

Abstract
Ribosomes are key to cellular self-fabrication and limit growth rate. While most enzymes are proteins, ribosomes consist of 1/3 protein and 2/3 ribonucleic acid (RNA) (in E. coli).
Here, we develop a mechanistic model of a self-fabricating cell, validated across diverse growth conditions. Through resource balance analysis (RBA), we explore the variation in maximum growth rate with ribosome composition, assuming constant kinetic parameters.
Our model highlights the importance of RNA instability. If we neglect it, RNA synthesis is always cheaper than protein synthesis, leading to an RNA-only ribosome at maximum growth rate. Upon accounting for RNA turnover, we find that a mixed ribosome composed of RNA and proteins maximizes growth rate. To account for RNA turnover, we explore two scenarios regarding the activity of RNases. In (a) degradation is proportional to RNA content. In (b) ribosomal proteins cooperatively mitigate RNA instability by protecting it from misfolding and subsequent degradation. In both cases, higher protein content elevates protein synthesis costs and simultaneously lowers RNA turnover expenses, resulting in mixed RNA-protein ribosomes. Only scenario (b) aligns qualitatively with experimental data across varied growth conditions.
Our research provides fresh insights into ribosome biogenesis and evolution, paving the way for understanding protein-rich ribosomes in archaea and mitochondria.
Subject terms: Computational models, Bioenergetics
Mechanistic modeling (resource balance analysis) links ribosome composition to growth rate. Only if the costs of ribosomal RNA stabilization are taken into account, maximum growth occurs for a mixed (RNA+protein) ribosome, as observed experimentally.
Introduction
The ribosome is at the core of any (known) self-replicating organism. In a process called translation, ribosomes read the instructions from messenger ribonucleic acids (mRNAs) to synthesize the corresponding proteins, including ribosomal proteins (rPs). This autocatalytic nature of ribosomes ultimately limits the doubling time of a cell to the period it takes a ribosome to synthesize itself1,2. In Escherichia coli this would be 6 min, assuming that the ribosome consists of a 55-protein complex of approximately 7400 amino acids (AAs) that is translated at a speed of 21 AA/sec3. In fact, even in growth-optimized E. coli, that doubling limit remains far from being reached4. Nonetheless, it has been proposed that ribosomes, not only in E. coli, have been subjected to strong selective pressure to minimize their own duplication time in order to speed up the production of all other proteins5. With this principle in mind, Reuveni et al.5 explain why ribosomes have many rPs of similar length.
Ribosomes are structures that have developed over time by adding ribosomal ribonucleic acid (rRNA) and rP around a central core6. This core is considered to be a leftover from ancient translation systems that evolved alongside the genetic code. Different types of ribosomes have evolved in bacteria, archaea, and eukaryotes, but their overall structures are similar within each kingdom7. For example, the mass of ribosomes in prokaryotes is made up of approximately 63% rRNA and 37% rPs8,9, whereas eukaryotic ribosomes have an equal mass distribution of rRNA and rPs5,10,11. Thus, the question arises whether there is an evolutionary advantage in having such a high RNA content.
It has been suggested that the ribosome composition can be understood as a competition for resources between rRNA synthesis and rP synthesis12,13. In particular, Kostinski and Reuveni12 derived two upper bounds on growth rate resulting from two autocatalytic loops, one for rP production, and one for RNA polymerase (RNAP) and rRNA production. By analyzing allocation data from E. coli, they concluded that maximum growth rate occurs at the current ribosome composition of 2/3 RNA and 1/3 protein. However, the specific processes that limit the two autocatalytic processes remained elusive.
Here we aim to provide a mechanistic understanding of these processes. We set up a small (coarse-grained) model of a self-replicating cell and perform resource balance analysis (RBA)14,15. In particular, we vary ribosome composition and ribosome allocations (fractions of ribosomes allocated to the synthesis of different proteins) and maximize growth rate. For simplicity, we assume a constant ribosome mass.
We focus on the primary role of ribosomal proteins, which is stabilizing rRNA (by preventing its degradation or misfolding). Ribosome protein content might also affect other parameters, such as translation rate. Proteins are generally better catalysts than RNA16, but the ribosome’s catalytic core is formed by rRNA17 and operates at a relatively slow catalytic rate compared to typical enzymes. This suggests that there is little evolutionary pressure to increase the catalytic rate. Furthermore, ribosomes with the lowest protein content, like the E. coli ribosome, exhibit the highest translation rates18–20. Therefore, we do not consider the impact on translation rate in this study.
We find that the costs of stabilizing rRNA strongly influence the optimal ribosome composition. If we neglect rRNA turnover, our predictions suggest the presence of RNA-only ribosomes (in contrast to experimental evidence). Taking RNA degradation into account increases its biosynthesis costs, and maximum growth occurs for a mixed (RNA+protein) ribosome.
Results
We introduce a (coarse-grained) mechanistic model of a self-fabricating cell and investigate optimal ribosome composition using RBA. That is, we maximize growth rate under several sets of constraints. We validate the model by predicting RNAP fluxes (rRNA synthesis fluxes), and RNA to protein ratios at different growth rates. Ultimately, we predict maximum growth rate at different ribosome compositions.
A small model of a self-fabricating cell
We consider the small (coarse-grained) model of a self-fabricating cell depicted in Fig. 1. The cell imports a carbon source (C) and has two types of metabolic enzymes, one synthesizing amino acids (AA) from the carbon source and the other one synthesizing nucleotides (NT) from the carbon source and amino acids. RNA polymerase (RNAP) uses nucleotides to form the ribosomal RNA (rRNA), while the ribosome (R) uses amino acids to synthesize all proteins, including the importer (IC), the metabolic enzymes (EAA, ENT), the RNA polymerase and optionally a ribonuclease (RNase), the ribosomal assembly factors (AF), and the ribosomal proteins (rP). Finally, the assembly factors build the ribosome from ribosomal RNA and protein. In a base model, we neglect RNA degradation, whereas in an extended model we consider the enzyme (RNase) that breaks down RNA into nucleotides. We now provide a more formal definition of the two models.
Fig. 1. A small model of a self-fabricating cell.

a The cell imports a carbon source (C) and has two types of metabolic enzymes synthesizing amino acids (AA) from the carbon source and nucleotides (NT) from the carbon source and amino acids. The RNA polymerase (RNAP) uses nucleotides to form the ribosomal RNA (rRNA), and the ribosome (R) uses amino acids to synthesize the importer (IC), the metabolic enzymes (EAA, ENT), the ribosomal assembly factors (AF), and the ribosomal protein (rP). Finally, the assembly factors build the ribosome from ribosomal RNA and protein. The processes above constitute the base model. In the extended model, RNase degrades ribosomal RNA (and is synthesized by the ribosome). The additional processes are shown in red. Created with BioRender.com. b The resulting stoichiometric matrix and the corresponding flux vector. Here, s is used for protein synthesis reactions (and w for the corresponding fluxes), and r is used for all other reactions (and v for the corresponding fluxes). Additional columns and rows for the extended model are shown in red.
Given the stoichiometric matrix N and the vector of molar masses ω, the dynamic model of cellular growth relates growth rate μ, the vector of (metabolite, RNA, protein, and ribosome) concentrations c, and the vector of fluxes (v for enzymatic reactions and w for protein synthesis) according to
At steady state, growth rate μ and concentrations c are determined by the fluxes v and w,
| 1 |
To take limited cellular resources into account, we consider capacity constraints for the enzymatic fluxes v, including transcription (and optionally RNA degradation),
| 2a |
| 2b |
| 2c |
Further, we consider the ribosome capacity constraint for the protein synthesis fluxes w,
| 2d |
Here, nrRNA is the number of nucleotides in rRNA, ni is the number of amino acids in protein i, is the corresponding enzyme turnover rate, and and are the effective transcription and translation elongation rates, respectively. is the fraction of actively transcribing RNAPs, and is the fraction of actively translating ribosomes. As mentioned above, RNase is synthesized optionally and hence put in brackets. By defining the ribosome allocations,
| 3 |
that is, the fraction of ribosomes translating a certain protein i, constraint (2d) can be written as
We refer to the model given by Equations (1) and (2a, b, d) as the base RBA model. Equations (1) and (2), including (2c), define the extended RBA model which additionally considers RNA degradation.
Throughout our study, we consider a fixed molar ribosome mass ωR, but variable rRNA and protein content,
and we study the influence of ribosome composition on the cell’s maximum growth rate, under the constraints specified above. Here, ωNT and ωAA are the molar masses of nucleotides and amino acids, respectively. For convenience, we define the ribosomal protein (mass) fraction
| 4 |
and express nrRNA and nrP by xrP,
In our analysis, we vary ribosomal protein fraction and maximize growth rate under given constraints. Modeling details can be found in section Methods/subsection Model, the stoichiometric, capacity, and (dry) mass constraints are summarized in Table 1, and the parameter values, variables and the units are given in Supplementary Table 1. We used parameters from E. coli grown in six different media. Three of them are rich media (Gly+AA, Glc+AA, LB) where amino acids (and nucleotides) are provided so cells only have to express the corresponding transporters instead of the synthesis pathways. In our model, the enzymes ENT and EAA represent lumped pathways for glycolysis and nucleotide/amino acid synthesis, and we only consider one type of transporter. Therefore, to model the changing nutrient quality of the different media (inspired by Scott et al.21), we assume that turnover numbers of EAA and ENT increase with growth rate.
Table 1.
Constraints used in the extended and base models (with and without RNA degradation), see Fig. 1.
| vIC | vEAA | vENT | vRNAP | vRNase | vAF | wIC | wEAA | wENT | wRNAP | wRNase | wAF | wrP | sign | rhs | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C | 1 | −nAA | −nNT | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | = | 0 |
| AA | 0 | 1 | −1 | 0 | 0 | 0 | −nIC | −nEAA | −nENT | −nRNAP | −nRNase | −nAF | −nrP | = | 0 |
| NT | 0 | 0 | 1 | −nrRNA | nrRNA | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | = | 0 |
| rRNA | 0 | 0 | 0 | 1 | −1 | −1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ≥ (=) | 0 |
| rP | 0 | 0 | 0 | 0 | 0 | −1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | ≥ | 0 |
| cap IC | −μ | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ≥ | 0 | |
| cap EAA | 0 | −μ | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ≥ | 0 | |
| cap ENT | 0 | 0 | −μ | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ≥ | 0 | |
| cap RNAP | 0 | 0 | 0 | −μnrRNA | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ≥ | 0 | |
| cap RNase | 0 | 0 | 0 | 0 | −μnrRNA | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ≥ | 0 | |
| cap AF | 0 | 0 | 0 | 0 | 0 | −μ | 0 | 0 | 0 | 0 | 0 | 0 | ≥ | 0 | |
| cap R | 0 | 0 | 0 | 0 | 0 | −μnIC | −μnEAA | −μnENT | −μnRNAP | −μnRNase | −μnAF | −μnrP | ≥ (=) | 0 | |
| min deg | 0 | 0 | 0 | 0 | μ | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ≥ (=) | 0 | |
| (dry) mass | ωC | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | = | μ |
In particular, stoichiometric constraints (for the metabolites C, AA, NT, rRNA, rP), capacity constraints (for the catalysts IC, EAA, ENT, RNAP, RNase, AF, R), and the (dry) mass constraint. The column sign indicates an equality (=) or inequality (≥) constraint, and the column rhs specifies the right-hand side (a homogeneous or inhomogeneous constraint). The columns vRNase, wRNase, and the rows cap RNase and min deg are only present in the extended model.
Base model recovers linear correlation of RNA to protein ratio with growth rate
With parameters for E. coli in different media (listed in Supplementary Table 1) and the experimentally observed ribosome composition (xrP = 36%), the base model correctly recovers the well-known linear dependence of the RNA to protein ratio and growth rate21, see Fig. 2a, but not the offset at zero growth rate, since our model does not contain any non-growth associated processes and we assume constant translation elongation rate as in Scott et al.21. At low growth rate, decreases, most likely because of the lower availability of the required substrates20,22. Interestingly, when we use variable , we observe a nonzero offset (Supplementary Fig. 1).
Fig. 2. Validation of the base model.

a The model predicts a linear relationship between RNA to protein ratio and growth rate. The points represent the predicted maximum growth rates in six experimental conditions (Supplementary Table 1). The line is a linear fit. b Alternative RNAP fluxes at different non-optimal growth rates in glucose minimal medium. These are obtained by varying the growth rate step by step from zero to maximum and enumerating all solutions (elementary growth vectors as defined in Müller et al.23) for each growth rate. The grey and blue lines are the alternative solutions. The blue line corresponds to solutions, where rRNA and ribosomes do not accumulate (constraints rRNA and cap R in Table 1 are limiting). Light green diamonds are experimental data from Bremer and Dennis20, black triangles are the data from Bremer and Dennis20 corrected for rRNA degradation28. Data was converted to mmol g−1 with E. coli dry masses from Bremer and Dennis20 and the number of nucleotides in rRNA (nrRNA) at xrP = 36%.
To further test the model, we predict RNAP fluxes (vRNAP) at various non-optimal growth rates in glucose minimal medium. In particular, we compute alternative solutions to the system of (in)equalities (1) and (2). (Technically, these solutions are elementary growth vectors as defined in Müller et al.23). We observe three lines (Fig. 2b). Two lines (in gray) correspond to solutions where either ribosomes or rRNA accumulate in excess of what is needed to support growth. In other words, constraints (2d) and (2b) (rows cap R and rRNA in Table 1) are not limiting. With increasing growth rate, the excess of rRNA and ribosome decreases, reaching zero at the maximum growth rate. The third line (in blue) corresponds to no accumulation of ribosomes or rRNA. In particular, the RNAP flux exactly matches the demand. At maximum growth rate, all lines converge to one optimal value.
For higher growth rates, experimental data are best fit by the line without accumulation of ribosomes or rRNA. In fact, the accumulation of free rRNA in a cell is biologically not realistic as it is bound by rPs already during transcription24. Furthermore, if rRNA is expressed in excess of rPs, it is rapidly degraded25. For the growth rates studied here (0.4–2.1 h−1), the fraction of inactive ribosomes stays roughly constant at 15% to 20%3,20,22. In our model, we have already incorporated this fraction using the effective translation elongation rate (). (However, below the growth rate of ≈0.5 h−1, the fraction of active ribosomes rapidly decreases22). Therefore, the disagreement between experimental and simulated data at lower growth rates is probably caused by neglecting other types of RNA. Indeed, RNAP allocation to the synthesis of different types of RNA changes with growth rate3,20.
Base model predicts maximal growth for RNA-only ribosomes
We study the dependence of maximum growth rate on the ribosomal protein fraction using the base model described above. We find that, for realistic parameters from E. coli (Supplementary Table 1), rRNA synthesis is cheaper than protein synthesis for all tested growth conditions (see Fig. 3a). Thus, according to our base model, ribosomes should consist of rRNA only. Indeed, it has been suggested that higher growth rates could be achieved if ribosomes were to consist only of rRNA5.
Fig. 3. Maximum growth rate and ribosome allocations as functions of ribosomal protein fraction xrP for the base model.

a Maximum growth rate for E. coli in six different conditions (see Supplementary Table 1). b Ribosome allocations as defined in Eqn. (3), for glucose minimal medium (Glc).
If we (hypothetically) adjust the parameters to make rRNA synthesis more expensive than protein synthesis (e.g. by decreasing or increasing ), then maximum growth rate is achieved for a protein-only ribosome (Supplementary Fig. 2). By a symbolic analysis, we can rigorously prove that maximum growth rate is generically attained at an exclusive ribosome composition, either at xrP = 0% or xrP = 100%, regardless of the parameters (see section Methods/subsection Symbolic analysis of growth rate maximization).
To conclude, RBA with standard capacity constraints does not explain mixed (RNA+ protein) ribosomes. Thus, additional constraints are needed.
rRNA instability leads to maximal growth for mixed ribosomes
As one potential explanation, we hypothesize that the different stabilities of rPs and rRNA affect the composition of the ribosome. While proteins are known to be highly stable26, rRNA is susceptible to degradation by RNases, which are ubiquitous in cells27. Even at maximum growth, about 10% of rRNA is still degraded, and, thus, cannot be incorporated into the ribosome27,28. Furthermore, rRNA can easily misfold, rendering it inactive and prone to degradation24,29.
To account for rRNA degradation, we introduce an RNase enzyme that breaks down rRNA into individual nucleotides (NT), via the reaction
see Fig. 1. Since RNases are essential for quality control, we assume some minimum activity and add a minimum degradation rate,
| 5 |
to the list of constraints (row min deg in Table 1). In the simplest case, this rate is directly proportional (with a constant ) to the rRNA concentration. The latter is given by the fraction of rRNA in the ribosome concentration, since there is no free rRNA in the cell3. Additionally, can be a (monotonically decreasing) function of xrP,
| 6 |
modeling the cooperative protection of rRNA by proteins, where n is Hill-factor and K is half-saturation constant. As for the other enzymes, we add a capacity constraint for the RNase to account for its cost,
| 7 |
where we use of an enzyme called RNase R30. The base RBA model together with RNA degradation, RNase synthesis, and constraints (5) and (7) constitutes the extended RBA model.
Taking rRNA degradation into account leads to maximum growth rates at mixed (RNA+protein) ribosome compositions (Fig. 4). As it turns out, the assumption of a constant in constraint (5) leads to a very shallow optimum (Fig. 4a). To account for the stabilizing influence of rPs on the folded structure, we introduce the non-linear (Hill-type) degradation term (Equation (6) with half-saturation K = 0.2 and Hill-factors n = 2 or n = 6), leading to a pronounced optimum, see Fig. 4b and c.
Fig. 4. Extended model. Accounting for RNA degradation leads to a mixed (RNA+protein) ribosome composition.

a–c Maximum growth rate of E. coli in six different conditions (see Supplementary Table 1) for three types of rRNA degradation. a No cooperativity of rPs. b Weak cooperativity of rPs. c Strong cooperativity of rPs. d–f Ribosome allocations in glucose minimal medium (Glc) for three types of rRNA degradation. At low protein fractions, rRNA degradation is high, and RNAP (light green) takes up a large amount of cellular resources.
In the following, we investigate how the optimal ribosome composition depends on growth conditions.
First, we study growth on glucose minimal medium and adjust such that the optimal ribosome composition matches the experimentally observed value of xrP = 0.36 for E. coli. We validate the model for the three types of degradation, and we correctly predict the linear dependence of the RNA to protein ratio on growth rate (Supplementary Fig. 3). However, RNAP flux predictions are only realistic when assuming strong cooperativity (n = 6). For the other two cases, rRNA degradation in the optimum is too high which leads to overestimated RNAP fluxes (Supplementary Fig. 4).
Second, we predict maximum growth rate as a function of the ribosomal protein fraction in six different growth media. We find that the more proteins cooperate, the less the optimal ribosome composition depends on the growth conditions, see Fig. 4a–c.
Using variable or constant has no impact on the predicted optimal ribosome composition. As in the base model, variable leads to predicted non-zero offset of RNA/protein ratio at zero growth rate (Supplementary Fig. 5).
Third, to further understand these results, we plot ribosome allocations for glucose minimal medium, see Fig. 4d–f. Interestingly, at low xrP, a large fraction of ribosomes is allocated to the production of RNAP, whereas with increasing xrP, this ribosome allocation rapidly drops. In the case of the highest cooperativity, allocations at the optimal xrP are comparable to the base RBA model (without RNA degradation), compare Fig. 4f with Fig. 3b.
Finally, we qualitatively predict that the fraction of degraded rRNA decreases with growth rate (Fig. 5), which is in agreement with experimental observations28. This effect gets stronger (and closer to experimental data) with higher rP cooperativity. The quantitative disagreement between the experimental and predicted values is probably due to the simplicity of our model. For example, it does not include other types of RNA or regulatory processes, both of which influence RNAP activity. If we consider RNAP allocation to rRNA (, where is the fraction of RNAP allocated to the synthesis of rRNA), the results get closer to the experimental data (Supplementary Fig. 6).
Fig. 5. Fraction of degraded rRNA at different growth rates.

The extended model recapitulates the experimentally observed decrease in the fraction of degraded RNA with increasing growth rate. The circles are the predicted ratios of RNAse fluxes to RNAP fluxes at different conditions. The triangles represent experimental data from Gausing28, extracted from the original plot with WebPlotDigitizer66. Panels (a–c) represent three types of rRNA degradation. a No cooperativity of rPs. b Weak cooperativity of rPs. c Strong cooperativity of rPs.
Based on these results, we conclude that accounting for RNA degradation and cooperative binding of rP can explain the mixed ribosome composition.
Extreme conditions increase the optimal protein fraction in (archaeal) ribosomes
As a straightforward extension, we explore whether the current model can be adapted to predict the ribosome composition of other organisms. For example, archaeal ribosomes contain 36–50% protein31, eukaryotic ribosomes 42–50% protein5,10,11, and mitochondrial ribosomes 51–89% protein32. We ask whether this variability can be explained by efficient resource allocation.
It has been hypothesized that the extra archaeal/eukaryotic ribosomal proteins primarily serve to stabilize the ribosomes33. This may be particularly important for archaea because they commonly live in extreme conditions, such as high temperatures or low pH, which may lead to higher (misfolding and) degradation of RNA. To mitigate this, archaea might need a higher protein content compared to bacteria. It has been shown that the initial steps in ribosome assembly of the thermophilic archaeon Sufolobus solfataricus do not require high temperature and likely involve core proteins that are also present in bacteria. However, completing the assembly requires high temperature, suggesting that these proteins have evolved to cope with such extreme conditions34,35.
We model this process by increasing which leads to a higher predicted protein content of the ribosome (Fig. 6). Similarly to E. coli, the higher the cooperativity, the lower the sensitivity of the optimum to the other parameters. Moreover, when using parameters from Thermococcus (see Supplementary Table 1) we observe an increase in ribosomal protein content, in accordance with experimental evidence31, and predict a decrease in growth rate.
Fig. 6. The model can be adjusted to predict archaeal protein-rich ribosome composition.

The model was adapted to archaea by increasing two-fold. The remaining parameters were either kept the same as in E. coli (red solid line), or parameters from Thermococcus (molecular masses of R and RNAP, transcription and translation rates, see Supplementary Table 1) were used (red dashed line). Panels (a–c) represent three types of rRNA degradation. a No cooperativity of rPs. b Weak cooperativity of rPs. c Strong cooperativity of rPs.
Discussion
The ribosome is a central player in cellular self-fabrication, placing an upper bound on growth rate. To grow faster, a cell needs more ribosomes which, in turn, requires even more ribosomes to produce themselves. While most catalysts and molecular machines within a cell are proteins, ribosomes stand out by having a substantial (mass) fraction of rRNA, playing a catalytic role. The mass fraction of rPs varies across kingdoms, ranging from approximately 36% in prokaryotes9 to around 50% in eukaryotes10, and even higher in eukaryotic mitochondria, reaching up to 89% in Trypanosoma brucei32,36. This prompts the question: what factors determine the ratio of RNA to protein in ribosomes?
The analysis of our base model (without RNA degradation) suggests that RNA-only ribosomes maximize growth rate (Fig. 3). This results from the lower cost of rRNA synthesis compared to rP synthesis. It remains true even when one accounts for the synthesis of inactive RNAP and enzymes required for nucleotide synthesis5, which suggests that the costs of rRNA synthesis and associated processes are underestimated in the base model.
In order to explain a mixed (RNA+protein) ribosome, we consider rRNA degradation in our extended model, thereby increasing the costs for RNA synthesis. While rRNA that is already integrated into a ribosome is stable, nascent RNA may be susceptible to degradation27. Indeed, it has been experimentally observed that even at maximum growth rate, 10% of newly synthesized rRNA is degraded28, and the degradation rate increases if ribosome assembly is delayed27. Furthermore, when rRNA is overexpressed in excess of rPs, it is rapidly degraded25. Due to the extremely high rates at which rRNA is synthesized, errors become inevitable, necessitating the action of quality control enzymes such as polynucleotide phosphorylase (PNPase) and RNase R to ensure ribosome integrity37. The absence of the RNases results in the accumulation of rRNA fragments, ultimately leading to cell death27,38. In contrast, protein turnover is negligible20, and most ribosomal proteins can exist without rRNA and can be reused39,40. Therefore, we do not consider protein degradation in our model.
In our resource balance approach, decreasing the RNA content of the ribosome saves resources by reducing RNA turnover. At the same time, protein synthesis costs increase, leading to a mixed (RNA+protein) ribosome at maximum growth rate.
We include RNA degradation in two scenarios. (a) RNA is degraded at a rate proportional to its concentration, or (b) RNA degradation rate decreases non-linearly with ribosomal protein content, since proteins cooperatively protect RNA from degradation29,41,42. Both versions of the model predict an optimal mixed (RNA+protein) ribosome. However, without considering cooperative protein binding, optimal ribosome compositions depend on growth conditions. Notably, the higher the cooperativity, the closer the predicted RNAP fluxes and the fraction of degraded rRNA are to experimental data. Yet, more experimental data is needed to decide whether ribosome composition in E. coli remains truly independent of growth conditions when the bacterium is evolutionarily adapted to a single environment. Based on these results and available experimental evidence for cooperative protein binding42, we conclude that scenario (b) is more likely.
Our simple model lumps ribosome assembly and RNA degardation and hence allows multiple explanations for the precise mechanism. On the one hand, proteins may stabilize RNA either by blocking the access of RNases to RNA or by preventing misfolding. Intuitively, this could be explained by the fact that RNA molecules are long, and in order to protect them from misfolding and degradation, a certain critical amount of proteins is needed. Folding intermediates can get trapped in misfolded states and are subsequently degraded as a part of quality control. Proteins may help RNA to avoid these kinetic traps24,29,43,44. On the other hand, proteins may increase the rate of ribosome assembly and thereby reduce the number of ribosome intermediates (pre-R in Fig. 7). Indeed, it was observed that rRNA can fold to near-native conformation45,46. Yet, this process is slower than the protein-supported one, especially for long molecules24,47.
Fig. 7. Potential mechanisms by which ribosomal proteins affect the biosynthesis of ribosomes.
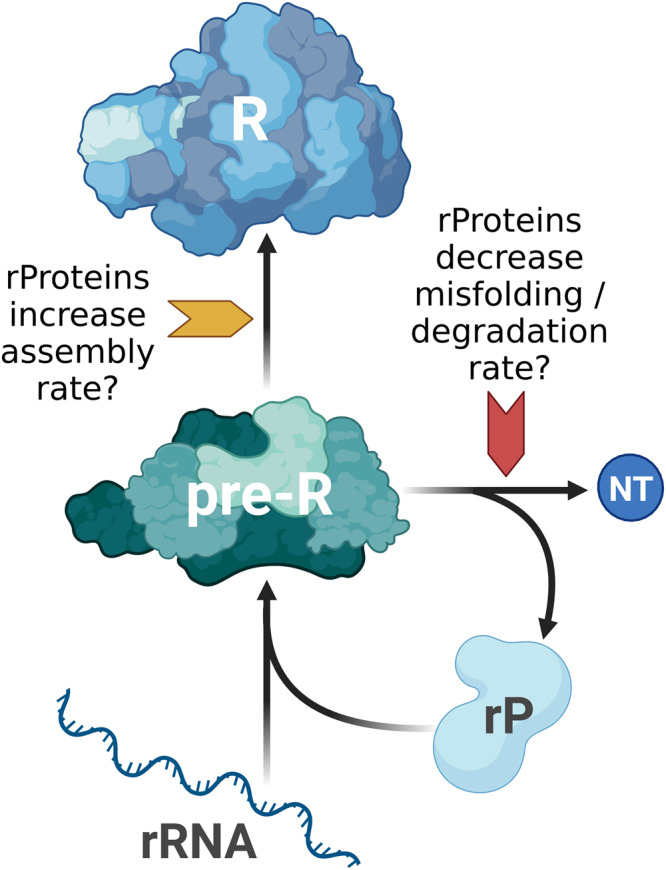
Ribosomal proteins may decrease the degradation rate or misfolding of ribosome assembly intermediates (pre-R). Alternatively, they may increase the assembly rate. Created with BioRender.com.
Throughout the manuscript, we make use of two simplifications:
As in Kostinski and Reuveni12, we consider ribosomes with different compositions, but equal mass. RNA enzymes, known as ribozymes, are generally smaller than proteins and require only a few nucleotides for catalytic activity48. However, such small ribozymes are also inefficient. Increasing their size often improves turnover number, but may impede folding16,47,49. Therefore, we consider the case of a large, hard-to-fold, but catalytically efficient RNA-only ribosome.
We do not consider the effect of protein content on catalytic rates of the ribosomes. Proteins are generally more efficient catalysts than ribozymes16, yet rRNA is still present in the peptidyl transferase center17, and translation rate does not increase in ribosomes from different species which have higher protein content18,19. Furthermore, despite the modest catalytic rate of peptide bond formation, it does not appear to be the rate-limiting step. Given the size of the substrate molecules (mRNA), diffusion may be the limiting factor16,50. Therefore, we assume that enhancing ribosome catalytic rate is not the main reason for the addition of proteins. However, it is possible that proteins stabilize the ribosome structure and thereby indirectly ensure efficient peptide bond formation16.
In future versions of the model, these assumptions can be relaxed. Furthermore, incorporating other types of RNA (mRNA, tRNA) and energy metabolism, or even constructing a genome-scale RBA model51, will likely lead to more quantitative predictions of fluxes and growth rate. A strong indication of this is that including a variable RNAP allocation into the model leads to quantitatively better predictions (see Supplementary Fig. 6). Therefore, in the future, we aim to model RNAP allocation mechanistically. This will involve for example adding other RNA species (mRNA, tRNA), and considering non-specifically bound RNAP which is a considerable fraction of RNAP52.
In our current model, we approximate the cellular rRNA concentration using the ribosome’s rRNA content, see Equation (5). However, rRNA degradation likely begins already during transcription and ribosome assembly. This aspect is not captured in standard RBA as concentrations of all non-catalysts approach zero at maximum growth rates.
In the future, we aim to use growth balance analysis53,54. Growth balance analysis allows the integration of nonlinear kinetics, depending not only on catalyst concentrations but also on substrate concentrations. This will enable us to model RNA degradation based on the concentrations of free rRNA or assembly intermediates. While this shift may alter quantitative predictions, such as RNA degradation fluxes and estimates of , the fundamental conclusions drawn from the model are expected to remain unchanged.
To better model protein-rich organisms such as archaea, the model could be expanded by including the temperature dependence of rRNA degradation and assembly in more detail. Apart from , other parameters (e.g. K or n in the Hill function) might change too to capture the effects of extreme conditions. Furthermore, the effects of other extreme conditions (such as pH and osmolarity), and the reasons for the variability of archaeal ribosome composition could also be investigated55,56. However, the predictions of our current model are in agreement with the naive expectation that more proteins are required to keep ribosomes stable in harsh conditions. More experimental data is needed to model the archaeal ribosomes realistically.
Additionally, some extremophilic organisms such as the bacteria Chloroflexus aurantiacus or Fervidobacterium islandicum, exhibit ribosomes with lower protein content (approximately 40%) compared to extremophilic archaea (50%). It has been suggested that protein-rich ribosomes can be traced back to the oldest phylogenetic lineages, with some ribosomal proteins being lost over time31,57. Organisms with lower protein content in their ribosomes may have evolved alternative strategies to thrive in extreme conditions. Examples of such strategies include the presence of specific rRNA sequence variants or base modifications, as recently discussed by Nissley et al.58.
Moreover, certain archaeal species, such as those from Methanobacteriales or Halobacteriales, have transitioned to milder environmental conditions and subsequently shed unnecessary ribosomal proteins31,57.
To gain a comprehensive understanding of ribosome evolution in response to changing conditions, a thorough phylogenetic analysis is warranted. This analysis should be complemented by measurements of growth rate, translation rate, RNA degradation rate, among other parameters, to delineate the order of protein loss or gain, and the emergence of sequence variations and base modifications.
The protein content of eukaryotic ribosomes in the cytoplasm (approximately 50%) is higher than in bacteria10. This is consistent with the lower growth rates seen in eukaryotes like yeast and mammalian cells. Mitochondrial ribosomes show an even higher protein content, ranging from approximately 50% to 89%32. This may be advantageous since rP are not made directly in mitochondria but are imported for free from the cytoplasm59. Indeed, when we allow a free import of rP in our model, we observe that the optimum moves towards a protein-rich ribosome (Fig. 8). However, to accurately model eukaryotic ribosomes, it is essential to include the synthesis of both cytoplasmic and mitochondrial ribosomes, several different types of RNAPs, transport between nucleus and cytoplasm, and the dynamic interaction between host cells and mitochondria. While the cytoplasm provides ribosomal proteins for mitochondria, mitochondria synthesize enzymes of oxidative phosphorylation and provide ATP back to the host cell.
Fig. 8. The model can be adjusted to predict mitochondrial protein-rich ribosome composition.

All parameters used for the simulation of mitochondria are the same as for E. coli in glucose minimal media, except a fraction of rPs can be imported for free from the cytoplasm and does not have to be synthesized. For simplicity, we assumed that 1/3 of rP are imported. (In reality, almost all rP are imported, but mitochondria make additional proteins to provide energy for the whole cell). Panels (a–c) represent three types of rRNA degradation. a No cooperativity of rPs. b Weak cooperativity of rPs. c Strong cooperativity of rPs.
We hypothesize that eukaryotic cells can afford a higher protein content in their cytoplasmic and mitochondrial ribosomes without affecting the growth rate, and thereby gain additional functionalities that might provide a fitness advantage. Ribosomal proteins participate in translation processes, for example, binding of translation factors, release of tRNA, and translocation. They may also affect the fidelity of translation60. Furthermore, they play roles in various cellular processes such as cell proliferation, apoptosis, DNA repair, cell migration, and others33. These additional functions might have conferred evolutionary fitness advantages. Nevertheless, the primary role of ribosomal proteins seems to be stabilization and folding of rRNA33,60.
There are still many open questions about ribosome biogenesis and evolution. Our model could guide future experiments. There are a few studies that assessed the effect of individual rP deletions in E. coli, for example, mutation in S10 increased RNA degradation61, and mutation in L6 led to disrupted ribosomal assembly62. A systematic knock-out screen of all ribosomal proteins could be done (as in63), complemented with quantification of RNA degradation and misfolding. In case of extremophilic organisms with protein-rich ribosomes, temperature sensitivity could also be assessed. We would expect that deletion of the extra proteins would cause growth defects only at high temperatures. Furthermore, after removal of proteins from archaeal protein-rich ribosomes, laboratory evolution could be performed to see whether growth rate increases beyond wild-type.
Comprehensive datasets, akin to the work of Bremer and Dennis in 2008 for E. coli, should be generated for non-standard organisms by measuring various parameters such as transcription and translation rates, ribosome and RNAP activities, and other relevant factors.
Finally, phylogenetic analysis or ribosome evolution across different species and environments could be done.
Formal comparison with Kostinski and Reuveni (2020)
Our analysis is motivated by the previous work of Kostinski and Reuveni12, who understand ribosome composition as a competition between two autocatalytic loops. One loop is responsible for synthesizing rRNA, while the other loop is responsible for rP synthesis, both competing for limited resources. These loops and their constraints, namely, the stoichiometric constraints for rRNA and rP and the capacity constraint for RNAP, are contained in our more detailed RBA model, see Table 1. In addition to these three conditions, Kostinski and Reuveni12 make two more assumptions: they fix the ribosome allocations and for the synthesis of rP and RNAP, defined in Eqn. (3).
The resulting upper limits on growth rate can be derived easily by considering the synthesis of rRNA and rP, separately.
- (rP) The stoichiometric constraint for rP is given by vAF ≤ wrP, see Table 1. Together with the definition of the corresponding ribosome allocation , this yields
8a - (rRNA) The stoichiometric constraint for rRNA and the capacity constraint for RNAP are given by vAF ≤ vRNAP and , see Table 1. By multiplication, they imply . Together with the definition of the ribosome allocation for the synthesis of RNAP, this yields
8b
These upper bounds (8) are Eqns. (2) and (5) in Kostinski and Reuveni12, after inserting the effective transcription and translation elongation rate constants and , respectively. Here, denotes the fraction of RNAP transcribing rRNA (which we assume to equal one in the rest of this work).
Using Eqn. (4), the two upper bounds (8) can be written as functions of the rP fraction xrP, namely as
| 9 |
with constants γrP, γRNAP > 0. For fixed ribosome allocations and , the two curves necessarily intersect at some , and is the maximum growth rate allowed by the constraints considered above, namely the stoichiometric constraints for rP and rRNA, and the RNAP capacity constraint.
Kostinski and Reuveni12 interpret Eqns. (9) as a trade-off between rRNA and rP production. This effect arises because they fix the ribosome allocations. In particular, Kostinski and Reuveni12 fix and to experimental values for E. coli (in multiple growth conditions), and find that maximum growth rate occurs close to the current rP fraction (xrP = 36%), and the resulting μ(xrP) is close to the experimental value. If we use their parameters (see Supplementary Table 1), we can exactly reproduce their results (see Supplementary Fig. 7). Our base model provides an explanation for the protein investment costs, giving a proper mechanistic interpretation to the argument presented by Kostinski and Reuveni12. Moreover, it is closer to an evolutionary scenario, where a cell can adjust both ribosome composition xrP and ribosome allocations ϕR. However, the base model predicts an optimal ribosome that is RNA-only (for realistic parameters), see Fig. 3a. This is possible because the ribosome allocations are adjusted according to demand. The ribosome allocations corresponding to varying ribosomal protein fraction are illustrated in Fig. 3b. Only the extended model with RNA degradation predicts a mixed (RNA+protein) ribosome at maximum growth rate.
Methods
Our analysis is based on the small model of a self-replicating cell depicted in Fig. 1 and described below. Constraints are listed in Table 1 and parameters in Supplementary Table 1. For an introduction to resource allocation in next-generation models of cellular growth, including the definition of elementary growth vectors, see Müller et al.23. Elementary growth vectors were enumerated using the package efmtool 0.2.064 in Python 3.8.13.
Figures 1a and 7 were created with BioRender.com and the remaining figures with R version 4.1.2.
Model details
We consider the small model of a self-fabricating cell depicted in Fig. 1 which contains metabolic reactions and macromolecular synthesis reactions. To take into account the limitation of cellular resources, we use three types of capacity constraints: enzyme capacity constraints limit the rate of metabolic reactions, the RNAP capacity constraint limits transcription rate, and the ribosome capacity constraint limits the synthesis rates of all proteins (including the ribosomal proteins).
The cell takes up a carbon source (C) via the reaction
catalyzed by the importer IC, and forms amino acids (AA), nucleotides (NT), and ribosomal rRNA (rRNA) via
catalyzed by the enzymes EAA, ENT, and the RNA polymerase (RNAP). Ultimately, the ribosome R is built from rRNA and ribosomal protein (rP) via
catalyzed by the assembly factors AF. The processes above are part of the base model. In an extended model, ribosomal RNA degrades via
catalyzed by the RNase. Finally, we consider the synthesis of all proteins (enzymes and ribosomal protein) via the reactions
catalyzed by the ribosome.
The resulting stoichiometric matrix and the corresponding flux vector are displayed in Fig. 1b, and parameter values are given in Supplementary Table 1. In fact, the stoichiometric matrix can be partitioned into two submatrices,
corresponding to the metabolites Met = {C, AA, NT, rRNA, rP} and the catalysts Cat = Enz ∪ {R} including the enzymes Enz = {IC, EAA, ENT, RNAP, (RNase), AF} and the ribosome. The (total) flux vector, vtot, can be partitioned into two subvectors,
corresponding to the enzymatic reactions r and the protein synthesis reactions s.
In general, comprehensive models of cellular growth lead to linear (in-)equality systems for the fluxes, and concentrations are determined by fluxes, see Box 1. In the example, we distinguish enzymatic reactions r and protein synthesis reactions s (with corresponding fluxes v and w), and further metabolites Met and catalysts Cat, see above. Explicitly, we study the inequality system
since rIC is the only exchange reaction.
In fact, only yields non-trivial constraints, since yields wi ≥ 0 for i ∈ Enz and vAF ≥ 0, already included in v ≥ 0, w ≥ 0. However, NCat determines the catalyst concentrations via or, explicitly,
| 10 |
(Recall that the ribosome is formed by the assembly factors).
Now, catalyst concentrations are used to formulate capacity constraints (for importer, metabolic enzymes, and assembly factors),
| 11a |
where are the corresponding enzyme turnover numbers. The capacity constraints for the RNA polymerase, (optionally the RNase), and the ribosome are given by
| 11b |
and
| 11c |
respectively. Here, nrRNA is the number of nucleotides in rRNA, and ni is the number of amino acids in protein i, cf. the stoichiometric coefficients in Fig. 1b. Further, and are the effective transcription and translation elongation rate constants, respectively, and is the RNA degradation rate constant.
Finally, catalyst concentrations are expressed by corresponding fluxes in all capacity constraints (11) via Eqns. (10). The stoichiometric, capacity, and (dry) mass constraints described so far are summarized in Table 1, and the parameter values are given in Supplementary Table 1.
In particular, after using (10), the ribosome capacity constraint (11c) takes the form
which suggests the definition of ribosome allocations (ribosome fractions translating certain proteins),
Obviously, . Instead of varying the protein synthesis fluxes w, one may vary vAF (the ribosome synthesis flux) and the (vector of) ribosome allocations ϕR.
Throughout this work, we consider a fixed ribosome mass, but variable ribosomal RNA and protein content,
where nrRNA and nrP are the numbers of nucleotides and amino acids in rRNA and rP, respectively. We define the ribosomal protein (mass) fraction
and express nrRNA and nrP by xrP,
For variable ribosomal protein fraction xrP (from 0 to 100%), we maximize growth rate (by varying fluxes under the given constraints).
Box 1 Comprehensive models of cellular growth.
Comprehensive models of cellular growth (as used in RBA) need not be genome-scale, but involve explicit synthesis reactions for all catalysts. This is in contrast to traditional metabolic models (as used in FBA) which involve an approximate biomass reaction, thereby fixing biomass composition.
At steady state, the dynamic model of cellular growth yields Nvtot = μ c together with the (dry) mass constraint ωTc = 1. Thereby, μ is growth rate, vtot is the vector of all fluxes, c is the vector of concentrations, and ω is the vector of molar masses.
In the constraint-based approach, we consider the (in-)equality system for the fluxes
Thereby, we assume that all reactions have a given direction, and we use the fact that growth rate is determined by the exchange reactions, cf. Müller et al. (2022)23.
Finally, concentrations are determined by fluxes via c = Nvtot/μ. In particular, concentrations of catalysts are used to formulate additional capacity constraints.
Converting constraints from equations/inequalities into matrix form
For convenience, we describe how to obtain the constraints in Table 1 from the equations/inequalities given in the subsection Model details above.
The capacity constraints for enzymes have the form
Concentrations ci can be expressed by synthesis fluxes wi via ci = wi/μ, and hence
Multiplying by μ and bringing all terms to one side, we get
or, in vector form,
The row vector is the non-zero part of the row in Table 1 that specifies the capacity constraint cap i, for i ∈ {IC, EAA, ENT, AF}.
For the RNAP and RNase capacity constraints (cap RNAP and cap RNase, respectively), we further have and . Hence,
and
Finally, for the ribosome capacity constraint, , ribosome concentration cR can be expressed by assembly flux vAF via cR = vAF/μ. Ultimately,
After expanding the sum, this yields the row in Table 1 that specifies cap R.
Minimum degradation rate is enforced by
As before, ribosome concentration is expressed by assembly flux,
which can be written as in Table 1,
As stated in Box 2, growth rate is determined by the exchange fluxes. Indeed, in the dry mass constraint ωTc = 1, concentrations c can be expressed by fluxes vtot via Nvtot = μc and hence ωTNvtot = μ. Since internal reactions are mass-balanced, only exchange reactions contribute to growth,
In our small model, the only exchange reaction is carbon import and hence
Box 2 The determinant method.
(In-)homogeneous linear equality and inequality constraints on a vector can be summarized by matrices and vectors as
After homogenization, one obtains
Assume that, for a particular x, all inequality constraints are active, that is, . Then,
If B is square (that is, if ), then
that is, its determinant is zero.
In the main text, we consider particular (sub-)sets of constraints on the vector of fluxes vtot in the form and assume that, at maximum growth rate, all constraints are active, and the resulting matrix B is square. We compute its determinant, set it to zero, and determine the maximum growth rate from the resulting (quadratic) equation.
Symbolic analysis of growth rate maximization
In order to confirm our numerical results, we also perform a symbolic analysis of growth rate maximization.
The base model involves five stoichiometric constraints (for the species C, AA, NT, RNAP, rP), six capacity constraints (for the reactions catalyzed by IC, EAA, ENT, RNAP, AF, R), and one (dry) mass constraint, cf. Table 1 (without the columns and rows indicated in the caption). They define a linear equality and inequality system with 12 constraints (either ≥ or = ) for 11 fluxes and 1 right-hand side.
We apply the determinant method introduced in Box 2 to the resulting matrix , and we find
Using ribosomal protein fraction and rescaling time,
we obtain a quadratic equation for maximum growth rate,
| 12 |
with
For fixed xrP ∈ [0, 1], the quadratic equation (12) has one positive solution . To show that it is monotone in xrP, we differentiate (12) and set . We get
which has the positive solution . Insertion into (12) yields
which does not depend on xrP. In fact, if ε = 0, then is constant. Otherwise, is strictly monotone in xrP (decreasing if ε > 0 and increasing if ε < 0).
For realistic parameters, is decreasing (and ).
Approximation
For realistic parameters, α ≪ β ≤ 1, and for all xrP ∈ [0, 1], we may set α = 0 in the quadratic equation (12): For xrP → 0, obviously α + (1 − xrP)β → α + β ≈ β. For xrP → 1, the crucial quantity , and the quadratic term can be neglected.
Numerical growth rate maximization
We fix growth rate and solve the system of equations (1) and (2) using efmtool 0.2.064 in Python 3.8.13. We use bisection search to find the highest growth rate that still enables a feasible solution.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Acknowledgements
This research was funded in part, by the Austrian Science Fund (FWF), Grant 10.55776/P33218.
Author contributions
Diana Széliová: conceptualization, data curation, formal analysis, investigation, methodology, software, validation, visualization, writing - original draft, writing - review & editing; Stefan Müller: conceptualization, formal analysis, funding acquisition, methodology, validation, writing - original draft, writing - review & editing; Jürgen Zanghellini: conceptualization, formal analysis, funding acquisition, methodology, project administration, resources, supervision, writing - original draft, writing - review & editing.
Peer review
Peer review information
Communications Biology thanks Jonas Cremer, Xiao-Pan Hu, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Manuel Breuer. A peer review file is available.
Data availability
All input data and simulation results are available in Zenodo with the identifier 10.5281/zenodo.1045641765.
Code availability
All code including the input files and parameters are available in Zenodo with the identifier 10.5281/zenodo.1045641765. The calculations were done using efmtool 0.2.064 in Python 3.8.13. Figures were generated with R version 4.1.2.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Diana Széliová, Stefan Müller, Jürgen Zanghellini.
Change history
4/11/2024
A Correction to this paper has been published: 10.1038/s42003-024-06082-z
Supplementary information
The online version contains supplementary material available at 10.1038/s42003-024-05815-4.
References
- 1.Dill KA, Ghosh K, Schmit JD. Physical limits of cells and proteomes. Proc. Natl Acad. Sci. 2011;108:17876–17882. doi: 10.1073/pnas.1114477108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shore D, Albert B. Ribosome biogenesis and the cellular energy economy. Curr. Biol. 2022;32:R611–R617. doi: 10.1016/j.cub.2022.04.083. [DOI] [PubMed] [Google Scholar]
- 3.Bremer H, Dennis P. Modulation of chemical composition and other parameters of the cell by growth rate. Escherichia coli and salmonella: cellular and molecular biology. Am. Soc. Microbiol. 1996;2:1553–1568. [Google Scholar]
- 4.Long CP, Gonzalez JE, Cipolla RM, Antoniewicz MR. Metabolism of the fast-growing bacterium Vibrio natriegens elucidated by 13C metabolic flux analysis. Metab. Engineer. 2017;44:191–197. doi: 10.1016/j.ymben.2017.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reuveni S, Ehrenberg M, Paulsson J. Ribosomes are optimized for autocatalytic production. Nature. 2017;547:293–297. doi: 10.1038/nature22998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petrov AS, et al. History of the ribosome and the origin of translation. Proc. Natl Acad. Sci. 2015;112:15396–15401. doi: 10.1073/pnas.1509761112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Melnikov S, Manakongtreecheep K, Söll D. Revising the structural diversity of ribosomal proteins across the three domains of life. Mol. Biol. Evol. 2018;35:1588–1598. doi: 10.1093/molbev/msy021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Melnikov S, et al. One core, two shells: bacterial and eukaryotic ribosomes. Nat. Structural Mol. Biol. 2012;19:560–567. doi: 10.1038/nsmb.2313. [DOI] [PubMed] [Google Scholar]
- 9.Kurland CG. Molecular characterization of ribonucleic acid from Escherichia coli ribosomes: I. Isolation and molecular weights. J. Mol. Biol. 1960;2:83–91. doi: 10.1016/S0022-2836(60)80029-0. [DOI] [Google Scholar]
- 10.Wilson DN, Cate JHD. The structure and function of the eukaryotic ribosome. Cold Spring Harbor Perspect. Biol. 2012;4:a011536. doi: 10.1101/cshperspect.a011536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verschoor A, Warner JR, Srivastava S, Grassucci RA, Frank J. Three-dimensional structure of the yeast ribosome. Nucleic Acids Res. 1998;26:655–661. doi: 10.1093/nar/26.2.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kostinski S, Reuveni S. Ribosome composition maximizes cellular growth rates in E. coli. Phys. Rev. Lett. 2020;125:028103. doi: 10.1103/PhysRevLett.125.028103. [DOI] [PubMed] [Google Scholar]
- 13.Klumpp S. Speed limit for cell growth. Physics. 2020;13:108. doi: 10.1103/Physics.13.108. [DOI] [Google Scholar]
- 14.Goelzer A, et al. Quantitative prediction of genome-wide resource allocation in bacteria. Metab. Engineer. 2015;32:232–243. doi: 10.1016/j.ymben.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 15.Molenaar D, Van Berlo R, De Ridder D, Teusink B. Shifts in growth strategies reflect tradeoffs in cellular economics. Mol. Syst. Biol. 2009;5:323. doi: 10.1038/msb.2009.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeffares DC, Poole AM, Penny D. Relics from the RNA world. J. Mol. Evol. 1998;46:18–36. doi: 10.1007/PL00006280. [DOI] [PubMed] [Google Scholar]
- 17.Tirumalai MR, Rivas M, Tran Q, Fox GE. The peptidyl transferase center: a window to the past. Microbiol. Mol. Biol. Rev. 2021;85:e00104–21. doi: 10.1128/MMBR.00104-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bonven B, Gulløv K. Peptide chain elongation rate and ribosomal activity in Saccharomyces cerevisiae as a function of the growth rate. Mol. General Genet. MGG. 1979;170:225–230. doi: 10.1007/BF00337800. [DOI] [PubMed] [Google Scholar]
- 19.Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat. Structural Mol. Biol. 2009;16:574–581. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- 20.Bremer H, Dennis PP. Modulation of chemical composition and other parameters of the cell at different exponential growth rates. EcoSal Plus. 2008;3:10–1128. doi: 10.1128/ecosal.5.2.3. [DOI] [PubMed] [Google Scholar]
- 21.Scott M, Gunderson CW, Mateescu EM, Zhang Z, Hwa T. Interdependence of cell growth and gene expression: origins and consequences. Science. 2010;330:1099–1102. doi: 10.1126/science.1192588. [DOI] [PubMed] [Google Scholar]
- 22.Dai X, et al. Reduction of translating ribosomes enables Escherichia coli to maintain elongation rates during slow growth. Nat. Microbiol. 2016;2:1–9. doi: 10.1038/nmicrobiol.2016.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Müller S, Széliová D, Zanghellini J. Elementary vectors and autocatalytic sets for resource allocation in next-generation models of cellular growth. PLoS Comput. Biol. 2022;18:e1009843. doi: 10.1371/journal.pcbi.1009843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodgers ML, Woodson SA. A roadmap for rRNA folding and assembly during transcription. Trends Biochem. Sci. 2021;46:889–901. doi: 10.1016/j.tibs.2021.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siehnel RJ, Morgan EA. Unbalanced rRNA gene dosage and its effects on rRNA and ribosomal-protein synthesis. J. Bacteriol. 1985;163:476–486. doi: 10.1128/jb.163.2.476-486.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Milo, R. & Phillips, R. Cell biology by the numbers (Garland Science, 2015).
- 27.Jain C. Role of ribosome assembly in Escherichia coli ribosomal RNA degradation. Nucleic Acids Res. 2018;46:11048–11060. doi: 10.1093/nar/gky808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gausing K. Regulation of ribosome production in Escherichia coli: synthesis and stability of ribosomal RNA and of ribosomal protein messenger RNA at different growth rates. J. Mol. Biol. 1977;115:335–354. doi: 10.1016/0022-2836(77)90158-9. [DOI] [PubMed] [Google Scholar]
- 29.Shajani Z, Sykes MT, Williamson JR. Assembly of bacterial ribosomes. Ann. Rev. Biochem. 2011;80:501–526. doi: 10.1146/annurev-biochem-062608-160432. [DOI] [PubMed] [Google Scholar]
- 30.Fazal FM, Koslover DJ, Luisi BF, Block SM. Direct observation of processive exoribonuclease motion using optical tweezers. Proc. Natl Acad. Sci. 2015;112:15101–15106. doi: 10.1073/pnas.1514028112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Acca M, et al. Updating mass and composition of archaeal and bacterial ribosomes. Archaeal-like features of ribosomes from the deep-branching bacterium Aquifex pyrophilus. Syst. Appl. Microbiol. 1993;16:629–637. doi: 10.1016/S0723-2020(11)80334-6. [DOI] [Google Scholar]
- 32.Moore PB. In which the deity attempts to make a ribose-free ribosome. Biochemistry. 2019;58:431–432. doi: 10.1021/acs.biochem.8b01191. [DOI] [PubMed] [Google Scholar]
- 33.Kisly I, Tamm T. Archaea/eukaryote-specific ribosomal proteins-guardians of a complex structure. Comput. Structural Biotechnol. J. 2023;21:1249–1261. doi: 10.1016/j.csbj.2023.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Altamura S, Caprini E, Sanchez M, Londei P. Early assembly proteins of the large ribosomal subunit of the thermophilic archaebacterium Sulfolobus. Identification and binding to heterologous rRNA species. J. Biol. Chem. 1991;266:6195–6200. doi: 10.1016/S0021-9258(18)38103-1. [DOI] [PubMed] [Google Scholar]
- 35.Londei P, Teuudò J, Acca M, Cammarano P, Amils R. Total reconstitution of active large ribosomal subunits of the thermoacidophilic archaebacterium Sulfolobus solfataricus. Nucleic Acids Res. 1986;14:2269–2285. doi: 10.1093/nar/14.5.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramrath DJ, et al. Evolutionary shift toward protein-based architecture in trypanosomal mitochondrial ribosomes. Science. 2018;362:eaau7735. doi: 10.1126/science.aau7735. [DOI] [PubMed] [Google Scholar]
- 37.Dos Santos RF, et al. Major 3′–5′ exoribonucleases in the metabolism of coding and non-coding RNA. Prog. Mol. Biol. Transl. Sci. 2018;159:101–155. doi: 10.1016/bs.pmbts.2018.07.005. [DOI] [PubMed] [Google Scholar]
- 38.Cheng Z-F, Deutscher MP. Quality control of ribosomal RNA mediated by polynucleotide phosphorylase and RNase R. Proc. Natl Acad. Sci. 2003;100:6388–6393. doi: 10.1073/pnas.1231041100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reier K, Lahtvee P-J, Liiv A, Remme J. A conundrum of r-protein stability: unbalanced stoichiometry of r-proteins during stationary phase in Escherichia coli. Mbio. 2022;13:e01873–22. doi: 10.1128/mbio.01873-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deutscher MP. Degradation of stable RNA in bacteria. J. Biol. Chem. 2003;278:45041–45044. doi: 10.1074/jbc.R300031200. [DOI] [PubMed] [Google Scholar]
- 41.Bowman JC, Hud NV, Williams LD. The ribosome challenge to the RNA world. J. Mol. Evol. 2015;80:143–161. doi: 10.1007/s00239-015-9669-9. [DOI] [PubMed] [Google Scholar]
- 42.Rodgers ML, Woodson SA. Transcription increases the cooperativity of ribonucleoprotein assembly. Cell. 2019;179:1370–1381.e12. doi: 10.1016/j.cell.2019.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bushhouse DZ, Choi EK, Hertz LM, Lucks JB. How does RNA fold dynamically? J. Mol. Biol. 2022;434:167665. doi: 10.1016/j.jmb.2022.167665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abeysirigunawardena SC, et al. Evolution of protein-coupled RNA dynamics during hierarchical assembly of ribosomal complexes. Nat. Commun. 2017;8:492. doi: 10.1038/s41467-017-00536-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lenz TK, Norris AM, Hud NV, Williams LD. Protein-free ribosomal RNA folds to a near-native state in the presence of Mg 2+ RSC Adv. 2017;7:54674–54681. doi: 10.1039/C7RA08696B. [DOI] [Google Scholar]
- 46.Adilakshmi T, Ramaswamy P, Woodson SA. Protein-independent folding pathway of the 16S rRNA 5’ Domain. J. Mol. Biol. 2005;351:508–519. doi: 10.1016/j.jmb.2005.06.020. [DOI] [PubMed] [Google Scholar]
- 47.Hyeon C, Thirumalai D. Chain length determines the folding rates of RNA. Biophys. J. 2012;102:L11–L13. doi: 10.1016/j.bpj.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bernhardt HS. The RNA world hypothesis: the worst theory of the early evolution of life (except for all the others) Biol. Direct. 2012;7:1–10. doi: 10.1186/1745-6150-7-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martick M, Scott WG. Tertiary contacts distant from the active site prime a ribozyme for catalysis. Cell. 2006;126:309–320. doi: 10.1016/j.cell.2006.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bernhardt HS, Tate WP. A ribosome without RNA. Front. Ecol. Evol. 2015;3:129. doi: 10.3389/fevo.2015.00129. [DOI] [Google Scholar]
- 51.Hu X-P, Dourado H, Schubert P, Lercher MJ. The protein translation machinery is expressed for maximal efficiency in Escherichia coli. Nat. Commun. 2020;11:5260. doi: 10.1038/s41467-020-18948-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klumpp S, Hwa T. Growth-rate-dependent partitioning of RNA polymerases in bacteria. Proc. Natl Acad. Sci. 2008;105:20245–20250. doi: 10.1073/pnas.0804953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dourado H, Liebermeister W, Ebenhöh O, Lercher MJ. Mathematical properties of optimal fluxes in cellular reaction networks at balanced growth. PLOS Comput. Biol. 2023;19:e1011156. doi: 10.1371/journal.pcbi.1011156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dourado H, Lercher MJ. An analytical theory of balanced cellular growth. Nat. Commun. 2020;11:1226. doi: 10.1038/s41467-020-14751-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Greber BJ, et al. Cryo-EM structure of the archaeal 50S ribosomal subunit in complex with initiation factor 6 and implications for ribosome evolution. J. Mol. Biol. 2012;418:145–160. doi: 10.1016/j.jmb.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Londei P, Ferreira-Cerca S. Ribosome biogenesis in archaea. Front. Microbiol. 2021;12:1476. doi: 10.3389/fmicb.2021.686977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Amils, R., Cammarano, P. & Londei, P. Chapter 13 Translation in archaea. In Kates, M., Kushner, D. & Matheson, A. (eds.) The Biochemistry of Archaea (Archaebacteria), vol. 26 of New Comprehensive Biochemistry, 393–438 (Elsevier, 1993). https://www.sciencedirect.com/science/article/pii/S0167730608602628.
- 58.Nissley AJ, Penev PI, Watson ZL, Banfield JF, Cate JH. Rare ribosomal RNA sequences from archaea stabilize the bacterial ribosome. Nucleic Acids Res. 2023;51:1880–1894. doi: 10.1093/nar/gkac1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Woellhaf MW, Hansen KG, Garth C, Herrmann JM. Import of ribosomal proteins into yeast mitochondria. Biochem. Cell Biol. 2014;92:489–498. doi: 10.1139/bcb-2014-0029. [DOI] [PubMed] [Google Scholar]
- 60.Nikolay, R., van den Bruck, D., Achenbach, J. & Nierhaus, K. H. Ribosomal proteins: role in ribosomal functions. eLS 1–12 (2015).
- 61.Kuwano M, Taniguchi H, Ono M, Endo H, Ohnishi Y. An Escherichia coli K12 mutant carrying altered ribosomal protein (S10) Biochem. Biophys. Res. Commun. 1977;75:156–162. doi: 10.1016/0006-291X(77)91303-1. [DOI] [PubMed] [Google Scholar]
- 62.Shigeno Y, Uchiumi T, Nomura T. Involvement of ribosomal protein L6 in assembly of functional 50S ribosomal subunit in Escherichia coli cells. Biochem. Biophys. Res. Commun. 2016;473:237–242. doi: 10.1016/j.bbrc.2016.03.085. [DOI] [PubMed] [Google Scholar]
- 63.Shoji S, Dambacher CM, Shajani Z, Williamson JR, Schultz PG. Systematic chromosomal deletion of bacterial ribosomal protein genes. J. Mol. Biol. 2011;413:751–761. doi: 10.1016/j.jmb.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Terzer M, Stelling J. Large-scale computation of elementary flux modes with bit pattern trees. Bioinformatics. 2008;24:2229–2235. doi: 10.1093/bioinformatics/btn401. [DOI] [PubMed] [Google Scholar]
- 65.Széliová, D., Müller, S. & Zanghellini, J. Costs of rRNA stabilization affect ribosome composition at maximum growth rate. [code, data set]. Zenodo (2023).
- 66.Rohatgi, A. Webplotdigitizer: Version 4.6 (2022). https://automeris.io/WebPlotDigitizer. Last accessed on 2023-6-7.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All input data and simulation results are available in Zenodo with the identifier 10.5281/zenodo.1045641765.
All code including the input files and parameters are available in Zenodo with the identifier 10.5281/zenodo.1045641765. The calculations were done using efmtool 0.2.064 in Python 3.8.13. Figures were generated with R version 4.1.2.