

Abstract
The widespread occurrence of sulfonamides raises significant concerns about the evolution and spread of antibiotic resistance genes. Biodegradation represents not only a resistance mechanism but also a clean-up strategy. Meanwhile, dynamic and diverse environments could influence the cellular function of individual sulfonamide-degrading strains. Here, we present Paenarthrobacter from different origins that demonstrated diverse growth patterns and sulfonamide-degrading abilities. Generally, the degradation performance was largely associated with the number of sadA gene copies and also relied on its genotype. Based on the survey of sad genes in the public database, an independent mobilization of transposon-borne genes between chromosome and plasmid was observed. Insertions of multiple sadA genes could greatly enhance sulfonamide-degrading performance. Moreover, the sad gene cluster and sadA transposable element showed phylogenetic conservation currently, being identified only in two genera of Paenarthrobacter (Micrococcaceae) and Microbacterium (Microbacteriaceae). Meanwhile, Paenarthrobacter exhibited a high capacity for genome editing to adapt to the specific environmental niche, opening up new opportunities for bioremediation applications.
Keywords: sulfonamide, biodegradation, Paenarthrobacter, sad genes, transposable element
Graphical Abstract
Graphical Abstract.
Introduction
Sulfonamides are among the most widely used antibiotics for both human and veterinary applications chiefly because of their broad inhibition ability for bacteria [1, 2]. However, there are growing concerns regarding their widespread distribution in various natural environments. In the past decades, sulfonamides have been frequently detected in wastewater [3, 4], surface water [5, 6], groundwater [6, 7], and drinking water [8, 9] around the world in concentrations from nanograms to micrograms per liter. In particular, sulfamethoxazole, sulfadiazine, and sulfamethazine have been observed with the highest reported human consumption and detection rates in aquatic environments [10-12]. The residual sulfonamide can accelerate the evolution and spread of antibiotic resistance genes [13], posing a great threat to ecosystems and global health.
Although the evolution of bacteria to gain antibiotic resistance has long been appreciated, our knowledge of the involved mechanisms has increased significantly in recent years. Well-documented antibiotic resistance mechanisms include prevention of access to the target (e.g. efflux pumps), changes in structure and modification of antibiotic targets (e.g. mutation), and direct modification of antibiotics (e.g. hydrolysis) [14]. In addition, antibiotic subsistence in bacteria is an alternative resistance mechanism, while importantly, it can inactivate antibiotics and reduce environmental concentrations, representing a cure for environmental resistance. In the past two decades, a wide variety of sulfonamide-catabolizing bacterial strains have been isolated, spanning diverse bacterial genera [15, 16]. Nevertheless, the sad gene cluster is the sole experimentally validated sulfonamide-degrading gene cluster so far [17, 18]. In Microbacterium spp. BR1 and CJ77, e.g. sulfonamides were initially attacked by the flavin-dependent monooxygenase encoded by the sadA gene (the homologous gene sulX in CJ77) with the generation of 4-aminophenol, the corresponding dead-end metabolites, and sulfite (Fig. S1). Subsequently, another flavin-dependent monooxygenase encoded by the sadB gene was responsible for the conversion of 4-aminophenol into 1,2,4-trihydroxybenzene. In this biotransformation pathway, flavin reductase encoded by the sadC (sulR) gene plays an auxiliary role in electron transport [17, 18]. The complete sulfonamide mineralization was then achieved by the interspecific interactions among sulfonamide degraders and other species, such as Pimelobacter [19] and Acidovorax [20]. Unlike the phylogenetical diversity of sulfonamide-catabolizing strains, the sad genes were only reported in a few sulfonamide degraders affiliated with the families of Microbacteriaceae and Micrococcaceae. The underlying propagation pattern of sulfonamide-degrading genes associated with the limited spread beyond the boundary of Microbacteriaceae and Micrococcaceae lineages remains to be elucidated.
Likewise, environmental niches not only shape the structure of microbial communities but also strain-level diversities, resulting in distinct functional performance and influencing intraspecific or interspecific interactions [21]. However, strain-level variation is frequently overlooked in surveys of community structure due to the limitations inherent in marker gene-based analysis. A finer-grained assessment of genetic diversity is heavily dependent on technological revolution. Recent advances in sequencing technologies have allowed microbiologists to determine the functions of individual strains according to the circular genome instead of just genetic fragments [22, 23]. Despite the fact that the strain-level differentiation of pollutant-degrading capacities among a single genus is of vital importance and significance in practical applications, relevant studies are very limited.
Our study focused on Paenarthrobacter, a bacterial genus first described in 2016 and previously known as Arthrobacter [24], which is the genus most of the reported sulfonamide degraders were affiliated with. We hypothesized that Paenarthrobacter would exhibit functional diversity despite the high genetic similarity of individual strains. In this study, we presented the genomic characteristics and growth kinetics of eight Paenarthrobacter strains isolated from sulfonamide-degrading enrichments seeded with activated sludge from different sewage treatment plants. Then, we conducted a phylogenetic analysis of the sad genes and degradation experiments to reveal the sulfonamide-degrading capability of four selected strains with different sad arrangements. Furthermore, a survey of sad genes in the public database was performed to investigate potential degraders and propagation patterns of sulfonamide-degrading genes.
Materials and methods
Chemicals and pure strains
Sulfadiazine (SDZ), sulfamethoxazole (SMX), sulfamethazine (SMZ), and formic acid were purchased from Sigma-Aldrich (USA). Liquid chromatography/mass spectrometry–grade acetonitrile was purchased from Fisher Chemicals (Pittsburgh, PA). Ultrapure water was produced by a Barnstead EASYpure UV/UF water purification system. As described in our previous studies [25, 26], the Paenarthrobacter spp. were isolated from eight SDZ-degrading enrichments seeded with different activated sludge from six local sewage treatment works.
Growth kinetics
Paenarthrobacter spp. used in this study were kept frozen at −80°C in Luria-Bertani (LB) medium with 20% (v/v) glycerol. Before the experiment, a 50-μl frozen suspension was inoculated into 30 ml of liquid LB medium and incubated at room temperature for 24 hours. The Paenarthrobacter sp. was purified by streaking on LB agar plates (1.5% agar). Then, a single colony was used to inoculate 30 ml of LB medium and incubate for ~48 hours (log phase based on observation). In the next step, the dense culture was inoculated in a ratio of 1:100 into a fresh 30 ml of LB medium in triplicate. Cell density was measured by optical density at 595 nm (OD595) with an iMark Microplate Absorbance Reader (Bio-Rad, Hercules, CA, USA). The logistic model was used to describe the growth pattern. The growth kinetics were determined in LB medium without sulfonamide (pre-antibiotic). In addition, the post-antibiotic replication rates of the isolates under sulfonamide pressure (100 mg/L SDZ) were estimated using the growth rate index (GRiD, v1.3) [27] in single mode.
Sulfonamide degradation experiment
Four Paenarthrobacter spp. were selected for degradation experiments based on their different sad gene arrangements. The Paenarthrobacter spp. were grown in fresh LB medium at room temperature with shaking at 180 rpm to exponential phase (OD595 of 1.0–1.5). The biomass was harvested by centrifugation at 3725 g for 20 min (Beckman Coulter Avant J-15R) and washed with mineral salt medium (MSM, Table S1) trice. The biodegradation experiment was carried out in triplicate in 250-ml Erlenmeyer flasks containing 150 ml of MSM amended with 100 mg/L sulfonamide as the sole carbon source. Inoculum of Paenarthrobacter sp. was added in a set of three flasks to a target initial OD595 of 0.1. The control treatment was inoculated with mixed sterilized cultures to monitor the abiotic loss of sulfonamides. All the flasks were sealed with sterile breathable sealing film and incubated at room temperature and 180 rpm. A 2-ml suspension of Paenarthrobacter sp. was withdrawn at designed intervals from three independent flasks and centrifuged at 20000 g for 2 min in a 4°C pre-chilled centrifuge. The supernatant was filtered with 0.22-μm polyvinylidene fluoride syringe filters (Millipore, Germany) and stored at 4°C until analysis. The modified Gompertz model was applied to fit the degradation data.
Sequencing and assembly of Paenarthrobacter spp.
To obtain the circular chromosome of our Paenarthrobacter spp., additional Nanopore sequencing for each isolate was performed. Briefly, the DNA of each isolate was extracted using a DNeasy PowerSoil Kit (Qiagen, Germany) following the manufacturer’s instructions. DNA purification was performed with a standard AMPure XP bead (Beckman Coulter) clean-up purification protocol. An equal amount of DNA from each isolate was used for library preparation with a ligation sequencing kit (SQK-LSK109) and purified following the standard AMPure XP bead clean-up protocol. Subsequently, the prepared library was loaded onto an R9 flow cell (FLO-MIN106) for sequencing on a GridION using MinKNOW (v21.10.8). Base calling was processed using Guppy (v5.0.17, Oxford Nanopore Technologies), and raw long reads were processed with Porechop (v0.2.4) (https://github.com/rrwick/Porechop) to remove adapter barcode sequences. The total additional sequencing amount is 14.09 Gb. Together with our previously sequenced Illumina short reads and Nanopore long reads (PRJNA669352), the genome of eight Paenarthrobacter spp. was hybrid assembled using Unicycler (v0.5.0) [28]. Whole-genome pairwise average nucleotide identity (ANI) and average amino acid identity (AAI) were calculated using fastANI (v1.32) [29] and CompareM (v0.0.23) AAI workflow (https://github.com/dparks1134/CompareM), respectively. The plasmids were determined by PlasFlow (v1.1) [30] with a 0.8 threshold. Phylogenetic tree construction and taxonomic reassignment were performed by GTDB-tk (v2.1.1) [31] based on Genome Taxonomy Database taxonomy R214. The phylogenetic tree was midpoint rooted and visualized in iTOL (v6) [32]. Subsequently, the open reading frames (ORFs) were predicted by Prodigal (v2.6.3) [33] and annotated by eggNOG-mapper (v2.1.9) [34] against the eggNOG v5.0 database.
sad gene identification and database mining
The GenBank flat files (.gbff) of 433 218 bacteria were downloaded from the NCBI database (updated in July 2023). Then, the nucleotide sequences were extracted by an in-house Python script, and the ORFs were predicted by Prodigal (v2.6.3) [33]. The sad genes were identified using DIAMOND (v2.0.8.147) [35] with strict criteria (>70% identity, >70% query length coverage, and <1e-5 e-value). Gene arrangement of sulfonamide-degrading gene clusters was visualized using the R package gggenes (v0.5.0) (https://github.com/wilkox/gggenes).
Analytical methods
The concentrations of sulfonamides were determined by ultraperformance liquid chromatography–tandem mass spectrometry (Acquity UPLC system, Waters) with positive ion mode ESI. In this method, the extract was gradient eluted from a BEH C18 column (2.1 × 50 mm, 1.7 μm, held at 50°C) using water (A) and acetonitrile (B), containing 0.1% formic acid. The gradient was initially 95% A for 2 min, linearly increased to 78% A at 6.5 min, and returned to 95% A at 6.6 min, with a flow rate of 0.4 ml/min. The sample injection volume was 10 μl. The desolvation and source temperatures were 400 and 120°C, respectively. The desolvation and cone gas (nitrogen) flow rates were 600 and 50 L/h, respectively. Argon was used as the collision gas at a flow rate of 0.15 ml/min. Instrument control and data acquisition were processed with MassLynx software (v4.2, Waters). Multiple reaction monitoring mode was used to monitor the m/z transition of sulfonamides and their major metabolites (Table S2).
Results and discussion
Taxonomy reassignment of eight isolates
Eight strains used in the present study were isolated from sulfonamide-degrading consortia fed with sulfadiazine as the sole carbon source (100 mg/L). They were formerly identified as Arthrobacter spp. In this study, based on additional sequencing data, four complete genomes (D2, D4, SK, and SL) and four draft genomes (SWH, ST, YL, and TP) were achieved with a genome size of 4.78 ± 0.09 Mb and a high average GC content of 63.4%. Each genome comprises a circular chromosome (4.58 ± 0.04 Mb) and multiple plasmids (Table S3). Consistent with the average number of 16S rRNA copies in the genus Paenarthrobacter [36], the eight isolates under study contain six copies of the 16S rRNA gene. In a genome-based phylogeny (Fig. S2), all eight isolates were placed together with Paenarthrobacter ureafaciens DSM 20126 as a monophyletic sister group within the clade Paenarthrobacter, a novel genus proposed in 2016 [24], implying that these strains were properly affiliated with Paenarthrobacter. Meanwhile, the isolates were similar to each other, with extremely high ANI and AAI values above 99.9% (Table S4), stimulating interest in investigating the underlying genetic impact on sulfonamide biodegradation at the strain level.
Phylogeny of sad genes
To date, sad gene diversity as well as different arrangements in identified sulfonamide degraders have been reported in a few studies [18, 37]. Here, the recovered sad genes together with reference sequences from the NCBI GenBank and RefSeq databases were used to construct the phylogenetic trees. As expected, sadA genes were only identified in Microbacteriaceae and Micrococcaceae. All the sadA genes retrieved from our isolates fell into Micrococcaceae sadA, being grouped with sadA sequences from other Paenarthrobacter spp. (Fig. 1A). These sadA genes could be further assigned into three clusters based on the topological structure. Specifically, sadA Type II shared 97.5% identity with Type III, whereas sadA Type I was homologous to Types II and III with 78.9% and 79.2% amino acid identities, respectively. Among the eight isolates, three different arrangements of sadA were found, namely, (i) Type I sadA and Type II sadA (Paenarthrobacter sp. D2), (ii) Type I sadA and Type III sadA (Paenarthrobacter spp. ST, SK, SL, SWH, YL, TP), and (iii) Type II sadA only (Paenarthrobacter sp. D4).
Figure 1.

Maximum likelihood trees of sadA (A), sadB (B), and sadC (C) based on amino acid sequences. The strains under study are highlighted.
In comparison, the diversity of sadB and sadC retrieved from our isolates was relatively lower. Analogous to sadA genes, sadB genes could be divided into Micrococcaceae sadB and Microbacteriaceae sadB according to their affiliated families. On the contrary, Micrococcaceae Type I sadB, adjacent to Type I sadA, displayed a high similarity with some Microbacteriaceae sadB genes, indicating that they were orthologous genes and originated from a common ancestor. Furthermore, Micrococcaceae Type II sadB was a plasmid-borne gene, only identified in Paenarthrobacter spp., sharing 73.2% AAI with Type I sadB. Nevertheless, the expression of the Type II sadB during sulfonamide biodegradation remains unclear as it is located remotely from the sad gene cluster. In addition to sadA-carrying strains, a sadB homogenous gene was also identified in a Nocardioidaceae bacterium (85.0% identity with Type I sadB), which was a metagenome-assembled genome (MAG) reconstructed from the activated sludge sample of the Sha Tin sewage treatment works [38]. The monooxygenase encoded by sadB could be involved in the conversion of 4-aminophenol, which entered the municipal wastewater by inevitable release from chemical processes or industries, or as a biotransformation intermediate of dyes and pharmaceuticals [39]. Besides, sadC is an accessory gene for sulfonamide biodegradation, which is highly conserved (>99.5% amino acid identity) and located near sadB in all experimentally verified degraders.
Growth kinetics of eight Paenarthrobacter spp.
In nutrition-rich conditions, the growth data fitted well with logistic regression (R2 > 0.89, Fig. 2). Paenarthrobacter sp. SK entered exponential growth after inoculation without an obvious lag phase and reached equilibrium after just 24 hours, implying a rapid growth rate of SK and an ability to survive and thrive in the microbial jungle. Meanwhile, the species with a fast growth rate may introduce some unique mutations, thereby enabling a rapid adaption to particular niches [40]. The other seven strains exponentially grew after an approximate 10-hour lag phase before asymptotically reaching equilibrium. Among them, SL and ST achieved lower saturation concentrations (K), but the maximum population size (carrying capacity) is independent of the initial value (P0). To reveal the growth-dependent behavioral difference between pre- and post-antibiotic, we estimated the GRiD of those strains that grew in the medium containing sulfonamide using a sequencing-based approach. Under sulfonamide pressure, SK, D2, and D4 have higher average growth rates (GRiD values) compared to the other five strains, generally agreeing with the result from cultivation in pre-antibiotic conditions. However, in a fluctuating environment, the advantages of short lag time and fast growth rate might average out [41], resulting in a coexistence of diverse sulfonamide-degrading strains in bacterial competition.
Figure 2.

Growth curves of eight Paenarthrobacter strains. The growth data was fitted with a logistic model, where P0 indicates the initial population, r defines the growth rate, and K is the maximum value that the population can reach. The growth rates of isolates under 100-mg/L SDZ pressure were estimated by GRiD (v1.3). Error bars represent the standard deviation from triplicates.
sad-dependent degradation performance of Paenarthrobacter spp. with strain-level diversity
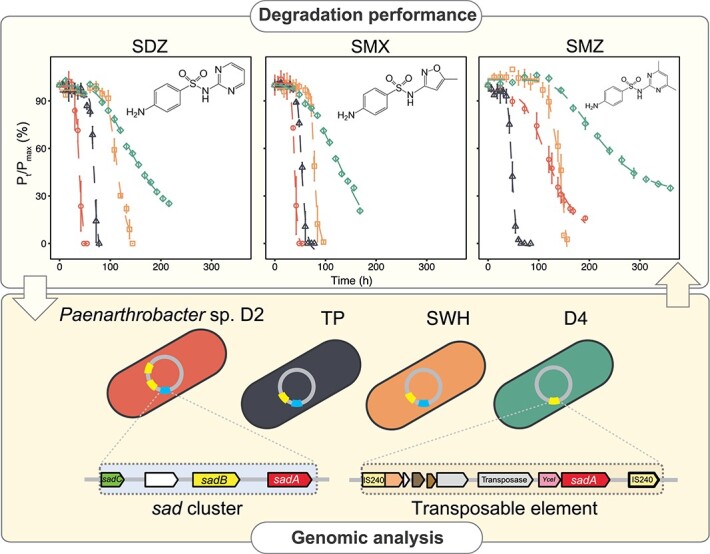
In the present study, four strains were selected for further degradation assay based on their composition of sad genes (Fig. S3B). Previous investigations have demonstrated that abiotic processes (e.g. sorption, hydrolysis, and volatility) play a minor role in sulfonamide elimination [42, 43], which is attributed to biotransformation to a large extent (Fig. 3). Herein, all four selected strains exhibited a capability of sulfonamide utilization, and the degradation data could be described well by the modified Gompertz model. Nevertheless, it is inadequate to only focus on parent compound removal, the metabolite generation and bioactivity removal are important as well. Judging from the low total organic carbon removal rate (data not shown), the degree of sulfonamide mineralization by Paenarthrobacter was low, which is consistent with the previous study reporting that a low fraction of 14CO2 could be recovered in a mass balance analysis [44]. Hence, the corresponding dead-end metabolites of sulfonamides were monitored subsequently. The accumulation of three major metabolites (2-aminopyrimidine for SDZ, 3-amino-5-methylisoxazole for SMX, and 4,6-dimethylpyrimidine-2-amine for SMZ) was observed synchronously (Fig. S4), indicating that the degradation was via the enzymatic reactions encoded by sad genes. In line with previous findings [37, 42, 45], SMX and SDZ were more easily degraded compared with SMZ (Fig. S5). The different heterocyclic moieties (five-member heterocyclic ring for SMX, different six-member heterocyclic rings for SMZ and SDZ) might be responsible for the distinct degradation behaviors [37, 45, 46]. The large-size heterocyclic structure, such as methyl groups on the six-member ring of SMZ, hinders sulfonamide binding to monooxygenase.
Figure 3.

Sulfonamide degradation curves of four selected Paenarthrobacter strains in MSM with 100 mg/L sulfonamide (SDZ, SMX, SMZ) under aerobic conditions. All experiments were conducted in triplicate at an initial OD595 of 0.1. The degradation data were described using a modified Gompertz model, where μm represents the maximum biodegradation rate, and λ indicates the lag phase time.
In detail, a similar degradation performance was observed between TP and SWH because they have an identical sad gene arrangement, namely, two sadA (Types I and III), two sadB (Types I and II), and one sadC. Nevertheless, SWH has the longest lag phase time for all sulfonamides (133.9, 71.2, and 94.5 hours for SMZ, SMX, and SDZ, respectively), probably owing to its relatively low growth rate (Fig. 2). It suggests a longer time is required for SWH to adapt its metabolism to the new environment compared with other strains. Meanwhile, a comparable long lag phase was also observed in D4, but D4 has relatively poor sulfonamide-degrading performance with incomplete parent compound removal due to the possession of only one Type II sadA transposable element and a plasmid-borne Type II sadB. However, parent compound biotransformation and the release of the major metabolite are only related to the enzymatic reaction involving sadA and sadC genes (Fig. S1). Moreover, flavin reductase encoded by sadC is not specific to the sulfonamide-degrading reaction [18], and therefore, a remotely located flavin reductase–coding gene could also participate in sulfonamide elimination as an electron transporter. In comparison, D2 possesses the most copies of sadA genes, including one Type I sadA gene (sad cluster) and two Type II sadA genes (sadA transposable element), achieving the best performance for SMX and SDZ biodegradation. D2 could utilize 100 mg/L SMX and SDZ completely with high biodegradation rates (9.8 and 6.7 mg/L-1·h-1) and short lag phases (33.2 and 30.7 hours). Also, TP could degrade sulfonamides efficiently within 72 hours and reach comparative μmax values for all three sulfonamides (i.e. 6.2, 6.9, and 6.6 mg/L-1·h-1 for SMX, SDZ, and SMZ, respectively). In addition, we found that both Type II and III sadA can contribute to SMZ elimination, whereas Type II sadA could only realize a limited SMZ consumption, even though D2 contains two Type II sadA genes. It implied that Type III sadA is the key driver of SMZ biodegradation.
Database mining of sad genes
Unlike natural antibiotics, sulfonamide antibiotics are produced via chemical synthesis, and the discovery of sulfonamide-degrading strains is relatively recent. The genes responsible for sulfonamide biodegradation are clustered, comprising two monooxygenases and one flavin reductase. The sad cluster originated from Microbacterium spp. on the chromosome (Fig. 4) [18, 47-49]. The cleavage of the S-N bond not only allowed the bacteria to utilize the benzene ring as a substrate but also destroyed the antibacterial activity, conferring the ability to survive in environments with sulfonamides. Then, the sadA gene, responsible for the first-step cleavage, was integrated into a composite transposon in the later discovered strains, enabling a potential rapid propagation of the target gene within genomes [50-52]. The sadA-carrying composite transposon on the chromosome was first reported in Paenarthrobacter spp. D2 and D4 (previously named Arthrobacter) in 2016 [25]. Subsequently, the convergence of advances in long-read sequencing makes it possible to complete the chromosome and resolve the arrangement of sad genes. Based on circular chromosomes, a similar gene arrangement, namely, one sad cluster and one sadA transposable element, was observed in six newly isolated Paenarthrobacter spp. and Paenarthrobacter ureafaciens YL1, obtained from activated sludge in 2020, which showed comparable sulfonamide degradation rates [26, 53]. The latest isolated SMX degrader, Paenarthrobacter ureafaciens SD-1 [54], revealed a sadA gene jumping from chromosome to plasmid, realizing an independent replication of the functional genes. Meanwhile, aggressive insertions of sadA were recently reported on a plasmid of Paenarthrobacter sp. R1 [37], including four sadA transposable elements and one sad gene cluster. The densely nested sadA transposable elements in Paenarthrobacter sp. R1 achieved an efficient utilization of sulfonamides, showing complete removal of 50 mg/L SDZ and SMX within 24 hours. Moreover, it indicated that the cleavage of the S-N bond is the rate-limiting step of sulfonamide biodegradation and multiple sadA genes could improve the degradation and inactivation performance. Those observations provided valuable insights into the construction of engineered strains as well as an early warning for the rapid evolution and potentially widespread. Currently, in strains with a complete genome, sad cluster and sadA transposable element were only identified on nonmobilizable plasmids (CP127116.1 in R1 and NZ_CP101187.1 in SD-1), which was conducive to independent replication but hardly processed horizontal gene transfer. It partially explained that the specialized capability for sulfonamide biodegradation was evolutionarily conserved in three genera, namely Paenarthrobacter (previously affiliated with Arthrobacter), Microbacterium, and Leucobacter, and not yet widespread. Additionally, sad cluster and sadA transposable element were only found in two genera except for Leucobacter (Candidatus Leucobacter sulfamidivorax GP). However, environments with high concentrations of sulfonamides, such as sulfonamide-degrading enrichments in lab-scale investigations, could facilitate the evolution of antibiotic-resistant microbes, posing a potential threat to ecosystems.
Figure 4.

Diverse gene arrangement of sulfonamide-degrading genes in Microbacteriaceae spp. and Micrococcaceae spp. The phylogenetic tree was rooted using midpoint rooting. Arthrobacter spp. M5 and M6 were reassigned to Paenarthrobacter by GTDB-tk (v2.1.1) [31] based on Genome Taxonomy Database taxonomy R214.
Apart from sulfonamide degraders, homologous genes of Type I sadB were identified in three MAGs (Table S5), namely Nocardioidaceae bacterium HKST-UBA51 (GCA_020438665.1, 85.0% identity) [38], uncultured Propionibacterium sp. SRR8859111_bin.135_CONCOCT_v1.1_MAG (GCA_937867175.1, 83.5% identity), and uncultured Micropruina sp. ERR3519522_bin.78_CONCOCT_v1.1_MAG (GCA_937867535.1, 70.2% identity) [55]. They were affiliated with Propionibacteriaceae and Nocardioidaceae within Propionibacteriales and were retrieved from freshwater sediment [56] and wastewater [57] samples, respectively. The sadB orthologous genes in Nocardioidaceae bacterium and uncultured Propionibacterium sp. shared 92.0% identity and were a paraphyletic sister to sadB Type I (Fig. S6).
Despite the fact that either sul or sad gene can confer the bacteria with sulfonamide resistance, the sul gene was identified in all sad-carrying sulfonamide degraders, which is consistent with a recent study proposing the co-occurrence of sad and sul genes as a coherent feature of efficient sulfonamide degraders [54]. However, even though Paenarthrobacter sp. SK contained a complete sad gene cluster, sadA transposable element, sul918, and sul1, it displayed a limited ability to utilize sulfonamide as the sole carbon or energy source [26]. This result was not fully in agreement with the conclusion that the lack of sul918 leads to the poor SMX-degrading performance of Paenarthrobacter ureafaciens SD-2, which contains a complete sad gene cluster, sadA transposable element, and sul1 gene [54]. Compared with other strains, the sadA transposable element of SK is incomplete (Fig. 4). The lack of the transposase may result in the low expression of nearby genes [58, 59], but more solid evidence is still needed to confirm this suspect. Generally, the current results were in accordance with the notion that the sul genes contributed to sulfonamide resistance, whereas sad genes were responsible for sulfonamide degradation, and the combination of sul and sad genes enhanced the biodegradation performance.
In the present study, we only monitored the parent compound degradation and the formation of corresponding dead-end metabolites, but mass balance analysis is still needed to resolve quantitative differences in sulfonamide-degrading performance among Paenarthrobacter strains. Moreover, transcriptomic analysis could be further applied to verify the contribution of Type III sadA to SMZ degradation and the involvement of Type II sadB in sulfonamide biodegradation. On the other hand, pure culture results generally cannot be extrapolated to mixed communities, and serial issues on biosafety of these rapid evolution strains remain to be addressed before practical application. Lastly, antibiotic use is a double-edged sword for bacteria, which kills them but also facilitates the evolution of resistance genes as well as degradation genes, reminding the relationship between nature and humans.
Conclusions
In light of the widespread use of sulfonamides, the evolutionary pressure for the emergence of antibiotic resistance is great. Under this circumstance, biodegradation plays a crucial role in sulfonamide elimination in natural environments. In the present study, we explored the strain-level differentiation in Paenarthrobacter on sulfonamide biodegradation. Our results revealed that SDZ and SMX were easy to degrade, whereas SMZ was relatively recalcitrant probably owing to its heterocyclic ring. The sulfonamide-degrading performance was generally related to sadA numbers, but SMZ biodegradation might rely on the sadA genotype. Based on thesurvey of sad genes in the public database, sad gene cluster and sadA transposable element were conserved in two genera (i.e. Paenarthrobacter [Micrococcaceae] and Microbacterium [Microbacteriaceae]). In addition, Paenarthrobacter demonstrated a high capacity for genome editing, propagating multiple sadA genes in both chromosome and plasmid and promoting sulfonamide biodegradation, which opened up new possibilities for engineering applications.
Supplementary Material
Acknowledgements
Y.H., Y.S., and Y.D. thank The University of Hong Kong for the postdoctoral research fellowship. The computations were performed using research computing facilities offered by Information Technology Services, The University of Hong Kong. Y.H. also appreciates in-depth discussions with Dr. Zhong Yu and the technical support from Ms. Vicky Fung.
Contributor Information
Yue Huang, Environmental Microbiome Engineering and Biotechnology Lab, Department of Civil Engineering, The University of Hong Kong, Pokfulam Road, Hong Kong SAR 999077, China.
Anxin Pan, Environmental Microbiome Engineering and Biotechnology Lab, Department of Civil Engineering, The University of Hong Kong, Pokfulam Road, Hong Kong SAR 999077, China.
Ying Song, Environmental Microbiome Engineering and Biotechnology Lab, Department of Civil Engineering, The University of Hong Kong, Pokfulam Road, Hong Kong SAR 999077, China.
Yu Deng, Environmental Microbiome Engineering and Biotechnology Lab, Department of Civil Engineering, The University of Hong Kong, Pokfulam Road, Hong Kong SAR 999077, China.
Alnwick Long-Hei Wu, Environmental Microbiome Engineering and Biotechnology Lab, Department of Civil Engineering, The University of Hong Kong, Pokfulam Road, Hong Kong SAR 999077, China.
Colin Shiu-Hay Lau, Environmental Microbiome Engineering and Biotechnology Lab, Department of Civil Engineering, The University of Hong Kong, Pokfulam Road, Hong Kong SAR 999077, China.
Tong Zhang, Environmental Microbiome Engineering and Biotechnology Lab, Department of Civil Engineering, The University of Hong Kong, Pokfulam Road, Hong Kong SAR 999077, China.
Conflicts of interest
The authors declare that they have no competing interests.
Funding
This study was financially supported by the Hong Kong Theme Based Research Scheme (T21-705/20-N).
Data availability
The raw data of short- and long-read sequences have been deposited in the NCBI database under project ID PRJNA669352. The reassembled genomes were deposited under BioSample Accession numbers SAMN38477253–SAMN38477260. Raw data of additional long-read sequences are available from the corresponding author on reasonable request.
References
- 1. Scozzafava A, Owa T, Mastrolorenzo Aet al. Anticancer and antiviral sulfonamides. Curr Med Chem 2003;10:925–53. 10.2174/0929867033457647. [DOI] [PubMed] [Google Scholar]
- 2. Sarmah AK, Meyer MT, Boxall AB. A global perspective on the use, sales, exposure pathways, occurrence, fate and effects of veterinary antibiotics (VAs) in the environment. Chemosphere 2006;65:725–59. 10.1016/j.chemosphere.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 3. Göbel A, McArdell CS, Joss Aet al. Fate of sulfonamides, macrolides, and trimethoprim in different wastewater treatment technologies. Sci Total Environ 2007;372:361–71. 10.1016/j.scitotenv.2006.07.039. [DOI] [PubMed] [Google Scholar]
- 4. Li B, Zhang T, Xu Zet al. Rapid analysis of 21 antibiotics of multiple classes in municipal wastewater using ultra performance liquid chromatography-tandem mass spectrometry. Anal Chim Acta 2009;645:64–72. 10.1016/j.aca.2009.04.042. [DOI] [PubMed] [Google Scholar]
- 5. Le TX, Munekage Y. Residues of selected antibiotics in water and mud from shrimp ponds in mangrove areas in Viet Nam. Mar Pollut Bull 2004;49:922–9. 10.1016/j.marpolbul.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 6. Lindsey ME, Meyer M, Thurman EM. Analysis of trace levels of sulfonamide and tetracycline antimicrobials in groundwater and surface water using solid-phase extraction and liquid chromatography/mass spectrometry. Anal Chem 2001;73:4640–6. 10.1021/ac010514w. [DOI] [PubMed] [Google Scholar]
- 7. Zainab SM, Junaid M, Xu Net al. Antibiotics and antibiotic resistant genes (ARGs) in groundwater: a global review on dissemination, sources, interactions, environmental and human health risks. Water Res 2020;187:116455. 10.1016/j.watres.2020.116455. [DOI] [PubMed] [Google Scholar]
- 8. Batt AL, Snow DD, Aga DS. Occurrence of sulfonamide antimicrobials in private water wells in Washington County, Idaho, USA. Chemosphere 2006;64:1963–71. 10.1016/j.chemosphere.2006.01.029. [DOI] [PubMed] [Google Scholar]
- 9. Cui C, Jin L, Han Qet al. Removal of trace level amounts of twelve sulfonamides from drinking water by UV-activated peroxymonosulfate. Sci Total Environ 2016;572:244–51. 10.1016/j.scitotenv.2016.07.183. [DOI] [PubMed] [Google Scholar]
- 10. Jia A, Hu J, Wu Xet al. Occurrence and source apportionment of sulfonamides and their metabolites in Liaodong Bay and the adjacent Liao River basin, North China. Environ Toxicol Chem 2011;30:1252–60. 10.1002/etc.508. [DOI] [PubMed] [Google Scholar]
- 11. Baran W, Adamek E, Ziemiańska Jet al. Effects of the presence of sulfonamides in the environment and their influence on human health. J Hazard Mater 2011;196:1–15. 10.1016/j.jhazmat.2011.08.082. [DOI] [PubMed] [Google Scholar]
- 12. Göbel A, Thomsen A, McArdell CSet al. Occurrence and sorption behavior of sulfonamides, macrolides, and trimethoprim in activated sludge treatment. Environ Sci Technol 2005;39:3981–9. 10.1021/es048550a. [DOI] [PubMed] [Google Scholar]
- 13. Amarasiri M, Sano D, Suzuki S. Understanding human health risks caused by antibiotic resistant bacteria (ARB) and antibiotic resistance genes (ARG) in water environments: current knowledge and questions to be answered. Crit Rev Environ Sci Technol 2020;50:2016–59. 10.1080/10643389.2019.1692611. [DOI] [Google Scholar]
- 14. Blair JM, Webber MA, Baylay AJet al. Molecular mechanisms of antibiotic resistance. Nat Rev Microbiol 2015;13:42–51. 10.1038/nrmicro3380. [DOI] [PubMed] [Google Scholar]
- 15. Deng Y, Li B, Zhang T. Bacteria that make a meal of sulfonamide antibiotics: blind spots and emerging opportunities. Environ Sci Technol 2018;52:3854–68. 10.1021/acs.est.7b06026. [DOI] [PubMed] [Google Scholar]
- 16. Chen J, Xie S. Overview of sulfonamide biodegradation and the relevant pathways and microorganisms. Sci Total Environ 2018;640:1465–77. [DOI] [PubMed] [Google Scholar]
- 17. Ricken B, Kolvenbach BA, Bergesch Cet al. FMNH2-dependent monooxygenases initiate catabolism of sulfonamides in Microbacterium sp. strain BR1 subsisting on sulfonamide antibiotics. Sci Rep 2017;7:15783. 10.1038/s41598-017-16132-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim D-W, Thawng CN, Lee Ket al. A novel sulfonamide resistance mechanism by two-component flavin-dependent monooxygenase system in sulfonamide-degrading actinobacteria. Environ Int 2019;127:206–15. 10.1016/j.envint.2019.03.046. [DOI] [PubMed] [Google Scholar]
- 19. Deng Y, Wang Y, Mao Yet al. Partnership of Arthrobacter and Pimelobacter in aerobic degradation of sulfadiazine revealed by metagenomics analysis and isolation. Environ Sci Technol 2018;52:2963–72. 10.1021/acs.est.7b05913. [DOI] [PubMed] [Google Scholar]
- 20. Qi M, Liang B, Zhang Let al. Microbial interactions drive the complete catabolism of the antibiotic sulfamethoxazole in activated sludge microbiomes. Environ Sci Technol 2021;55:3270–82. 10.1021/acs.est.0c06687. [DOI] [PubMed] [Google Scholar]
- 21. Bakker DP, Postmus BR, Busscher HJet al. Bacterial strains isolated from different niches can exhibit different patterns of adhesion to substrata. Appl Environ Microbiol 2004;70:3758–60. 10.1128/AEM.70.6.3758-3760.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Leventhal GE, Boix C, Kuechler Uet al. Strain-level diversity drives alternative community types in millimetre-scale granular biofilms. Nat Microbiol 2018;3:1295–303. 10.1038/s41564-018-0242-3. [DOI] [PubMed] [Google Scholar]
- 23. Gushgari-Doyle S, Lui LM, Nielsen TNet al. Genotype to ecotype in niche environments: adaptation of Arthrobacter to carbon availability and environmental conditions. ISME Commun 2022;2:32. 10.1038/s43705-022-00113-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Busse H-J. Review of the taxonomy of the genus Arthrobacter, emendation of the genus Arthrobacter sensu lato, proposal to reclassify selected species of the genus Arthrobacter in the novel genera Glutamicibacter gen. nov., Paeniglutamicibacter gen. nov., Pseudoglutamicibacter gen. nov., Paenarthrobacter gen. nov. and Pseudarthrobacter gen. nov., and emended description of Arthrobacter roseus. Int J Syst Evol Microbiol 2016;66:9–37. 10.1099/ijsem.0.000702. [DOI] [PubMed] [Google Scholar]
- 25. Deng Y, Mao Y, Li Bet al. Aerobic degradation of sulfadiazine by Arthrobacter spp.: kinetics, pathways, and genomic characterization. Environ Sci Technol 2016;50:9566–75. 10.1021/acs.est.6b02231. [DOI] [PubMed] [Google Scholar]
- 26. Deng Y, Huang Y, Che Yet al. Microbiome assembly for sulfonamide subsistence and the transfer of genetic determinants. ISME J 2021;15:2817–29. 10.1038/s41396-021-00969-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Emiola A, Oh J. High throughput in situ metagenomic measurement of bacterial replication at ultra-low sequencing coverage. Nat Commun 2018;9:4956. 10.1038/s41467-018-07240-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wick RR, Judd LM, Gorrie CLet al. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol 2017;13:e1005595. 10.1371/journal.pcbi.1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jain C, Rodriguez-R LM, Phillippy AMet al. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun 2018;9:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Krawczyk PS, Lipinski L, Dziembowski A. PlasFlow: predicting plasmid sequences in metagenomic data using genome signatures. Nucleic Acids Res 2018;46:e35. 10.1093/nar/gkx1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chaumeil P-A, Mussig AJ, Hugenholtz Pet al. GTDB-Tk: A Toolkit to Classify Genomes with the Genome Taxonomy Database. Bioinformatics 2020;36:1925–1927. 10.1093/bioinformatics/btz848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Letunic I, Bork P. Interactive tree of life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 2007;23:127–8. 10.1093/bioinformatics/btl529. [DOI] [PubMed] [Google Scholar]
- 33. Hyatt D, Chen G-L, LoCascio PFet al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010;11:119. 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cantalapiedra CP, Hernández-Plaza A, Letunic Iet al. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol Biol Evol 2021;38:5825–9. 10.1093/molbev/msab293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods 2015;12:59–60. 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 36. Klappenbach JA, Saxman PR, Cole JRet al. rrndb: the ribosomal RNA operon copy number database. Nucleic Acids Res 2001;29:181–4. 10.1093/nar/29.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen J, Ke Y, Zhu Yet al. Deciphering of sulfonamide biodegradation mechanism in wetland sediments: from microbial community and individual populations to pathway and functional genes. Water Res 2023;240:120132. 10.1016/j.watres.2023.120132. [DOI] [PubMed] [Google Scholar]
- 38. Liu L, Wang Y, Yang Yet al. Charting the complexity of the activated sludge microbiome through a hybrid sequencing strategy. Microbiome 2021;9:205. 10.1186/s40168-021-01155-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Karimi-Maleh H, Darabi R, Karimi Fet al. State-of-art advances on removal, degradation and electrochemical monitoring of 4-aminophenol pollutants in real samples: a review. Environ Res 2023;222:115338. 10.1016/j.envres.2023.115338. [DOI] [PubMed] [Google Scholar]
- 40. Hibbing ME, Fuqua C, Parsek MRet al. Bacterial competition: surviving and thriving in the microbial jungle. Nat Rev Microbiol 2010;8:15–25. 10.1038/nrmicro2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fuentes DAF, Manfredi P, Jenal Uet al. Pareto optimality between growth-rate and lag-time couples metabolic noise to phenotypic heterogeneity in Escherichia coli. Nat Commun 2021;12:3204. 10.1038/s41467-021-23522-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li B, Zhang T. Biodegradation and adsorption of antibiotics in the activated sludge process. Environ Sci Technol 2010;44:3468–73. 10.1021/es903490h. [DOI] [PubMed] [Google Scholar]
- 43. Alvarino T, Nastold P, Suarez Set al. Role of biotransformation, sorption and mineralization of 14C-labelled sulfamethoxazole under different redox conditions. Sci Total Environ 2016;542:706–15. 10.1016/j.scitotenv.2015.10.140. [DOI] [PubMed] [Google Scholar]
- 44. Achermann S, Bianco V, Mansfeldt CBet al. Biotransformation of sulfonamide antibiotics in activated sludge: the formation of pterin-conjugates leads to sustained risk. Environ Sci Technol 2018;52:6265–74. 10.1021/acs.est.7b06716. [DOI] [PubMed] [Google Scholar]
- 45. Ricken B, Corvini PF, Cichocka Det al. Ipso-hydroxylation and subsequent fragmentation: a novel microbial strategy to eliminate sulfonamide antibiotics. Appl Environ Microbiol 2013;79:5550–8. 10.1128/AEM.00911-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Boreen AL, Arnold WA, McNeill K. Photochemical fate of sulfa drugs in the aquatic environment: sulfa drugs containing five-membered heterocyclic groups. Environ Sci Technol 2004;38:3933–40. 10.1021/es0353053. [DOI] [PubMed] [Google Scholar]
- 47. Topp E, Chapman R, Devers-Lamrani Met al. Accelerated biodegradation of veterinary antibiotics in agricultural soil following long-term exposure, and isolation of a sulfamethazine-degrading Microbacterium sp. J Environ Qual 2013;42:173–8. 10.2134/jeq2012.0162. [DOI] [PubMed] [Google Scholar]
- 48. Tappe W, Herbst M, Hofmann Det al. Degradation of sulfadiazine by Microbacterium lacus strain SDZm4, isolated from lysimeters previously manured with slurry from sulfadiazine-medicated pigs. Appl Environ Microbiol 2013;79:2572–7. 10.1128/AEM.03636-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bouju H, Ricken B, Beffa Tet al. Isolation of bacterial strains capable of sulfamethoxazole mineralization from an acclimated membrane bioreactor. Appl Environ Microbiol 2012;78:277–9. 10.1128/AEM.05888-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wells JN, Feschotte C. A field guide to eukaryotic transposable elements. Annu Rev Genet 2020;54:539–61. 10.1146/annurev-genet-040620-022145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Feschotte C. Transposable elements and the evolution of regulatory networks. Nat Rev Genet 2008;9:397–405. 10.1038/nrg2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Siguier P, Gourbeyre E, Chandler M. Bacterial insertion sequences: their genomic impact and diversity. FEMS Microbiol Rev 2014;38:865–91. 10.1111/1574-6976.12067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yu L, Wang Y, Su Xet al. Biodiversity, isolation and genome analysis of sulfamethazine-degrading bacteria using high-throughput analysis. Bioprocess Biosyst Eng 2020;43:1521–31. 10.1007/s00449-020-02345-1. [DOI] [PubMed] [Google Scholar]
- 54. Wu T, Guo S-Z, Zhu H-Zet al. The sulfonamide-resistance dihydropteroate synthase gene is crucial for efficient biodegradation of sulfamethoxazole by Paenarthrobacter species. Appl Microbiol Biotechnol 2023;107:5813–27. 10.1007/s00253-023-12679-x. [DOI] [PubMed] [Google Scholar]
- 55. Saary P, Kale V, Finn R. Large-scale analysis reveals the distribution of novel cellular microbes across multiple biomes and kingdoms. 2022. Research Square preprint: not peer reviewed. 10.21203/rs.3.rs-1441815/v1. [DOI]
- 56. Das BK, Behera BK, Chakraborty HJet al. Metagenomic study focusing on antibiotic resistance genes from the sediments of River Yamuna. Gene 2020;758:144951. 10.1016/j.gene.2020.144951. [DOI] [PubMed] [Google Scholar]
- 57. Perry MR, Lepper HC, McNally Let al. Secrets of the hospital underbelly: patterns of abundance of antimicrobial resistance genes in hospital wastewater vary by specific antimicrobial and bacterial family. Front Microbiol 2021;12:703560. 10.3389/fmicb.2021.703560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Barkan A, Martienssen RA. Inactivation of maize transposon Mu suppresses a mutant phenotype by activating an outward-reading promoter near the end of Mu1. Proc Natl Acad Sci U S A 1991;88:3502–6. 10.1073/pnas.88.8.3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hudson ME, Lisch DR, Quail PH. The FHY3 and FAR1 genes encode transposase-related proteins involved in regulation of gene expression by the phytochrome A-signaling pathway. Plant J 2003;34:453–71. 10.1046/j.1365-313X.2003.01741.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data of short- and long-read sequences have been deposited in the NCBI database under project ID PRJNA669352. The reassembled genomes were deposited under BioSample Accession numbers SAMN38477253–SAMN38477260. Raw data of additional long-read sequences are available from the corresponding author on reasonable request.