

Key Points
-
•
The combination of ibrutinib and venetoclax induced deep and durable responses in treatment-naïve patients with Waldenström macroglobulinemia.
-
•
Planned study therapy was stopped early due to a higher-than-expected occurrence of ventricular arrhythmia in 4 of the 45 participants.
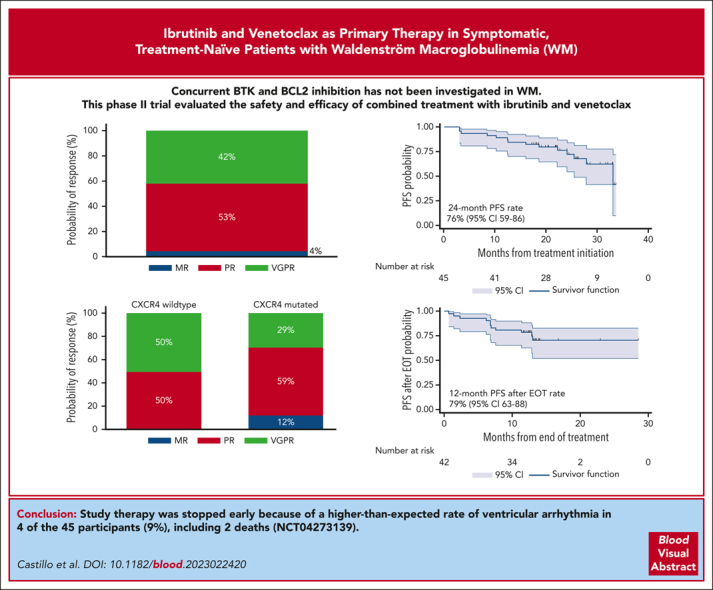
Visual Abstract
Abstract
Concurrent Bruton tyrosine kinase and BCL2 inhibition has not yet been investigated in Waldenström macroglobulinemia (WM). We performed an investigator-initiated trial of ibrutinib and venetoclax in symptomatic treatment-naïve patients with MYD88-mutated WM. Patients received ibrutinib 420 mg once daily (cycle 1), followed by a ramp-up of venetoclax to 400 mg daily (cycle 2). The combination was then administered for 22 additional 4-week cycles. The attainment of very good partial response (VGPR) was the primary end point. Forty-five patients were enrolled in this study. The median baseline characteristics were as follows: age 67 years, serum IgM 43 g/L, and hemoglobin 102 g/L. Seventeen patients (38%) carried CXCR4 mutations. Nineteen patients (42%) achieved VGPR. Grade 3 or higher adverse events included neutropenia (38%), mucositis (9%), and tumor lysis syndrome (7%). Atrial fibrillation occurred in 3 (9%), and ventricular arrhythmia in 4 (9%) patients that included 2 grade 5 events. With a median follow-up of 24.4 months, the 24-month progression-free survival (PFS) and overall survival (OS) rates were 76% and 96%, respectively, and were not impacted by CXCR4 mutations. The median time on therapy was 10.2 months, and the median time after the end of therapy (EOT) was 13.3 months. Eleven of the 12 progression events occurred after EOT, and the 12-month PFS rates after EOT were 79%; 93% if VGPR was attained, and 69% for other patients (P = .12). Ibrutinib and venetoclax induced high VGPR rates and durable responses after EOT, although they were associated with a higher-than-expected rate of ventricular arrhythmia in patients with WM, leading to early study treatment termination. This trial was registered at www.clinicaltrials.gov as #NCT04273139.
Castillo et al report on an investigator-initiated trial of ibrutinib and venetoclax in MYD88-mutated Waldenström macroglobulinemia. The primary end point of very good partial response was achieved in 42% of patients, with progression-free and overall survival of 76% and 96%, respectively. Unfortunately, the trial was stopped prematurely because of a surprisingly high rate of ventricular arrhythmia occurring in 4 patients, including 2 deaths.
Introduction
Waldenström macroglobulinemia (WM) is an indolent lymphoma characterized by malignant accumulation of immunoglobulin M (IgM)-secreting lymphoplasmacytic cells in the bone marrow (BM) and other organs.1 Somatic mutations in MYD88 and CXCR4 have been detected in 90% to 95% and 30% to 40% of patients with WM, respectively.2, 3, 4, 5, 6, 7, 8 Clinically, CXCR4 mutations are associated with higher serum IgM levels, higher rates of symptomatic hyperviscosity, and acquired von Willebrand disease.9, 10, 11
Because WM is incurable with available therapies and patients with WM can survive for decades,12 more treatment options are needed. Current frontline treatment options include chemoimmunotherapy or Bruton tyrosine kinase (BTK) inhibitors, which have proven safe and effective in patients with WM.13 Chemoimmunotherapy provides the option of finite-duration therapy, but potential downsides include parenteral administration, IgM flare, development of secondary myeloid malignancies, and significant infection risk.14, 15, 16, 17 BTK inhibitors, on the other hand, provide ease of oral administration; however, they have an indefinite therapy duration and cumulative risks of cardiac arrhythmia, hypertension, and bleeding.18, 19, 20, 21, 22, 23
The first-in-class BTK inhibitor ibrutinib is highly effective in treating WM and has been associated with a very good partial response (VGPR) rate of 30% in treatment-naïve patients with WM.22,24 CXCR4 mutations adversely impact the time to response, VGPR rate, and progression-free survival (PFS) with ibrutinib.11,25 The BCL2 antagonist venetoclax, administered orally for 24 months, showed high efficacy in previously treated patients with WM and induced a VGPR rate of 19%.26 CXCR4 mutations impacted the VGPR rate to venetoclax but not PFS. Although half of the patients who experienced disease progression did so within a year of stopping venetoclax therapy, a sustained response after therapy completion was observed.26
In our preclinical experiments, the combination of ibrutinib and venetoclax synergistically induced higher death rates in WM and primary cell lines.27 Furthermore, the combination of ibrutinib and venetoclax is safe and effective in other B-cell malignancies, such as chronic lymphocytic leukemia (CLL) and mantle cell lymphoma (MCL).28, 29, 30 Therefore, we executed a prospective study to evaluate the safety and efficacy of an orally administered, chemotherapy-free, finite-duration combination of ibrutinib and venetoclax for primary treatment of patients with WM.
Methods
Study design and oversight
This study was an investigator-initiated, multicenter, single-arm, prospective phase 2 trial of a combination of ibrutinib and venetoclax in patients with previously untreated WM. The study enrolled patients at the Dana-Farber Cancer Institute (DFCI), Massachusetts General Hospital, and Beth Israel Deaconess Medical Center in Boston, Massachusetts. Enrollment began in July 2020 and was closed in January 2022. The date of data cutoff was 31 May 2023. This study was approved by the DFCI's institutional review board. All the patients provided written informed consent for participation. Pharmacyclics and AbbVie provided the research funding and study drugs. The trial was registered under ClinicalTrials.gov #NCT04273139.
Study patients
Individuals aged 18 years or older with a clinicopathological diagnosis of WM, per the second International Workshop for WM (IWWM2) criteria,31 were eligible if they were previously untreated, had an Eastern Cooperative Oncology Group performance ≤2, adequate BM function (ie, absolute neutrophil count ≥1.0 × 109/L, platelet count ≥50 × 109/L, and hemoglobin ≥80 g/L), creatinine clearance ≥50 mL/min, adequate hepatic function, and met the IWWM2 criteria to receive therapy.32 MYD88 wild-type mutational status, central nervous system involvement, active HIV, hepatitis B or hepatitis C infection, concurrent therapy for other cancers, clinically significant cardiovascular disease, ongoing use of warfarin or medications that could prolong the QT interval, or current pregnancy or breastfeeding were excluded from the study. A CONSORT diagram is shown in Figure 1.
Figure 1.

CONSORT diagram.
Study treatment
All the treatments were administered orally in an outpatient setting. The cycles were administered every 28 days. Cycle 1 consisted of ibrutinib 420 mg once daily. In cycle 2, ibrutinib was continued and venetoclax was administered once daily at 100 mg for 1 week, 200 mg for 1 week, and 400 mg for 2 weeks. Three days before initiating venetoclax, participants started allopurinol 300 mg orally once daily and 2-liter-per-day oral hydration for tumor lysis syndrome prophylaxis. If oral hydration was not optimal, 2 liters of normal saline was administered intravenously in the clinic before the dose of venetoclax. Allopurinol was continued for cycle 2. From cycles 3 to 24, participants received ibrutinib 420 mg and venetoclax 400 mg once daily, unless there was disease progression or unacceptable toxicity. Granulocyte colony-stimulating factor support was permitted for an absolute neutrophil count <1.0 K/μL. A temporary hold of ibrutinib before and after invasive procedures was recommended to minimize bleeding risk. Dose reductions for both drugs were allowed for toxicity.
Assessments
Complete blood counts, a comprehensive metabolic panel, and serum immunoglobulin levels were measured at baseline, once monthly for the first 3 months, and quarterly thereafter for the study duration. Tumor lysis syndrome monitoring with a complete metabolic panel, lactate dehydrogenase, phosphorus, and uric acid levels took place at 0, 8, and 24 hours on day 1 of each venetoclax dose increase. BM aspiration and biopsies were obtained at baseline and at 6, 12, and 24 months, and were evaluated at each participating institution. Computerized tomography scans with intravenous contrast were obtained at baseline and repeated at 6, 12, and 24 months if there was evidence of extramedullary disease at screening.
The primary objective was to determine the VGPR rate, defined as a decrease in serum IgM level of ≥90% compared with baseline or normalization of serum IgM level with a persistent IgM monoclonal protein in serum immunofixation electrophoresis through 24 months of therapy. Despite the observed clinical benefits with minor and partial responses (PRs) in routine clinical care, VGPR was selected as the primary outcome for research purposes, given its use in recent practice-changing randomized clinical trials and growing evidence associating VGPR with longer PFS in WM.23,33, 34, 35 Secondary objectives included determining the overall response rate (ORR, minor response or better), major response (PR or better), time to minor response, time to major response, time to VGPR, PFS, time to next treatment (TTNT), overall survival (OS), and drug safety. Categorical responses were defined according to the modified IWWM6 criteria.36 Adverse events were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 5.
MYD88 L265P mutations were assessed using an allele-specific polymerase chain reaction (AS-PCR) assay in CD19-selected BM cells,2 as well as an in-house, clinically validated next-generation sequencing (NGS) panel (Rapid Heme Panel) and an AS-PCR assay in unselected BM cells.37 CXCR4 mutations were assessed as previously described using an AS-PCR assay and Sanger sequencing in CD19-selected BM cells6 and the Rapid Heme Panel in unselected BM cells.37
Statistical analysis
A sample size of 50 participants was estimated using a 2-sided alpha level of 0.03 and a power of 80%, assuming a null VGPR rate of ≤25% and an alternative VGPR rate of ≥45%, based on previously reported VGPR rates to ibrutinib and venetoclax separately.
Continuous variables were dichotomized. Descriptive statistics such as proportions, medians, and ranges were calculated and reported. Subset analyses were performed to identify groups with differential outcomes and were meant to be hypothesis-generating, although with a special interest in evaluating differences according to CXCR4 mutational status. Differences between group the categories were assessed using Fisher exact test. Logistic regression models were used to investigate the association between baseline characteristics and attainment of VGPR, and outcomes were reported as odds ratio (OR) with 95% confidence interval (CI). The Kaplan-Meier method was used for time-to-event analyses with censoring, and the log-rank test was used to assess differences between groups. Cox proportional hazard regression models were fitted to evaluate factors associated with time-to-event and outcomes reported as hazard ratio with 95% CI. P-values <.05 indicate statistical significance. Statistical analyses and graphs were performed using STATA 17.0 (College Station, TX).
Results
Baseline characteristics
Forty-five participants with MYD88-mutated WM were enrolled. The indications for treatment included symptomatic anemia (n = 27; 60%), symptomatic hyperviscosity (n = 12; 27%), constitutional symptoms (n = 11; 24%), symptomatic extramedullary disease (n = 4; 9%), and peripheral neuropathy (n = 3; 7%). Median baseline characteristics were as follows: age 67 years (range, 38-81); serum IgM level 43 g/L (range, 5.7-92); hemoglobin 102 g/L (range, 75-153); platelet count 274 × 109/L (range, 75-596); and BM involvement 60% (range, 5-90). CXCR4 mutations were detected in 17 patients (38%), of which 10 were nonsense and 7 were frameshift mutations. In 14 participants, CXCR4 mutation detection was concordant between AS-PCR and NGS results. However, in 3 participants, CXCR4 mutations were detected by AS-PCR but not in the NGS panel. Categorized baseline characteristics according to CXCR4 mutational status are shown in Table 1. Patients with CXCR4 mutations were more likely to have acquired von Willebrand disease (41% vs 7%; P = .02), defined as von Willebrand antigen, ristocetin cofactor, and factor VIII activity levels of <30%. The median serum IgM level in participants with acquired von Willebrand disease was 64 g/L (range, 44-92). There were no differences in baseline characteristics between participants with nonsense and frameshift CXCR4 mutations (supplemental Table 1, available on the Blood website).
Table 1.
Baseline characteristics of participants according to CXCR4 mutational status
| Characteristic | All participants (n = 45) | CXCR4 WT (n = 28) | CXCR4 MUT (n = 17) | P value |
|---|---|---|---|---|
| Age >65 y | 24 (53%) | 15 (54%) | 9 (53%) | .60 |
| Male sex | 30 (67%) | 19 (68%) | 11 (65%) | .54 |
| Hemoglobin level <115 g/L | 33 (73%) | 20 (71%) | 13 (76%) | .50 |
| Platelet count <100 × 109/L | 3 (7%) | 0 (0%) | 3 (18%) | .05 |
| Serum β2-microglobulin ≥3 mg/L | 28 (64%) | 20 (74%) | 8 (47%) | .11 |
| Serum IgM level ≥40 g/L | 26 (58%) | 14 (50%) | 12 (71%) | .22 |
| Serum IgM level ≥70 g/L | 6 (13%) | 3 (11%) | 3 (18%) | .66 |
| BM involvement ≥60% | 26 (58%) | 16 (57%) | 10 (59%) | .58 |
| Adenopathy ≥1.5 cm | 24 (53%) | 18 (64%) | 6 (35%) | .07 |
| Splenomegaly ≥15 cm | 12 (27%) | 10 (36%) | 2 (12%) | .08 |
| Acquired von Willebrand disease | 9 (20%) | 2 (7%) | 7 (41%) | .02 |
| Cryoglobulinemia | 3 (7%) | 2 (7%) | 1 (6%) | .67 |
| Low IPSSWM score | 9 (20%) | 3 (11%) | 6 (35%) | .06 |
| Intermediate IPSSWM score | 16 (36%) | 13 (48%) | 3 (18%) | |
| High IPSSWM score | 19 (43%) | 11 (40%) | 8 (47%) | |
| ≥12 mo from WM diagnosis | 18 (40%) | 11 (39%) | 7 (41%) | .57 |
IPSSWM, International Prognostic Scoring System for WM; MUT, mutated; WT, wild-type.
Response to therapy
At the best response, 19 participants (42%) attained a VGPR, 24 (53%) a PR, and 2 (4%) a minor response (Figure 2A), for an ORR of 100% and a major response rate of 96%. There were no complete responses, although 1 participant with CXCR4 WT disease attained normalization of blood counts, serum IgM levels, serum electrophoresis and immunofixation, and clearance of BM infiltrate but had persistent lymphadenopathy ≥1.5 cm at 1 site. CXCR4 mutations were associated with a numerically, but not statistically significantly, lower VGPR rate (29% vs 50%; P = .15; Figure 2B). There was no difference in VGPR between participants with frameshift and nonsense CXCR4 mutations (P = .59). The univariate and multivariate logistic regression models for VGPR attainment are shown in supplemental Table 2.
Figure 2.

Response to therapy. Categorical response rates in all patients (A) and according to the CXCR4 mutation status (B). MR, minor response.
The median time to a minor response was 0.9 months (95% CI, 0.9-1), the median time to a major response was 1.8 months (95% CI, 1.4-1.9), and the median time to VGPR was not reached, with 6-, 12- and 18-month VGPR rates of 33% (95% CI, 22-49), 40% (95% CI, 27-56), and 43% (95% CI, 30-60), respectively. CXCR4 mutations were not associated with a longer time to VGPR (not reached vs 17.3 months; hazard ratio, 0.51; 95% CI, 0.19-1.43; P = .20). There was no difference in the time to VGPR between patients with frameshift and nonsense CXCR4 mutations (P = .41). The univariate and multivariate Cox proportional hazard regression models for the time to VGPR are shown in supplemental Table 3.
At best response, hemoglobin levels normalized in all participants who presented with anemia (n = 27). Symptoms resolved in all participants who presented with hyperviscosity (n = 12). Neuropathic symptoms remained stable in the 3 participants who presented with neuropathy. Of the 24 participants with lymphadenopathy (≥1.5 cm), 16 (67%) showed resolution and 4 (17%) showed a reduction in lymphadenopathy after treatment. There was an increase in lymphadenopathy in 2 (8%) participants, including 1 who transformed to diffuse large B-cell lymphoma and 1 with mesenteritis unrelated to WM. There was no follow-up imaging in 2 participants. Of the 12 participants with splenomegaly (≥15 cm), 11 (92%) showed normalized splenic size after treatment. There was no follow-up imaging in 1 participant. After treatment, the median BM disease involvement decreased from 60% to 5% (range, 0%-70%; P < .001).
Survival analysis
At a median follow-up time of 24.4 months (95% CI, 21.8-26.2), there were 12 progression events and 2 deaths. Five patients started a new therapy. The 24-month PFS rate was 76% (95% CI, 59-86; Figure 3A). CXCR4 mutations did not impact PFS (P = .89; Figure 3B). There was no difference in PFS between patients with frameshift and nonsense CXCR4 mutations (P = .43). The univariate and multivariate Cox proportional hazard regression models for PFS are shown in supplemental Table 4. The 24-month next-treatment-free rate was 89% (95% CI, 72-96; Figure 3C). The 24-month OS rate was 96% (95% CI, 84-99; Figure 3D). CXCR4 mutations did not impact TTNT (P = .43) or OS (P = .27).
Figure 3.

PFS, TTNT, and OS analysis. Kaplan-Meier estimates of PFS in all participants (A) and according to CXCR4 mutational status (B), TTNT in all participants (C), and OS in all participants (D).
The median number of cycles administered was 10 (range, 2-22), with a median time on therapy of 10.2 months (range, 1.9-20.8 months). The median time after the end of therapy (EOT) was 13.3 months (95% CI, 12.9-13.5). Eleven progression events occurred after EOT, and the 12-month PFS rate after EOT was 79% (95% CI, 63-88; Figure 4A). CXCR4 mutations did not impact the 12-month PFS rate after EOT (84% vs 71%; P = .35; Figure 4B). Time on therapy of ≥12 vs <12 months did not impact the 12-month PFS rate after EOT (77% vs 81%; P=.76; Figure 4C). The 12-month PFS rate after EOT in participants who attained VGPR was 93% and 69% in those who did not (P = .12; Figure 4D).
Figure 4.

PFS after end-of-treatment analysis. Kaplan-Meier estimates of PFS in all participants after the end of treatment (A) and according to CXCR4 mutational status (B), time on treatment (C), and VGPR attainment (D).
Thirty-one participants underwent a BM biopsy 12 months after EOT. The median BM disease burden was 8% (range, 0%-80%). No emergent mutations in BTK or TP53 were observed.
Safety
Grade 2 or higher adverse events associated with ibrutinib and venetoclax therapy are shown in Table 2. The most common grade ≥3 adverse events observed in more than 1 participant were neutropenia, mucositis, diarrhea, laboratory tumor lysis syndrome, and atrial fibrillation. Grade 4 neutropenia events (n = 4) were resolved with granulocyte-colony stimulating factor therapy, prompting dose reduction of venetoclax in 1 participant. Mucositis and diarrhea (n = 7) responded to a temporary hold of study therapy, prompting dose reduction of ibrutinib in 2, venetoclax in 2, and both ibrutinib and venetoclax in 1 participant. None of the patients in whom the doses were reduced progressed after the EOT. All tumor lysis syndrome events (n = 3) were laboratory-based and managed in an outpatient setting with rasburicase and normal saline. There was no clinical tumor lysis syndrome. The 3 participants who developed atrial fibrillation were managed medically and continued the study therapy without disruption.
Table 2.
Grade 2 or higher adverse events associated with ibrutinib and venetoclax therapies
| Adverse event | Grade 2 | Grade 3 | Grade 4 | Grade 5 |
|---|---|---|---|---|
| Alanine aminotransferase increase | 1 | |||
| Anemia | 1 | |||
| Anorexia | 1 | |||
| Arthralgia | 5 | 1 | ||
| Atrial fibrillation | 1 | 2 | ||
| Bruising | 2 | |||
| Diarrhea | 11 | 3 | ||
| Fatigue | 2 | 1 | ||
| Gastroesophageal reflux disease | 12 | |||
| Headache | 1 | |||
| Hematoma | 1 | |||
| Hematuria | 1 | |||
| Hyperphosphatemia | 8 | |||
| Hypertension | 2 | 1 | ||
| Hyponatremia | 1 | |||
| Intracranial hemorrhage | 1 | |||
| Lung infection | 2 | |||
| Malaise | 1 | |||
| Mucositis | 9 | 4 | ||
| Myalgia | 3 | |||
| Nausea | 5 | |||
| Neutropenia | 2 | 13 | 4 | |
| Platelet decrease | 1 | |||
| Skin rash | 5 | |||
| Soft tissue infection | 2 | 1 | ||
| Tumor lysis syndrome | 3 | |||
| Upper respiratory infection | 4 | |||
| Urinary tract infection | 5 | |||
| Ventricular arrhythmia | 1 | 1 | 2 |
Two grade 5 and 1 grade 4 ventricular arrhythmia events occurred, leading to termination of the study therapy in the latter case. The accrual was stopped after the second event of grade 5 ventricular arrhythmia. All the remaining participants were tested for cardiac enzyme levels and underwent electrocardiography, echocardiography, and cardiac stress tests to assess cardiovascular health. The study therapy was stopped in all participants after a grade 2 ventricular arrhythmia event occurred in a participant undergoing a cardiac stress test. The 3 participants with grade ≥3 ventricular arrhythmias were male, older than 65 years, and had significant, though well-controlled, cardiac comorbidities including a history of arrhythmia, coronary artery disease, hypertension, hypercholesterolemia, diabetes, or obesity. Three participants stopped treatment before early termination of the study therapy. One because of intracranial bleeding possibly related to study therapy in a patient with a previously unknown benign brain lesion, 1 because of transformation to diffuse large B-cell lymphoma, and 1 because of grade 4 ventricular arrhythmia.
The median number of expected doses of ibrutinib and venetoclax was 307 (range, 58-625). The dose intensity for ibrutinib was 97.6% (range, 83%-100%), and that for venetoclax was 98.3% (range, 83%-100%).
The median serum IgG level decreased from 5.7 g/L (range, 1.1-83 g/L) at baseline to 4.4 g/L (range, 1.1-32 g/L) during follow-up (P = .01). The median serum IgA level decreased from 0.4 g/L (range, 0.1-5.2) at baseline to 0.3 g/L (range, 0.05-2.1) during follow-up (P = .005). To date, none of the participants have required intravenous immunoglobulin therapy.
Discussion
Herein, we present the results of an investigator-initiated prospective phase 2 study evaluating concurrent BTK and BCL2 inhibition in patients with WM. We combined ibrutinib and venetoclax to provide chemotherapy-free, all-oral, finite-duration treatment, which is supported by previous preclinical and clinical evidence. The outcome of interest in this study was the attainment of VGPR or better within 24 months of fixed-duration therapy. With a median follow-up time of 24 months and a median of 10 cycles of therapy, 19 (42%) patients attained VGPR. However, the study therapy was stopped early because of a higher-than-expected rate of ventricular arrhythmia.
The VGPR rate reported here was higher than that expected for both ibrutinib monotherapy in the frontline setting and venetoclax monotherapy in the relapsed setting, previously reported by our group at 30% at 50 months of active therapy22 and 19% at 33 months of follow-up.26 However, the proportion of patients with CXCR4 mutations differed between these studies, with a higher proportion of CXCR4 mutations in ibrutinib and venetoclax monotherapy studies, which could partly account for the observed differences. The VGPR rate observed here is comparable to the 10% to 40% reported with rituximab-containing regimens as frontline treatment in previous prospective clinical trials.38, 39, 40 Overall, our findings strongly suggest a likely additive effect of combining BTK and BCL2 inhibitors in WM, similar to that reported for CLL and MCL.
Clinically, the combination of BTK and BCL2 inhibition induced normalization of hemoglobin levels and resolution of hyperviscosity symptoms and extramedullary disease in most patients exposed to these agents. The responses were fast, with a median time to minor and major response of 0.9 and 1.8 months, respectively, comparable to ibrutinib monotherapy.24 Interestingly, the BM disease burden decreased from 60% at baseline to 5% at best response. The BM clearance was better than that reported with ibrutinib monotherapy and similar to that reported with venetoclax monotherapy. Again, the different proportions of patients with CXCR4 mutations may have affected the results. Therefore, concurrent BTK and BCL2 inhibition provides fast responses with effective BM clearance, building on the best features of ibrutinib and venetoclax.
The presence of CXCR4 mutations have been associated with a longer response time, lower VGPR rates, and shorter PFS in patients with WM treated with BTK inhibitors.20, 21, 22 CXCR4 mutations also impacted the VGPR rate but not PFS, to venetoclax monotherapy in WM.26 This study showed a trend toward fewer VGPRs in patients with CXCR4 mutations. However, although the follow-up duration was relatively short, CXCR4 mutations did not appear to impact PFS, TTNT, or OS with ibrutinib and venetoclax. In our previous reports, a difference in PFS between patients with and without CXCR4 mutations was evident within 12 months of ibrutinib monotherapy.21,22 Despite the small sample size, our findings suggest that concurrent BTK and BCL2 inhibition may partially overcome the impact of CXCR4 mutations on patient with WM outcomes.
There was a higher-than-expected rate of ventricular arrhythmia at 9%, including 2 deaths, prompting us to stop accrual and study therapy. The rate of ventricular events has not been previously observed in studies evaluating the combination of ibrutinib and venetoclax for CLL or MCL. Four prospective studies evaluated the combination of ibrutinib and venetoclax as a frontline treatment in patients with CLL.30,41, 42, 43 In all studies, the doses of ibrutinib and venetoclax were similar to those used in our study. In addition, 3 studies evaluated this combination in patients with MCL.28,44,45 In these studies, the doses of ibrutinib and venetoclax were higher.
The etiology of the cardiovascular events observed in our study remains to be determined, as postmortem evaluations were not performed per the patient’s family request, and pharmacokinetic samples from participants were not obtained as part of this study. All events occurred within cycles 3 and 7 when patients were on full doses of both agents and at a time when electrolyte derangements due to tumor lysis syndrome were unlikely. Potential hypothetical explanations for our observation include an unknown underlying cardiac involvement by amyloid or paraprotein deposition; a higher risk of cardiac events in the participants given the personal history of coronary artery disease or risk factors such as hypertension, hypercholesterolemia, atrial fibrillation, diabetes, or obesity; a potential pharmacological interaction between ibrutinib and venetoclax exacerbated by the underlying paraproteinemia of WM; or a random occurrence independent of the intervention or the participants. There was no clinical suspicion of systemic light-chain amyloidosis in these patients, although formal evaluations for amyloidosis were not performed at the baseline.
The durability of the off-therapy response was a secondary outcome of interest in this study. Previous studies have reported that half of the patients experienced disease progression within 4 weeks of stopping ibrutinib monotherapy,46 supporting indefinite-duration therapy. In the case of venetoclax, half of the patients experienced disease progression within 1 year of stopping therapy, with most progression events occurring in the first 5 months off therapy.26 After a median time off therapy of 13 months, the 12-month PFS rate after EOT was 79%, which appeared to be more durable than that observed with ibrutinib or venetoclax monotherapy. Of importance was the absence of acquired mutations in BTK and TP53, which have been reported with long-term use of BTK inhibitors and exposure to chemotherapy, respectively. A longer follow-up is needed to better understand the response durability of the combination of ibrutinib and venetoclax in WM.
Based on the serious and unexpected cardiac toxicity reported here, the combination of ibrutinib and venetoclax is not recommended for the treatment or further investigation of WM. A study combining ibrutinib, venetoclax, and rituximab (NCT04840602) was suspended, given these findings. However, concurrent BTK and BCL2 inhibition induced a high VGPR rate in patients with WM. Moreover, our findings suggested durable responses off-therapy in many participants, supporting further investigation of concurrent BTK and BCL2 inhibition. The emergence of second-generation covalent and novel noncovalent BTK inhibitors with lower rates of cardiac events and minimal pharmacological interactions with BCL2 antagonists could provide safer combinations for patients with WM.35,47,48 Based on this hypothesis, we initiated a phase 2 study combining pirtobrutinib and venetoclax in previously treated patients with WM, given the rare incidence of arrhythmias reported with pirtobrutinib (NCT05734495). A careful selection of participants is warranted in future studies that combine BTK and BCL2 inhibitors in WM, including a complete cardiac disease history, electrocardiogram, echocardiogram, and stress test, as deemed clinically necessary.
Conflict-of-interest disclosure: J.J.C. received research funds and honoraria from AbbVie, AstraZeneca, BeiGene, Cellectar, Janssen, Kite, LOXO, Mustang Bio, and Pharmacyclics. S.S. received research funds and/or honoraria from ADC Therapeutics, BeiGene, and Cellectar. A.R.B. received research funds and/or honoraria from Adaptive, BeiGene, CSL Behring, Genzyme, Karyopharm, Pharmacyclics, and Sanofi. D.S. received research funds and honoraria from AbbVie, Genmab, and Syros. S.P.T. received research funds and/or honoraria from AbbVie/Pharmacyclics, BeiGene, Bristol-Myers Squibb, Eli Lilly, Janssen, Ono, Parexel and X4. The remaining authors declare no competing financial interests.
Acknowledgments
The authors thank all study participants and their family members.
The authors gratefully acknowledge the generous support of Peter Bing, the David and Janet Bingham Research Fund of the International Waldenström Macroglobulinemia Foundation, the Yang Family Research Fund of the International Waldenström Macroglobulinemia Foundation, the Leukemia and Lymphoma Society (grant R6507-18), the NIH SPORE in Multiple Myeloma (grant 2P50CA100707-16A1), the Edward and Linda Nelson Fund for WM Research, the Kerry Robertson Fund for WM Research, the Bauman Family Trust, the Siegel Family Fund for WM, the Lee and Michael Bell Waldenström Macroglobulinemia Fellowship, the Fred and Michele Brettschneider Family Research Fund, the Bayer Family Research Fund, the Kaplan Family Fund for Waldenström Macroglobulinemia, and the WMR Fund. This study was supported by Pharmacyclics and AbbVie (grant funding and drugs).
Authorship
Contribution: J.J.C., S.S., and S.P.T. designed the study; K.M., M.L., K.S., T.W., and C.J.P. provided administrative support; J.J.C., S.S., A.R.B., D.S., and C.A.F. provided care to patients; A.C., M.L.G., A.K., S.L., X.L., K.R., N.T., and Z.R.H. performed the genomic analyses; J.J.C., S.S., A.R.B., D.S., and S.P.T. analyzed the data; J.J.C. drafted the initial manuscript; and all the authors critically reviewed the manuscript and approved the final version.
Footnotes
Portions of this research were presented at the 11th International Workshop for Waldenström Macroglobulinemia, Madrid, Spain, 29 October 2022; the 64th annual meeting of the American Society of Hematology, New Orleans, LA, 10 December 2022; and the 65th annual meeting of the American Society of Hematology, San Diego, CA, 9 December 2023.
Ethically appropriate data requests should be made to the corresponding author, Jorge J. Castillo (jorgej_castillo@dfci.harvard.edu). Deidentified data will be available immediately after print publication.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Supplementary Material
References
- 1.Swerdlow SH, Cook JR, Sohani AR, et al. In: WHO Classification of Tumours of Hematopoietic and Lymphoid Tissues. Swerdlow SH, Campo E, Harris NL, et al., editors. IARC; 2017. Lymphoplasmacytic lymphoma; pp. 232–235. [Google Scholar]
- 2.Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenstrom's macroglobulinemia. N Engl J Med. 2012;367(9):826–833. doi: 10.1056/NEJMoa1200710. [DOI] [PubMed] [Google Scholar]
- 3.Jiménez C, Sebastián E, Chillón MC, et al. MYD88 L265P is a marker highly characteristic of, but not restricted to, Waldenstrom's macroglobulinemia. Leukemia. 2013;27(8):1722–1728. doi: 10.1038/leu.2013.62. [DOI] [PubMed] [Google Scholar]
- 4.Poulain S, Roumier C, Decambron A, et al. MYD88 L265P mutation in Waldenstrom macroglobulinemia. Blood. 2013;121(22):4504–4511. doi: 10.1182/blood-2012-06-436329. [DOI] [PubMed] [Google Scholar]
- 5.Varettoni M, Arcaini L, Zibellini S, et al. Prevalence and clinical significance of the MYD88 (L265P) somatic mutation in Waldenstrom's macroglobulinemia and related lymphoid neoplasms. Blood. 2013;121(13):2522–2528. doi: 10.1182/blood-2012-09-457101. [DOI] [PubMed] [Google Scholar]
- 6.Hunter ZR, Xu L, Yang G, et al. The genomic landscape of Waldenstrom macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood. 2014;123(11):1637–1646. doi: 10.1182/blood-2013-09-525808. [DOI] [PubMed] [Google Scholar]
- 7.Schmidt J, Federmann B, Schindler N, et al. MYD88 L265P and CXCR4 mutations in lymphoplasmacytic lymphoma identify cases with high disease activity. Br J Haematol. 2015;169(6):795–803. doi: 10.1111/bjh.13361. [DOI] [PubMed] [Google Scholar]
- 8.Poulain S, Roumier C, Venet-Caillault A, et al. Genomic landscape of CXCR4 mutations in Waldenstrom macroglobulinemia. Clin Cancer Res. 2016;22(6):1480–1488. doi: 10.1158/1078-0432.CCR-15-0646. [DOI] [PubMed] [Google Scholar]
- 9.Treon SP, Cao Y, Xu L, Yang G, Liu X, Hunter ZR. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenstrom macroglobulinemia. Blood. 2014;123(18):2791–2796. doi: 10.1182/blood-2014-01-550905. [DOI] [PubMed] [Google Scholar]
- 10.Castillo JJ, Gustine JN, Meid K, et al. Low levels of von Willebrand markers associate with high serum IgM levels and improve with response to therapy, in patients with Waldenstrom macroglobulinaemia. Br J Haematol. 2019;184(6):1011–1014. doi: 10.1111/bjh.15200. [DOI] [PubMed] [Google Scholar]
- 11.Castillo JJ, Xu L, Gustine JN, et al. CXCR4 mutation subtypes impact response and survival outcomes in patients with Waldenstrom macroglobulinaemia treated with ibrutinib. Br J Haematol. 2019;187(3):356–363. doi: 10.1111/bjh.16088. [DOI] [PubMed] [Google Scholar]
- 12.Castillo JJ, Olszewski AJ, Kanan S, Meid K, Hunter ZR, Treon SP. Overall survival and competing risks of death in patients with Waldenstrom macroglobulinaemia: an analysis of the Surveillance, Epidemiology and End Results database. Br J Haematol. 2015;169(1):81–89. doi: 10.1111/bjh.13264. [DOI] [PubMed] [Google Scholar]
- 13.Castillo JJ, Advani RH, Branagan AR, et al. Consensus treatment recommendations from the tenth International Workshop for Waldenstrom macroglobulinaemia. Lancet Haematol. 2020;7(11):e827–e837. doi: 10.1016/S2352-3026(20)30224-6. [DOI] [PubMed] [Google Scholar]
- 14.Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet. 2013;381(9873):1203–1210. doi: 10.1016/S0140-6736(12)61763-2. [DOI] [PubMed] [Google Scholar]
- 15.Martin P, Chen Z, Cheson BD, et al. Long-term outcomes, secondary malignancies and stem cell collection following bendamustine in patients with previously treated non-Hodgkin lymphoma. Br J Haematol. 2017;178(2):250–256. doi: 10.1111/bjh.14667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paludo J, Abeykoon JP, Shreders A, et al. Bendamustine and rituximab (BR) versus dexamethasone, rituximab, and cyclophosphamide (DRC) in patients with Waldenstrom macroglobulinemia. Ann Hematol. 2018;97(8):1417–1425. doi: 10.1007/s00277-018-3311-z. [DOI] [PubMed] [Google Scholar]
- 17.Laribi K, Poulain S, Willems L, et al. Bendamustine plus rituximab in newly-diagnosed Waldenstrom macroglobulinaemia patients. A study on behalf of the French Innovative Leukaemia Organization (FILO) Br J Haematol. 2019;186(1):146–149. doi: 10.1111/bjh.15718. [DOI] [PubMed] [Google Scholar]
- 18.Abeykoon JP, Zanwar S, Ansell SM, et al. Ibrutinib monotherapy outside of clinical trial setting in Waldenstrom macroglobulinaemia: practice patterns, toxicities and outcomes. Br J Haematol. 2020;188(3):394–403. doi: 10.1111/bjh.16168. [DOI] [PubMed] [Google Scholar]
- 19.Castillo JJ, Gustine JN, Meid K, et al. Response and survival outcomes to ibrutinib monotherapy for patients with Waldenström macroglobulinemia on and off clinical trials. Hemasphere. 2020;4(3):e363. doi: 10.1097/HS9.0000000000000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tam CS, Opat S, D'Sa S, et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenstrom macroglobulinemia: the ASPEN study. Blood. 2020;136(18):2038–2050. doi: 10.1182/blood.2020006844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Treon SP, Meid K, Gustine J, et al. Long-term follow-up of ibrutinib monotherapy in symptomatic, previously treated patients with Waldenstrom macroglobulinemia. J Clin Oncol. 2021;39(6):565–575. doi: 10.1200/JCO.20.00555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castillo JJ, Meid K, Gustine JN, et al. Long-term follow-up of ibrutinib monotherapy in treatment-naive patients with Waldenstrom macroglobulinemia. Leukemia. 2022;36(2):532–539. doi: 10.1038/s41375-021-01417-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buske C, Tedeschi A, Trotman J, et al. Ibrutinib plus rituximab versus placebo plus rituximab for Waldenstrom's macroglobulinemia: final analysis from the randomized phase III iNNOVATE Study. J Clin Oncol. 2022;40(1):52–62. doi: 10.1200/JCO.21.00838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Treon SP, Gustine J, Meid K, et al. Ibrutinib monotherapy in symptomatic, treatment-naive patients with Waldenstrom macroglobulinemia. J Clin Oncol. 2018;36(27):2755–2761. doi: 10.1200/JCO.2018.78.6426. [DOI] [PubMed] [Google Scholar]
- 25.Castillo JJ, Sarosiek SR, Gustine JN, et al. Response and survival predictors in a cohort of 319 patients with Waldenstrom macroglobulinemia treated with ibrutinib monotherapy. Blood Adv. 2022;6(3):1015–1024. doi: 10.1182/bloodadvances.2021006106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Castillo JJ, Allan JN, Siddiqi T, et al. Venetoclax in previously treated Waldenstrom macroglobulinemia. J Clin Oncol. 2022;40(1):63–71. doi: 10.1200/JCO.21.01194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao Y, Yang G, Hunter ZR, et al. The BCL2 antagonist ABT-199 triggers apoptosis, and augments ibrutinib and idelalisib mediated cytotoxicity in CXCR4 Wild-type and CXCR4 WHIM mutated Waldenstrom macroglobulinaemia cells. Br J Haematol. 2015;170(1):134–138. doi: 10.1111/bjh.13278. [DOI] [PubMed] [Google Scholar]
- 28.Tam CS, Anderson MA, Pott C, et al. Ibrutinib plus venetoclax for the treatment of mantle-cell lymphoma. N Engl J Med. 2018;378(13):1211–1223. doi: 10.1056/NEJMoa1715519. [DOI] [PubMed] [Google Scholar]
- 29.Jain N, Keating M, Thompson P, et al. Ibrutinib and venetoclax for first-line treatment of CLL. N Engl J Med. 2019;380(22):2095–2103. doi: 10.1056/NEJMoa1900574. [DOI] [PubMed] [Google Scholar]
- 30.Jain N, Keating M, Thompson P, et al. Ibrutinib plus venetoclax for first-line treatment of chronic lymphocytic leukemia: a nonrandomized phase 2 trial. JAMA Oncol. 2021;7(8):1213–1219. doi: 10.1001/jamaoncol.2021.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Owen RG, Treon SP, Al-Katib A, et al. Clinicopathological definition of Waldenstrom's macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom's Macroglobulinemia. Semin Oncol. 2003;30(2):110–115. doi: 10.1053/sonc.2003.50082. [DOI] [PubMed] [Google Scholar]
- 32.Kyle RA, Treon SP, Alexanian R, et al. Prognostic markers and criteria to initiate therapy in Waldenstrom's macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom's macroglobulinemia. Semin Oncol. 2003;30(2):116–120. doi: 10.1053/sonc.2003.50038. [DOI] [PubMed] [Google Scholar]
- 33.Treon SP, Yang G, Hanzis C, et al. Attainment of complete/very good partial response following rituximab-based therapy is an important determinant to progression-free survival, and is impacted by polymorphisms in FCGR3A in Waldenstrom macroglobulinaemia. Br J Haematol. 2011;154(2):223–228. doi: 10.1111/j.1365-2141.2011.08726.x. [DOI] [PubMed] [Google Scholar]
- 34.Treon SP, Tripsas CK, Meid K, et al. Carfilzomib, rituximab, and dexamethasone (CaRD) treatment offers a neuropathy-sparing approach for treating Waldenstrom's macroglobulinemia. Blood. 2014;124(4):503–510. doi: 10.1182/blood-2014-03-566273. [DOI] [PubMed] [Google Scholar]
- 35.Dimopoulos MA, Opat S, D'Sa S, et al. Zanubrutinib versus ibrutinib in symptomatic Waldenstrom macroglobulinemia: final analysis from the randomized phase III ASPEN Study. J Clin Oncol. 2023 doi: 10.1200/JCO.22.02830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Treon SP, Tripsas CK, Meid K, et al. Ibrutinib in previously treated Waldenstrom's macroglobulinemia. N Engl J Med. 2015;372(15):1430–1440. doi: 10.1056/NEJMoa1501548. [DOI] [PubMed] [Google Scholar]
- 37.Kluk MJ, Lindsley RC, Aster JC, et al. Validation and implementation of a custom next-generation sequencing clinical assay for hematologic malignancies. J Mol Diagn. 2016;18(4):507–515. doi: 10.1016/j.jmoldx.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Treon SP, Ioakimidis L, Soumerai JD, et al. Primary therapy of Waldenstrom macroglobulinemia with bortezomib, dexamethasone, and rituximab: WMCTG clinical trial 05-180. J Clin Oncol. 2009;27(23):3830–3835. doi: 10.1200/JCO.2008.20.4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kastritis E, Gavriatopoulou M, Kyrtsonis MC, et al. Dexamethasone, rituximab, and cyclophosphamide as primary treatment of Waldenstrom macroglobulinemia: final analysis of a phase 2 study. Blood. 2015;126(11):1392–1394. doi: 10.1182/blood-2015-05-647420. [DOI] [PubMed] [Google Scholar]
- 40.Rummel MJ, Lerchenmüller C, Hensel M, et al. Two years rituximab maintenance vs. observation after first line treatment with bendamustine plus rituximab (B-R) in patients with Waldenström’s macroglobulinemia (MW): results of a prospective, randomized, multicenter phase 3 study (the StiL NHL7-2008 MAINTAIN trial) [abstract] Blood. 2019;134(suppl 1):343. [Google Scholar]
- 41.Wierda WG, Allan JN, Siddiqi T, et al. Ibrutinib plus venetoclax for first-line treatment of chronic lymphocytic leukemia: primary analysis results from the Minimal Residual Disease Cohort of the Randomized Phase II CAPTIVATE Study. J Clin Oncol. 2021;39(34):3853–3865. doi: 10.1200/JCO.21.00807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huber H, Edenhofer S, von Tresckow J, et al. Obinutuzumab (GA-101), ibrutinib, and venetoclax (GIVe) frontline treatment for high-risk chronic lymphocytic leukemia. Blood. 2022;139(9):1318–1329. doi: 10.1182/blood.2021013208. [DOI] [PubMed] [Google Scholar]
- 43.Eichhorst B, Niemann CU, Kater AP, et al. First-line venetoclax combinations in chronic lymphocytic leukemia. N Engl J Med. 2023;388(19):1739–1754. doi: 10.1056/NEJMoa2213093. [DOI] [PubMed] [Google Scholar]
- 44.Le Gouill S, Morschhauser F, Chiron D, et al. Ibrutinib, obinutuzumab, and venetoclax in relapsed and untreated patients with mantle cell lymphoma: a phase 1/2 trial. Blood. 2021;137(7):877–887. doi: 10.1182/blood.2020008727. [DOI] [PubMed] [Google Scholar]
- 45.Wang M, Ramchandren R, Chen R, et al. Concurrent ibrutinib plus venetoclax in relapsed/refractory mantle cell lymphoma: the safety run-in of the phase 3 SYMPATICO study. J Hematol Oncol. 2021;14(1):179. doi: 10.1186/s13045-021-01188-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gustine JN, Meid K, Dubeau T, et al. Ibrutinib discontinuation in Waldenstrom macroglobulinemia: etiologies, outcomes, and IgM rebound. Am J Hematol. 2018;93(4):511–517. doi: 10.1002/ajh.25023. [DOI] [PubMed] [Google Scholar]
- 47.Palomba ML, Patel MR, Eyre TA, et al. Efficacy of pirtobrutinib, a highly selective, non-covalent (reversible) BTK inhibitor in relapsed / refractory Waldenström macroglobulinemia: results from the phase 1/2 BRUIN Study [abstract] Blood. 2022;140(suppl 1):557–560. [Google Scholar]
- 48.Mato AR, Woyach JA, Brown JR, et al. Pirtobrutinib after a covalent BTK inhibitor in chronic lymphocytic leukemia. N Engl J Med. 2023;389(1):33–44. doi: 10.1056/NEJMoa2300696. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.