

Abstract
Neuronal activity drives alterations in gene expression within neurons, yet how it directs transcriptional and epigenomic changes in neighboring astrocytes in functioning circuits is unknown. We found that neuronal activity induces widespread transcriptional upregulation and downregulation in astrocytes, highlighted by the identification of a neuromodulator transporter Slc22a3 as an activity-inducible astrocyte gene regulating sensory processing in the olfactory bulb. Loss of astrocytic Slc22a3 reduced serotonin (5HT) levels in astrocytes, leading to alterations in histone serotonylation. Inhibition of histone serotonylation in astrocytes reduced expression of GABA biosynthetic genes and GABA release, culminating in olfactory deficits. Our study revealed that neuronal activity orchestrates transcriptional and epigenomic responses in astrocytes, while illustrating new mechanisms for how astrocytes process neuromodulatory input to gate neurotransmitter release for sensory processing.
Astrocytes are intimately associated with neuronal activity and neurotransmission, participating in a host of essential roles that facilitate synaptic function (1–5). Cell intrinsic activation of Gq- or Gi- signaling and associated calcium activity in astrocytes can influence a multitude of behavioral outputs (6–10). However, in native behavioral states astrocyte activation results from cell extrinsic sources, usually in response to a neuronal stimulus, and therefore typically follows neuronal activation (11). In neurons, heightened activity induces expression of ‘immediate early genes’, which are predominately transcription factors (TF’s) that modify gene expression programs and activity-dependent epigenomic states, ultimately regulating circuit activity, plasticity, and associated behavioral outputs (12). Astrocytic TF’s can also regulate region specific-circuits in the adult brain (13, 14), indicating that transcriptional states in astrocytes can influence neuronal activity. Evidence for the reciprocal relationship can be found in the developing cortex (15) and in vitro (16, 17), where alterations in neuronal activity influences expression of synaptogenic genes in differentiating astrocytes. Single-cell transcriptomic studies have shown non-neuronal cells undergo transcriptional changes in response to neuronal activity driven by visual stimuli (18). However, whether heightened neuronal activity induces an analogous ‘immediate early gene-like’ response in mature astrocytes and how this sculpts astrocytic transcriptional and epigenomic responses to regulate circuit function remains unclear.
A central component of astrocyte-neuron communication is synaptic neurotransmitter signaling, as astrocytes express receptors and transporters for both glutamate and GABA, activation of which can elicit calcium-dependent responses that modify circuit activity (5, 19). Maintenance of extracellular glutamate levels via astrocytic glutamate transporters are required for drug-seeking behaviors, circadian rhythms, and avoidance behaviors, among others (20–23). Similarly, disruption of GABA signaling in astrocytes leads to repetitive behaviors, impaired motor function, and deficits in learning and memory (7, 24–26). Astrocytes also express a host of neuromodulator receptors and respond to noradrenaline, acetylcholine, and serotonin with intracellular calcium elevations (16). Manipulations of cholinergic inputs to hippocampal astrocytes influence sleep states, while noradrenaline primes cortical astrocyte responses to local neuronal activity during locomotion, and dopamine promotes astrocyte-mediated depression of excitatory synapses, which influences drug-induced locomotor activity (27–29). Despite direct roles in gating responses to neuromodulators, the signaling mechanisms utilized by astrocytes to respond- and process- neuromodulatory cues remain unclear. Furthermore, how astrocytes coordinate the integration of neuromodulator with neurotransmitter signaling to regulate circuit function is unclear.
Epigenomic regulation of gene expression participates in encoding information in the central nervous system (CNS), where alterations in synaptic activity induce epigenetic modifications that influence behavioral outcomes (12). For example, alterations in histone acetylation are directly linked to memory storage and synaptic plasticity in hippocampal neurons after learning (30). Among neuromodulators, serotonin and dopamine can be incorporated into histones, with histone serotonylation enabling permissive gene expression states in serotonergic neurons (31) and histone dopaminylation regulating cocaine seeking behaviors in rodents (32). Nevertheless, several aspects of this biology are still unclear, including the extent and significance of these histone modifications throughout the brain and the fundamental mechanisms of how neuromodulators are transported to the nucleus and added to histones. Furthermore, how these and other epigenomic mechanisms are utilized by astrocytes to regulate gene expression, circuit function, and associated behavioral responses are unknown.
Neuronal activity directs transcriptional responses in astrocytes
To understand how neuronal activation influences transcriptional responses in astrocytes we employed chemogenetics to activate neurons in Aldh1l1-GFP reporter mice (Fig. 1A–B). We used intraventricular injection of viral expression vector (pAAV) containing hM3Dq DREADD (Gq-DREADD) under the pan-neuronal synapsin promoter, which allowed widespread neuronal Gq-DREADD expression throughout the brain. When mice reached early adulthood (~8 weeks) we activated neuronal Gq signaling via intraperitoneal injection of clozapine-N-oxide (CNO) (0.3 mg/kg) or saline (represented as Gq-Saline or Gq-CNO herein), and harvested brains 30 minutes after treatment (Fig. 1C). We confirmed enhanced neuronal activity in Gq-CNO treated samples using electrophysiology in brain slice preparations, coupled with expression of neuronal immediate early gene marker c-Fos (Fig. S1A–C). To identify neuronal activity-dependent gene expression changes in astrocytes, we utilized the presence of distinct reporters in Gq-DREADD expressing neurons (mCherry) and astrocytes (GFP) to isolate these two cell types from the cortex (CX), hippocampus (HP), and olfactory bulb (OB) (Fig. 1D) by fluorescent activated cell sorting (FACS) and performed RNA-sequencing (RNA-Seq) (Fig. S1D). Analysis of Gq-CNO vs. Gq-Saline cohorts from each region revealed widespread changes in gene expression in astrocytes, identifying 267, 300 and 256 astrocyte-specific differentially expressed genes (DEGs) in the CX, HP, and OB, respectively (Fig.1E–F). These DEGs were specific to astrocytes, demonstrating that immediate early responses in neurons are distinct from those in astrocytes (Fig. S2A–C). Gene ontology (GO) analyses revealed distinct terms associated with astrocytes and neurons: top astrocytic GO terms included protein phosphorylation, calcium ion transport and serotonin transport, in the CX, HP, and OB, respectively (Fig. S2D–F; Table S1). To control for possible transcriptional artifacts due to CNO treatment, we treated Aldh1l1-GFP mice with 0.3 mg/kg CNO (represented as CNO-control herein), harvested their OBs after 30 minutes, and FACS isolated GFP-expressing astrocytes, which was followed by RNA-Seq (Fig. S3A). Comparing RNA-Seq profiles between untreated OB astrocytes and CNO-controls identified widespread changes in gene expression (Fig. S3B). However, these CNO-induced changes do not correspond to any of the GO groups we identified in our activity-based profiling experiments (Fig. S3C). Comparison of the activity-induced DEGs with the CNO-associated DEG’s revealed miniscule overlap. Amongst the 256 DEGs induced in OB astrocytes by neuronal activity only 7 also demonstrated altered expression in CNO-treated control Aldh1l1-GFP mice (Fig. S3D–E).
Fig. 1. Neuronal activity directs Sox9-regulated transcriptional responses in astrocytes.
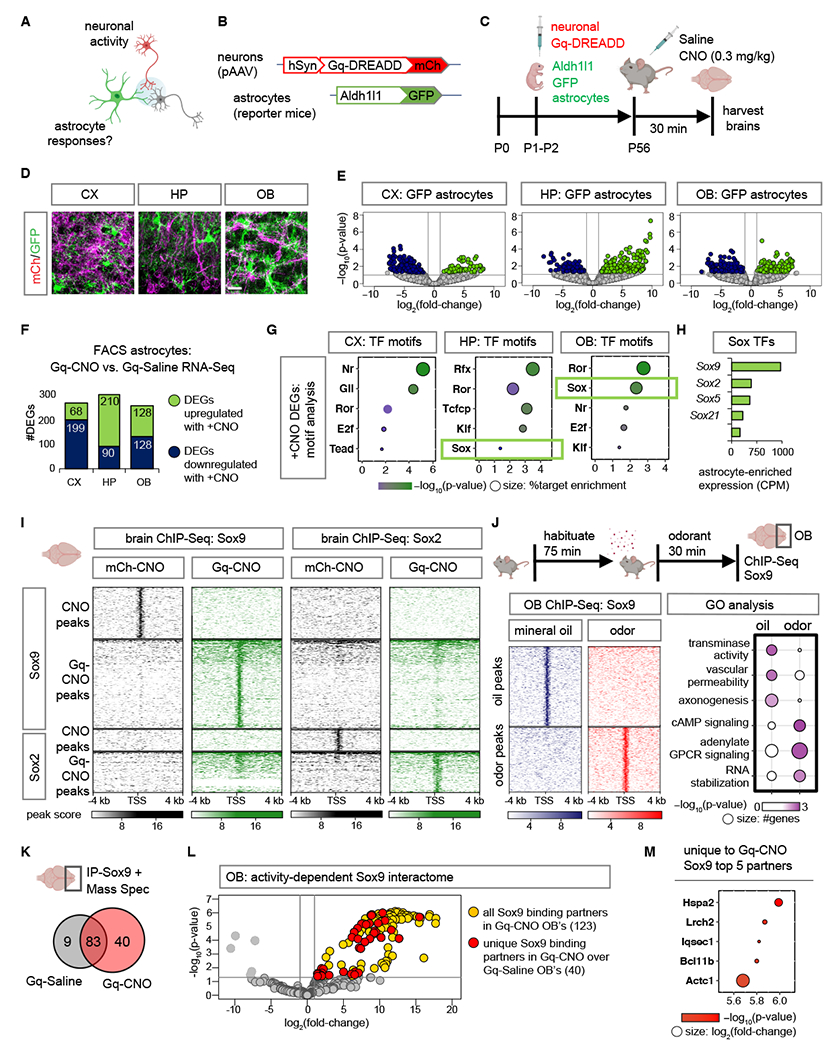
(A-C) Schematic illustrating chemogenetic neuronal activation experimental design. (D) Sections showing distinct labeling of astrocytes (GFP) and Gq-DREADD neurons (mCh) in CX, cortex; HP, hippocampus; OB, olfactory bulb. Scale bar: 25 µm. (E) Volcano plots depicting RNA-Seq from GFP astrocytes comparing Gq-CNO vs. Gq-Saline. (F) Number of DEGs that are upregulated or downregulated in GFP astrocytes in Gq-CNO vs. Gq-Saline (n= 3/cohort, p< 0.05, log2fold-change 1). (G) Significant transcription factor (TF) motifs (p< 0.05) enriched in these DEGs and exhibiting astrocyte-specific expression. (H) Average transcript expression of Sox family TF’s in GFP astrocytes (CPM: counts per million). (I) Comparison showing heatmaps of ChIP-Sox9 and ChIP-Sox2 at 4 kb from peak center in Gq-CNO vs. mCh-CNO control (n= 3/cohort). (J) Schematic for odor evoked neuronal activation in the OB. Left panel: heatmaps of ChIP-Sox9 at 4 kb from peak center in mineral oil vs. odor exposed mice. Right panel: enriched gene ontology terms associated with these peaks (n= 6/cohort). (K) Sox9 binding partners in OB’s from Gq-CNO vs. Gq-Saline. (L) Volcano plot depicting IP-MS (IP-Mass Spectrometry) data of Sox9 interactome in Gq-CNO. Fold change was calculated over control lysates incubated with beads only without antibody. Sox9 binding partners unique to Gq-CNO vs. Gq-Saline are highlighted in red (n= 9–12/cohort, p< 0.05, log2fold-change >1). (M) Top 5 Sox9 interactors unique to Gq-CNO (p< 0.0001, log2fold-change >8.5). Color code represent p-values (x-axis), and size represent log2fold-change with larger circles denoting greater binding affinity.
To dissect the transcriptional mechanisms that underly these changes in astrocyte gene expression, we used TF motif analysis on the astrocyte DEGs. Filtering for TFs that also exhibit enriched expression in astrocytes (Fig. S4A) identified a cohort of TFs with motifs associated with activity-dependent DEGs in astrocytes, including the Sox-family of TFs in both the HP and OB (Fig. 1G). The Sox family has multiple members exhibiting elevated expression in astrocytes, with Sox9 and Sox2 being the most significant (Fig. 1H). Sox9 is critical for olfactory circuit processing (13), while Sox2 plays a role in responses to brain injury (33). We next evaluated Sox9 and Sox2 protein levels after neuronal activation and observed no significant changes in protein expression measured by both immunostaining (Fig. S4B) and immunoblot blot (Fig. S4C–D). Therefore, we asked if these Sox TFs undergo changes in their transcriptional activity after neuronal activation. We performed chromatin immunoprecipitation coupled with next generation sequencing (ChIP-Seq) of Sox9 and Sox2 in CNO treated Gq-DREADD (Gq-CNO) expressing mice in comparison to control pAAV-mCh expressing mice (represented as mCh-CNO herein), harvesting brains 30 minutes after CNO treatment. Although both Sox9 and Sox2 are equally immunoprecipitated (IP) in Gq-CNO and mCh-CNO control lysates (Fig. S5A), we found that both of these Sox TFs exhibit >1.5-fold increase in DNA binding capacity in Gq-CNO brains compared to mCh-CNO controls (Fig. 1I; Fig. S5B–C). Differential ChIP-Seq peak analysis between Gq-CNO treatment vs. controls revealed 295 Sox9-specific peaks, 139 Sox2-specific peaks and 117 Sox9 and Sox2 shared peaks (Fig. S5D). GO analysis revealed activity-dependent peaks were associated with dendrite development (Sox9-specific), proteoglycans (Sox2-specific), while Sox9 and Sox2 shared peaks were enriched in metabolic and mRNA-processing (Fig. S5E).
We next examined whether such changes in TF DNA binding would also occur after native stimulation. Because Sox9 exhibited greater DNA binding capacity, and astrocytic Sox9 is critical for olfactory sensory circuit function in the OB (13), we examined whether olfactory sensory input driven neuronal activation influences Sox9 transcriptional activity. In the OB, Sox9 expression is restricted to astrocytes (>96% Aldh1l1-GFP+ cells) and has miniscule expression in the oligodendroglial lineage (2.79% Olig2+ cells) (Fig. S6A–B). To examine Sox9 transcriptional activity after native olfactory driven neuronal stimuli, we performed Sox9 ChIP-Seq on OBs from mice sacrificed after exposure to odorant isoamyl acetate or mineral oil control for 30 minutes, revealing largely distinct Sox9 peaks (Figure 1J; Figure S6C). Associated GO’s showed that while Sox9 peaks in mineral oil exposed OBs were enriched in transaminase activity GO terms, peaks in odor exposed OBs were enriched in cAMP signaling GO terms (Fig. 1J). To assess if these changes in Sox9 DNA binding capacity after neuronal activation are due to changes in Sox9 binding partners, we performed Sox9-IP coupled with mass spectrometry and observed a >4-fold increase in Sox9 interactions after neuronal activation (Fig. 1K; Table S2), identifying 40 proteins that specifically interact with Sox9 in the Gq-CNO treated OBs in comparison to Gq-Saline control (Fig. 1L). We observed overlap between the top activity-specific Sox9 interacting proteins (Fig. 1M) and TFs identified from motif analysis of activity-specific ChIP-Sox9 peaks in the OB (Fig. S6D).
Slc22a3 is an activity-dependent gene target of Sox9 in OB astrocytes
To identify astrocyte genes that are induced after neuronal activity, we compared the activity-dependent astrocyte-specific DEGs in the OB (Fig. 1E), with existing RNA-Seq data from OB astrocytes from Sox9 conditional knockout mouse (Sox9-cKO) (13), where Sox9 is deleted specifically from OB astrocytes. We identified 62 genes that are activity-dependent and Sox9 regulated, representing 24.2% of activity-dependent DEGs in OB astrocytes (Fig. S7A). Among these, the 34 genes demonstrating decreased expression in the Sox9-cKO (Fig. 2A; Fig. S7B), were further filtered based on Sox9 motif binding score at their promoters (Fig. 2B). In a ChIP-PCR screen, we immunoprecipitated DNA bound to Sox9, and performed PCR (Fig. S7C) using primers spanning Sox9 motifs at promoter regions of candidate genes. Among the candidates demonstrating Sox9 promoter occupancy in the OB specifically after Gq-based neuronal stimulation was Slc22a3 (Fig. 2C–D; Fig. S7D). To validate Sox9-regulation of Slc22a3, we evaluated its protein expression in mice where Sox9 was specifically knocked out in adult astrocytes (Sox9-cKO; Fig. S8A) (13), and found a 76% reduction of its expression in astrocytes from the OB (Fig. 2E–F), but not CX or HP (Fig. S8B–E).
Fig. 2. Neuronal activation induces Slc22a3 in OB astrocytes.
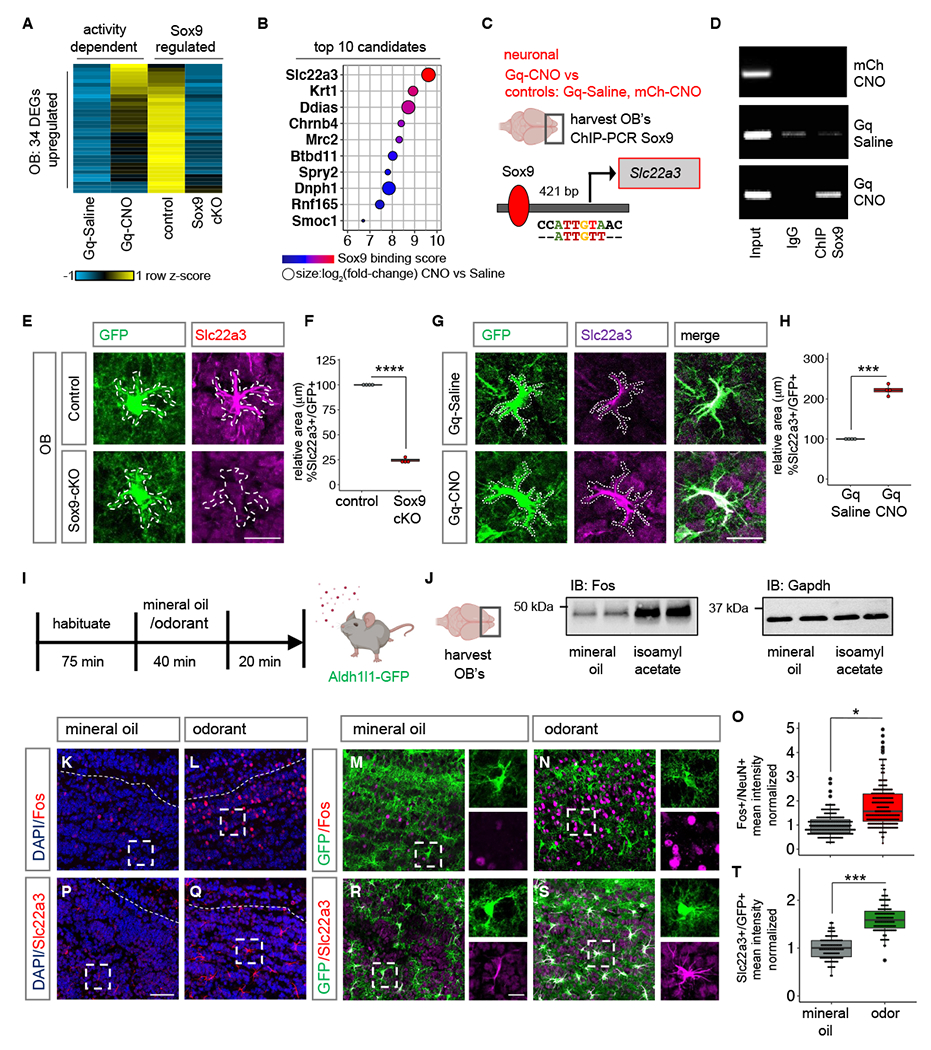
(A-B) Heatmap depicting the 34 neuronal activity-dependent and Sox9 regulated DEGs (n= 3, p< 0.05), and the top 10 candidates filtered by Sox9 motif binding score 1000 bp from transcription start site of candidates. (C-D) ChIP-PCR screen identifying activity-dependent Sox9 regulation at Slc22a3 promoter (n= 4-6 OB). (E-H) Slc22a3 expression in control vs. Sox9-cKO OB astrocytes and in Gq-Saline vs. Gq-CNO; and box plots depicting area covered by Slc22a3 in GFP astrocytes (average of 116-122 cells/cohort, ****p= 1.93E-08; ***p= 0.00013, unpaired Student’s two-tailed t-test on n= 4 mice/cohort); Scale bar: 20 µm. (I) Schematic illustrating odor-evoked neuronal activation experimental design. (J) Immunoblots of Fos and loading control from OB lysates (n= 4/cohort). (K-O) Immunostaining of Fos in Aldh1l1-GFP mice and quantification of mean fluorescence intensity in NeuN+ neurons (244-250 cells/cohort, *p= 0.0285, Wilcoxon rank sum test on n= 4 mice/cohort). (P-T) Immunostaining of Slc22a3 in Aldh1l1-GFP mice and quantification of mean fluorescence intensity in GFP+ astrocytes (130-131 cells/cohort, ***p= 0.00039, unpaired Student’s two-tailed t-test on n= 4 mice/cohort). Scale bar: 50 µm; inset: 10 µm. Dashed line represents boundary of granule cell layer. See Table S3 for data summary.
We examined whether astrocytic Slc22a3 is regulated by neuronal activity by evaluating its protein and transcript expression in our Gq-DREADD based paradigm. Its expression was upregulated 2.22-fold in Gq-CNO treated OB astrocytes in comparison to Gq-Saline and CNO-control OBs (Fig 2G–H; Fig. S9A–E). We investigated whether neuronal activation from olfactory sensory input promotes Slc22a3 expression in the OB. We exposed habituated Aldh1l1-GFP mice to mineral oil or the odorant isoamyl acetate. Exposure was maintained for 40 minutes and after removal of oil or odorant, mice were kept in the chamber for an additional 20 minutes to allow protein turnover (Fig. 2I). We evaluated Fos expression, a marker of neuronal activity, revealing increased protein expression in lysates obtained from the OB (Fig. 2J; Fig. S10A) and we observed a significant 88.8% increase in neurons of odor exposed OBs compared to mineral oil control, but not in astrocytes (Fig. 2K–O; Fig. S10B). Next, we assessed Slc22a3, observing a significant 58.9% increase in its astrocytic expression in odor exposed mice compared to mineral oil and non-habituated controls (Fig. 2P–T; Fig S10C–E). These analyses were performed in the granular cell layer of the OB given both Fos and Slc22a3 enrichment in this sub-region of the OB. Moreover, we observed a small increase in the number of astrocytes that express Slc22a3 (Fig. S10F) and increased levels of Slc22a3 expression was observed only in astrocytes and not in neurons as evaluated by co-labeling with NeuN (Fig. S10G).
Astrocytic Slc22a3 is required for olfactory sensory processing
Slc22a3 is a membrane transport protein and is involved in both release and uptake of the catecholamine family of chemicals, which include neuromodulators serotonin, dopamine and noradrenaline (34–38). Slc22a3 is expressed in astrocytes (36, 37) (Fig. S11A–B), however, its contributions to astrocyte function and associated brain circuits are unknown. We determined whether Slc22a3 contributes to OB astrocyte function and olfactory circuits. To achieve selective knockout of Slc22a3 we generated a transgenic mouse line containing a floxed Slc22a3 allele (Slc22a3-FF), in combination with OB injection of pAAV containing Cre that is driven by the astrocyte specific Gfap promoter (Fig. S11C–D). Injections occurred at P60, mice were harvested at P90, which enabled region- and cell type- specific deletion of Slc22a3 in OB astrocytes (Fig. S11E–F), but not in neurons (Fig. S11G–I). To quantify Slc22a3 knockout efficiency in astrocytes, we generated Slc22a3-FF; Aldh1l1-GFP, a double transgenic mouse line obtained by crossing Slc22a3-FF and Aldh1l1-GFP mice. For control experiments, we injected Gfap-mCherry and for knockout experiments, we injected Gfap-Cre-RFP viral vectors; as before mice were injected at P60 and harvested at P90 (Fig. 3A). Quantification of Slc22a3 knockout efficiency across all Aldh1l1-GFP OB astrocytes revealed a significant 58.34% reduction in Slc22a3 expression in astrocytes (Fig. 3B–C; Fig. S12A–D), but not in neurons (Fig. 3D–E). Finally, we did not observe any changes in the overall numbers of astrocytes and neurons after Slc22a3 deletion in the OB (Fig. S12E–F).
Fig. 3. Astrocytic Slc22a3 regulates olfactory circuit function.
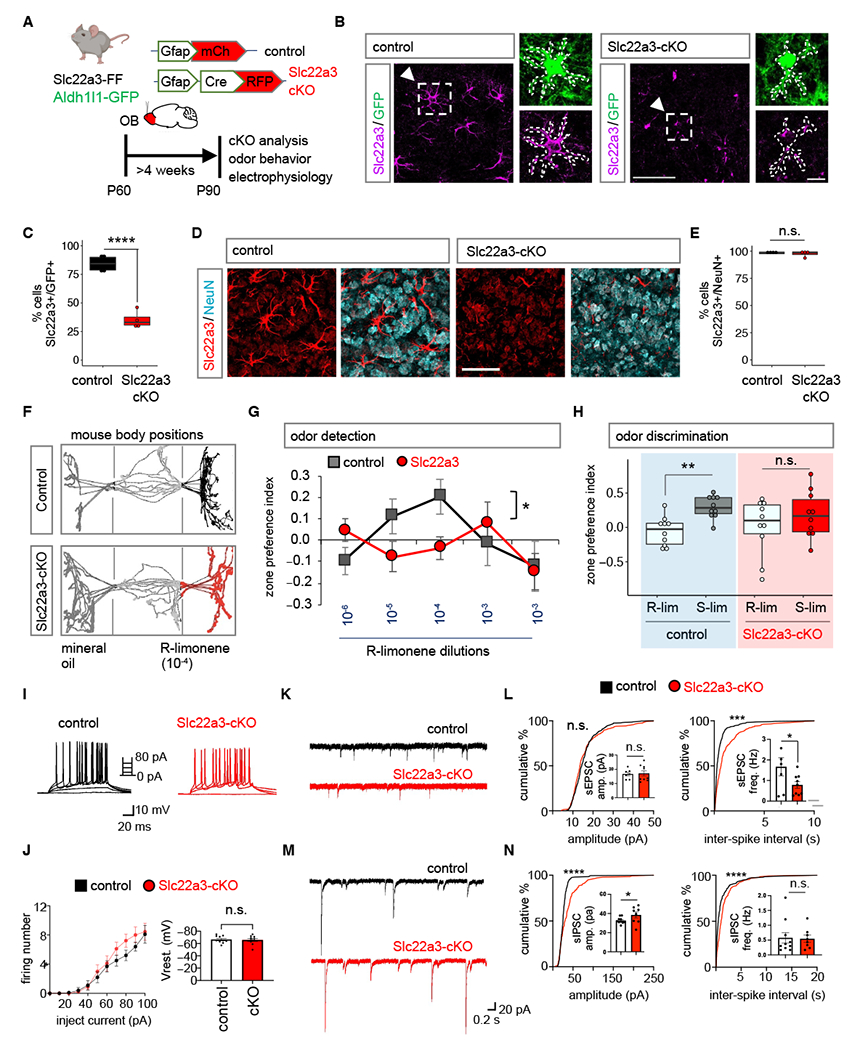
(A) Schematic illustrating viral vectors used for Slc22a3 conditional deletion from OB astrocytes. (B-E) Immunostaining and quantification of Slc22a3 in GFP+ astrocytes (average of 83-93 cells/cohort, ****p= 9.59E-05, unpaired Student’s two-tailed t-test on n= 4 mice/cohort); and in NeuN+ neurons (average of 350-363 cells/cohort, p= 0.5828, unpaired Student’s two-tailed t-test on n= 4 mice/cohort). Scale bar: 50 µm, inset: 10 µm. (F) Schematic illustrating live mouse tracking in three-chamber assay for odor detection. Top represent a control mouse exploring R-limonene (R-lim) at dilution of 10−4, while bottom represent Slc22a3-cKO mouse showing no preference for same R-lim concentration. (G) Quantification of odor detection in control and Slc22a3-cKO mice (n= 10/cohort, *p= 0.0211; two-way repeated measures ANOVA with Sidak multiple comparison). (H) Quantification of odor discrimination between R-lim and S-limonene (S-lim) from the same cohorts of mice (n= 10/cohort, **p= 0.0052; two-way repeated measures ANOVA with Sidak multiple comparisons). (I-J) Whole-cell patch clamp electrophysiology of granule cells firing number from stepped current injections (n= 3, 10 cells/cohort, p= 0.0578, two-way ANOVA with Sidak’s multiple comparison correction). (K-L) Traces and summary data of amplitude and frequency from sEPSC recordings (7-9 cells/cohort, sEPSC amplitude p= 0.7546; sEPSC frequency *p= 0.0473, unpaired Student’s two-tailed t-test on n= 3 mice/cohort; ****p< 0.0001); Kolmogorov-Smirnov (K-S) test. (M-N) Traces and quantification of amplitude and frequency from sIPSC recordings (8-10 cells/cohort, sIPSC amplitude *p= 0.0164, sIPSC frequency p= 0.8095, unpaired Student’s two-tailed t-test on n= 3 mice/cohort ****p< 0.0001 K-S test). All recordings in (L, N) are in granule cells and data is presented as mean ± SEM. See Table S3 for data summary.
To determine how loss of Slc22a3 in OB astrocytes affects olfactory behaviors, we used Slc22a3-cKO and controls (Fig. 3A). We performed behavioral assays on these mice to evaluate potential alterations in odor behaviors (Figure S13A). In a 3-chamber place preference assay mice were exposed to a novel odorant (R)-limonene in an increasing concentration series (Fig. S13B). Odor detection was calculated based on the time spent investigating the odorant containing chamber in comparison to mineral oil chamber and represented as zone preference index. Slc22a3-cKO required a higher concentration of odorant to exhibit preference for R-limonene, compared to control, thus demonstrating a significant reduction in olfactory detection (Fig. 3F–G). Next, we evaluated odor discrimination, using a similar paradigm, where we compared the preference for structurally similar odorants (Fig. S13B). While control mice spent more time in the new odorant containing chamber, thus demonstrating odor preference, Slc22a3-cKO mice showed no significant difference in preference, suggesting Slc22a3-cKO mice are deficient in their ability to discriminate between chemically similar, but distinct odorants (Fig. 3H). Given the observed defects in olfactory behaviors, we examined whether the electrophysiological properties of OB neurons are impacted by astrocytic knockout of Slc22a3. We performed whole-cell recordings from granule cells in the OB and found that excitability of granular cell layer neurons was unaffected (Fig. 3I–J). Examination of their synaptic properties revealed a significant 52.78% decrease in sEPSC frequency (Fig. 3K–L), coupled with a 17.78% increase in sIPSC amplitude (Fig. 3M–N) in Slc22a3-cKO mice.
Slc22a3 regulates astrocytic calcium responses to neuromodulators
The forgoing results indicate that loss of astrocytic Slc22a3 disrupts olfactory function, leading us to examine how its loss impacts core features of astrocytes in the OB. We performed RNA-Seq on FACS isolated astrocytes from control and Slc22a3-cKO OBs to identify molecular features impacting core astrocyte properties after loss of Slc22a3 (Fig. 4A). Slc22a3 deletion (Fig. S13C) affected astrocyte transcriptomics: 565 upregulated and 1049 downregulated genes were differentially expressed in Slc22a3-cKO RFP astrocytes in comparison to control mCh injected control astrocytes (Fig. 4B, Table S4). GO analysis of these DEG’s revealed synaptic transmission as the most impacted category, in addition to cell-cell adhesion, interferon response, neurotransmitter transport, potassium ion transport and calcium ion binding. Constituent genes of all these categories were mostly downregulated. Only interferon response genes were upregulated (Fig. 4C–D). Given that synaptic transmission is affected by astrocyte-neuron communication, which is reliant upon their proximity, and facilitated by complex and elaborate astrocyte morphologies, we evaluated how loss of Slc22a3 impacts astrocyte morphology. Furthermore, Slc22a3-cKO astrocytes demonstrate downregulation of several morphology-related genes (Fig 4E). We performed high resolution confocal imaging and three-dimensional reconstruction of OB astrocytes using GFP reporter in control (Aldh1l1-GFP; Slc22a3-FF + pAAV-Gfap-mCh) and Slc22a3-cKO (Aldh1l1-GFP; Slc22a3-FF + pAAV-Gfap-Cre-RFP) OB’s (Fig. 4F). Sholl analysis of astrocytes revealed decreased morphological complexity in Slc22a3-cKO in comparison to controls (Fig. 4G) based on a host of parameters including number of process intersections as a function of distance from soma (Fig. 4H) (82.96% reduction), total process length (48.25% reduction), number of branches (21.23% reduction) and terminal points (54.24% reduction) (Fig. 4I).
Fig. 4. Astrocytic Slc22a3 regulates astrocyte morphology and calcium activity.
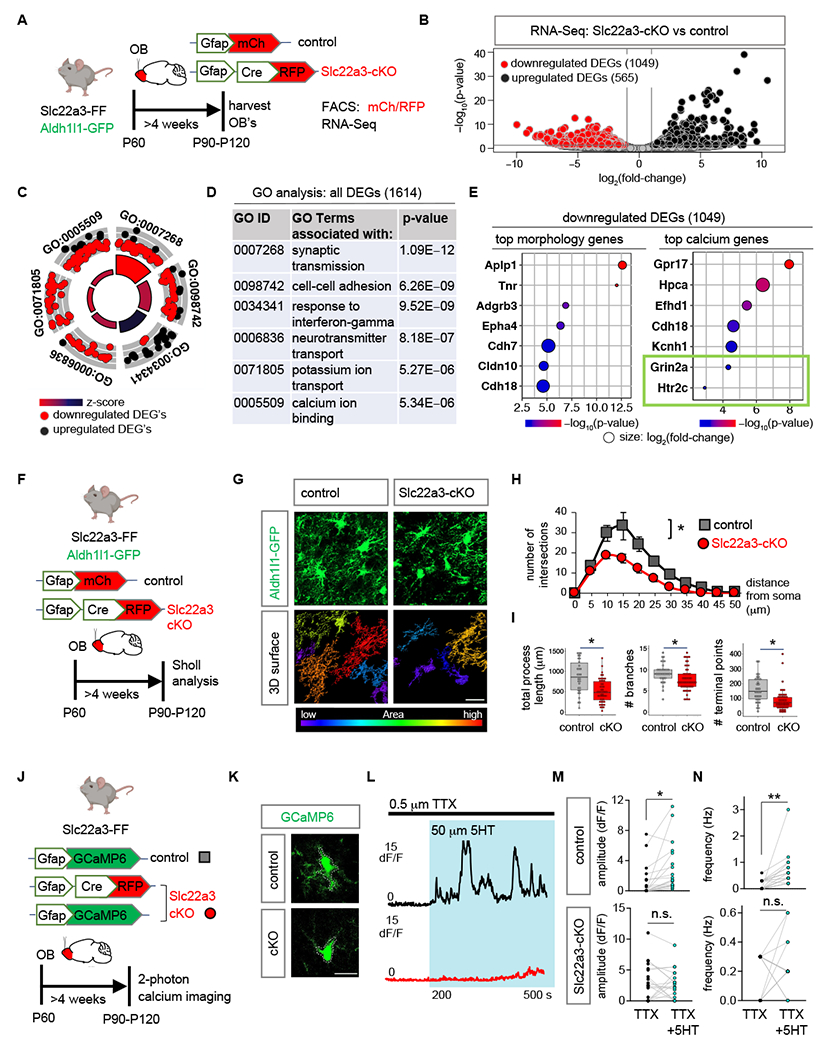
(A) Schematic illustrating viral vectors and timelines for RNA-Seq experiment. (B) Volcano plots depicting RNA-Seq from FACS sorted astrocytes comparing Slc22a3-cKO vs. control samples. (C-D) Gene Ontology circle plot and table, showing the top GO terms found in DEGs shown in (B). (E) Top morphology and calcium associated genes (p< 0.01) in downregulated DEGs in the Slc22a3-cKO OB astrocytes. (F) Schematic illustrating viral vectors and timelines for evaluation of astrocyte morphology. (G) High-magnification confocal images of Aldh1l1-GFP from control and Slc22a3-cKO mice and 3D surface rendering of the same showing reduced astrocyte morphological complexity in Slc22a3-cKO OB astrocytes. Scale bar: 20 µm. (H) Sholl analysis of astrocyte complexity (n= 4, average of 34-54 cells/cohort, *p= 0.0117; two-way repeated measures ANOVA with Sidak correction). Data presented as mean ± SEM. (I) Quantification of total process length (*p= 0.0285, Wilcoxon rank sum test), branch number (*p= 0.0198, unpaired Student’s two-tailed t-test), and terminal points (*p= 0.0177, unpaired Student’s two-tailed t-test) in control and Slc22a3-cKO OB astrocytes (34-54 cells/cohort, statistics on n= 4 mice/cohort). (J) Schematic illustrating mice, viral vectors, and timelines for expression of optical calcium sensor in OB astrocytes. (K-L) Traces from two photon, slice imaging of GCaMP6 activity from OB astrocyte soma in control and Slc22a3-cKO in the presence of TTX (0.5 µM) and serotonin (5HT, 50 µM). Scale bar: 10 µm. (M-N) Quantification of amplitude and frequency from 5HT induced calcium activity from astrocyte soma (19-20 cells/cohort, control amplitude *p= 0.0346; Slc22a3-cKO amplitude p= 0.6992; control frequency **p= 0.0029; Slc22a3-cKO frequency p= 0.5588; paired Student’s two-tailed t-test on n= 4 mice/cohort). See Table S3 for data summary.
Astrocyte calcium activity is considered a proxy for the physiological activities of astrocytes and has been linked to astrocyte regulation of neuronal function. Furthermore, our RNA-Seq studies identified several genes linked to calcium activity demonstrating significant downregulation in Slc22a3-cKO astrocytes (Fig. 4E). To assess astrocytic calcium, we used pAAV containing Gfap promoter driven GCaMP6 encoding a fluorescent calcium optical sensor, which we injected into OBs of control (Slc22a3-FF) and Slc22a3-cKO (Slc22a3-FF + pAAV-Gfap-Cre) mice (Fig. 4J). We generated OB slices from these mice and performed two-photon imaging to assess calcium dynamics under basal conditions from astrocytes located in the granular cell layer of the OB. We detected no change in soma spontaneous calcium amplitude and frequency between control and Slc22a3-cKO OB astrocytes (Fig. S14A–C).
Astrocyte calcium activity is modified in response to neurotransmitters and neuromodulators providing an indirect measure of their interactions with neurons (16). We observed a significant downregulation of the ionotropic glutamate receptor Grin2a and the metabotropic serotonin receptor Htr2c in Slc22a3-cKO OB astrocytes (Fig. 4E). We therefore assessed calcium activity in astrocytes from OB slices after application of glutamate and serotonin. We bath-applied serotonin or glutamate onto OB slices while monitoring calcium elevations in astrocytes (with TTX in the solution). Furthermore, because Slc22a3 has been implicated in maintenance of extracellular serotonin levels in the brain (38), we were especially interested in serotonin induced calcium dynamics. We performed two-photon imaging in control and Slc22a3-cKO mice, while TTX (0.5 μM) was applied to block neuronal activity for 200s before application of glutamate (300 μM) or serotonin (50 μM) for an additional 300s. Quantification of fluorescence from astrocytic soma revealed a significant 66.56% reduction in serotonin-induced amplitude and 72.27% reduction in frequency of calcium events in Slc22a3-cKO OB astrocytes in comparison to controls (Fig. 4K–N). In contrast, we observed no significant difference between amplitude of glutamate-induced calcium activity in Slc22a3-cKO astrocytes in comparison to controls, whereas frequency showed a small reduction in Slc22a3-cKO (Fig. S14D–F). Analysis of astrocyte microdomain calcium activity in response to serotonin or glutamate revealed reduced amplitude and frequency of calcium events for both in Slc22a3-cKO (Fig. S15A–D).
Slc22a3 regulates histone serotonylation in astrocytes
We next examined intracellular serotonin levels within astrocytes in the OB. (Fig. S16A). Immunostaining for serotonin (5-HT) revealed a decrease in its intensity within Slc22a3-cKO astrocytes (Fig. S16B–D), suggesting a defect in serotonin transport. In neurons serotonin can be added to histones and serotonylation of histones directly potentiates epigenetic mechanisms of gene regulation (31). While both astrocyte morphology and calcium have been linked to a host of behavioral outcomes (39), the role of astrocytic histone modifications in the regulation of circuits and behavior remains unknown. Furthermore, Slc22a3 is a serotonin transporter in the central nervous system and the role of serotonin in olfactory bulb astrocytes remains relatively unexplored. We first asked whether the observed reduction in intracellular serotonin in Slc22a3-cKO astrocytes impacts histone serotonylation. Using the Aldh1l1-GFP reporter mouse, we performed immunostaining with antibodies specific for the serotonylated histone mark on histone H3, H3K4me3Q5ser (H3-5HT) and found that 86% of GFP+ OB astrocytes contain H3-5HT (Fig. 5A; Fig. S17A). Next, we immunostained for H3-5HT in Slc22a3-cKO (Aldh1l1-GFP; Slc22a3-FF + pAAV-Gfap-Cre) and control (Aldh1l1-GFP; Slc22a3-FF + pAAV-Gfap-mCh) OB astrocytes (Fig. 5B). We found a 18% reduction in the levels of H3-5HT in Slc22a3-cKO OB astrocytes (Fig. 5C–D), but not in neurons of Slc22a3-cKO OB’s (Fig. S17B). We next performed ChIP-Seq of H3-5HT on control and Slc22a3-cKO OBs to determine whether patterns of H3-5HT epigenetic modification are affected after astrocytic Slc22a3 loss and observed a 2.3-fold reduction in H3-5HT peaks in Slc22a3-cKO OBs (Fig. 5E).
Fig. 5. Slc22a3 regulates histone serotonylation in OB astrocytes.
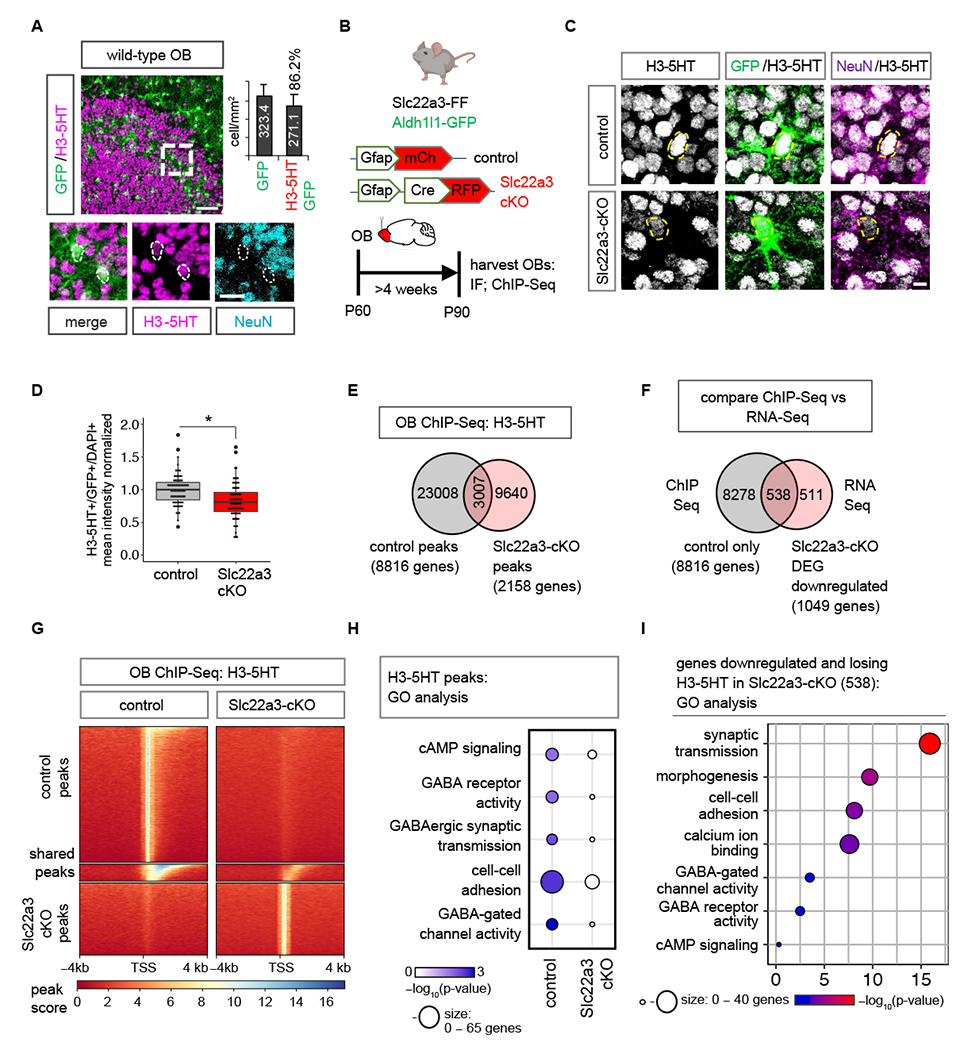
(A) Immunostaining of H3-5HT in OBs of Aldh1l1-GFP mouse and quantification of GFP+/H3-5HT+ co-labeling (n= 3, 25-45 cells). (B) Schematic illustrating viral vectors and timelines for H3-5HT quantification and ChIP-Seq. (C-D) H3-5HT immunostaining and quantification in control and Slc22a3-cKO OB astrocytes (74-79 cells/cohort, *p= 0.0377; unpaired Student’s two-tailed t-test on n= 4 mice/cohort). Scale bar: 5 µm. (E) Venn diagram depicting number of H3-5HT ChIP-Seq peaks unique and shared between control and Slc22a3-cKO OB’s (n=4 OBs/cohort). (F) Venn diagram depicting number of genes that both lose H3-5HT peaks and are downregulated in Slc22a3-cKO. (G) Heatmaps comparing ChIP H3-5HT at 4 kb from peak center in control vs Slc22a3-cKO OB’s. (H) GO analysis of genes at H3-5HT peaks revealing loss of H3-5HT regulation at GABA-associated pathways in Slc22a3-cKO, and (I) of the 538 overlapping genes shown in Figure 5F. See Table S3 for data summary.
Prior studies demonstrating that histone serotonylation activates gene expression (31), led us to cross-compare our ChIP-Seq results with the RNA-Seq data obtained from Slc22a3-cKO. Of the 1049 downregulated DEGs in the Slc22a3-cKO, 538 genes (51% of the DEGs) also lose histone serotonylation modifications in the Slc22a3-cKO (Fig. 5F–G). In comparison, of the 565 upregulated DEGs in Slc22a3-cKO, only 26 genes (4.6% of the DEGs) acquire H3-5HT modifications in the Slc22a3-cKO (Fig. S17C). We next interrogated GO and pathways exhibiting differential H3-5HT modification in the OB from Slc22a3-cKO mice. GO analysis between control and Slc22a3-KO H3-5HT peaks revealed that a prevalence of GO terms linked to GABAergic signaling are lost after astrocytic Slc22a3 deletion (Fig. 5H). Among these genes are various receptor subtypes for GABA, and GABA biosynthetic enzymes Maob and Aldh1a1 (Fig. S17D). Further analysis of the GO terms from the 538 genes that lose H3-5HT and are downregulated in Slc22a3-cKO revealed categories of synaptic transmission, morphology, cell-cell adhesion and calcium ion binding, GO terms that were also observed in the Slc22a3-cKO RNA-Seq (Fig 4D). GABA signaling pathway GO categories were also maintained in this smaller subset of filtered astrocytic genes (Fig. 5I).
Because the GABA pathway represents one of the top GO terms demonstrating a loss of histone serotonylation modifications in the Slc22a3-cKO (Fig. 5H) we focused on the GABA synthesis pathway. Assessing astrocyte-specific expression of GABA and protein expression of Maob and Aldh1a1 using localization with GFP in control and Slc22a3-cKO, we found that both MAOB and astrocytic GABA were significantly downregulated (31.5% and 30.2%, respectively) in OB astrocytes from Slc22a3-cKO mice, while Aldh1a1 was unaffected (Fig. 6A–D; Fig. S18A–B). Similar analysis evaluating GABA and MAOB levels in neurons with NeuN colabeling revealed no significant differences (Fig. 6E–H). However, we observed an overall reduction in mean intensity of GABA in whole sections, but not of MAOB (Fig S18C–D). Given that astrocytes are capable of synthesizing and releasing GABA (25, 40) and our results suggest a general decrease in astrocytic GABA in Slc22a3-cKO astrocytes, we used slice electrophysiology recordings to measure tonic GABA release from OB astrocytes in control and Slc22a3-cKO mice (Fig. 6I). We found a 86.71% decrease in tonic GABA current from granular cell layer neurons of the OB in Slc22a3-cKO. To exclude the possibility that the tonic GABA current is derived from neurons, we performed control recordings in the presence of TTX (0.5 μM) to dampen neuronal activity. This revealed a 75.24% decrease in tonic current in Slc22a3-cKO OBs, confirming that the observed reduction in tonic GABA current is driven by astrocytes, and not neurons (Fig. 6J–L). Finally, to determine whether this reduction in tonic GABA currents was due to a decrease in high-affinity neuronal extrasynaptic GABA-A receptor function or expression, we recorded tonic GABA currents in the presence of GABA (5 μM), a saturating concentration for extrasynaptic GABA-A receptors. We identified no differences between control and Slc22a3-cKO groups (Fig. 6M–N), further indicating that the observed reduction in tonic GABA currents (Fig. 6J–L) results from a decrease in GABA release from astrocytes.
Fig. 6. Slc22a3 regulates tonic GABA release from OB astrocytes.
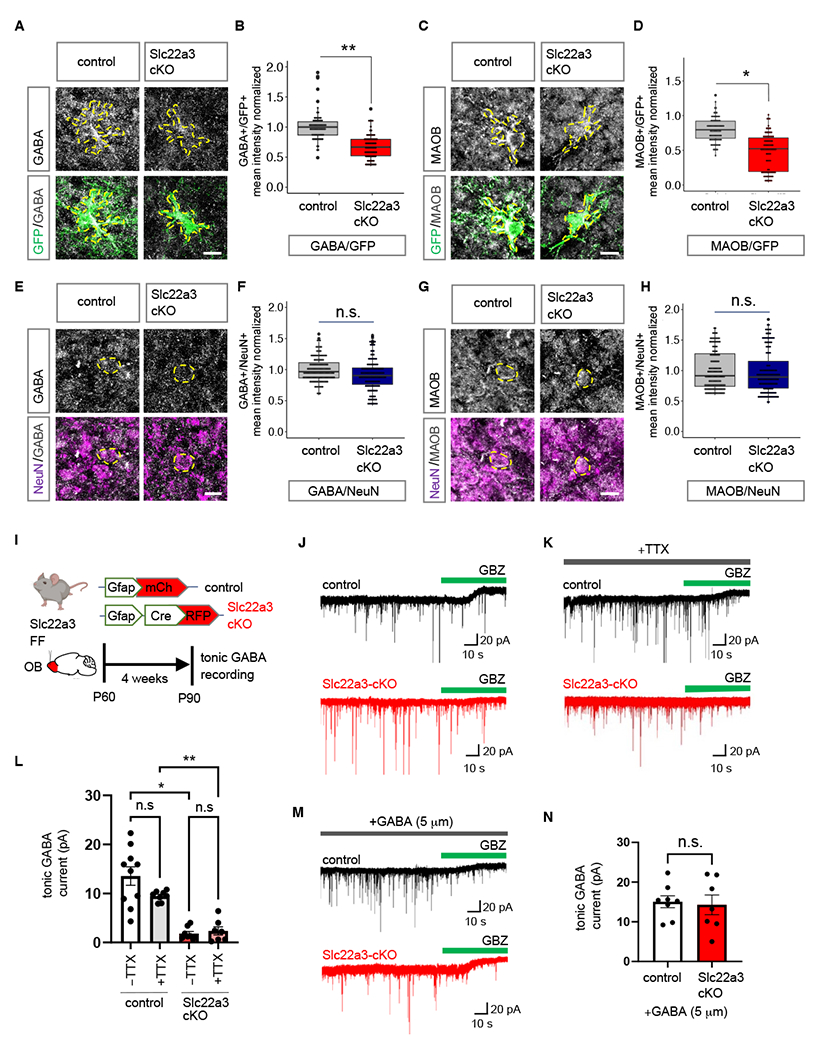
(A-H) Immunostaining and quantification of GABA and MAOB in control vs. Slc22a3-cKO (A-D) GFP+ astrocytes (73-88 cells/cohort; GABA **p= 0.0032, MAOB *p= 0.0121) and (E-H) NeuN+ neurons in the OB (144 cells/cohort; GABA p= 0.2119, MAOB p= 0.9355); unpaired Student’s two-tailed t-test on n= 4 mice/cohort. Scale bar: 10 µm. (I) Schematic illustrating viral vectors and timelines for tonic GABA current measurement experiments. (J-K) Traces of tonic GABA currents in granule cells in OBs from control and Slc22a3-cKO treated with gabazine (GBZ, 20 μM), with or without TTX (0.5 μM). (L) Quantification of tonic GABA current (7-10 cells/cohort, −TTX *p= 0.0331, +TTX **p= 0.0056) (M-N) Traces and quantification of measurement of tonic GABA current in presence of GABA (7-8 cells/cohort, p= 0.8483); unpaired Student’s two-tailed t-test on n= 3 mice/cohort. Data presented as mean ± SEM. See Table S3 for data summary.
Inhibition of H3-5HT in OB astrocytes disrupts sensory processing
To examine whether H3-5HT modifications in OB astrocytes directly contribute to olfactory sensory processing, we utilized a dominant negative mutant H3.3Q5A, where the histone glutamine residue that is modified by serotonin is mutated to an alanine to attenuate H3-5HT (31). The use of histone variant 3.3 (H3.3) ensures histone incorporation to replace canonical H3 during histone protein turnover (41). We generated pAAV containing control H3.3 and mutant H3.3Q5A under control of the Gfap promoter and introduced these into the OBs of wild-type mice (Fig. 7A). Astrocyte-specific expression of H3.3 and H3.3Q5A was confirmed by colocalization of the GFP reporter on H3.3 constructs with astrocyte marker Sox9, but not neuronal marker NeuN (Fig. S19A–B). We observed no differences in cell numbers of GFP+ astrocytes and NeuN+ neurons in OB’s expressing these H3.3 constructs (Fig. S19C–D). Quantification of H3-5HT revealed a significant 46.3% reduction of H3-5HT in H3.3Q5A expressing OB astrocytes in comparison to H3.3 controls (Fig. 7B–C), but not in surrounding neurons (Fig. S19E). We next assessed astrocyte morphology and neuronal electrophysiology. High resolution confocal imaging and Sholl analysis revealed decreased morphological complexity in H3.3Q5A-expressing astrocytes compared to H3.3 controls (Fig. 7D) as evaluated by number of process intersections as a function of distance from soma (59.5% reduction) (Fig. 7E), total process length (58.73% reduction), number of branches (26.44% reduction) and terminal points (63.03% reduction) (Fig. 7F). Astrocytes exhibit extensive structural plasticity in perisynaptic astrocyte processes (PAPs) (42, 43). We therefore measured the number of astrocyte terminal process points and determined distance between PAPs labeled by ezrin (Ezr) and postsynaptic marker PSD95. There was a significant reduction in the number of PSD95-expressing puncta localized to ezrin expressing terminal astrocytic processes in H3.3Q5A-expressing astrocytes compared to control (Figure S20A–C), suggesting reduced interactions between astrocyte processes and neuronal synapses.
Fig. 7. Inhibition of H3-5HT in OB astrocytes disrupts astrocyte morphology.
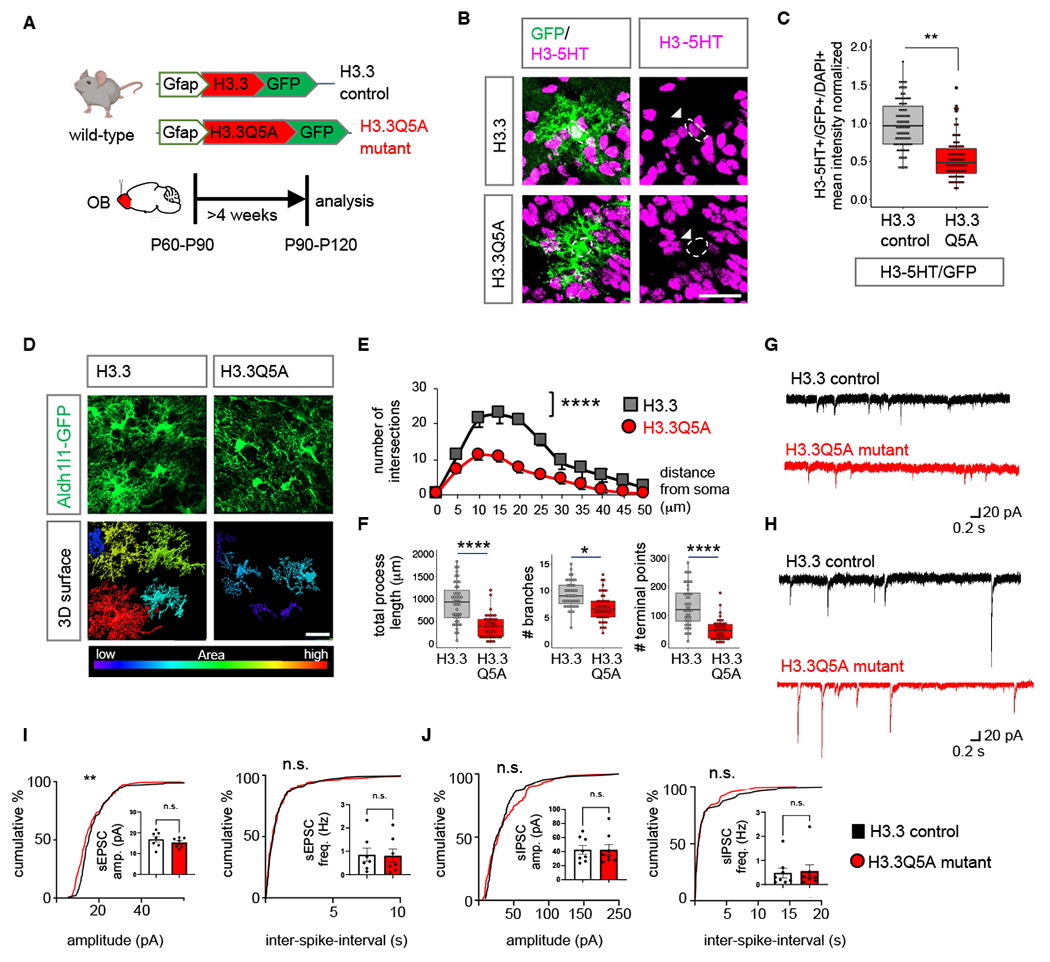
(A) Schematic illustrating viral vectors used for H3.3 and H3.3Q5A expression in OB astrocytes. (B) H3-5HT and GFP co-labeling in H3.3 and H3.3Q5A expressing OBs and (C) box plots depicting quantification of astrocytic H3-5HT (99-110 cells/cohort, **p= 0.0092; unpaired Student’s two-tailed t-test on n= 4 mice/cohort). Scale bar: 25 µm. (D) High-magnification confocal images of H3.3-GFP and 3D surface rendering of the same showing reduced astrocyte morphological complexity in H3.3Q5A OB astrocytes. Scale bar: 20 µm. (E) Sholl analysis of astrocyte complexity (n= 4, average of 44 cells/cohort, ****p= 1.9e-05; two-way repeated measures ANOVA with Sidak correction). Data presented as mean ± SEM. (F) Quantification of total process length, branch number, and terminal points (44 cells/cohort, ****p= 2.75e-05, *p= 0.0122, ****p= 8.73e-06, unpaired Student’s two-tailed t-test on n= 4 mice/cohort). (G-H) Traces and (I-J) summary data of amplitude and frequency from (I) sEPSC recordings (7 cells/cohort, sEPSC amplitude p= 0.2386; sEPSC frequency p= 0.7917; unpaired two-tailed Student’s t-test on n= 3 mice/cohort, **p= 0.0059 K-S test); and from (J) sIPSC recordings (8 cells/cohort, sIPSC amplitude p= 0.8277, sIPSC frequency p= 0.7128, unpaired two-tailed Student’s t-test on n= 3 mice/cohort. All recordings are in granule cells from H3.3 and H3.3Q5A OB’s and data is presented as mean ± SEM. See Table S3 for data summary.
Examination of basal properties of excitatory (Fig. 7G) and inhibitory (Fig. 7H) postsynaptic currents revealed no changes in sEPSC/sIPSC frequency and amplitudes (Fig. 7I–J). Because Slc22a3-cKO showed a significant reduction in H3-5HT epigenomic regulation of GABA pathways, we assessed expression of MAOB and GABA. We found a significant reduction of both (MAOB: 17.3%; GABA: 23.5%) in OB astrocytes expressing H3.3Q5A in comparison with H3.3 control (Fig. 8A–C), but not in neurons (Fig. S21A–B). We also observed an overall reduction in GABA mean intensity in whole fields (Fig. S21C–D). We performed control experiments to confirm that expression of Gfap-H3.3 alone does not alter levels of Slc22a3, GABA and MAOB, in comparison to Aldh1l1-GFP animals (Fig. S21E–G). We used electrophysiology to measure GABA release from OB astrocytes expressing H3.3Q5A mutant or H3.3 controls and saw a 92.97% reduction in tonic GABA current in H3.3Q5A condition (Fig. 8D–F). Control recordings were performed in the presence of TTX (0.5 μM), revealing a 86.09% decrease in tonic current in H3.3Q5A-expressing OBs consistent with alterations astrocyte-derived tonic GABA currents. Control recordings in the presence of saturating GABA (5 μM) further corroborate reduced tonic GABA currents due to a reduction of tonic GABA release from OB astrocytes expressing H3.3Q5A (Fig. S22A–B).
Fig. 8. Inhibition of H3-5HT disrupts astrocytic tonic GABA release and sensory processing.
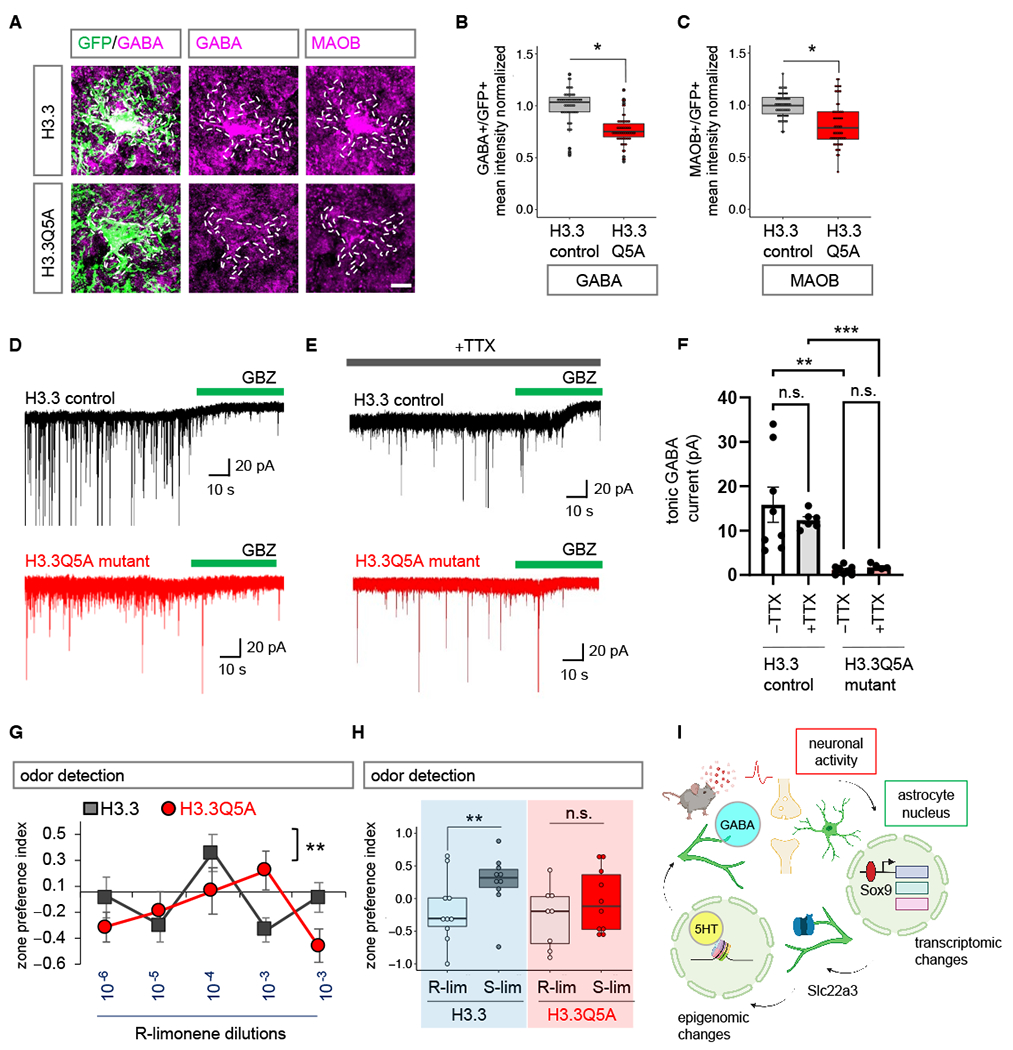
(A-C) Immunostaining and box plots depicting quantification of astrocytic GABA and MAOB in H3.3 and H3.3Q5A OBs (56-58 cells/cohort, GABA: *p= 0.0117, unpaired two-tailed Student’s t-test on n= 4 mice/cohort; MAOB *p= 0.0285, Wilcoxon rank sum test on n= 4 mice/cohort). Scale bar: 10 µm. (D-E) Traces of tonic GABA currents in granule cells in OBs from H3.3 and H3.3Q5A treated with gabazine (GBZ, 20 μM), with or without TTX (0.5 μM). (F) Quantification of tonic GABA currents (5-8 cells/cohort, –TTX **p= 0.0017, +TTX ***p= 0.0007; unpaired two-tailed Student’s t-test on n= 3 mice/cohort). Data presented as mean ± SEM. (G) Quantification of odor detection in H3.3 and H3.3Q5A mice (n=10/cohort, **p= 0.0072; two-way repeated measures ANOVA with Sidak multiple comparison). (H) Quantification of odor discrimination between R-lim and S-lim from the same cohorts of mice (n=10/cohort, **p= 0.0058; two-way repeated measures ANOVA with Sidak multiple comparisons). See Table S3 for data summary. (I) Model figure integrating activity dependent transcriptional changes in astrocytes, with Slc22a3 function in olfactory circuits, and histone serotonylation regulation of GABA in astrocytes.
Given the links between GABA signaling and neuronal circuit function in the OB, we examined how inhibition of H3-5HT impacts olfactory sensory processing. We performed odor detection and odor discrimination behavioral assays as previously described (Fig. 3). First, we compared odor behaviors of mice expressing H3.3 in OB astrocytes with wild-type control mice and found that astrocytic H3.3 expression alone has no significant impact on odor detection limit and odor discrimination ability (Fig. S22C–D). Comparing odor behaviors of H3.3 control- and H3.3Q5A mutant- expressing mice revealed reduced odor detection and impaired odor discrimination in mice expressing H3.3Q5A in OB astrocytes (Fig. 8G–H). Mice expressing astrocytic H3.3Q5A recapitulated the phenotypes of impaired tonic GABA and olfactory behaviors observed in the Slc22a3-cKO (Figs. 3–4, 6). Collectively, these findings suggest that the neuronal activity-dependent target Slc22a3 in OB astrocytes facilitates serotonin mediated epigenetic regulation of GABA synthesis, which contributes to sensory processing of olfaction (Fig. 8I).
Discussion
Neuronal circuits and associated behaviors are intimately linked to astrocytes, yet how neuronal activation sculpts astrocyte transcriptional responses to support circuit activities is unclear. We identified neuronal activity-dependent transcriptional changes in astrocytes, highlighted by alterations in DNA-binding capacity of the TF Sox9 and induction of a prospective immediate early gene, the neuromodulator transporter Slc22a3. Further examination of Slc22a3 in astrocytes revealed that it is required to maintain olfactory circuit function. Our study pinpointed Sox-family TFs as key components of activity-dependent responses in astrocytes in the OB. However, given that astrocyte function is regulated by region-specific transcriptional dependencies (13, 14, 44), there are likely region-specific astrocyte TFs that mediate these responses and responses associated with injury and disease (45). It is also likely that different forms of neuronal input (excitatory, inhibitory, etc.) or behavioral states influence these transcriptional responses in astrocytes. These findings illustrate that astrocytes exhibit a form of plasticity in response to neuronal activity and this plasticity is reliant upon a combination of inputs from neuronal circuits and region-specific TFs. These observations also raise the question of how this form of transcriptional plasticity in astrocytes is regulated. The increase in Sox9-DNA binding in the presence of heightened neuronal activity is likely the result of activity-dependent protein interactions. Our Mass-Spec studies identified a cohort of 40 proteins that interact with Sox9 in the OB specifically after neuronal activation. These findings indicate that neuronal input influences the protein constituency of transcriptional complexes and suggests that these inputs shape protein interactions that drive gene expression. Understanding how neuronal input remodels transcriptional complexes and the specific roles of activity-dependent protein interactions in astrocyte-neuron communication are important areas of future investigations. From these findings a model emerges, where neuronal activity orchestrates transcriptional responses in astrocytes to meet the demands of a functioning circuit.
We identified Slc22a3 as a prospective immediate early gene that is specifically induced in OB astrocytes after exposure to odor, and analysis of OB astrocytes from Slc22a3-cKO mouse revealed a host of phenotypes including reduced morphological complexity, reduced calcium activity, decreased tonic GABA release, altered histone serotonylation, and impaired olfactory detection. These observations suggest that in addition to an acute function after odor exposure or neuronal activity, Slc22a3 also plays a chronic, longer-term role in astrocyte function and communication with neurons. The chronic role is likely due to epigenomic changes that result in increased MAOB expression, GABA production, and altered morphology. This chronic role is likely to reflect a homeostatic function for Slc22a3 and/or serotonin transport in maintaining physiological activities of OB astrocytes. It is also possible that these two roles are interdependent, where the changes in GABA release from astrocytes influences neuronal activity in a way that impacts astrocyte morphology. Alternatively, we must consider the possibility that shorter astrocytic processes may lead to longer diffusion time for GABA to reach synaptic GABA receptors, ultimately leading to reduced tonic GABA current. Another possibility is that there is overall less tonic GABA release from astrocytes, as suggested from our data of reduced astrocytic GABA biosynthetic enzyme MAOB and astrocytic GABA levels. Slc22a3 functions as a monamine transporter that regulates the transport of serotonin (38). Because the OB is densely innervated by serotonergic fibers, which activate interneuron granule cells (46), it raises the question of whether serotonin is a factor that modulates inhibitory outputs and the subsequent impact on odor discrimination. Our finding that odor-evoked neuronal activation leads to enrichment of cAMP signaling GO terms in astrocytes, suggests that neuronal activation leads to astrocytic activation of Gs or Gi GPCRs, including astrocytic serotonergic receptors. In turn, this leads to upregulated expression of serotonin transporter Slc22a3 in astrocytes via Sox9. Our data demonstrating serotonin-induced calcium signaling is reduced in Slc22a3-deficient astrocytes is further supported by our observation of reduced astrocytic serotonergic receptors in Slc22a3-cKO. These collective findings indicate that astrocytes utilize serotonin through Slc22a3 to control tonic GABA inhibition, highlighting a new mechanism of serotonergic modulation of inhibitory outputs through astrocytes, which ultimately impacts olfactory sensory processing.
Our observations that Slc22a3-cKO astrocytes display both reduced morphological complexity and reduced calcium activity raises questions regarding the relationship between astrocyte morphology and astrocyte calcium. Since the elaborate processes of astrocytes enables close proximity with synapses, it is likely that astrocyte morphology directly influences calcium. It is also likely, that increases in astrocyte calcium can affect gene expression or local protein translation to increase structural complexity to enhance synaptic contact. However, whether astrocyte morphology and calcium directly influence each other is context-dependent. For example, a recent study has shown that hippocampal astrocytes from socially isolated animals have altered astrocyte calcium activity, however no changes in astrocyte morphological complexity was observed (47). Therefore, additional studies are required to better understand if there is any direct relationship between morphology and calcium.
Epigenetic mechanisms of gene regulation play a key role in all facets of cell physiology, but how these processes influence roles for astrocytes in circuits remains undefined. We found that serotonin is added to histones in astrocytes and this modification is utilized to regulate gene expression and olfactory sensory processing. Mechanistically, Slc22a3 regulates serotonin levels in astrocytes, which influences the extent of histone serotonylation, reflecting a long-term change in transcriptional activity that ultimately regulates the expression of GABA-synthesis components. This represents a new mechanism of epigenomic regulation in astrocytes, while also highlighting that a host of epigenomic phenomenon remain undefined in astrocytes. A recent study demonstrated that Slc22a3 may have roles in noradrenergic signaling at astrocyte nuclear membranes, highlighting another way for Slc22a3 to modulate nuclear processes (48). Synaptic changes in neurons affect chromatin accessibility (30, 49, 50), and it is likely that classic histone acetylation and methylation states are also modified in astrocytes by neuronal activity. Indeed, Sox9 has been implicated in regulating epigenomic states in brain tumors (51). In olfactory circuits, a few studies have investigated the serotonergic system in modulation of olfactory processing (52, 53). Our findings reveal a new role for serotonin in astrocytes, wherein it gets deposited in the genome and indirectly regulates astrocytic release of GABA, demonstrating how astrocytes use serotonin to gate GABA in the olfactory bulb. Studies in mouse and other species have established links between neuromodulator and neurotransmitter signaling (28, 29, 54, 55), mediated by astrocytes. Here, we identified an epigenomic intermediatory. It will be critical to decipher the extent to which these neuromodulatory signaling mechanisms in astrocytes directly regulate neurotransmitter signaling or go through an epigenomic intermediatory and the nature of this form of transcriptional regulation under different behavioral states.
Supplementary Material
ACKNOWLEDGMENTS
We thank Anna Yu-Szu Huang for providing training in stereotaxic injection and Kevin Ung for comments regarding odor behavioral assays. Images in schematics were created using Biorender.com.
Funding:
This work was supported by grants from the NIH (NINDS R01-NS071153 to B.D., R01-AG071687 to B.D., NIDCD 1K99-DC019668 to D.S.). The Baylor College of Medicine Mass Spectrometry Proteomics Core is supported by a Dan L. Duncan Comprehensive Cancer Center NIH award (P30 CA125123), a CPRIT Core Facility Award (RP210227), and an NIH High End Instrument award (S10OD026804).
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability:
All data are available in the manuscript or the supplementary material. The RNA-Seq and ChIP-Seq datasets generated in this study will be made available at NCBI GEO.
REFERENCES AND NOTES
- 1.Cheng YT, Woo J, Deneen B, Neuroscientist, 1–16 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kofuji P, Araque A, Annu. Rev. Neurosci. 44, 49–67 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khakh B, Deneen B, Annu. Rev. Neurosci. 42, 187–207 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Dallérac G, Zapata J, Rouach N, Nat. Rev. Neurosci. 19, 729–743 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Bazargani N, Attwell D, Nat. Neurosci. 19, 182–189 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Kol A, et al. , Nat. Neurosci. 23, 1229–1239 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagai J, et al. , Cell 177, 1280–1292 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adamsky A, et al. , Cell 174, 59–71 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Martin-Fernandez M, et al. , Nat. Neurosci. 20, 1540–1548 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perea G, Yang A, Boyden ES, Sur M, Nat. Commun. 5, 3262 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stobart JL, et al. , Neuron 98, 726–735 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Yap EL, Greenberg ME, Neuron 100, 330–348 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ung K, et al. , Nat. Commun. 12, 5230 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang AY, et al. , Neuron 106, 992–1008 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farhy-Tselnicker I, et al. , Elife 10, e70514 (2021).34494546 [Google Scholar]
- 16.Sardar D, et al. , Int. J. Mol. Sci. 22, 3975 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hasel P, et al. , Nat. Commun. 8, 15132 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hrvatin S, et al. , Nat. Neurosci. 21, 120–129 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khakh BS, McCarthy KD, Csh. Perspect. Biol. 7, a020404 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer KD, Houston ACW. G. V. Rebec, J. Neurosci. 33, 9319–9327 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brancaccio M, Patton AP, Chesham JE, Maywood M ES. H. Hastings, Nat. Neurosci. 21, 120–129 (2018).29230054 [Google Scholar]
- 22.Gomez JA, et al. , Nat. Commun. 10, 1455 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scofiled MD, Kalivas PW, Neurosci. 20, 610–622 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu X, et al. , Neuron 99, 1170–1187 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woo J, et al. , Proc. Natl. Acad. Sci. 115, 5004–5009 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mederos S, et al. , Nat. Neurosci. 24, 82–92 (2021). [DOI] [PubMed] [Google Scholar]
- 27.Paukert M, et al. , Nat. Neuron 82, 1263–1270 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Papouin T, Dunphy JM, Tolman M, Dineley KT, Haydon PG, Neuron 94, 840–854 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Corkrum M, et al. , Neuron 105, 1036–1047 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mews P, et al. , Nature, 546, 381–386 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Farrelly LA, et al. , Nature 567, 535–539 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lepack AE, et al. , Science 368, 197–201 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen C, et al. , Cereb. Cortex 29, 54–69 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petrelli F, et al. , Mol. Psychiatr. 25, 732–749 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song W, et al. , PLoS Biol. 17, e2006571 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cui M, et al. , Proc. Natl. Acad. Sci. 106, 8043–8048 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vialou V, et al. , J. Neurochem. 106, 1471–1482 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Baganz NL, et al. , Proc. Natl. Acad. Sci. 105, 18976–18981 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oliveira JF, Sardinha VM, Guerra-Gomez S, Araque A, Sousa N, Trends Neurosci. 38, 535–549 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Lee S, et al. , Science 330, 790–796 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Maze I, et al. , Neuron 87, 77–94 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bernardinelli Y, et al. , Curr. Biol. 24, 1679–1688 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Perez-Alvarez A, Navarrete M, Covelo A, Martin E, Araque A, J. Neurosci 34, 12738–12744 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chai H, et al. , Neuron 95, 531–549 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burda JE, et al. , Nature 606, 557–564 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brunert D, Tsuno Y, Rothermel M, Shipley MT, Wachowiak M, J. Neurosci 36, 6820–6835 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng Y-T, et al. , Neuron, 111, 1–15 (2023). [DOI] [PubMed] [Google Scholar]
- 48.Benton KC, et al. , Glia 70, 1777–1794 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Su Y, et al. , Nat. Neurosci. 20, 476–483 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen L-F, et al. , Cell Rep. 26, 1174–1188.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sardar D, et al. , Proc. Natl. Acad. Sci. 119, (2022). [Google Scholar]
- 52.Petzold GC, Hagiwara A, Murthy VN, Nat. Neurosci. 12, 784–791 (2009). [DOI] [PubMed] [Google Scholar]
- 53.Kapoor V, Provost A, Agarwal P, Murthy VN, Nat. Neurosci. 19, 271–282 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ma Z, Stork T, Bergles DE, Freeman MR, Nature 539, 428–432 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mu Y, et al. , Cell 178, 27–43 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the manuscript or the supplementary material. The RNA-Seq and ChIP-Seq datasets generated in this study will be made available at NCBI GEO.