Wang S, and Kaufman RJ (2012). The impact of the unfolded protein response on human disease. J. Cell Biol. 197, 857–867. 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Devanathan V, Jakovcevski I, Santuccione A, Li S, Lee HJ, Peles E, Leshchyns’ka I, Sytnyk V, and Schachner M (2010). Cellular form of prion protein inhibits Reelin-mediated shedding of Caspr from the neuronal cell surface to potentiate Caspr-mediated inhibition of neurite outgrowth. J. Neurosci. 30, 9292–9305. 10.1523/JNEUROSCI.5657-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rios JC, Melendez-Vasquez CV, Einheber S, Lustig M, Grumet M, Hemperly J, Peles E, and Salzer JL (2000). Contactin-associated protein (Caspr) and contactin form a complex that is targeted to the paranodal junctions during myelination. J. Neurosci. 20, 8354–8364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scherer SS (2006). Inherited neuropathies: new genes don’t fit old models. Neuron 51, 672–674. 10.1016/j.neuron.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 55.Kramarz C, and Rossor AM (2022). Neurological update: hereditary neuropathies. J. Neurol. 269, 5187–5191. 10.1007/s00415-022-11164-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frasquet M, and Sevilla T (2022). Hereditary motor neuropathies. Curr. Opin. Neurol. 35, 562–570. 10.1097/WCO.0000000000001087. [DOI] [PubMed] [Google Scholar]
- 57.Phillips JP, Warner LE, Lupski JR, and Garg BP (1999). Congenital hypomyelinating neuropathy: two patients with long-term follow-up. Pediatr. Neurol. 20, 226–232. 10.1016/s0887-8994(98)00138-6. [DOI] [PubMed] [Google Scholar]
- 58.Harati Y, and Butler IJ (1985). Congenital hypomyelinating neuropathy. J. Neurol. Neurosurg. Psychiatry 48, 1269–1276. 10.1136/jnnp.48.12.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Garel P, Lesca G, Ville D, Poulat AL, Chatron N, Sanlaville D, Des Portes V, Arzimanoglou A, and Lion-François L (2022). CNTNAP1-encephalopathy: Six novel patients surviving the neonatal period. Eur. J. Paediatr. Neurol. 37, 98–104. 10.1016/j.ejpn.2022.01.015. [DOI] [PubMed] [Google Scholar]
- 60.Takeya H, Itai S, Kimura H, Kurashina Y, Amemiya T, Nagoshi N, Iwamoto T, Sato K, Shibata S, Matsumoto M, et al. (2023). Schwann cell-encapsulated chitosan-collagen hydrogel nerve conduit promotes peripheral nerve regeneration in rodent sciatic nerve defect models. Sci. Rep. 13, 11932. 10.1038/s41598-023-39141-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shepilov D, Osadchenko I, Kovalenko T, Yamada C, Chereshynska A, Smozhanyk K, Ostrovska G, Groppa S, Movila A, and Skibo G (2023). Maternal antibiotic administration during gestation can affect the memory and brain structure in mouse offspring. Front. Cell. Neurosci. 17, 1176676. 10.3389/fncel.2023.1176676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alexandris AS, Lee Y, Lehar M, Alam Z, Samineni P, Tripathi SJ, Ryu J, and Koliatsos VE (2023). Traumatic axonopathy in spinal tracts after impact acceleration head injury: Ultrastructural observations and evidence of SARM1-dependent axonal degeneration. Exp. Neurol. 359, 114252. 10.1016/j.expneurol.2022.114252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schuster KH, Zalon AJ, Zhang H, DiFranco DM, Stec NR, Haque Z, Blumenstein KG, Pierce AM, Guan Y, Paulson HL, and McLoughlin HS (2022). Impaired Oligodendrocyte Maturation Is an Early Feature in SCA3 Disease Pathogenesis. J. Neurosci. 42, 1604–1617. 10.1523/JNEUROSCI.1954-20.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Park HJ, Tsai E, Huang D, Weaver M, Frick L, Alcantara A, Moran JJ, Patzig J, Melendez-Vasquez CV, Crabtree GR, et al. (2022). ACTL6a coordinates axonal caliber recognition and myelination in the peripheral nerve. iScience 25, 104132. 10.1016/j.isci.2022.104132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nguyen NP, Lopez S, Smith CL, Lever TE, Nichols NL, and Bunyak F (2022). Axon and Myelin Sheath Segmentation in Electron Microscopy Images using Meta Learning. IEEE Appl. Imag. Pattern Recognit. Workshop 2022. 10.1109/aipr57179.2022.10092238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McKee KK, and Yurchenco PD (2022). Amelioration of muscle and nerve pathology of Lama2-related dystrophy by AAV9-laminin-alphaLN linker protein. JCI Insight 7, e158397. 10.1172/jci.insight.158397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Elazar N, Vainshtein A, Rechav K, Tsoory M, Eshed-Eisenbach Y, and Peles E (2019). Coordinated internodal and paranodal adhesion controls accurate myelination by oligodendrocytes. J. Cell Biol. 218, 2887–2895. 10.1083/jcb.201906099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shi Q, Chang C, Saliba A, and Bhat MA (2022). Microglial mTOR Activation Upregulates Trem2 and Enhances beta-Amyloid Plaque Clearance in the 5XFAD Alzheimer’s Disease Model. J. Neurosci. 42, 5294–5313. 10.1523/JNEUROSCI.2427-21.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shi Q, Saifetiarova J, Taylor AM, and Bhat MA (2018). mTORC1 Activation by Loss of Tsc1 in Myelinating Glia Causes Downregulation of Quaking and Neurofascin 155 Leading to Paranodal Domain Disorganization. Front. Cell. Neurosci. 12, 201. 10.3389/fncel.2018.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Taylor AM, Saifetiarova J, and Bhat MA (2017). Postnatal Loss of Neuronal and Glial Neurofascins Differentially Affects Node of Ranvier Maintenance and Myelinated Axon Function. Front. Cell. Neurosci. 11, 11. 10.3389/fncel.2017.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saifetiarova J, Liu X, Taylor AM, Li J, and Bhat MA (2017). Axonal domain disorganization in Caspr1 and Caspr2 mutant myelinated axons affects neuromuscular junction integrity, leading to muscle atrophy. J. Neurosci. Res. 95, 1373–1390. 10.1002/jnr.24052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Green JA, Yang J, Grati M, Kachar B, and Bhat MA (2013). Whirlin, a cytoskeletal scaffolding protein, stabilizes the paranodal region and axonal cytoskeleton in myelinated axons. BMC Neurosci. 14, 96. 10.1186/1471-2202-14-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data, including mouse lines, DNA constructs and original western blot and confocal images, reported in this paper can be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.