

Abstract
The prefrontal cortex is a crucial regulator of alcohol drinking, and dependence, and other behavioral phenotypes associated with AUD. Comprehensive identification of cell-type specific transcriptomic changes in alcohol dependence will improve our understanding of mechanisms underlying the excessive alcohol use associated with alcohol dependence and will refine targets for therapeutic development. We performed single nucleus RNA sequencing (snRNA-seq) and Visium spatial gene expression profiling on the medial prefrontal cortex (mPFC) obtained from C57BL/6 J mice exposed to the two-bottle choice-chronic intermittent ethanol (CIE) vapor exposure (2BC-CIE, defined as dependent group) paradigm which models phenotypes of alcohol dependence including escalation of alcohol drinking. Gene co-expression network analysis and differential expression analysis identified highly dysregulated co-expression networks in multiple cell types. Dysregulated modules and their hub genes suggest novel understudied targets for studying molecular mechanisms contributing to the alcohol dependence state. A subtype of inhibitory neurons was the most alcohol-sensitive cell type and contained a downregulated gene co-expression module; the hub gene for this module is Cpa6, a gene previously identified by GWAS to be associated with excessive alcohol consumption. We identified an astrocytic Gpc5 module significantly upregulated in the alcohol-dependent group. To our knowledge, there are no studies linking Cpa6 and Gpc5 to the alcohol-dependent phenotype. We also identified neuroinflammation related gene expression changes in multiple cell types, specifically enriched in microglia, further implicating neuroinflammation in the escalation of alcohol drinking. Here, we present a comprehensive atlas of cell-type specific alcohol dependence mediated gene expression changes in the mPFC and identify novel cell type-specific targets implicated in alcohol dependence.
Keywords: Alcohol dependence, Chronic intermittent ethanol exposure, Alcohol dependence cell-type specific responses, Single nucleus RNA sequencing, Spatial transcriptomics, Multimodal data integration, Gene co-expression networks
1. Introduction
Prefrontal cortex (PFC) is involved in executive function and reward circuitry (Ball et al., 2011), and is a crucial regulator of escalation of alcohol drinking and dependence (Abernathy et al., 2010; Heilig et al., 2017). We and others have shown transcriptomic changes in postmortem brain samples from alcohol-dependent individuals (Warden and Dayne Mayfield, 2017; Flatscher-Bader et al., 2005) as well as animal models of alcohol consumption and dependence (Ferguson et al., 2019; Osterndorff-Kahanek et al., 2015). Growing evidence shows cell-type specific responses and roles in alcohol dependence (E. K. Erickson et al., 2021; Warden et al., 2020, Warden et al., 2021). Recent studies from our group identified cell-type specific changes in post-mortem brain samples from alcohol-dependent individuals (Brenner et al., 2020). The results indicated that previous bulk sequencing approaches did not have the resolution to identify transcripts in specific cell types such as microglia. In addition, we used snRNA-seq to identify an alcohol withdrawal-sensitive subtype of protein kinase C delta-expressing GABAergic neurons in rat central amygdala (Dilly et al., 2022).
Alcohol dependence is characterized by escalation of alcohol consumption (Borgonetti et al., 2023a, 2023b; Varodayan et al., 2023; Patel et al., 2019; Warden et al., 2020; Cruz et al., 2023). Understanding cell-type specific mechanisms underlying excessive alcohol consumption associated with alcohol dependence and comparing it to transcriptomic signatures observed in individuals with AUD allows for identifying translationally relevant target networks. Here, we used the well-established two-bottle choice-chronic intermittent ethanol (CIE) vapor exposure (2BC-CIE), an alcohol dependence model, which results in the escalation of voluntary alcohol consumption in mice (Lopez and Becker, 2005; Patel et al., 2021; Borgonetti et al., 2023a, 2023b; Varodayan et al., 2023; Patel et al., 2022; Patel et al., 2019), and results in neuro-biological and behavioral adaptations similar to humans with AUD (Kimbrough et al., 2017; Ehlers et al., 2018).
We performed single nucleus RNA sequencing (snRNA-seq) on medial prefrontal cortex (mPFC) obtained from mice exposed to 2BC-CIE paradigm, an alcohol dependence model, and Visium Spatial Gene Expression on coronal brain sections obtained from the same animals. Differential gene expression analysis identified alcohol-dependence responsive genes in each cell type. Gene co-expression network analysis in each cell type identified alcohol-dependence sensitive modules. Spatial Visium Gene Expression analysis identified highly alcohol-dependence susceptible regions within the coronal brain sections.
Bioinformatics approaches have been developed to integrate single-cell RNA sequencing data and spatial transcriptomics data to deconvolute the information of overlapping cells in spatial transcriptomics data (Longo et al., 2021). Thus, allowing for the inference of the location of every cell type in the brain region of interest. We integrated the snRNA-seq and spatial gene expression data to map the spatial location of different excitatory and inhibitory neurons subtypes.
Here, we sought to determine the cell-type specific gene expression changes in the mPFC of a mouse model of alcohol dependence and identify alcohol-sensitive gene co-expression networks.
2. Methods
2.1. 2BC-CIE model and brain harvesting
12 weeks-old C57/BL6J male mice underwent the two-bottle choice-chronic intermittent ethanol (CIE) vapor exposure (2BC-CIE) procedure described in (Patel et al., 2021). 2BC-CIE is a well-established model used to generate ethanol dependent mice that are exposed to CIE vapor and display excessive (escalated) 2BC ethanol intake as well as a control group (defined as non-dependent) that voluntarily consumes ethanol in the same 2BC paradigm, but does not receive passive ethanol vapor exposure (Borgonetti et al., 2023a, 2023b; Varodayan et al., 2023; Patel et al., 2019; Warden et al., 2020). Briefly, mice were acclimated to 2BC testing, i.e., given a bottle of water and a bottle of ethanol (15% ethanol/water) for 2 h, 5 days per week for 3 weeks. Mice were then exposed to ethanol (n = 6) or air (n = 8) in vapor chambers for four days (16 h vapor on, 8 h vapor off). Following a 72-h abstinence period, both treatment groups were given 2BC testing for 5 days. These weeks of 2BC and vapor exposure were repeated for a total of 5 cycles. Brains were harvested while the mice were still intoxicated (<1 h) after the last vapor exposure as in our previous studies (Borgonetti et al., 2023a, 2023b; Varodayan et al., 2023; Patel et al., 2019; Warden et al., 2020). This study was performed in male mice, future studies are warranted on female mice to confirm the generalizability of its conclusions.
2.2. Brain sectioning and mPFC micropunches
Whole brains were embedded in OCT and 300-μm coronal sections were then obtained. At Bregma 2.96, two 300-μm sections were placed on cleaned glass slides, followed by a 10-μm section, which was placed on a Visium Spatial Gene Expression slide (10× Genomics 1000184, described later in the methods). Then two more 300-μm sections were collected. ~ 8 micropunches/sample were obtained for single nuclei isolation from the 300-μm sections using a 1.5 mm diameter micropuncher as shown in (Supplementary Fig. 1).
2.3. Single nucleus isolation and library preparation
Micropunches were homogenized in 1.5 ml Nuclei EZ Lysis Buffer (Sigma # NUC101) supplemented with 0.2 U/ul RNAse inhibitor (NEB # ML314L) and 1X protease inhibitor (Sigma, 05892791001) in a pre-chilled 2mls KIMBLE Dounce tissue grinder (Sigma Aldrich D8938) until no tissue was visible. The lysate was filtered through a 35-μm cell strainer and centrifuged at 900 xg for 5 min at 4 °C. The cell pellet was resuspended in wash/resuspension buffer (2% BSA in 1X PBS supplemented with 0.2 U/ul RNAse inhibitor) and mixed with Optiprep medium (60% iodixanol, Sigma-Aldrich # D1556) to a 25% final concentration. The suspension was carefully layered on to a 29% iodixanol cushion and centrifuged at 13,000 xg for 30 min at 4 °C. The supernatant was carefully removed without disturbing the pellet and the pellet was resuspended in 100 μl wash and resuspension buffer. The nuclei were stained with DAPI and counted using a Countess Automated Cell Counter (Thermo Fisher Scientific). Single nuclei libraries were prepared using the Chromium Next GEM Single Cell 3’ Kit v3.1 (10× Genomics, PN-1000128) with a target cell count of 10,000 cells. Libraries were sequenced on a NovaSeq 6000 using an S4 flow cell. snRNA-seq Fastq files were processed using CellRanger (v6.1.2) using the reference genome mm10–2020-A with introns included. Sequencing depth provided an average of 38,000 reads per cell exceeding the optimal of one read per cell per gene (Zhang et al., 2020).
CellRanger outputs were imported to Seurat (v4.3.0), gene by feature count matrix was constructed, mitochondrial and ribosomal genes were eliminated, log normalization was performed, and the log normalized values were used for downstream analysis.
The supervised clustering pipeline scSorter (Guo and Li, 2021) was used to classify the nuclei into 19 distinct cell types (6 excitatory neurons subtypes, 6 inhibitory neurons subtypes and 7 non-neuronal cell types based on markers from (Guo and Li, 2021; Tran et al., 2021).
Differential Gene Expression.
The R package Libra was used to access multiple differential expression pipelines (Patel et al., 2021; Squair et al., 2021). We chose four pipelines with varying in the implemented statistical methods to identify differentially expressed genes for treatment groups in each cell type. We used a single cell-based method MAST (Finak et al., 2015) and pseudobulking methods edgeR (Robinson et al., 2010) and DESeq2 (v1.30.1, (Love et al., 2014). Additionally, we applied the DEsingle method (v1.6.0, (Miao et al., 2018) which employs a Zero-Inflated Negative Binomial model to estimate the proportion of real and dropout zeros to address the limitation of the sparsity of snRNA-seq data. The overlapping differentially expressed genes identified using at least three pipelines in each cell type and edgeR calculated fold changes were used for subsequent analyses.
2.4. Weighted correlation network analysis
Weighted correlation network analysis, also known as weighted gene co-expression network analysis (WGCNA, v1.72–1) (Langfelder and Horvath, 2008) was performed on control cells for each cell type. Gene co-expression modules were constructed using blockwiseModules, using minimum module size of 30 genes, and unsigned topological overlap matrix. Soft Threshold (power) was determined by selecting the lowest power for which the scale-free topology fit index curve reached a value of 0.9. Resulting gene dendrograms were used for module detection using the dynamic tree cut method (minimum module size = 30). The hub gene for each module was detected using chooseTopHubI-nEachModule. Within each module, we calculated the percentage of genes that were determined to be differentially expressed in the alcohol-dependent samples to identify alcohol dysregulated gene co-expression modules (Fig. 2b).
Fig. 2.
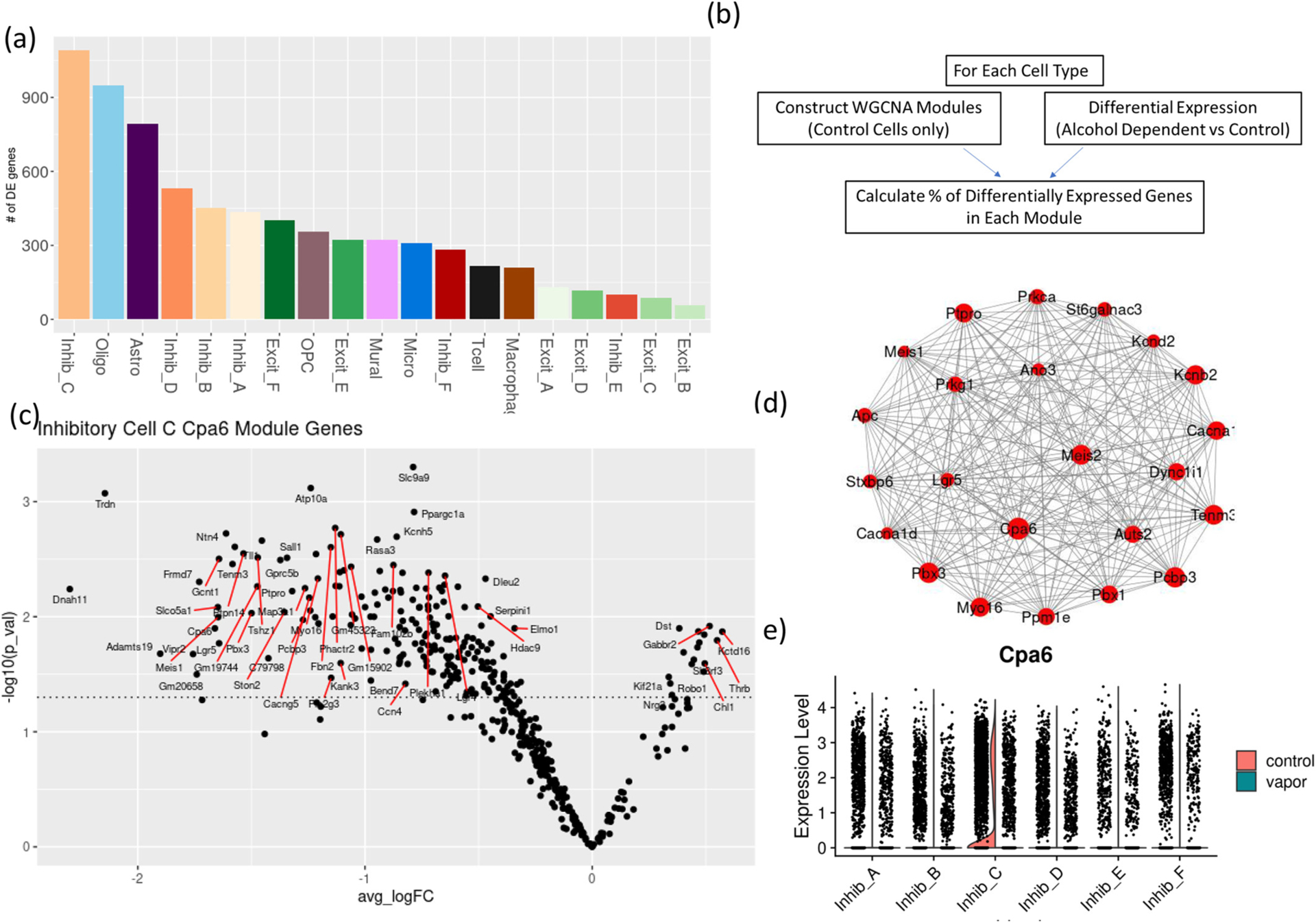
2BC-CIE results in cell-type specific transcriptomic changes and disruption of gene expression networks:
(a) number of differentially expressed genes identified by at least 3 differential expression pipelines in each cell type.
(b) schematic of the identification of 2BC-CIE exposure-dysregulated gene co-expression modules.
(c) Average log2 fold change (x-axis) and – log 10 (p-value) of gene members of Inhibitory Cells Subtype C Cpa6 module.
(d) Top connected genes in inhibitory cell subtype C Cpa6 module (e) Violin plot showing the expression of Cpa6 gene in inhibitory cells subtypes in air control and alcohol dependent samples.
Additionally, we used hdWGCNA (Morabito et al., 2022; Morabito et al., 2021), a weighted gene co-expression network analysis pipeline optimized for single-cell RNA-seq experiments, to test the stability of WGCNA findings in select cell types. Additionally, we removed predicted genes (Gm and Riken genes) from the hdWGCNA analysis to focus only on named gene annotations, and to test if the modules constructed by WGCNA were not heavily influenced by predicted genes (Gm and Riken annotations). Briefly, hdWGCNA constructs meta cells, through collapsing highly similar cells thus reducing spurious gene-gene correlations resulting from the sparsity of the data. The pipeline applies the WGCNA analysis on the metacells resulting in gene co-expression modules and hub genes identification. We used the pipeline to compute module eigengenes for each module and to calculate module differential expression between alcohol-dependent samples and air controls. Meta cells were constructed on each cell type using the following parameters: reduction method = tsne, nearest neighbors parameter k = 25 and maximum number of shared cells between two meta cells = 10. Enrichr analysis, a component of the hdWGCNA pipeline, was performed on genes of each module to determine associated biological pathways and functions.
2.5. Visium spatial gene expression library preparation
Visium Spatial Expression slides contain within each capture area 4992 oligonucleotide barcoded spots (capture spots), each spot is 55 μm in diameter. Barcoded capture spots allow for tagging the transcripts from the tissue section within each spot therefore enabling the identification of each transcript’s spatial location. Slides containing 10-μm coronal sections (described above) were fixed for 30 min in ice-cold methanol at −20 °C, followed by H&E fixation and imaging as described in the manufacturer protocol (10× Genomics, 1000187). Sections were permeabilized for 18 min, followed by cDNA synthesis, second strand synthesis, cDNA amplification, and library preparation as described in the manufacturer protocol. Libraries were sequenced on NovaSeq 6000 using S4 flow cell. Visium Fastq files were processed using Space Ranger (v1.3.1) and reference genome mm10–2020-A. Folds or damaged regions were removed from downstream analysis using the manual draw tool in Loupe Browser. Barcodes of valid sections were exported to the Seurat package (v4.3.0) followed by unsupervised clustering of the capture spots. The FindAllMarkers function was used to identify each cluster’s top enriched markers (Supplementary Table 1). Differential expression analysis was performed on clusters represented in all the samples, comparing alcohol-dependent samples to air-treated controls to identify region-specific transcriptomic changes in response to CIE treatment. Average expression of each gene within each cluster in each sample was calculated, t-tests were run comparing alcohol vapor samples to air treated controls. Genes with average expression within the lower quartile of gene expression values were eliminated to avoid differential gene expression signals produced by technical noise.
2.6. Integration of snRNA-seq data and spatial transcriptomics
A challenge with Visium Gene Expression data is the lack of single cell resolution. A single capture spot (55 μm) can contain multiple cell types. We used the Seurat integration pipeline (Stuart et al., 2019), which applies an anchor-based integration workflow, to integrate the spatial transcriptomics and snRNA-seq data, to mitigate the lack of the single cell resolution on the Visium capture spots. The pipeline identifies anchor genes between a reference annotated dataset (snRNA-seq) and a query data set (Visium Spatial Gene Expression data), followed by the probabilistic transfer of annotations from the reference to the query set resulting in a prediction score for snRNA-seq for each cell type contained in each Visium capture spot.
3. Results
3.1. CIE exposure escalates voluntarily ethanol consumption
Mice treated with either ethanol or air had similar baseline levels of 2BC ethanol consumption. 2BC ethanol drinking escalated in CIE-treated mice after each cycle compared to baseline drinking levels, while there was no escalation in air-exposed controls compared to baseline (repeated measures ANOVA, interaction effect between exposure group * drinking session, F (5, 60) = 5.222, p-value = 0.0005, Fig. 1b). Average weekly alcohol consumption in each of the alcohol-dependent and air-control groups and average blood alcohol levels in alcohol dependent group after each vapor cycle are shown in (Table 1). Average blood alcohol level in the alcohol dependent group before euthanization was 188.4 +/−5.38 mg/dl.
Fig. 1.
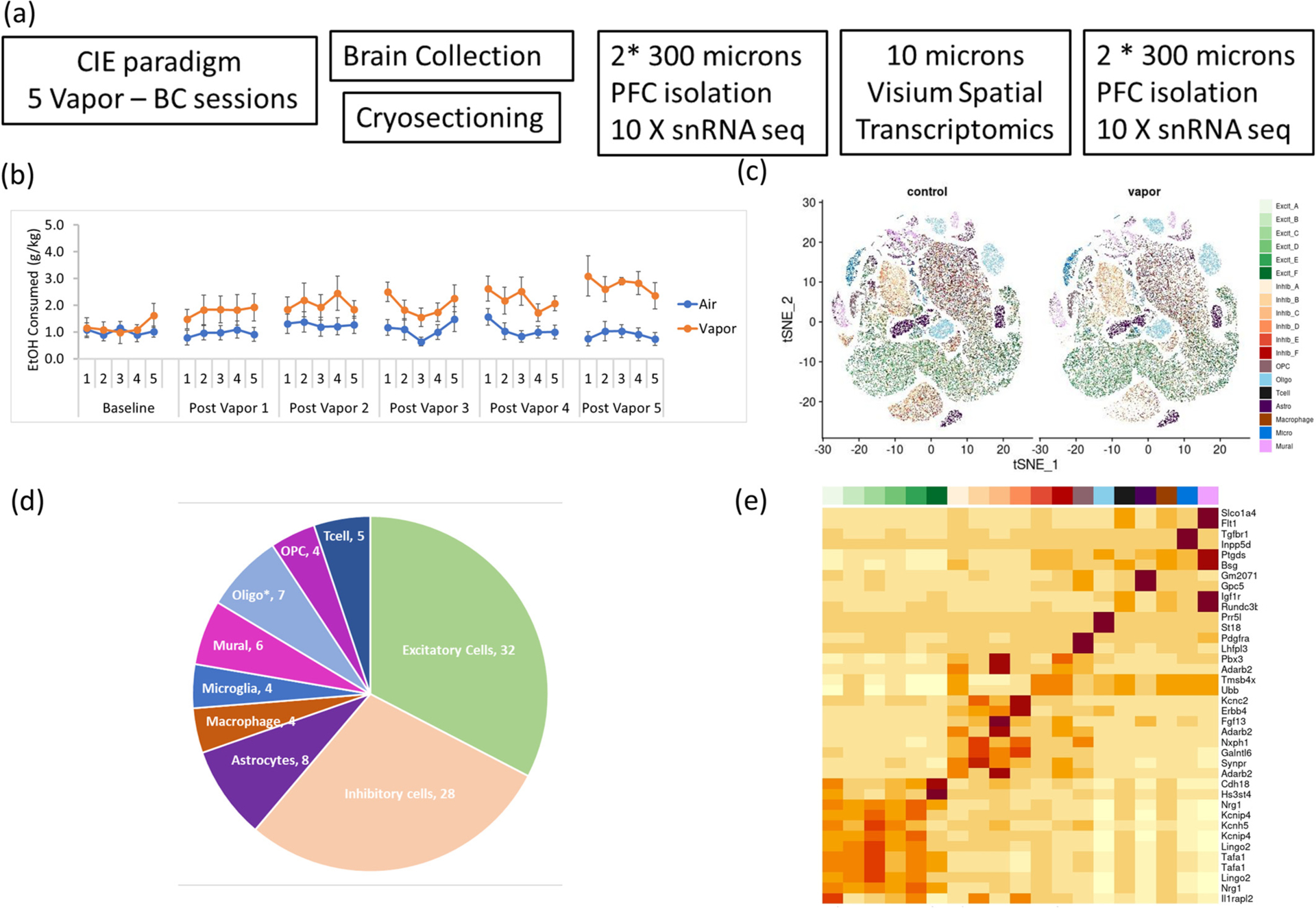
Single nucleus RNA sequencing of mPFC from 2BC-CIE paradigm treated animals:
(a) Schematic diagram showing the experimental design, animals were subjected to chronic intermittent ethanol or air (control) followed by 2 h 2 bottle choice (ethanol/water), after 5 CIE sessions, brains were collected, flash frozen, mounted in OCT, cryosectioned into 300 μm section to obtain PFC punches for single nucleus RNA sequencing, one 10 μm section followed by two more 300 μm section for snRNA-seq, was collected for Visium Spatial transcriptomics.
(b) Amount of ethanol consumed (g/Kg) during the 2-h 2 bottle choice sessions after each alcohol vapor/air control exposure session.
(c) tSNE plot showing single nuclei clustering from alcohol vapor and air control samples, color coded by cell types.
(d) Percentage of nuclei assigned to each cell type. (*oligo: oligodendrocytes).
(e) Heatmap showing average expression of the top enriched markers (rows) in each cell type (columns)
Table 1.
average consumption (+/−SEM) of 15% ethanol in 2 h across each 5-day 2 bottle choice testing cycle and average blood alcohol levels in alcohol dependent group on the third day of each four-day alcohol vapor cycle.
| Average Air-Control Group Consumption (gm/Kg/2 h) | Average Alcohol-Dependent Group Consumption (gm/Kg/2 h) | Average Blood Alcohol Level (Alcohol dependent group) (mg/dl) | |
|---|---|---|---|
| Baseline (final week of 3) | 1.01 +/− 0.23 | 1.18 +/− 0.35 | |
| Post Vapor 1 | 0.94 +/− 0.28 | 1.78 +/− 0.5 | 206.44 +/− 12.3 |
| Post Vapor 2 | 1.27 +/− 0.35 | 2.05 +/− 0.51 | 151.50 +/− 12.24 |
| Post Vapor 3 | 1.08 +/− 0.32 | 1.97 +/− 0.38 | 171.77 +/− 8.19 |
| Post Vapor 4 | 1.08 +/− 0.26 | 2.22 +/− 0.42 | 162.14 +/− 7.97 |
| Post Vapor 5 | 0.89 +/− 0.27 | 2.76 +/− 0.46 | 224.59 +/− 6.11 |
3.2. Identification of major cell types in mPFC
We sequenced libraries from ~150,000 single nuclei from the mPFC of 6 CIE-treated and 8 air-treated control mice. The supervised clustering pipeline scSorter (Guo and Li, 2021) was used to classify the nuclei into 19 distinct cell types (6 excitatory neurons subtypes, 6 inhibitory neurons subtypes and 7 non-neuronal cell types based on markers from (Guo and Li, 2021; Tran et al., 2021) (Fig. 1c). The full set of cell type markers is presented in Supplementary Table 2. Excitatory and inhibitory cell types comprised 32% and 28% of the identified cells, respectively, while astrocytes and oligodendrocytes comprised 8% and 7% of all cells respectively, while each of microglia, macrophage, and oligodendrocyte precursor cells (OPC) comprised 4% of the identified cell types (Fig. 1d).
The FindMarkers function in the Seurat pipeline was used to identify genes enriched in each cell type over all other cell types. Two inhibitory cells subtypes (C & F) were Adarb2 positive suggesting they are Lamp5/Pax6 or VIP interneurons, inhibitory cell subtypes (B & D) are Adarbd2 negative suggesting they are PVALB or SST interneurons (Hodge et al., 2019) (Supplementary Fig. 3d). In contrast to inhibitory cells, sub-clustering of excitatory neurons did not show robust and distinct subtype markers, indicating that the differences between the excitatory cells’ subtypes are subtle (Fig. 1e). Distinct layer information of excitatory neurons is discussed later in the results section (integration of snRNA-seq and spatial gene expression results).
Non-neuronal cell types were enriched in known cell-type markers validating the supervised clustering assignments. For example, astrocytes were enriched in Gpc5 (Lau et al., 2020) and Slc1a3 (Lau et al., 2020; Achicallende et al., 2022), microglia in Tgfbr1 (Butovsky et al., 2014) and Apbb1ib, oligodendrocytes in St18 (Zhao et al., 2022), Prr5l (Falcão et al., 2018) and Mbp (Galiano et al., 2006), and OPCs were enriched in Pdgfra (Ellison and de Vellis, 1994) and Lhfpl3 (Huang et al., 2020) (Fig. 1e). Additionally, we identified enriched genes in each cell type that can serve as novel cell type markers; the lncRNA Gm20713 was among the top enriched genes in astrocytes, Rnf220 is enriched in oligodendrocytes, Zfhx3 and Lrmda in microglia (Supplementary Fig. 2d).
A cell-type diversity statistic (CTDS) was calculated for each sample (Karagiannis et al., 2022) to determine if CIE exposure altered the overall cell-type composition. The CTDS did not differ between the two sample groups (t-test, p-value = 0.12), indicating no differences in cell-type composition after CIE exposure. Furthermore, the percentage of cells assigned to each cell type in each sample was not significantly different between treatment groups (Supplementary Table 3), except for excitatory cells subtype F showed a trend towards a higher percentage in CIE-treated mice (nominal p-value = 0.012) and OPCs that showed a trend towards a lower percentage in CIE-treated mice compared to controls (nominal p-value<0.05, Supplementary Fig. 2a, b). This result shows that CIE did not result in significant changes in mPFC cell-type composition, loss of a cell type, or the emergence of a CIE-specific cell type, a result consistent with those reported from human postmortem brain snRNA-seq from individuals with AUD (Van Den et al., 2023; Brenner et al., 2020).
3.3. Cell-type specific differential expression in mPFC of CIE-treated mice
We applied four different pipelines, two based on pseudobulk data which account for biological replicates (edgeR and DESeq2), and two using single cell-based methods (MAST, and DEsingle). MAST identified the greatest number of differentially expressed genes, while DESeq2 was the most conservative pipeline. For our analysis, a gene was determined to be differentially expressed for subsequent downstream analysis if it was identified by at least three of these pipelines. The full sets of differentially expressed genes within each cell subtype are listed in Supplementary Table 4.
We identified a group of genes that were dysregulated across the majority of identified cell types (at least 15 clusters). For example, Gm47283, Pbx3 and Scgb3a1 were dysregulated in all 19 cell types; Gm21887, Lcn2, Lgr5, Scg2 and Tenm3 in 18 cell types; and Gad1, Pcbp3, Ptgds and Ptpro in 17 cell types (Supplementary Fig. 3b). The number of differentially expressed genes was greatest within specific cell type clusters including inhibitory subtype C neurons, oligodendrocytes, and astrocytes (Fig. 2a). The full list of cell specific differentially expressed genes is shown in Supplementary Table 5. The Metascape gene enrichment tool (Y. Zhou et al., 2019) was used to identify gene ontology (GO) terms and biological pathways. Differentially expressed genes in inhibitory subtype C neurons were significantly enriched in Go Biological processes: learning and memory (37 genes), regulation of membrane potential (56 genes), and synaptic signaling (55 genes); Reactome gene sets: GABA receptor activation (11 genes); and KEGG pathways neuroactive ligand-receptor interaction (34 genes) and morphine addiction (19 genes). Enrichment of oligodendrocyte differentially expressed genes included GO Biological processes: regulation of membrane potential (64 genes), potassium ion transport (28 genes), cell junction organization (57 genes), cell-cell adhesion (45 genes), regulation of synapse organization (33 genes) and trans-synaptic signaling (47 genes). Enrichment of differentially expressed genes in astrocyte included GO Biological processes: angiogenesis (32 genes), extracellular matrix organization (31 genes), modulation of chemical synaptic transmission (53 genes), cell-cell adhesion (39 genes); and the Reactome Gene Sets: SLC-mediated transmembrane transport (22 genes, including the astrocyte enriched genes Slc1a3 and Slc1a2). Metascape full results are presented in Supplementary Table 6.
Previous snRNA-seq in human postmortem brain identified numerous cell specific changes in neuroinflammatory-related genes (Brenner et al., 2020); thus, we investigated CIE-mediated enrichment of differentially expressed neuroinflammation genes in each cell type. A comprehensive list of neuroimmune related genes (757 genes) utilized by the Nanostring nCounter Neuroinflammation Panel (NanoString Technologies nCounter®) was used as an input for the enrichment analysis, we removed cell marker genes and neuronal signaling genes from the panel gene list, remaining genes (501 genes) were used as input for neuroinflammation related genes enrichment calculations. Differentially expressed in microglia were enriched in neuroinflammation related genes (30 neuroinflammation genes, p-value of enrichment = 5.23 * 10−5). 15 of the 30 genes were related to activated microglia, 7 genes to adaptive immune response, 7 genes to growth factor signaling, and 4 genes to matrix remodeling (Table 2). The lncRNA Neat1, an inflammation associated lncRNA (Pan et al., 2022) was upregulated in oligodendrocytes from alcohol-dependent samples, a result consistent with bulk RNA sequencing data from mPFC of alcohol dependent mice (Farris et al., 2020). A subset of neuroinflammation genes were dysregulated in multiple cell types. For example, Lcn2 was downregulated in 18 cell types, Spp1 downregulated in 15 cell types, Apoe and Eomes were downregulated in 14 cell types. CIE-mediated upregulated neuroinflammation genes included Cd8a in T Cells, Ccl5 in Excitatory cells subtype B, Top2a in OPCs, Cd86 and Cd33 in microglia.
Table 2.
Overlap between microglial DE genes and neuroinflammation genes.
| Genes | Neuroinflammation Category |
|---|---|
| Apoe, Abcc3, Asb2, Blnk, Cd33, Cd86, Chn2, Epsti1, Fgf13, Lst1, Mertk, P2ry12, Slc2a5, Spp1 and Tmcc3 | Activated Microglia |
| Asb2, Blnk, Cd33, Cd74, Cd86, Inpp5d and Lyn | Adaptive Immune Response |
| Cd86, Fgf13, Hdac2, Lyn, Spp1 and Tgfbr1 | Growth Factor Signaling |
| Cntnap2, Reln, Cd86 and Spp1 | Matrix remodeling |
3.4. CIE induced changes in neuron-neuron communications
We performed NeuronChat (W. Zhao et al., 2023) to investigate neuron-neuron communications. Briefly, neuron-neuron communication is defined as the significant expression of the ligand from a sender cell type and the receptor from a receiver cell type as compared to the expression of those genes in a permutated dataset. NeuronChat identified incoming and outgoing communication patterns to and from each cell type based on the expression of ligands and receptors identified from manually curated neural signaling interactions. Outgoing communication patterns (each cell type as a sender/secreting cells) show astrocytes, inhibitory E, F, microglia, mural, oligodendrocytes to be contributing to gap junction and Cck_cckbr signaling (Pattern2), excitatory cells subtypes B and D to be contributing to glutamate signaling (Pattern 1), subtypes A, E and F to be contributing to pattern 4 (CO_gucy1a1). Inhibitory cells B, C, D and OPC contribute to Nrxn_Nlgn signaling (Pattern 3, Supplementary Fig. 3c). Comparing neuron-neuron communications between control and alcohol dependent samples showed decreased number of significant communication links between GABA and different types of GABA receptors and increased excitatory connections, Glutamate - Grin receptors and Neurexin-Neuroligin connections in alcohol-dependent samples (Supplementary Fig. 3d).
3.5. Cell type specific gene co-expression modules
We identified WGCNA gene co-expression modules that were significantly enriched with differentially expressed genes and were confirmed by hdWGCNA analysis (Table 3 and Supplementary Table 7). The greatest enrichment was found in modules from oligodendrocytes and inhibitory cells. Excitatory cells subtypes were not enriched in differentially expressed genes. Interestingly, Cpa6, a gene significantly expressed at higher levels in inhibitory cells subtype C (Fig. 2e), was identified as a hub gene in WGCNA in oligodendrocyte and inhibitory cell subtypes A and C modules. In addition, Cpa6 modules in these three subtypes were significantly enriched in differentially expressed genes (Table 3). The oligodendrocyte module containing Cpa6 included 266 genes, 77% of which were differentially expressed compared to alcohol-dependent samples (odds ratio = 30.26, p-value = 3*10−138, Fisher’s Exact test, Supplementary Fig. 4a). Modules within Inhibitory cell types A and C that included Cpa6 as a hub gene contained 149 (55% of which are differentially expressed; odds ratio = 38.73, p-value = 6.7*10−79, Fisher’s Exact test) and 447 genes (52% of which are differentially expressed; odds ratio = 11.21, p-value = 1.69*10−109, Fisher’s Exact test), respectively (Supplementary Figs. 4d). Cpa6 was identified as hub gene in inhibitory cells subtype C module 3 identified by hdWGCNA (Supplementary Fig. 4b), differential expression analysis of module eigengenes shows this module to be significantly downregulated in alcohol vapor samples (Supplementary Fig. 4c). GO enrichment analysis shows this module to be enriched in heparan sulfate proteoglycan biosynthesis and positive regulation of adenylate cyclase biological processes (Supplementary Fig. 4d). Cpa6 is connected to critical genes in the highly dysregulated inhibitory cells subtype C (Fig. 2d). For example, Cacna1ac, encoding for Calcium channels, the Pbx and Meis genes which are transcription factors interacting in transcriptional activation complexes (Y. Liu et al., 2001) and CPA6 was identified in GWAS as a gene involved in the regulation of alcohol consumption (Schumann et al., 2011). Cpa6 modules in oligodendrocytes, inhibitory cells subtype A & C shared significant overlap between their gene members (p-value<9e−05) (Supplementary Fig. 4e). The enriched pathways for Cpa6 module genes include neurovascular coupling, synaptogenesis signaling pathway, opioid, and endocannabinoid signaling pathway (Supplementary Fig. 4f).
Table 3.
WGCNA modules in select cell types and percent of genes differentially expressed in alcohol-dependent samples.
| Module color | Hub Gene | Number of module genes | % of DE genes | |
|---|---|---|---|---|
| Inhibitory Cells subtype A | Blue | Cpa6 | 149 | 55%* |
| Brown | Grip1 | 106 | 19%* | |
| Turquoise | Fgf14 | 839 | 8.5% | |
| Yellow | Npas3 | 66 | 13.6%* | |
| Inhibitory Cells subtype C | Blue | Tmem163 | 87 | 47%* |
| Turquoise | Cpa6 | 447 | 49%* | |
| Oligodendrocytes | Blue | Pde4b | 122 | 43%* |
| Turquoise | Cpa6 | 266 | 77%* | |
| Astrocytes | Blue | Gm12510 | 80 | 0% |
| Turquoise | Gpc5 | 484 | 22%* |
Indicates the module is significantly enriched in differentially expressed genes. (Full list of modules, modules gene members, hub genes and percent differential expressed genes in all cell types are presented in Supplementary Table 5).
WGCNA identified an additional highly dysregulated co-expression module in oligodendrocytes, consisting of 122 genes, 43% of which were upregulated in alcohol-dependent samples (odds ratio = 5.54, p-value = 2.63*10−17, Fisher’s Exact test, Supplementary Fig. 4a), and Pde4b as a hub gene. Pde4b, a gene of interest in alcohol research (Avila et al., 2017; Blednov et al., 2014; Wen et al., 2012; Wen et al., 2012), was identified in our differential expression analysis as an upregulated gene in oligodendrocytes (Supplementary Fig. 4g). Pde4b module genes were enriched in myelination signaling, synaptogenesis signaling, cAMP mediated signaling pathways (Supplementary Fig. 4g). This result is consistent with hdWGCNA oligodendrocyte module 5 showing Pde4b, St18, and Plc11 as hub genes (Supplementary Fig. 4h). Oligodendrocyte module 5 module genes were enriched in organelle organization and positive regulation of transcription biological pathways. Differential module eigengene analysis shows this module to be significantly upregulated in alcohol vapor samples (Supplementary Fig. 4i).
WGCNA identified an astrocyte module with Gpc5 as a hub gene. This module was enriched in differentially expressed genes (22% of the 484 module genes are differentially expressed, p-value = 4.26*10−29, Table 3). 102 of the Gpc5 module genes were upregulated in alcohol-dependent astrocytes, while 3 were downregulated (Supplementary Fig. 4a). Slc1a3 a gene upregulated in human AUD astrocytes (Brenner et al., 2020), is a member of Gpc5 module, and is upregulated in alcohol vapor treated animals’ astrocytes. hdWGCNA identified the same module (Supplementary Fig. 4k), confirming the stability of this module using different modes of module construction, differential expression analysis of module eigengenes confirmed the upregulation of this module in alcohol-dependent samples (Supplementary Fig. 4l). Gene enrichment analysis of Gpc5 modules shows enrichment in amino acid: sodium symporter activity, L-glutamate transporter activity and l-aspartate transmembrane transport.
Module enrichment of neuroinflammation genes (NanoString Technologies nCounter®) was examined. Six modules were significantly enriched in neuroinflammation genes (Table 4), one of which (microglia Blue Module; p-value<7.5*10−64) included alcohol-dependence differentially expressed genes. 19% of the genes in this microglial module, were differentially expressed in vapor-treated samples (p-value<7.5*10−64, Supplementary Fig. 4m), all but one of the differentially expressed genes in this module were upregulated in alcohol vapor samples. The hub gene of this module is Inpp5d.
Table 4.
Neuroinflammation enriched modules: * indicates the module is significantly enriched in differentially expressed genes.
| Cell type | Module | Module size | Hub gene | Neuroinflammation genes | % DE genes |
|---|---|---|---|---|---|
| Microglia | Blue | 376 | Inpp5d | 60 | 19%* |
| Excitatory Cells E | Pink | 53 | Fyb | 11 | 0% |
| Microglia | Yellow | 60 | Itgal | 8 | 2% |
| Excitatory Cells A | Pink | 31 | Gm20703 | 5 | 0% |
| Excitatory Cells C | Cyan | 31 | 8430423G03Rik | 5 | 0% |
| Inhibitory Cells B | Green | 41 | Calm1 | 5 | 5% |
This analysis identifies CIE-sensitive gene co-expression modules in the cell types with the highest number of differentially expressed genes and connects known alcohol related targets to dysregulated cell-type specific gene regulatory networks in an alcohol dependence model.
3.6. Oligodendrocyte subclusters exhibit unique transcriptomic responses to CIE
t-SNE plots and unsupervised clustering identified multiple clusters of oligodendrocytes (Fig. 1c). Unsupervised clustering of the oligodendrocytes at resolution = 0.1 identified 4 subclusters (Fig. 3b) with distinct enriched markers (Fig. 3e). WGCNA identified Cpa6 as a hub gene. Only a subset of oligodendrocytes, subcluster 2 showed a high expression level of Cpa6 (Fig. 3d), a WGCNA identified hub gene of a highly dysregulated module in oligodendrocytes (Fig. 3a).
Fig. 3.
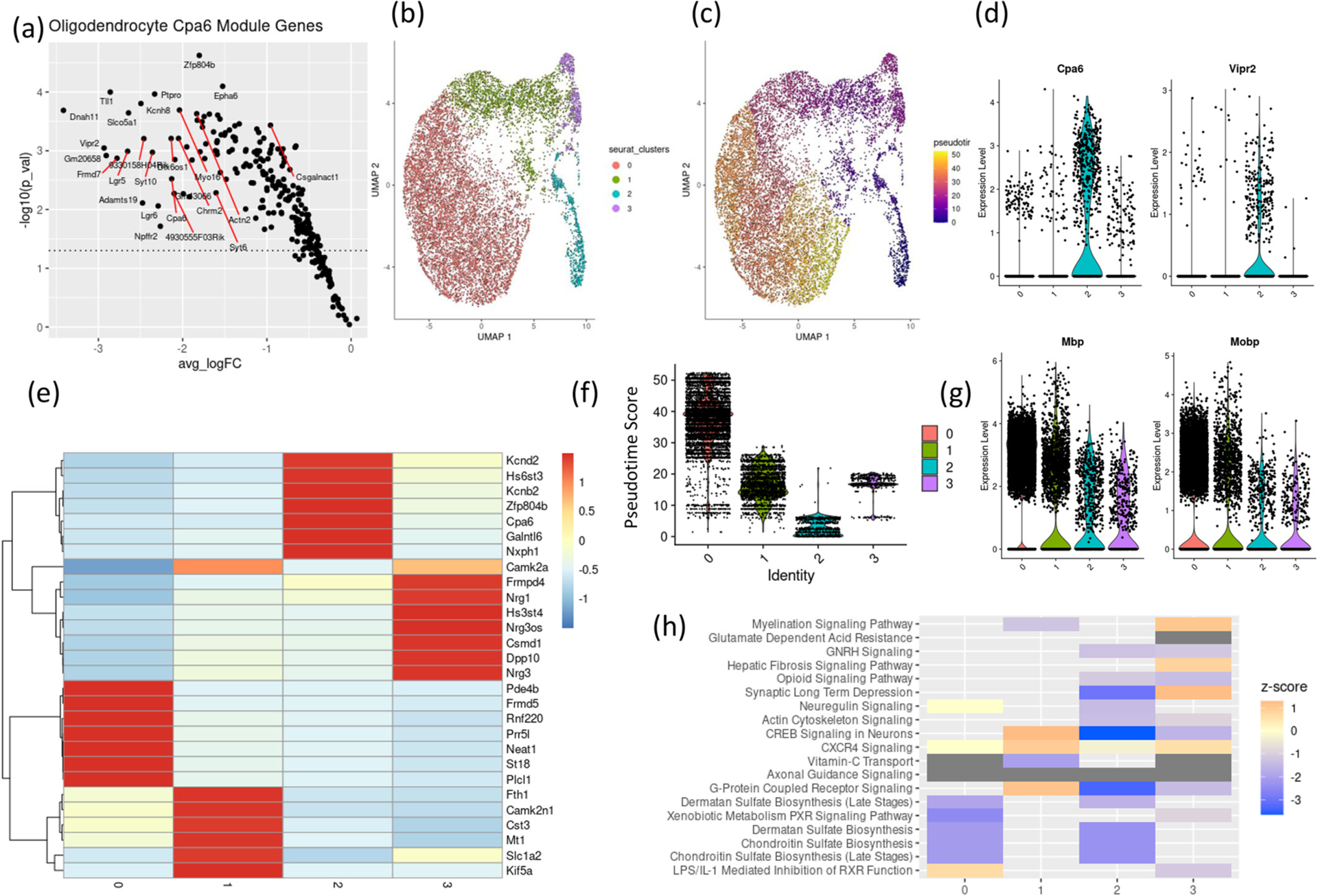
Oligodendrocytes sub clustering into transcriptionally distinct subtypes.
(a) Average log2 fold change (x-axis) and – log 10 (p-value) of gene members of oligodendrocytes Cpa6 module. UMAP plots of oligodendrocytes colored by (b) unsupervised clustering assignment and (c) pseudotime score.
(d) expression of Cpa6 and Vipr2 in oligodendrocyte subclusters.
(e) heatmap showing scaled expression values of seven top expressed genes in each oligodendrocyte subclusters.
(f) violin plots showing pseudotime scores in each oligodendrocyte clusters (g) expression of myelin related genes, Mbp & Mobp, in oligodendrocyte subclusters.
(h) z-score of select predicted pathways enriched in differentially expressed genes in oligodendrocytes subclusters, positive z-score = predicted activation, negative z-score = predicted inhibition.
We performed pseudotime analysis (Trapnell et al., 2014) to order oligodendrocyte cells and identify if they can be ordered across a continuum. This analysis identified genes that vary across the pseudotime continuum. Myelin basic protein (Mbp) and myelin associated oligodendrocyte basic protein (Mobp) were highly expressed at the higher pseudotime values (Supplementary Fig. 5b). Cluster 0 exhibited higher pseudotime (Fig. 3f) with higher expression of Mbp & Mobp (Fig. 3g) suggesting that higher pseudotime scores correspond to more mature myelinating oligodendrocytes. Genes varying across the oligodendrocyte pseudotime trajectory included Potassium channel genes Kcnb2 and Kcnd2 which were highly expressed at lower pseudotime values (Supplementary Fig. 5b). Potassium channels were shown to be prominent in OPCs which are known to be important for differentiation to oligodendrocytes, and their expression is subsequently downregulated in mature oligodendrocytes (Cherchi et al., 2021). Vipr2 was enriched in the oligodendrocytes subcluster 2 cluster, the same subcluster expressing Cpa6, suggesting an association of this subset of oligodendrocytes with the Cpa6 positive VIP neurons (Fig. 3d).
A cell-type diversity statistic (CTDS) was calculated for each sample (Karagiannis et al., 2022) to test if subtypes of oligodendrocytes are changed between the treatment groups. There was a significant difference (p-value = 0.028) between the alcohol vapor treated samples and air control samples suggesting that oligodendrocyte subtype composition differs between groups. We found a higher percentage of subcluster 0 oligodendrocytes in alcohol vapor-treated samples (subcluster 0) compared to air-treated controls, while subcluster 2 oligodendrocytes showed a lower oligodendrocytes percentage total oligodendrocytes compared to air-control samples (Supplementary Fig. 5a). Differential expression analysis comparing alcohol vapor samples to air control samples for each subcluster identified uniquely dysregulated genes in each oligodendrocyte subtype (Supplementary Fig. 5b). We performed Ingenuity Pathway Analysis (IPA) on differentially expressed genes from each subcluster. Pathways significantly enriched in differentially expressed genes from cluster 0 and 2 overlapped, included dermatan sulfate and chondroitin sulfate biosynthesis pathways, dermatan and chondroitin sulfate are proteoglycans critical for synaptogenesis and axon-guidance (Schwartz and Domowicz, 2018) and are modulators of the microenvironment of extracellular matrix (Yamada et al., 2022). Opioid signaling pathway and synaptic long-term depression were significant in clusters 2 and 3 (Fig. 3h).
This data identifies a Cpa6 enriched oligodendrocyte subcluster with immature characteristics. Alcohol vapor samples show fewer cells belonging to this subcluster compared to air control samples. Oligodendrocyte subtypes show distinct differential expression responses.
3.7. Integration of snRNA and Spatial Transcriptomics Shows Regional Specificity of Neuronal Cell Subtypes
Unsupervised clustering of gene expression profiles across the capture spots of Visium Spatial Gene Expression data (described in methods) identified 18 unique spatially defined clusters (Fig. 4b). The Seurat FindAllMarkers function was used to identify each cluster’s top enriched genes (Fig. 4a row names, Supplementary Table 8). The top gene markers of each cluster are consistent with gene expression patterns of corresponding major brain regions illustrated in the Allen Brain Atlas (Lein et al., 2007). Clusters 1, 8, 0, 3, 12 and 6 (listed in order of location, lateral to medial) occupied the dorsal regions of the section and were patterned in distinct layers consistent with specific cortical layers. Clusters 8 and 0 mapped to cortical layers 2/3 with cluster 8 showing enrichment in Tmem215, Ddit4l, and Igfn1, and cluster 0 enrichment in Tnnc1 and Pamr1. These 2 clusters potentially differentiate layers 2 and 3 based solely on transcriptomic profiles. Cluster 3 mapped with cortical layer 5 and showed enrichment of Ighm and Igfbp4 compared to other clusters.
Fig. 4.
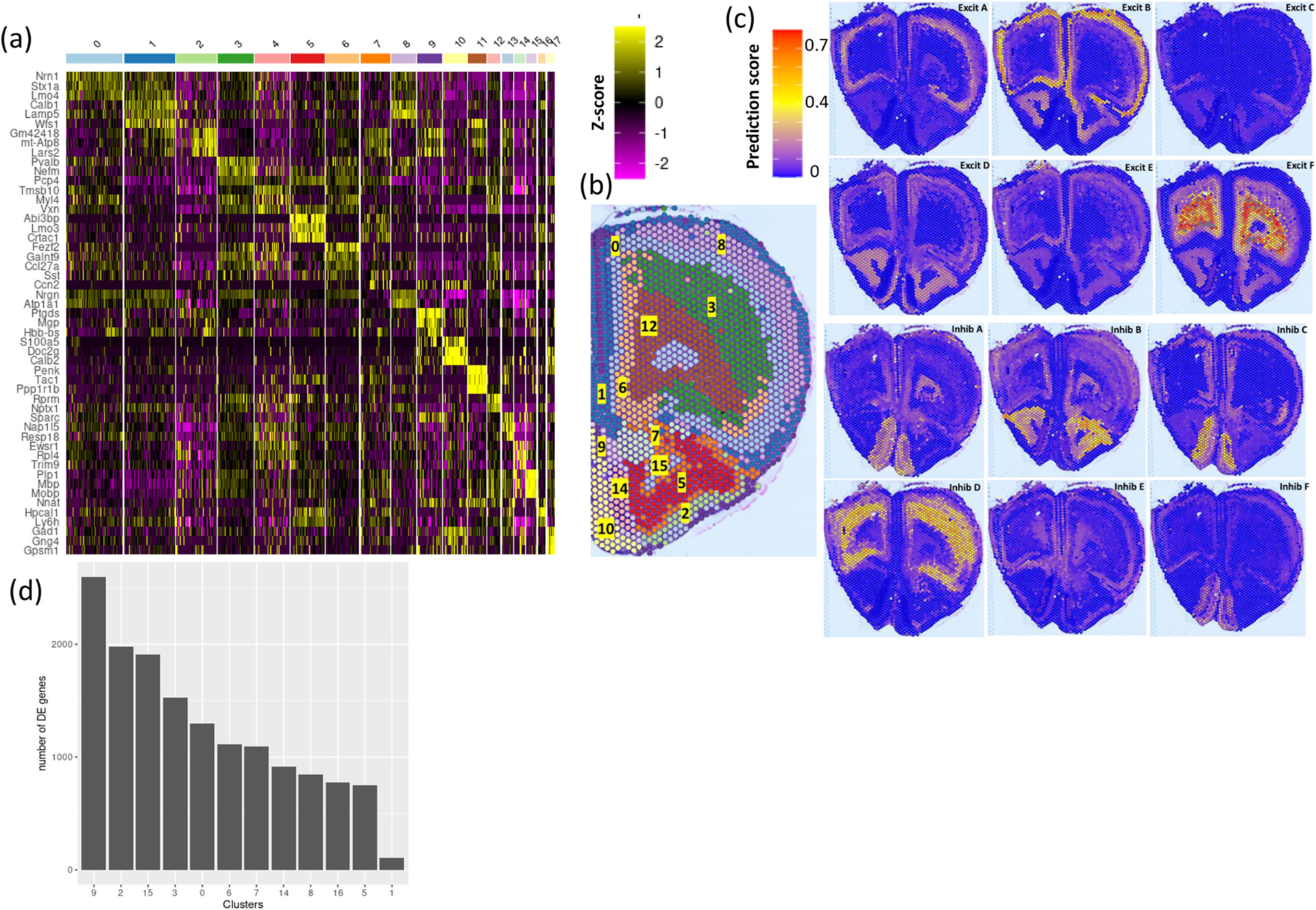
Visium Spatial Transcriptomics Identify Gene Expression Signatures of Spatially Defined Regional Clusters.
(a) heatmap showing the top enriched genes in each of the unsupervised clusters identified in the Visium sections.
(b) the spatial location of each of the unsupervised clusters in the brain section.
(c) prediction scores of each of excitatory and inhibitory cells subtypes.
(d) number of alcohol-dependence differentially expressed genes in each spatial cluster.
Cluster 15 overlapped with white matter tracts, top expressed markers in this cluster include myelin related genes (Mbp and Mobp). Clusters 2, 5, 10 and 14 were located in the ventral region of the brain section. Cluster 5’s location is consistent with the anterior olfactory nucleus; markers of this cluster included Crtac1 and Lmo3. Clusters 10 and 14 locations are suggestive of representing the main olfactory bulb nucleus. Cluster 10 exclusively expresses tyrosine hydroxylase (Th). This data provides comprehensive transcriptomic atlas for various cortical and olfactory bulb layers. The top enriched markers of each cluster confirmed regional specificity when compared to corresponding ISH data from Allen Brain Atlas.
Integration of snRNA-seq and Visium Spatial Gene Expression data (detailed in methods) derived scores which enable the identification of the spatial location of each of the 19 cell types identified by snRNA-seq. This analysis deciphered the spatial location of the different subtypes of excitatory and inhibitory cells (Fig. 4c). snRNA-seq data shows that the excitatory cell subtypes, in contrast to inhibitory cell subtypes, are closely related in their enriched markers (Fig. 1e), prediction scores of the 6 excitatory cell subtypes show regional specificity of each of the subtypes (Fig. 4c). Integration analysis shows the location of the highly CIE susceptible inhibitory neurons subtype C, to be specific to layer 2, differentiating it from the closely related subtype (Inhibitory neurons subtype F), which is shown by the integration analysis to overlap with spatial cluster 0 location, suggesting layer 3 specificity (Fig. 4c). The number of differentially expressed genes per cluster was quantified (Fig. 4d). Cluster 9 and 2 showed the highest number of differentially expressed genes, the two clusters’ capture spots occupied a fine outer-most lining of the sections, the top enriched gene was Ptgds, with spatial distribution matching ISH data from the Allen Brain Atlas. Cluster 15 was among the highest alcohol-dependent dysregulated clusters, cluster 15 overlapped with white matter tract and were enriched in myelin related genes (Fig. 4a). These region-specific gene expression changes validate the susceptibility of oligodendrocytes to CIE treatment. (Differentially expressed genes in each spatial cluster are presented in Supplementary Table 9).
4. Discussion
In this study, we provide a comprehensive cell-type specific brain region specific transcriptional responses atlas in a mouse model of alcohol dependence. We performed snRNA-seq on ~150,000 nuclei from the medial prefrontal cortex (mPFC) obtained from mice exposed to the two-bottle choice-chronic intermittent ethanol exposure (2BC-CIE) paradigm, an alcohol dependence model, as well as control non-dependent mice. Additionally, we generated Visium Spatial Gene Expression profiling on coronal brain sections obtained from the same animals. snRNA-seq does not retain spatial information of the sequenced nuclei, Visium Spatial Gene Expression profiling retains the spatial information of the sequenced genes however it lacks single cell resolution. Integrating brain cell-type specific and spatially defined transcriptomic data from alcohol-dependent mice allowed for registering cells from the snRNA-seq into anatomical locations based on the data obtained from the spatial transcriptomics analyses. We utilized the Visium Spatial Gene Expression platform to: 1) identify spatially defined regional transcriptomic clusters, 2) to decipher the localization of cell types of interest identified by snRNA-seq by integrating cell-type specific and spatial gene expression data and 3) identify region-specific transcriptomic changes in response to CIE treatment.
Alcohol use disorders cell-type specific changes were identified in human postmortem PFC (Brenner et al., 2020) and nucleus accumbens (Van Den et al., 2023). A novel population of cells in central amygdala associated with alcohol withdrawal was identified in a rat model of alcohol withdrawal (Van Den et al., 2023; Dilly et al., 2022). Our group showed cell-type specific responses and roles in alcohol dependence models, specifically glial cells (Erickson et al., 2021; Warden et al., 2020; Warden et al., 2021). In our human postmortem PFC snRNA-seq study glial cells were shown to be the most affected in alcohol use disorders (Brenner et al., 2020). Although cell-type specific responses in alcohol dependence have been shown, comprehensive transcriptomic profiling of cell-type specific changes in mouse models of alcohol dependence has been lacking.
In this study, cell diversity analysis showed that alcohol-dependent samples did not exhibit altered cell type composition compared to air-control samples, indicating that alcohol did not result in the depletion or enrichment of a specific cell type. snRNA-seq enables the identification of cell type specific CIE- transcriptomic changes (Supplementary Fig. 3a). We utilized multiple pipelines for snRNA-seq differential expression analysis for each cell type. These pipelines differ in their principles, assumptions, and whether they account for variation between biological replicates (Squair et al., 2021), pipelines designed for single cell analysis identified the greatest number of differentially expressed genes, while pseudobulking methods were the most conservative pipelines. For our analysis, a gene was determined to be differentially expressed for subsequent downstream analysis if it was identified by at least three of these pipelines.
WGCNA allows for construction of modules of co-expressed genes, allowing the study of gene regulation on network basis not individual gene basis. Combining network information with differential expression information enables the study of alcohol-associated network dysregulation and enables the identification of critical hub genes of the dysregulated modules. We constructed gene co-expression modules and identified cell-type specific modules dysregulated in the ethanol dependent model. We applied two pipelines for module construction, WGCNA (Langfelder and Horvath, 2008) and a similar pipeline modified for single-cell sequencing data (hdWGCNA, (Morabito et al., 2021, 2022). In the WGCNA, we used all the genes, including transcripts with no official gene name (Gm and Riken genes). Those unannotated transcripts can include functional protein-coding genes and long non-coding RNA that are not yet studied or annotated (Su et al., 2004), thus they were not eliminated from our differential expression results or module construction. To validate that the modules are not influenced by potential transcriptional noise from unannotated genes, we omitted those genes from the hdWGCNA module construction. Modules of interest identified in the WGCNA were consistent with modules identified in the hdWGCNA, indicating that signal from the unannotated genes did not “override” the overall module structure.
Inhibitory cell subtypes were the most sensitive neuronal cell types to the CIE paradigm, with inhibitory neurons subtype C being the most susceptible. Cortical layer-specific dissection followed by snRNA-seq identified caudal ganglionic eminence (CGE)-derived inhibitory neurons (Lamp5/Pax6 and VIP interneurons) to be enriched in Adarb2 and Gad1 and to be enriched in layer I-III (Hodge et al., 2019). Our data shows inhibitory cells subtypes C and F to be Adarb2 and Gad1 positive, suggesting their CGE origin. Importantly, since inhibitory cell type C and F showed transcriptional similarity, we integrated the snRNA-seq and spatial datasets to show that these cell types have distinctly different spatial locations. We identified Cpa6 as a hub gene of highly dysregulated gene co-expression modules in two inhibitory cell subtypes and oligodendrocytes. Cpa6 was significantly enriched in inhibitory cells subtype C. Our data show low average expression of Cpa6 in oligodendrocytes; however, being a hub gene of a highly dysregulated module, suggests that it might be expressed in only a subcluster of oligodendrocytes. Unsupervised clustering of oligodendrocytes identified a subset enriched in Cpa6, we utilized pseudotime analysis to order oligodendrocytes on a continuum of maturity stages. Cpa6 expressing oligodendrocytes are shown in pseudotime analysis to be of immature nature. This subset of oligodendrocytes was also enriched in Vipr2 suggesting an association of this subset of oligodendrocytes with the Cpa6 positive VIP neurons. Our study identifies Cpa6 as a novel transcriptomic target for AUD research, to our knowledge, there is no research on its role in mediating excessive alcohol drinking associated with dependence; however, GWAS studies identified CPA6 and AUTS2 to be associated with alcohol consumption at genome-wide significance (Schumann et al., 2011). Cpa6 has been linked to epilepsy (Salzmann et al., 2012; Sapio et al., 2012, 2015) and neurodevelopment (Lyons et al., 2010; Wilfert et al., 2021), GWAS studies linked it to Alzheimer’s disease (Adewuyi et al., 2022), and unipolar depression (Thalamuthu et al., 2022). We show Auts2 to be highly connected to the hub gene Cpa6 in the highly dysregulated gene co-expression module in inhibitory cells subtype C, suggesting an important network in the alcohol use disorders context. In contrast to inhibitory cells, excitatory neurons subtypes did not show distinct gene markers, indicating subtle transcriptomic differences between the subtypes, integration of snRNA sequencing and spatial transcriptomics data showed regional specificity of each subtype. This indicates that the subtypes are brain region driven rather than functionally driven.
Human postmortem snRNA-seq studies identified oligodendrocytes as the most susceptible cell type in the mPFC of individuals with AUD (Brenner et al., 2020). This current study confirms this finding and validates the translational relevance of the CIE paradigm to human alcohol dependence. We identified Pde4b as a critical hub gene of a highly dysregulated module in oligodendrocytes; differential expression analysis shows upregulation of Pde4b in alcohol-dependent mice, all dysregulated genes in the Pde4b module were upregulated, indicating that upregulation of the hub genes resulted in upregulation of the module. Nonselective PDE4 inhibitors decrease ethanol intake in mice (Blednov et al., 2014, 2018, 2020, 2022), rats (Wen et al., 2012; Franklin et al., 2015), and in non-treatment seeking individuals with alcohol use disorders (Grigsby et al., 2023); thus, the current work may shed additional light on the molecular and cellular mechanisms associated with this class of drugs. In addition, PDE4B was recently identified as significantly associated with genetic liability to substance use disorders (Hatoum et al., 2023; Clarke et al., 2017; Zhou et al., 2020; Liu et al., 2019). These results further strengthen the link between Pde4b and AUD and highlight oligodendrocytes’ connection with this target and offer insight into its downstream genes.
Our data identifies Gpc5 as a gene enriched in astrocytes and a hub gene of an astrocyte upregulated module. Gpc5, a cell surface heparan sulfate proteoglycan, was shown to be linked with alcohol consumption in humans and modulated behavioral response to ethanol in Drosophila (Joslyn et al., 2011). Our data identifies Gpc5 as a potential gene of interest in studying astrocytes’ role in excessive alcohol drinking associated with alcohol dependence. A secondary analysis of our published data from dlPFC from individuals with AUD (Brenner et al., 2020) also identified an astrocytic gene expression module with GPC5 as a hub gene. This module was upregulated in astrocytes from AUD individuals, and gene members of the human GPC5 module significantly overlapped with gene members of mouse Gpc5 modules (Supplementary Fig. 4p&q). This cross-species analysis shows the conservation between dysregulated modules in a mouse model of alcohol dependence and human AUD data emphasizing the validity and translational value of the identified signals.
Our results link alcohol-dependence dysregulated genes to neuroinflammation. Microglia was the only cell type with differentially expressed genes showing enrichment in neuroinflammation genes, however multiple neuroinflammation genes were dysregulated in other cell types in alcohol-dependent samples. Lcn2, a downregulated gene in multiple cell types in alcohol-dependent samples, was shown to be a chemokine inducer in the CNS, to accelerate recruitment of astrocytes during inflammatory conditions (Lee et al., 2011), and to protect the brain during LPS induced inflammation (Kang et al., 2018). Other data showed its role in neurotoxic glial activation under chronic inflammatory conditions (M. Jin et al., 2014). Lcn2 was upregulated in our RNAseq data from astrocytes and microglia from alcohol dependence model (Emma K. Erickson et al., 2019). This suggests that Lcn2 expression might be dynamic depending on the context of neuroinflammation. Neat1, a lncRNA upregulated in alcohol-dependent treated animals’ oligodendrocytes (Supplementary Fig. 4n), has been shown to mediate and promote inflammation in cell culture and in animal models. It was shown to promote LPS induced inflammation in macrophages (Dai et al., 2021; Li et al., 2020; Zhang et al., 2019). Neat1 was upregulated after middle cerebral artery occlusion (MCOA), an experimental model of stroke, and it was a hub gene positively correlated with activated microglia, further linking the lncRNA to neuroinflammatory responses (F. Jin et al., 2021). Our WGCNA data shows Neat1 as a member of the upregulated Pde4b module.
Bank1, a gene shown to be associated with multiple substance use disorders in a multivariate genome-wide association meta-analysis of over 1 million subjects (Hatoum et al., 2023), is upregulated in alcohol-dependent microglia (Supplementary Fig. 4o). Bank1 was shown to be associated with critical immune signaling pathways including, Tlr7 pathway (Le Berre et al., 2021).
Collectively our data identifies cell-type specific changes in alcohol dependence. Dysregulated networks can be a detrimental consequence of alcohol dependence or a compensatory mechanism to restore normal PFC functions. Our data identifies a susceptible upregulated network in astrocytes; cortical astrocytes have been shown to regulate acute stimulatory and sedative-hypnotic ethanol-induced phenotypes (Erickson et al., 2021). We additionally identified a downregulated module in the most susceptible inhibitory cell type, Cpa6 the hub gene of this module was shown in GWAS to be associated with alcohol dependence. Our analysis of neuronal cell communication suggests decreased inhibitory signaling and increase in excitatory and neurexin-neuroligin signaling, a shift that may contribute to alcohol-dependence induced neuro-adaptations (Constance et al., 2018; Fish et al., 2022). While possible cellular damage after CIE treatment can influence some of the transcriptomic changes we identified, our differentially expressed genes are not enriched in cell death or cellular damage associated pathways.
Overall, this study identifies new alcohol-related genes, sensitive mPFC cell types, and dysregulated gene co-expression networks in a mouse model of alcohol dependence. Cell-type specific signatures in the mouse model of alcohol dependence allow for mechanistic and functional studies of changes identified in human data through targeting common human-mouse dysregulated mechanisms. Spatial transcriptomics enabled the deciphering of spatial location of transcriptionally similar cell subtypes; spatial location of cells or transcripts are not retained in snRNA-seq.
Supplementary Material
Acknowledgments
This project was funded by NIAAA/NIH grants: K00 AA029955 (NAS), R01 AA012404 (RDM), U01 AA020926 (RDM), U01AA013498 (MR & AJR), R01 AA021491 (MR & AJR). Chromium Single Cell 3’ RNA Sequencing and Visium Spatial Gene Expression library preparations and sequencing were performed by the Genomic Sequencing and Analysis Facility at UT Austin, Center for Biomedical Research Support. RRID#: SCR_021713
Footnotes
Credit author statement
NAS, AJR, MR and RDM conceived the project and designed the experiments. AJR performed the CIE experiments. NAS, LM and OP performed tissue sectioning & nuclei isolations. NS & MWK performed the bioinformatic analysis. All authors drafted, edited, and approved the final version of the manuscript.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2023.106361.
Data availability
The raw FASTQ files and processed data generated for this study have been deposited in the Gene Expression Omnibus (GEO) database (accession number GSE233763). The processed data can be accessed and visualized using the Shiny app: https://wcaar.shinyapps.io/CIE_PFC_snRNA/
References
- Abernathy Kenneth, Judson Chandler L, Woodward John J., 2010. Alcohol and the prefrontal cortex. Int. Rev. Neurobiol 91, 289–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achicallende Svein, Río Itziar Bonilla-Del, Serrano Maitane, Mimenza Amaia, Lekunberri Leire, Anaut-Lusar Ilazki, Puente Nagore, Gerrikagoitia Inmaculada, Grandes Pedro, 2022. GLAST versus GFAP as Astroglial marker for the subcellular study of cannabinoid CB receptors in astrocytes. Histochem. Cell Biol 158 (6), 561–569. [DOI] [PubMed] [Google Scholar]
- Adewuyi Emmanuel O., O’Brien Eleanor K., Nyholt Dale R., Porter Tenielle, Laws Simon M., 2022. A large-scale genome-wide cross-trait analysis reveals shared genetic architecture between Alzheimer’s disease and gastrointestinal tract disorders. Communications Biology 5 (1), 691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila Diana V., Myers Scott A., Zhang Jingwen, Kharebava Giorgi, McClain Craig J., Kim Hee-Yong, Whittemore Scott R., Gobejishvili Leila, Barve Shirish, 2017. Phosphodiesterase 4b expression plays a major role in alcohol-induced neuroinflammation. Neuropharmacology 125 (October), 376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball Gareth, Stokes Paul R., Rhodes Rebecca A., Bose Subrata K., Rezek Iead, Wink Alle-Meije, Lord Louis-David, Mehta Mitul A., Grasby Paul M., Turkheimer Federico E., 2011. Executive functions and prefrontal cortex: A matter of persistence? Front. Syst. Neurosci 5 (January), 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berre Le, Ludmilla Mélanie Chesneau, Danger Richard, Dubois Florian, Chaussabel Damien, Garand Mathieu, Brouard Sophie, 2021. Connection of BANK1, tolerance, regulatory B cells, and apoptosis: perspectives of a reductionist investigation. Front. Immunol 12 (March), 589786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov Yuri A., Benavidez Jillian M., Black Mendy, Adron Harris R, 2014. Inhibition of phosphodiesterase 4 reduces ethanol intake and preference in C57BL/6J mice. Front. Neurosci 8 (May), 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov Yuri A., Da Costa Adriana J., Tarbox Tamara, Ponomareva Olga, Messing Robert O., Adron Harris R, 2018. Apremilast alters behavioral responses to ethanol in mice: I. Reduced consumption and preference. Alcohol. Clin. Exp. Res 42 (5), 926–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov Yuri A., Borghese Cecilia M., Dugan Michael P., Pradhan Swetak, Thodati Thanvi M., Kichili Nikhita R., Adron Harris R, Messing Robert O., 2020. Apremilast regulates acute effects of ethanol and other GABAergic drugs via protein kinase A-dependent signaling. Neuropharmacology 178 (November), 108220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov Yuri A., Costa Da, Adriana Mason, Sonia Mayfield, Jody Moss, Stephen J, Messing Robert O., 2022. Apremilast-induced increases in acute ethanol intoxication and decreases in ethanol drinking in mice involve PKA phosphorylation of GABAA β3 subunits. Neuropharmacology 220 (December), 109255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgonetti Vittoria, Roberts Amanda J., Bajo Michal, Galeotti Nicoletta, Roberto Marisa, 2023a. Chronic alcohol induced mechanical allodynia by promoting Neuroinflammation: A mouse model of alcohol-evoked neuropathic pain. Br. J. Pharmacol 180 (18), 2377–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgonetti Vittoria, Roberts Amanda J., Bajo Michal, Galeotti Nicoletta, Roberto Marisa, 2023b. Chronic alcohol induced mechanical allodynia by promoting Neuroinflammation: A mouse model of alcohol-evoked neuropathic pain. Br. J. Pharmacol 10.1111/bph.16091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner Eric, Tiwari Gayatri R., Kapoor Manav, Liu Yunlong, Brock Amy, Dayne Mayfield R, 2020. Single cell transcriptome profiling of the human alcohol-dependent brain. Hum. Mol. Genet 29 (7), 1144–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky Oleg, Jedrychowski Mark P., Moore Craig S., Cialic Ron, Lanser Amanda J., Gabriely Galina, Koeglsperger Thomas, et al. , 2014. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat. Neurosci 17 (1), 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherchi Federica, Bulli Irene, Venturini Martina, Pugliese Anna Maria, Coppi Elisabetta, 2021. Ion channels as new attractive targets to improve re-myelination processes in the brain. Int. J. Mol. Sci 22 (14) 10.3390/ijms22147277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke T-K, Adams MJ, Davies G, Howard DM, Hall LS, Padmanabhan S, Murray AD, et al. , 2017. Genome-wide association study of alcohol consumption and genetic overlap with other health-related traits in UK biobank (N=112 117). Mol. Psychiatry 22 (10), 1376–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constance William D., Mukherjee Amrita, Fisher Yvette E., Pop Sinziana, Blanc Eric, Toyama Yusuke, Williams Darren W., 2018. Neurexin and Neuroligin-based adhesion complexes drive axonal arborisation growth independent of synaptic activity. eLife 7 (March). 10.7554/eLife.31659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz Bryan, Borgonetti Vittoria, Bajo Michal, Roberto Marisa, 2023. Sex-dependent factors of alcohol and Neuroimmune mechanisms. Neurobiol. Stress 26 (100562), 100562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Wenyu, Wang Manyi, Wang Peiqi, Wen Ji, Wang Jiangyue, Cha Sa, Xiao Xueling, He Yiruo, Shu Rui, Bai Ding, 2021. lncRNA NEAT1 ameliorates LPS-induced inflammation in MG63 cells by activating autophagy and suppressing the NLRP3 Inflammasome. Int. J. Mol. Med 47 (2), 607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilly Geoffrey A., Kittleman Cory W., Kerr Tony M., Messing Robert O., Dayne Mayfield R, 2022. Cell-type specific changes in PKC-Delta neurons of the central amygdala during alcohol withdrawal. Transl. Psychiatry 12 (1), 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers Cindy L., Sanchez-Alavez Manuel, Wills Derek, 2018. Effect of gabapentin on sleep and Delta and Theta EEG power in adult rats exposed to chronic intermittent ethanol vapor and protracted withdrawal during adolescence. Psychopharmacology 235 (6), 1783–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison JA, de Vellis J, 1994. Platelet-derived growth factor receptor is expressed by cells in the early oligodendrocyte lineage. J. Neurosci. Res 37 (1), 116–128. [DOI] [PubMed] [Google Scholar]
- Erickson Emma K., Blednov Yuri A., Adron Harris R, Dayne Mayfield R, 2019. Glial gene networks associated with alcohol dependence. Sci. Rep 9 (1), 10949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson EK, DaCosta AJ, Mason SC, Blednov YA, Mayfield RD, Harris RA, 2021. Cortical astrocytes regulate ethanol consumption and intoxication in mice. Neuropsychopharmacology vol. 46 (3), 500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcão Ana Mendanha, van Bruggen David, Marques Sueli, Meijer Mandy, Jäkel Sarah, Agirre Eneritz, Samudyata, et al. , 2018. Disease-specific oligodendrocyte lineage cells Arise in multiple sclerosis. Nat. Med 24 (12), 1837–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farris SP, Tiwari GR, Ponomareva O, Lopez MF, Mayfield RD, Becker HC, 2020. Transcriptome analysis of alcohol drinking in non-dependent and dependent mice following repeated cycles of forced swim stress exposure. Brain Sci. 10 (5) 10.3390/brainsci10050275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson Laura B., Zhang Lingling, Kircher Daniel, Wang Shi, Mayfield, Dayne R, Crabbe John C., Morrisett Richard A., Adron Harris R, Ponomarev Igor, 2019. Dissecting brain networks underlying alcohol binge drinking using a systems genomics approach. Mol. Neurobiol 56 (4), 2791–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak Greg, McDavid Andrew, Yajima Masanao, Deng Jingyuan, Gersuk Vivian, Shalek Alex K., Slichter Chloe K., et al. , 2015. MAST: A flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16 (1), 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish Kenneth N., Max E, 2022. Targeting prefrontal cortex GABAergic microcircuits for the treatment of alcohol use disorder. Front. Synaptic Neurosci 14, 936911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flatscher-Bader Traute, van der Brug Marcel, Hwang John W., Gochee Peter A., Matsumoto Izuru, Niwa Shin-Ichi, Wilce Peter A., 2005. Alcohol-responsive genes in the frontal cortex and nucleus Accumbens of human alcoholics. J. Neurochem 93 (2), 359–370. [DOI] [PubMed] [Google Scholar]
- Franklin Kelle M., Hauser Sheketha R., Lasek Amy W., McClintick Jeanette, Ding Zheng-Ming, McBride William J., Bell Richard L., 2015. Reduction of alcohol drinking of alcohol-preferring (P) and high-alcohol drinking (HAD1) rats by targeting Phosphodiesterase-4 (PDE4). Psychopharmacology 232 (13), 2251–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galiano MR, Andrieux A, Deloulme JC, Bosc C, Schweitzer A, Job D, Hallak ME, 2006. Myelin basic protein functions as a microtubule stabilizing protein in differentiated oligodendrocytes. J. Neurosci. Res 84 (3), 534–541. [DOI] [PubMed] [Google Scholar]
- Grigsby Kolter B., Mangieri Regina A., Roberts Amanda J., Lopez Marcelo F., Firsick Evan J., Townsley Kayla G., Beneze Alan, et al. , 2023. Preclinical and clinical evidence for suppression of alcohol intake by Apremilast. J. Clin. Invest 133 (6) 10.1172/JCI159103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Hongyu, Li Jun, 2021. scSorter: assigning cells to known cell types according to marker genes. Genome Biol. 22 (1), 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatoum Alexander S., Colbert Sarah M.C., Johnson Emma C., Huggett Spencer B., Deak Jose, Pathak Gita A., Jennings Mariela V., et al. , 2023. Multivariate genome-wide association Meta-analysis of over 1 million subjects identifies loci underlying multiple substance use disorders. Nature Mental Health 1 (3), 210–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig M, Barbier E, Johnstone AL, Tapocik J, Meinhardt MW, Pfarr S, Wahlestedt C, Sommer WH, 2017. Reprogramming of mPFC transcriptome and function in alcohol dependence. Genes Brain Behav. 16 (1), 86–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge Rebecca D., Bakken Trygve E., Miller Jeremy A., Smith Kimberly A., Barkan Eliza R., Graybuck Lucas T., Close Jennie L., et al. , 2019. Conserved cell types with divergent features in human versus mouse cortex. Nature 573 (7772), 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Wei, Bhaduri Aparna, Velmeshev Dmitry, Wang Shaohui, Wang Li, Rottkamp Catherine A., Alvarez-Buylla Arturo, Rowitch David H., Kriegstein Arnold R., 2020. Origins and proliferative states of human oligodendrocyte precursor cells. Cell 182 (3), 594–608.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Myungwon, Jang Eunha, Suk Kyoungho, 2014. Lipocalin-2 acts as a Neuroinflammatogen in lipopolysaccharide-injected mice. Experimental Neurobiology 23 (2), 155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Fa, Weiyang Ou, Wei Boyang, Fan Haiyan, Wei Chengcong, Fang Dazhao, Li Guangxu, Liu Wenchao, Liu Jiahui, Jin Lei, 2021. Transcriptome-wide analysis to identify the inflammatory role of lncRNA Neat1 in experimental ischemic stroke. Journal of Inflammation Research 14, 2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joslyn Geoff, Wolf Fred W., Brush Gerry, Lianqun Wu, Schuckit Marc, White Raymond L., 2011. Glypican gene GPC5 participates in the behavioral response to ethanol: evidence from humans. Mice, and Fruit Flies G3 1 (7), 627–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SS, Ren Y, Liu CC, Kurti A, Baker KE, Bu G, Asmann Y, Fryer JD, 2018. Lipocalin-2 protects the brain during inflammatory conditions. Mol. Psychiatry 23 (2), 344–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagiannis Tanya T., Monti Stefano, Sebastiani Paola, 2022. Cell type diversity statistic: an entropy-based metric to compare overall cell type composition across samples. Front. Genet 13 (April), 855076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimbrough Adam, de Guglielmo Giordano, Kononoff Jenni, Kallupi Marsida, Zorrilla Eric P., George Olivier, 2017. CRF receptor-dependent increases in irritability-like behavior during abstinence from chronic intermittent ethanol vapor exposure. Alcohol. Clin. Exp. Res 41 (11), 1886–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder Peter, Horvath Steve, 2008. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau Shun-Fat, Cao Han, Fu Amy K.Y., Nancy Y.Ip., 2020. Single-nucleus transcriptome analysis reveals dysregulation of Angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A 117 (41), 25800–25809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Shinrye, Kim Jong-Heon, Kim Jae-Hong, Seo Jung-Wan, Han Hyung-Soo, Lee Won-Ha, Mori Kiyoshi, Nakao Kazuwa, Barasch Jonathan, Suk Kyoungho, 2011. Lipocalin-2 is a chemokine inducer in the central nervous system: ROLE OF CHEMOKINE LIGAND 10 (CXCL10) IN LIPOCALIN-2-INDUCED CELL MIGRATION *. J. Biol. Chem 286 (51), 43855–43870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein Ed S., Hawrylycz Michael J., Ao Nancy, Ayres Mikael, Bensinger Amy, Bernard Amy, Boe Andrew F., et al. , 2007. Genome-wide atlas of gene expression in the adult mouse brain. Nature 445 (7124), 168–176. [DOI] [PubMed] [Google Scholar]
- Li Yanhui, Guo Wei, Cai Yeping, 2020. NEAT1 promotes LPS-induced inflammatory injury in macrophages by regulating miR-17–5p/TLR4. Open Medicine 15 (1), 38–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, MacDonald RJ, Swift GH, 2001. DNA binding and transcriptional activation by a PDX1.PBX1b.MEIS2b trimer and cooperation with a pancreas-specific basic Helix-loop-Helix complex. J. Biol. Chem 276 (21), 17985–17993. [DOI] [PubMed] [Google Scholar]
- Liu Mengzhen, Jiang Yu, Wedow Robbee, Li Yue, Brazel David M., Chen Fang, Datta Gargi, et al. , 2019. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat. Genet 51 (2), 237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo Sophia K., Guo Margaret G., Ji Andrew L., Khavari Paul A., 2021. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nat. Rev. Genet 22 (10), 627–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez Marcelo F., Becker Howard C., 2005. Effect of pattern and number of chronic ethanol exposures on subsequent voluntary ethanol intake in C57BL/6J mice. Psychopharmacology 181 (4), 688–696. [DOI] [PubMed] [Google Scholar]
- Love Michael I., Huber Wolfgang, Anders Simon, 2014. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 15 (12), 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons Peter J., Ma Leung-Hang, Baker Robert, Fricker Lloyd D., 2010. Carboxypeptidase A6 in zebrafish development and implications for VIth cranial nerve pathfinding. PloS One 5 (9), e12967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Zhun, Deng Ke, Wang Xiaowo, Zhang Xuegong, 2018. DEsingle for detecting three types of differential expression in single-cell RNA-Seq data. Bioinformatics 34 (18), 3223–3224. [DOI] [PubMed] [Google Scholar]
- Morabito Samuel, Miyoshi Emily, Michael Neethu, Shahin Saba, Martini Alessandra Cadete, Head Elizabeth, Silva Justine, Leavy Kelsey, Perez-Rosendahl Mari, Swarup Vivek, 2021. Single-nucleus chromatin accessibility and transcriptomic characterization of Alzheimer’s disease. Nat. Genet 10.1038/s41588-021-00894-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morabito Samuel, Reese Fairlie, Rahimzadeh Negin, Miyoshi Emily, Swarup Vivek, 2022. High dimensional co-expression networks enable discovery of transcriptomic drivers in complex biological systems. bioRxiv. 10.1101/2022.09.22.509094. [DOI] [Google Scholar]
- Osterndorff-Kahanek Elizabeth A., Becker Howard C., Lopez Marcelo F., Farris Sean P., Tiwari Gayatri R., Nunez Yury O., Adron Harris R, Dayne Mayfield R, 2015. Chronic ethanol exposure produces time- and brain region-dependent changes in gene Coexpression networks. PloS One 10 (3), e0121522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Wang T, Zhao Z, Wei W, Yang X, Wang X, Xin W, 2022. Novel insights into the emerging role of Neat1 and its effects downstream in the regulation of inflammation. J. Inflamm. Res 15, 557–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel Reesha R., Khom Sophia, Steinman Michael Q., Varodayan Florence P., Kiosses William B., Hedges David M., Vlkolinsky Roman, et al. , 2019. IL-1β expression is increased and regulates GABA transmission following chronic ethanol in mouse central amygdala. Brain Behav. Immun 75 (January), 208–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel Reesha R., Wolfe Sarah A., Bajo Michal, Abeynaike Shawn, Pahng Amanda, Borgonetti Vittoria, D’Ambrosio Shannon, et al. , 2021. IL-10 normalizes aberrant amygdala GABA transmission and reverses anxiety-like behavior and dependence-induced escalation of alcohol intake. Prog. Neurobiol 199 (April), 101952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel Reesha R., Wolfe Sarah A., Borgonetti Vittoria, Gandhi Pauravi J., Rodriguez Larry, Snyder Angela E., D’Ambrosio Shannon, et al. , 2022. Ethanol withdrawal-induced adaptations in prefrontal Corticotropin releasing factor receptor 1-expressing neurons regulate anxiety and conditioned rewarding effects of ethanol. Mol. Psychiatry 27 (8), 3441–3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson Mark D., McCarthy Davis J., Smyth Gordon K., 2010. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26 (1), 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzmann Annick, Guipponi Michel, Lyons Peter J., Fricker Lloyd D., Sapio Matthew, Lambercy Carmen, Buresi Catherine, et al. , 2012. Carboxypeptidase A6 gene (CPA6) mutations in a recessive familial form of febrile seizures and temporal lobe epilepsy and in sporadic temporal lobe epilepsy. Hum. Mutat 33 (1), 124–135. [DOI] [PubMed] [Google Scholar]
- Sapio Matthew R., Salzmann Annick, Vessaz Monique, Crespel Arielle, Lyons Peter J., Malafosse Alain, Fricker Lloyd D., 2012. Naturally occurring carboxypeptidase A6 mutations: effect on enzyme function and association with epilepsy. J. Biol. Chem 287 (51), 42900–42909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapio Matthew R., Vessaz Monique, Thomas Pierre, Genton Pierre, Fricker Lloyd D., Salzmann Annick, 2015. Novel carboxypeptidase A6 (CPA6) mutations identified in patients with juvenile myoclonic and generalized epilepsy. PloS One 10 (4), e0123180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann Gunter, Coin Lachlan J., Lourdusamy Anbarasu, Charoen Pimphen, Berger Karen H., Stacey David, Desrivières Sylvane, et al. , 2011. Genome-wide association and genetic functional studies identify autism susceptibility candidate 2 gene (AUTS2) in the regulation of alcohol consumption. Proc. Natl. Acad. Sci. U. S. A 108 (17), 7119–7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz Nancy B., Domowicz Miriam S., 2018. Proteoglycans in brain development and pathogenesis. FEBS Lett. 592 (23), 3791–3805. [DOI] [PubMed] [Google Scholar]
- Squair Jordan W., Gautier Matthieu, Kathe Claudia, Anderson Mark A., James Nicholas D., Hutson Thomas H., Hudelle Ŕemi, et al. , 2021. Confronting false discoveries in single-cell differential expression. Nat. Commun 12 (1), 5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart Tim, Butler Andrew, Hoffman Paul, Hafemeister Christoph, Papalexi Efthymia, Mauck William M. 3rd, Hao Yuhan, Stoeckius Marlon, Smibert Peter, Satija Rahul, 2019. Comprehensive integration of single-cell data. Cell 177 (7), 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Andrew I., Wiltshire Tim, Batalov Serge, Lapp Hilmar, Ching Keith A., Block David, Zhang Jie, et al. , 2004. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. U. S. A 101 (16), 6062–6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalamuthu Anbupalam, Mills Natalie T., Berger Klaus, Minnerup Heike, Grotegerd Dominik, Dannlowski Udo, Meinert Susanne, et al. , 2022. Genome-wide interaction study with major depression identifies novel variants associated with cognitive function. Mol. Psychiatry 27 (2), 1111–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran Matthew N., Maynard Kristen R., Spangler Abby, Huuki Louise A., Montgomery Kelsey D., Sadashivaiah Vijay, Tippani Madhavi, et al. , 2021. Single-nucleus transcriptome analysis reveals cell-type-specific molecular signatures across reward circuitry in the human brain. Neuron 109 (19), 3088–3103.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell Cole, Cacchiarelli Davide, Grimsby Jonna, Pokharel Prapti, Li Shuqiang, Morse Michael, Lennon Niall J., Livak Kenneth J., Mikkelsen Tarjei S., Rinn John L., 2014. The dynamics and regulators of cell fate decisions are revealed by Pseudotemporal ordering of single cells. Nat. Biotechnol 32 (4), 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den, Oord Edwin J.C.G., Xie Lin Y., Zhao Min, Aberg Karolina A., Clark Shaunna L., 2023. A single-nucleus transcriptomics study of alcohol use disorder in the nucleus Accumbens. Addict. Biol 28 (1), e13250. [DOI] [PubMed] [Google Scholar]
- Varodayan FP, Pahng AR, Davis TD, Gandhi P, Bajo M, Steinman MQ, Kiosses WB, et al. , 2023. Chronic ethanol induces a pro-inflammatory switch in interleukin-1β regulation of GABAergic signaling in the medial prefrontal cortex of male mice. Brain Behav. Immun 110 (May), 125–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warden Anna S., Dayne Mayfield R, 2017. Gene expression profiling in the human alcoholic brain. Neuropharmacology. 10.1016/j.neuropharm.2017.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warden Anna S., Wolfe Sarah A., Khom Sophia, Varodayan Florence P., Patel Reesha R., Steinman Michael Q., Bajo Michal, et al. , 2020. Microglia control escalation of drinking in alcohol-dependent mice: genomic and synaptic drivers. Biol. Psychiatry 88 (12), 910–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warden Anna S., Triplett Todd A., Lyu Aram, Grantham Emily K., Azzam Moatasem M., DaCosta Adriana, Mason Sonia, et al. , 2021. Microglia depletion and alcohol: transcriptome and behavioral profiles. Addict. Biol 26 (2), e12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Rui-Ting, Zhang Min, Qin Wang-Jun, Liu Qing, Wang Wei-Ping, Lawrence Andrew J., Zhang Han-Ting, Liang Jian-Hui, 2012. The Phosphodiesterase-4 (PDE4) inhibitor Rolipram decreases ethanol seeking and consumption in alcohol-preferring fawn-hooded rats. Alcohol. Clin. Exp. Res 36 (12), 2157–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilfert Amy B., Turner Tychele N., Murali Shwetha C., Hsieh Pinghsun, Sulovari Arvis, Wang Tianyun, Coe Bradley P., et al. , 2021. Recent ultra-rare inherited variants implicate new autism candidate risk genes. Nat. Genet 53 (8), 1125–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Momona, Iwase Miho, Sasaki Binri, Suzuki Nobuharu, 2022. The molecular regulation of oligodendrocyte development and CNS myelination by ECM proteins. Frontiers in Cell and Developmental Biology 10 (September), 952135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Pengfei, Cao Limian, Zhou Rongbin, Yang Xiaolu, Mian Wu., 2019. The lncRNA Neat1 promotes activation of Inflammasomes in macrophages. Nat. Commun 10 (1), 1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Martin Jinye, Ntranos Vasilis, Tse David, 2020. Determining sequencing depth in a single-cell RNA-Seq experiment. Nat. Commun 11 (1), 774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Yan, Xie Yong-Zhi, Liu You-Shuo, 2022. Accelerated aging-related transcriptome alterations in neurovascular unit cells in the brain of Alzheimer’s disease. Front. Aging Neurosci 14 (August), 949074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Wei, Johnston Kevin G., Ren Honglei, Xiangmin Xu, Nie Qing, 2023. Inferring neuron-neuron communications from single-cell transcriptomics through NeuronChat. Nat. Commun 14 (1), 1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Yingyao, Zhou Bin, Pache Lars, Chang Max, Khodabakhshi Alireza Hadj, Tanaseichuk Olga, Benner Christopher, Chanda Sumit K., 2019. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun 10 (1), 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Hang, Sealock Julia M., Sanchez-Roige Sandra, Clarke Toni-Kim, Levey Daniel F., Cheng Zhongshan, Li Boyang, et al. , 2020. Genome-wide Meta-analysis of problematic alcohol use in 435,563 individuals yields insights into biology and relationships with other traits. Nat. Neurosci 23 (7), 809–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw FASTQ files and processed data generated for this study have been deposited in the Gene Expression Omnibus (GEO) database (accession number GSE233763). The processed data can be accessed and visualized using the Shiny app: https://wcaar.shinyapps.io/CIE_PFC_snRNA/