

Abstract
Premise
A family‐specific probe set for sunflowers, Compositae‐1061, enables family‐wide phylogenomic studies and investigations at lower taxonomic levels, but may lack resolution at genus to species levels, especially in groups complicated by polyploidy and hybridization.
Methods
We developed a Hyb‐Seq probe set, Compositae‐ParaLoss‐1272, that targets orthologous loci in Asteraceae. We tested its efficiency across the family by simulating target enrichment sequencing in silico. Additionally, we tested its effectiveness at lower taxonomic levels in the historically complex genus Packera. We performed Hyb‐Seq with Compositae‐ParaLoss‐1272 for 19 Packera taxa that were previously studied using Compositae‐1061. The resulting sequences from each probe set, plus a combination of both, were used to generate phylogenies, compare topologies, and assess node support.
Results
We report that Compositae‐ParaLoss‐1272 captured loci across all tested Asteraceae members, had less gene tree discordance, and retained longer loci than Compositae‐1061. Most notably, Compositae‐ParaLoss‐1272 recovered substantially fewer paralogous sequences than Compositae‐1061, with only ~5% of the recovered loci reporting as paralogous, compared to ~59% with Compositae‐1061.
Discussion
Given the complexity of plant evolutionary histories, assigning orthology for phylogenomic analyses will continue to be challenging. However, we anticipate Compositae‐ParaLoss‐1272 will provide improved resolution and utility for studies of complex groups and lower taxonomic levels in the sunflower family.
Keywords: Asteraceae, double‐capture, Hyb‐Seq, MarkerMiner, Packera, polyploidy, target enrichment
The sunflower family, also known as the daisy family, Asteraceae, or Compositae, is one of the largest flowering plant families, making up roughly 10% of all angiosperms. This large and diverse group has presented many challenges for resolving evolutionary relationships and studying diversifications through time and space. Recent phylogenetic work in the family has employed various methods to reconstruct family‐level phylogenies to better understand the evolutionary history and relationships of Asteraceae. For example, Huang et al. (2016) used transcriptome data, Zhang et al. (2021) used a combination of transcriptome and whole‐genome sequence data, while Mandel et al. (2019) used target enrichment sequencing with a custom probe set designed to enrich for conserved gene sequences in Asteraceae (Mandel et al., 2014, 2017). This probe set has become popular among researchers studying members of Asteraceae and has enabled investigations at lower taxonomic levels, especially understudied groups (e.g., Siniscalchi et al., 2019, 2023; Lichter‐Marck et al., 2020; Thapa et al., 2020; de Lima Ferreira et al., 2022).
Targeted sequence probe sets have grown in popularity over the past 10 years with sets designed to target loci across large plant groups: bryophytes (i.e., mosses: Liu et al., 2019), pteridophytes (i.e., ferns: Wolf et al., 2018), and angiosperms (i.e., Johnson et al., 2019), as well as for specific plant families (i.e., Asteraceae: Mandel et al., 2014, 2017; Fabaceae: Chapman, 2015; Ochnaceae: Shah et al., 2021; Orchidaceae: Eserman et al., 2021). Typically, low‐coverage genome skimming and/or transcriptome data have been used to design probe sets (Straub et al., 2012; Weitemier et al., 2014; Folk et al., 2015; Fonseca and Lohmann, 2020); however, genome skimming is generally not as effective for designing a probe set for nuclear genes, as low‐coverage genome skimming data typically enrich for organellar genomes and other high‐copy genomic sequences in plants (Stull et al., 2013). These genomic regions are often highly conserved and repetitive and are thus less useful for resolving relationships in some groups. Using transcriptome data offers the potential to sequence and select from thousands of loci, enabling the survey of genomic regions with different rates of molecular evolution.
Several tools have recently become available to design targeted sequence probe sets using transcriptome data more easily, such as OrthoFinder (Emms and Kelly, 2019) and MarkerMiner (Chamala et al., 2015). OrthoFinder is a pipeline that identifies orthogroups and/or orthologs in transcriptomes based on sequence similarities across many species (Emms and Kelly, 2015). In return, the output returns a list of exons usable for probe design. One disadvantage to OrthoFinder, and ultimately the transcriptome‐only approach, is that without knowledge of intron–exon topology, probes could overlap boundaries and thus would not be effective at sequence capture (McKain et al., 2018). Alternatively, the identification of intron–exon boundaries is straightforward using the MarkerMiner tool, which aligns transcriptome data to reference angiosperm genome sequences and returns intron‐masked multiple sequence alignments (Chamala et al., 2015; McKain et al., 2018). The general workflow for MarkerMiner compares user‐provided transcriptome sequences against reference genomes with known single‐copy orthologous genes (e.g., Arabidopsis thaliana (L.) Heynh.), drastically reducing the number of paralogous sequences, or “paralogs,” retained for each gene. Probe sets designed using this approach have yielded greater phylogenetic resolution in some groups at the family level (e.g., Cactaceae: Acha and Majure, 2022) and genus/species level (e.g., Euphorbia L.: Villaverde et al., 2018; Zanthoxylum L.: Reichelt et al., 2021). Retaining only single‐copy orthologs as a result of MarkerMiner can greatly improve species tree inference, as paralogs complicate phylogeny building by causing gene tree heterogeneity. If not accounted for properly, this heterogeneity can lead to misleading phylogeny construction and an incorrect interpretation of species relationships (Smith and Hahn, 2021).
In this study, we used 48 transcriptomes to generate a new probe set for sequencing orthologous sequences in Asteraceae utilizing MarkerMiner. Our sampling included 45 Asteraceae taxa and three outgroups from across the order Asterales: Calyceraceae, Campanulaceae, and Goodeniaceae. Although Compositae‐1061 has been shown to be efficient at higher and some lower taxonomic levels within the family, it generally lacks resolution at the genus to species level. Therefore, we designed this probe set with the aim to provide higher resolution at lower taxonomic levels and help tackle challenges associated with paralogy, especially among complex groups. To do this, we tested the compatibility and efficiency of this new probe set across the entire family by simulating target enrichment sequencing in silico in six Compositae members spanning across the family. We then used members of the genus Packera Á. Löve & D. Löve as a model system to directly test the efficacy of the probe set by sequencing 16 Packera and three outgroup taxa using this newly designed probe set, named Compositae‐ParaLoss‐1272, and the Compositae‐1061 probe set. Additionally, we combined the Compositae‐1061 and Compositae‐ParaLoss‐1272 sequence data to represent an in silico double‐capture method. We then generated phylogenetic trees, compared their topologies, and assessed node support to determine whether Compositae‐ParaLoss‐1272 provided greater resolution at the genus and/or species level compared to Compositae‐1061.
METHODS
Probe development
To identify single‐copy nuclear loci and select regions for target enrichment probe design, transcriptome data from 48 taxa spanning Asterales were compiled from the 1KP initiative (One Thousand Plant Transcriptomes Initiative, 2019), Sunflower Genome database (https://sunflowergenome.org/), or generated de novo (Appendix S1; see Supporting Information with this article). Four specimens were collected from the Memphis Botanic Garden live collection, of which we did not make herbarium vouchers. All 48 samples were used as input for MarkerMiner version 1.0 (Chamala et al., 2015) using default settings with both A. thaliana and Vitis vinifera L. as reference genomes. MarkerMiner is a freely available bioinformatic workflow that compares user‐provided transcriptomes against reference angiosperm genomes with known single‐copy orthologous genes that can be used to design primers or probes for targeted sequencing. Orthologous genes are classified as single copy in the reference genomes if they are present across 17 genomes that were previously annotated as part of a systematic survey on duplication‐resistant genes (De Smet et al., 2013). We aimed for this new probe set to have no gene overlap with Compositae‐1061 (Mandel et al., 2014, 2017) and Angiosperms353 (Johnson et al., 2019). Therefore, if a gene present in our new probe set was in either Compositae‐1061 or Angiosperms353, we removed it from our targeted gene list, e.g., if AT3G47610 was included in the Angiosperms353 gene list and ours, we removed this gene from our list and did not design probes for it.
Exons with lengths ranging from 120 to 1000 bp and a minimum variability of two single‐nucleotide polymorphisms (SNPs) were selected using a custom Python script (https://github.com/ClaudiaPaetzold/MarkerMinerFilter). The resulting 3853 exonic regions, spanning 1925 genes around 1112–85,780 bp long (Appendix S2), were further processed by MyBaits at Arbor Biosciences (Ann Arbor, Michigan, USA) to produce a set of 120‐mer tiled baits that overlap every 60 bases and share an 80% identity when possible, similar to methods used to develop the MyBaits Compositae‐1061 kit (Mandel et al., 2014), hereafter referred to as Comp‐1061. Additional filtering steps were implemented as follows: (1) sequence clusters containing five or more taxa not targeting lineage‐specific genes or clusters were retained, (2) clusters containing only the reference sequence data were removed, (3) probes with at least three sequences that covered the alignment were retained, and (4) probes with high similarities (80% or 90%) representing only one or two species were collapsed. Finally, two additional loci were added to the probe design: the MADS‐box transcription factor LEAFY (LFY; Weigel et al., 1992) and the transmembrane pseudokinase CORYNE (CRN; Müller et al., 2008), two conserved single‐copy genes that regulate flower development and meristem size, respectively, in angiosperms. Gene sequences for LFY were identified using the TBLASTX plug‐in in Geneious Prime version 2023.0.4 (https://www.geneious.com) with custom Bidens ferulifolia (Jacq.) Sweet (cv. Compact Yellow) leaf transcriptome and Lactuca sativa L. genome assembly (v.8) BLAST databases, respectively. The CRN gene sequence (AT5G13290) came directly from A. thaliana using The Arabidopsis Information Resource (TAIR; https://www.arabidopsis.org/).
The resulting MyBaits target enrichment kit contains 60,158 120‐bp‐long, in‐solution, biotinylated baits based on target sequence information. The final bait panel, Compositae‐ParaLoss‐1272, consisted of 13,117 probes and 1272 loci after filtering (Table 1).
Table 1.
A summary of the major differences between the sunflower‐family‐specific probe sets, Compositae‐ParaLoss‐1272 (Comp‐ParaLoss‐1272) and Compositae‐1061 (Comp‐1061), and the angiosperm‐wide probe set, Angiosperms353 (Angio‐353).
| Characteristic | Comp‐ParaLoss‐1272 | Comp‐1061 | Angio‐353 |
|---|---|---|---|
| No. of loci | 1272 | 1061 | 353 |
| No. of baits | 60,158 | 9678a | 75,151b |
| No. of overlapping loci | 0 | 30 (with Angio‐353)c | 30 (with Comp‐1061)c |
| No. of species | 48 | 3 | 42 |
| Input data | Transcriptomes | Expressed sequence tags (EST) | Transcriptomes |
| Tool | MarkerMiner | BLAST | k‐medoid clustering |
Note: No. of loci = number of targeted loci; No. of baits = number of baits in probe set; No. of overlapping loci = number of loci that overlap with another probe set indicated within parentheses; No. of species = number of species used to develop probe set; Input data = input data type to develop probe set; Tool = tool used to develop probe set.
Mandel et al., 2014.
Johnson et al., 2019.
Siniscalchi et al., 2021.
These methods are compared to Comp‐1061, which was developed via BLAST searches of expressed sequence tag (EST) data from three species within the sunflower family (Helianthus annuus L. [sunflower], Lactuca sativa [lettuce], and Carthamus tinctorius L. [safflower]) to a set of previously identified A. thaliana single‐copy genes. This resulted in 1061 genes, for which 9678 biotinylated baits were designed (Mandel et al., 2014, 2017). Refer to Table 1 for a comparison between Compositae‐ParaLoss‐1272 and Comp‐1061.
Simulating capture sequencing across Compositae
We simulated a target enrichment sequencing run in silico on six published genomes spanning Asteraceae (Figure 1) using Compositae‐ParaLoss‐1272, hereafter referred to as Comp‐ParaLoss‐1272, and Comp‐1061 in the software CapSim (Cao et al., 2018) to investigate the efficiency of this new probe set for recovering loci across the sunflower family. CapSim is a tool that simulates a sequence run in silico with a given genome sequence and probe set as input. The simulated data can be used for evaluating the performance of the analysis pipeline, as well as the efficiency of the probe design.
Figure 1.
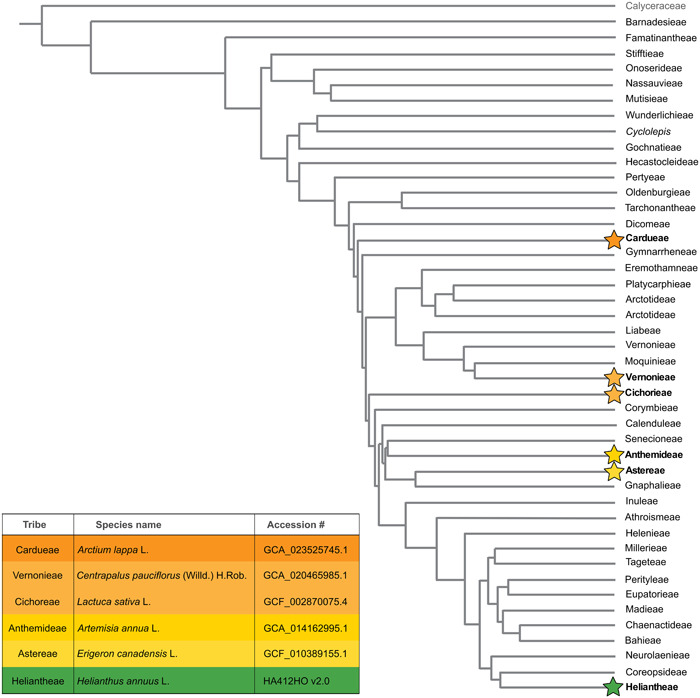
Phylogeny of Asteraceae tribes and the family's proposed sister group, Calyceraceae, modified from Mandel et al. (2019). Stars at branch tips indicate a specimen from that tribe was used for in silico sequencing analyses utilizing CapSim. The colors of stars relate to the table in the bottom left containing NCBI sequence accession numbers, excluding Helianthus annuus, which came from Badouin et al. (2017; https://sunflowergenome.org/assembly-data/).
Prior to running CapSim, an index file was generated, and probes were aligned to the six genomes using Bowtie2 version 2.3.5.1 (Langmead and Salzberg, 2012; Langmead et al., 2019). After the alignment, the sequence alignment/map (SAM) files were sorted and indexed into binary alignment map (BAM) files using SAMtools version 1.9 (Danecek et al., 2021). The resulting BAM files were then used as input in CapSim using the jsa.sim.capsim command with the following settings: median fragment size at shearing (‐‐fmedian) set to 250, MiSeq simulated (‐‐miseq), Illumina read length (‐‐illen) set to 150, and the number of fragments (‐‐num) set to 50,000,000. The resulting FASTQ files were used as input in the HybPiper version 2.0.1 (Johnson et al., 2016) pipeline to map simulated sequences against the probe set. Summary and paralog statistics were recovered using the ‘stats’ and ‘paralog_retriever’ options in HybPiper.
Specimen collection
An Illumina sequence run was performed using the new probe set on a selection of 19 total taxa—16 Packera and three outgroup taxa—that were previously sequenced with the Comp‐1061 probe set (Moore‐Pollard and Mandel, 2023a). Packera taxa were selected to be representative across the entire Packera phylogenetic tree from Moore‐Pollard and Mandel (2023a). One outgroup taxon, Packera loratifolia (Greenm.) W. A. Weber & Á. Löve, was included in this analysis as an outgroup instead of an ingroup because previous studies have shown it is likely misclassified in Packera and instead should be in Senecio (Barkley, 1985; Bain and Jansen, 1995; Bain and Golden, 2000; Pelser et al., 2007; Moore‐Pollard and Mandel, 2023a). A complete list of sampled species, herbarium vouchers, and National Center for Biotechnology Information (NCBI) accession numbers can be found in Table 2.
Table 2.
Voucher specimens for the Illumina sequence run. Publication status and authorities are according to the International Plant Names Index (IPNI).
| NCBI accession | ||||||||
|---|---|---|---|---|---|---|---|---|
| Species | Location | Collector and voucher no. (Herbarium)a | Collection date | Sheet barcode or ID number | Raw reads (paired)b | Reads mappedb | Comp‐1061 | Comp‐ParaLoss‐1272 |
| Emilia fosbergii Nicolson | USA; Florida, Osceola County | Wayne D. Longbottom, David H. Williams, Holly L. Williams 14545 (NY) | 18‐Nov‐2010 | 02074297 | 1,572,629,062 | 10,414,762 | SRR22543392 | SRR24860889 |
| Packera aurea (L.) Á. Löve & D. Löve | USA; Tennessee, Campbell County | Floden 866 (TENN) | s.d. | N/A | 3,009,238,834 | 19,928,734 | SRR22543326 | SRR24860888 |
| Packera cana (Hook.) W. A. Weber & Á. Löve | USA; Idaho, Adams County | Don Knoke 2101 (WTU) | 25‐Jun‐2011 | 406472 | 4,989,136,942 | 33,040,642 | SRR24862023 | SRR24860878 |
| Packera candidissima (Greene) W. A. Weber & Á. Löve | Mexico; Sierra Madre Occidental | Robert A. Bye 9680 (ASU) | 26‐May‐1980 | 121438 | 2,880,272,150 | 19,074,650 | SRR22543387 | SRR24860877 |
| Packera castoreus (S. L. Welsh) Kartesz | USA; Utah, Piute County | Alan Taye 3674 (OSC) | 20‐Sep‐1987 | 172202 | 2,269,567,448 | 15,030,248 | SRR22543385 | SRR24860876 |
| Packera crocata (Rydb.) W. A. Weber & Á. Löve | USA; Colorado, Jackson County | Mary Damm 38 (OSC) | 29‐Jul‐2002 | 244322 | 6,132,282,442 | 40,611,142 | SRR22543379 | SRR24860875 |
| Packera cynthioides (Greene) W. A. Weber & Á. Löve | USA; New Mexico, Grant County | Darrell E. Ward 80‐010 (NY) | 6‐Sep‐1980 | 03088483 | 2,917,414,224 | 19,320,624 | SRR22543377 | SRR24860874 |
| Packera dubia (Spreng.) Trock & Mabb. | USA; North Carolina, Chesapeake County | J. Brandon Fuller (NCU) | 29‐Jun‐2020 | N/A | 2,167,035,730 | 14,351,230 | SRR22543313 | SRR24860880 |
| Packera franciscana (Greene) W. A. Weber & Á. Löve | USA; Arizona, Coconino County | J. Resinger 1577 (ARIZ) | 14‐Jul‐1976 | 233800 | 4,604,239,452 | 30,491,652 | SRR22543368 | SRR24860873 |
| Packera glabella (Poir.) C. Jeffrey | USA; Tennessee, Bradley County | DeSelm 06‐04 (TENN) | s.d. | N/A | 3,641,082,026 | 24,113,126 | SRR22543366 | SRR24860872 |
| Packera greenei (A. Gray) W. A. Weber & Á. Löve | USA; California, Trinity County | E. R. Moore 8 (MEM) | 27‐Jun‐2019 | 20904 | 2,943,301,060 | 19,492,060 | SRR22543365 | SRR24860871 |
| Packera layneae (Greene) W. A. Weber & Á. Löve | USA; California, El Dorado County | Kathryn A. Beck 200310 (WTU) | 30‐Apr‐2003 | 375035 | 5,681,052,766 | 37,622,866 | SRR22543356 | SRR24860887 |
| Packera loratifolia (Greenm.) W. A. Weber & Á. Löve | Mexico; Sierra La Viga | J. A. Villarreal, J. Valdes R 5163 (ASU) | 16‐Sep‐1989 | 182928 | 2,487,875,698 | 16,475,998 | SRR22543355 | SRR24860886 |
| Packera musiniensis (S. L. Welsh) Trock | USA; Utah, Sanpete County | D. Atwood 21259 (ARIZ) | 9‐Aug‐1996 | 334839 | 2,988,242,284 | 19,789,684 | SRR22543346 | SRR24860885 |
| Packera porteri (Greene) C. Jeffrey | USA; Oregon | Coll. Wm. Cusick 2308 (OSC) | 8/3/1899 | 97915 | 4,421,594,684 | 29,282,084 | SRR22543334 | SRR24860884 |
| Packera pseudaurea (Rydb.) W. A. Weber & Á. Löve | USA; Idaho, Valley County | James F. Smith 9147 (OSC) | 29‐Jul‐2010 | 228940 | 3,922,950,102 | 25,979,802 | SRR22543332 | SRR24860883 |
| Packera streptanthifolia (Greene) W. A. Weber & Á. Löve | USA; Oregon, Grant County | Sharon Birks 2010‐42 (OSC) | 16‐Jul‐2010 | 255384 | 13,606,754,356 | 90,110,956 | SRR22543319 | SRR24860882 |
| Packera texensis O'Kennon & Trock | USA; Texas, Gillespie County | B. L. Turner 24‐75 (TEX) | 10‐Apr‐2004 | 00211804 | 4,920,515,898 | 32,586,198 | SRR22543316 | SRR24860881 |
| Roldana gilgii (Greenm.) H. Rob. & Brettell | Mexico; Chiapas | D. E. Breedlove 24411 (TEX) | 5‐Mar‐1972 | 00062617 | 2,082,647,568 | 13,792,368 | SRR22543307 | SRR24860879 |
Note: N/A = not available; s.d. = sine datum (without date).
Herbarium acronyms per Index Herbariorum (Thiers, 2024).
Indicates a report for only the Compositae‐ParaLoss‐1272 probe set.
DNA extraction and sequencing
DNA extraction and sequencing methods for the 19 taxa utilizing the Comp‐ParaLoss‐1272 probe set followed steps outlined by Moore‐Pollard and Mandel (2023a). Briefly, dried leaf tissue collected from herbarium specimens was used to extract DNA. DNA length was assessed by running a 1% agarose gel in 1× TBE and GelRed 3× (Biotium, Fremont, California, USA), with a target DNA length of 400–500 bp. If DNA fragments appeared larger than 500 bp, up to 1 µg of DNA was sheared via sonication with a QSonica machine (amp: 20%; pulse: 10 seconds on, 10 seconds off) (QSonica, Newtown, Connecticut, USA). Sheared DNA was then used to generate barcoded libraries utilizing NEBNext Ultra II DNA Library Prep Kit (New England Biolabs, Ipswich, Massachusetts, USA). Libraries produced followed the NEBNext Ultra II Version 5 protocol with size selection on DNA fragments at a range of 300–400 bp but were adjusted by halving the amount of reagents and DNA. Targeted sequence capture was performed on the libraries using the newly designed probe set, Comp‐ParaLoss‐1272, from Arbor Biosciences described above, following manufacturer's protocols (version 4.01). Captured targets were amplified and quantified using KAPA library quantification kits (Kapa Biosystems, Wilmington, Massachusetts, USA). Quality and quantity checks were performed throughout using a NanoDrop 2000 (Thermo Fisher Scientific, Waltham, Massachusetts, USA) and Qubit High Sensitivity Assay (Thermo Fisher Scientific), respectively. The pooled libraries were sequenced on an Illumina NovaSeq 6000 (Illumina, San Diego, California, USA) at HudsonAlpha Institute for Biotechnology (Huntsville, Alabama, USA). Data for the Comp‐1061 taxa were obtained from Moore‐Pollard and Mandel (2023a) and are available at NCBI (BioProject: PRJNA907383; see Data Availability Statement).
Phylogenetic analyses
Raw sequence reads from Comp‐1061 and Comp‐ParaLoss‐1272 were cleaned and trimmed of adapters using Trimmomatic version 0.36 (Bolger et al., 2014), implementing the Sliding Window quality filter (illuminaclip 2:30:10, leading 20, trailing 20, sliding window 5:20). Cleaned reads were retained if they had a minimum length of 36 bp and were then mapped against the corresponding loci targeted in the Comp‐1061 (Mandel et al., 2014) or Comp‐ParaLoss‐1272 probe sets using the HybPiper pipeline. A combined reference/de novo assembly was performed using BWA version 0.7.17 (Li and Durbin, 2009) and SPAdes version 3.5 (Bankevich et al., 2012), respectively, with specified k‐mer lengths: 21, 33, 55, 77, and 99. The resulting sequences were then aligned using MAFFT version 7.407 (Katoh and Standley, 2013). Maximum likelihood trees were built in RAxML version 8.1.3 (Stamatakis, 2014) with 1000 bootstrap replicates under the GTR+I+Γ model. Species trees were generated from each resulting RAxML gene matrix using ASTRAL‐III version 5.7.3 (Zhang et al., 2018), a pseudo‐coalescent tree building method. Local posterior probability (LPP) values were generated at each node to indicate the probability that the resulting branch is the true branch given the set of input gene trees. LPP is considered a more reliable clade support measure than bootstrapping because it is computed based on a quartet score (Sayyari and Mirarab, 2016) and assumes incomplete lineage sorting (Zhang et al., 2018).
The sequence data from Comp‐1061 and Comp‐ParaLoss‐1272 were also combined, hereafter referred to as Comp‐1061 + Comp‐ParaLoss‐1272, and a phylogenetic tree was built following the methods above. The resulting species trees—Comp‐1061, Comp‐ParaLoss‐1272, and Comp‐1061 + Comp‐ParaLoss‐1272—were then visualized using the package phytools (Revell, 2012) in R version 4.0.5 (R Core Team, 2016; RStudio Team, 2020).
Measuring phylogenomic discordance
To determine if Comp‐ParaLoss‐1272 increased node resolution across Packera, Quartet Sampling (Pease et al., 2018) was used to assess the confidence, consistency, and informativeness of internal tree relationships. Quartet Sampling provides a more comprehensive support value estimate than LPP by calculating four scores, three at each node (quartet concordance [QC], quartet differential [QD], and quartet informativeness [QI]) and one at the tip (quartet fidelity [QF]), to determine if the internal relationships are caused by a lack of data, underlying biological processes, or rogue taxa. QC specifies how often a concordant quartet is inferred over other discordant quartets as a range from −1 to 1; −1 indicates that the quartets are more often discordant than concordant, and 1 indicates that all quartets are concordant. QD reveals how skewed the discordant quartets are as a range from 0 (high skew) to 1 (low skew). QI suggests how informative the quartets are as a range from 0 (none are informative) to 1 (all are informative). Each terminal branch is then given a QF score, which reports how often a taxon is included in the concordant topology given a range of 0 (taxon is present in none) to 1 (taxon is present in all). Quartet Sampling requires a concatenated nucleotide matrix and a rooted species tree. The concatenated matrices were generated using FASconCAT‐G version 1.02 (Kück and Longo, 2014) into a PHYLIP format. The input phylogeny was then rooted using the pxrr command in Phyx (Brown et al., 2017).
PhyParts version 0.0.1 (Smith et al., 2015) was then used to quantify and visualize discordance in the final phylogenies. PhyParts summarizes and visualizes conflict among gene trees given the resulting species tree topology by performing a bipartition analysis, which helps determine if the node support values are misleading because of underlying discordance. This tool requires a rooted final species tree and rooted gene trees as input. Thus, these trees were rooted to the three outgroup taxa, Roldana gilgii (Greenm.) H. Rob. & Brettell, Emilia fosbergii Nicolson, and Packera loratifolia. The script “phypartspiecharts.py” (available at https://github.com/mossmatters/MJPythonNotebooks) was then used to map pie charts onto the nodes in the final species tree, detailing whether there is one dominant topology in the gene trees with not much conflict, if there is one frequent alternative topology, or many low‐frequency topologies.
To estimate similarity scores between the Comp‐1061 and Comp‐ParaLoss‐1272 tree topologies, we calculated the adjusted Robinson–Foulds (RFadj) distance as outlined by Moore‐Pollard and Mandel (2023a) between the two trees using the RF.dist function in package phangorn (Schliep, 2011) in R. Unrooted ASTRAL‐III trees were used as input with the “normalize” argument set to TRUE. RFadj calculates the distance between two unrooted trees, with resulting RFadj values closer to zero indicating that the tree topologies are similar, and values closer to one indicating complete dissimilarity. Parsimony informativeness was calculated between matrices of Comp‐1061 and Comp‐ParaLoss‐1272 using MEGA‐X version 10.2.5 (Kumar et al., 2018). Heatmaps to compare sequence lengths of retained loci between probe sets were generated in R using the package ggplot2 (Wickham, 2016). Additionally, the average and standard deviation of locus lengths were calculated using the mean and sd functions in base R.
RESULTS
CapSim
CapSim results showed that both the Comp‐1061 and Comp‐ParaLoss‐1272 probe sets were successful across a broad range of Asteraceae members as both probe sets retained a moderate number of loci. The Comp‐1061 probe set generally retained more loci than Comp‐ParaLoss‐1272, with an average of about 551 loci retained using the Comp‐1061 probe set and an average of 453 loci with the Comp‐ParaLoss‐1272 probe set (Table 3). Even so, the average length of the loci was much longer in the Comp‐ParaLoss‐1272 probe set with genes averaging 1922 bp long, and the Comp‐1061 probe set produced genes averaging 403 bp long (Appendix S3). Additionally, Comp‐ParaLoss‐1272 produced fewer paralog warnings than Comp‐1061, with a range of 0–2 paralogs retained per sample with the Comp‐ParaLoss‐1272 probe set and a range of 96–250 paralogs per sample with Comp‐1061 (Table 3). A full list of statistics can be found in Appendix S3.
Table 3.
Summary statistics of the CapSim run after running the ‘stats’ function in HybPiper.
| Comp‐1061 | Comp‐ParaLoss‐1272 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Species | Reads mapped | % on target | Genes mapped | % genes retained | Paralog warnings | Reads mapped | % on target | Genes mapped | % genes retained | Paralog warnings |
| Artemisia annua L. | 93,739,367 | 93.7% | 407 | 38.4% | 108 | 97,421,399 | 97.4% | 433 | 34.0% | 1 |
| Helianthus annuus L. | 97,351,903 | 97.4% | 750 | 70.7% | 250 | 97,357,378 | 97.4% | 403 | 31.7% | 1 |
| Centrapalus pauciflorus (Willd.) H. Rob. | 94,823,613 | 94.8% | 466 | 43.9% | 101 | 97,708,408 | 97.7% | 468 | 36.8% | 0 |
| Lactuca sativa L. | 98,218,579 | 98.2% | 749 | 70.6% | 223 | 97,532,753 | 97.5% | 519 | 40.8% | 2 |
| Erigeron canadensis L. | 95,987,418 | 96.0% | 548 | 51.6% | 96 | 97,231,893 | 97.2% | 500 | 39.3% | 1 |
| Arctium lappa L. | 92,956,716 | 93.0% | 388 | 36.6% | 103 | 97,530,647 | 97.5% | 399 | 31.4% | 1 |
Packera sequence statistics
Illumina sequencing utilizing the Comp‐ParaLoss‐1272 probe set resulted in a total of 501 million reads and 76 billion sequences across the 19 newly sequenced taxa. Additionally, the minimum and maximum number of reads ranged from 10.4 million in Emilia fosbergii to 90.1 million in Packera streptanthifolia (Greene) W. A. Weber & Á. Löve (Table 2). The Comp‐1061 sequence data from Moore‐Pollard and Mandel (2023a) totaled 142 million reads and 21 billion sequences, with the minimum and maximum number of reads ranging from 1.2 million in P. musiniensis (S. L. Welsh) Trock to 15 million in P. dubia (Spreng.) Trock & Mabb., respectively.
The HybPiper pipeline retained 1049 genes (out of 1061) when using the Comp‐1061 probe set and 1213 genes (out of 1272) with the Comp‐ParaLoss‐1272 probe set. The number of loci recovered for each taxon ranged from 923 in Packera musiniensis to 1051 in Roldana gilgii using the Comp‐1061 probe set and from 1258 in P. musiniensis to 1271 in P. streptanthifolia using the Comp‐ParaLoss‐1272 probe set. The number of loci retained was proportionally higher in Comp‐ParaLoss‐1272 compared to Comp‐1061 (Figure 2B), although the Comp‐1061 alignment contained less missing data (Comp‐1061: 34.89%; Comp‐ParaLoss‐1272: 35.05%) and was more parsimony informative (Comp‐1061: 11.7%; Comp‐ParaLoss‐1272: 8.3%) than Comp‐ParaLoss‐1272 (Appendix S4). Alternatively, the Comp‐ParaLoss‐1272 probe set recovered drastically fewer paralogous sequences (“paralogs”) than the Comp‐1061 probe set, with only about 5% of the recovered loci reporting as paralogous, compared to 59% with the Comp‐1061 probe set (Figure 2A). The number of paralog warnings ranged from 35–407 genes per sample with the Comp‐1061 probe set, compared to 0–14 in the Comp‐ParaLoss‐1272 probe set (Table 4). Additionally, Comp‐ParaLoss‐1272 recovered much longer loci compared to Comp‐1061 (MeanComp‐1061 = 292.13, SDComp‐1061 = 146.18; MeanComp‐ParaLoss‐1272 = 1192.02, SDComp‐ParaLoss‐1272 = 809.5; Figure 3). Using the combined probe set, Comp‐1061 + Comp‐ParaLoss‐1272, resulted in a species tree made from 2182 loci (out of 2333). A full compilation of statistics is provided in Appendix S4.
Figure 2.
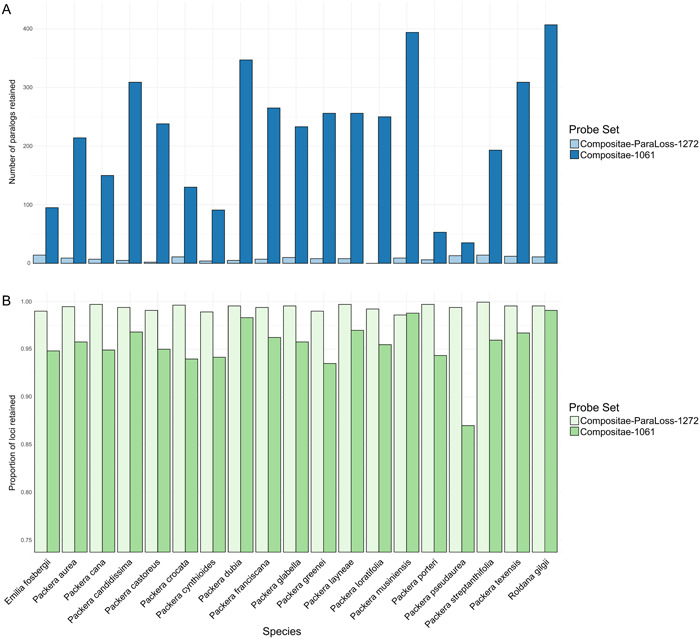
Barplots showing (A) the number of flagged paralogs and (B) the proportion of loci retained for each species depending on the probe set used. Lighter colors represent the Compositae‐ParaLoss‐1272 probe set, while darker colors represent the Compositae‐1061 probe set as indicated by the keys to the right of the plots. Barplots were generated using base R.
Table 4.
Summary statistics of the Illumina sequencing run after running the ‘stats’ function in HybPiper.
| Comp‐1061 | Comp‐ParaLoss‐1272 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Species | Reads mapped | % on target | Genes mapped | % genes retained | Paralog warnings | Reads mapped | % on target | Genes mapped | % genes retained | Paralog warnings |
| Emilia fosbergii | 1,185,704 | 59% | 1006 | 94.8% | 95 | 11,236,129 | 65% | 1259 | 99.0% | 14 |
| Packera aurea | 5,184,671 | 53% | 1016 | 95.8% | 214 | 4,438,388 | 26% | 1265 | 99.4% | 9 |
| Packera cana | 1,532,039 | 21% | 997 | 94.0% | 130 | 8,297,634 | 35% | 1268 | 99.7% | 7 |
| Packera candidissima | 558,742 | 36% | 999 | 94.2% | 91 | 5,690,438 | 43% | 1264 | 99.4% | 5 |
| Packera castoreus | 1,654,718 | 37% | 1043 | 98.3% | 347 | 2,912,785 | 32% | 1260 | 99.1% | 2 |
| Packera crocata | 2,361,884 | 36% | 1021 | 96.2% | 265 | 10,762,526 | 35% | 1267 | 99.6% | 11 |
| Packera cynthioides | 1,171,793 | 29% | 1007 | 94.9% | 150 | 2,064,556 | 36% | 1258 | 98.9% | 4 |
| Packera dubia | 4,514,739 | 39% | 1016 | 95.8% | 233 | 2,775,445 | 26% | 1266 | 99.5% | 5 |
| Packera franciscana | 1,573,692 | 41% | 992 | 93.5% | 256 | 9,169,648 | 45% | 1264 | 99.4% | 7 |
| Packera glabella | 1,972,057 | 34% | 1029 | 97.0% | 256 | 6,012,371 | 31% | 1266 | 99.5% | 10 |
| Packera greenei | 2,024,706 | 34% | 1013 | 95.5% | 250 | 4,102,840 | 27% | 1259 | 99.0% | 8 |
| Packera layneae | 2,814,096 | 35% | 1048 | 98.8% | 394 | 8,240,509 | 26% | 1268 | 99.7% | 8 |
| Packera loratifolia | 511,859 | 43% | 1001 | 94.3% | 53 | 2,435,806 | 35% | 1262 | 99.2% | 0 |
| Packera musiniensis | 68,064 | 9% | 923 | 87.0% | 35 | 6,518,518 | 44% | 1254 | 98.6% | 9 |
| Packera porteri | 1,510,836 | 39% | 1018 | 95.9% | 193 | 5,896,137 | 40% | 1268 | 99.7% | 6 |
| Packera pseudaurea | 3,914,039 | 41% | 1027 | 96.8% | 309 | 7,423,481 | 41% | 1264 | 99.4% | 13 |
| Packera streptanthifolia | 2,695,188 | 38% | 1026 | 96.7% | 309 | 23,885,890 | 39% | 1271 | 99.9% | 14 |
| Packera texensis | 2,516,755 | 33% | 1008 | 95.0% | 238 | 9,026,502 | 38% | 1266 | 99.5% | 12 |
| Roldana gilgii | 1,545,552 | 28% | 1051 | 99.1% | 407 | 2,105,459 | 34% | 1266 | 99.5% | 11 |
Figure 3.
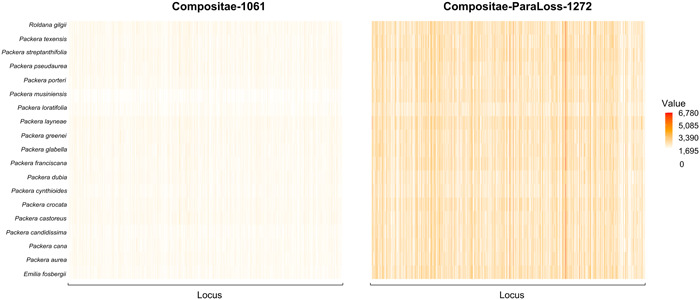
Heatmap of retained locus length in the Compositae‐1061 (left) and Compositae‐ParaLoss‐1272 (right) analyses for each locus (x‐axis) of each species in the analysis (y‐axis). The longest loci are indicated by vertical red lines, and the shortest loci are indicated by vertical orange to white lines. Loci not retained are shown as white. Heatmaps were generated in R.
Discordance of Packera taxa
A higher number of gene trees were represented in the final Comp‐ParaLoss‐1272 species tree compared to the Comp‐1061 tree (normalized quartet score = 0.461 and 0.424, respectively), with the Comp‐1061 + Comp‐ParaLoss‐1272 species tree having an intermediate value (normalized quartet score = 0.436). Additionally, the Comp‐ParaLoss‐1272 probe set provided higher resolution at internal nodes compared to Comp‐1061, with 13 of the 17 internal nodes having LPP values greater than or equal to 0.97, eight of those being fully supported (1.0 LPP). In comparison, the Comp‐1061 probe set had only eight nodes greater than or equal to 0.97 LPP, seven of those with 1.0 LPP (Figure 4), while Comp‐1061 + Comp‐ParaLoss‐1272 had 12 nodes greater than or equal to 0.97 LPP, nine of which were 1.0 LPP (Appendix S5). Additionally, the level of discordance of internal Packera relationships varied between both trees. Quartets are more often discordant than concordant in the Comp‐1061 tree, with four internal nodes having negative QC values, compared to only one node (between Packera pseudaurea (Rydb.) W. A. Weber & Á. Löve and P. aurea (L.) Á. Löve & D. Löve, QC = −0.3) in the Comp‐ParaLoss‐1272 tree (Figure 5).
Figure 4.
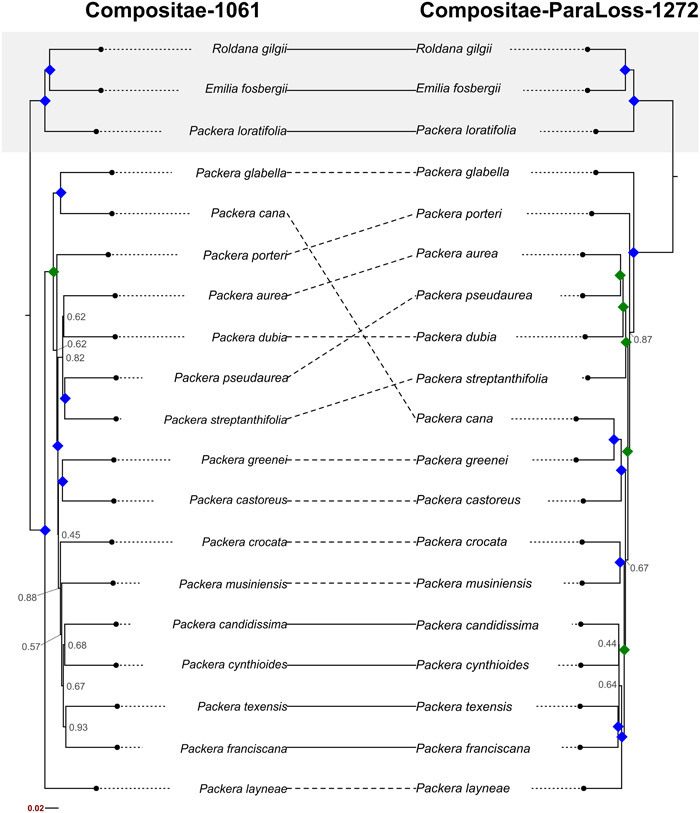
Tanglegram comparing species topologies when phylogenies were developed using the Compositae‐1061 probe set (left) or the Compositae‐ParaLoss‐1272 probe set (right). Topologies representing the same relationship are indicated with a solid line, differing relationships are indicated by a dashed line. Local posterior probability (LPP) values of 1.0 LPP are indicated by a blue diamond at the node. LPP values ranging from 0.97–0.99 are indicated by a green diamond. LPP values lower than 0.97 are shown at the corresponding node in gray font. Outgroup species are highlighted with a gray shadow box.
Figure 5.
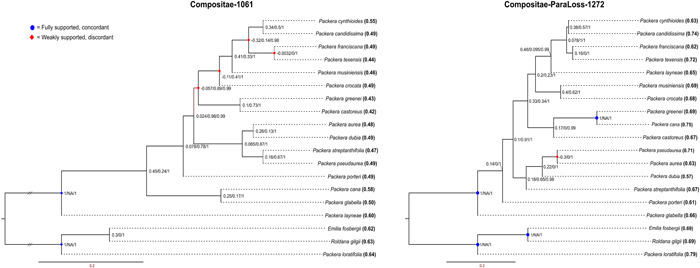
Discordance and support values in the Compositae‐1061 (left) and Compositae‐ParaLoss‐1272 (right) trees indicated by Quartet Sampling. At each node, three values are represented: quartet concordance (QC), quartet differential (QD), and quartet informativeness (QI), shown as QC/QD/QI. Blue circles at the node indicate fully supported and concordant quartets; red diamonds indicate weakly supported and discordant quartets as indicated by Quartet Sampling. Quartet fidelity (QF) scores are at each tip label in parentheses and bolded.
The resulting Comp‐1061 and Comp‐ParaLoss‐1272 species tree topologies were moderately incongruent with each other (RFadj = 0.625). Of the taxon relationships that remained the same in both trees, Comp‐ParaLoss‐1272 showed more concordant and strongly supported relationships compared to Comp‐1061 (Figures 5 and 6). For example, both tree topologies have P. cynthioides (Greene) W. A. Weber & Á. Löve and P. candidissima (Greene) W. A. Weber & Á. Löve as sister, and P. franciscana (Greene) W. A. Weber & Á. Löve and P. texensis O'Kennon & Trock as sister; all four within the same smaller clade (Figure 5). However, the node between P. franciscana and P. texensis and the node joining the two sister groups were majorly discordant in the Comp‐1061 tree (QC = −0.0032 and −0.32, respectively), while the same relationships in the Comp‐ParaLoss‐1272 tree were less discordant (QC = 0.16 and 0.078, respectively). Even so, the internal relationships were still not strongly supported.
Figure 6.
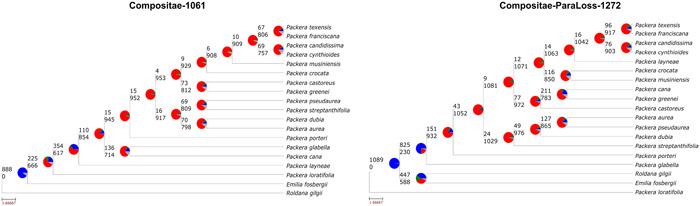
PhyParts results between the Compositae‐1061 probe set (left) and Compositae‐ParaLoss‐1272 probe set (right). Pie charts at nodes show the percentage of gene tree discordance or concordance when compared to the final species tree. The color scheme reveals the percentage of gene trees that are: concordant (blue), the top alternative bipartition (green), all other alternative bipartitions (red), or uninformative at that node (gray). Numbers above and below the branch indicate the number of concordant (blue) and conflicting (red) gene trees, respectively.
The outgroup relationships and monophyly of Packera were fully supported in the Comp‐ParaLoss‐1272 tree (Figure 5). Alternatively, the Comp‐1061 tree showed the monophyly of Packera with full support; however, the relationship between the outgroup taxa, Emilia fosbergii and Roldana gilgii, showed weak support with a discordant skew (QS score at node: 0.3/0/1; Figure 5). Quartet fidelity scores were generally higher in the Comp‐ParaLoss‐1272 tree than the Comp‐1061 tree, which ranged from 0.57–0.79 and 0.42–0.64, respectively (Figure 5), indicating a higher percentage of quartet topologies involving the tested taxa were concordant with the focal tree branch in the Comp‐ParaLoss‐1272 tree.
DISCUSSION
In this study, we designed and tested a complementary Compositae‐specific probe set, Compositae‐ParaLoss‐1272, that provided higher resolution at the lower taxonomic levels of species in our Packera test case. The new probe set dramatically reduced the number of paralogs recovered, retained longer gene sequences, and was likely important for improving the resolution in our Packera comparison. Also, this new probe set successfully retained genes across all tested members of Asteraceae and recovered more and longer orthologous genes than Comp‐1061 (Appendix S3), as well as retained a substantially lower number of paralogs than Comp‐1061 (Table 3) when tested in silico. Finally, it is possible to perform a double sequence capture because the genes associated with Comp‐1061 and Angiosperms353 are not included in the Comp‐ParaLoss‐1272 probe design (Table 1).
While our results showed that Comp‐1061 retained a higher number of genes in silico (Table 3), the Illumina sequencing run of the Comp‐ParaLoss‐1272 probe set shows much higher locus retention and greater resolution than the Comp‐1061 probe set (Table 3). We hypothesize that the low loci retention in silico is a relic of read simulators not always capturing the variances of Illumina‐sequenced data because they cannot perfectly model noise or sequencing technology biases (May et al., 2022; Duncavage et al., 2023). Additionally, we suspect that having longer gene sequences in the probe set influences read simulator results, although we cannot confirm the validity of these suspicions.
Comp‐ParaLoss‐1272 contained more missing data and was considered slightly less parsimony informative (PI) than Comp‐1061 (Appendix S4); however, the differences were minimal (PIComp‐ParaLoss‐1272 = 23.4%, PIComp‐1061 = 24.1%). Interestingly, similar results were found in a previous study that generated a Fabaceae‐specific probe set using MarkerMiner and compared the results to other probe design methods (Vatanparast et al., 2018). This study found that MarkerMiner produced fewer paralogous loci than other design methods, but also was not as parsimony informative as other methods, following our results.
When comparing the Comp‐1061 and Comp‐ParaLoss‐1272 tree topologies to the larger Packera phylogeny (Moore‐Pollard and Mandel, 2023a), the evolutionary relationships of the Comp‐ParaLoss‐1272 tree were in slightly higher agreement with the whole‐genus phylogeny (RFadj = 0.6) as compared to Comp‐1061 (RFadj = 0.667) (Appendix S6), potentially indicating this new probe set is more robust to species sampling compared to Comp‐1061. For example, our Comp‐1061 tree places P. layneae (Greene) W. A. Weber & Á. Löve as sister to the remaining core Packera species. This relationship differs from both the Comp‐ParaLoss‐1272 and Moore‐Pollard and Mandel (2023a) trees, which have P. layneae placed more deeply nested and with other California‐endemic species (Figure 4; Moore‐Pollard and Mandel, 2023a). Additionally, the placement of P. glabella (Poir.) C. Jeffrey in the Comp‐1061 tree differs from past phylogenomic studies, as well as the Comp‐ParaLoss‐1272 tree in this study, which place it as sister to all remaining Packera taxa (Freeman, 1985; Barkley, 1988; Trock, 1999; Bain and Golden, 2000; Schilling and Floden, 2015). While this is promising, further studies are needed to investigate whether the new probe set is more robust to taxon sampling.
The resulting tree topologies were moderately incongruent between Comp‐1061 and Comp‐ParaLoss‐1272 (RFadj = 0.625; Figure 4), indicating that species relationships varied depending on the probe set used. We suggest that these differences can be explained by (1) the different gene sets used to make the phylogeny, (2) the differences in paralog retention, or (3) the underlying biological processes present within Packera. First, given that this new probe set was complemented against Comp‐1061 during production, there is no overlap of gene sequences between probe sets; consequently, only unique gene sequences, which have their own evolutionary histories, were used to generate each phylogeny. Therefore, the tree topologies and species relationships could differ as the Comp‐ParaLoss‐1272 phylogeny may be reflecting unique gene histories not shared with Comp‐1061, and vice versa. Next, having fewer paralogs, as is seen in Comp‐ParaLoss‐1272, resulted in species relationships that may better reflect the underlying evolutionary histories and not as much gene heterogeneity (Smith and Hahn, 2021; Zhou et al., 2021). Finally, biological processes, such as hybridization, reticulation, or incomplete lineage sorting, may be influencing our results as these processes are known to cause complications in phylogenetic construction (Arnold, 1997; Maddison, 1997; Alberts et al., 2002; Nussbaum et al., 2007).
Although only marginal, the Comp‐ParaLoss‐1272 tree had lower levels of discordance, indicating that Comp‐ParaLoss‐1272 provides more concordant nodes than Comp‐1061, although the nodes are still highly discordant (Figures 5 and 6). It is reasonable to consider that the underlying biological processes discussed above may be influencing the level of discordance in our phylogeny, as Packera members have a long history of reticulation (e.g., Bremer, 1994; Bain et al., 1997) and hybridizing in the wild (e.g., Fernald, 1943; Barkley, 1962; Chapman and Jones, 1971; Uttal, 1984; Bain, 1988; Trock, 1999; Gramling, 2006; Weakley et al., 2011). Similar conclusions have been found in other groups (e.g., Sessa et al., 2012; Vargas et al., 2017; Morales‐Briones et al., 2018). Interestingly, a recent study in Packera showed that low support or discordant clades may be the result of ancient reticulation events in Packera's history (Moore‐Pollard and Mandel, 2023b), ultimately influencing the relationships and support within the species trees. We hypothesize that using Comp‐ParaLoss‐1272 will not only directly reduce issues associated with polyploidy, but also reduce issues from hybridization even if not addressed directly. Another possible explanation for the low node resolution is that only a subset of taxa (16 out of 88 Packera taxa) were used to generate these phylogenies. Having such low species sampling could influence species relationships and node support values given a lack of data (Heath et al., 2008; Sanderson et al., 2010).
Combining the sequence data from Comp‐1061 with Comp‐ParaLoss‐1272, Comp‐1061 + Comp‐ParaLoss‐1272 resulted in a topology that differed more substantially from the phylogeny generated using the Comp‐1061 probe set (RFadj = 0.625) compared to the Comp‐ParaLoss‐1272 probe set (RFadj = 0) (Appendix S5). Additionally, Comp‐1061 + Comp‐ParaLoss‐1272 resulted in a more resolved phylogeny than using Comp‐1061 and Comp‐ParaLoss‐1272 alone (Appendix S5). For example, only three nodes had low support in the Comp‐1061 + Comp‐ParaLoss‐1272 tree compared to four nodes in the Comp‐ParaLoss‐1272‐only tree, and eight in the Comp‐1061‐only tree (Appendix S5). Even so, one of the discordant nodes in the combined tree had the lowest reported LPP value (LPP = 0.19), potentially indicating that underlying biological processes, such as hybridization or polyploidy, may be complicating the relationships at that node.
Ultimately, the most notable difference between the Comp‐ParaLoss‐1272 and Comp‐1061 probe sets is the number of paralogs retained per individual, which was far fewer in the Comp‐ParaLoss‐1272 probe set than the Comp‐1061 probe set. We predict this difference may result from (1) performing stricter filtering in the probe design process, (2) using more data to generate the probe set, e.g., Comp‐1061 used ESTs that were designed using low‐coverage transcriptomes vs. Comp‐ParaLoss‐1272 which used complete transcriptomes, and (3) using more sequences across the phylogenetic breadth of the family, e.g., a single‐copy gene in one lineage may be a multi‐copy gene in a different lineage; therefore, using limited sampling when generating the Comp‐1061 probe set (only three taxa in probe design) very likely missed some duplications that Comp‐ParaLoss‐1272 (48 taxa in probe design) was able to detect. While removing paralogs from a data set may alleviate issues associated with ortholog determination in phylogenomic studies, it is important to note that paralogs are still reflective of the true evolutionary history of genes within some groups, including Packera. For example, hybridization and polyploidy are common in Packera, with around 40% of all Packera members exhibiting polyploidy (Trock, 1999; Moore‐Pollard and Mandel, 2023a, 2023b), and thus paralogs are expected in the data set as it reflects the true evolutionary history of the group. Therefore, removing paralogs can remove full gene histories, impacting the ability to accurately model processes like reticulation and polyploidy. Combining sequence data from both Comp‐1061 and Comp‐ParaLoss‐1272 may be ideal if investigating clades for signals of reticulation or gene and genome duplication events. Additionally, new methods have been developed to better address these processes (Yang and Smith, 2014; Morales‐Briones et al., 2021; Nauheimer et al., 2021; Zhang and Mirarab, 2022; Jackson et al., 2023), so we anticipate our combined probe set data will be useful for researchers who are interested in exploring their data in new ways. Even so, the Comp‐1061 and Comp‐ParaLoss‐1272 probe sets are still comparable options for target enrichment sequencing in lower taxonomic members of Compositae.
Overall, the low paralog retention of the Comp‐ParaLoss‐1272 probe set can be very advantageous when dealing with groups known to be complicated by polyploidy because polyploidy is typically associated with higher paralog retention (Lynch and Conery, 2000; Wolfe, 2001; Veitia, 2005). More attention is being focused on polyploidy in non‐model plant groups (e.g., Lim et al., 2008; Bellinger et al., 2022; Fernández et al., 2022), and the underlying challenges associated with it are becoming more well known (see Rothfels, 2021). Being able to address these challenges early in the phylogenomic pipeline can improve phylogenetic reconstructions and provide more confidence in data interpretations. We therefore anticipate that future work will test this probe set across different taxonomic levels, given that this study only tested it at the generic level, and provide additional support for the utility of this probe set in complex groups in the sunflower family. We hope this design approach will be seen as a model for other complex systems.
AUTHOR CONTRIBUTIONS
E.R.M.P. designed the probe set, generated and analyzed data, and wrote the manuscript. J.R.M. helped design the probe set. D.S.J. provided transcriptome data for probe design and funds for sequencing. J.R.M. and D.S.J. provided edits to the manuscript. All authors approved the final version of the manuscript.
Supporting information
Appendix S1. Voucher specimens used to develop the Compositae‐ParaLoss‐1272 probe set using MarkerMiner. Species names and authorities are according to the International Plant Names Index (IPNI).
Appendix S2. List of 1925 targeted loci in the Compositae‐ParaLoss‐1272 probe set and information about their associated functions in Arabidopsis thaliana (source: The Arabidopsis Information Resource [TAIR]; https://www.arabidopsis.org/tools/bulk/genes/index.jsp). Vitis vinifera–specific genes that have no known function (n = 17) are included.
Appendix S3. HybPiper summary statistics for the six Asteraceae genomes from the CapSim run.
Appendix S4. General and full HybPiper statistics of the Illumina sequence run.
Appendix S5. Tanglegrams comparing the relationships between the combined data set, Compositae‐1061 + Compositae‐ParaLoss‐1272, against the individual data sets: Compositae‐1061 (A) and Compositae‐ParaLoss‐1272 (B). Lines between the taxa at the tips compare relationships: solid lines indicate the same relationship; dashed lines indicate differing relationships. Local posterior probability (LPP) values are represented at each node, with full support (1.0 LPP) in blue, moderate support (0.9–0.99 LPP) in green, and low support (≤0.89 LPP) in red.
Appendix S6. Tanglegrams comparing the relationships between a pruned‐down version of the Moore‐Pollard and Mandel (2023a) tree now containing the 19 taxa used in this study, compared to the Compositae‐1061 (A) and Compositae‐ParaLoss‐1272 (B) trees generated in this study. Lines between the taxa at the tips compare relationships: solid lines indicate the same relationship; dashed lines indicate differing relationships.
Appendix S7. Compositae‐ParaLoss‐1272 gene set file for bioinformatic analyses.
ACKNOWLEDGMENTS
The authors thank Matthew D. Pollard (University of Memphis) for his bioinformatic help; Brian Brunelle at Arbor Biosciences for his assistance and expertise with probe design; the University of Memphis High‐Performance Cluster (HPC) administrators, Eric Spangler and Kristian Skjervold, for their assistance with the HPC and overall willingness to provide support; and Jane Grimwood at HudsonAlpha.
Moore‐Pollard, E. R. , Jones D. S., and Mandel J. R.. 2024. Compositae‐ParaLoss‐1272: A complementary sunflower‐specific probe set reduces paralogs in phylogenomic analyses of complex systems. Applications in Plant Sciences 12(1): e11568. 10.1002/aps3.11568
Contributor Information
Erika R. Moore‐Pollard, Email: moore.erika.r@gmail.com.
Jennifer R. Mandel, Email: jmandel@memphis.edu.
DATA AVAILABILITY STATEMENT
Raw sequence data are available in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (BioProjects PRJNA978591, PRJNA907383, and PRJNA994483).
REFERENCES
- Acha, S. , and Majure L. C.. 2022. A new approach using targeted sequence capture for phylogenomic studies across Cactaceae. Genes 13: 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberts, B. , Johnson A., Lewis J., Raff M., Roberts K., and Walte P.. 2002. Molecular biology of the cell, 4th ed. Garland Science, New York, New York, USA. [Google Scholar]
- Arnold, M. L. 1997. Natural hybridization and evolution. Oxford University Press, New York, New York, USA. [Google Scholar]
- Badouin, H. , Gouzy J., Grassa C. J., Murat F., Staton S. E., Cottret L., Lelandais‐Brière C., et al. 2017. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 546: 148–152. [DOI] [PubMed] [Google Scholar]
- Bain, J. F. 1988. Taxonomy of Senecio streptanthifolius Greene. Rhodora 90: 277–312. [Google Scholar]
- Bain, J. F. , and Jansen R. K.. 1995. A phylogenetic analysis of the aureoid Senecio (Asteraceae) complex based on ITS sequence data. Plant Systematics and Evolution 195: 209–219. [Google Scholar]
- Bain, J. F. , Tyson B. S., and Bray D. F.. 1997. Variation in pollen wall ultrastructure in New World Senecioneae (Asteraceae), with special reference to Packera . Canadian Journal of Botany 75: 730–735. [Google Scholar]
- Bain, J. F. , and Golden J. L.. 2000. A phylogeny of Packera (Senecioneae; Asteraceae) based on internal transcribed spacer region sequence data and a broad sampling of outgroups. Molecular Phylogenetics and Evolution 16: 331–338. [DOI] [PubMed] [Google Scholar]
- Bankevich, A. , Nurk S., Antipov D., Gurevich A. A., Dvorkin M., Kulikov A. S., Lesin V. M., et al. 2012. SPAdes: A new genome assembly algorithm and its applications to single‐cell sequencing. Journal of Computational Biology 19: 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkley, T. M. 1962. A revision of Senecio aureus . Transactions of the Kansas Academy of Science 65: 318–364. [Google Scholar]
- Barkley, T. M. 1985. Infrageneric groups in Senecio, S.L., and Cacalia, S.L. (Asteraceae: Senecioneae) in Mexico and Central America. Brittonia 37: 211–218. [Google Scholar]
- Barkley, T. M. 1988. Variation among the Aureoid Senecios of North America: A geohistorical interpretation. Botanical Review 54: 82–106. [Google Scholar]
- Bellinger, M. R. , Datlof E. M., Selph K. E., Gallaher T. J., and Knope M.L.. 2022. A genome for Bidens hawaiensis: A member of a hexaploid Hawaiian plant adaptive radiation. Journal of Heredity 113: 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A. M. , Lohse M., and Usadel B.. 2014. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremer, K. 1994. Tribe Senecioneae. In Asteraceae: Cladistics and classification, 479–520. Timber Press, Portland, Oregon, USA. [Google Scholar]
- Brown, J. W. , Walker J. F., and Smith S. A.. 2017. Phyx: Phylogenetic tools for unix. Bioinformatics 33: 1886–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, M. D. , Ganesamoorthy D., Zhou C., and Coin L. J. M.. 2018. Simulating the dynamics of targeted capture sequencing with CapSim. Bioinformatics 34: 873–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamala, S. , García N., Godden G. T., Krishnakumar V., Jordon‐Thaden I. E., De Smet R., Barbazuk W. B., et al. 2015. MarkerMiner 1.0: A new application for phylogenetic marker development using angiosperm transcriptomes. Applications in Plant Sciences 3: 1400115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman, G. C. , and Jones S. B.. 1971. Hybridization between Senecio smallii and S. tomentosus (Compositae) on the granitic flatrocks of the Southeastern United States. Brittonia 23: 209–216. [Google Scholar]
- Chapman, M. A. 2015. Transcriptome sequencing and marker development for four underutilized legumes. Applications in Plant Sciences 3: 1400111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek, P. , Bonfield J. K., Liddle J., Marshall J., Ohan V., Pollard M. O., Whitwham A., et al. 2021. Twelve years of SAMtools and BCFtools. Gigascience 10: giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lima Ferreira, P. , Batista R., Andermann T., Groppo M., Bacon C. D., and Antonelli A.. 2022. Target sequence capture of Barnadesioideae (Compositae) demonstrates the utility of low coverage loci in phylogenomic analyses. Molecular Phylogenetics and Evolution 169: 107432. [DOI] [PubMed] [Google Scholar]
- De Smet, R. , Adams K. L., Vandepoele K., Van Montagu M. C. E., Maere S., and Van De Peer Y.. 2013. Convergent gene loss following gene and genome duplications creates single‐copy families in flowering plants. Proceedings of the National Academy of Sciences, USA 110: 2898–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncavage, E. J. , Coleman J. F., de Baca M. E., Kadri S., Leon A., Routbort M., Roy S., et al. 2023. Recommendations for the use of in silico approaches for next‐generation sequencing bioinformatic pipeline validation: A joint report of the Association for Molecular Pathology, Association for Pathology Informatics, and College of American Pathologists. Journal of Molecular Diagnostics 25: 3–16. [DOI] [PubMed] [Google Scholar]
- Emms, D. M. , and Kelly S.. 2015. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biology 16: 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms, D. M. , and Kelly S.. 2019. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biology 20: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eserman, L. A. , Thomas S. K., Coffey E. E. D., and Leebens‐Mack J. H.. 2021. Target sequence capture in orchids: Developing a kit to sequence hundreds of single‐copy loci. Applications in Plant Sciences 9: 11416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernald, M. L. 1943. Virginia botanizing under restrictions. Rhodora 45: 485–511. [Google Scholar]
- Fernández, P. , Hidalgo O., Juan A., Leitch I. J., Leitch A. R., Palazzesi L., Pegoraro L., et al. 2022. Genome insights into autopolyploid evolution: A case study in Senecio doronicum (Asteraceae) from the Southern Alps. Plants 11: 1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folk, R. A. , Mandel J. R., and Freudenstein J. V.. 2015. A protocol for targeted enrichment of intron‐containing sequence markers for recent radiations: A phylogenomic example from Heuchera (Saxifragaceae). Applications in Plant Sciences 3: 1500039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca, L. H. M. , and Lohmann L. G.. 2020. Exploring the potential of nuclear and mitochondrial sequencing data generated through genome‐skimming for plant phylogenetics: A case study from a clade of neotropical lianas. Journal of Systematics and Evolution 58: 18–32. [Google Scholar]
- Freeman, C. C. 1985. A revision of the aureiod species of Senecio (Asteraceae: Senecioneae) in Mexico, with a cytogeographic and phylogenetic interpretation of the aureoid complex. Ph.D. dissertation, Kansas State University, Manhattan, Kansas, USA.
- Gramling, A. 2006. A conservation assessment of Packera millefolium, a Southern Appalachian endemic. M.S. thesis, University of North Carolina, Chapel Hill, North Carolina, USA.
- Heath, T. A. , Hedtke S. M., and Hillis D. M.. 2008. Taxon sampling and the accuracy of phylogenetic analyses. Journal of Systematics and Evolution 48: 239–257. [Google Scholar]
- Huang, C. H. , Zhang C., Liu M., Hu Y., Gao T., Qi J., and Ma H.. 2016. Multiple polyploidization events across Asteraceae with two nested events in the early history revealed by nuclear phylogenomics. Molecular Biology and Evolution 33: 2820–2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, C. , McLay T., and Schmidt‐Lebuhn A. N.. 2023. hybpiper‐nf and paragone‐nf: Containerization and additional options for target capture assembly and paralog resolution. Applications in Plant Sciences 11: 11532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, M. G. , Gardner E. M., Liu Y., Medina R., Goffinet B., Shaw A. J., Zerega N. J. C., and Wickett N. J.. 2016. HybPiper: Extracting coding sequence and introns for phylogenetics from high‐throughput sequencing reads using target enrichment. Applications in Plant Sciences 4: 1600016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, M. G. , Pokorny L., Dodsworth S., Botigué L. R., Cowan R. S., Devault A., Eiserhardt W. L., et al. 2019. A universal probe set for targeted sequencing of 353 nuclear genes from any flowering plant designed using k‐medoids clustering. Systematic Biology 68: 594–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. , and Standley D. M.. 2013. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Molecular Biology and Evolution 30: 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kück, P. , and Longo G. C.. 2014. FASconCAT‐G: Extensive functions for multiple sequence alignment preparations concerning phylogenetic studies. Frontiers in Zoology 11: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S. , Stecher G., Li M., Knyaz C., and Tamura K.. 2018. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution 35: 1547–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. , and Salzberg S. L.. 2012. Fast gapped‐read alignment with Bowtie 2. Nature Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. , Wilks C., Antonescu V., and Charles R.. 2019. Scaling read aligners to hundreds of threads on general‐purpose processors. Bioinformatics 35: 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , and Durbin R.. 2009. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichter‐Marck, I. H. , Freyman W. A., Siniscalchi C. M., Mandel J. R., Castro‐Castro A., Johnson G., and Baldwin B. G.. 2020. Phylogenomics of Perityleae (Compositae) provides new insights into morphological and chromosomal evolution of the rock daisies. Journal of Systematics and Evolution 58: 853–880. [Google Scholar]
- Lim, K. Y. , Soltis D. E., Soltis P. S., Tate J., Matyasek R., Srubarova H., Kovarik A., et al. 2008. Rapid chromosome evolution in recently formed polyploids in Tragopogon (Asteraceae). PLoS ONE 3: e3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , Johnson M. G., Cox C. J., Medina R., Devos N., Vanderpoorten A., Hedenäs L., et al. 2019. Resolution of the ordinal phylogeny of mosses using targeted exons from organellar and nuclear genomes. Nature Communications 10: 1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M. , and Conery J. S.. 2000. The evolutionary fate and consequences of duplicate genes. Science 290: 1151–1155. [DOI] [PubMed] [Google Scholar]
- Maddison, W. P. 1997. Gene trees in species trees. Systematic Biology 46: 523–536. [Google Scholar]
- Mandel, J. R. , Dikow R. B., Funk V. A., Masalia R. R., Staton S. E., Kozik A., Michelmore R. W., et al. 2014. A target enrichment method for gathering phylogenetic information from hundreds of loci: An example from the Compositae. Applications in Plant Sciences 2: 1300085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel, J. R. , Barker M. S., Bayer R. J., Dikow R. B., Gao T. G., Jones K. E., Keeley S., et al. 2017. The Compositae Tree of Life in the age of phylogenomics. Journal of Systematics and Evolution 55: 405–410. [Google Scholar]
- Mandel, J. R. , Dikow R. B., Siniscalchi C. M., Thapa R., Watson L. E., and Funk V.A.. 2019. A fully resolved backbone phylogeny reveals numerous dispersals and explosive diversifications throughout the history of Asteraceae. Proceedings of the National Academy of Sciences, USA 116: 14083–14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May, V. , Koch L., Fischer‐Zirnsak B., Horn D., Gehle P., Kornak U., Beule D., and Holtgrewe M.. 2022. ClearCNV: CNV calling from NGS panel data in the presence of ambiguity and noise. Bioinformatics 38: 3871–3876. [DOI] [PubMed] [Google Scholar]
- McKain, M. R. , Johnson M. G., Uribe‐Convers S., Eaton D., and Yang Y.. 2018. Practical considerations for plant phylogenomics. Applications in Plant Sciences 6: 1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore‐Pollard, E. R. , and Mandel J. R.. 2023a. Resolving evolutionary relationships in the groundsels: Phylogenomics, divergence time estimates, and biogeography of Packera (Asteraceae: Senecioneae). bioRxiv [Preprint]. Available at 10.1101/2023.07.18.549592 [posted 19 July 2023; accessed 3 January 2024]. [DOI] [Google Scholar]
- Moore‐Pollard, E. R. , and Mandel J. R.. 2023b. From paralogy to hybridization: Investigating causes of underlying phylogenomic discordance using the complex genus Packera (Senecioneae; Asteraceae). bioRxiv [Preprint]. Available at 10.1101/2023.08.14.553290 [posted 15 August 2023; accessed 3 January 2024]. [DOI] [Google Scholar]
- Morales‐Briones, D. F. , Liston A., and Tank D. C.. 2018. Phylogenomic analyses reveal a deep history of hybridization and polyploidy in the Neotropical genus Lachemilla (Rosaceae). New Phytologist 218: 1668–1684. [DOI] [PubMed] [Google Scholar]
- Morales‐Briones, D. F. , Kadereit G., Tefarikis D. T., Moore M. J., Smith S. A., Brockington S. F., Timoneda A., et al. 2021. Disentangling sources of gene tree discordance in phylogenomic data sets: Testing ancient hybridizations in Amaranthaceae s.l. Systematic Biology 70: 219–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, R. , Bleckmann A., and Simon R.. 2008. The receptor kinase CORYNE of Arabidopsis transmits the stem cell‐limiting signal CLAVATA3 independently of CLAVATA1 . The Plant Cell 20: 934–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauheimer, L. , Weigner N., Joyce E., Crayn D., Clarke C., and Nargar K.. 2021. HybPhaser: A workflow for the detection and phasing of hybrids in target capture data sets. Applications in Plant Sciences 9: 11441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussbaum, S. , McInnes R. R., and Willard H. F.. 2007. Genetics in medicine. Elsevier, Philadelphia, Pennsylvania, USA. [Google Scholar]
- One Thousand Plant Transcriptomes Initiative. 2019. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574: 679–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pease, J. B. , Brown J. W., Walker J. F., Hinchliff C. E., and Smith S. A.. 2018. Quartet sampling distinguishes lack of support from conflicting support in the green plant tree of life. American Journal of Botany 105: 385–403. [DOI] [PubMed] [Google Scholar]
- Pelser, P. B. , Nordenstam B., Kadereit J. W., and Watson L. E.. 2007. An ITS phylogeny of tribe Senecioneae (Asteraceae) and a new delimitation of Senecio L. Taxon 56: 1077–1104. [Google Scholar]
- Core Team R. 2016. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. Website: http://www.R-project.org/ [accessed 3 January 2024].
- Reichelt, N. , Wen J., Pätzold C., and Appelhans M. S.. 2021. Target enrichment improves phylogenetic resolution in the genus Zanthoxylum (Rutaceae) and indicates both incomplete lineage sorting and hybridization events. Annals of Botany 128: 497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revell, L. J. 2012. phytools: An R package for phylogenetic comparative biology (and other things). Methods in Ecology and Evolution 3: 217–223. [Google Scholar]
- Rothfels, C. J. 2021. Polyploid phylogenetics. New Phytologist 230: 66–72. [DOI] [PubMed] [Google Scholar]
- RStudio Team . 2020. RStudio: Integrated development for R. RStudio, PBC, Boston, Massachusetts, USA.
- Sanderson, M. J. , McMahon M. M., and Steel M.. 2010. Phylogenomics with incomplete taxon coverage: The limits to inference. BMC Evolutionary Biology 10: 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayyari, E. , and Mirarab S.. 2016. Fast coalescent‐based computation of local branch support from quartet frequencies. Molecular Biology and Evolution 33: 1654–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling, E. E. , and Floden A.. 2015. Barcoding the Asteraceae of Tennessee, tribe Cichorieae. Phytoneuron 19: 1–8. [Google Scholar]
- Schliep, K. P. 2011. phangorn: Phylogenetic analysis in R. Bioinformatics 27: 592–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa, E. B. , Zimmer E. A., and Givnish T. J.. 2012. Reticulate evolution on a global scale: A nuclear phylogeny for New World Dryopteris (Dryopteridaceae). Molecular Phylogenetics and Evolution 64: 563–581. [DOI] [PubMed] [Google Scholar]
- Shah, T. , Schneider J. V., Zizka G., Maurin O., Baker W., Forest F., Brewer G. E., et al. 2021. Joining forces in Ochnaceae phylogenomics: A tale of two targeted sequencing probe kits. American Journal of Botany 108: 1201–1216. [DOI] [PubMed] [Google Scholar]
- Siniscalchi, C. M. , Loeuille B., Funk V. A., Mandel J. R., and Pirani J. R.. 2019. Phylogenomics yields new insight into relationships within Vernonieae (Asteraceae). Frontiers in Plant Science 10: 01224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siniscalchi, C. M. , Hidalgo O., Palazzesi L., Pellicer J., Pokorny L., Maurin O., Leitch I. J., et al. 2021. Lineage‐specific vs. universal: A comparison of the Compositae1061 and Angiosperms353 enrichment panels in the sunflower family. Applications in Plant Sciences 9: 11422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siniscalchi, C. M. , Ackerfield J., and Folk R. A.. 2023. Diversification and biogeography of North American thistles (Cirsium: Carduoideae: Compositae): Drivers of a rapid continent‐wide radiation. International Journal of Plant Sciences 184: 322–341. [Google Scholar]
- Smith, M. L. , and Hahn M. W.. 2021. New approaches for inferring phylogenies in the presence of paralogs. Trends in Genetics 37: 174–187. [DOI] [PubMed] [Google Scholar]
- Smith, S. A. , Moore M. J., Brown J. W., and Yang Y.. 2015. Analysis of phylogenomic datasets reveals conflict, concordance, and gene duplications with examples from animals and plants. BMC Evolutionary Biology 15: 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis, A. 2014. RAxML version 8: A tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics 30: 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub, S. C. K. , Parks M., Weitemier K., Fishbein M., Cronn R. C., and Liston A.. 2012. Navigating the tip of the genomic iceberg: Next‐generation sequencing for plant systematics. American Journal of Botany 99: 349–364. [DOI] [PubMed] [Google Scholar]
- Stull, G. W. , Moore M. J., Mandala V. S., Douglas N. A., Kates H., Qi X., Brockington S. F., et al. 2013. A targeted enrichment strategy for massively parallel sequencing of angiosperm plastid genomes. Applications in Plant Sciences 1: 1200497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thapa, R. , Bayer R. J., and Mandel J. R.. 2020. Phylogenomics resolves the relationships within Antennaria (Asteraceae, Gnaphalieae) and yields new insights into its morphological character evolution and biogeography. Systematic Botany 45: 387–402. [Google Scholar]
- Thiers, B. 2024. Index Herbariorum. Website: http://sweetgum.nybg.org/science/ih/ [accessed 4 January 2024].
- Trock, D. K. 1999. A revisionary synthesis of the genus Packera (Asteraceae: Senecioneae). Ph.D. dissertation, Kansas State University, Manhattan, Kansas, USA.
- Uttal, L. J. 1984. Senecio millefolium T. & G. (Asteraceae) and its introgressants. SIDA Contributions to Botany 10: 216–222. [Google Scholar]
- Vargas, O. M. , Ortiz E. M., and Simpson B. B.. 2017. Conflicting phylogenomic signals reveal a pattern of reticulate evolution in a recent high‐Andean diversification (Asteraceae: Astereae: Diplostephium). New Phytologist 214: 1736–1750. [DOI] [PubMed] [Google Scholar]
- Vatanparast, M. , Powell A., Doyle J. J., and Egan A. N.. 2018. Targeting legume loci: A comparison of three methods for target enrichment bait design in Leguminosae phylogenomics. Applications in Plant Sciences 6: 1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veitia, R. A. 2005. Paralogs in polyploids: One for all and all for one? Plant Cell 17: 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villaverde, T. , Pokorny L., Olsson S., Rincón‐Barrado M., Johnson M. G., Gardner E. M., Wickett N. J., et al. 2018. Bridging the micro‐ and macroevolutionary levels in phylogenomics: Hyb‐Seq solves relationships from populations to species and above. New Phytologist 220: 636–650. [DOI] [PubMed] [Google Scholar]
- Weakley, A. S. , LeBlond R. J., Sorrie B. A., Witsell C. T., Estes L. D., Gandhi K., Mathews K. G., and Ebihara A.. 2011. New combinations, rank changes, and nomenclatural and taxonomic comments in the vascular flora of the Southeastern United States. Journal of the Botanical Research Institute of Texas 5: 437–455. [Google Scholar]
- Weigel, D. , Alvarez J., Smyth D. R., Yanofsky M. F., and Meyerowitz E. M.. 1992. LEAFY controls floral meristem identity in Arabidopsis. Cell 69: 843–859. [DOI] [PubMed] [Google Scholar]
- Weitemier, K. , Straub S. C. K., Cronn R. C., Fishbein M., Schmickl R., McDonnell A., and Liston A.. 2014. Hyb‐Seq: Combining target enrichment and genome skimming for plant phylogenomics. Applications in Plant Sciences 2: 1400042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham, H. 2016. ggplot2: Elegant graphics for data analysis. Springer‐Verlag, New York, New York, USA. [Google Scholar]
- Wolf, P. G. , Robison T. A., Johnson M. G., Sundue M. A., Testo W. L., and Rothfels C. J.. 2018. Target sequence capture of nuclear‐encoded genes for phylogenetic analysis in ferns. Applications in Plant Sciences 6: 01148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe, K. H. 2001. Yesterday's polyploids and the mystery of diploidization. Nature Reviews Genetics 2: 333–341. [DOI] [PubMed] [Google Scholar]
- Yang, Y. , and Smith S. A.. 2014. Orthology inference in nonmodel organisms using transcriptomes and low‐coverage genomes: Improving accuracy and matrix occupancy for phylogenomics. Molecular Biology and Evolution 31: 3081–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, C. , Rabiee M., Sayyari E., and Mirarab S.. 2018. ASTRAL‐III: Polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics 19: 15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, C. , Huang C.‐H., Liu M., Hu Y., Panero J. L., Luebert F., Gao T., and Ma H.. 2021. Phylotranscriptomic insight into Asteraceae diversity, polyploidy, and morphological innovation. Journal of Integrative Plant Biology 63: 1273–1293. [DOI] [PubMed] [Google Scholar]
- Zhang, C. , and Mirarab S.. 2022. ASTRAL‐Pro 2: Ultrafast species tree reconstruction from multi‐copy gene family trees. Bioinformatics 38: 4949–4950. [DOI] [PubMed] [Google Scholar]
- Zhou, W. , Soghigian J., and Xiang Q. Y.. 2021. A new pipeline for removing paralogs in target enrichment data. Systematic Biology 71: 410–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Voucher specimens used to develop the Compositae‐ParaLoss‐1272 probe set using MarkerMiner. Species names and authorities are according to the International Plant Names Index (IPNI).
Appendix S2. List of 1925 targeted loci in the Compositae‐ParaLoss‐1272 probe set and information about their associated functions in Arabidopsis thaliana (source: The Arabidopsis Information Resource [TAIR]; https://www.arabidopsis.org/tools/bulk/genes/index.jsp). Vitis vinifera–specific genes that have no known function (n = 17) are included.
Appendix S3. HybPiper summary statistics for the six Asteraceae genomes from the CapSim run.
Appendix S4. General and full HybPiper statistics of the Illumina sequence run.
Appendix S5. Tanglegrams comparing the relationships between the combined data set, Compositae‐1061 + Compositae‐ParaLoss‐1272, against the individual data sets: Compositae‐1061 (A) and Compositae‐ParaLoss‐1272 (B). Lines between the taxa at the tips compare relationships: solid lines indicate the same relationship; dashed lines indicate differing relationships. Local posterior probability (LPP) values are represented at each node, with full support (1.0 LPP) in blue, moderate support (0.9–0.99 LPP) in green, and low support (≤0.89 LPP) in red.
Appendix S6. Tanglegrams comparing the relationships between a pruned‐down version of the Moore‐Pollard and Mandel (2023a) tree now containing the 19 taxa used in this study, compared to the Compositae‐1061 (A) and Compositae‐ParaLoss‐1272 (B) trees generated in this study. Lines between the taxa at the tips compare relationships: solid lines indicate the same relationship; dashed lines indicate differing relationships.
Appendix S7. Compositae‐ParaLoss‐1272 gene set file for bioinformatic analyses.
Data Availability Statement
Raw sequence data are available in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (BioProjects PRJNA978591, PRJNA907383, and PRJNA994483).