

Abstract
Shiga toxin (Stx) follows a complex intracellular pathway in order to kill susceptible cells. After binding to cell surface glycolipids, the toxin is internalized and trafficked in retrograde fashion to the endoplasmic reticulum (ER). From the ER lumen, the toxin must gain access to the cytoplasm, where it enzymatically inactivates the 28S rRNA, inhibiting protein synthesis. The host molecules involved in this pathway and the mechanisms utilized by the toxin to access the cytoplasm from the ER are largely unknown. We found that Stx is capable of energy-dependent transport across the ER lumen, as has recently been demonstrated for the cholera and ricin toxins. Genetic screening for molecules involved in Shiga toxin trafficking yielded a cDNA encoding a prematurely truncated protein. Characterization of this cDNA revealed that it encodes a novel Hsp40 chaperone, designated HEDJ or ERdj3, localized to the ER lumen, where it interacts with BiP, a molecule known to be involved in protein retrotranslocation out of the ER. We demonstrated that within the ER lumen Stx interacts with HEDJ and other chaperones known to be involved in retrotranslocation of proteins across the ER membrane. Moreover, sequential immunoprecipitation revealed that Shiga toxin was present in a complex that included HEDJ and Sec61, the translocon through which proteins are retrotranslocated to the cytoplasm. These findings suggest that HEDJ is a component of the ER quality control system and that Stx utilizes HEDJ and other ER-localized chaperones for transport from the ER lumen to the cytosol.
Bacterial toxins, by their very nature, are remarkably adept at exploiting host cell biology (7, 17, 20). Shiga toxins (Stx), which are responsible for thousands of cases of hemorrhagic colitis annually and are the causative agents of hemolytic-uremic syndrome, provide a dramatic example. Intracellular trafficking of Stx appears to take advantage of two recently described quality control pathways. After binding to receptors on the cell surface, Shiga toxin is internalized, and rather than being routed to the lysosome for degradation, the toxin follows a novel retrograde pathway through the Golgi apparatus to the lumen of the endoplasmic reticulum (ER), presumably riding a preexisting membrane recycling pathway (25, 26). From the lumen of the ER, the A subunit of Stx (StxA) must gain access to the cytoplasm. Unlike some other bacterial toxins (such as diphtheria toxin and the anthrax toxins), Shiga toxin has no intrinsic ability to cross host membranes and therefore is presumed to utilize host machinery to cross intracellular membranes. It has recently been proposed that such toxins gain access to the cytoplasm via a retrograde pathway normally utilized for export of misfolded host proteins from the ER (4, 23, 27).
The ER is an important site of protein quality control. Molecular chaperones such as BiP, an Hsp70 chaperone, assist in protein import and folding within the ER lumen (12-14, 32). Nascent proteins that do not fold correctly are transported for degradation from the ER lumen to the cytosol via a translocon, Sec61, in a process that also requires BiP (6, 18, 19). In Saccharomyces cerevisiae, this process is also known to require members of the Hsp40 family (16). The mechanisms by which proteins are recognized as misfolded and then recruited to Sec61 for reverse translocation are currently unknown. However, these processes are under intense scrutiny because several human diseases result either from pathological retention of misfolded proteins in the ER or degradation of proteins, such as CFTRΔF508, that are functional but never reach the cell surface because of misfolding and degradation by quality control systems in the ER (10, 37, 38).
Recent evidence has implicated the ER quality control system in the intracellular trafficking of some bacterial toxins. For example, the cholera and ricin toxins are trafficked in retrograde fashion to the ER lumen. Like Stx, these toxins lack pore-forming activity. Instead, both have recently been demonstrated to undergo reverse translocation through the Sec61 translocon in order to reach the cytoplasm (26, 27, 29, 33, 35). It has been proposed that StxA, like the ricin and cholera toxins, is able to mimic the structure of a misfolded protein and thereby utilize the host translocation machinery for transport from the ER to the cytosol.
We demonstrate here that the Stx A subunit, like the subunits of the cholera and ricin toxins, is capable of retrotranslocation across the ER membrane. A screening approach was utilized to identify host genes involved Shiga toxin trafficking. Through toxin selection and sequential enrichment, this approach yielded cDNA encoding a novel, truncated protein. We subsequently isolated the entire gene, which encoded a protein that we designated HEDJ (also known as ERdj3), and demonstrated that the protein is an ER-localized Hsp40 chaperone (36). Transfection of Vero cells with a truncated cDNA encoding only the amino-terminal 126 amino acids of HEDJ resulted in diminished Stx susceptibility. In order to determine whether HEDJ was capable of interacting with Stx within the ER lumen, an in vitro assay was utilized. Within the ER lumen and prior to transport, the toxin was found to interact with HEDJ, BiP, and Grp94. Sequential immunoprecipitation demonstrated that a portion of the toxin associated with HEDJ was also associated with Sec61. Our results demonstrated that there was an interaction with HEDJ prior to its transport across the ER membrane. Based on similarities between HEDJ and related molecules in the yeast ER, we suggest here that the normal role of HEDJ may be recruitment of misfolded proteins to the Sec61 translocon and that Stx is capable of exploiting this function of HEDJ for its own transport across the ER membrane.
MATERIALS AND METHODS
Cell lines and antibodies.
Vero cells (African green monkey adult kidney cells; ATCC CCL-81; American Type Culture Collection, Bethesda, Md.) were maintained in Dulbecco's modified Eagle's medium containing 10% fetal calf serum (Gibco BRL, Grand Island, N.Y.).
Plasmid pHED7, which includes 92 bp of 5′untranslated region and encodes the first 121 amino acids of HEDJ fused to the V5 epitope and a histidine tag, was created by amplifying the HEDJ gene with primers GFP-1 and Hsp-4 (5′CCTGTTCTTGCTGACGAGGGGTTCC-3′) and cloning in frame into plasmid pCR3.1V5/His/Topo (Invitrogen).
In order to generate affinity-purified antiserum, peptides corresponding to regions of StxA and HEDJ were synthesized by Biosynthesis Inc. (Galveston, Tex.) and were purified by high-performance liquid chromatography. A cysteine residue was added at either the amino or carboxyl terminus of each peptide. The corresponding peptide sequences were CCEAREGIKQLLKQGSVQKVYNGLQGY for HEDJ and CDVRGIDPEEGRFNNLR for StxA. A peptide consisting of amino acids PGPTPSGTNC, derived from Sec61β and described previously (34), was also synthesized. Peptides were conjugated to maleimide-activated keyhole limpet hemocyanin (Pierce Corp.), dialyzed against phosphate-buffered saline (PBS), and then used to immunize two rabbits (Cocalico Biolgicals). After the third booster dose, 25 ml of serum was passed over peptide immobilized on Sulfolink beads (Pierce Corp.). After washing, affinity-purified antibodies were eluted with 100 mM glycine (pH 2.5), neutralized, and then dialyzed against PBS. Polyclonal rabbit antiserum was raised against purified StxB, as previously described (2).
Shiga toxin preparation.
Semipurified Stx-1 was prepared from Escherichia coli harboring the genes encoding Shigella dysenteriae Stx-1 on plasmid pNAS13 (generously provided by Alison O'Brien), which contained the operon on a 3.4-kb NcoI fragment from pNAS4 (30). Two-liter cultures of the bacteria were grown overnight at 37°C. Periplasmic extraction was performed by osmotic shock (15). The periplasmic extract was concentrated to 10 ml and then subjected to anion-exchange chromatography on a Q-Sepharose column by using a gradient from 20 mM Tris (pH 7.5) to 20 mM Tris (pH 7.0)-1 M NaCl. Fractions containing StxA, as judged by dot blotting, were pooled and purified further by gel filtration by using a Superdex 75 HR 10/30 column equilibrated with PBS. Immunoreactive fractions were again pooled and stored frozen. Western blotting revealed the presence of StxA and StxB in the pooled sample. Coomassie blue staining of the pooled sample indicated that the protein concentration was approximately 5 μg/ml and the toxin purity was approximately 30%. Serial dilution of the semipurified Stx revealed cytotoxic activity against Vero cells even at a 1:10,000 dilution.
Generation of a Vero cell cDNA library.
mRNA was isolated from approximately 1 × 108 Vero cells. First-strand cDNA was synthesized by addition of random hexamer primers and avian myeloblastosis virus reverse transcriptase. Second-strand synthesis was performed, the ends were blunted with T4 DNA polymerase, nonpalindromic BStxI adapters (Invitrogen) were ligated to the cDNA ends, and the resulting cDNA was then size selected (>500 bp) by agarose gel electrophoresis. The adapter-ligated cDNA was recovered and ligated into BstxI-digested expression plasmid pcDNAI (Invitrogen). In this bidirectional library, each cDNA could have been ligated in the sense or antisense orientation. The ligated cDNA library was then electroporated into E. coli strain MC1061/P3 and plated on 30 15-cm petri dishes containing ampicillin and tetracycline. The next day, bacterial colonies were harvested, aliquoted, and stored at −20°C.
Cloning of full-length HEDJ cDNA.
In order to identify the 3′ transcriptional termination site of the HEDJ gene, 3′ rapid amplification of cDNA ends was performed. cDNA that was obtained from human skeletal muscle and was ligated to adapters whose sequences were known was used in the reaction (Clontech). A PCR was performed with a primer internal to the sequence of HEDJ (GFP-1; 5′-GGCCTCACAGGGCCGGGTGGGCTGG-3′) and another primer purchased from Clontech that recognized the ligated adapter (AP-1). A second round of PCR was performed with nested primers, which again consisted of a gene-specific primer (HSP-5; 5′-GCTTCATC-CCGACCGGAACCCTGAT-3′) and another primer that recognized the adapter (AP-2; Clontech). The product was subcloned, and the nucleotide sequence, which contained the putative stop codon, was determined. As a complementary means of identifying the start and stop codons, iterative BLAST searches were performed against the human EST database. By successive walking in both directions, several overlapping sequences in the human EST database were identified. No additional putative start codons were identified 5′ to the codon already identified, and in the other direction the entire coding sequence could be identified. This sequence matched that obtained by 3′ rapid amplification of cDNA ends. A primer complementary to and overlapping the stop codon (5′-TCACTCTCAATATCCT-TGCAGTCCATTGTATACC-3′) was used along with primer GFP-1 to amplify the entire HEDJ coding region from skeletal muscle cDNA. As described previously, nucleotide sequencing of the PCR product revealed the presence of an 1,175-bp insert encoding a protein consisting of 359 amino acids (36). We subsequently demonstrated that the cDNA encoded a putative signal peptide, a highly conserved J domain, and other features of Hsp40 chaperones (36).
Shiga toxin cytotoxicity assays.
For cytotoxicity studies, approximately 5 × 105 Vero cells were transfected with either empty pCR3.1 vector (wild-type control) or plasmid pHED-7 encoding amino acid residues 1 to 126 of HEDJ and bearing a V5 epitope and a six-His tag (36). Transfections were performed by using 50 μl of lipofectamine (Gibco BRL) and a suspension containing 3 μg of plasmid DNA. Forty-eight hours later, neomycin (Gibco BRL) was added to the medium to a concentration of 1,000 μgm/ml, and neomycin-resistant clones were allowed to proliferate for 14 days.
Shiga toxin inhibits protein synthesis in susceptible cells (17). In order to determine the effect of Stx on wild-type and pHED-7-transfected cells, protein synthesis assays were carried out. Wild-type or pHED-7-transfected cells were suspended in triplicate in 400 μl of replete medium, and 2.5 × 104 cells were placed in each well in a 24-well dish. The following day, cells were either left untreated or exposed for 4 h to Stx at a 1:100 dilution in complete medium. Cell monolayers were then washed two times with PBS and incubated in Cys/Met-free medium containing [35S]Cys/Met (ICN Biomedicals, Inc., Costa Mesa, Calif.) at a concentration of 10 μCi/ml. The cells were incubated for 30 min at 37°C and then washed six times in PBS and lysed by addition of 500 μl of 0.2% sodium dodecyl sulfate (SDS) in 50 mM Tris-HCl (pH 7.5). Trichloroacetic acid (TCA) was added to a concentration of 10%, and the samples were incubated on ice for 30 min. After a 10-min centrifugation at 14,000 × g, the supernatant was removed, and the pellet was washed in 10% TCA and then transferred to a scintillation tube. Incorporation of [35S]Cys/Met, as an indication of de novo protein synthesis after toxin exposure, was determined by scintillation counting of precipitated lysates. In order to determine whether the [35S]Cys/Met incorporation in untreated and treated cells was significantly different, a statistical analysis was performed by using the paired two-tailed t test.
In vitro transcription and translation of Shiga toxin.
In order to translate the wild-type StxA gene, we utilized an E. coli coupled transcription-translation system (Promega Corp., Madison, Wis.) rather than the more commonly used rabbit reticulocyte lysate because we found that the de novo-synthesized toxin was very active and inactivated rabbit ribosomes efficiently and the toxin yields were extremely poor. Moreover, in order to allow toxin translocation into the ER lumen during translation, the StxA signal peptide was replaced with a eukaryotic signal peptide known to be cleaved by signal peptidase during protein import into the ER (derived from the immunoglobulin G gene). This was done by PCR by using 5′ primer ST-17 (5′-AATATGGGTTGGTCATGTATTATTCTGTTCCTGGTAGCAACTGCAACTGGGGTTCATTCGCAGGTTCAGCTGAAGGAATTTACCTTAGACTTC-3′), which was optimized for E. coli codon usage, and primer ST-9 (5′-GCAGGCGGCCGCCCTCAACTGCTAATAGTTCTGCGC-3′). The resulting product, encoding the signal peptide from the immunoglobulin G chain, the signal peptidase cleavage site, and the mature Shiga toxin A subunit, was ligated into plasmid pT7-7 to create plasmid pST17/9 and allowed in vitro transcription with T7 RNA polymerase.
Microsomes (containing ER fragments) were prepared by scraping approximately 5 × 106 Vero cells into 3 ml of PBS. The cells were sedimented by centrifugation, resuspended in 1 ml of isotonic buffer (25 mM HEPES, 110 mM KCl; pH 6.8), and subjected on ice to 30 strokes in a Dounce homogenizer. Nuclei were removed by centrifugation at 2,000 × g for 5 min, and the microsomal fraction was then pelleted either by centrifugation at 14,000 × g for 5 min or by addition of MgCl2 to a concentration of 20 mM, incubation on ice for 10 min, and centrifugation at 6,000 × g for 5 min. Microsomal pellets were resuspended in 50 μl of isotonic buffer.
Coupled in vitro transcription and translation were carried out by using the E. coli T7 S30 extract system (Promega Corp.) according to the manufacturer's recommendations. Each 50-μl reaction mixture contained 1 μg of plasmid pST17/9, 5 μl of microsomes, and 30 μCi of [35S]methionine.
Protease protection and luciferase assays.
Protease protection assays were carried out as described previously (36). Coupled transcription and translation reactions were carried out in the presence of 5 μl of microsomes by using an E. coli T7 S30 extract as described above. After this, the microsomes were harvested by centrifugation and then washed once in 100 μl of PBS by centrifugation at 10,000 × g for 5 min and removal of the supernatant. The microsomal pellet was resuspended in 50 μl of PBS alone or PBS containing 1% Triton X-100. To two of the samples, proteinase K was added to a final concentration of 100 μg/ml. The samples were incubated on ice for 30 min and then boiled and subjected to SDS-polyacrylamide gel electrophoresis (PAGE). The gels were fixed, soaked in Enlightening solution (NEN DuPont), and dried as described above. StxA was detected by fluorography.
In order to determine whether translated StxA within microsomes was enzymatically active, protein synthesis inhibition assays were carried out. First, StxA was translated into microsomes as described above. Control reaction mixtures contained either no plasmid or empty pCRT7 cloning vector. After coupled transcription and translation as described above, microsomes were harvested by centrifugation at 10,000 × g for 5 min. The microsomal pellet was resuspended in 100 μl of PBS containing 0.1% Triton X-100. One microliter of each of the preparations was added to a second in vitro transcription and translation reaction mixture in which rabbit ribosomes were used to transcribe and translate the luciferase protein. The reactions were performed with the TNT T7 coupled reticulocyte lysate system (Promega Corp.) and the control luciferase expression plasmid, as described in the manufacturer's recommendations. However, before the luciferase expression plasmid was added, reticulocyte lysates were treated for 15 min at 37°C with 1 μl of the microsomal extract derived from StxA or control translation reactions. After this, luciferase plasmid DNA was added, and the reactions were allowed to proceed for 30 min. Five microliters of each reaction product was added to 100 μl of luciferase substrate (luciferase assay system; Promega Corp.), and the light output was quantified with a luminometer.
Shiga toxin transport assays.
Transport assays were carried out by using a modification of in vitro assays for cholera toxin translocation (27). Radioactive Stx was loaded into the ER lumen by in vitro transcription and translation in the presence of 5 μl of microsomes. The toxin-loaded microsomes were then washed in PBS and resuspended in 100 μl of transport buffer containing ATP and an ATP-regenerating system (2 mM MgCl2, 2 mM ATP, 20 mM creatine phosphate, 400 μg of creatine kinase per ml in PBS) on ice. For control reactions, the loaded microsomes were resuspended in PBS lacking ATP or an ATP-regenerating system. The transport reaction mixtures were incubated for various times ranging from 0 to 120 min at 37°C. Samples were centrifuged at 14,000 × g for 5 min, and the supernatant (containing transported toxin) was removed from the microsomal pellet. The samples were boiled with Laemmli buffer, subjected to SDS-PAGE, and imaged by fluorography.
Coimmunoprecipitation of Stx with ER-localized chaperones.
In order to examine the interaction of Stx with various ER-localized chaperones, coimmunoprecipitation reactions were carried out with antibodies against HEDJ, Sec61, BiP, protein disulfide isomerase (PDI), and Grp94. Immunoprecipitation was performed by pelleting microsomes by centrifugation after in vitro transcription and translation. Prior to immunoprecipitation, one-half of a sample was subjected to cross-linking by resuspending it in 2 mM dithiobis(succinimidylpropionate) (DSP) in PBS and incubating the preparation on ice for 30 min. Cross-linked and uncross-linked microsomes were washed with cold PBS and lysed in 0.5 ml of lysis buffer (1% Triton X-100, 0.5% NaDoC, 10 mM EDTA in PBS). For the cross-linked product, 25 mM glycine was added. The samples were precleared by addition of 0.15 ml of protein G in bovine serum albumin wash buffer (1% bovine serum albumin, 1% SDS, 1% Triton X-100, 0.5% NaDoC in PBS). After incubation for 1 h at 4°C, the protein G was removed by centrifugation. Approximately 2 μg of the appropriate anti-chaperone antibody was added, and each sample was incubated overnight at 4°C. The next day, protein G (0.15 ml) was added, and after incubation for 1 h at 4°C, immunoprecipitated antigen and protein G were recovered by centrifugation, washed several times in bovine serum albumin wash buffer, and then resuspended in gel loading buffer and subjected to SDS-PAGE. The gel was fixed, soaked in Enlightening solution (NEN Dupont), dried, and exposed to film to detect coimmunoprecipitated radiolabeled Stx. Control experiments demonstrated that none of the anti-chaperone antibodies cross-reacted with in vitro-translated StxA (data not shown).
RESULTS
Phenotypic cloning of HEDJ.
In an attempt to identify host genes involved in Shiga toxin trafficking, we used an approach that should have disrupted the normal trafficking pathway and thereby rendered cells relatively resistant to the toxin. Whereas cell death requires up to 48 h of toxin exposure, we hoped to use a more rapid and quantitative assay for toxin resistance. Since Stx inhibits protein synthesis (20), we anticipated that treatment of cells expressing a destabilized green fluorescent protein (dGFP), designated d2EGFP, with Stx would result in decreased de novo synthesis of d2EGFP. Existing protein would be degraded, and in the absence of ongoing protein synthesis, fluorescence would be lost from toxin-susceptible cells. In contrast, toxin-resistant cells were expected to continue dGFP synthesis in the presence of Stx and thereby remain fluorescent. Resistant cells could be separated from susceptible cells by fluorescence-activated cell sorting (FACS).
Vero cells were stably transfected with plasmid pd2EGFP-N1,l and FACS and immunofluorescence were used to select cells that had cytoplasmic expression of green fluorescent protein. In order to demonstrate that inhibition of protein synthesis resulted in loss of fluorescence over time, control experiments with cycloheximide were performed. Vero:d2EGFP cells were treated with cycloheximide (final concentration, 10 mM) and examined either by fluorescence microscopy or by FACS at various times. Maximal loss of fluorescence occurred after exposure of cells for 12 to 18 h to cycloheximide. The abilities of cycloheximide and Shiga toxin to decrease the fluorescence of d2EGFP:Vero cells were then compared. After 12 h of exposure to either Shiga toxin or cycloheximide, fluorescence was markedly decreased in d2EGFP-expressing cells (Fig. 1B).
FIG. 1.

(A) Schematic drawing of the phenotypic cloning approached used to isolate HEDJ. Vero cells stably expressing destabilized green fluorescent protein (Vero:dGFP) were transfected with a Vero cell cDNA library and then exposed to toxin. After 18 h of incubation, toxin-resistant cells were separated from susceptible cells by FACS. Plasmid DNA was recovered from enriched cells by Hirt extraction and were subjected to another two rounds of transfection and enrichment. (B) FACS profile demonstrating the baseline fluorescence of untreated Vero:dGFP cells and cells treated for 18 h either with cycloheximide or with Shiga toxin. The region gated by M1 included less than 1% Vero cells lacking green fluorescent protein expression. (C) Schematic representation of the HEDJ gene product. The residues enriched by phenotypic cloning and included in plasmid pHED-7 are indicated by the dotted line. The locations of the J domain, the glycine- and phenylalanine-rich domain (G/F region), and the cysteine-rich domain found in most Hsp40 chaperones (36) are indicated.
In order to randomly impair gene expression or function, a cDNA library was created in the sense and antisense orientations. The library was calculated to contain 1.4 × 106 independent cDNA clones. Restriction digestion of 20 randomly selected clones revealed that the average insert size was approximately 0.8 kb. It was anticipated that high-level expression of an antisense mRNA might decrease the amount of an endogenous mRNA that encoded a product involved in toxin trafficking. In addition, since the library was created from random primed cDNA, it was anticipated that the cDNA library contained predominantly truncated messages. Expression of a truncated protein or antisense message might have a dominant negative effect on an endogenous protein involved in toxin internalization or trafficking and thereby enable toxin resistance. By either mechanism, transfecting cells with the random primed, bidirectional cDNA library might have conferred toxin resistance on a small number of cells.
In order to enrich for cDNA clones that enabled toxin resistance, the contents of 10 100-mm3 dishes, each containing approximately 1 × 106 Vero:dGFP cells, were transfected with the Vero cDNA library (2.5 μg) by using lipofectamine (50 μl). The library was cotransfected with plasmid pSVE (0.4 μg), which encodes the simian virus 40 large T antigen, driving high-level replication of plasmids containing the Vero cDNA library.
Approximately 48 h after transfection, cells were harvested and replated in the presence of Shiga toxin (1:100 dilution of semipurified extract; approximately 15 ng of toxin per ml). Eighteen hours later the cells were harvested and subjected to fluorescence-activated cell sorting such that the 1% of cells that retained the most fluorescence was separated. Plasmid DNA was isolated by Hirt extraction (5) from the FACS-sorted Vero:dGFP cells and then electroporated into E. coli MC1061/P3 and plated onto medium containing ampicillin and tetracycline (which did not allow replication of transformants containing the pSVE large T plasmid, which encodes only ampicillin resistance). Plasmid DNA, representing cDNA clones enriched through toxin selection, was used in a second round of transfection of Vero:dGFP cells, toxin exposure, and sorting by FACS as described above. A second Hirt extract was obtained from enriched fluorescent cells, electroporated, and used for a third round of transfection and enrichment. However, after the third transfection, cells were incubated in the presence of toxin (1:100 dilution of semipurified toxin) for 48 h. The medium was changed, and fresh toxin (1:100 dilution) was added in order to select for cells that were stably transfected with cDNAs putatively encoding toxin resistance. After several days, very few viable cells were evident. The cells that were present appeared to be vacuolated, grew slowly, and did not form large colonies as cells became rounded and detached almost at the same rate that cells divided and replaced dying cells. This phenotype persisted for more than 48 h after toxin was removed from the medium, likely indicating a toxic effect of the expressed cDNA.
Hirt extraction could not be used to isolate DNA from these stably transfected cells, as the plasmid DNA was presumed to have integrated into the genome. Instead, in order to determine whether these Stx-resistant cells contained a Vero cDNA insert, the cells were harvested, centrifuged, and resuspended in 50 μl of water. Five microliters of the total cell extract was subjected to PCR by using primers T7 and Sp6, which flanked inserts in plasmid pcDNAI. Two bands at approximately 600 and 200 bp were visualized. As a control, cells transfected with pSVE, which lacked T7 and Sp6 priming sites, were subjected to PCR with the same primers. No product was visualized when these cells were used as the DNA source. The PCR products from Stx-resistant cells were directly subcloned into vector pCR3.1/Topo (Invitrogen). After transformation into E. coli, approximately 30 colonies were isolated. Plasmid DNA was isolated from six of the colonies, and the nucleotide sequences were determined.
When the sequences were compared with the GenBank database by using the program BLAST, there were no matches. However, when two of the resulting clones were compared with the human EST database, two entries matched with very high sequence identities. We previously described an initial characterization of the gene product, which we designated HEDJ and has also been called ERdj3 (1, 28).
Expression of a truncated HEDJ results in decreased susceptibility to Stx.
The HEDJ cDNA isolated from Stx-resistant cells consisted of the start codon and sequences encoding the signal peptide, the J domain, and a portion of the linker region, but it did not encode the full-length protein. In order to demonstrate the effect of truncated HEDJ on Stx susceptibility in naïve cells, Vero cells were transfected with an epitope-tagged, truncated HEDJ. The truncated molecule used here was nearly identical to that isolated in the original screening analysis and lacked the carboxyl terminus, which in other Hsp40 chaperones is thought to be the substrate binding portion. The J domain was expressed here intact and is known to be able to bind BiP (36). After transfection with truncated HEDJ, cells were selected with neomycin for 2 weeks. Control cells were transfected with the cloning vector alone and likewise were selected with neomycin for 2 weeks. Notably, the cells expressing truncated HEDJ had a phenotype similar to that of the cells isolated after the third round of cDNA enrichment with toxin. In particular, within individual colonies, some cells became rounded and detached, resulting in a lower rate of proliferation and sparse colonies. Moreover, we were not able to select stable cells that expressed the truncated HEDJ encoded by plasmid pHED-7, since after approximately 2 weeks the viability of the cells was markedly compromised as cells proliferated but lacked detectable HEDJ expression, as judged by immunofluorescence and Western blotting. Instead, we utilized cells approximately 14 days after pHED-7 transfection in the susceptibility assays.
Cells were then assayed for toxin susceptibility by using inhibition of protein synthesis as a quantitative indicator. Wild-type Vero cells, which are exquisitely susceptible to Stx (11), exhibited an 82% decrease in protein synthesis upon exposure to Shiga toxin. In contrast, cells transfected with truncated HEDJ were significantly less susceptible, exhibiting a decrease in protein synthesis of only 24% (Fig. 2). These results demonstrated that expression of truncated HEDJ resulted in reduced susceptibility to Stx and were consistent the hypothesis that truncated HEDJ acts in a dominant-negative manner to inhibit the function of native HEDJ during toxin transport from the ER. In order to directly corroborate a role for HEDJ in toxin trafficking, we attempted to decrease HEDJ mRNA expression by RNA inhibition. Despite using a stable inhibitory RNA expression system, we observed only a transient decrease in HEDJ mRNA, and there was only a minimal effect on the HEDJ protein levels, as judged by Western blotting. Features of HEDJ expression and protein stability apparently conspired to make RNA inhibition unsuccessful, as the expression of HEDJ mRNA is inducible under ER stress conditions and the HEDJ protein has a long half-life (approximately 36 h) (N. Marcus and D. B. Haslam, unpublished data). Instead, in support of a role for this chaperone in toxin trafficking, we examined the interaction between Stx and HEDJ within the ER after first demonstrating that the toxin was capable of translocation across the ER membrane.
FIG. 2.

Cells transfected with pHED-7 exhibit reduced susceptibility to Shiga toxin. Vero cells were transfected with either plasmid alone (WT) or plasmid a containing truncated HEDJ cDNA (pHED-7). Neomycin-resistant cells were selected for 2 weeks and then exposed to Shiga toxin for 4 h. The cells were pulsed with [35S]methionine for 30 min in methionine-free medium. After several washes, the cells were lysed, TCA was added to a concentration of 10%, and the amount of radioactivity incorporated into protein was quantified by scintillation counting of the TCA precipitate. Two asterisks indicate that the difference in [35S]methionine incorporation between untreated and toxin-treated wild-type cells is statistically significant (P < 0.05).
StxA translocation from the endoplasmic reticulum.
In order to determine whether Stx, like the cholera and ricin toxins, is capable of translocation across the ER membrane, an in vitro assay was developed. The in vitro approach used by other investigators to study ER translocation of misfolded host proteins (such as factor H, CFTRΔF508, and alpha-1-antitrypsin) was modified in order to examine Stx transport. Radiolabeled StxA was incorporated into the ER lumen by in vitro transcription and translation. Microsomes loaded with radioactive StxA were washed after translation and then resuspended in transport buffer containing an ATP-regenerating system (or in the absence of ATP, as a control for nonspecific loss of toxin from microsomes). The microsomes were incubated at 37°C for various times, and microsomes were separated from supernatants (which contained toxin translocated out of the ER) by centrifugation. In most experiments, transported toxin appeared as a doublet. The higher-molecular-weight form corresponded to full-length toxin, whereas the lower-molecular-weight form likely represented the cleaved form of the toxin (the A1 form). The A1 form, which is the product of proteolytic cleavage prior to transport out of the ER of living cells, has a molecular mass of 27.5 kDa (3, 9).
Whereas essentially no toxin was translocated in the absence of ATP, as expected, toxin was detected the supernatant in the presence of ATP (Fig. 3). The amount of toxin transported into the supernatant was time and ATP dependent. The amount of toxin translocated in these in vitro assays (approximately 5% over 2 h) was not markedly different than the amount reported for cholera toxin under similar conditions (27). In summary, we demonstrated in vitro here for the first time that StxA, like the cholera and ricin toxins, is capable of energy-dependent retrotranslocation across the ER membrane.
FIG. 3.

StxA is transported across the ER membrane in an ATP-dependent manner. StxA was synthesized by coupled in vitro transcription and translation in the presence of ER extracts and 35S-labeled methionine and cysteine as described previously. Microsomes were then washed in PBS and resuspended in buffer lacking ATP or containing ATP and an ATP-regenerating system. After 30, 60, or 90 min of incubation at 37°C, microsomes were separated from the supernatant, and all samples were boiled and subjected to SDS-PAGE. The toxin remaining in the microsomes (Retained) and the toxin transported into the supernatant (Transported) were detected by fluorography.
Interaction of Stx with ER-localized chaperones.
In order to determine whether StxA was capable of interaction with HEDJ or other intraluminal chaperones, an in vitro assay for toxin translocation was utilized. This assay allowed us to examine events that occurred in the ER separate from earlier stages in intoxication, including binding and trafficking to the ER. Similar assays have enabled researchers to obtain direct evidence for the involvement of Sec61 in retrotranslocation of misfolded human proteins and are currently used to study ER-associated translocation and degradation of the cystic fibrosis transmembrane regulator, alpha-1-antitrypsin, prion proteins, and other proteins involved in human disease. Recently, a similar in vitro approach was used to demonstrate that cholera toxin is exported from the ER via Sec61 (27).
Transport of nascent proteins into the ER lumen during translation depends on the presence of a eukaryotic signal peptide. In order to import the Shiga toxin A subunit into microsomes in the in vitro system, the bacterial signal peptide was replaced with a eukaryotic signal peptide. In addition, a signal peptidase cleavage site was placed adjacent to the signal peptide, so that the mature StxA translated in vitro would have a sequence identical to that of native mature StxA expressed in E. coli. The signal peptide and cleavage site derived from human immunoglobulin G were added to the StxA mature peptide by PCR by using a 5′ primer that encoded the 22 amino acids of the signal peptide and cleavage site. Initial in vitro translation experiments were plagued by poor toxin yields; however, when the sequence encoding the signal peptide was revised and optimized for E. coli codon usage, the resulting construct enabled abundant in vitro translation of recombinant StxA.
In most other studies in which proteins are cotranslationally translocated into microsomes, rabbit reticulocyte lysate is used in the translation reaction. However, StxA potently inactivates eukaryotic ribosomes, and rabbit reticulocyte lysate could not be used in these experiments, a feature that we utilized later to demonstrate that in vitro-translated toxin was enzymatically active. Instead, E. coli lysate containing T7 RNA polymerase was found to efficiently transcribe and translate the Shiga toxin A subunit. Although not previously reported to our knowledge, E. coli ribosomes were able to efficiently mediate cotranslational import of translated protein into the mammalian ER lumen.
In order to demonstrate that StxA translated in this system was efficiently incorporated into the ER lumen, protease protection assays were performed. StxA was synthesized by coupled in vitro transcription and translation in the presence of 35S-labeled methionine and cysteine and ER extracts as described above. The microsomal extract was aliquoted into three tubes and exposed either to no proteinase K or detergent, to both proteinase K and Triton X-100, or to proteinase K without detergent. After 30 min of incubation, the reaction mixtures were subjected to SDS-PAGE and fluoroscopy to detect residual toxin. Whereas there was minimal degradation of StxA in the absence of detergent, addition of detergent resulted in permeabilization of the ER membrane and complete proteolysis of the toxin (Fig. 4A). These results indicate that the recombinant StxA translated in vitro was protected from protease and that the toxin was present in the ER lumen.
FIG. 4.

Demonstration that StxA is incorporated into the ER lumen and is active. (A) StxA was synthesized by coupled in vitro transcription and translation in the presence of 35S-labeled methionine and cysteine and ER extracts. The microsomal extract was aliquoted into three tubes and exposed either to no proteinase K or detergent, to both proteinase K and detergent, or to proteinase K without detergent. After 30 min of incubation, the reaction mixtures were subjected to SDS-PAGE and fluoroscopy to detect residual toxin. (B) StxA was synthesized by coupled in vitro transcription and translation in the presence of ER extracts. Microsomes were then permeabilized with 0.1% Triton X-100 and diluted 100-fold with PBS, and 1-μl portions were added to rabbit reticulocyte lysates. As a control, an in vitro transcription-translation reaction mixture containing empty expression plasmid was used as the microsome source. After reticulocyte lysates were treated with Stx or control extracts, a plasmid encoding luciferase was added, and a second in vitro transcription and translation reaction was carried out. The amount of luciferase produced was quantified by adding 5 μl of the translation reaction mixture to luciferase substrate, and the light output was quantified with a luminometer. RLU, relative light units.
During intoxication of human cells, Stx is presumed to be folded and in its enzymatically active state upon entry into the ER. Unlike host proteins that are translocated across the ER membrane by chaperones in the ER quality control system, Stx apparently can subvert this pathway for its own translocation, even when it is correctly folded. In order to determine whether the intraluminal toxin was folded correctly in our assays, the ability of in vitro-translated StxA to inhibit rabbit ribosomes was examined. StxA was translated into microsomes by using E. coli lysate as described above. Control reactions were carried out with E. coli lysate, microsomes, and either no plasmid DNA or plasmid lacking the StxA gene. After translation, microsomes were washed and then resuspended in PBS containing 0.1% Triton X-100 to destabilize the membrane and allow leakage of StxA into the supernatant. After residual microsomal membranes were removed by centrifugation, the supernatant was added at a 1:100 dilution to rabbit reticulocyte lysate. After 15 min of incubation at 37°C, during which active toxin should have inactivated ribosomes, a plasmid encoding firefly luciferase (Promega Corp.) was added. Translation was carried out for 30 min, and the resulting luciferase activity was determined by adding 5 μl of the translation product to 100 μl of luciferase substrate and quantifying the light output with a luminometer. Compared to supernatant derived from the control reactions, supernatant derived from microsomes containing recombinant StxA markedly inhibited luciferase translation (Fig. 4B).
Coimmunoprecipitation of StxA with HEDJ and other ER-localized chaperones.
In yeast, the retrotranslocation of several misfolded proteins requires the BiP homologue (Kar2) and luminal Hsp40 chaperones (6, 16, 18). In order to determine whether HEDJ, a luminal Hsp40 in human cells, played a similar role in Stx translocation, coimmunoprecipitation experiments were performed. After in vitro transcription and translation of toxin into the ER lumen, immunoprecipitation was performed with antibodies against HEDJ. In addition, coimmunoprecipitation experiments were performed with antibodies against the intraluminal chaperones BiP, GRP94, Erp57, and calnexin, all of which are known to interact with at least a subset of misfolded proteins (8, 12, 13, 32). Our laboratory has recently demonstrated that BiP and Grp94 coimmunoprecipitate with HEDJ from ER extracts of untreated Vero, HepG2, and HTO cells (Marcus and Haslam, unpublished), suggesting that these chaperones are capable of forming a complex in the ER. We were therefore particularly interested in seeing if these chaperones interacted with StxA within the ER lumen.
In support of the hypothesized role for HEDJ in toxin translocation, this chaperone was found to interact with StxA both in the presence and in the absence of a cross-linker (Fig. 5). Likewise, StxA was coimmunoprecipitated with BiP and Grp94 both in the presence and in the absence of a cross-linker (Fig. 5). Calnexin was an important negative control in these experiments, as this chaperone interacted with carbohydrate moieties on misfolded glycoproteins and therefore was not expected to interact directly with StxA, which was not glycosylated in these experiments, as demonstrated by endoglycanase digestion of the translated product (data not shown). Therefore, the small amount of toxin coimmunoprecipitated with anti-calnexin antibodies after DSP cross-linking may have represented nonspecific binding of toxin to protein G Sepharose beads. Notably, StxA did not coimmunoprecipitate with the luminal PDI-like disulfide isomerase Erp57, either in the presence or in the absence of a cross-linker. Together, the results of the coimmunoprecipitation experiments demonstrated that Stx associates within the ER lumen with HEDJ and, to a lesser extent, with other luminal chaperones.
FIG. 5.

StxA coimmunoprecipitates with HEDJ and other luminal ER chaperones. StxA was synthesized by coupled in vitro transcription and translation in the presence of 35S-labeled methionine and cysteine and ER extracts. Microsomes were aliquoted and either exposed to a cross-linker (DSP) (+) or not cross-linked (−). The microsomes were then washed and subjected to immunoprecipitation with antibodies against various ER chaperones. The immunoprecipitated material was boiled and subjected to SDS-PAGE. Coimmunoprecipitated toxin was detected by fluorography.
Next, in order to determine whether HEDJ-associated StxA was found in a complex with Sec61 or other chaperones, sequential immunoprecipitation experiments were performed. StxA was translated into microsomes, cross-linked with DSP, and subjected to immunoprecipitation with anti-HEDJ as described above. Following the anti-HEDJ immunoprecipitation, the pellet was resuspended in lysis buffer, heated, and subjected to a second round of immunoprecipitation with antibodies against Sec61, BiP, and Grp94 or with no second antibody as a control. The amount of StxA isolated after the second immunoprecipitation represented the toxin that was associated both with HEDJ and the second targeted chaperone. As expected, the control experiment, in which no second antibody was added, yielded no detectable toxin. In contrast, radiolabeled StxA was detected after the second immunoprecipitation with the anti-BiP and anti-Grp94 antibodies (Fig. 6). These results are consistent with recent findings of Meunier et al., who demonstrated that HEDJ/ERdj3 that is associated with misfolded substrate proteins can be found in association with a preformed complex that includes BiP and Grp94 (14). Moreover, a significant amount of HEDJ-bound toxin was associated with Sec61. This result demonstrated that Stx interacts with HEDJ, and at least a portion of the HEDJ-interacting toxin was closely associated with the translocon, Sec61.
FIG. 6.
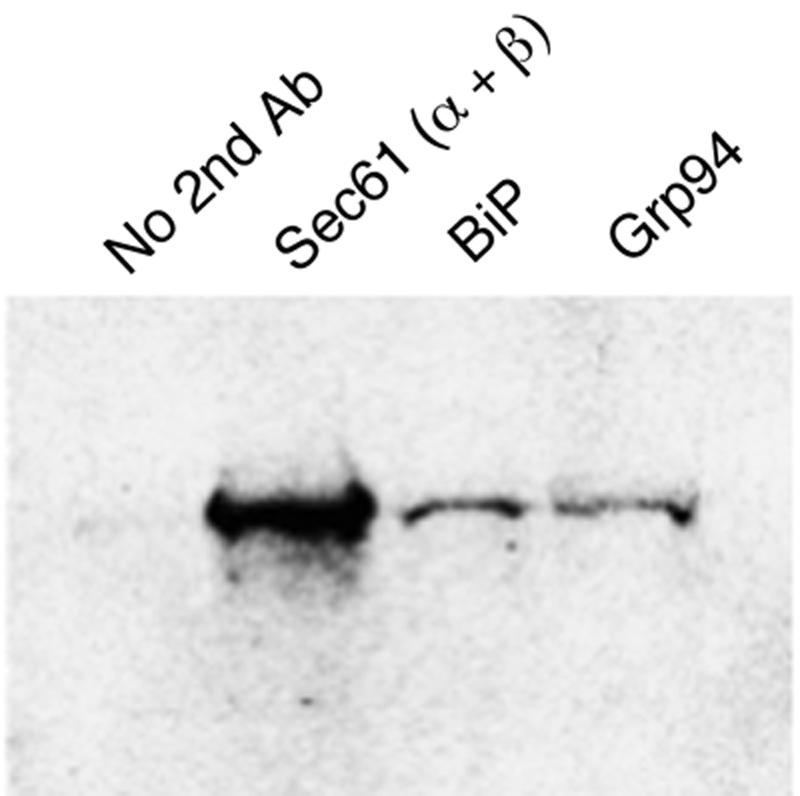
StxA is found in a complex that includes HEDJ and Sec61. StxA was synthesized by coupled in vitro transcription and translation in the presence of 35S-labeled methionine and cysteine and ER extracts as described previously. After cross-linking with DSP, the first immunoprecipitation was carried out with antibodies against HEDJ. The immunoprecipitated material was heated to 95°C for 5 min and then subjected to a second round of immunoprecipitation with either no second antibody (Ab) or antibody against Sec61, BiP, or Grp94.
DISCUSSION
In an attempt to characterize the components of the Stx trafficking pathway, genetic screening for molecules involved in intracellular toxin trafficking was performed. Cells were transfected with a Vero cell cDNA library and then selected for Stx resistance. Plasmid DNA that was presumed to encode a factor that allowed resistance to the toxin was recovered from resistant cells. One clone was recovered after three rounds of transfection and enrichment and was found to contain a cDNA encoding the first 124 amino acids of a previously undescribed protein. Further characterization and cloning of the full-length cDNA revealed that this gene encodes a novel Hsp40 chaperone. An initial characterization demonstrated that this molecule is localized to the ER, where it is able to interact with BiP and other luminal chaperones (36). The canine and human homologues of this molecule have been designated Erdj3, and it has been shown that they also interact with BiP and misfolded substrate proteins (1, 28). In analogy with the yeast ER-associated Hsp40 chaperones, HEDJ likely functions in recognizing proteins as being misfolded and recruits aberrantly folded molecules to Sec61 for reverse translocation.
Shiga toxin, like ricin and cholera toxin, has no intrinsic pore-forming activity, yet all of these toxins must gain access to the cytosol (22, 24). These toxins require trafficking in a circuitous route to the ER, unlike toxins that enter the cytosol directly from the plasma membrane or from the endosomal compartment. Lacking the ability to cross membranes, Shiga toxin and related toxins apparently utilize existing host machinery to enter the cytosol. It has recently been proposed that these toxins are able to mimic the structure of an unfolded protein and thereby utilize the host translocation machinery for transport from the ER to the cytosol (4, 21).
The process of protein retrotranslocation across the ER membrane has been studied extensively in yeast. Temperature-sensitive mutants have revealed central roles for the integral membrane translocon, Sec61, as well as an Hsp70 known as BiP (or Kar2), in protein translocation from the ER to the cytosol. In addition, Jem1p and Scj1p, two Hsp40 family members, have been shown to be required for retrotranslocation of misfolded proteins (16). The identification of HEDJ in a screening analysis for toxin resistance and the demonstration that the toxin interacts with this chaperone within the ER suggest that HEDJ plays a similar role in translocation of misfolded proteins, as well as Stx, from the mammalian ER.
Hsp40 chaperones are involved in nearly all aspects of protein synthesis and secretion. Using the genetic screening method described here, we isolated the first Hsp40 cochaperone localized to the ER lumen of human cells. The requirement for ER-localized chaperones in retrotranslocation of bacterial toxins has not been examined, except for the recent discovery that transport of cholera toxin requires its unfolding by a chaperone known as protein disulfide isomerase (33). Whether Shiga toxin and ricin interact with PDI is not known. Moreover, the mechanism by which all three of these toxins are apparently targeted to the Sec61 pore remains unknown. Presumably, the upstream events include interaction with ER-localized chaperones that recognize these toxins as misfolded host proteins and target them for retrotranslocation. Recently, evidence has accumulated that both ricin and cholera toxin utilize the Sec61 pore to access the cytoplasm from the ER (16, 27, 29, 33). Based on its similar intracellular trafficking and inability to cross host membranes, Shiga toxin is also likely to utilize the Sec61 translocon. Our data suggest that Stx may be recruited to the Sec61 apparatus by HEDJ and other luminal ER chaperones.
The structural features of toxins that allow translocation have not been determined, nor has the role of ER-localized chaperones in this process been determined. Exposed hydrophobic domains have been proposed to be an important signal for interaction of Hsp70 chaperones with misfolded proteins. With this in mind, it has been proposed that a hydrophobic domain located near the carboxyl terminus of StxA, cholera toxin, and ricin is exposed upon dissociation of the B subunits and is responsible for recognition of the toxin by host chaperones. In support of a role for this region in toxin translocation, it has been demonstrated that mutations in the hydrophobic domain of StxA result in diminished cytotoxicity (31). Whether this region is crucial for retrotranslocation of any toxin across the ER membrane has not been tested yet. Targeted mutagenesis of toxin and HEDJ should allow molecular dissection of the residues involved in their interaction and should be facilitated by the in vitro binding and transport assays described here. Ultimately, this information may allow rational design of pharmacologic agents that inhibit toxin binding to HEDJ and transport to the host cell cytoplasm.
Acknowledgments
This work was supported by grant R01AI47900 from the NIAID and by grant 0050644N from the American Heart Association. D.H. is the recipient of an Investigator in Microbial Pathogenesis Award from the Burroughs Wellcome Foundation.
We appreciate the helpful comments and suggestions of Linda Hendershot and Ying Shen.
Editor: A. D. O'Brien
REFERENCES
- 1.Bies, C., S. Guth, K. Janoschek, W. Nastainczyk, J. Volkmer, and R. Zimmermann. 1999. A Scj1p homolog and folding catalysts present in dog pancreas microsomes. Biol. Chem. 380:1175-1182. [DOI] [PubMed] [Google Scholar]
- 2.Elliott, S. P., M. Yu, H. Xu, and D. B. Haslam. 2003. Forssman synthetase expression results in diminished Shiga toxin susceptibility: a role for glyocolipids in determining host-microbe interactions. Infect. Immun. 71:6543-6552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garred, O., B. van Deurs, and K. Sandvig. 1995. Furin-induced cleavage and activation of Shiga toxin. J. Biol. Chem. 270:10817-10821. [DOI] [PubMed] [Google Scholar]
- 4.Hazes, B., and R. J. Read. 1997. Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry 36:11051-11054. [DOI] [PubMed] [Google Scholar]
- 5.Hirt, B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 26:365-369. [DOI] [PubMed] [Google Scholar]
- 6.Holkeri, H., E. Paunola, E. Jamsa, and M. Makarow. 1998. Dissection of the translocation and chaperoning functions of yeast BiP/Kar2p in vivo. J. Cell Sci. 111:749-757. [DOI] [PubMed] [Google Scholar]
- 7.Johannes, L., and B. Goud. 1998. Surfing on a retrograde wave: how does Shiga toxin reach the endoplasmic reticulum? Trends Cell Biol. 8:158-162. [DOI] [PubMed] [Google Scholar]
- 8.Kuznetsov, G., L. B. Chen, and S. K. Nigam. 1997. Multiple molecular chaperones complex with misfolded large oligomeric glycoproteins in the endoplasmic reticulum. J. Biol. Chem. 272:3057-3063. [DOI] [PubMed] [Google Scholar]
- 9.Lea, N., J. M. Lord, and L. M. Roberts. 1999. Proteolytic cleavage of the A subunit is essential for maximal cytotoxicity of Escherichia coli O157:H7 Shiga-like toxin-1. Microbiology 145:999-1004. [DOI] [PubMed] [Google Scholar]
- 10.Lee, A. S. 2001. The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem. Sci. 26:504-510. [DOI] [PubMed] [Google Scholar]
- 11.Lingwood, C. A. 1993. Verotoxins and their glycolipid receptors. Adv. Lipid Res. 25:189-211. [PubMed] [Google Scholar]
- 12.Linnik, K. M., and H. Herscovitz. 1998. Multiple molecular chaperones interact with apolipoprotein B during its maturation. J. Biol. Chem. 273:21368-21373. [DOI] [PubMed] [Google Scholar]
- 13.Melnick, J., J. L. Dul, and Y. Argon. 1994. Sequential interaction of the chaperones BiP and GRP94 with immunoglobulin chains in the endoplasmic reticulum. Nature 370:373-375. [DOI] [PubMed] [Google Scholar]
- 14.Meunier, L., Y.-K. Usherwood, K. Chung, and L. Hendershot. 2002. A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteins. Mol. Biol. Cell 13:4456-4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neu, H. C., and L. A. Heppel. 1965. The release of enzymes from Escherichia coli by osmotic shock and during the formation of spheroplasts. J. Biol. Chem. 240:3685-3692. [PubMed] [Google Scholar]
- 16.Nishikawa, S. I., S. W. Fewell, Y. Kato, J. L. Brodsky, and T. Endo. 2001. Molecular chaperones in the yeast endoplasmic reticulum maintain the solubility of proteins for retrotranslocation and degradation. J. Cell Biol. 153:1061-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Obrig, T. G. 1994. 1994. Toxins that inhibit host protein synthesis. Methods Enzymol. 235:647-656. [DOI] [PubMed] [Google Scholar]
- 18.Pilon, M., K. Romisch, D. Quach, and R. Schekman. 1998. Sec61p serves multiple roles in secretory precursor binding and translocation into the endoplasmic reticulum membrane. Mol. Biol. Cell 9:3455-3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pilon, M., R. Schekman, and K. Romisch. 1997. Sec61p mediates export of a misfolded secretory protein from the endoplasmic reticulum to the cytosol for degradation. EMBO J. 16:4540-4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Proulx, F. 2001. Pathogenesis of Shiga toxin-associated hemolytic uremic syndrome. Pediatr. Res. 50:163-171. [DOI] [PubMed] [Google Scholar]
- 21.Rapak, A., P. O. Falnes, and S. Olsnes. 1997. Retrograde transport of mutant ricin to the endoplasmic reticulum with subsequent translocation to cytosol. Proc. Natl. Acad. Sci. USA 94:3783-3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sandvig, K., and B. van Deurs. 1994. Endocytosis and intracellular sorting of ricin and Shiga toxin. FEBS Lett. 346:99-102. [DOI] [PubMed] [Google Scholar]
- 23.Sandvig, K., and B. van Deurs. 1999. Endocytosis and intracellular transport of ricin: recent discoveries. FEBS Lett. 452:67-70. [DOI] [PubMed] [Google Scholar]
- 24.Sandvig, K., and B. van Deurs. 1996. Endocytosis, intracellular transport, and cytotoxic action of Shiga toxin and ricin. Physiol. Rev. 76:949-966. [DOI] [PubMed] [Google Scholar]
- 25.Sandvig, K., and B. van Deurs. 2000. Entry of ricin and Shiga toxin into cells: molecular mechanisms and medical perspectives. EMBO J. 19:5943-5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sandvig, K., and B. van Deurs. 2002. Transport of protein toxins into cells: pathways used by ricin, cholera toxin and Shiga toxin. FEBS Lett. 529:49-53. [DOI] [PubMed] [Google Scholar]
- 27.Schmitz, A., H. Herrgen, A. Winkeler, and V. Herzog. 2000. Cholera toxin is exported from microsomes by the Sec61p complex. J. Cell Biol. 148:1203-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen, Y., L. Meunier, and L. M. Hendershot. 2002. Identification and characterization of a novel endoplasmic reticulum (ER) DnaJ homologue, which stimulates ATPase activity of BiP in vitro and is induced by ER stress. J. Biol. Chem. 277:15947-15956. [DOI] [PubMed] [Google Scholar]
- 29.Simpson, J. C., L. M. Roberts, K. Romisch, J. Davey, D. H. Wolf, and J. M. Lord. 1999. Ricin A chain utilises the endoplasmic reticulum-associated protein degradation pathway to enter the cytosol of yeast. FEBS Lett. 459:80-84. [DOI] [PubMed] [Google Scholar]
- 30.Strockbine, N. A., M. P. Jackson, L. M. Sung, R. K. Holmes, and A. D. O'Brien. 1988. Cloning and sequencing of the genes for Shiga toxin from Shigella dysenteriae type 1. J. Bacteriol. 170:1116-1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suhan, M. L., and C. J. Hovde. 1998. Disruption of an internal membrane-spanning region in Shiga toxin 1 reduces cytotoxicity. Infect. Immun. 66:5252-5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tatu, U., and A. Helenius. 1997. Interactions between newly synthesized glycoproteins, calnexin and a network of resident chaperones in the endoplasmic reticulum. J. Cell Biol. 136:555-565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsai, B., C. Rodighiero, W. I. Lencer, and T. A. Rapoport. 2001. Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell 104:937-948. [DOI] [PubMed] [Google Scholar]
- 34.Tyedmers, J., M. Lerner, C. Bies, J. Dudek, M. H. Skowronek, I. G. Haas, N. Heim, W. Nastainczyk, J. Volkmer, and R. Zimmermann. 2000. Homologs of the yeast Sec complex subunits Sec62p and Sec63p are abundant proteins in dog pancreas microsomes. Proc. Natl. Acad. Sci. USA 97:7214-7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wesche, J., A. Rapak, and S. Olsnes. 1999. Dependence of ricin toxicity on translocation of the toxin A-chain from the endoplasmic reticulum to the cytosol. J. Biol. Chem. 274:34443-34449. [DOI] [PubMed] [Google Scholar]
- 36.Yu, M., R. H. Haslam, and D. B. Haslam. 2000. HEDJ, an Hsp40 co-chaperone localized to the endoplasmic reticulum of human cells. J. Biol. Chem. 275:24984-24992. [DOI] [PubMed] [Google Scholar]
- 37.Zhang, Y., S. Michaelis, and J. L. Brodsky. 2002. CFTR expression and ER-associated degradation in yeast. Methods Mol. Med. 70:257-265. [DOI] [PubMed] [Google Scholar]
- 38.Zhang, Y., G. Nijbroek, M. L. Sullivan, A. A. McCracken, S. C. Watkins, S. Michaelis, and J. L. Brodsky. 2001. Hsp70 molecular chaperone facilitates endoplasmic reticulum-associated protein degradation of cystic fibrosis transmembrane conductance regulator in yeast. Mol. Biol. Cell 12:1303-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]