

Abstract
Natural killer (NK) effector functions can be triggered by inflammatory cytokines and engagement of activating receptors. NK cell production of IFN-γ, an important immunoregulatory cytokine, exhibits activation-specific IFN-γ regulation. Resting murine NK cells exhibit activation-specific metabolic requirements for IFN-γ production, which are reversed for activating receptor-mediated stimulation following IL-15 priming. While both cytokine and activating receptor stimulation leads to similar IFN-γ protein production, only cytokine stimulation upregulates Ifng transcript, suggesting that protein production is translationally regulated after receptor stimulation. Based on these differences in IFN-γ regulation, we hypothesized that ex vivo IL-15 priming of murine NK cells allows a switch to IFN-γ transcription upon activating receptor engagement. Transcriptional analysis of primed NK cells compared to naïve cells or cells cultured with low-dose IL-15 demonstrated that primed cells strongly upregulated Ifng transcript following activating receptor stimulation. This was not due to chromatin accessibility changes in the Ifng locus or changes in ITAM signaling, but was associated with a distinct transcriptional signature induced by ITAM stimulation of primed compared to naïve NK cells. Transcriptional analyses identified a common signature of c-Myc (Myc) targets associated with Ifng transcription. While Myc marked NK cells capable of Ifng transcription, Myc itself was not required for Ifng transcription using a genetic model of Myc deletion. This work highlights altered regulatory networks in IL-15 primed cells, resulting in distinct gene expression patterns and IFN-γ regulation in response to activating receptor stimulation.
Introduction
Natural killer (NK) cells are innate lymphocytes important for the immune responses to infections and tumors. They are aptly named after their ability to recognize and eliminate infected, “stressed”, or malignant cells through directed release of their cytotoxic granule content or death receptor-mediated apoptosis (1, 2). NK cells are a major producer of interferon-gamma (IFN-γ) prior to antigen-specific T cell responses, which is important for modulation of the immune response, supporting optimal anti-viral and anti-tumor responses through effects such as enhanced macrophage function and upregulation of antigen presentation-related proteins (3–6).
NK effector functions are controlled by cytokines and the integration of activating and inhibitory signals from germline-encoded receptor interactions with ligands on potential target cells (7). The common gamma cytokine interleukin (IL)-15 supports NK cell proliferation, survival and in vivo persistence, homeostasis, cytotoxicity and cytokine production (8, 9). Administration of IL-15 in vivo in murine models enhances anti-tumor activity and anti-viral responses (10–12). In vitro culture systems have delineated effects of low versus high dose IL-15 in both human (13–15) and murine systems (16, 17). At low concentrations, IL-15 supports NK survival. High dose IL-15 induces proliferation, acquisition of enhanced cytotoxic capacity (16), and metabolic reprogramming through upregulation of mTOR and glycolysis (18, 19). Upregulation of mTOR and glycolysis supports optimal responses to MCMV infection, including antigen-specific killing, degranulation, and granzyme B expression (12, 18, 20, 21). Due to the effect of IL-15 on NK cells, IL-15-based therapies have been developed and are undergoing testing in clinical trials, where they have shown promise in enhancing antitumor responses, with enhanced NK cell persistence and expansion without stimulating regulatory T cells (22–24).
We previously reported that IFN-γ production by freshly isolated (naïve) murine NK cells has distinct metabolic requirements dependent on the route of activation. IFN-γ production following activating receptor stimulation, mediated by ITAM signaling, has a high metabolic requirement, and both glycolysis and mitochondrial oxidative phosphorylation (OXPHOS) are needed. By contrast, cytokine-stimulated IFN-γ production remains intact with metabolic inhibition, demonstrating metabolic flexibility (19). The strict metabolic requirement for ITAM signaling can be reversed by culturing NK cells with high dose IL-15 for 72 hours (priming) in vitro (19), and in vivo by administration of IL-15 leading to rescue of otherwise fatal infection with murine cytomegalovirus (MCMV) when glycolysis is inhibited (12).
In this study, we characterized the effects of IL-15 priming on NK cells, focusing on the changes in IFN-γ regulation downstream of activating receptor stimulation. We previously demonstrated that while cytokine stimulation induces rapid upregulation of Ifng transcript followed by protein, ITAM-based stimulation leads to similar IFN-γ protein production but without upregulation of transcript (19). Based on these differences in metabolic requirements and Ifng expression, we hypothesized that transcriptional regulation of Ifng is related to the metabolic requirement for protein production, with new transcription/translation having metabolic flexibility. Here, we identify that IL-15 priming alters chromatin accessibility and leads to altered transcriptional responses to ITAM-associated activating receptors, including transcriptional regulation of IFN-γ production in response to ITAM-signaling. Acquisition of metabolic flexibility is associated with induction of c-Myc, which marks cells capable of Ifng transcription, but is not itself required for transcription. Overall, this study provides insight into mechanisms of IL-15 priming and has implications for the design of therapeutic approaches to optimize NK cell effector functions in metabolically challenging environments in vivo (21, 25).
Materials and Methods
Mice and NK cell enrichment
All mice were maintained in specific pathogen-free conditions and studies were approved by the Washington University Animal Studies Committee. Wild-type mice were purchased from Charles River (C57BL/6NCrl, #556) and used for studies between 6 and 14 weeks of age. Splenic NK cells were enriched using a negative selection magnetic bead kit (STEMCELL Technologies). NK cell purity was 95% or higher, and the majority of contaminating cells were CD3 negative. Unless otherwise specified, controls were C57BL/6NCrl. Heterozygous ΔARE mice were generated and obtained from Howard A. Young’s lab and bred in our animal facility with WT mice. These mice have a targeted substitution of a conserved 162 nucleotide AU-rich sequence in the 3’ untranslated region (3’UTR) of the IFN-γ (26).
Reporter mice were obtained from The Jackson Laboratory. IFN-γ YFP reporter mice on a C57BL/6 background (B6.129S4-Ifngtm3.1Lky/J, JAX stock #017581) have an IRES-eYFP reporter cassette inserted between the translational stop codon and 3’ UTR/polyA tail of the interferon gamma (Ifng) gene (referred to as GREAT mice (27)). Myc reporter mice (MycEGFP) (B6;129-Myctm1Slek/J, JAX stock #021935) express an N-terminal fusion EGFP/MYC protein (28). MycEGFP mice with NK1.1 expression comparable to a C57BL/6 background were used for experiments.
An NK cell and ILC1-specific inducible model of c-Myc deletion, Ncr1-MycΔ/Δ, was generated by crossing mice with yellow fluorescent protein (YFP) Cre-reporter and tamoxifen-inducible iCre recombinase driven by Ncr1 (Ncr1-iCreERT2) (29) with mice carrying loxP sites flanking exons 2 and 3 of c-Myc (30) (generated by the Alt lab (30) and generously shared by Dr. Ruoning Wang at Nationwide Children’s Hospital). Ncr1-iCreERT2 heterozygous mice (Ncr1-WT) were used as controls for experiments where Myc deletion was induced with tamoxifen chow or 4-OHT. The controls were housed in the same facility but were not littermates or co-housed.
Cell culture
Complete media (“R10”) contained RPMI 1640 (Corning), 10% fetal bovine serum (FBS) (Sigma Aldrich), L-glutamine (Sigma Aldrich), non-essential amino acids (Corning), penicillin/streptomycin (Sigma Aldrich). For glucose-free media, the following substitutions/replacements were made: glucose-free RPMI (Corning), 15% PBS (Corning), and 10% dialyzed FBS. For 72 hour cultures, media also contained 50 μM β-mercaptoethanol (Sigma Aldrich) and 10 ng/ml (LD) or 100 ng/ml (HD) IL-15 (Peprotech). For experiments with 4-hydroxytamoxifen (4-OHT), 0.8 uM was the final 4-OHT concentration in vitro. Metabolic or pathway inhibitors were used at the following final concentrations: 2DG (1mM, Sigma Aldrich), rapamycin (10 nM, 100 nM), oligomycin (10 or 100 nM).
Human NK cells
Cryopreserved human peripheral blood mononuclear cells (PBMCs) from anonymous healthy platelet donors (de-identified Leukoreduction System chambers, non-human subjects) were thawed and rested overnight in complete media (10% human AB serum). Cells were then washed with PBS and resuspended in either complete or glucose-free media. The cells were stimulated with IL-12+IL-18 (10 ng/ml, Peprotech, and 50 ng/ml, MBL, respectively) or plate-bound anti-CD16 (1 mg/ml) for 6 hours.
Flow cytometry
A live/dead stain was included in most assays to measure viability (Zombie Yellow, BioLegend). After murine NK cell enrichment, which consistently resulted in a ~95% CD3−NK1.1+ or CD3−NKp46+ population, cells were gated on NKp46 to identify NK cells. For human NK cells, cells were gated based on Zombie Yellow to identify live cells, followed by CD3 and CD56 to identify NK cells. Murine cells were blocked with anti-FcγRIII (2.4G2) prior to surface staining. Human cells were blocked with mouse IgG prior to surface staining. Cells were fixed with 1% paraformaldehyde for surface analysis, Cytofix/Cytoperm (BD Bioscience) for intracellular staining, or eBioscience™ Foxp3/Transcription Factor Staining Buffer set (Thermo Fisher Scientific) for c-Myc detection, according to the manufacturer’s instructions. For phospho flow assays, cells were fixed with 2% paraformaldehyde, permeabilized with methanol, and stained overnight at 4C with phospho antibodies. Data was acquired on a Cytek-modified FACScan (BD and Cytek) or LSRFortessa (BD); data was analyzed using FlowJo software. The following fluorochrome-conjugated antibodies were used: CD3ε (145-2C11, BD/BioLegend), CD19 (1D3, BioLegend), CD56 (NKH-1-RD1, Beckman Coulter), ERK1/2 (T202/Y204) (6B8B69, BD), anti-human IFN-γ (B27, BD), anti-mouse IFN-γ (XMG1.2, BioLegend), NFkB p65 (S536) (93H1, BD), NK1.1 (PK136, BD), NKp46 (29A1.4, BD), p38 MAPK (T180/Y182) (36/p38, BD), PLCg2 (Y759) (K86-689.37, BD), ZAP70 (Y319)/Syk(Y352) (XMG1.2, BD), c-Myc/N-Myc (D3N8F).
NK cell stimulation and IFN-γ production
For cytokine stimulation, NK cells were cultured for 6 hours in the presence of 1 ng/ml murine IL-12 (Peprotech) and 1 ng/ml murine IL-18 (MBL). NK1.1 was engaged by culturing NK cells in plates coated with 20 μg/ml purified anti-NK1.1 (PK136; BioXcell). Where indicated, PMA (Sigma Aldrich) and calcimycin (Sigma Aldrich) were used at 10 ng/ml and 500 ng/ml, respectively. For experiments with actinomycin, freshly isolated splenic NK cells were stimulated in the presence of the indicated doses of actinomycin (Sigma Aldrich). For phospho-flow experiments, cells were incubated with anti-NK1.1 (PK136) and anti-NKp46 (29A1.4) for 10-15 min, followed by the addition of goat anti-mouse antibody (BioLegend) to begin the NK1.1 stimulation. For assays measuring intracellular IFN-γ production, GolgiPlug™ (BD Biosciences, Brefeldin A) was added after 1 h of stimulation to inhibit IFN-γ secretion. BD Cytofix/Cytoperm™ (BD Biosciences) was used as recommended for intracellular staining.
Quantitative PCR
NK cells (~2-3x10^5) were activated with plate-bound anti-NK1.1 (20 μg/ml) and harvested at the indicated time points. RNA was isolated with the RNeasy Plus Micro kit (Qiagen) and cDNA was generated using random hexamers (Promega), following the GoScript Reverse Transcription System protocol (Promega). Primers for Actb (Mm00607939, Thermo Fisher Scientific), Ifng ((31), IDT) and Myc (Mm00487804_m1, Thermo Fisher Scientific) were used to evaluate gene expression. Copy numbers of Ifng and Actb transcript were determined by standard curves generated with plasmid clones of Ifng and Actb amplicons and quantified via real-time quantitative PCR (TaqMan, QuantStudio 3 instrument from Thermo Fischer Scientific). To normalize across conditions, Ifng copy number relative to Actb was calculated. For Myc expression, data were analyzed using the 2−ΔΔCt method (32).
ATAC-seq
Aliquots of approximately 75,000 cells (naïve, LD/HD IL-15) were used as input and processed as previously described (33). Briefly, the cells were washed in cold PBS and lysed in cold lysis buffer (10mM Tris-HCl pH 7.4, 10mM NaCl, 3 mM MgCl2, 0.1 % IGEPAL CA-630). Transposase reaction occurred at 37C for 30 minutes. DNA was purified with the Qiagen MinElute PCR purification kit and amplified for 5 cycles. The appropriate number of additional PCR cycles was determined by real-time PCR. The final product was cleaned using AMPure XP beads (Beckman Coulter). Quality control of the generated libraries was performed using the Agilent TapeStation system. Libraries were sequenced on a NovaSeq6000.
ATAC-seq data were first processed using the AIAP (34) pipeline. Briefly, reads were aligned to the mm10 genome using bwa; locations of Tn5 insertion events were inferred from properly paired, non-PCR duplicate reads with MAPQ >= 10; each Tn5 insertion event was extended from insertion site up and down 75 bp to generate a 150 bp “pseudo read”; pseudo reads were piled up to generate an ATAC signal profile, which was subsequently normalized against both sequencing depth and sample quality, similar to the getGroupBW() function of the ArchR package (35). Briefly, sample quality was calculated as the fraction of reads mapped to promoters (transcription start sites ± 1 Kb. frac.pro); a sequencing depth scaling factor (depth.fac) was calculated for each sample as the ratio of the sequencing depth of that sample over the median sequencing depth of all samples; each ATAC-seq profile (bigWig track) was then divided by frac.pro and depth.fac to obtain normalized signals; normalized ATAC-seq signals were divided by 3 because the typical value of depth.fac is 0.3 - 0.4. ATAC-seq data were visualized using WashU Epigenome Browser (36).
ATAC-seq peaks for each sample were called as a part of the AIAP pipeline, using macs2 (37) with FDR cutoff at 0.01. After manual inspection of the peaks called, we further filtered the resulting narrowPeak files for peaks with −log10FDR >= 8. To call differentially accessible regions (DARs), we merged all overlapping peaks to create a union peakset. We then formed a peak-sample matrix with each entry corresponding to the number of Tn5 insertion sites in each peak for each sample. We filtered out peaks where any sample has no reads. DARs were called using DESeq2 (38) (v1.26.0), with the design formula ~ replicate + type. “Type” represents high or low dose IL15 treatment. The cutoff for DARs were set at log2FoldChange > 1 & FDR < 0.05.
For motif enrichment in DARs, up (high dose > low dose) and down (low dose > high dose) DARs were separately ranked by p value and the top 1000 peaks in each category were used for motif enrichment. For each DAR used for motif enrichment, its 25 closest non-DARs in the Euclidean space constructed from low dose samples were selected without replacement as background peaks. We used HOMER (v4.11.1) (39) for motif enrichment in DARs relative to background peaks (findMotifsGenome.pl up_or_down_DAR.bed mm10.fa out_dir -bg up_or_down_DAR_background.bed -nomotif -size given).
RNA-seq analysis
Enriched NK cells (>95% purity) were either cultured for 72 hours in media containing LD or HD IL-15, or used immediately after enrichment. Where indicated, cells were stimulated with anti-NK1.1 or IL-12 and IL-18 as described above. RNA was isolated using Dynabeads mRNA DIRECT Kit (Invitrogen), and then reverse transcribed using the AffinityScript Multiple Temperature cDNA Synthesis Kit (Agilent Technologies) to synthesize cDNA. After first strand synthesis, samples were pooled together based on ACTB qPCR values, and RNA-DNA hybrids degraded using consecutive acid-alkali treatment. A second sequencing linker (AGATCGGAAGAGCACACGTCTG) was ligated using T4 ligase (NEB) followed by SPRI clean-up. The mixtures were then PCR enriched for 14 (IL-15 dataset) or 17-20 (activation dataset) cycles and SPRI purified to yield final strand specific RNA-seq libraries. Final library QC on pooled libraries was performed using the Agilent TapeStation system. Libraries were sequenced on HiSeq 2500 (Illumina) using 40 bp X 10 bp paired-end sequencing. Second mate was used for sample demultiplexing, generating individual single-end fastq files.
Reads were aligned to the mouse genome GRCm38/mm10 primary assembly (GENCODE) and gene annotation Ver.M16 with STAR 2.5.4a. The raw read counts were generated by featureCounts (40) and normalized with DESeq2 (38) package from R/Bioconductor. The top 12,000 genes ranked by average gene expression were selected for differential expression analysis using the DESeq2 (38). The cutoff for determining significantly differentially expressed genes was an FDR-adjusted p value less than 0.05. Differential expression analysis was performed with the limma package via Phantasus, a web-based application (41). Bubble plots and GSEA (42) plots were generated in R with the enrichplot package (43).
For the IFN-γ reporter (GREAT) dataset, cells were primed as above and stimulated via plate-bound anti-NK1.1 for 6 hours, then sorted on a FACSAria Fusion (BD Biosciences) based on YFP. RNeasy Plus Micro kit (Qiagen) was used for RNA extraction. Samples were prepared according to the library kit manufacturer’s protocol, indexed, pooled, and sequenced on an Illumina NovaSeq 6000. Basecalls and demultiplexing were performed with Illumina’s bcl2fastq software and a custom python demultiplexing program with a maximum of one mismatch in the indexing read. RNA-seq reads were then aligned to the Ensembl release 76 primary assembly with STAR version 2.7.9a (44). Gene counts were derived from the number of uniquely aligned unambiguous reads by Subread:featureCount version 2.0.3 (40). Sequencing performance was assessed for the total number of aligned reads, total number of uniquely aligned reads, and features detected. The ribosomal fraction, known junction saturation, and read distribution over known gene models were quantified with RSeQC version 4.0 (45).
All gene counts were then imported into the R/Bioconductor package EdgeR (46) and TMM normalization size factors were calculated to adjust for samples for differences in library size. Ribosomal genes and genes not expressed in the smallest group size minus one samples greater than one count-per-million were excluded from further analysis. The TMM size factors and the matrix of counts were then imported into the R/Bioconductor package Limma (47). Weighted likelihoods based on the observed mean-variance relationship of every gene and sample were then calculated for all samples and the count matrix was transformed to moderated log 2 counts-per-million with Limma’s voomWithQualityWeights (48). The performance of all genes was assessed with plots of the residual standard deviation of every gene to their average log-count with a robustly fitted trend line of the residuals. Differential expression analysis was then performed to analyze for differences between conditions and the results were filtered for only those genes with Benjamini-Hochberg false-discovery rate adjusted p-values less than or equal to 0.05. Gene set co-regulation analysis (GESECA) was performed using the fgsea package (49).
Data Availability
RNA and ATAC sequencing data generated during this study is available at Gene Expression Omnibus with accession number GSE227894 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE227894).
Results
Regulation of IFN-γ in naïve and IL-15 primed NK cells
We previously demonstrated that IL-12+IL-18 activation of IFN-γ protein production is transcriptionally regulated in freshly isolated (naïve) murine NK cells, whereas ITAM-based (anti-NK1.1) activation leads to similar protein production, but not IFN-γ transcript (19). To investigate how IFN-γ protein production is regulated by cytokine or activating receptor stimulation, we first assessed the contribution of transcription to IFN-γ production after IL-12+IL-18 or plate-bound anti-NK1.1 stimulation by culturing the cells in the presence of the transcription inhibitor actinomycin D. While the percentage of IFN-γ+ NK cells decreased with both stimulations, the decrease was significantly more pronounced in the cytokine stimulated cells due to the drastic reduction of Ifng transcript (Fig. 1A, Supplemental Fig. 1A). This supports the hypothesis that IFN-γ is predominantly translated from pre-formed transcript when NK cells are activated via ITAM-bearing receptors, while cytokine (IL-12/18) stimulation is highly dependent on new transcription.
Figure 1. Regulation of IFN-γ production in naïve and IL-15 primed NK cells.
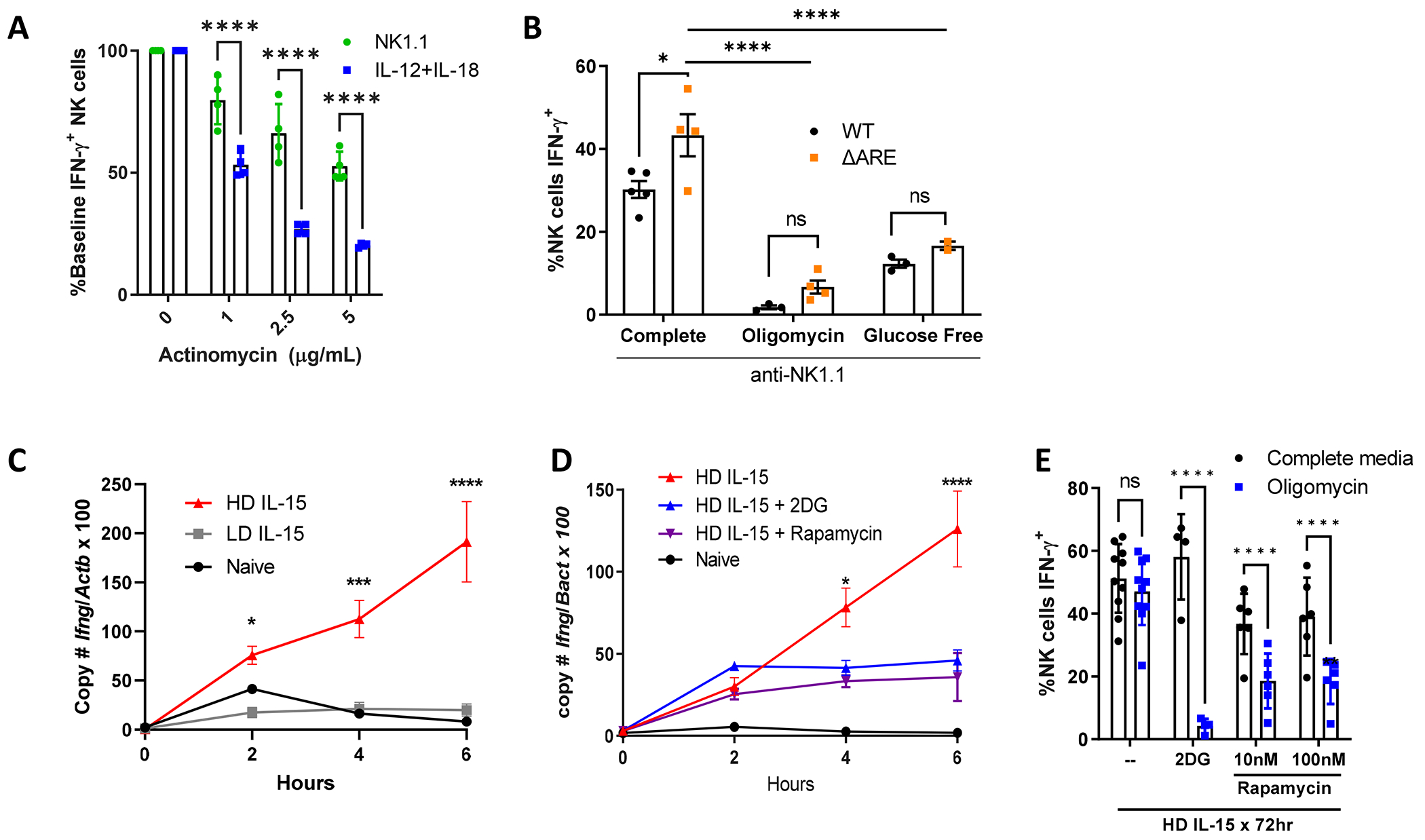
A) Freshly isolated NK cells stimulated with plate-bound anti-NK1.1 (PK136) or low dose IL-12+IL-18 (1 ng/ml each) in the presence of indicated doses of actinomycin. % maximal IFN-γ+ NK cells shown compared to baseline (no actinomycin). 2 independent experiments, duplicate wells; B) NK cells from WT mice or mice with deleted 3’ AU rich elements (ARE) of Ifng were cultured in complete media +/− 100nM oligomycin or in glucose-free media during the 6 hour anti-NK1.1 stimulation. n=4-5 mice per genotype, 2 independent experiments. C-E) NK cells were freshly isolated (naïve) or cultured for 72 hours with 10 ng/ml (LD) or 100 ng/ml (HD) IL-15, followed by anti-NK1.1 stimulation for the indicated time, up to 6 hours. C) Absolute copy number of Ifng and Actb (beta-actin) were quantified by real-time PCR. Results represent the ratio of Ifng to Actb x 100 and the mean +/− SEM of duplicate wells from 2 independent experiments. D-E) NK cells were primed in HD IL-15 in the absence or presence of 2DG (1 mM) or rapamycin (100 nM) prior to NK1.1 stimulation. D) Ratio of Ifng to Actb x 100 and the mean +/− SEM from 1-3 independent experiments, duplicate wells. E) % IFN-γ+ NK cells were measured by flow cytometry after 6h anti-NK1.1 stimulation, 2-6 independent experiments, 4-10 mice per condition. Statistical analysis: 2way ANOVA (A, C-E), Ordinary one-way ANOVA (B).
In addition to differences in transcriptional or post-transcriptional regulation of IFN-γ production, we also previously demonstrated that there are distinct metabolic requirements for IFN-γ production. Activating receptor-stimulated IFN-γ production is highly sensitive to metabolic inhibitors of both OXPHOS and glycolysis. In contrast, IL-12+IL-18 cytokine-stimulated IFN-γ protein production, is metabolism-independent (19). This dependence on a secondary metabolic signal for IFN-γ production downstream of ITAM signaling was also observed in human NK cells (Supplemental Fig. 1B). However, we previously demonstrated that culturing murine NK cells with high dose (HD, 100ng/ml) IL-15 for 72 hours (IL-15 priming) leads to metabolic flexibility upon activating receptor stimulation, with intact IFN-γ production in the presence of metabolic inhibitors, similar to IL-12+IL-18 stimulated naive NK cells (19). The mechanism of this metabolism-independent receptor-stimulated IFN-γ production is relevant when considering how to prepare NK cells for metabolically challenging microenvironments, for example with NK cell adoptive immunotherapy (25).
One potential mechanism for metabolic regulation of IFN-γ protein production may be through GAPDH acting as an IFN-γ translation repressor as demonstrated in CD4+ T cells. In T cells, GAPDH binds to AU-rich elements (ARE) in the 3’ untranslated region (UTR) of IFN-γ mRNA (50). Upon glycolysis upregulation, which engages GAPDH enzymatic activity, IFN-γ translation is no longer inhibited in CD4+ T cells. To test if a 3’UTR-dependent mechanism also occurs in naïve NK cells, we evaluated the effect of metabolic inhibition on IFN-γ production in mice with a 162 nucleotide ARE region substitution in the 3’UTR of the IFN-γ gene (ΔARE) (51). NK cells in ΔARE mice have been shown to have elevated IFN-γ protein production at baseline (26), and we observed this with receptor stimulation (Fig. 1B complete). In this genetic model, metabolic inhibitors, 2-DG to inhibit glycolysis or oligomycin to inhibit mitochondrial OXPHOS, both inhibited activating-receptor induced IFN-γ production similar to wild-type (WT), suggesting that the mechanism of metabolic dependence for activating receptor stimulation in NK cells is 3’UTR-independent (Fig. 1B).
Based on the observed differences in IFN-γ regulation, we hypothesized that IL-15 priming of NK cells allows a switch to IFN-γ transcriptional regulation upon receptor activation, thus allowing for metabolic flexibility. To determine the effect of IL-15 priming on NK1.1-mediated Ifng transcription, Ifng copy number was measured in anti-NK1.1 stimulated NK cells that were either freshly isolated (naïve) or primed for 72 hours with a high-dose IL-15 (HD, 100 ng/ml - primed cells) or cultured in the presence of low-dose (LD, 10 ng/ml), which does not lead to metabolic flexibility (19). At baseline (time 0), Ifng copy number by qPCR was similar across conditions, demonstrating that IL-15 priming itself does not upregulate Ifng transcript. Over the course of the 6 hour anti-NK1.1 stimulation, Ifng significantly increased only in NK cells previously cultured in HD IL-15. By contrast, cells cultured in LD IL-15, similar to naïve NK cells, expressed very low Ifng transcript levels that did not change over the course of the 6 hour anti-NK1.1 stimulation (Fig. 1C). These results demonstrate that IL-15 primed cells switch to transcriptional regulation of IFN-γ and that transcriptional regulation of IFN-γ is dependent on IL-15 dose and associated with metabolic flexibility.
Culturing NK cells in HD IL-15 promotes proliferation and increases metabolic activity with a prominent upregulation of glycolysis (18, 19). Undivided cells produce similar amounts of IFN-γ protein after HD IL-15 priming, suggesting that the change in metabolic requirements is not related to proliferation (19). High dose IL-15 activates mTOR and results in increased glucose uptake (18). In short term culture experiments, IL-15-induced mTOR is important for NK IFN-γ production (52). To evaluate the contribution of glycolysis and mTOR to the IL-15 priming effect on IFN-γ transcription, NK cells were primed with HD-IL-15 in the presence of 2-DG or rapamycin followed by activating receptor stimulation (anti-NK1.1). Cells primed in the presence of these inhibitors had impaired upregulation of Ifng transcript production, with reduced protein production that was most prominent in cells cultured with 2DG (Fig. 1D, 1E). This suggests that the ability to upregulate glycolysis, and mTORC1, is critical during the priming phase for the IL-15 priming effect.
IL-15 priming induces chromatin accessibility changes
To study the effect of IL-15 priming on the epigenome and regulatory network of NK cells, ATAC-seq was performed on primed NK cells. Differentially accessible chromatin regions were compared with NK cells cultured with LD IL-15, which do not upregulate Ifng transcript and lack metabolic flexibility, to account for changes associated with in vitro culture. There were 5120 differentially accessible regions (DARs), 3845 of which were more accessible and 1275 of which were less accessible in primed NK cells. For example, one of the genes with several regions of increased chromatin accessibility was Spp1 (Fig. 2A, 2C). Spp1 encodes for secreted phosphoprotein 1 (osteopontin), which is upregulated downstream of IL-15 signaling (53). The more accessible (HD>LD) or “up” DARs displayed a strong enrichment for motifs bound by transcription factors Fos, JunB, Fra1, Fra2, and BATF, sharing a common binding sequence (5’ - TGA(C/G)TCA - 3’) (Fig. 2B). With these transcription factors representing members of the AP-1 transcription factor family, the results suggest that IL-15 primed NK cells may be more responsive to activation of the MAPK pathway, and subsequent activation of AP-1 family members.
Figure 2. IL-15 priming induces chromatin accessibility changes.
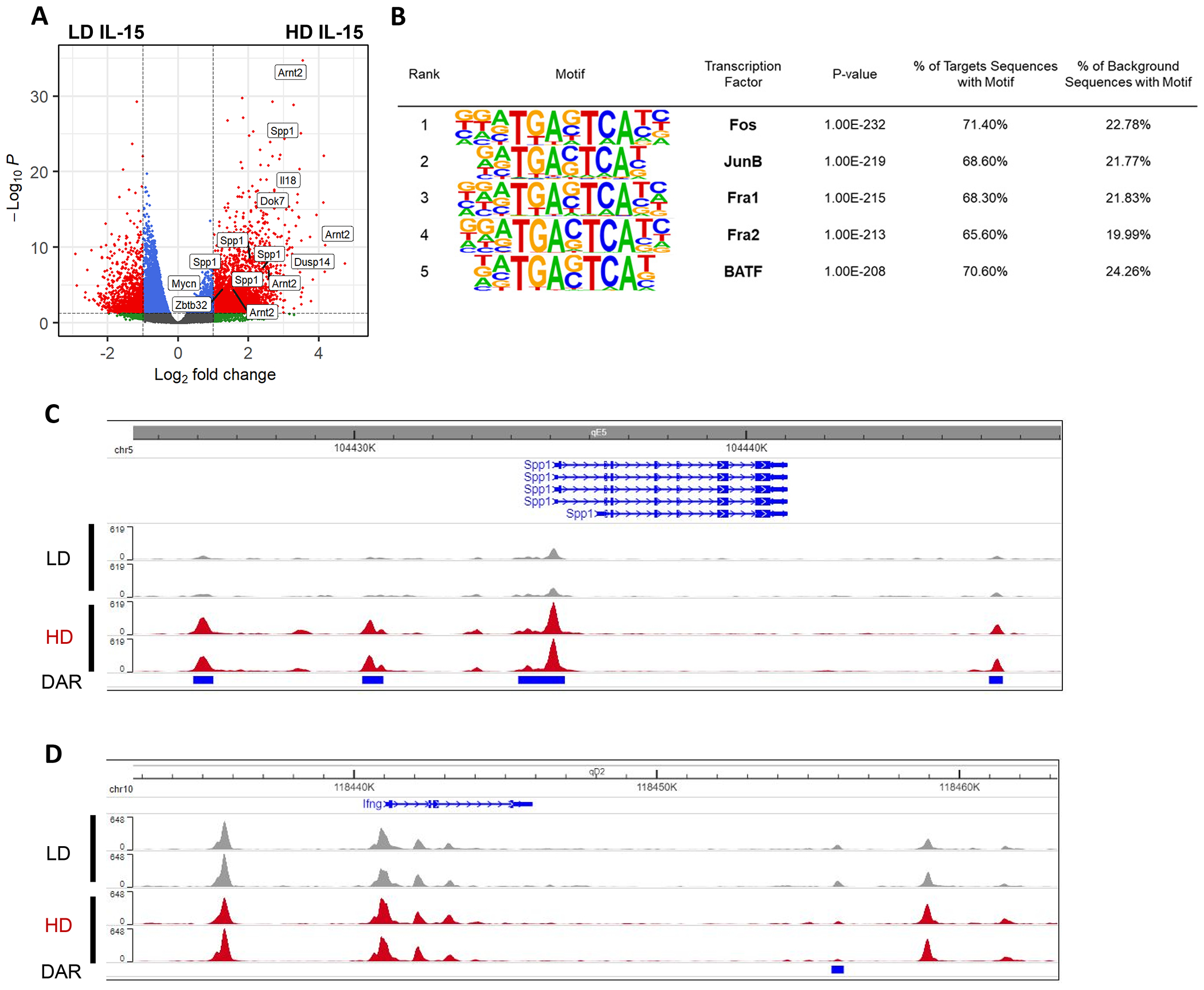
ATAC-seq analysis comparing LD vs HD IL-15 cultured NK cells, after 72 hours. One independent experiment, 2 mice per group. A) Volcano plot showing the differentially accessible (log2FoldChange > 1, FDR < 0.05) chromatin regions assigned to genes. B) Motif enrichment at regions of open chromatin as defined by ATAC-seq in primed NK cells compared to NK cells cultured in LD IL-15. C-D) Genome browser visualization of peaks near Spp1 (C) and Ifng (D).
Given the time-dependent effect of IL-15 priming on Ifng regulation, we hypothesized that priming might induce chromatin accessibility changes in the Ifng locus, including conserved non-coding sequences that contribute regulatory functions to the Ifng locus, potentially creating new sites for transcription factors to occupy and modulate Ifng transcription. However, the Ifng region remains almost unchanged, with the exception of one DAR located approximately 15kb from the TSS (Fig. 2D). This DAR is modestly less accessible in IL-15 primed NK cells and has an undetermined biological significance. Therefore, chromatin accessibility changes, directly at regulatory elements in the Ifng locus, are unlikely to play a part in the switch to Ifng transcriptional regulation. Collectively, this data demonstrates that culturing NK cells in high dose IL-15 for 72 hours predominantly increases chromatin accessibility across the genome in murine NK cells, but not in the Ifng locus.
IL-15 priming does not alter canonical ITAM signaling in a dose-dependent manner
With the knowledge that there are no up DARs in the Ifng locus induced by IL-15 priming, an alternative hypothesis for transcriptional regulation of Ifng with priming was that strength of ITAM signaling is altered in by IL-15 priming. Based on current knowledge of ITAM-based signaling in NK cells (Fig. 3A) (7, 54), phosphorylation of relevant proximal and distal signaling molecules including Zap70, Syk, PLCγ2, MAPK Erk and p38, and p65 (RelA) following anti-NK1.1 stimulation was measured by flow cytometry (Fig. 3B–F). There were no differences in the mean fluorescence intensity (MFI) of these phosphorylated proteins in NK cells cultured with LD IL-15 versus HD IL-15 (primed) (Fig. 3B–F). Similarly, the presence of oligomycin, or culture in glucose-free media during the stimulation did not alter ITAM signaling (Supplemental Fig. 1C–E). These results demonstrate that there are no significant differences in canonical ITAM signaling that correlate with the switch to transcriptional regulation Ifng after IL-15 priming.
Figure 3. IL-15 priming does not alter ITAM signaling manner.
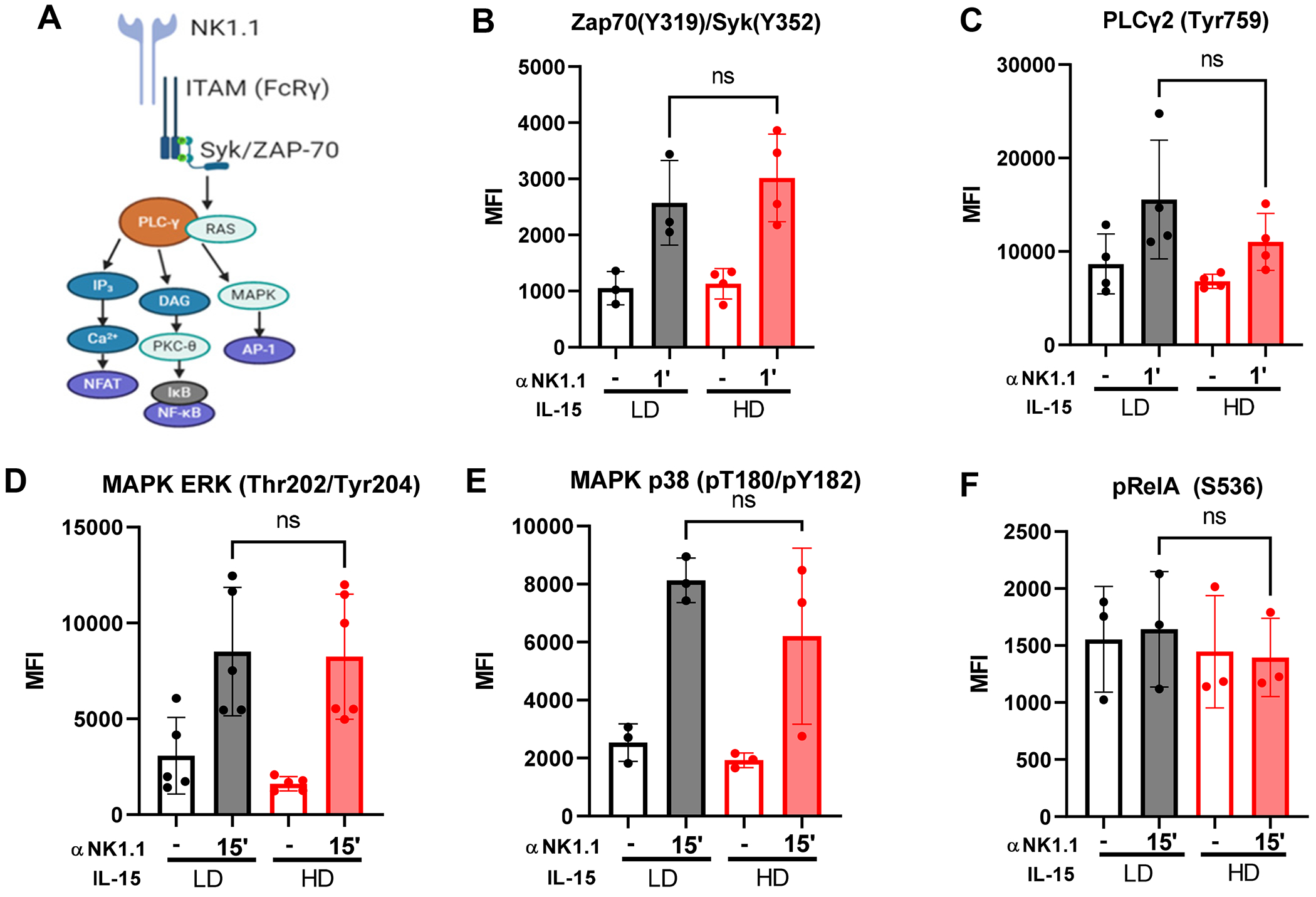
A) ITAM signaling pathways downstream of NK1.1. B-F) Murine NK cells were cultured for 72 hours in LD (10 ng/ml) or HD (100 ng/ml) IL-15. Cells were washed, then cultured for 30-60 min in complete media (+/− 100 nM oligomycin) or glucose-free media. Results shown are from a minimum of 3 independent experiments, 1 mouse per experiment. Each data point represents an independent experiment. Phosphorylation of Zap70/SYK (B), PLCγ2 (C), ERK (D), MAPK p38 (E), and RelA (F) was quantified by flow cytometry. Statistical analysis: Wilcoxon test (paired t-test, non-parametric)
Transcriptional changes associated with Ifng transcription in IL-15 primed NK cells
While ITAM signaling itself appears unchanged, the regulation of IFN-γ is different in primed NK cells. Given that chromatin accessibility changes, we hypothesized that altered chromatin accessibility in primed NK cells leads to a differential transcriptional response in how NK cells receive those signals. To determine if primed NK cells have a different transcriptional response to activating receptor stimulation, RNA-seq was performed to compare the responses of naïve, LD IL-15 and primed NK cells (Fig. 4A). First, to examine transcriptional changes associated with IL-15 priming, unstimulated naïve and HD IL-15 treated NK cells were compared. As expected, and consistent with the reported biology of IL-15 and NK cells, primed NK cells upregulated genes and pathways associated with proliferation, glycolysis, and cytotoxicity (16, 18) (Fig. 4B). Glycolysis (Aldoc, Pgam1, Eno1, Pkm, Ldha; log2FC>2) and cell cycle (Aurka, Ube2c, Cdkn3, Cdk1; log2FC>3) genes were upregulated, as well as several granzyme genes, with Gzmd, Gzme, and Gzmc representing the highest fold changes (log2FC>9) (Fig. 4C).
Fig. 4. Transcriptional changes associated with Ifng transcription.
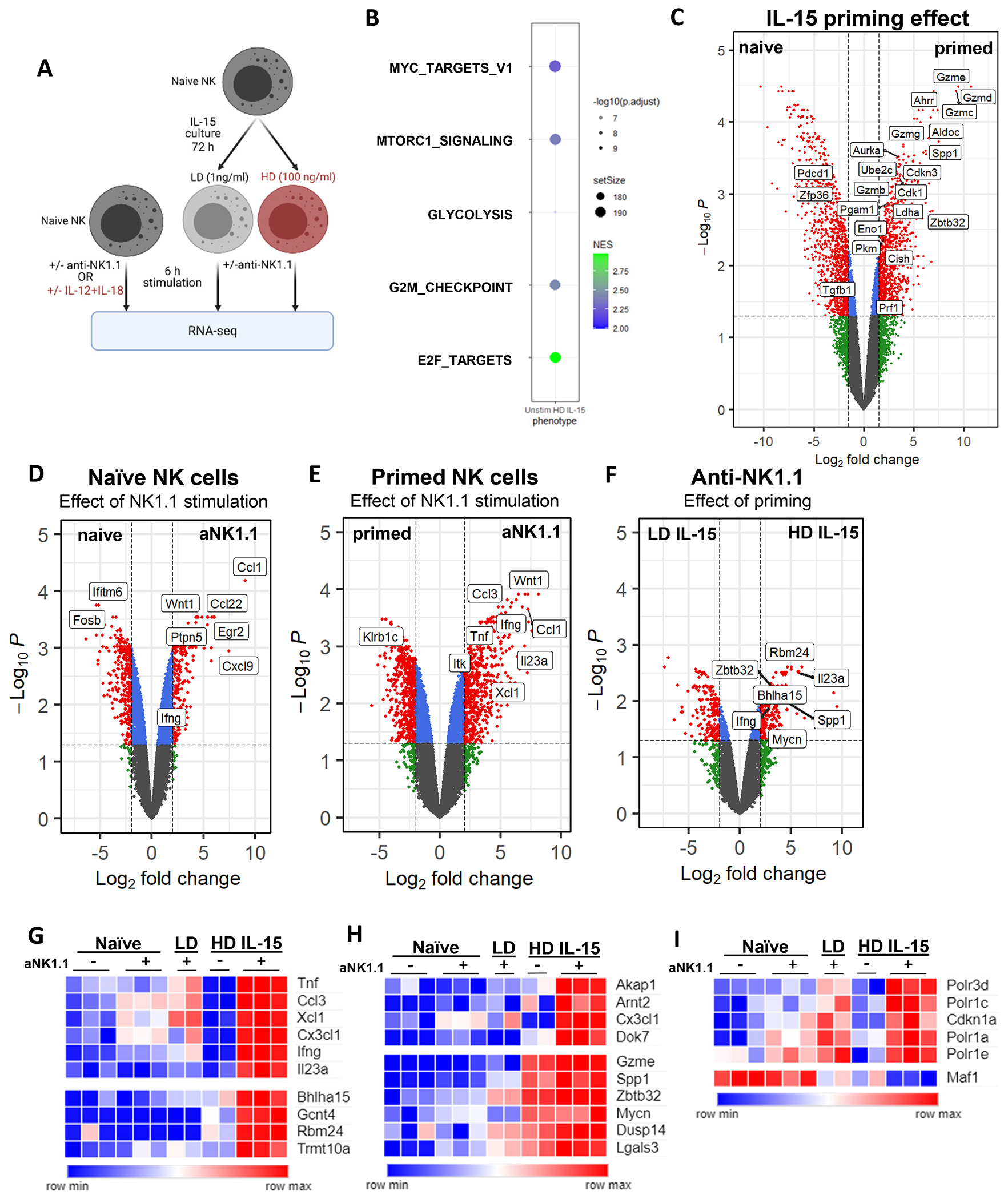
A) Experiment schema. NK cells from the spleens of 15 male mice were pooled for each dataset (naïve NK +/−stimulation, naïve and IL-15 cultured NK cells +/− anti-NK1.1). One independent experiment, 2-3 technical replicates per condition. Red text indicates conditions with metabolic flexibility for IFN-γ production. B) Bubbleplot of gene set enrichment analysis (GSEA) select pathways upregulated in HD unstim compared to naive unstim. C) Volcano plot showing differentially expressed genes from the naïve unstim vs HD IL-15 unstim. D-F) Volcano plots showing differentially expressed genes from the following comparisons: (D) naïve unstimulated vs aNK1.1, (E) HD IL-15 unstimulated vs aNK1.1, and (F) LD IL-15 aNK1.1 vs HD IL-15 aNK1.1. G-I) Heatmap highlighting select genes that are differentially regulated in (G) HD+aNK1.1, (H) have both increased chromatin accessibility and gene expression in primed NK cells +/− aNK1.1, and (I) select Myc targets differentially regulated in HD IL-15+aNK1.1.
Next, NK cell responses to ITAM signaling via anti-NK1.1 in the naïve state versus primed state were evaluated. NK1.1 stimulation of naïve cells strongly upregulated chemokine genes such as Ccl1 and Ccl22 rather than Ifng (Fig. 4D). In contrast, NK1.1 stimulation of primed cells strongly upregulated cytokine genes such as Ifng and Tnf (Fig. 4E). The distinct transcriptional response of primed NK cells to ITAM stimulation was not solely due to in vitro culture, as primed cells and LD IL-15 cultured NK cells also exhibited distinct gene expression patterns after stimulation (Fig. 4F, Supplemental Fig. 1H). Consistent with our prior data (Fig. 1C), Ifng transcript was highly upregulated only in NK1.1-activated primed NK cells (Fig. 4D–G).
Based on the hypothesis that ITAM signaling leads to a distinct transcriptional signature in IL-15 primed NK cells, allowing for metabolic flexibility, k-means clustering was performed, and genes uniquely upregulated in the NK1.1-stimulated primed NK cells were identified (Supplemental Fig. 1F, Supplemental table 1). The cluster with upregulated genes only in NK1.1-activated primed cells contained Ifng, cytokine subunit Il23a, and genes associated with protein synthesis (Gcnt4, Polr3d, Trmt10a) and secretion (Bhlha15) compared to NK1.1-stimulated LD IL-15 cultured cells (Fig. 4G). Several of these uniquely upregulated genes were also identified to have increased chromatin accessibility in the ATAC-seq dataset, including Akap1, Arnt2, Cx3cl1, Dok7 (Fig. 2A, 4H). Additionally, some of the genes with increased chromatin accessibility in primed cells also exhibited increased gene expression compared to naïve unstimulated cells, and their expression further increased in receptor-stimulated primed cells (Dusp14, Gzme, Lgals3, Mycn, Spp1, Zbtb32) (Fig. 4H). Lgals3, Spp1, and Zbtb32 have been previously reported to play a role in the regulation of various NK cell functions (53, 55–57). Like Ifng expression patterns, genes encoding several cytokines (Tnf, Il23a) and chemokines (Ccl3, Cx3cl1, Xcl1) were also upregulated only in NK1.1-activated primed NK cells, although in a different cluster than Ifng (Fig. 4G). Collectively, these data support that altered chromatin accessibility leads to a differential transcriptional response to ITAM signaling by IL-15 primed NK cells.
Among the genes uniquely upregulated in the NK1.1-stimulated primed NK cells were several Myc targets, including genes encoding for RNA polymerase I and III subunits (58, 59). Polr3d and Polr1a,c,e, to a lesser extent, were uniquely upregulated in NK1.1-stimulated primed cells (Fig. 4I). Additionally, Maf1, a known negative regulator of polymerase III-mediated transcription (60), was uniquely downregulated. Cdkn1a, which Maf1 has been reported to repress through a Pol III-dependent mechanism (61), is also uniquely upregulated in NK1.1-stimulated primed cells (Fig. 4I). Thus, IL-15 primed NK cells exhibit unique and distinct transcriptional changes in response to NK1.1 activation.
With the knowledge that Ifng is transcriptionally regulated with both IL12/18 and primed NK cells with ITAM stimulation, we hypothesized that these conditions might share a common pathway of transcriptional regulation. Among the 159 genes that overlapped between those conditions, we again identified Ifng, Myc, and several known Myc targets (Polr3d, Polr1e, Fosl1, Mybbp1a, etc.) (Fig. 5A, Supplemental table 2).
Figure 5. Myc marks NK cells capable of Ifng transcription.
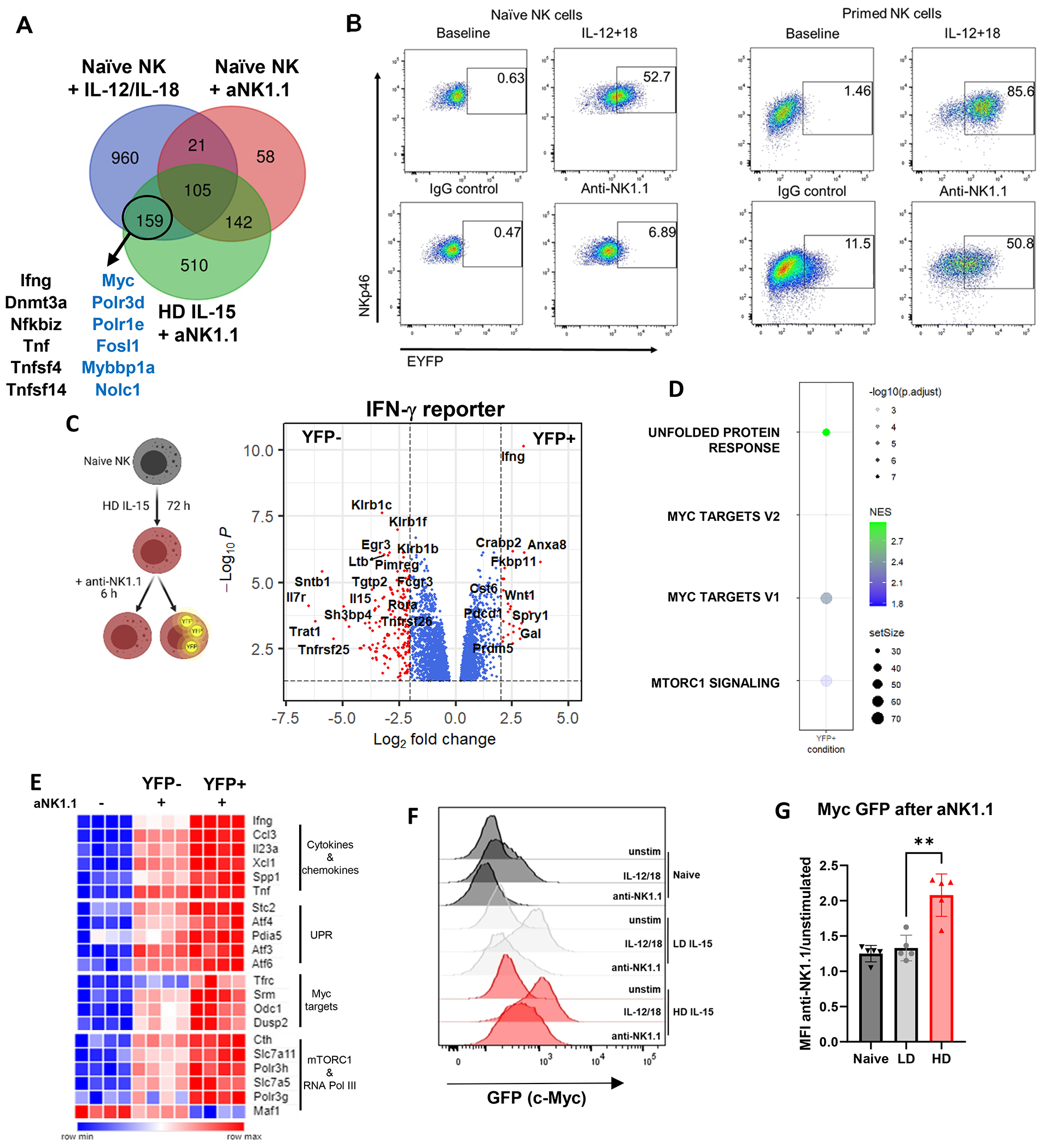
A) Comparison of genes upregulated in naïve NK cells with IL-12/IL-18 (blue), anti-NK1.1 (red), or in HD IL-15 primed NK cells with anti-NK1.1 (green, compared to HD IL-15 alone). Blue text marks Myc targets. (although Dnmt3a has been shown experimentally to be a Myc target). B) Representative flow cytometry plots from naïve or primed NK cells from Ifng reporter mice, +/− 6 h stimulation. C-E) NK cells from 5 GREAT mice (3 males, 2 pooled females) were primed with IL-15, then stimulated via plate-bound anti-NK1.1 for 6 hours. Cells were then sorted based on YFP. One independent experiment, 4 biological replicates. C) Volcano plot shows differentially expressed genes from the IFN-γ+ (YFP+) and IFN-γ− (YFP−) comparison. D) Bubbleplot of GSEA analysis pathways upreg in IFN-γ+ compared to IFN-γ−. E) Heatmap highlighting select genes that are differentially expressed in YFP+ cells compared to YFP− cells. F-G) NK cells from c-Myc EGFP reporter mice were either primed prior to stimulation or directly stimulated for 3 hours. EGFP was measured by flow cytometry. F) Histogram from a representative experiment. G) EGFP quantification shown as mean fluorescence intensity of the stimulated condition relative to their respective unstimulated. 5 independent experiments, 3 mice per experiment.
While Ifng transcription is induced in primed NK cells (Fig. 1C–D), ~50% of cells produce IFN-γ protein after 6 hours of activating receptor stimulation (Fig. 1E). To more specifically evaluate changes associated with Ifng transcription, RNA-seq was performed using primed NK cells from an Ifng reporter mouse (GREAT mice, (27)) (Fig. 5B–C). These mice serve as a faithful reporter for transcription of Ifng (Supplemental Fig. 1G), and consistent with our prior data, naïve NK cells from GREAT mice increase YFP expression only after IL-12/18 stimulation, with minimal change with anti-NK1.1 stimulation (Fig. 5B). Primed NK cells, however, express YFP after anti-NK1.1 stimulation, indicating new Ifng transcript production (Fig. 5B). For RNA-seq, primed cells were stimulated with anti-NK1.1 and sorted based on YFP expression. Ifng transcript was the most significant gene upregulated in the YFP+ population, demonstrating the utility of this reporter in murine NK cells (Fig. 5C). Several pathways were upregulated in the YFP+ (IFN-γ-producing) population compared to the YFP− population, including the unfolded protein response (Pdia5, Stc2, Atf3, Atf4, Atf6), Myc targets (Srm, Odc1, Tfrc, Dusp2), and mTORC1 signaling (Cth, Slc7a11, Slc7a5, Polr3g) (Fig. 5D–E). Among Myc targets, RNA polymerase III genes Polr3h and Polr3g were upregulated, while negative regulator of RNA polymerase III Maf1 was downregulated only in IFN-γ+ cells, supporting that downregulation of this gene is specific to those cells actively transcribing Ifng (Fig. 5E). Additionally, Tnf, Il23a, Spp1, Ccl3 and Xcl1 were upregulated (Fig. 5E), indicating that the expression of these genes identified in the previous dataset correlates with Ifng transcript expression.
c-Myc marks NK cells capable of Ifng transcription
Myc (c-Myc) is a basic helix-loop-helix transcription factor that forms a heterodimer with Myc associated factor X (Max) to bind E-box sequences and modulate expression of target genes. In NK cells, cytokine (IL-2+IL-12, IL-12+IL-18, or IL-15) stimulation upregulates c-Myc expression (62–65), which, in turn, has been linked to metabolic and effector function changes (62, 64, 65). Given the convergence of transcriptional evidence here and existing literature suggesting a link between Myc and Ifng transcription, we further characterized the potential relationship between Myc and Ifng in our system. To assess the expression of c-Myc in NK cells after priming, mice expressing a N-terminal fusion EGFP/MYC protein (28) (c-MycEGFP) were evaluated. IL-12+IL-18 stimulation (3 hours) upregulated Myc protein (marked by GFP expression) in naïve, LD IL-15 cultured, and HD IL-15 primed NK cells. In contrast, ITAM-based anti-NK1.1 stimulation resulted in upregulation of c-Myc only in HD IL-15 primed NK cells (Fig. 5F, 5G). Thus, c-Myc expression is upregulated in stimulated NK cells capable of Ifng transcription.
One hypothesis is that c-Myc may directly regulate Ifng transcription in primed NK cells. The Ifng locus features several E-box sequences and degenerate sequences, supporting the possibility of Myc binding and regulating Ifng transcription. To determine if Myc transcription factor activity regulates Ifng production, a pharmacological approach to inhibit Myc-Max dimerization, and therefore abrogate Myc transcription factor function, was utilized. Primed NK cells were treated with KJ-Pyr-9 (66) during the 6 hour anti-NK1.1 (Fig. 6A) or anti-Ly49D stimulation (Supplemental Fig. 2A). KJ-Pyr-9 interferes with Myc-Max complex formation in cells and reduces MYC-driven transcriptional signature (66, 67). Treatment with KJ-Pyr-9 led to reduced Ifng transcript upregulation upon activating receptor stimulation in primed NK cells, and a reversion to metabolic dependence for IFN-γ production (Fig. 6A–B, Supplemental Fig. 2A–B). Myc inhibition did not affect cytokine-stimulated Ifng transcription, but did inhibit transcription when cytokine-stimulated cells were cultured with the OXPHOS inhibitor oligomycin (Supplemental Fig. 2C–D). This suggests that Myc-Max dimerization may be essential for activating receptor-stimulated Ifng transcription, but only becomes important for cytokine-stimulated Ifng transcription with metabolic inhibition.
Fig 6. Myc inhibition alters IFN-γ regulation.
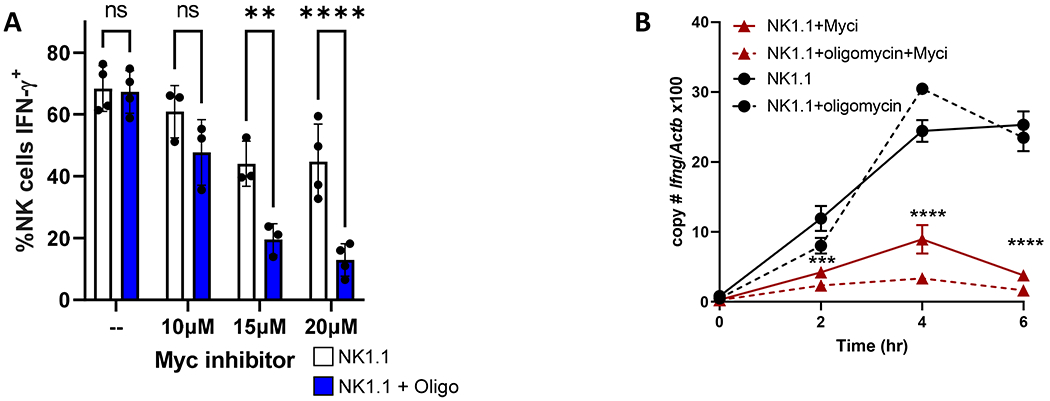
Purified mouse NK cells were cultured for 72 hours in 100 ng/ml IL-15, washed to remove the cytokine, then stimulated with anti-NK1.1 (A,B) +/− Myc inhibitor KJ-Pyr-9 (Myci) +/− OXPHOS inhibitor oligomycin. A) % IFN-γ+ NK cells were measured by flow cytometry, n=8 mice, 4 independent experiments; 2way ANOVA, Sidak’s correction for multiple comparison test. B) Ifng transcript was measured by quantitative RT-PCR normalized to beta-actin (Actb). Representative experiment with 1 technical and 1 biological replicate (2 mice), out of 3 independent experiments. Statistical analysis: 2-way ANOVA; * denotes significance from NK1.1 vs. NK1.1+Myci comparison.
Next, a genetic approach in which Myc was conditionally deleted in NK cells (Ncr1-MycΔ/Δ) in vitro with hydroxytamoxifen (4-OHT) was used to determine if Myc is required for Ifng upregulation (Fig. 7A–B). In this model, iCre recombinase nuclear activity was induced by tamoxifen and marked by YFP expression, allowing for detection and temporal control of Myc deletion, without affecting NK development and homeostasis. Enriched NK cells were primed (72 hours, 100 ng/ml IL-15) in the presence of 4-hydroxytamoxifen (48-72 hours, 0.8 uM 4-OHT). Myc deletion was confirmed by real-time PCR comparison of cells marked by Cre activity (YFP+) or not (YFP−) (Fig. 7B), and by flow cytometry (Fig. 7C). In contrast to Myc inhibitor data, there was no significant change in the Ifng transcript or the percentage of IFN-γ+ NK cells between YFP+ and YFP− cells after 6 hours of anti-NK1.1 stimulation, regardless of the presence or absence of oligomycin (Fig. 7D–E). Furthermore, c-Myc was not required during the IL-15 priming phase for metabolic flexibility for activating receptor stimulated IFN-γ production (Fig. 7F). Therefore, c-Myc marks NK cells capable of Ifng transcription, but it is not required itself for Ifng transcription in IL-15 primed NK cells. There are several possibilities that could explain the contradictory results observed with chemical Myc inhibition, including lack of inhibitor specificity, affecting other leucine zipper-based dimerization of transcription factors, and compensatory activity of another Myc family member in the genetic model.
Figure 7. Myc deletion does not affect Ifng transcription.
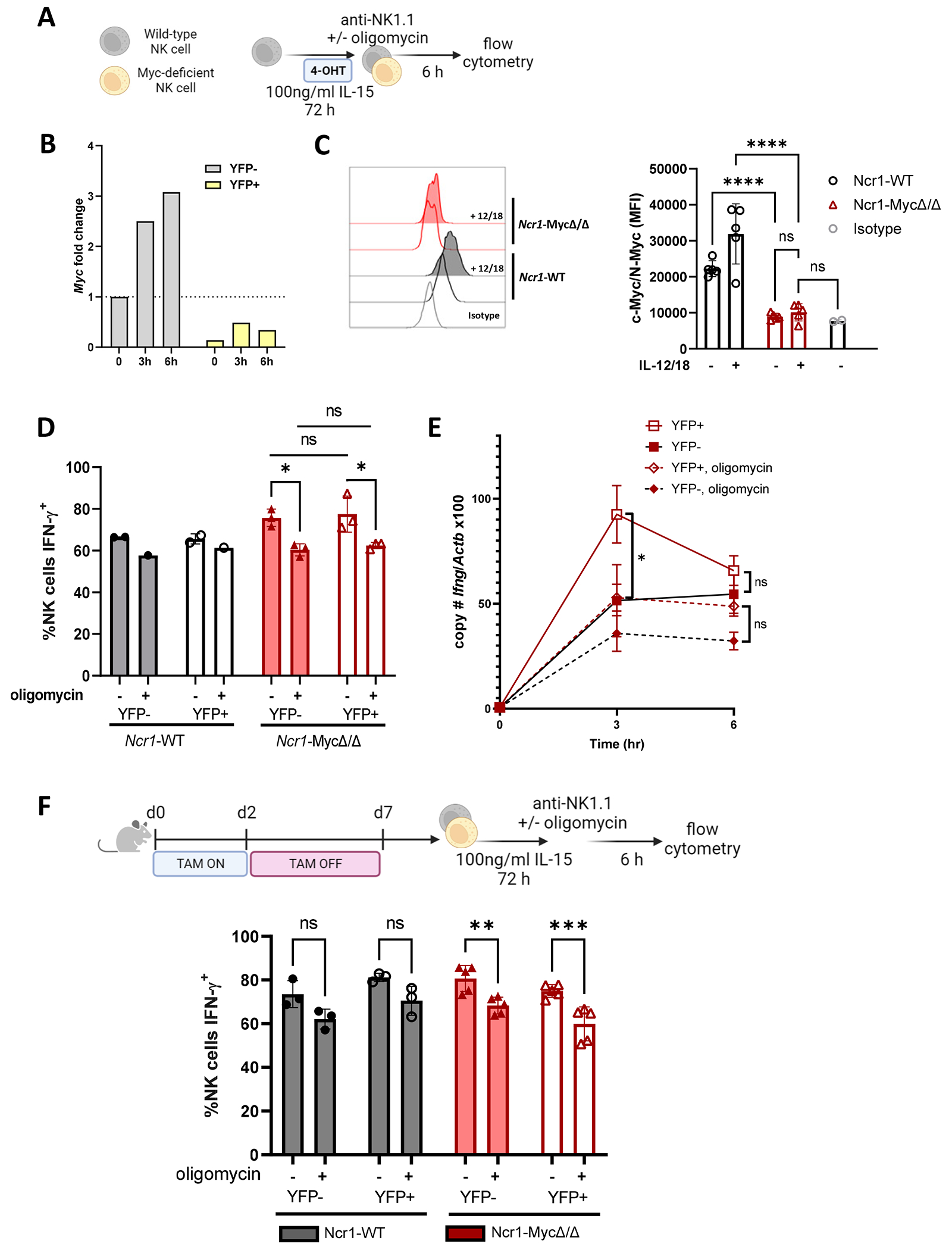
A) Experimental schema. Enriched splenic NK cells were cultured in 100 ng/ml IL-15. 4-OHT was added after 6-24 hours at a final concentration of 0.8 uM for the remainder of the 72 hours. Cells were washed and then stimulated via IL-12+IL-18 (1ng/ml each) or plate-bound anti-NK1.1 +/− 100 nM oligomycin. B) Myc transcript (day 3) normalized to unstimulated YFP− cells from Ncr1-MycΔ/Δ; representative experiment. C) Confirmation of c-Myc protein deletion by flow cytometry, cells were primed with 100 ng/ml IL-15 with 4-OHT as in (A), and c-Myc protein measured without or with IL-12+IL-18 (1ng/ml each, 6hr). Representative histogram and median fluorescence intensity in the YFP+ populations, 2 independent experiments, 2-5 mice per group. D) % IFN-γ+ NK cells; Ncr1-WT were used as controls. 2 independent experiments, 2-3 mice per experiment. E) NK cells from Ncr1-MycΔ/Δ mice were primed and stimulated with anti-NK1.1 for up to 6 hours. Absolute copy number of Ifng and Actb were quantified by real-time PCR for sorted YFP+ and YFP− populations. Results represent the ratio of Ifng to Actb x 100 and the mean +/− SEM from 3 independent experiments. F) Experimental schema and % IFN-γ+ NK cells measured by flow cytometry. 2 independent experiments with replicate wells, 2-6 mice per group. Statistical analysis: 2 way ANOVA.
DISCUSSION
In this study, we demonstrate that IL-15 priming leads to changes in murine NK regulatory networks, altering IFN-γ regulation and providing metabolic flexibility upon activating receptor stimulation. Such priming has the potential to impart NK cells with enhanced function in metabolically challenging environments such as the tumor microenvironment. Furthermore, IL-15 based therapies have been developed, with improved antitumor activity in preclinical models, especially when combined with immune checkpoint inhibition strategies (22).
Naïve splenic NK cells constitutively express Ifng transcript, but not protein, at levels greater than naïve CD4 T cells sorted from lymph nodes in C57BL/6 mice (68). This suggests a translational repression mechanism occurs in resting murine NK cells. In contrast to IFN-γ regulation in CD4+ T cells, where GAPDH acts as a translation repressor by binding to the Ifng 3’UTR (50), we demonstrate here that metabolic regulation of IFN-γ in naïve murine NK cells is 3’UTR-independent. Furthermore, our results suggest the main mode of IFN-γ production in cytokine stimulated naïve NK cells is by production of new Ifng transcript, rather than production of the protein from pre-formed Ifng transcript, as is the case for activating receptor stimulation.
IL-15 primed NK cells exhibited metabolic flexibility and a switch to transcriptional regulation for activating receptor stimulated IFN-γ production. Glycolysis, and, to a lesser extent, mTORC1, are required during the priming phase for this effect. To investigate mechanisms responsible for the switch to IFN-γ transcriptional regulation in primed NK cells, we tested several hypotheses. ATAC-seq analysis demonstrated that while IL-15 priming leads to broad changes in chromatin accessibility, it does not alter accessibility at the Ifng itself. In NK cells, the IFN-γ locus is already accessible and primed for transcription when compared to T cells (68–72). However, it is possible that methylation and acetylation changes modulate transcription factor occupancy, which is something not explored here. DNA hypomethylation in the IFNG locus has been reported during terminal differentiation of human NK cells and in an HCMV adaptive subset with a greater capacity to produce IFN-γ (73, 74).
Canonical ITAM signaling strength was not different between primed and LD IL-15 cultured NK cells for several of the signaling molecules assessed, including MAPK p38, which has been linked to increased IFNG transcript stability in human NK cells (75, 76). However, IL-15 priming led to a new transcriptional profile upon activating receptor stimulation. We hypothesize that changes in chromatin accessibility induced by IL-15 priming fundamentally alter how NK cells receive and respond to signaling downstream from ITAM receptor activation, either directly by chromatin accessibility or potentially through upregulation of pathways that allow for non-canonical signaling. For example, IL-15 priming led to a strong enrichment for AP-1 family motifs in the peaks with increased chromatin accessibility, and these motifs are also enriched in memory (Ly49H+) NK cells in mice (77) and human HCMV adaptive NK cells (78). This suggests IL-15 induces an epigenetic signature associated with enhanced function.
A significant number of published NK cell studies rely on IL-15 or IL-2, a cytokine with similar effects and shared receptor subunits, to maintain or expand NK cells in culture. High dose IL-15 in vitro significantly alters NK cell metabolism and functional capacity, and, as we show here, fundamentally alters regulation of IFN-γ production (16, 19, 79). Therefore, it is important to consider how cells are treated ex vivo when trying to extrapolate in vitro findings to what might be occurring in vivo. We demonstrate that NK1.1 activation of IL-15 primed NK cells generated a distinct transcriptional profile compared to LD IL-15 cultured or naïve NK cells and marked a switch to Ifng transcriptional regulation. RNA-seq studies identified c-Myc and its targets to be positively correlated with Ifng transcription. In T cells, Myc is upregulated upon TCR stimulation, and protein levels are then sustained with the help of cytokines (such as IL-2 or IL-15) (80) and amino acid transporter Slc7a5 (81). Similarly, another study demonstrated that in NK cells, IL-2+IL-12 stimulation leads to c-Myc upregulation, as well as Slc7a5 expression and activity, which in turn is required for c-Myc expression at later time points (62). Additionally, loss of c-Myc protein in vitro reduced the percentage of IFN-γ+ NK cells and the amount of IFN-γ produced per cell after IL-2+IL-12 (62). While activating receptor stimulation leads to c-Myc upregulation in primed NK cells, loss of c-Myc in these cells did not alter the percentage of IFN-γ+ NK cells with ITAM stimulation. We did observe, however, striking results with Myc:Max heterodimer formation inhibitor KJ-Pyr-9. Ifng transcript and the percentage of IFN-γ+ NK cells were reduced, with further reduction in the latter upon OXPHOS inhibition. The differences between the results obtained with the inhibitor and those obtained with the genetic model may simply indicate off-target effects of the inhibitor or a lack of specificity for disruption of Myc:Max dimer formation. Inhibition of oncogenic activity of N-MYC, v-jun and PIK3CA-H1047R (gain of function) has been reported with the inhibitor used in the studies here (66), supporting the possibility of off-target effects. In addition, a separate but not mutually exclusive mechanism may involve other Myc family members compensating for c-Myc’s absence. Mycn, encoding for n-Myc, is not normally expressed in NK cells but Mycn transcript was upregulated in primed NK cells (Fig. 4H). This, together with KJ-Pyr-9 inhibition of N-MYC oncogenic activity (66), offers a potential explanation for the discrepancies between the inhibitor and genetic model results that will require further investigation.
Another pathway observed to be specifically upregulated in IFN-γ+ cells here was the unfolded protein response. This pathway has been linked to production of an active transcriptional activator, spliced XBP1 (XBP1s) (82, 83), which acts downstream of IL-15 to support human NK cell survival (84) and can interact with T-bet to support GZMB (84) and Ifng/IFNG transcription (84, 85). Further studies of XBP1s function in NK cells may determine its role in Ifng regulation in IL-15 primed NK cells. The UPR pathway has also been linked to IL23A regulation and subsequent IL-23 production in myeloid cells (86–88), and may explain the Il23a upregulation observed in IFN-γ+ cells.
Overall, this study highlights that IL-15 dose and culture time can dramatically alter chromatin accessibility and regulatory networks in murine NK cells. Functionally, primed NK cells switch to Ifng transcriptional regulation in response to ITAM signaling via NK1.1, and no longer require a secondary, metabolism-derived signal for optimal IFN-γ production. Therefore, primed cells have distinct properties from freshly isolated NK cells or NK cells cultured in low dose IL-15, and interpretation of results from NK cell studies should take into account cell culture conditions such as IL-15 dose and time. While IL-15 priming confers this advantage, supporting IL-15 use for immunotherapeutic purposes, IL-15 use must be carefully considered and calibrated in light of reported detrimental effects (22, 89, 90).
Supplementary Material
Key points.
IL-15 priming of NK cells enables metabolic flexibility for IFN-γ production.
IL-15 priming alters transcriptional response to activating receptor stimulation.
Myc marks NK cells capable of Ifng transcription, but is not required.
Acknowledgements
The authors would like to thank Kelsey Trammel and Katherine Owens for mouse colony management and technical assistance, and Prabhakar Sairam Andhey for assistance with data processing. We thank Eric Tycksen and the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine for help with genomic analysis.
Figures 3A, 4A, 5C, 7A, and part of 7F were created with BioRender.com.
Work in the Cooper laboratory was supported by NIH R01AI127752. The Genome Technology Access Center GTAC is partially supported by NCI Cancer Center Support Grant #P30 CA91842 to the Siteman Cancer Center and by ICTS/CTSA Grant# UL1 TR000448 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. This publication is solely the responsibility of the authors and does not necessarily represent the official view of NCRR or NIH. This research was supported, in part, by the intramural Research Program of the NIH, National Cancer Institute.
References
- 1.Prager I, and Watzl C. 2019. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J Leukoc Biol 105: 1319–1329. [DOI] [PubMed] [Google Scholar]
- 2.Hsu HT, Mace EM, Carisey AF, Viswanath DI, Christakou AE, Wiklund M, Onfelt B, and Orange JS. 2016. NK cells converge lytic granules to promote cytotoxicity and prevent bystander killing. J Cell Biol 215: 875–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alspach E, Lussier DM, and Schreiber RD. 2019. Interferon gamma and Its Important Roles in Promoting and Inhibiting Spontaneous and Therapeutic Cancer Immunity. Cold Spring Harb Perspect Biol 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orange JS, Wang B, Terhorst C, and Biron CA. 1995. Requirement for natural killer cell-produced interferon gamma in defense against murine cytomegalovirus infection and enhancement of this defense pathway by interleukin 12 administration. J Exp Med 182: 1045–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelly JM, Darcy PK, Markby JL, Godfrey DI, Takeda K, Yagita H, and Smyth MJ. 2002. Induction of tumor-specific T cell memory by NK cell-mediated tumor rejection. Nat Immunol 3: 83–90. [DOI] [PubMed] [Google Scholar]
- 6.Borst K, Flindt S, Blank P, Larsen PK, Chhatbar C, Skerra J, Spanier J, Hirche C, Konig M, Alanentalo T, Hafner M, Waibler Z, Pfeffer K, Sexl V, Sutter G, Muller W, Graalmann T, and Kalinke U. 2020. Selective reconstitution of IFN-gamma gene function in Ncr1+ NK cells is sufficient to control systemic vaccinia virus infection. PLoS Pathog 16: e1008279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Long EO, Kim HS, Liu D, Peterson ME, and Rajagopalan S. 2013. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol 31: 227–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooper MA, Bush JE, Fehniger TA, VanDeusen JB, Waite RE, Liu Y, Aguila HL, and Caligiuri MA. 2002. In vivo evidence for a dependence on interleukin 15 for survival of natural killer cells. Blood 100: 3633–3638. [DOI] [PubMed] [Google Scholar]
- 9.Becknell B, and Caligiuri MA. 2005. Interleukin-2, interleukin-15, and their roles in human natural killer cells. Adv Immunol 86: 209–239. [DOI] [PubMed] [Google Scholar]
- 10.Yajima T, Nishimura H, Wajjwalku W, Harada M, Kuwano H, and Yoshikai Y. 2002. Overexpression of interleukin-15 in vivo enhances antitumor activity against MHC class I-negative and -positive malignant melanoma through augmented NK activity and cytotoxic T-cell response. Int J Cancer 99: 573–578. [DOI] [PubMed] [Google Scholar]
- 11.Munger W, DeJoy SQ, Jeyaseelan R Sr., Torley LW, Grabstein KH, Eisenmann J, Paxton R, Cox T, Wick MM, and Kerwar SS. 1995. Studies evaluating the antitumor activity and toxicity of interleukin-15, a new T cell growth factor: comparison with interleukin-2. Cell Immunol 165: 289–293. [DOI] [PubMed] [Google Scholar]
- 12.Mah AY, Rashidi A, Keppel MP, Saucier N, Moore EK, Alinger JB, Tripathy SK, Agarwal SK, Jeng EK, Wong HC, Miller JS, Fehniger TA, Mace EM, French AR, and Cooper MA. 2017. Glycolytic requirement for NK cell cytotoxicity and cytomegalovirus control. JCI Insight 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carson WE, Giri JG, Lindemann MJ, Linett ML, Ahdieh M, Paxton R, Anderson D, Eisenmann J, Grabstein K, and Caligiuri MA. 1994. Interleukin (IL) 15 is a novel cytokine that activates human natural killer cells via components of the IL-2 receptor. J Exp Med 180: 1395–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conlon KC, Lugli E, Welles HC, Rosenberg SA, Fojo AT, Morris JC, Fleisher TA, Dubois SP, Perera LP, Stewart DM, Goldman CK, Bryant BR, Decker JM, Chen J, Worthy TA, Figg WD Sr., Peer CJ, Sneller MC, Lane HC, Yovandich JL, Creekmore SP, Roederer M, and Waldmann TA. 2015. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol 33: 74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carson WE, Fehniger TA, Haldar S, Eckhert K, Lindemann MJ, Lai CF, Croce CM, Baumann H, and Caligiuri MA. 1997. A potential role for interleukin-15 in the regulation of human natural killer cell survival. J Clin Invest 99: 937–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fehniger TA, Cai SF, Cao X, Bredemeyer AJ, Presti RM, French AR, and Ley TJ. 2007. Acquisition of murine NK cell cytotoxicity requires the translation of a pre-existing pool of granzyme B and perforin mRNAs. Immunity 26: 798–811. [DOI] [PubMed] [Google Scholar]
- 17.Lucas M, Schachterle W, Oberle K, Aichele P, and Diefenbach A. 2007. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 26: 503–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marcais A, Cherfils-Vicini J, Viant C, Degouve S, Viel S, Fenis A, Rabilloud J, Mayol K, Tavares A, Bienvenu J, Gangloff YG, Gilson E, Vivier E, and Walzer T. 2014. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat Immunol 15: 749–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keppel MP, Saucier N, Mah AY, Vogel TP, and Cooper MA. 2015. Activation-specific metabolic requirements for NK Cell IFN-gamma production. J Immunol 194: 1954–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sheppard S, Santosa EK, Lau CM, Violante S, Giovanelli P, Kim H, Cross JR, Li MO, and Sun JC. 2021. Lactate dehydrogenase A-dependent aerobic glycolysis promotes natural killer cell anti-viral and anti-tumor function. Cell Rep 35: 109210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cimpean M, and Cooper MA. 2023. Metabolic regulation of NK cell antiviral functions during cytomegalovirus infection. J Leukoc Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma S, Caligiuri MA, and Yu J. 2022. Harnessing IL-15 signaling to potentiate NK cell-mediated cancer immunotherapy. Trends Immunol 43: 833–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Romee R, Cooley S, Berrien-Elliott MM, Westervelt P, Verneris MR, Wagner JE, Weisdorf DJ, Blazar BR, Ustun C, DeFor TE, Vivek S, Peck L, DiPersio JF, Cashen AF, Kyllo R, Musiek A, Schaffer A, Anadkat MJ, Rosman I, Miller D, Egan JO, Jeng EK, Rock A, Wong HC, Fehniger TA, and Miller JS. 2018. First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood 131: 2515–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosario M, Liu B, Kong L, Collins LI, Schneider SE, Chen X, Han K, Jeng EK, Rhode PR, Leong JW, Schappe T, Jewell BA, Keppel CR, Shah K, Hess B, Romee R, Piwnica-Worms DR, Cashen AF, Bartlett NL, Wong HC, and Fehniger TA. 2016. The IL-15-Based ALT-803 Complex Enhances FcgammaRIIIa-Triggered NK Cell Responses and In Vivo Clearance of B Cell Lymphomas. Clin Cancer Res 22: 596–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sohn H, and Cooper MA. 2023. Metabolic regulation of NK cell function: implications for immunotherapy. Immunometabolism (Cobham) 5: e00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hodge DL, Berthet C, Coppola V, Kastenmuller W, Buschman MD, Schaughency PM, Shirota H, Scarzello AJ, Subleski JJ, Anver MR, Ortaldo JR, Lin F, Reynolds DA, Sanford ME, Kaldis P, Tessarollo L, Klinman DM, and Young HA. 2014. IFN-gamma AU-rich element removal promotes chronic IFN-gamma expression and autoimmunity in mice. J Autoimmun 53: 33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reinhardt RL, Liang HE, and Locksley RM. 2009. Cytokine-secreting follicular T cells shape the antibody repertoire. Nat Immunol 10: 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang CY, Bredemeyer AL, Walker LM, Bassing CH, and Sleckman BP. 2008. Dynamic regulation of c-Myc proto-oncogene expression during lymphocyte development revealed by a GFP-c-Myc knock-in mouse. Eur J Immunol 38: 342–349. [DOI] [PubMed] [Google Scholar]
- 29.Wagner JA, Wong P, Schappe T, Berrien-Elliott MM, Cubitt C, Jaeger N, Lee M, Keppel CR, Marin ND, Foltz JA, Marsala L, Neal CC, Sullivan RP, Schneider SE, Keppel MP, Saucier N, Cooper MA, and Fehniger TA. 2020. Stage-Specific Requirement for Eomes in Mature NK Cell Homeostasis and Cytotoxicity. Cell Rep 31: 107720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Alboran IM, O’Hagan RC, Gartner F, Malynn B, Davidson L, Rickert R, Rajewsky K, DePinho RA, and Alt FW. 2001. Analysis of C-MYC function in normal cells via conditional gene-targeted mutation. Immunity 14: 45–55. [DOI] [PubMed] [Google Scholar]
- 31.Overbergh L, Giulietti A, Valckx D, Decallonne R, Bouillon R, and Mathieu C. 2003. The use of real-time reverse transcriptase PCR for the quantification of cytokine gene expression. J Biomol Tech 14: 33–43. [PMC free article] [PubMed] [Google Scholar]
- 32.Livak KJ, and Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 33.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu S, Li D, Lyu C, Gontarz PM, Miao B, Madden PAF, Wang T, and Zhang B. 2021. AIAP: A Quality Control and Integrative Analysis Package to Improve ATAC-seq Data Analysis. Genomics Proteomics Bioinformatics 19: 641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Granja JM, Corces MR, Pierce SE, Bagdatli ST, Choudhry H, Chang HY, and Greenleaf WJ. 2021. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat Genet 53: 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li D, Purushotham D, Harrison JK, Hsu S, Zhuo X, Fan C, Liu S, Xu V, Chen S, Xu J, Ouyang S, Wu AS, and Wang T. 2022. WashU Epigenome Browser update 2022. Nucleic Acids Res 50: W774–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Love MI, Huber W, and Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liao Y, Smyth GK, and Shi W. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930. [DOI] [PubMed] [Google Scholar]
- 41.Zenkova D,KV, Sablina R, Artyomov M, Sergushichev A. 2018. Phantasus: visual and interactive gene expression analysis. [DOI] [PMC free article] [PubMed]
- 42.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu G 2020. enrichplot: Visualization of Functional Enrichment Result.
- 44.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang L, Wang S, and Li W. 2012. RSeQC: quality control of RNA-seq experiments. Bioinformatics 28: 2184–2185. [DOI] [PubMed] [Google Scholar]
- 46.Robinson MD, McCarthy DJ, and Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK. 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43: e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu R, Holik AZ, Su S, Jansz N, Chen K, Leong HS, Blewitt ME, Asselin-Labat ML, Smyth GK, and Ritchie ME. 2015. Why weight? Modelling sample and observational level variability improves power in RNA-seq analyses. Nucleic Acids Res 43: e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, and Sergushichev A. 2021. Fast gene set enrichment analysis. 060012. [Google Scholar]
- 50.Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, Weber JD, Pearce EJ, Jones RG, and Pearce EL. 2013. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153: 1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hodge DL, Martinez A, Julias JG, Taylor LS, and Young HA. 2002. Regulation of nuclear gamma interferon gene expression by interleukin 12 (IL-12) and IL-2 represents a novel form of posttranscriptional control. Mol Cell Biol 22: 1742–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nandagopal N, Ali AK, Komal AK, and Lee SH. 2014. The Critical Role of IL-15-PI3K-mTOR Pathway in Natural Killer Cell Effector Functions. Front Immunol 5: 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leavenworth JW, Verbinnen B, Wang Q, Shen E, and Cantor H. 2015. Intracellular osteopontin regulates homeostasis and function of natural killer cells. Proc Natl Acad Sci U S A 112: 494–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lanier LL 2008. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol 9: 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chaudhari AD, Gude RP, Kalraiya RD, and Chiplunkar SV. 2015. Endogenous galectin-3 expression levels modulate immune responses in galectin-3 transgenic mice. Mol Immunol 68: 300–311. [DOI] [PubMed] [Google Scholar]
- 56.Brittoli A, Fallarini S, Zhang H, Pieters RJ, and Lombardi G. 2018. “In vitro” studies on galectin-3 in human natural killer cells. Immunol Lett 194: 4–12. [DOI] [PubMed] [Google Scholar]
- 57.Beaulieu AM, Zawislak CL, Nakayama T, and Sun JC. 2014. The transcription factor Zbtb32 controls the proliferative burst of virus-specific natural killer cells responding to infection. Nat Immunol 15: 546–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gomez-Roman N, Grandori C, Eisenman RN, and White RJ. 2003. Direct activation of RNA polymerase III transcription by c-Myc. Nature 421: 290–294. [DOI] [PubMed] [Google Scholar]
- 59.Kenneth NS, Ramsbottom BA, Gomez-Roman N, Marshall L, Cole PA, and White RJ. 2007. TRRAP and GCN5 are used by c-Myc to activate RNA polymerase III transcription. Proc Natl Acad Sci U S A 104: 14917–14922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bonhoure N, Praz V, Moir RD, Willemin G, Mange F, Moret C, Willis IM, and Hernandez N. 2020. MAF1 is a chronic repressor of RNA polymerase III transcription in the mouse. Sci Rep 10: 11956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee YL, Li YC, Su CH, Chiao CH, Lin IH, and Hsu MT. 2015. MAF1 represses CDKN1A through a Pol III-dependent mechanism. Elife 4: e06283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Loftus RM, Assmann N, Kedia-Mehta N, O’Brien KL, Garcia A, Gillespie C, Hukelmann JL, Oefner PJ, Lamond AI, Gardiner CM, Dettmer K, Cantrell DA, Sinclair LV, and Finlay DK. 2018. Amino acid-dependent cMyc expression is essential for NK cell metabolic and functional responses in mice. Nat Commun 9: 2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dong H, Adams NM, Xu Y, Cao J, Allan DSJ, Carlyle JR, Chen X, Sun JC, and Glimcher LH. 2019. The IRE1 endoplasmic reticulum stress sensor activates natural killer cell immunity in part by regulating c-Myc. Nat Immunol 20: 865–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Khameneh HJ, Fonta N, Zenobi A, Niogret C, Ventura P, Guerra C, Kwee I, Rinaldi A, Pecoraro M, Geiger R, Cavalli A, Bertoni F, Vivier E, Trumpp A, and Guarda G. 2023. Myc controls NK cell development, IL-15-driven expansion, and translational machinery. Life Sci Alliance 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cichocki F, Hanson RJ, Lenvik T, Pitt M, McCullar V, Li H, Anderson SK, and Miller JS. 2009. The transcription factor c-Myc enhances KIR gene transcription through direct binding to an upstream distal promoter element. Blood 113: 3245–3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hart JR, Garner AL, Yu J, Ito Y, Sun M, Ueno L, Rhee JK, Baksh MM, Stefan E, Hartl M, Bister K, Vogt PK, and Janda KD. 2014. Inhibitor of MYC identified in a Krohnke pyridine library. Proc Natl Acad Sci U S A 111: 12556–12561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Raffeiner P, Rock R, Schraffl A, Hartl M, Hart JR, Janda KD, Vogt PK, Stefan E, and Bister K. 2014. In vivo quantification and perturbation of Myc-Max interactions and the impact on oncogenic potential. Oncotarget 5: 8869–8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stetson DB, Mohrs M, Reinhardt RL, Baron JL, Wang ZE, Gapin L, Kronenberg M, and Locksley RM. 2003. Constitutive cytokine mRNAs mark natural killer (NK) and NK T cells poised for rapid effector function. J Exp Med 198: 1069–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chang S, and Aune TM. 2005. Histone hyperacetylated domains across the Ifng gene region in natural killer cells and T cells. Proc Natl Acad Sci U S A 102: 17095–17100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Balasubramani A, Mukasa R, Hatton RD, and Weaver CT. 2010. Regulation of the Ifng locus in the context of T-lineage specification and plasticity. Immunol Rev 238: 216–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tato CM, Martins GA, High FA, DiCioccio CB, Reiner SL, and Hunter CA. 2004. Cutting Edge: Innate production of IFN-gamma by NK cells is independent of epigenetic modification of the IFN-gamma promoter. J Immunol 173: 1514–1517. [DOI] [PubMed] [Google Scholar]
- 72.Hatton RD, Harrington LE, Luther RJ, Wakefield T, Janowski KM, Oliver JR, Lallone RL, Murphy KM, and Weaver CT. 2006. A distal conserved sequence element controls Ifng gene expression by T cells and NK cells. Immunity 25: 717–729. [DOI] [PubMed] [Google Scholar]
- 73.Luetke-Eversloh M, Cicek BB, Siracusa F, Thom JT, Hamann A, Frischbutter S, Baumgrass R, Chang HD, Thiel A, Dong J, and Romagnani C. 2014. NK cells gain higher IFN-gamma competence during terminal differentiation. Eur J Immunol 44: 2074–2084. [DOI] [PubMed] [Google Scholar]
- 74.Schlums H, Cichocki F, Tesi B, Theorell J, Beziat V, Holmes TD, Han H, Chiang SC, Foley B, Mattsson K, Larsson S, Schaffer M, Malmberg KJ, Ljunggren HG, Miller JS, and Bryceson YT. 2015. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity 42: 443–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mavropoulos A, Sully G, Cope AP, and Clark AR. 2005. Stabilization of IFN-gamma mRNA by MAPK p38 in IL-12- and IL-18-stimulated human NK cells. Blood 105: 282–288. [DOI] [PubMed] [Google Scholar]
- 76.Ochayon DE, Ali A, Alarcon PC, Krishnamurthy D, Kottyan LC, Borchers MT, and Waggoner SN. 2020. IL-33 promotes type 1 cytokine expression via p38 MAPK in human NK cells. J Leukoc Biol 107: 663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lau CM, Adams NM, Geary CD, Weizman OE, Rapp M, Pritykin Y, Leslie CS, and Sun JC. 2018. Epigenetic control of innate and adaptive immune memory. Nat Immunol 19: 963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ruckert T, Lareau CA, Mashreghi MF, Ludwig LS, and Romagnani C. 2022. Clonal expansion and epigenetic inheritance of long-lasting NK cell memory. Nat Immunol 23: 1551–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wagner JA, Rosario M, Romee R, Berrien-Elliott MM, Schneider SE, Leong JW, Sullivan RP, Jewell BA, Becker-Hapak M, Schappe T, Abdel-Latif S, Ireland AR, Jaishankar D, King JA, Vij R, Clement D, Goodridge J, Malmberg KJ, Wong HC, and Fehniger TA. 2017. CD56bright NK cells exhibit potent antitumor responses following IL-15 priming. J Clin Invest 127: 4042–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Preston GC, Sinclair LV, Kaskar A, Hukelmann JL, Navarro MN, Ferrero I, MacDonald HR, Cowling VH, and Cantrell DA. 2015. Single cell tuning of Myc expression by antigen receptor signal strength and interleukin-2 in T lymphocytes. EMBO J 34: 2008–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, and Cantrell DA. 2013. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol 14: 500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yoshida H, Matsui T, Yamamoto A, Okada T, and Mori K. 2001. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107: 881–891. [DOI] [PubMed] [Google Scholar]
- 83.Lee AH, Iwakoshi NN, and Glimcher LH. 2003. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 23: 7448–7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang Y, Zhang Y, Yi P, Dong W, Nalin AP, Zhang J, Zhu Z, Chen L, Benson DM, Mundy-Bosse BL, Freud AG, Caligiuri MA, and Yu J. 2019. The IL-15-AKT-XBP1s signaling pathway contributes to effector functions and survival in human NK cells. Nat Immunol 20: 10–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ma S, Han J, Li Z, Xiao S, Zhang J, Yan J, Tang T, Barr T, Kraft AS, Caligiuri MA, and Yu J. 2023. An XBP1s-PIM-2 positive feedback loop controls IL-15-mediated survival of natural killer cells. Sci Immunol 8: eabn7993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Goodall JC, Wu C, Zhang Y, McNeill L, Ellis L, Saudek V, and Gaston JS. 2010. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc Natl Acad Sci U S A 107: 17698–17703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Marquez S, Fernandez JJ, Teran-Cabanillas E, Herrero C, Alonso S, Azogil A, Montero O, Iwawaki T, Cubillos-Ruiz JR, Fernandez N, and Crespo MS. 2017. Endoplasmic Reticulum Stress Sensor IRE1alpha Enhances IL-23 Expression by Human Dendritic Cells. Front Immunol 8: 639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mogilenko DA, Haas JT, L’Homme L, Fleury S, Quemener S, Levavasseur M, Becquart C, Wartelle J, Bogomolova A, Pineau L, Molendi-Coste O, Lancel S, Dehondt H, Gheeraert C, Melchior A, Dewas C, Nikitin A, Pic S, Rabhi N, Annicotte JS, Oyadomari S, Velasco-Hernandez T, Cammenga J, Foretz M, Viollet B, Vukovic M, Villacreces A, Kranc K, Carmeliet P, Marot G, Boulter A, Tavernier S, Berod L, Longhi MP, Paget C, Janssens S, Staumont-Salle D, Aksoy E, Staels B, and Dombrowicz D. 2019. Metabolic and Innate Immune Cues Merge into a Specific Inflammatory Response via the UPR. Cell 178: 263. [DOI] [PubMed] [Google Scholar]
- 89.Donnelly RP, Loftus RM, Keating SE, Liou KT, Biron CA, Gardiner CM, and Finlay DK. 2014. mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J Immunol 193: 4477–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Felices M, Lenvik AJ, McElmurry R, Chu S, Hinderlie P, Bendzick L, Geller MA, Tolar J, Blazar BR, and Miller JS. 2018. Continuous treatment with IL-15 exhausts human NK cells via a metabolic defect. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA and ATAC sequencing data generated during this study is available at Gene Expression Omnibus with accession number GSE227894 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE227894).