

Abstract
Idelalisib targets PI3Kδ in the BCR pathway generating only a partial response in CLL patients, indicating that the leukemic cells may have evolved escape signals. Indeed, we detected increased activation of AKT accompanied by upregulation of MYC/BCL2 in post-therapy CLL cells from patients treated with idelalisib/ofatumumab. To unravel the mechanism of increased AKT-activation, we studied the impact of idelalisib on a CLL-derived cell line, MEC1, as a model. After an initial inhibition, AKT-activation level was restored in idelalisib-treated MEC1 cells in a time-dependent manner. As BCAP (B-cell adaptor for PI3K) and CD19 recruit PI3Kδ to activate AKT upon BCR-stimulation, we examined if idelalisib-treatment altered PI3Kδ-recruitment. Immunoprecipitation of BCAP/CD19 from idelalisib-treated MEC1 cells showed increased recruitment of PI3Kδ in association with PI3Kβ, but not PI3Kα or PI3Kγ and that, targeting both PI3Kδ with PI3Kβ inhibited AKT-reactivation. We detected similar, patient-specific recruitment pattern of PI3K-isoforms by BCAP/CD19 in post-idelalisib CLL cells with increased AKT-activation. Interestingly, a stronger inhibitory effect of idelalisib on P-AKT (T308) than S473 was discernible in idelalisib-treated cells despite increased recruitment of PI3Kδ/PI3Kβ and accumulation of phosphatidylinositol-3,4,5-triphosphate; which could be attributed to reduced PDK1 activity. Thus, administration of isoform-specific inhibitors may prove more effective strategy for treating CLL patients.
INTRODUCTION
Signaling from the B cell receptor (BCR) is essential for normal B cell development and controls several cellular functions such as proliferation, apoptosis, differentiation and cell migration [1, 2]. Constitutive activation of BCR is a common hallmark of B cell malignancies, including chronic lymphocytic leukemia (CLL), an observation that has led to the design of a novel panel of inhibitors targeting kinases responsible for BCR signal transduction. CLL arises from mature B cells, expressing functional BCRs, mainly of immunoglobulin M (IgM) and IgD isotypes with variable clinical courses [3]. Despite significant therapeutic progress in recent years, CLL still remains an incurable disease due, in part, to the persistence of residual leukemia cells after therapy.
Class-I PI3Ks are a group of heterodimeric molecules comprised of a regulatory and catalytic subunit. There are four catalytic isoforms of class-I PI3Ks (class-IA: p110α, p110β, p110δ, and class-IB: p110γ). While p110α and p110β are ubiquitously expressed, the p110γ/p110δ isoforms are predominantly expressed in hematopoietic cell types [4]. In the absence of activating signals, the regulatory p85 subunit interacts with the PI3K110 catalytic subunit and inhibits kinase activity [5].
The BCR signal is mediated partly by the activation of the PI3Kδ-isoform [6] which subsequently activates AKT and mammalian target of rapamycin (mTOR) impacting cell migration, metabolism, proliferation, differentiation and survival [7, 8]. AKT is a key component of the phospholipid phosphatidylinositol-[3–5]-triphosphate (PIP3) signaling network. Activation of PI3K by growth factor receptor stimulation or mutation drives phosphatidylinositol-[4, 5]-biphosphate (PIP2) to PIP3 conversion [9]. In the classical model of AKT regulation, phospholipid PIP3 recruits AKT via its pleckstrin homology (PH) domain to the plasma membrane where it is acted upon by two protein kinases, mTOR kinase from the mTOR complex 2 (mTORC2) and phosphoinositide-dependent kinase-1 (PDK1) phosphorylating AKT on its C terminus at serine-473 (S473) and activation loop at threonine-308 (T308), respectively [10–12]. Dual phosphorylation of AKT leads to its enhanced kinase activity that targets protein substrates including GSK3 and Foxo1/3a [13].
Idelalisib is a potent, selective, small molecule inhibitor of PI3Kδ with in vitro IC50 concentrations for PI3Kδ, PI3Kα, PI3Kβ and PI3Kγ that are 2.5, 820, 565 and 89 nmol/L, respectively [14]. Although blockade of PI3Kδ-signaling is toxic to CLL cells [15], patients treated with idelalisib reached only partial remissions [16]. Given the role of PI3Kδ in B-cell malignancies, we hypothesized that prolonged administration of idelalisib may activate pathway-based cell survival signals in post-therapy CLL cells, allowing escape from detrimental effects of the agent. To pursue this, we analyzed the activation status of multiple signal mediators in post-therapy CLL cells from patients treated with idelalisib/ofatumumab. We detected increased activation of AKT in post-idelalisib CLL cells mediated by increased recruitment of PI3Kδ and/or PI3Kβ to the BCR signalosome. In addition, our findings indicated that idelalisib-treatment could also inhibit PDK1 catalytic activity by reducing its protein level via an undefined mechanism in post-therapy CLL cells.
MATERIALS AND METHODS
CLL patients
Pre-/post-therapy CLL cells were obtained from the CLL patients (n = 20) treated with idelalisib-based therapies (Supplementary Table S1) at the Dana-Farber Cancer Institute, Boston. Supplementary Table S1 is showing available prognostic parameters of the CLL patients including tumor burden (CD5+/CD19+ leukemic CLL cells in blood). Samples were randomly selected for each experiment based on cell availability and viability, but not based on any patient characteristics or experiment outcomes. All patients provided written informed consent according to the Declaration of Helsinki to the DFCI and OUHSC Institutional Review Boards, which approved these studies. RosetteSep human B cell enrichment kit was used to purify CLL cells (≥95%) as needed.
Reagents
All antibodies were purchased from Cell Signaling Technologies except the antibodies to CD19, BCAP, GAPDH (Santa Cruz Biotechnologies) and PIP3 (Eschelon). Idelalisib and a PI3Kβ-inhibitor, GSK2636771 were purchased from Selleckchem. Details of all the reagents used in this study are summarized in Supplementary Table S2.
Flow cytometry
Leukemic tumor burden (CD5+/CD19+) in CLL PBMCs or purity of CLL cells was determined by flow cytometric analysis after staining the cells with chromogen conjugated antibodies to CD5 and CD19. In addition, pre-/post-idelalisib CLL cells from a few CLL patients were analyzed to determine P-AKT (T308) levels by flow cytometry using a phospho-specific phycoerythrin (PE)-conjugated antibody (BD Biosciences).
Treatment and immunoprecipitation/western blot
A CLL cell line, MEC1 [17], was maintained in appropriate growth media and tested for Mycoplasma negativity using a PCR-based assay kit (ABM, Richmond, Canada) prior to treatment with idelalisib (0.2 µM) or GSK2636771 (0.2 µM) as a single agent or in combination for different time intervals. BCAP/CD19 were immunoprecipitated from lysates of MEC1 or purified CLL cells (CD5+/CD19+ ≥ 95%) and western blotted [18] for the class I PI3K isoforms and BCAP/CD19 using specific antibodies.
To validate the specificity of the immunoprecipitating antibodies, CD19 or BCAP was pulled down from the lysates of MEC1 or purified CLL cells obtained from CLL patients (P48, P54) using the anti-CD19 or anti-BCAP mouse monoclonal antibody mentioned above, respectively, or the isotype control antibody (mouse IgG1). The immune complexes were analyzed for CD19 or BCAP in western blots.
Immunofluorescence study
MEC1 cells treated with idelalisib for different time intervals or pre-/post-idelalisib CLL cells were stained with an antibody to PIP3 after permeabilizing the cells, followed by incubation with a chromogen-conjugated secondary antibody (Invitrogen). Cells were counterstained with DAPI (Vectashield) and analyzed under confocal microscope (Leica). The intensity of the stain in cells was calculated using the Leica Application Suite X (LAS X) software and presented as the average of five random fields by bar diagrams with standard deviations.
Semi-quantitative/quantitative RT-PCR
TRIZOL reagent (Invitrogen) was used to isolate RNA from MEC1 and CLL cells collected at different time points. One microgram of RNA was used for cDNA synthesize using iScript First Strand Synthesis kit (BioRad). Semi-quantitative polymerase chain reaction (PCR) was performed to detect PDK1 expression using a specific set of primers. GAPDH was used as internal control. PCR products were analyzed on 2% agarose gel and images were captured. Quantitative RT-PCR was also performed in triplicate for MYC, BCL2 or PDK1 in pre-/post-therapy CLL cells using specific primers and iTaq Universal SYBR Supermix (BioRad). GAPDH was used for cDNA normalization. Relative expression was calculated using the comparative Ct-method and presented as “fold expression”[19]. Please see Supplementary Table S3 for the primer sequences.
Statistical analysis
PRIZM GraphPad software was used for paired t-test analysis as needed.
RESULTS
Idelalisib-based therapy induces activation of multiple kinases
To interrogate if CLL cells could escape from the cytotoxic effects of idelalisib-based therapies by activating cell survival pathways, pre-/post-therapy CLL cell lysates were analyzed for the activation status of multiple intracellular kinases using a human phosphokinase antibody array blot containing 43 different kinases (R&D Systems). Pre-therapy samples were collected on day 1 before initiation of therapy while post-therapy CLL cells were obtained after 1 month or 4 months of therapy from patient (P)1, and 1 month or 2 months after therapy from P2. Densitometric analyses were performed to determine “fold changes” of individual kinase-activation levels after normalization in post-therapy CLL cells relative to the pre-therapy samples according to the manufacturer’s instructions. Indeed, we detected increased activation of multiple intracellular kinases (Supplementary Table S4), including AKT (phosphorylation of S473/T308 was elevated in post-therapy CLL cells from P1, while there was no alteration or a subtle decrease of P-AKT at T308 in post-therapy CLL cells from P2), HCK (SRC family kinase), WNK1 (serine-threonine kinase), p70S6K and STAT5A/B in post-idelalisib CLL cells (Fig. 1).
Fig. 1. Post-idelalisib CLL cells express aberrantly activated signal mediators.
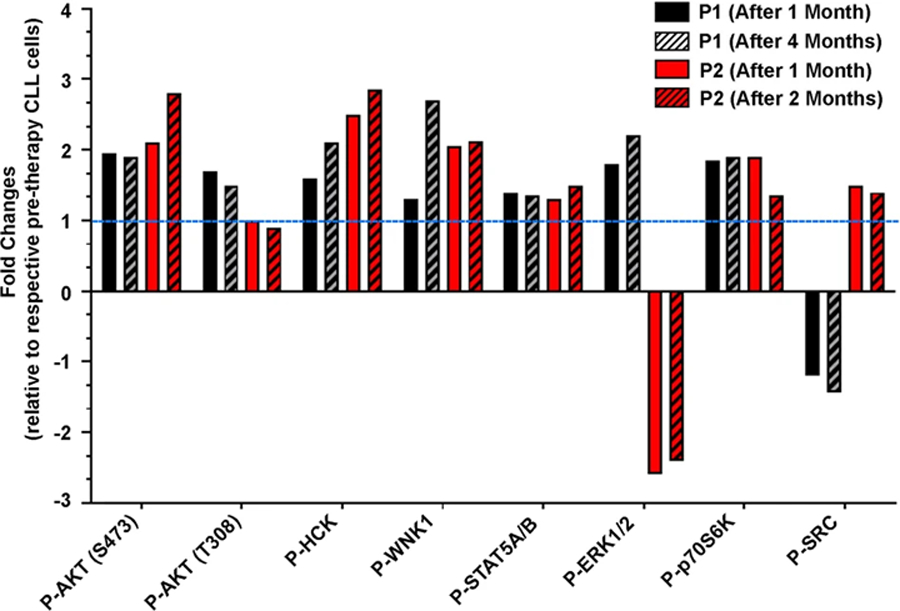
Lysates of pre-and post-therapy, follow-up CLL cells (as indicated) from patients (n = 2) treated with idelalisib-based therapies were analyzed for the activation of intracellular kinases using human phosphokinase antibody array blots following the manufacturer’s instructions. Densitometric analyses were performed and results of selected kinases are presented after normalization as per the manufacturer’s instructions as “fold-changes” relative to pre-therapy cells (value of which was taken as 1). The dotted horizontal line indicates P-AKT levels of the respective pre-therapy CLL cells.
Idelalisib-based therapy-induced increased AKT-activation in post-therapy CLL cells
Despite the fact that the PI3K/AKT signaling axis is a primary target of idelalisib therapy, we detected increased activating phosphorylation of AKT (S473) in post-idelalisib CLL cells obtained from two CLL patients (P1/P2) (Fig. 1). Interestingly, while increase of P-AKT (T308) levels in post-therapy CLL cells of P1 was discernible, we detected no effect or a subtle decrease of P-AKT (T308) in post-idelalisib CLL clones of P2 (Fig. 1). Thus, we validated these findings in pre-/post-therapy CLL cells obtained from a cohort of short-term follow-up CLL patients (n = 17) treated with idelalisib-based therapies including P1/P2 in western blot analysis. Indeed, increased activation of AKT at S473 and at T308 was discernible in post-therapy CLL cells from 11 of 17 and 8 of 15 short-term (up to 143 days) follow-up CLL patients, respectively (Fig. 2A, B). Interestingly, while cells from P4 showed inhibition of P-AKT at S473 and T308 by ~40% and ~80%, respectively, relative to pre-therapy cells; post-idelalisib CLL cells from P2, P9, P14 and P15 showed increased levels of P-AKT at S473 but either inhibition or no alteration of P-AKT levels at T308 (Fig. 2A).
Fig. 2. Post-idelalisib CLL cells show increased activation of AKT.

A Activating phosphorylation status of AKT (S473 and T308) was analyzed in lysates of pre-/post-therapy CLL cells from CLL patients treated with idelalisib-based therapies in western blots using specific antibodies to further validate the findings of increased AKT-activation shown in Fig. 1. Densitometric values of P-AKT (S473/T308) vs. total AKT in post-therapy CLL cells are presented as “fold-activation” relative to pre-therapy cells. “N/A” indicates that the lysates were not sufficient to analyze the activation status of P-AKT (T308). B AKT activation levels in pre-/post-therapy CLL cells of individual paired samples are plotted and presented. C Pre-/post-therapy CLL cells from a few CLL patients were further analyzed for the P-AKT (T308) by flow cytometric analysis using a phycoerythrin-conjugated specific antibody or an isotype control antibody. Results are presented as mean fluorescent intensity (MFI). C: Cycle of idelalisib therapy; D: Day.
In contrast to an earlier study which demonstrated inhibition of P-AKT (T308) levels in CLL cells collected at 8 and 28 days post-idelalisib therapy by flow cytometric analysis [16], we detected increased activation of AKT at both S473/T308 in post-therapy CLL cells at day-15, day-30 and beyond from majority of CLL patients treated with idelalisib in western blot analysis. Thus, phosphorylation status of AKT at T308 residue was further analyzed in pre-/post-therapy CLL cells from a few CLL patients (n = 3) using a specific-antibody to P-AKT (T308) (BD Biosciences) by flow cytometry. Results of flow cytometric assessment of P-AKT (T308) levels in post-idelalisib CLL clones supported our western blot findings (Fig. 2C). In addition, increased activation of AKT in some post-therapy CLL cells was also accompanied by elevated levels of P-Erk1/2 (Supplementary Fig. S1).
Analysis of post-therapy CLL clones from few long-term (≥72 weeks) follow-up CLL patients (P18 – P20) receiving idelalisib showed a similar pattern of increased activation of AKT (S473) and Erk1/2 (Fig. 3A). Additionally, a subtle to moderate increase of MYC and BCL2 expression levels was also discernible in few short-term (P7, P9, P11) and long-term (P18–P20), follow-up, post-idelalisib CLL cells as determined by western blot/ densitometric analyses (Fig. 3B). These findings were further supported by the increased levels of MYC/BCL2 mRNA in these post-idelalisib CLL clones, as assessed by qRT-PCR (Supplementary Fig. S2). Next, we interrogated why some post-therapy CLL cells showed increased activating phosphorylation of AKT despite continued treatment with idelalisib-based therapies.
Fig. 3. Impact of idelalisib-based treatment on AKT activation status in long-term follow-up CLL patients and its downstream targets, MYC and BCL2.
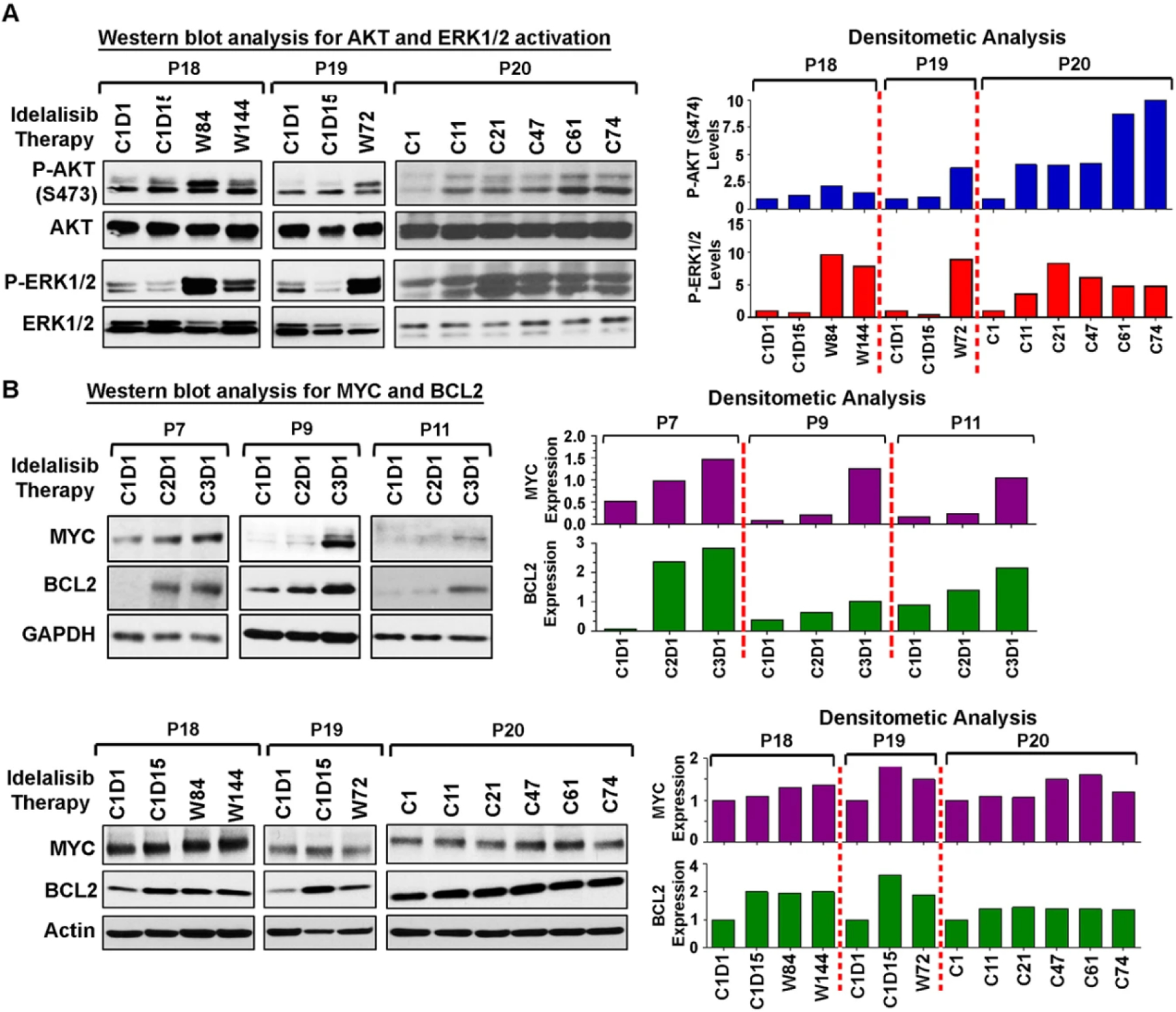
A AKT remains activated in long-term follow-up post-therapy CLL cells. Lysates of CLL cells obtained from pre- and long-term follow-up, post-idelalisib CLL patients (P18–P20) were analyzed to determine the activation status of AKT (S473) and Erk1/2 in western blots using phospho-specific antibodies. Total AKT and Erk1/2 were used as loading controls for normalization. Densitometric analyses were performed to determine the activation levels of AKT (S473) and Erk1/2 relative to total proteins (right panels). B MYC and BCL2 proteins are elevated in post-idelalisib CLL cells. Pre-/post-therapy CLL cell lysates used in Fig. 2 (P7, P9, P11) and above panel (P18–P20) were further analyzed for the expression of MYC and BCL2 in western blots using specific antibodies. GAPDH or Actin was used as loading control. Densitometric analyses were performed and results are presented as ratio of MYC or BCL2: GAPDH (P7, P9, P11) and “fold changes” (P18–P20) relative to pre-therapy cells (right panels). C: Cycle of idelalisib therapy; D: Day; W: Week.
Idelalisib treatment induced increased recruitment of PI3Kδ and PI3Kβ to the BCR signalosome via BCAP/CD19
To unravel the mechanism of increased AKT activation in post-idelalisib CLL cells, we, first, assessed the impact of idelalisib on AKT-activation status in MEC1 cells as a model. Indeed, idelalisib-treatment (0.2 µM) of MEC1 cells restored AKT-activation levels at S473 within 2–4 h after an initial inhibition (1–2 h), accompanied by activation of Erk1/2 after a transient inhibition with upregulation of MYC/BCL2 (Fig. 4A). A higher dose of idelalisib (1.0 µM) showed a similar pattern of P-AKT dynamics in MEC1 cells in a time-dependent manner (Supplementary Fig. S3A, upper panel). As P-AKT or P-Erk1/2 levels may alter during in vitro culture with time, we analyzed P-AKT levels in vehicle (DMSO)-treated MEC1 cells at different time points. While we did not find any significant alteration of P-AKT levels, a moderate increase of P-Erk1/2 levels was discernible in DMSO-treated MEC1 cells with time (Supplementary Fig. S3A, bottom panel). Additionally, MEC1 cells were also treated with 0.2 µM idelalisib for a shorter time intervals till 24 h under similar experimental conditions. Results demonstrate that idelalisib may exert its inhibitory effect on P-AKT within 30 min of treatment in MEC1 cells (Supplementary Fig. S3B). In total, these results corroborated well with our findings in post-therapy CLL cells from patients treated with idelalisib (Figs. 2 and 3).
Fig. 4. Idelalisib treatment induces increased recruitment of PI3Kδ and PI3Kβ to the BCR signalosome.
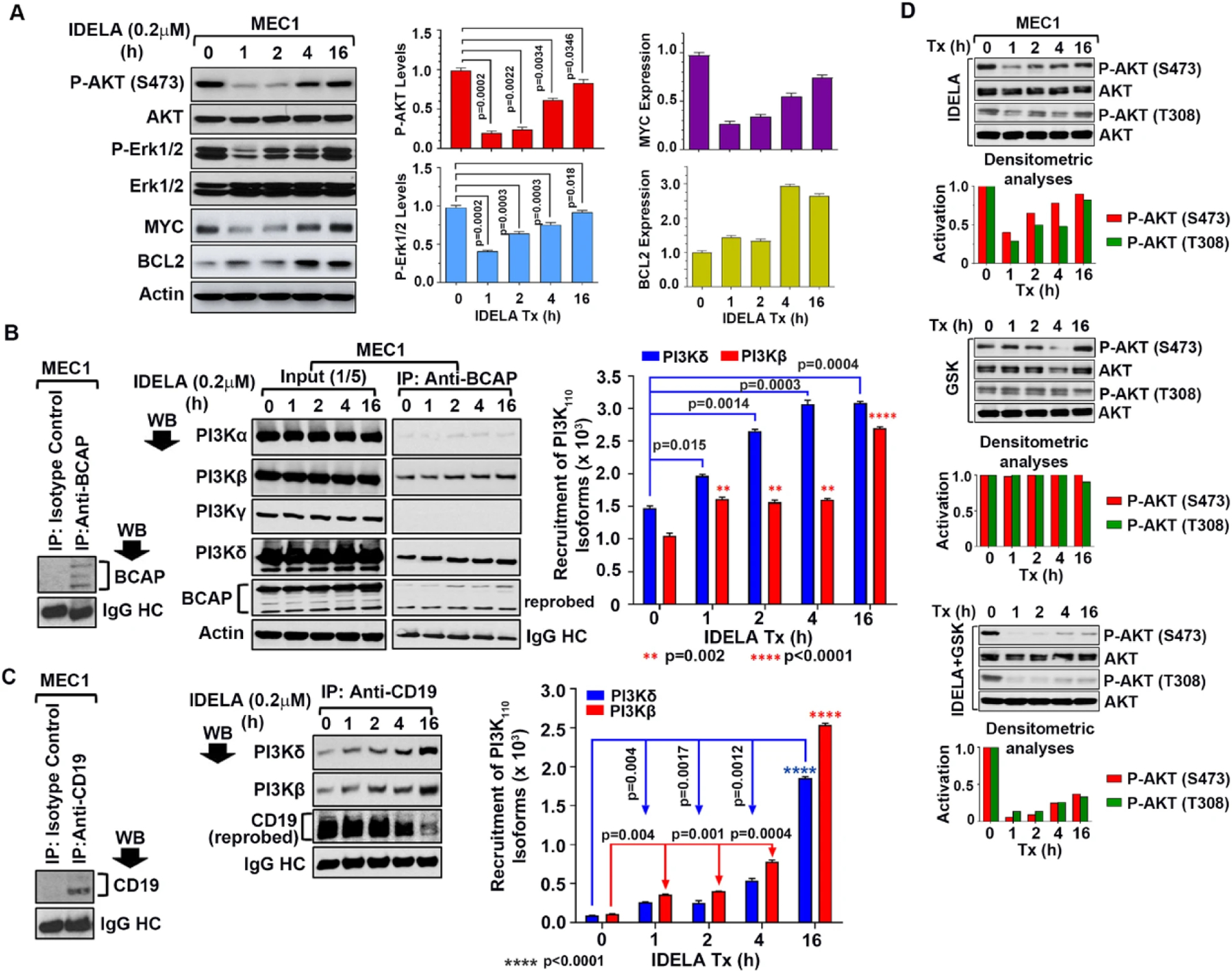
A Impact of idelalisib-treatment on AKT/Erk1/2 activation status and MYC/BCL2 expression levels in MEC1 cells. (Left panel) Synchronized MEC1 cells were treated with idelalisib (0.2 µM) for the indicated time periods. Cell lysates were analyzed for P-AKT (S473), P-Erk1/2 and expression levels of MYC/BCL2 in western blots using specific antibodies. Total AKT, Erk1/2 and actin were used as loading controls. Densitometric analyses for P-AKT/P-Erk1/2 levels and expression of MYC/BCL2 were also performed. Mean±SD values of 3 experiments are presented on right and bottom of the blots. B BCAP recruits increased levels of PI3Kδ and PI3Kβ in idelalisib-treated MEC1 cells. BCAP was immunoprecipitated from the idelalisib-treated MEC1 cell lysates used above (panel A) using a specific antibody to BCAP or an isotype control (IgG1), followed by western blot analysis of the immune complex to detect PI3Kα, PI3Kβ, PI3Kγ and PI3Kδ using specific antibodies. The blot was stripped and reprobed with an antibody to BCAP. IgG heavy chain (HC) was used as loading control. Densitometric analyses were performed to determine the levels of PI3K isoforms recruited by BCAP in idelalisib-treated MEC1 cells (right panel). Expression status of all the class-I PI3K isoforms and BCAP in idelalisib-treated MEC1 cells as determined in western blots is also shown on the left. C CD19 recruits increased levels of PI3Kδ and PI3Kβ in idelalisib-treated cells in a time-dependent manner. CD19 was immunoprecipitated from idelalisib-treated MEC1 cells used above using a specific antibody or an isotype control (IgG1), followed by western blot analysis of the immune complex to detect class-I PI3K-isoforms described in panel B. The blot was stripped and reprobed with an antibody to CD19. IgG HC was used as loading control. Recruitment status of PI3Kδ and PI3Kβ by CD19 was assessed by densitometric analyses. Results of a representative of 3 experiments are shown. D Combined inhibition of PI3Kδ and PI3Kβ reduced reactivation of AKT. MEC1 cells were treated with idelalisib (0.2 µM) or a high-affinity inhibitor of PI3Kβ (GSK2636771) as a single agent (0.2 µM) or in combination for the indicated time intervals. Cell lysates were analyzed for activation of AKT (S473/T308) in western blot analysis using phospho-specific antibodies. The blots were stripped and probed with an antibody to AKT and used as loading control. Desitometric analyses were performed to evaluate time-dependent activation status of AKT (P-AKT:AKT).
Both BCAP, a downstream adaptor molecule of the BCR, and CD19 participate in BCR-mediated activation of the PI3K/AKT signaling axis via recruitment of PI3Kδ [20, 21]. Thus, to investigate if idelalisib-induced AKT-reactivation was a result of increased recruitment of PI3Kδ or other PI3K-isoforms to the BCR, BCAP was immunoprecipitated from idelalisib-treated MEC1 cell lysates, followed by western blots to detect PI3Kα, PI3Kβ, PI3Kγ or PI3Kδ using specific antibodies. Indeed, increased recruitment of PI3Kδ together with the PI3Kβ-isoform, but not PI3Kα or PI3Kγ, by BCAP was detected in a time-dependent manner while expression of these PI3K-isoforms remained unaltered in idelalisib-treated MEC1 cells (Fig. 4B). Similarly, CD19 immunoprecipitation from the same idelalisib-treated MEC1 cell lysates showed increased association of CD19 only with PI3Kδ and PI3Kβ (Fig. 4C). However, isotype control antibody failed to immunoprecipitate BCAP or CD19 from the MEC1 cell lysates (Fig. 4B, C; left panels). Further analysis suggested that idelalisib did not have any impact on BCAP expression (Fig. 4B, Input panel and Supplementary Fig. S4) or CD19 levels in MEC1 cells (Supplementary Fig. S4). These findings indicated that time-dependent recruitment of PI3Kδ together with PI3Kβ to the BCR in response to idelalisib might be playing a part in restoring AKT-activation level in MEC1 cells. Thus, simultaneous targeting of PI3Kδ and PI3Kβ might inhibit AKT-reactivation. Indeed, we found that targeting PI3Kδ in combination with a PI3Kβ-inhibitor (GSK2636771) inhibited idelalisib-induced AKT-reactivation in MEC1 cells, while PI3Kβ-inhibitor didn’t exert any inhibitory effect on P-AKT levels as a single agent (Fig. 4D). Dual-inhibition of PI3Kδ and PI3Kγ by duvelisib, on the other hand, showed a similar pattern of AKT-reactivation as detected in cells treated with idelalisib as a single agent (Supplementary Fig. S5). It is also noteworthy that we did not find induction of any significant amount of cell death in idelalisib-treated MEC1 cells with or without PI3Kβ-inhibitor, even after 24 h. We detected >90% cell viability as determined by Trypan blue exclusion at all-time points.
To examine if we could find a similar pattern of PI3K-isoform recruitment in pre-/post-therapy CLL cells, BCAP or CD19 was immunoprecipitated from the lysates of CLL cells showing increased activation (P1, P2) or inhibition of P-AKT (P4, P5) in post-idelalisib clones (Fig. 2A), followed by western blots for different PI3K-isoforms. BCAP in post-therapy CLL cells from P1 showed significantly increased recruitment (C1D1 vs. C3D1; p = 0.0014) of PI3Kδ while reduced association with PI3Kβ (Fig. 5A). In post-idelalisib CLL cells from P2, BCAP was found to be associated with increased levels of both PI3Kδ and PI3Kβ, however the level of PI3Kδ-recruitment was significantly (C1D1 vs. C3D1; p = 0.0032) higher (Fig. 5A). On the other hand, CD19 in post-therapy CLL cells of P1 did not show any significant alteration of PI3Kδ/PI3Kβ recruitment relative to pre-therapy, except cells at C2D1 where increased association of CD19 with PI3Kβ was discernible (Fig. 5B). Similar to BCAP, CD19 also showed increased association primarily with PI3Kδ in post-idelalisib CLL cells of P2 (Fig. 5B). In contrast, recruitment of PI3Kδ by BCAP in post-therapy CLL cells (C3D1) of P4 was reduced significantly (p < 0.0001) as compared to that in pre-therapy leukemic cells (C1D1) (Fig. 5A); while CD19 recruited slightly higher amounts of both PI3Kδ (C3 vs. C1; p = 0.0216) and PI3Kβ (C3 vs. C1; p = 0.006) in post-therapy CLL clones relative to pre-therapy cells (Fig. 5B). As both CD19 and BCAP mediated recruitment of total amount of PI3K110 is critical for the activation level of the enzyme, it is tempting to speculate that the observed inhibition of P-AKT may be due to reduced amount of combined recruitment of PI3K-isoforms to the BCR in post-therapy CLL cells (C3D1) as compared to that in pre-therapy CLL clones (C1D1) resulting in its reduced activity. On the other hand, recruitment of PI3kδ/PI3Kβ by both BCAP (Fig. 5A, bottom panel) and CD19 (Fig. 5B, bottom panel) was reduced significantly in post-therapy vs. pre-therapy CLL cells of P5. Of note, the specificity of anti-CD19 and anti-BCAP antibodies used in these immunoprecipitation/ western blot analyses was validated using lysates of purified primary CLL cells and an isotype control antibody (Fig. 5A, B). Although the expression levels of class-I PI3K-isoforms remained unaltered in pre-/post-therapy CLL cells from individual patients (P1,P2,P4), relatively higher levels of PI3Kδ and PI3Kβ were detectable in these clones compared to PI3Kα or PI3Kγ (Fig. 5C). However, some pre-/post-therapy CLL cells (P5–P7) expressed relatively higher levels of PI3Kδ and PI3Kγ than PI3Kβ or PI3Kα (Supplementary Fig. S6), suggesting that these PI3K-isoforms may be differentially regulated in CLL cells. Together, these findings suggest that enhanced combined recruitment of PI3K-isoforms by BCAP/CD19 to the BCR signalosome may regulate AKT-activation in post-idelalisib CLL cells, at least in part.
Fig. 5. Idelalisib therapy promotes increased recruitment of PI3Kδ and PI3Kβ to the BCR signalosome in post-therapy primary CLL cells.
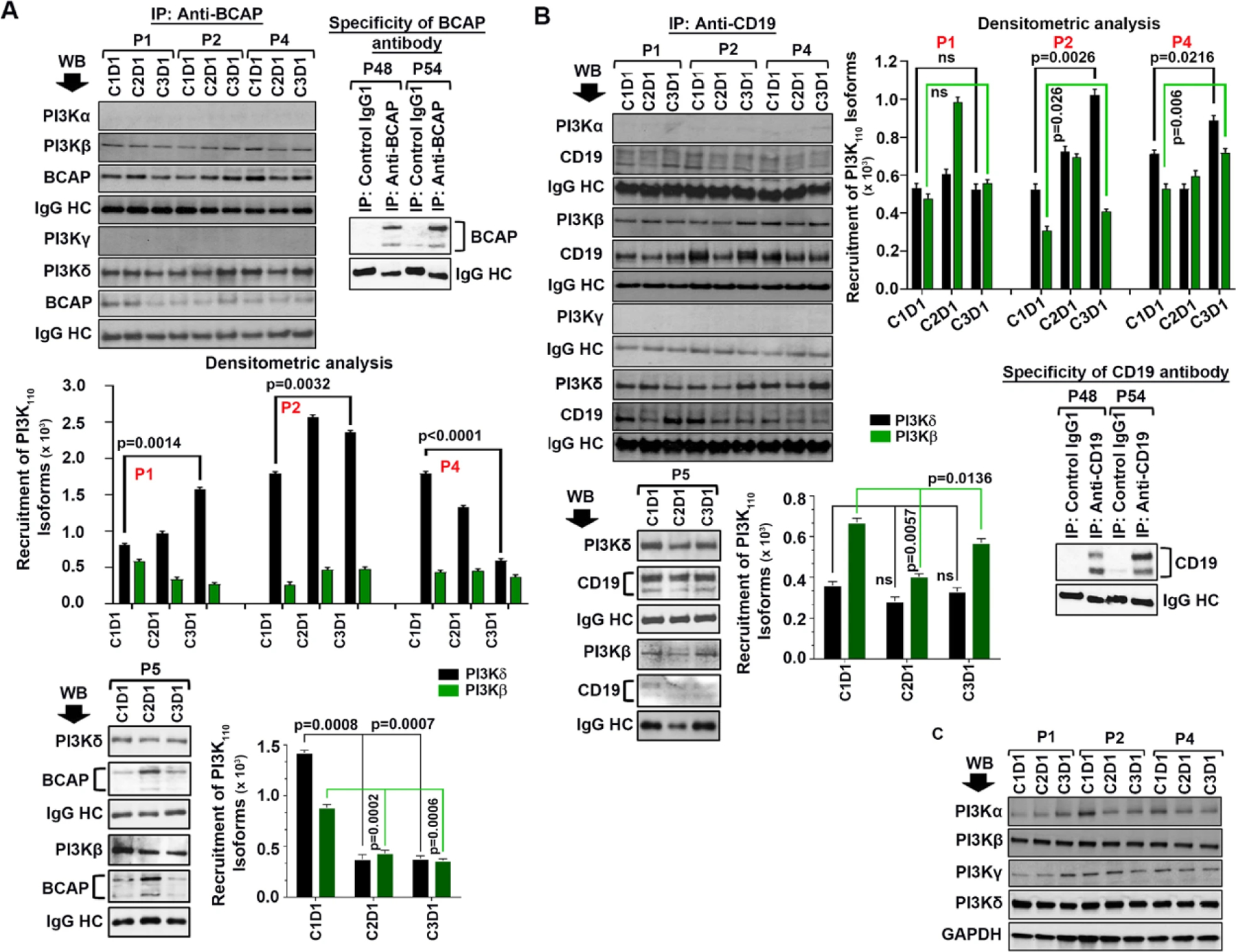
A BCAP recruits both PI3Kδ and PI3Kβ to the BCR in primary, post-idelalisib CLL cells. BCAP was immunoprecipitated from the pre- and post-idelalisib CLL cell lysates obtained from the CLL patients (P1, P2, P4, and P5) as indicated using an antibody to BCAP. To determine the specificity of the antibody, BCAP was also pulled-down from lysates of previously untreated CLL patients (P48, P54) using the same antibody or an isotype control antibody (IgG1), followed by western blot analysis to detect BCAP (right panel). Immune complexes were analyzed to detect all four class-I PI3K-isoforms in western blots as described above. The blots were stripped and reprobed with an antibody to BCAP. IgG HC was used as loading control. Amount of PI3K-isoforms (PI3Kδ and PI3Kβ) recruited by BCAP was quantified by densitometry and shown in the bottom panels with p-values as appropriate. B CD19 primarily recruits PI3Kδ and PI3Kβ to the BCR, not PI3Kα or PI3Kγ, in post-idelalisib CLL cells. As described in panel A above, CD19 was also immunoprecipitated from the same CLL cell lysates and performed western blot analyses to detect all four class-I PI3K-isofoms in the immune complexes. The blots were stripped and reprobed with an antibody to CD19. IgG HC was used as loading control. Specificity of the antibody was tested by immunoprecipitating CD19 from CLL cell lysates (P48, P54) used above in panel A or an isotype control antibody, followed by western blot analysis to detect CD19 (right bottom panel). Densitometric analyses were also performed to assess recruitment levels of PI3Kδ/PI3Kβ isofoms by CD19 in pre-/post-therapy CLL cells (right panels) with p-values as appropriate. C Expression of class-I PI3K-isoforms in pre-/post-therapy CLL clones remained unaltered. Pre-/post-idelalisib CLL cell lysates used above (P1, P2 and P4) were further analyzed to detect expression levels of PI3Kα, PI3Kβ, PI3Kγ and PI3Kδ in western blots using specific antibodies. GAPDH was used as loading control.
Idelalisib maintains prolonged inhibitory effect on P-AKT at T308, but not at S473
Further analysis of the results presented in Fig. 2 suggests that the inhibitory effect of idelalisib on P-AKT appears to be more pronounced on T308 phosphorylation than that on S473 in some post-idelalisib CLL cells (P2, P4, P9 and P14). To validate this observation, we treated MEC1 cells with idelalisib (0.2 µM) as described in Fig. 4, and determined the relative impact of the agent on P-AKT at S473 and T308 in western blot/densitometric analyses. Indeed, our results demonstrated that idelalisib exerted stronger inhibitory effect on P-AKT (T308) and maintained it for a relatively longer period of time than on P-AKT (S473) in MEC1 cells (Fig. 6A). Accumulation of PIP3 after being converted from PIP2 by PI3K recruits AKT to the plasma membrane where it is phosphorylated by mTORC2 and PDK1 at S473 and T308, respectively [22]. Thus, given our findings of increased recruitment of PI3Kδ/PI3Kβ to the BCR signalosome by BCAP/CD19 in idelalisib-treated cells, we further interrogated if PIP3 levels were also modulated during idelalisib-treatment. For this, MEC1 cells treated with idelalisib (used in panel A above) were stained with an antibody to PIP3 and subjected to confocal microscopy for detection. Indeed, we observed that regeneration of PIP3 started within 2–4 h of idelalisib-treatment after an initial inhibition and continued to increase in a time-dependent manner (Fig. 6B), parallel to the phosphorylation status of AKT at S473, but not with P-AKT at T308 (Fig. 6A).
Fig. 6. Measurement of PIP3 levels in idelalisib treated MEC1 cells.

A To determine the functional consequence of increased recruitment of PI3Kδ/PI3Kβ to the BCR complex (Fig. 4), MEC1 cells were treated with 0.2 µM idelalisib as described in Fig. 4A for the indicated time intervals. Cell lysates were analyzed for the activation status of AKT (S473/T308) in western blots using specific antibodies. The blots were stripped and probed with an antibody to AKT and used as loading control. Impact of idelalisib on AKT activation is shown by denistometric analyses (P-AKT: AKT) on right. B Accumulation of PIP3 in idelalisib-treated MEC1 cells was measured by staining the cells collected at the indicated time intervals following idelalisib-treatment with a specific antibody to PIP3, followed by incubation with an Alexa Fluor-594 conjugated secondary antibody. Cells were analyzed under confocal microscope (Leica) after counterstaining the cells with DAPI. Accumulation of PIP3 in idelalisib-treated MEC1 cells was quantified using the Leica Application Suite X (LAS X) software and presented the mean ± standard deviation (SD) of five random fields as relative fluorescent units (RFU) by bar diagrams. Magnification: x400.
Next, we determined PIP3 accumulation status in pre-/post-idelalisib, primary CLL cells from few randomly selected CLL patients (P6, P8, P9) as described above. Quantification of PIP3 levels demonstrated increased accumulation of the lipid in post-idelalisib CLL cells obtained from all three patients (Fig. 7). While increased AKT activation at both S473 and T308 was discernible in the post-idelalisib CLL cells obtained from P6 and P8, post-therapy cells from P9 showed increased activation of AKT at S473 with reduced phosphorylation at T308 despite elevated levels of PIP3. Together, these findings posed the questions: (i) how did idelalisib exert stronger inhibitory effect on AKT-phosphorylation at T308 in some CLL clones and (ii) why reactivation of AKT at T308 was delayed in idelalisib-treated MEC1 cells despite increased PIP3 accumulation?
Fig. 7. Post-idelalisib CLL cells showed increased accumulation of PIP3.
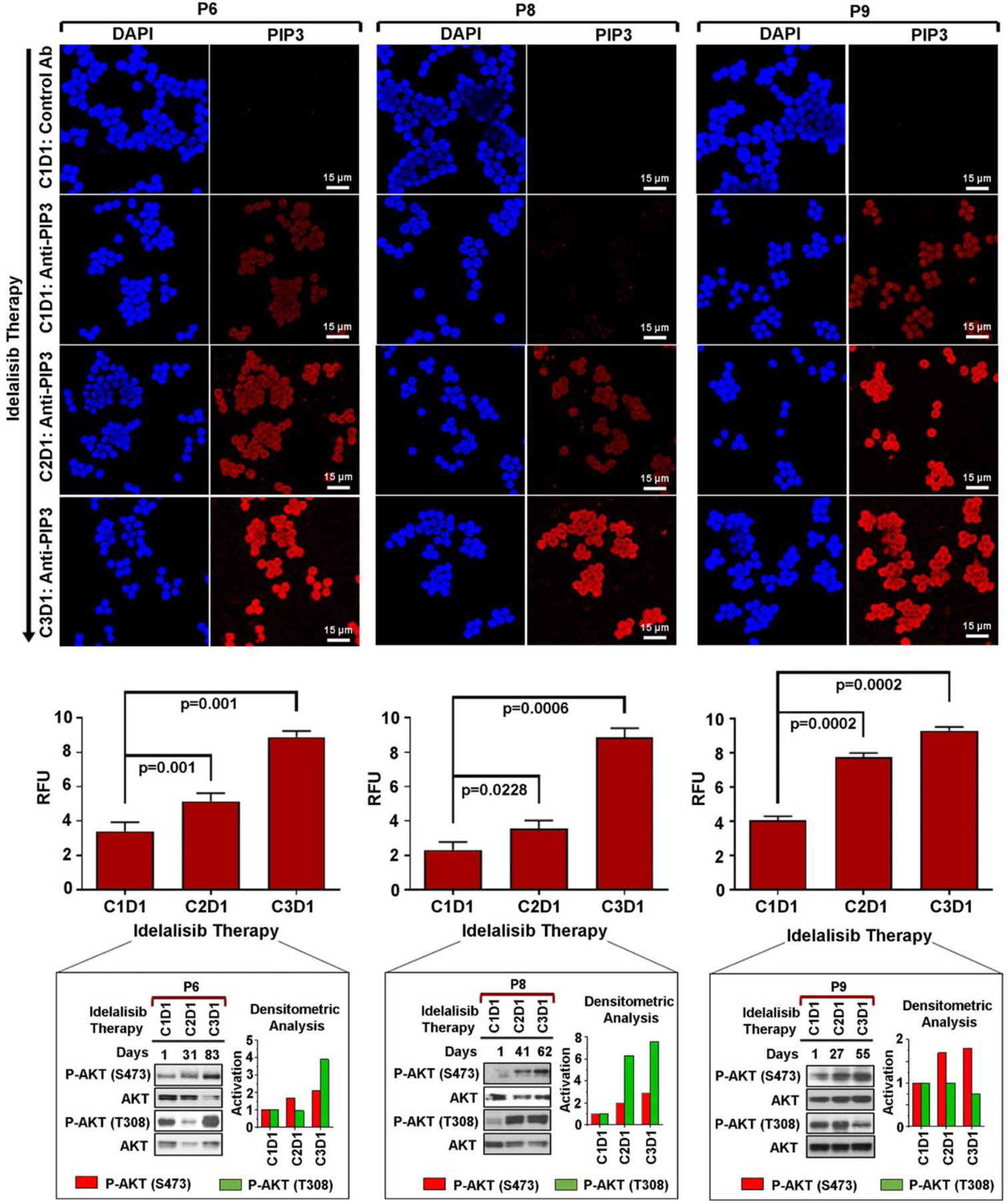
Pre-/post-idelalisib CLL cells from CLL patients (P6, P8, P9) treated with idelalisib-based therapies were stained with an antibody to PIP3 as described in Fig. 6B. Nuclear staining was performed using DAPI (blue). Cells were analyzed under the confocal microscope (Leica; Magnification: x400). Accumulation of PIP3 (red color) in pre-/post-idelalisib CLL cells was quantified using the LAS X software and presented the mean value ± SD of five random fields as relative fluorescent units (RFU) by bar diagrams (middle panels). P-values were determined to show differences of PIP3 accumulation in pre- vs. post-idelalisib CLL cells. Activation status of AKT (S473/T308) in these CLL cells is also shown with densitometric analyses (obtained from Fig. 2A) for comparison. C: Cycle of idelalisib; D: Day.
Idelalisib treatment reduces PDK1 protein level
To address the above questions, we hypothesized that constitutively active PDK1 (S241) could be another target of idelalisib. Thus, lysates of MEC1 cells treated with idelalisib (used in Fig. 6A) were further analyzed for the catalytic activation status of PDK1 in western blot using a phospho-PDK1 (S241) antibody. Additionally, P-mTOR (S2448) levels were also determined. Densitometric quantification showed a time-dependent inhibition of both P-PDK1 (S241) (relative to GAPDH) and P-mTOR (relative to total mTOR) in idelalisib-treated MEC1 cells (Fig. 8A). Interestingly, idelalisib-treatment reduced PDK1 protein levels (as obtained after stripping and reprobing the P-PDK1 blot with a specific antibody to PDK1, and quantified as relative to GAPDH). Thus, reduction of PDK1 level upon idelalisib treatment might be causing its reduced catalytic activity in MEC1 cells; while total mTOR expression levels remained unaltered under the similar experimental conditions (Fig. 8A). To rule out the possibility that the observed reduction of PDK1 protein levels was not due to an incomplete stripping/probing of the P-PDK1 blot, we further analyzed PDK1 protein level in idelalisib-treated MEC1 cell lysates used in panel A in a separate western blot experiment. Indeed, we detected substantial reduction of PDK1 protein levels in MEC1 cells upon idelalisib-treatment in a time-dependent manner (Supplementary Fig. S7A) as observed in panel A. To further analyze the dynamics of PDK1 expression in response to idelalisib, lysates of MEC1 cells treated with idelalisib (0.2 μM) for a shorter period of time till 24 h (used in Supplementary Fig. S3B) were examined for PDK1 expression in western blot. We detected reduction of PDK1 level in MEC1 cells as early as 30 min following idelalisib-treatment (Supplementary Fig. S7B). Taken together, our findings suggest that idelalisib may also target PDK1 affecting directly P-AKT (T308) level.
Fig. 8. Idelalisib reduces PDK1 protein level.

A Idelalisib-treated MEC1 cell lysates (obtained at different time intervals) used in Fig. 6A were further analyzed for the levels of P-PDK1 (S241) and P-mTOR (S2448) in western blots using phospho-specific antibodies. The blots were stripped and reprobed with an antibody to PDK1 or mTOR, and used as loading control. GAPDH was used as an internal control. Levels of P-PDK1 with respect to total PDK1 were calculated and presented below the PDK1 blot. In addition, levels of P-PDK1 and PDK1 expression with respect to GAPDH were also determined by densitometry and presented the mean ± SD values of 3 experiments by bar diagram with statistical analyses (upper right). P-mTOR levels were calculated as P-mTOR:mTOR and mean ± SD values are presented by bar diagram with statistical analyses (lower right). Similarly, total mTOR levels were determined by the ratio of mTOR and GAPDH. B Activation and expression levels of PDK1 were similarly analyzed in the lysates of pre-/post-idelalisib CLL cells from CLL patients (P1, P4, P6, P7, P9, P10, P14) treated with idelalisib-based therapies (used in Fig. 2) in western blots. Levels of P-PDK1 (P-PDK1:GAPDH) and PDK1 (PDK1:GAPDH) are shown by densitometric analyses below individual patient’s panel (bar diagrams). C Semi-quantitative RT-PCR was performed in idelalisib-treated MEC1 cells used in panel A to determine PDK1 mRNA levels using a specific set of primers. GAPDH was used as internal control. Densitometric values of PDK1 mRNA in idelalisib-treated MEC1 cells relative to GAPDH are shown at the bottom. D Pre-/post-idelalisib CLL cells from a few CLL patients (P4, P9, P10) used in panel C were also analyzed to determine PDK1 mRNA levels by quantitative RT-PCR. GAPDH was used as internal control. ΔCt values were calculated. Results are presented as “fold changes” relative to pre-therapy cells (C1D1).
Similarly, post-idelalisib CLL cells from P4 and P14 showed reduction of both P-PDK1 and PDK1, albeit at variable levels, (Fig. 8B) with reduced phosphorylation of AKT at T308 (Fig. 2A), while the cells from other patients (P1, P6, P7, P10) did not show any significant alteration of P-PDK1 levels (Fig. 8B). Of note, post-therapy leukemic cells from these later CLL patients showed increased activation of P-AKT (S473/T308) (Fig. 2A). To further evaluate if idelalisib-mediated reduction of PDK1 protein levels in MEC1 cells was as a result of down-regulation of its transcription, a semi-quantitative RT-PCR for PDK1 mRNA levels was performed. Interestingly, we did not find any significant modulation of PDK1 transcript levels in idelalisib-treated MEC1 cells (Fig. 8C). PDK1 mRNA levels in pre-/post-idelalisib CLL cells also remained unaltered as determined by qRT-PCR analysis (Fig. 8D). Together, these findings suggest that idelalisib may target PDK1 protein synthesis, not transcription, which warrants further investigation.
DISCUSSION
Cancer cells are quite adept at rewiring signaling pathways in response to targeted therapies in order to keep PI3K/AKT or other signaling axes active for their extended survival. Idelalisib mediated inhibition of PI3Kδ in CLL cells induces apoptosis and suppresses AKT-phosphorylation [15, 23]. In addition to blocking the BCR signal, PI3Kδ-inhibition interrupts CXCR4/CXCR5 signaling and subsequently, CLL cell homing, causing redistribution of these cells into the circulation, removal from the lymph node microenvironment’s pro-survival signals and sensitization to apoptosis [23]. While mutations associated with resistance to BTK inhibitors have been identified, limited data are available on mechanisms of resistance to PI3Kδ inhibitors. Upon BCR ligation, LYN and SYK are brought into proximity with the receptor to phosphorylate CD19 and/or BCAP, thus creating a SRC-homology 2 (SH2)-binding domain capable of binding SH2-domain proteins [7, 24, 25] including the regulatory subunit of PI3K and allowing activation of class-IA PI3K [26]. Interestingly, in activated B-cell-like diffuse large B-cell lymphoma cells, idelalisib-mediated PI3Kδ-inhibition bypassed PI3Kδ-engagement and instead, recruited the PI3Kα-isoform via BCAP and CD19 to reactivate AKT [27]. While in CLL cells, we detected that idelalisib-induced AKT-activation did not bypass PI3Kδ-recruitment to BCAP or CD19 but rather increased significantly the recruitment levels of PI3Kδ along with PI3Kβ to the BCR complex in a time-dependent manner. Another explanation of AKT-reactivation in MEC1 cells following idelalisib-treatment in vitro could be a decrease in the concentration of active drug at later time points. However, this later hypothesis could be argued by the findings that ex vivo primary, post-idelalisib CLL cells showed increased activation of AKT despite continuation of idelalisib therapy. Moreover, re-challenge of post-idelalisib MEC1 cells with fresh drug did not alter AKT-reactivation status (Supplementary Fig. S8), suggesting that reactivation of AKT upon idelalisib-therapy may be due to increased mobilization of PI3Kδ/PI3Kβ to BCR, at least in part. Structural studies have suggested that compared with p110α, PI3Kβ is less inhibited by the p85-subunit supplying a basal PIP3 level [28–30], and may explain why wild-type PI3Kβ can be oncogenic when it is overexpressed [31, 32].
The most common regulatory p85 subunit of the heterodimeric enzyme PI3K controls the catalytic activity of the p110 subunit of PI3K, and mediates its interactions with various receptor tyrosine kinases through a physical recognition between its two SH2 domains (the N-SH2 and C-SH2 domains, separated by an interSH2 coiled coil region) and a consensus pYxxM sequence [33]. The inhibitory activity of p85 upon the catalytic subunit PI3K110 can be relieved via tyrosine phosphorylation of the N-SH2 domain of p85 by phosphorylated tyrosine. Thus, we further examined idelalisib-treated MEC1 cells and pre-/post-idelalisib CLL cells for the p85 tyrosine phosphorylation status in western blots using a P-p85 (Y467) antibody. We did not find any significant alteration of P-p85 levels in idelalisib-treated MEC1 cells over time (Supplementary Fig. S9A). However, pre-/post-idelalisib CLL cells showed variable levels of P-p85 independent of their AKT activation status (Supplementary Fig. S9B), suggesting that increased activation of AKT in response to idelalisib may not depend on p85-phosphorylation levels. In-depth investigation may shed light on precise role of tyrosine phosphorylation of p85 in regulating PI3K110 catalytic activity in CLL cells. Interestingly, aberrant activation of Erk1/2 in some post-idelalisib CLL clones was attributed to upregulation of IGF1Rβ in a recent study [34] and to pathway activating mutations [35].
Anti-CD20 antibody therapies may also induce a BCR-like signaling including activation of AKT [36]. In addition, CD20 molecule has been shown to be directly involved in BCR signaling pathway [37]. However, induction of increased activation of AKT and Erk1/2 in post-therapy CLL cells seems to be a result of compensatory pathway activation by idelalisib. This statement is supported by the findings that increased activation of AKT/Erk1/2 occurred in some post-therapy CLL cells from patients treated with either idelalisib as a single agent or in combination with ofatumumab (Supplementary Table S1). In addition, MEC1 cells treated with idelalisib alone showed similar pattern of AKT/Erk1/2 activation.
A recent study showed increase of P-AKT (S473) levels in CLL cells treated with another BCR inhibitor, ibrutinib, providing evidence of a similar compensatory mechanism although the underlying mechanism might be different [38]. Using MEC1 cells as a model, we observed reactivation of AKT via increased phosphorylation at both S473 and T308 residues after an initial inhibition following in vitro treatment with idelalisib in a time-dependent manner. Further analysis suggested that idelalisib-treatment had stronger inhibitory effect on P-AKT (T308) than at S473, despite a time-dependent, enhanced accumulation of PIP3 in MEC1 cells. Indeed, elevated levels of PIP3 in post-idelalisib CLL cells (C2D1/C3D1) of P9 showed increase of P-AKT (S473) level with no alteration or inhibition of P-AKT levels at T308. Together, these findings prompted us to postulate that idelalisib might also target PDK1 catalytic activity to directly modulate the phosphorylation status of AKT at T308. Further analysis of idelalisib-treated MEC1 cells or some post-idelalisib primary CLL cells showed reduction of PDK1 protein levels compared to that in pre-therapy cells, which could lead to reduced catalytic activity of PDK1 resulting in relatively prolonged inhibitory effect on P-AKT (T308), despite PIP3 accumulation. As the mRNA levels of PDK1 in idelalisib-treated MEC1 cells or post-idelalisib CLL cells remained unaltered, we believe that idelalisib may inhibit PDK1 protein synthesis likely via time-dependent inhibition of the AKT/mTORC1/p70S6K/S6K signaling axis. Yang et al. previously detected significant decreases of global protein synthesis with idelalisib treatment both in mantel cell lymphoma (MCL) cell lines and primary cells [39]. This inhibitory effect on protein translation was likely mediated by PI3K/AKT/mTOR and MAPK pathways as the primary targets of idelalisib in MCL cells [39]. Thus, reduction of PDK1 protein levels in idelalisib-treated MEC1 cells or post-idelalisib primary CLL cells may likely be a consequence of the inhibitory effect of idelalisib on translational signaling machineries. However, further study is needed to unravel the mechanism of specific inhibition of PDK1 protein level in CLL cells.
A crucial node of the PI3K pathway is represented by the serine/threonine kinase PDK1. PDK1 contains a phosphorylation site within the activation loop (serine 241), which is constitutively phosphorylated by an autophosphorylation reaction in trans [40]. PDK1 kinase is therefore considered constitutively active. However, the regulation of PDK1-activated signaling pathways is based on different mechanisms [41]. The first mechanism is phosphorylation of AKT activation loop (T308). PDK1 localizes at the plasma membrane due to the interaction of its PH domain with PIP3 produced by PI3K and thus, physically interacts with and phosphorylates AKT [42]. The second mechanism of activation for substrates lacking a PH domain (p70S6K, serum and glucocorticoid-inducible kinase, p90RSK and PKCs) is PIP3-independent. On the kinase domain, PDK1 possesses a hydrophobic pocket, termed the PDK1 interacting fragment (PIF) pocket, which allows its interaction with the phosphorylated hydrophobic motif of the targeted kinases and the consequent phosphorylation of their activation loop [43–45]. Thus, PDK1 plays a critical role in normal cellular physiology and development. Our findings that idelalisib-treatment reduces PDK1 protein levels in CLL cells directly or indirectly may result in inhibition of cancer cell growth and survival via inhibition of multiple kinase signals including that of protein synthesis.
In summary, we discovered increased activation of AKT in post-idelalisib CLL cells from majority of CLL patients. This observation came as a surprise as these patients were on idelalisib-based therapies. Idelalisib-induced activation of AKT in post-therapy CLL cells may likely be as a result of increased recruitment of PI3Kδ and/or PI3Kβ to the BCR complex via BCAP/CD19. These findings are supported by increased accumulation of PIP3 which acts as a lipid second messenger recruiting AKT, PDK1 and mTORC2 to the membrane via interaction with the PH domain. However, our data also suggest that idelalisib-treatment may reduce PDK1 protein level resulting in reduced activity of PDK1 and thus, relatively stronger inhibition on P-AKT (T308) independent of PIP3 level in CLL cells (Supplementary Fig. S10). In total, given our findings we believe that combined inhibition of PI3Kδ and PI3Kβ in CLL cells using pharmacologic targeted-agents may prove beneficial for improving durable response in CLL patients.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
We thank all the CLL patients who participated in this study. This work was supported partly by a research fund from the National Cancer Institute (CA170006) and a Presbyterian Health Foundation (C5126601) Bridge grant to AKG. We also acknowledge the support received from the NCI Cancer Center Support Grant (P30 CA225520). In addition, this study was also supported in part by the Oklahoma Tobacco Settlement Endowment Trust awarded to the University of Oklahoma//Stephenson Cancer Center. JRB was supported by NIH R01 CA 213442.
Footnotes
COMPETING INTERESTS
JRB has served as a consultant for Abbvie, Acerta, Astra-Zeneca, Beigene, Catapult, Dynamo Therapeutics, Eli Lilly, Juno/Celgene, Kite, MEI Pharma, Nextcea, Novartis, Octapharma, Pfizer, Rigel, Sunesis, TG Therapeutics, Verastem; received honoraria from Janssen; received research funding from Gilead, Loxo, Sun and Verastem; and served on data safety monitoring committees for Invectys. All other authors declare that there are no competing financial interests.
ADDITIONAL INFORMATION
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41375-022-01595-0.
Reprints and permission information is available at http://www.nature.com/reprints
REFERENCES
- 1.Burger JA, Chiorazzi N. B cell receptor signaling in chronic lymphocytic leukemia. Trends Immunol 2013;34:592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maffei R, Fiorcari S, Martinelli S, Potenza L, Luppi M, Marasca R. Targeting neoplastic B cells and harnessing microenvironment: the “double face” of ibrutinib and idelalisib. J Hematol Oncol 2015;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ten Hacken E, Gounari M, Ghia P, Burger JA. The importance of B cell receptor isotypes and stereotypes in chronic lymphocytic leukemia. Leukemia 2019;33:287–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lucas CL, Chandra A, Nejentsev S, Condliffe AM, Okkenhaug K. PI3Kdelta and primary immunodeficiencies. Nat Rev Immunol 2016;16:702–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer 2015;15:7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herman SE, Johnson AJ. Molecular pathways: Targeting phosphoinositide 3-kinase p110-delta in chronic lymphocytic leukemia. Clin Cancer Res 2012;18:4013–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okkenhaug K, Vanhaesebroeck B. PI3K in lymphocyte development, differentiation and activation. Nat Rev Immunol 2003;3:317–30. [DOI] [PubMed] [Google Scholar]
- 8.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet 2006;7:606–19. [DOI] [PubMed] [Google Scholar]
- 9.Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell 2017;170:605–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, et al. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 1996;15:6541–51. [PMC free article] [PubMed] [Google Scholar]
- 11.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol 1997;7:261–9. [DOI] [PubMed] [Google Scholar]
- 12.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005;307:1098–101. [DOI] [PubMed] [Google Scholar]
- 13.Manning BD, Toker A. AKT/PKB signaling: Navigating the network. Cell 2017;169:381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lannutti BJ, Meadows SA, Herman SE, Kashishian A, Steiner B, Johnson AJ, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood 2011;117:591–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herman SE, Gordon AL, Wagner AJ, Heerema NA, Zhao W, Flynn JM, et al. Phosphatidylinositol 3-kinase-delta inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood 2010;116:2078–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown JR, Byrd JC, Coutre SE, Benson DM, Flinn IW, Wagner-Johnston ND, et al. Idelalisib, an inhibitor of phosphatidylinositol 3 kinase p110delta, for relapsed/refractory chronic lymphocytic leukemia. Blood 2014;123:3390–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stacchini A, Aragno M, Vallario A, Alfarano A, Circosta P, Gottardi D, et al. MEC1 and MEC2: Two new cell lines derived from B-chronic lymphocytic leukaemia in prolymphocytoid transformation. Leuk Res 1999;23:127–36. [DOI] [PubMed] [Google Scholar]
- 18.Mahmud H, Mendez M, Mukhopadhyay B, Holter-Chakrabarty J, Ghosh AK. HSP90 overexpression potentiates the B-cell receptor and fibroblast growth factor receptor survival signals in chronic lymphocytic leukemia cells. Oncotarget 2020;11:2037–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maiti GP, Sinha S, Mahmud H, Boysen J, Mendez MT, Vesely SK, et al. SIRT3 overexpression and epigenetic silencing of catalase regulate ROS accumulation in CLL cells activating AXL signaling axis. Blood. Cancer J 2021;11:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rickert RC. New insights into pre-BCR and BCR signalling with relevance to B cell malignancies. Nat Rev Immunol 2013;13:578–91. [DOI] [PubMed] [Google Scholar]
- 21.Aiba Y, Kameyama M, Yamazaki T, Tedder TF, Kurosaki T. Regulation of B-cell development by BCAP and CD19 through their binding to phosphoinositide 3-kinase. Blood 2008;111:1497–503. [DOI] [PubMed] [Google Scholar]
- 22.Chu N, Salguero AL, Liu AZ, Chen Z, Dempsey DR, Ficarro SB, et al. Akt kinase activation mechanisms revealed using protein semisynthesis. Cell 2018;174:897–907.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoellenriegel J, Meadows SA, Sivina M, Wierda WG, Kantarjian H, Keating MJ, et al. The phosphoinositide 3’-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood 2011;118:3603–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Niiro H, Clark EA. Regulation of B-cell fate by antigen-receptor signals. Nat Rev Immunol 2002;2:945–56. [DOI] [PubMed] [Google Scholar]
- 25.Dal Porto JM, Gauld SB, Merrell KT, Mills D, Pugh-Bernard AE, Cambier J. B cell antigen receptor signaling 101. Mol Immunol 2004;41:599–613. [DOI] [PubMed] [Google Scholar]
- 26.Geering B, Cutillas PR, Vanhaesebroeck B. Regulation of class IA PI3Ks: Is there a role for monomeric PI3K subunits? Biochem Soc Trans 2007;35:199–203. [DOI] [PubMed] [Google Scholar]
- 27.Pongas GN, Annunziata CM, Staudt LM. PI3Kdelta inhibition causes feedback activation of PI3Kalpha in the ABC subtype of diffuse large B-cell lymphoma. Oncotarget 2017;8:81794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dbouk HA, Pang H, Fiser A, Backer JM. A biochemical mechanism for the oncogenic potential of the p110beta catalytic subunit of phosphoinositide 3-kinase. Proc Natl Acad Sci USA 2010;107:19897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang X, Vadas O, Perisic O, Anderson KE, Clark J, Hawkins PT, et al. Structure of lipid kinase p110beta/p85beta elucidates an unusual SH2-domain-mediated inhibitory mechanism. Mol Cell 2011;41:567–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vogt PK. PI3K p110beta: More tightly controlled or constitutively active? Mol Cell 2011;41:499–501. [DOI] [PubMed] [Google Scholar]
- 31.Kang S, Denley A, Vanhaesebroeck B, Vogt PK. Oncogenic transformation induced by the p110beta, -gamma, and -delta isoforms of class I phosphoinositide 3-kinase. Proc Natl Acad Sci USA 2006;103:1289–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Denley A, Kang S, Karst U, Vogt PK. Oncogenic signaling of class I PI3K isoforms. Oncogene 2008;27:2561–74. [DOI] [PubMed] [Google Scholar]
- 33.Visconti L, Malagrino F, Toto A, Gianni S. The kinetics of folding of the NSH2 domain from p85. Sci Rep 2019;9:4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scheffold A, Jebaraj BMC, Tausch E, Bloehdorn J, Ghia P, Yahiaoui A, et al. IGF1R as druggable target mediating PI3K-delta inhibitor resistance in a murine model of chronic lymphocytic leukemia. Blood 2019;134:534–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ishwarya M, Siddha K, Aishath N, Svitlana T, Jasneet KK, Emily MT, et al. Activating MAPK pathway mutations mediate primary resistance to PI3K inhibitors in Chronic Lymphocytic Leukemia (CLL). Blood 2018;132(Supplement 1):587.29884741 [Google Scholar]
- 36.Edelmann J, Dokal AD, Vilventhraraja E, Holzmann K, Britton D, Klymenko T, et al. Rituximab and obinutuzumab differentially hijack the B cell receptor and NOTCH1 signaling pathways. iScience 2021;24:102089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pavlasova G, Borsky M, Svobodova V, Oppelt J, Cerna K, Novotna J, et al. Rituximab primarily targets an intra-clonal BCR signaling proficient CLL subpopulation characterized by high CD20 levels. Leukemia 2018;32:2028–31. [DOI] [PubMed] [Google Scholar]
- 38.Seda V, Vojackova E, Ondrisova L, Kostalova L, Sharma S, Loja T, et al. FoxO1-GAB1 axis regulates homing capacity and tonic AKT activity in chronic lymphocytic leukemia. Blood 2021;138:758–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang Q, Chen LS, Ha MJ, Do KA, Neelapu SS, Gandhi V. Idelalisib impacts cell growth through inhibiting translation-regulatory mechanisms in mantle cell lymphoma. Clin Cancer Res 2017;23:181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wick MJ, Ramos FJ, Chen H, Quon MJ, Dong LQ, Liu F. Mouse 3-phosphoinositide-dependent protein kinase-1 undergoes dimerization and trans-phosphorylation in the activation loop. J Biol Chem 2003;278:42913–9. [DOI] [PubMed] [Google Scholar]
- 41.Mora A, Komander D, van Aalten DM, Alessi DR. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol 2004;15:161–70. [DOI] [PubMed] [Google Scholar]
- 42.Currie RA, Walker KS, Gray A, Deak M, Casamayor A, Downes CP, et al. Role of phosphatidylinositol 3,4,5-trisphosphate in regulating the activity and localization of 3-phosphoinositide-dependent protein kinase-1. Biochem J 1999;337:575–83. [PMC free article] [PubMed] [Google Scholar]
- 43.Balendran A, Casamayor A, Deak M, Paterson A, Gaffney P, Currie R, et al. PDK1 acquires PDK2 activity in the presence of a synthetic peptide derived from the carboxyl terminus of PRK2. Curr Biol 1999;9:393–404. [DOI] [PubMed] [Google Scholar]
- 44.Biondi RM, Cheung PC, Casamayor A, Deak M, Currie RA, Alessi DR. Identification of a pocket in the PDK1 kinase domain that interacts with PIF and the C-terminal residues of PKA. EMBO J 2000;19:979–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Collins BJ, Deak M, Arthur JS, Armit LJ, Alessi DR. In vivo role of the PIF-binding docking site of PDK1 defined by knock-in mutation. EMBO J 2003;22:4202–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.