

Key Points
Question
Does targeting hepatic angiotensinogen synthesis with the RNA interference therapeutic zilebesiran reduce blood pressure in adults with mild to moderate hypertension?
Findings
In a phase 2 trial, subcutaneous zilebesiran doses of 150, 300, or 600 mg every 6 months or 300 mg every 3 months significantly decreased systolic blood pressure at 3 and 6 months vs placebo. Nonserious drug-related adverse events occurred in 16.9% of zilebesiran-treated patients, principally injection site reactions and mild hyperkalemia.
Meaning
Single subcutaneous doses of zilebesiran significantly lower blood pressure for up to 6 months, suggesting potential for use as an effective antihypertensive with quarterly or biannual dosing.
Abstract
Importance
Angiotensinogen is the most upstream precursor of the renin–angiotensin–aldosterone system, a key pathway in blood pressure (BP) regulation. Zilebesiran, an investigational RNA interference therapeutic, targets hepatic angiotensinogen synthesis.
Objective
To evaluate antihypertensive efficacy and safety of different zilebesiran dosing regimens.
Design, Setting, and Participants
This phase 2, randomized, double-blind, dose-ranging study of zilebesiran vs placebo was performed at 78 sites across 4 countries. Screening initiation occurred in July 2021 and the last patient visit of the 6-month study occurred in June 2023. Adults with mild to moderate hypertension, defined as daytime mean ambulatory systolic BP (SBP) of 135 to 160 mm Hg following antihypertensive washout, were randomized.
Interventions
Randomization to 1 of 4 subcutaneous zilebesiran regimens (150, 300, or 600 mg once every 6 months or 300 mg once every 3 months) or placebo (once every 3 months) for 6 months.
Main Outcomes and Measures
The primary end point was between-group difference in least-squares mean (LSM) change from baseline to month 3 in 24-hour mean ambulatory SBP.
Results
Of 394 randomized patients, 377 (302 receiving zilebesiran and 75 receiving placebo) comprised the full analysis set (93 Black patients [24.7%]; 167 [44.3%] women; mean [SD] age, 57 [11] years). At 3 months, 24-hour mean ambulatory SBP changes from baseline were −7.3 mm Hg (95% CI, −10.3 to −4.4) with zilebesiran, 150 mg, once every 6 months; −10.0 mm Hg (95% CI, −12.0 to −7.9) with zilebesiran, 300 mg, once every 3 months or every 6 months; −8.9 mm Hg (95% CI, −11.9 to −6.0) with zilebesiran, 600 mg, once every 6 months; and 6.8 mm Hg (95% CI, 3.6-9.9) with placebo. LSM differences vs placebo in change from baseline to month 3 were −14.1 mm Hg (95% CI, −19.2 to −9.0; P < .001) with zilebesiran, 150 mg, once every 6 months; −16.7 mm Hg (95% CI, −21.2 to −12.3; P < .001) with zilebesiran, 300 mg, once every 3 months or every 6 months; and −15.7 mm Hg (95% CI, −20.8 to −10.6; P < .001) with zilebesiran, 600 mg, once every 6 months. Over 6 months, 60.9% of patients receiving zilebesiran had adverse events vs 50.7% patients receiving placebo and 3.6% had serious adverse events vs 6.7% receiving placebo. Nonserious drug-related adverse events occurred in 16.9% of zilebesiran-treated patients (principally injection site reactions and mild hyperkalemia) and 8.0% of placebo-treated patients.
Conclusions and Relevance
In adults with mild to moderate hypertension, treatment with zilebesiran across a range of doses at 3-month or 6-month intervals significantly reduced 24-hour mean ambulatory SBP at month 3.
Trial Registration
ClinicalTrials.gov Identifier: NCT04936035
This phase 2 randomized trial examines the antihypertensive efficacy and safety of different zilebesiran dosing regimens.
Introduction
Despite the availability of a broad range of effective pharmacologic therapies, hypertension remains the leading risk factor worldwide for cardiovascular mortality and progression of kidney disease.1,2,3,4 Global estimates suggest that up to 80% of patients with hypertension do not meet guideline-recommended blood pressure (BP) targets.5,6 Furthermore, even patients who appear to have well-controlled hypertension during periodic office visits may experience significant between-visit variability, which is associated with residual risk for cardiovascular events.7,8,9,10,11,12 Although a broad range of patient and clinician factors contribute to inadequate BP control, inconsistent adherence to complex, multidrug oral treatment regimens that require daily dosing may be an important driver.13,14
Zilebesiran is an investigational RNA interference therapeutic targeting hepatic synthesis of angiotensinogen, the predominant precursor for angiotensin peptides and a key regulator of systemic BP.15,16 In a phase 1 study of patients with mild to moderate hypertension, treatment with single subcutaneous doses of zilebesiran was associated with dose-dependent reductions in serum angiotensinogen and 24-hour ambulatory BP that were sustained for 24 weeks.17 The randomized KARDIA-1 phase 2 study was conducted to evaluate the antihypertensive efficacy and safety of 4 different quarterly and biannual zilebesiran dosing regimens compared with placebo in patients with mild to moderate hypertension.
Methods
Study Design
KARDIA-1 (NCT04936035) was a phase 2, randomized, double-blind, placebo-controlled, dose-ranging study conducted at 78 sites in Canada, Ukraine, the UK, and the US between July 2021 and June 2023 (eFigure 1 in Supplement 1). The protocol was approved by the institutional review board or ethics committee from each site before enrollment of the first patient and all patients provided written informed consent. The trial was conducted in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice guidelines from the International Conference on Harmonization.
Study Participants
Eligible patients included adults aged 18 to 75 years with hypertension who were either untreated or treated with a stable regimen of up to 2 antihypertensive therapies and had a daytime mean ambulatory systolic BP (SBP) between 135 mm Hg and 160 mm Hg following washout of background antihypertensive medication. Those with secondary hypertension, orthostatic hypotension, a serum potassium concentration greater than 5 mEq/L (5 mmol/L), or estimated glomerular filtration rate (eGFR) of 30 mL/min/1.73 m2 or lower (based on the Modification of Diet in Renal Disease formula18) were excluded. Patients with type 1 diabetes, poorly controlled type 2 diabetes (glycated hemoglobin A1c [HbA1c] >9.0% [75 mmol/mol]), or laboratory evidence of diabetes during screening (HbA1c ≥7.0% [53 mmol/mol]) without known diagnosis were also excluded. Detailed eligibility criteria are provided in the study protocol, available in Supplement 2.
Study Procedures
Following a washout period of at least 2 weeks, or 4 weeks for long-acting antihypertensives (a full list of antihypertensives and required washout period is provided in Supplement 1), eligible patients were randomized in a double-blind fashion and in equal proportions using interactive response technology to receive 1 of 4 dosing regimens of subcutaneous zilebesiran (150 mg once every 6 months, 300 mg once every 6 months, 300 mg once every 3 months, or 600 mg once every 6 months) or subcutaneous placebo once every 3 months. Patients randomized to receive zilebesiran once every 6 months received subcutaneous placebo at month 3 to maintain blinding. Randomization was stratified by race (Black vs other) and by baseline 24-hour mean ambulatory SBP (<145 or ≥145 mm Hg). No additional antihypertensive treatment was permitted before month 3, but oral antihypertensive medications could subsequently be reinitiated at the discretion of the investigator. Any additional antihypertensive medications were discontinued at month 5 to permit assessment of the isolated effect of zilebesiran vs placebo at month 6. At month 6, patients in the placebo group were rerandomized to 1 of the 4 zilebesiran groups for an extension phase of the study. This report presents results from the 6-month placebo-controlled treatment period.
End Points and Assessments
The primary end point was change from baseline to month 3 in 24-hour mean ambulatory SBP for each zilebesiran dose group vs placebo. Key secondary end points were change from baseline to month 3 in office SBP, change from baseline to month 6 in 24-hour mean ambulatory SBP, change from baseline to month 6 in office SBP, and the percentage of patients meeting the following response criteria at month 6: 24-hour mean ambulatory SBP of less than 130 mm Hg and/or reduction of 20 mm Hg or more from baseline without additional hypertensive medication intervention (binary end point). Other secondary end points included changes from baseline in 24-hour mean ambulatory diastolic BP (DBP), office DBP, serum angiotensinogen levels, daytime BP (6:00 am to 9:59 pm, from ambulatory BP monitoring), and nighttime BP (10:00 pm to 5:59 am) through 6 months. For the efficacy analyses of end points assessed at month 3, the zilebesiran 300 mg groups (once every 3 months and once every 6 months) were combined because both had received the same zilebesiran dose.
Ambulatory BP monitoring assessments were conducted using an automated device (Suntech Oscar 2M25019,20) that permitted measurement of BP and collected within a 24-hour measurement period every 20 minutes during the day and every 30 minutes during the night. Measurements were conducted initially during screening (following a 2- or 4-week antihypertensive therapy washout period) and again at months 1, 3, and 6. Automated office BP measurements (Microlife WBP Office 2G21) were performed during the screening period (following washout) and at day 1, week 2, and monthly thereafter through 6 months. Further details of the automated BP measurement methods are provided in Supplement 1.
The pharmacodynamic end point was change in serum angiotensinogen levels from baseline to month 6. Serum angiotensinogen levels were assessed at a central laboratory using validated enzyme-linked immunosorbent assays. The safety end point was frequency of adverse events (AEs) through month 6. Safety events were reported based on the Medical Dictionary for Regulatory Activities classifications. Assessment of safety included recording of AEs, measurements of body weight, and laboratory test results.
Analyses were also conducted to assess the consistency of treatment effect in various prespecified subgroups defined by the following baseline characteristics: age (<65 or ≥65 years), sex, race (Black or other), baseline 24-hour mean ambulatory SBP (<145 or ≥145 mm Hg), and eGFR (<60 or ≥60 mL/min/1.73 m2). Race and ethnicity were self-reported by patients and were included because it is known that race and ethnicity impact prevalence/severity of hypertension and response to renin–angiotensin–aldosterone system inhibition. In particular, the phenomenon of suppressed renin–angiotensin–aldosterone system activity is well-established in Black patients. Given the mechanism of action of zilebesiran (upstream targeting of the renin–angiotensin–aldosterone system via angiotensinogen), it was important to stratify randomization by race and incorporate it as a fixed variable in the model to ensure equal balance among the treatment groups and to specifically elucidate the impact of race on treatment response.
Statistical Analysis
Assuming an SD of 20 mm Hg for the primary end point and a 15% rate of loss to follow-up, a sample size of 375 (75 patients per treatment group) was calculated to provide at least 84% power to detect a reduction of 10 mm Hg in ambulatory SBP from baseline to month 3 among zilebesiran doses and placebo, with a 2-sided type I error of .05 (Supplement 3). The power calculation was based on the Dunnett test. The full analysis set included all patients who received at least 1 dose of the study drug (excluding 16 patients randomized in Ukraine before the onset of war in February 2022). BP assessments while patients were receiving, or less than 2 weeks after stopping, any rescue hypertension medication were censored to evaluate the estimated treatment effect of zilebesiran. A mixed model for repeated measures (MMRM) using a restricted maximum likelihood method was used to analyze the primary end point and missing data were addressed assuming missing at random. The model included treatment, visit, treatment × visit interaction, and race (Black vs other) as fixed factors and baseline 24-hour mean ambulatory SBP as a covariate. An unstructured covariance matrix was used.
Similar MMRMs were used for secondary end points (except the binary SBP response end point and change in serum angiotensinogen). The 95% CIs for least-squares mean (LSM) change from baseline and P values and 95% CIs for LSM difference (hereafter referred to as “difference”) between zilebesiran and placebo were calculated. Multiplicity adjustment using the Dunnett test was applied to test the primary hypothesis. After significant primary hypothesis testing, a multiplicity procedure was applied for treatment comparisons between each individual zilebesiran dose group and placebo for key secondary end points, which were tested in order with the primary end point. If results of a test were significant, the next test was evaluated; otherwise all hypotheses in the given and subsequent tests were deemed not statistically significant. Overall type I error was controlled at .05.
For analysis of the binary SBP response end point, a logistic regression model adjusted for treatment, race (Black vs other), and baseline 24-hour mean ambulatory SBP was used with summary of treatment comparisons as odds ratios. Frequency of AEs, percent change from baseline in serum angiotensinogen levels by visit, and changes in body weight were summarized by treatment group using descriptive statistics. Subgroup analyses of the primary end point using MMRM were summarized by LSM estimates of treatment effect and 95% CIs for each evaluated subgroup. All analyses were performed using SAS statistical software, version 9.4 (SAS Institute).
Results
Study Population
Of 1517 patients initially screened for enrollment, 394 were randomized (79 to receive placebo; 79 to receive zilebesiran, 150 mg, once every 6 mo; 78 to receive zilebesiran, 300 mg, every 6 mo; 79 to receive zilebesiran, 300 mg, every 3 mo; and 79 to receive zilebesiran, 600 mg, every 6 mo). All but 1 patient (n = 393) received at least 1 dose of the study drug (Figure 1). Owing to challenges with data collection related to the ongoing war, data from 16 patients randomized in Ukraine were excluded from the analyses. Accordingly, the full analysis set included 377 patients (302 assigned to receive zilebesiran and 75 assigned to receive placebo), of whom 347 patients (92.0%) completed the 6-month placebo-controlled treatment period.
Figure 1. Flow of Participants in the KARDIA-1 Trial of Subcutaneous Zilebesiran for Hypertension.
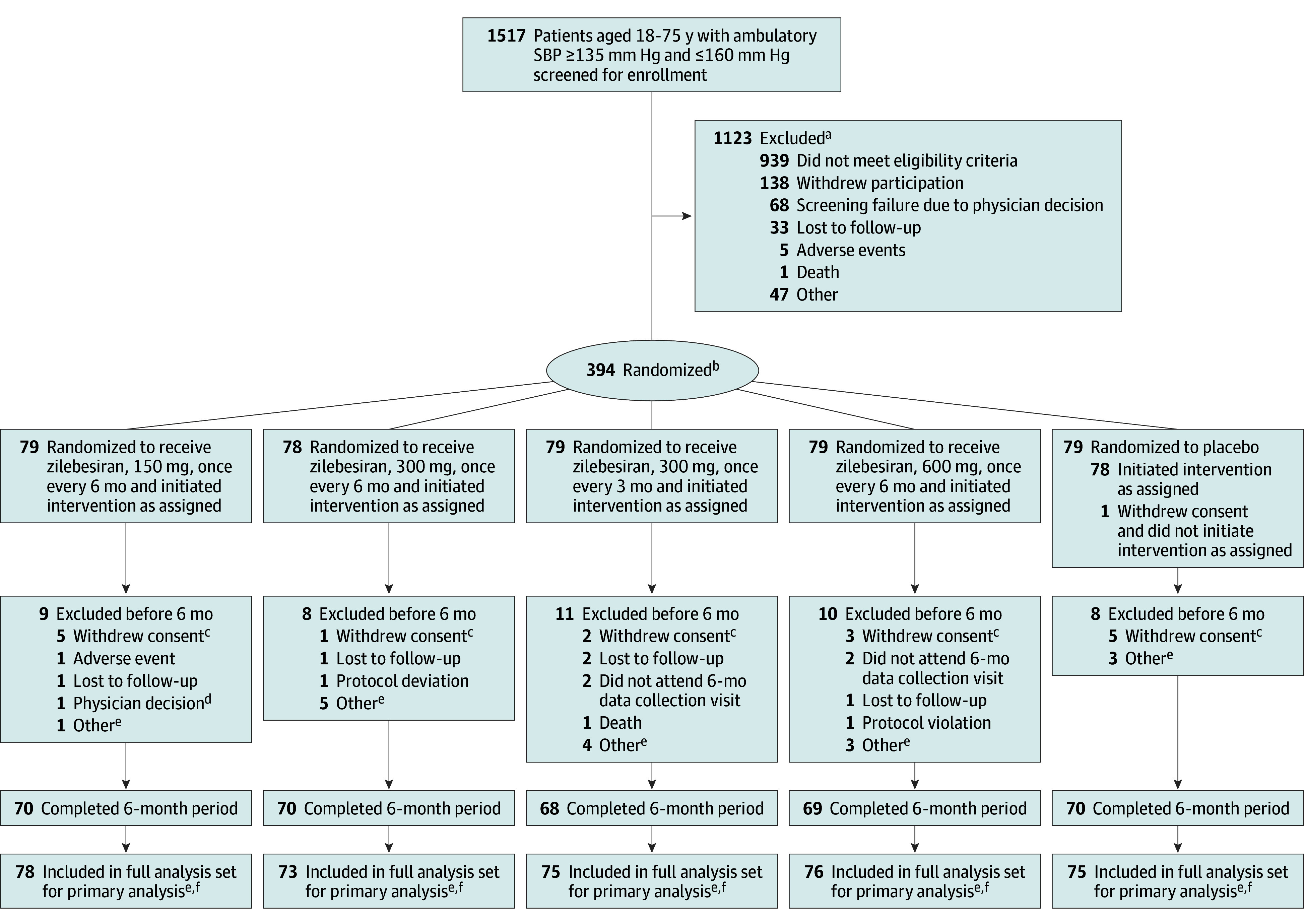
aA patient could have multiple reasons for screen failure and was counted for each reason separately.
bRandomization was stratified by race (Black or other) and baseline mean 24-hour systolic blood pressure (<145 or ≥145 mm Hg).
cPatients withdrew consent because of work-related reasons (n = 4), distance to the site (n = 3), no disclosed reason (n = 2), principal care clinician’s advice (n = 2), time constraints (n = 1), the study not being worth their time (n = 1), personal reasons (n = 1), unwillingness to adhere to ambulatory blood pressure monitoring requirements (n = 1), and adverse events (n = 1).
dPatient was incarcerated during study.
ePatients enrolled at sites in Ukraine (n = 16) were excluded after randomization because war prevented continued data collection.
fPrimary analysis was carried out in the full analysis set, which included all randomized patients who received any amount of study drug.
Baseline characteristics according to treatment assignment are summarized in Table 1. Of the analyzed population, 167 patients (44.3%) were women and 93 (24.7%) were Black. Mean (SD) age at baseline was 56.8 (10.6) years and, at baseline, 24-hour mean ambulatory SBP was 142 (8) mm Hg and DBP was 82 (8) mm Hg.
Table 1. Patient Baseline Characteristics Among the Full Analysis Seta.
| Characteristic | Zilebesiran | Placebo (n = 75) | |||
|---|---|---|---|---|---|
| 150 mg every 6 mo (n = 78) | 300 mg every 6 mo (n = 73) | 300 mg every 3 mo (n = 75) | 600 mg every 6 mo (n = 76) | ||
| Age, mean (SD), y | 55.5 (10.6) | 56.4 (10.3) | 57.7 (10.6) | 57.4 (10.2) | 56.8 (11.2) |
| Women, No. (%) | 39 (50) | 29 (40) | 30 (40) | 31 (41) | 38 (51) |
| Men, No. (%) | 39 (50) | 44 (60) | 45 (60) | 45 (59) | 37 (49) |
| Race, No. (%)b | |||||
| American Indian or Alaska Native | 1 (1) | 0 | 0 | 0 | 0 |
| Asian | 4 (5) | 2 (3) | 7 (9) | 5 (7) | 5 (7) |
| Black or African American | 20 (26) | 17 (23) | 19 (25) | 19 (25) | 18 (24) |
| Native Hawaiian or Pacific Islander | 0 | 0 | 1 (1) | 0 | 0 |
| White | 53 (68) | 54 (74) | 48 (64) | 52 (68) | 52 (69) |
| Ethnicity, No. (%)b | |||||
| Hispanic or Latino | 19 (24) | 16 (22) | 10 (13) | 20 (26) | 9 (12) |
| Not Hispanic or Latino | 59 (76) | 57 (78) | 65 (87) | 56 (74) | 66 (88) |
| Body mass index ≥30, No. (%) | 46 (59) | 46 (63) | 40 (53) | 45 (59) | 37 (49) |
| Type 2 diabetes, No. (%)c | 14 (18) | 11 (15) | 17 (23) | 16 (21) | 10 (13) |
| Receiving ≥1 hypertensive agent before study enrollment, No. (%)d | 43 (55) | 55 (75) | 57 (76) | 63 (83) | 55 (73) |
| 24-h ambulatory blood pressure, mean (SD), mm Hg | |||||
| Systolic | 140.6 (8.5) | 142.5 (8.8) | 141.6 (7.7) | 143.1 (9.0) | 141.1 (7.9) |
| Diastolic | 81.7 (8.3) | 82.3 (8.7) | 82.0 (8.6) | 81.4 (8.3) | 81.7 (7.8) |
| Mean office BP, mm Hg (SD) | |||||
| Systolic | 142.0 (10.9) | 143.0 (11.3) | 140.0 (11.0) | 140.8 (10.6) | 143.1 (13.3) |
| Diastolic | 87.4 (9.6) | 88.8 (8.8) | 85.3 (9.1) | 85.6 (8.8) | 87.9 (10.5) |
| eGFR, mean (SD), mL/min/1.73 m2 | 81.7 (16.5) | 82.0 (14.5) | 80.2 (18.3) | 81.9 (19.4) | 78.7 (21.0) |
| eGFR ≥60 mL/min/1.73 m2, No. (%) | 68 (87) | 70 (96) | 69 (92) | 68 (90) | 64 (85) |
| Serum angiotensinogen concentration, mean (SD), ng/mL | 22.1 (5.9) | 23.2 (7.8) | 20.8 (4.9) | 21.7 (5.9) | 23.9 (10.9) |
Abbreviation: eGFR, estimated glomerular filtration rate.
All randomized patients who received any amount of study drug. Patients enrolled at sites in Ukraine (n = 16) were excluded from the analysis populations.
Race and ethnicity were self-reported from patient to the investigator in closed categories. For ethnicity, categories were Hispanic or Latino, not Hispanic or Latino, not reported, or unknown. For race, categories were American Indian or Alaska Native, Asian, Black or African American, Native Hawaiian or Other Pacific Islander, White, other (please specify), or not reported.
Patients who met the study inclusion criteria with at least 1 of the following: medical history of type 2 diabetes, glycated hemoglobin A1c >7% before first study drug dose, or taking diabetes medication before first study drug dose.
Safety analysis set included all patients who received any amount of study drug, grouped according to the treatment actually received.
Primary End Point
Primary end point data were available for 68 patients in the group assigned to receive zilebesiran, 150 mg, every 6 months; 137 patients receiving zilebesiran, 300 mg, every 6 months or every 3 months; 65 patients receiving zilebesiran, 600 mg, every 6 months; and 60 patients receiving placebo. LSM changes from baseline to month 3 in 24-hour mean ambulatory SBP were −7.3 mm Hg (95% CI, −10.3 to −4.4) for zilebesiran, 150 mg, every 6 months; −10.0 mm Hg (95% CI, −12.0 to −7.9) for zilebesiran, 300 mg, every 3 months or every 6 months; −8.9 mm Hg (95% CI, −11.9 to −6.0) for zilebesiran, 600 mg, every 6 months; and 6.8 mm Hg (95% CI, 3.6-9.9) for placebo. LSM differences in the change from baseline to month 3 in 24-hour mean ambulatory SBP between zilebesiran and placebo were −14.1 mm Hg (95% CI, −19.2 to −9.0) for zilebesiran, 150 mg, every 6 months; −16.7 mm Hg (95% CI, −21.2 to −12.3) for zilebesiran, 300 mg, every 3 months or every 6 months; and −15.7 mm Hg (95% CI, −20.8 to −10.6) for zilebesiran, 600 mg, every 6 months (P < .001 for all comparisons) (Figure 2; Table 2). Treatment differences were consistent across prespecified subgroups (eFigure 2 in Supplement 1).
Figure 2. Ambulatory Systolic Blood Pressure (SBP) Among the Full Analysis Seta.
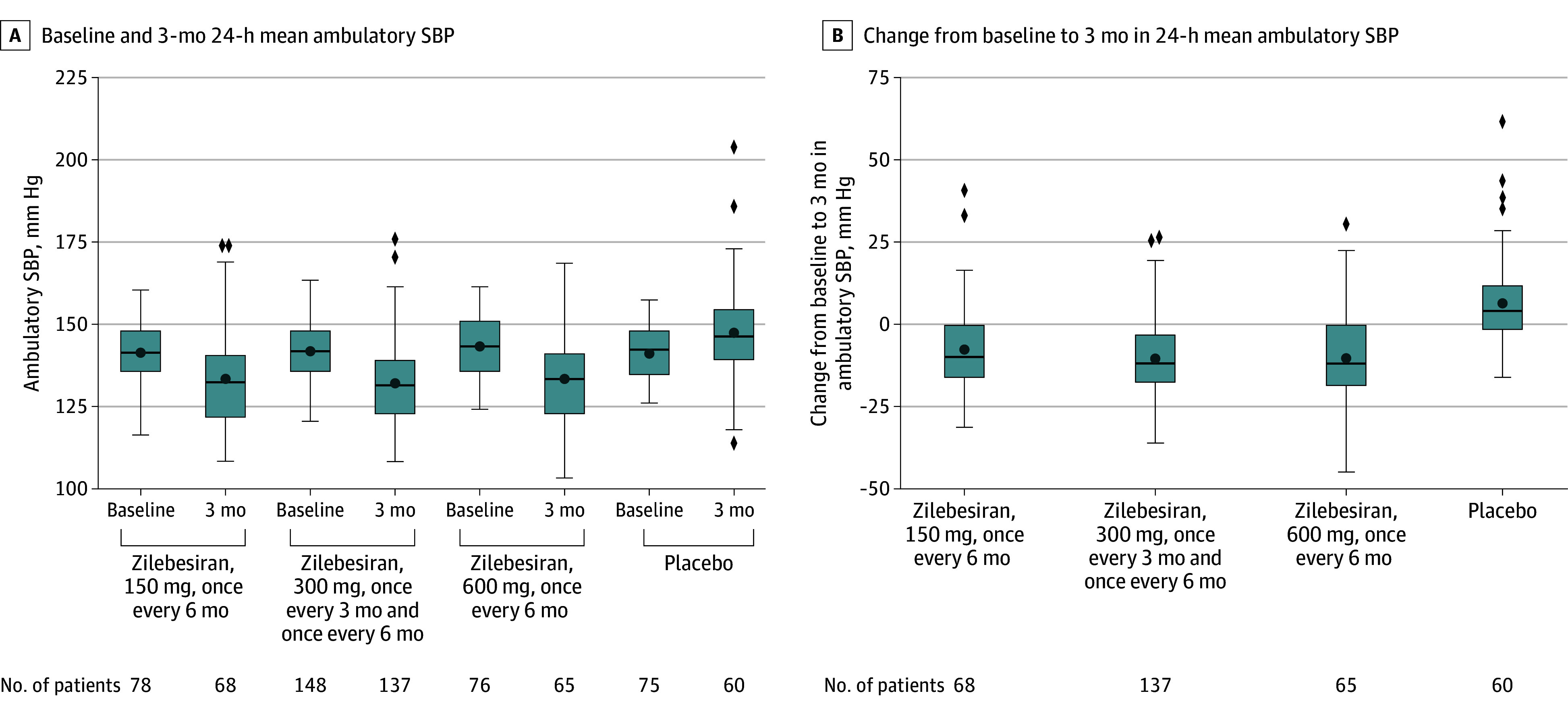
aAll randomized patients who received any amount of study drug, analyzed according to randomized treatment.
Box plots demonstrate median (thick horizontal line), mean (circle), IQR (box top and bottom), highest and lowest values within 1.5 × the IQR (whiskers), and more extreme values (diamonds). For the efficacy analyses of end points assessed at month 3, the zilebesiran 300 mg and 600 mg groups were combined because both had received the same zilebesiran dose.
Table 2. Change From Baseline to Month 3 or 6 in 24-Hour Mean Ambulatory and Office Systolic Blood Pressure (SBP) Among the Full Analysis Seta.
| Outcome | Zilebesiran | Placebo (n = 75) | ||||
|---|---|---|---|---|---|---|
| 150 mg every 6 mo (n = 78) | 300 mg every 6 mo (n = 73) | 300 mg every 3 mo (n = 75) | 300 mg every 3 mo or every 6 mo (n = 148) | 600 mg every 6 mo (n = 76) | ||
| Ambulatory SBP at baseline, patients with data, mean (SD), mm Hg | 140.6 (8.5) | 142.5 (8.8) | 141.6 (7.7) | 142.0 (8.3) | 143.1 (9.0) | 141.1 (7.9) |
| Office SBP at baseline, mean (SD), mm Hg | 142.0 (10.9) | 143.0 (11.3) | 140.0 (11.0) [n = 74] | 141.5 (11.2) [n = 147] | 140.8 (10.6) | 143.1 (13.3) |
| Primary end point | ||||||
| Ambulatory SBP at month 3, mean (SD), mm Hg | 133.2 (14.7) [n = 68] | 131.8 (12.8) [n = 137] | 133.1 (14.9) [n = 65] | 147.3 (15.2) [n = 60] | ||
| LSM change from baseline (95% CI), mm Hg | −7.3 (−10.3 to −4.4) | −10.0 (−12.0 to −7.9) | −8.9 (−11.9 to −6.0) | 6.8 (3.6 to 9.9) | ||
| LSM difference vs placebo (95% CI), mm Hgb | −14.1 (−19.2 to −9.0) | −16.7 (−21.2 to −12.3) | −15.7 (−20.8 to −10.6) | |||
| P valueb | <.001 | <.001 | <.001 | |||
| Secondary end point | ||||||
| Office SBP at month 3, mean (SD), mm Hg | 131.8 (13.6) [n = 68] | 129.1 (13.8) [n = 134] | 131.3 (15.9) [n = 64] | 141.4 (12.6) [n = 60] | ||
| LSM change from baseline (95% CI), mm Hg | −9.7 (−12.6 to −6.8) | −12.1 (−14.2 to −10.0) [n = 133] | −9.2 (−12.1 to −6.2) | −0.1 (−3.2 to 3.0) | ||
| LSM difference vs placebo (95% CI), mm Hgb | −9.6 (−13.8 to −5.3) | −12.0 (−15.7 to −8.3) [n = 133] | −9.1 (−13.4 to −4.8) | |||
| P value | <.001 | <.001 | <.001 | |||
| Ambulatory SBP at month 6, mean (SD), mm Hg | 134.4 (15.0) [n = 62] | 132.2 (13.8) [n = 68] | 131.6 (12.2) [n = 60] | 131.7 (16.8) [n = 63] | 144.6 (15.0) [n = 54] | |
| LSM change from baseline (95% CI), mm Hg | −6.5 (−9.7 to −3.3) | −9.9 (−13.0 to −6.8) | −9.5 (−12.8 to −6.3) | −9.6 (−12.8 to −6.4) | 4.6 (1.2 to 8.0) | |
| LSM difference vs placebo (95% CI), mm Hgb | −11.1 (−15.8 to −6.4) | −14.5 (−19.1 to −9.9) | −14.1 (−18.9 to −9.4) | −14.2 (−18.9 to −9.5) | ||
| P value | <.001 | <.001 | <.001 | <.001 | ||
| Office SBP at month 6, mean (SD), mm Hg | 133.7 (13.6) [n = 65] | 131.2 (16.1) [n = 68] | 127.4 (14.5) [n = 58] | 128.9 (15.6) [n = 62] | 140.6 (12.4) [n = 57] | |
| LSM change from baseline (95% CI), mm Hg | −8.2 (−11.5 to −4.8) | −11.1 (−14.4 to −7.8) | −12.8 (−16.3 to −9.2)[n = 57] | −10.8 (−14.2 to −7.4) | −0.6 (−4.2 to 2.9) | |
| LSM difference vs placebo (95% CI), mm Hgb | −7.5 (−12.4 to −2.7) | −10.5 (−15.3 to −5.7) | −12.1 (−17.2 to −7.1) [n = 57] | −10.2 (−15.1 to −5.3) | ||
| P value | .003 | <.001 | <.001 | <.001 | ||
Abbreviations: LSM, least-squares mean; SBP, systolic blood pressure.
All randomized patients who received any amount of study drug, analyzed according to randomized treatment.
Mixed model for repeated measures adjusted for race and corresponding baseline SBP. P values and 95% CIs for the primary end point are adjusted based on the Dunnett test.
Key Secondary End Points
At month 3, LSM change from baseline in office SBP was −9.7 mm Hg (95% CI, −12.6 to −6.8) for zilebesiran, 150 mg, every 6 months; −12.1 mm Hg (95% CI, −14.2 to −10.0) for zilebesiran, 300 mg, every 3 months or every 6 months; −9.2 mm Hg (95% CI, −12.2 to −6.2) for zilebesiran, 600 mg, every 6 months; and −0.1 mm Hg (95% CI, −3.2 to 3.0) for placebo (Figure 2B). Differences between zilebesiran and placebo were −9.6 mm Hg (95% CI, −13.8 to −5.3) for zilebesiran, 150 mg, every 6 months; −12.0 mm Hg (95% CI, −15.7 to −8.3) for zilebesiran, 300 mg, every 3 months or every 6 months; and −9.1 mm Hg (95% CI, −13.4 to −4.8) for zilebesiran, 600 mg, every 6 months (P < .001 for all comparisons). Similar changes from baseline and differences vs placebo were observed at month 6 for 24-hour mean ambulatory and office SBP (Table 2). At month 6, the percentage of patients who met the response criteria, and the odds ratio of meeting response criteria, were higher for all zilebesiran groups than for placebo (eTable 1 in Supplement 1).
Other Secondary End Points
Changes in both 24-hour mean ambulatory DBP and office DBP from baseline to month 3 and month 6 were consistent with observed changes in SBP (eTable 3 in Supplement 1).
The change from baseline to month 3 and month 6 in serum angiotensinogen was greater in patients who received any dose of zilebesiran than placebo, with a decrease of more than 90% from baseline persisting to month 6 after single 300-mg or 600-mg doses of zilebesiran (eFigure 3 in Supplement 1).
At month 3 and month 6, reductions from baseline SBP were largely consistent for each hour of the 24-hour diurnal cycle in zilebesiran-treated patients (eFigure 4 and eTables 3 and 4 in Supplement 1) and were greater in magnitude than placebo regardless of zilebesiran dose.
Exploratory End Points
During the 6-month double-blind treatment period, the percentage of patients requiring rescue antihypertensives was higher in the placebo group than in the zilebesiran groups (52.0% vs 20.5%-32.1%). The most frequently prescribed classes of antihypertensives in all study groups were calcium channel blockers and diuretics (eTable 5 in Supplement 1). Changes from baseline in serum creatinine, eGFR, and glucose levels over the 6-month treatment period were similar between zilebesiran groups and the placebo group (eTable 6 in Supplement 1).
Safety
Serious AEs were reported in 5 patients (6.7%) in the placebo group and 11 patients (3.6%) in the zilebesiran groups, including 1 death due to cardiopulmonary arrest 5 days after treatment with zilebesiran, 300 mg, none of which were considered by the investigators to be related to the study drug (Table 3). Drug-related AEs were all mild to moderate in severity; those reported in more than 5% of patients in the zilebesiran group were injection site reaction in 19 patients (6.3%) and hyperkalemia in 16 patients (5.3%). More patients in the group that received zilebesiran every 3 months experienced injection site reactions than those who received zilebesiran every 6 months; all events were mild or moderate in severity and transient, and the most common injection site reaction symptoms were pain and erythema. Three patients had drug-related AEs leading to an investigator decision to interrupt further study drug dosing during the double-blind period. Four patients had drug-related AEs leading to an investigator decision to discontinue further study drug dosing during the double-blind period (orthostatic hypotension [n = 2], BP elevation [n = 1], and injection site reaction [n = 1]). Clinically relevant AEs of acute kidney failure were reported in 4 patients (1.3%) receiving zilebesiran vs 0 receiving placebo, hepatic AEs were reported in 9 patients (3.0%) receiving zilebesiran vs 1 (1.3%) receiving placebo, hypotension was reported in 13 (4.3%) receiving zilebesiran vs 1 (1.3%) receiving placebo, and hyperkalemia was reported in 19 (6.3%) patients receiving zilebesiran vs 2 (2.7%) receiving placebo.
Table 3. Summary of Adverse Events (AEs)a.
| Adverse event | Patients, No. (%) | |||||
|---|---|---|---|---|---|---|
| Zilebesiran | Zilebesiran total (n = 302) | Placebo (n = 75) | ||||
| 150 mg every 6 mo (n = 78) | 300 mg every 6 mo (n = 73) | 300 mg every 3 mo (n = 75) | 600 mg every 6 mo (n = 76) | |||
| At least 1 AE | 45 (58) | 44 (60) | 46 (61) | 49 (64) | 184 (61) | 38 (51) |
| Related to study drugb | 12 (15) | 12 (16) | 14 (19) | 13 (17) | 51 (17) | 6 (8) |
| At least 1 SAEc | 0 | 1 (1) | 4 (5) | 6 (8) | 11 (4) | 5 (7) |
| Related to study drugb | 0 | 0 | 0 | 0 | 0 | 0 |
| At least 1 study drug–related AE leading to study drug interruptionb,d | 1 (1) | 0 | 1 (1) | 1 (1) | 3 (1) | 0 |
| At least 1 study drug–related AE leading to study drug discontinuationb,e | 1 (1) | 1 (1) | 1 (1) | 1 (1) | 4 (1) | 0 |
| Death | 0 | 0 | 1 (1) | 0 | 1 (0) | 0 |
| Study drug-related AEs occurring in at least 5% of patientsb | ||||||
| Hyperkalemia | 4 (5) | 3 (4) | 4 (5) | 5 (7) | 16 (5) | 1 (1) |
| Injection site reaction | 3 (4) | 4 (5) | 8 (11) | 4 (5) | 19 (6) | 0 |
| Additional treatment-emergent AE of clinical interest (any relatedness) | ||||||
| Potential hypotensionf | 6 (8) | 6 (8) | 5 (7) | 7 (9) | 24 (8) | 5 (7) |
| Hyperkalemiag | 5 (6) | 4 (5) | 5 (7) | 5 (7) | 19 (6) | 2 (3) |
| Hypotensionh | 3 (4) | 3 (4) | 3 (4) | 4 (5) | 13 (4) | 1 (1) |
| Hepatic AEi | 2 (3) | 2 (3) | 4 (5) | 1 (1) | 9 (3) | 1 (1) |
| Acute kidney failurej | 1 (1) | 1 (1) | 1 (1) | 1 (1) | 4 (1) | 0 |
AEs are defined per Medical Dictionary for Regulatory Activities (MedDRA) terminology.
Related to study drug indicates a reasonable possibility that the event may have been caused by the study drug as evaluated by the investigator.
Serious AEs (SAEs) are any untoward medical occurrence that, at any dose, results in death; is life-threatening (an event that places the patient at immediate risk of death from the event as it occurred but does not include an event that had it occurred in a more severe form might have caused death); requires inpatient hospitalization or prolongation of existing hospitalization; results in persistent or significant disability or incapacity; is a congenital anomaly or birth defect; is an important medical event that may not be immediately life-threatening or result in death or hospitalization but may jeopardize the patient and may require intervention to prevent one of the other outcomes listed in the definition above.
Study drug interruption refers to a pause of further study drug dosing (including placebo dosing) with potential to resume.
Study drug discontinuation is the stopping of further study drug dosing (including placebo dosing) without potential to resume.
Include decreased blood pressure, hypotension, orthostatic hypotension, dizziness, syncope, and orthostasis.
Include hyperkalemia, increased serum potassium, and abnormal serum potassium.
Include additional terms of decreased blood pressure, hypotension, and orthostatic hypotension.
Include AEs mapped to the standardized MedDRA query drug-related hepatic disorders, both narrow and broad terms. Terms include but are not limited to alanine aminotransferase increased; aspartate aminotransferase increased; serum alkaline phosphatase increased; serum bilirubin increased; gamma-glutamyltransferase increased; liver function test abnormal; liver function test increased; and transaminases increased.
Acute kidney failure includes events of increased serum creatinine, increased blood urea, decreased glomerular filtration rate, and acute kidney injury.
There were no serious hepatic AEs. The majority of liver function test result elevations were transient and resolved while receiving treatment. Across all zilebesiran groups, 7 patients (2.3%) experienced elevations of alanine aminotransferase or aspartate aminotransferase of at least 3-fold the upper limit of normal, all of which resolved spontaneously without treatment. No clear differences were identified between the treatment groups. No patients experienced total bilirubin elevation of at least 2-fold the upper limit of normal during the 6-month period.
Hypotension AEs were mild or moderate in severity, nonserious, and transient in nature, with a single event in the zilebesiran, 300 mg, every 3 months group requiring treatment with normal saline. Hyperkalemia AEs were mild and none were associated with acute kidney injury or led to study drug discontinuation. One event of acute kidney injury was reported by the investigator in a patient in the zilebesiran, 300 mg, every 3 months group at the month 6 visit. The event was deemed unrelated to zilebesiran; urinary tract infection was reported as the possible etiology by the investigator. On the start date of the acute kidney injury, BP measurements were 121/75 mm Hg.
At month 6, mean (SD) change in body weight was −0.04 (2.87) kg among patients receiving zilebesiran, 150 mg, every 6 months; 0.57 (3.20) kg among patients receiving zilebesiran, 300 mg, every 6 months; −0.03 (3.40) kg among those receiving zilebesiran, 300 mg, every 3 months; and 0.48 (4.83) kg among those receiving zilebesiran, 600 mg, every 6 months; and 0.35 (3.07) kg among those receiving placebo. On laboratory evaluation, 17 patients (5.6%) assigned to receiving zilebesiran had a serum potassium level greater than 5.5 mmol/L on at least 1 occasion over the 6-month treatment period compared with none assigned to receive placebo. Of these 17 patients, 2 (0.7%) had a serum potassium level greater than 6 mmol/L, which resolved on repeated measurement. Levels of greater than 5.5 mmol/L were confirmed on repeated measurement in 2 additional patients (0.7%) out of 17. Three patients assigned to receive zilebesiran received treatment for hyperkalemia with potassium binders during the study, although no hyperkalemia events led to study drug discontinuation during the 6-month treatment period.
Discussion
In this randomized, dose-ranging study of patients with mild to moderate hypertension, treatment with single subcutaneous doses of zilebesiran was associated with significant reductions in 24-hour mean ambulatory SBP at month 3 compared with placebo. These data support the potential for quarterly or biannual dosing of subcutaneous zilebesiran in achieving a consistent pharmacodynamic effect and effective BP reduction through 6 months. This targeted approach to reduce hepatic angiotensinogen levels through RNA interference is novel for the treatment of hypertension; the long-term safety profile of zilebesiran, either alone or in combination with other antihypertensive agents, requires further study. The study benefited from the use of continuous 24-hour ambulatory BP monitoring, which is considered the criterion-standard of BP assessment.
The magnitude of the placebo-adjusted difference in change from baseline in 24-hour mean ambulatory SBP (up to 16.7 mm Hg) was in part related to dose, but was largely consistent at doses of 300 mg or greater. Differences in both SBP and DBP were maintained over the full diurnal cycle, with consistently lower daytime BP, nighttime BP, and office BP in zilebesiran-treated patients. BP reductions associated with a single dose of zilebesiran persisted to month 6, particularly at 300-mg and 600-mg doses, consistent with pharmacodynamic data demonstrating angiotensinogen reduction. Although mild to moderate drug-related AEs, including injection site reactions and hyperkalemia, were more commonly reported in those receiving zilebesiran than placebo, most were mild and transient and did not require treatment or discontinuation of subsequent drug dosing, and no hyperkalemia AEs were classified as serious.
These data regarding the efficacy, safety, and pharmacodynamic effects of zilebesiran amplify and extend the preliminary findings from the phase 1 study of zilebesiran by confirming effective reduction of serum angiotensinogen associated with lowering of BP over the full 24-hour interval and persisting 6 months after treatment with zilebesiran. Notably, durable reductions of angiotensinogen levels (by greater than 90%) were achieved at month 6 in this study with doses of 300 mg or greater, rather than the 800-mg dose seen at month 6 in the previous phase 1 study, signifying the potential for efficacy at lower doses than suggested in the phase 1 study. Although dose-related reductions in serum angiotensinogen levels were observed, with some variability in duration of effect by dose and dosing interval, the observation that maximal SBP reduction occurred with doses of 300 mg or higher suggests that angiotensinogen reduction of greater than 90% may be sufficient for optimal and sustained lowering of BP. Consistent clinical and pharmacodynamic effects of 300 mg every 3 months, 300 mg every 6 months, and 600 mg every 6 months doses indicate that these regimens achieve effective BP reduction, although this needs to be confirmed in future studies and over longer duration of follow-up.
The between-group difference in change from baseline to month 3 in 24-hour mean ambulatory SBP was contributed to, in part, by an increase from baseline in ambulatory SBP in the placebo group, perhaps as a consequence of delayed BP recovery after the required withdrawal of antihypertensive therapy before randomization. However, a corresponding BP rebound in patients in the zilebesiran groups might similarly have resulted in underestimation of the observed treatment effect from baseline. Importantly, a similar treatment effect from baseline with zilebesiran was seen in the analysis of office SBP despite a lesser observed increase in the placebo group, suggesting that these overall results are a reasonable approximation of treatment effects that might be expected in clinical practice.
These data highlight an opportunity to provide durable BP reductions with biannual dosing of zilebesiran, which could be augmented with dosing of other antihypertensive agents. This may be particularly important in light of substantial data suggesting that challenges with adherence to prescribed oral antihypertensive therapies may be an important contributor to inadequate BP control to guideline-recommended targets in clinical practice.13,14 It is conceivable that dosing on this schedule could be accomplished in the clinical setting for many patients as part of routine scheduled follow-up, reducing the complexity of oral drug regimens (which often include as many as 3 or 4 antihypertensive drugs)22 and limiting the potential for inadvertent missed doses. Moreover, tonic BP control with consistent reduction during daytime and nighttime lasting for 6 months may prove to be a strategy for addressing the residual risk associated with between-visit variability that complicates current oral antihypertensive treatment.
A potential concern with durable reduction of angiotensinogen with zilebesiran is the risk of refractory hypotension, either as a direct adverse effect of treatment or as a consequence of impaired renin–angiotensin system activation during hemodynamic stress from unanticipated volume depletion, bleeding, infection, or cardiac injury. In this regard, it is reassuring that there were no serious or severe related AEs of hypotension or orthostasis observed during zilebesiran treatment in this study. In instances in which blood pressure needs to be restored while receiving zilebesiran, it is likely that standard intervention will be sufficient. Data from the phase 1 study17 have shown that some of the antihypertensive effects of zilebesiran can be attenuated in the short-term by administration of a high-sodium diet. Moreover, residual renin–angiotensin–aldosterone system activity and mechanisms, such as sympathetic nervous system activation, that are left intact by the targeted nature of angiotensinogen inhibition with zilebesiran should allow compensatory BP regulation in the event of hypotensive crisis episodes as shown in preclinical models.23 In addition, reversal agents targeted at RNA interference therapeutics are being considered for development. More frequent AEs of injection site reaction and hyperkalemia were observed in zilebesiran-treated patients compared with placebo, but most injection site reactions were mild, and analysis of serial laboratory testing suggested that the majority of hyperkalemia events were mild, transient, did not require therapeutic intervention, and resolved spontaneously on subsequent testing despite ongoing pharmacologic effect of zilebesiran. More research will be needed to establish the long-term efficacy and safety of zilebesiran. Drug-related AEs will be further evaluated in longer-term safety follow-up, and whether the safety profile of zilebesiran is modified during combination treatment with other therapies will be addressed in the ongoing KARDIA-2 study (NCT05103332), which will investigate the efficacy of zilebesiran as add-on therapy in patients with hypertension that is inadequately controlled with olmesartan, amlodipine, or indapamide.
Limitations
The results of this study should be viewed in the context of important limitations. First, the effects of zilebesiran treatment in a population with mild to moderate hypertension following withdrawal of antihypertensive treatment may not accurately represent the effects in a broader population of unselected patients including those treated with antihypertensives or those with more severe BP elevations. Second, follow-up in this report was limited to 6 months, during which patients received 1 to 2 doses of treatment, so it is unclear whether effects of zilebesiran will endure after repeated administration beyond 6 months or whether changes to its safety profile might arise. Data regarding the impact of longer-term exposure to zilebesiran will be evaluated from the ongoing extension phase of KARDIA-1, during which patients assigned to receive placebo during the initial 6-month period were rerandomized to one of the zilebesiran arms, and all patients will be followed for at least an additional 6 months. Third, although reductions in SBP of the magnitude observed in this study would be expected to reduce cardiovascular events based on extrapolation from prior studies, definitive data regarding the impact on long-term outcomes awaits an adequately powered clinical trial with a longer follow-up period.
Conclusions
In this study of patients with mild to moderate hypertension, treatment with single doses of subcutaneous zilebesiran was associated with clinically meaningful reductions in SBP, compared with placebo, sustained to 6 months. These data support further investigation of zilebesiran as a therapeutic strategy for patients with hypertension.
Educational Objective: To identify the key insights or developments described in this article.
-
What is the mechanism of action of zilebesiran?
A long-lived protein, it mimics angiotensinogen and so selectively inhibits downstream mediators.
It directly inhibits smooth muscle vascular constriction.
It interferes with hepatic synthesis of angiotensinogen, a precursor of angiotensin peptides and a regulator of systemic blood pressure.
-
What were the outcomes across the multiple doing regimens for the primary end point of change in 24-hour mean ambulatory systolic blood pressure at 3 months?
Cohorts treated at lower doses of zilbesiran demonstrated no change in blood pressure while those treated at the highest dose experienced a decrease of 10 mm Hg.
Treated cohorts experienced a significant decrease in blood pressure of 7 to 10 mm Hg while the placebo group experienced an increase in blood pressure.
Zilbesiran did not significantly alter blood pressure.
-
How do the authors interpret the results of this trial?
Adverse events that surfaced during the trial limit zilebesiran’s potential as a therapy for hypertension.
Data support the potential for every 3- or 6-month subcutaneous injections for the treatment of mild to moderate hypertension.
Twenty-four–hour blood pressure monitoring, a strength of the trial, revealed hourly variation in blood pressure that was not anticipated with this long-acting therapy.
eMethods and eResults
Trial protocol
Statistical analysis plan
Nonauthor collaborators
Data sharing statement
References
- 1.Danaei G, Ding EL, Mozaffarian D, et al. The preventable causes of death in the United States: comparative risk assessment of dietary, lifestyle, and metabolic risk factors. PLoS Med. 2009;6(4):e1000058. doi: 10.1371/journal.pmed.1000058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Forouzanfar MH, Liu P, Roth GA, et al. Global burden of hypertension and systolic blood pressure of at least 110 to 115 mm Hg, 1990-2015. JAMA. 2017;317(2):165-182. doi: 10.1001/jama.2016.19043 [DOI] [PubMed] [Google Scholar]
- 3.Global Burden of Metabolic Risk Factors for Chronic Diseases Collaboration . Cardiovascular disease, chronic kidney disease, and diabetes mortality burden of cardiometabolic risk factors from 1980 to 2010: a comparative risk assessment. Lancet Diabetes Endocrinol. 2014;2(8):634-647. doi: 10.1016/S2213-8587(14)70102-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou D, Xi B, Zhao M, Wang L, Veeranki SP. Uncontrolled hypertension increases risk of all-cause and cardiovascular disease mortality in US adults: the NHANES III Linked Mortality Study. Sci Rep. 2018;8(1):9418. doi: 10.1038/s41598-018-27377-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.World Health Organization . Hypertension. Published March 16, 2023. Accessed December 20, 2023. https://www.who.int/news-room/fact-sheets/detail/hypertension
- 6.Million Hearts Foundation . Estimated hypertension prevalence, treatment, and control among US adults: tables. Accessed February 7, 2024. https://millionhearts.hhs.gov/files/Estimated-Hypertension-Prevalence-tables-508.pdf [Google Scholar]
- 7.Parati G, Ochoa JE, Lombardi C, Bilo G. Assessment and management of blood-pressure variability. Nat Rev Cardiol. 2013;10(3):143-155. doi: 10.1038/nrcardio.2013.1 [DOI] [PubMed] [Google Scholar]
- 8.Rothwell PM, Howard SC, Dolan E, et al. ; ASCOT-BPLA and MRC Trial Investigators . Effects of beta blockers and calcium-channel blockers on within-individual variability in blood pressure and risk of stroke. Lancet Neurol. 2010;9(5):469-480. doi: 10.1016/S1474-4422(10)70066-1 [DOI] [PubMed] [Google Scholar]
- 9.Verdecchia P, Angeli F, Mazzotta G, et al. Day-night dip and early-morning surge in blood pressure in hypertension: prognostic implications. Hypertension. 2012;60(1):34-42. doi: 10.1161/HYPERTENSIONAHA.112.191858 [DOI] [PubMed] [Google Scholar]
- 10.Messerli FH, Hofstetter L, Rimoldi SF, Rexhaj E, Bangalore S. Risk factor variability and cardiovascular outcome: JACC review topic of the week. J Am Coll Cardiol. 2019;73(20):2596-2603. doi: 10.1016/j.jacc.2019.02.063 [DOI] [PubMed] [Google Scholar]
- 11.Kario K, Hoshide S, Mizuno H, et al. ; JAMP Study Group . Nighttime blood pressure phenotype and cardiovascular prognosis: practitioner-based nationwide JAMP study. Circulation. 2020;142(19):1810-1820. doi: 10.1161/CIRCULATIONAHA.120.049730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Havenon A, Fino NF, Johnson B, et al. Blood pressure variability and cardiovascular outcomes in patients with prior stroke: a secondary analysis of PRoFESS. Stroke. 2019;50(11):3170-3176. doi: 10.1161/STROKEAHA.119.026293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hill MN, Miller NH, Degeest S, et al. ; American Society of Hypertension Writing Group . Adherence and persistence with taking medication to control high blood pressure. J Am Soc Hypertens. 2011;5(1):56-63. doi: 10.1016/j.jash.2011.01.001 [DOI] [PubMed] [Google Scholar]
- 14.Vrijens B, Vincze G, Kristanto P, Urquhart J, Burnier M. Adherence to prescribed antihypertensive drug treatments: longitudinal study of electronically compiled dosing histories. BMJ. 2008;336(7653):1114-1117. doi: 10.1136/bmj.39553.670231.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morgan ES, Tami Y, Hu K, et al. Antisense inhibition of angiotensinogen with IONIS-AGT-LRx: results of phase 1 and phase 2 studies. JACC Basic Transl Sci. 2021;6(6):485-496. doi: 10.1016/j.jacbts.2021.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cruz-López EO, Ye D, Wu C, et al. Angiotensinogen suppression: a new tool to treat cardiovascular and renal disease. Hypertension. 2022;79(10):2115-2126. doi: 10.1161/HYPERTENSIONAHA.122.18731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Desai AS, Webb DJ, Taubel J, et al. Zilebesiran, an RNA interference therapeutic agent for hypertension. N Engl J Med. 2023;389(3):228-238. doi: 10.1056/NEJMoa2208391 [DOI] [PubMed] [Google Scholar]
- 18.Froissart M, Rossert J, Jacquot C, Paillard M, Houillier P. Predictive performance of the modification of diet in renal disease and Cockcroft-Gault equations for estimating renal function. J Am Soc Nephrol. 2005;16(3):763-773. doi: 10.1681/ASN.2004070549 [DOI] [PubMed] [Google Scholar]
- 19.Jones SC, Bilous M, Winship S, Finn P, Goodwin J. Validation of the OSCAR 2 oscillometric 24-hour ambulatory blood pressure monitor according to the International Protocol for the validation of blood pressure measuring devices. Blood Press Monit. 2004;9(4):219-223. doi: 10.1097/00126097-200408000-00007 [DOI] [PubMed] [Google Scholar]
- 20.Food and Drug Administration . Oscar 2 (model 250) 501k Marketing Notification. Accessed December 20, 2023. https://www.accessdata.fda.gov/cdrh_docs/pdf15/K151520.pdf
- 21.Kollias A, Ntineri A, Kyriakoulis KG, et al. Validation of the professional device for blood pressure measurement Microlife WatchBP Office in adults and children according to the American National Standards Institute/Association for the Advancement of Medical Instrumentation/International Organization for Standardization standard. Blood Press Monit. 2018;23(2):112-114. doi: 10.1097/MBP.0000000000000307 [DOI] [PubMed] [Google Scholar]
- 22.Unger T, Borghi C, Charchar F, et al. 2020 International Society of Hypertension global hypertension practice guidelines. Hypertension. 2020;75(6):1334-1357. doi: 10.1161/HYPERTENSIONAHA.120.15026 [DOI] [PubMed] [Google Scholar]
- 23.Uijl E, Mirabito Colafella KM, Sun Y, et al. Strong and sustained antihypertensive effect of small interfering RNA targeting liver angiotensinogen. Hypertension. 2019;73(6):1249-1257. doi: 10.1161/HYPERTENSIONAHA.119.12703 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eMethods and eResults
Trial protocol
Statistical analysis plan
Nonauthor collaborators
Data sharing statement