

Abstract
Parkinson's disease is a progressive neurological disorder, characterized by prominent movement dysfunction. The past two decades have seen a rapid expansion of our understanding of the genetic basis of Parkinson's, initially through the identification of monogenic forms and, more recently, through genome-wide association studies identifying common risk variants. Intriguingly, a number of cellular pathways have emerged from these analysis as playing central roles in the aetiopathogenesis of Parkinson's. In this review, the impact of data deriving from genome-wide analyses for Parkinson's upon our functional understanding of the disease will be examined, with a particular focus on examples of endo-lysosomal and mitochondrial dysfunction. The challenges of moving from a genetic to a functional understanding of common risk variants for Parkinson's will be discussed, with a final consideration of the current state of the genetic architecture of the disorder.
This article is part of a discussion meeting issue ‘Understanding the endo-lysosomal network in neurodegeneration’.
Keywords: Parkinson's disease, genome-wide association, functional genomics, endo-lysosomal
1. Parkinson's disease—a complex and heterogeneous disorder
Parkinson's disease (PD) is a progressive and chronic neurodegenerative disorder, first described by James Parkinson in 1817 [1,2]. It is characterized by motor symptoms, such as resting tremor, slowness of movement (bradykinesia), postural instability, gait impairment and limb rigidity [3–5]. There are also substantial non-motor symptoms, including memory and cognitive impairment, apathy, anhedonia, insomnia, fatigue, urogenital issues, dysfunction of the autonomic nervous system and loss of facial expressions. Some of the non-motor symptoms, such as constipation, depression, rapid eye movement sleep behaviour disorder (RBD) and loss of smell (hyposmia), can emerge well before the motor symptoms [6]. The neuropathology of PD is defined by the loss of dopaminergic neurons, predominantly (although not exclusively) within the substantia nigra pars compacta, and by the accumulation of intracellular inclusions called Lewy bodies, primarily made up of an aggregated form of the protein α-synuclein [7,8]. Although PD is characterized by these pathological hallmarks, there is a remarkable heterogeneity in the aetiology and pathogenesis of the disorder [5,9]. The hetereogeneity can manifest as variation in age of onset, disease progression, clinical phenotypes, cellular pathways, neurotransmitter systems, epigenetics and underlying genetic risks [10,11].
It is estimated that over 10 million people are living with PD across the world [12], with males displaying 1.5 times higher risk than females of developing the disease [12]. There are several methodologies to subtype PD depending on genetic, phenotypic or clinical features [13,14]. For example, ageing is a significant factor contributing to the risk of PD and the age of onset itself varies owing to the underlying genetic, environmental and pathophysiological factors. PD can be categorized according to the age of onset as: juvenile- (less than 20 years), young- (21–49 years), middle- (50–69 years) or late-onset PD (greater than 70 years) [13]. Together, the juvenile- and young-onset PD form the early-onset category of PD. The broader spectrum of PD can also be sub-classified as: PD (where movement dysfunction predominates), Parkinson's disease with dementia (PDD), or dementia with Lewy bodies (DLB), according to neurological diagnoses but differing in their secondary symptoms [3]. At present, there are no disease-modifying treatments available, with therapies limited to symptomatic interventions such as levodopa and deep brain stimulation [9].
It is evident that PD is a complex disorder, and the increasing number of people developing the disorder demands a greater understanding of the mechanisms that underlie the aetiology and pathogenesis to provide the foundations for novel avenues of drug discovery.
2. Mendelian and monogenic forms of Parkinson's disease
The identification and characterization of genetic variants that cause or predispose individuals to PD has provided important insights into the cellular pathways that result in neuronal death. A pivotal moment was the identification of a mutation (an alanine to threonine coding change at codon 53, A53T) in the SNCA gene causative for an autosomal dominant form of PD in 1997, which ushered in a new era of human molecular genetics for PD. SNCA encodes α-synuclein, the major constituent of Lewy bodies, and a central player in the aetiology of PD (box 1) [15–20].
Box 1. α-Synuclein.
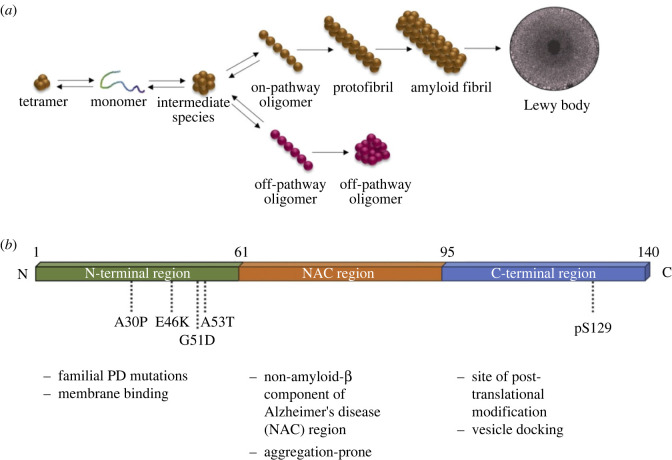
α-Synuclein aggregation is one of the most recognizable pathological features of Parkinson's disease (PD). α-Synuclein in complex with other pathological proteins such as tau or amyloid-β along with lysosomes, lipids and other proteins forms pathological lesions known as Lewy bodies (box figure (a)) [21,22]. The presence of Lewy bodies in the brainstem or the limbic region is a hallmark of PD, and the progression of those lesions to the neocortex defines PDD and DLB; however, some heterogeneity exists in disease manifestation owing to additional genetic or neuronal factors [23]. Despite its clear pathophysiological role in PD, the physiological function of α-synuclein is still debated. α-Synuclein can bind lipid membranes and sense their curvature, which has led to the hypothesis that it plays a direct role in synaptic vesicle trafficking and neurotransmitter release [24]. Additionally, α-synuclein has been reported to be involved in transcription and translation [25], and plays a role in the fission and fusion of mitochondria in nigrostriatal dopaminergic neurons [26]. Further, synergistic interactions have been suggested between α-synuclein, dopamine and calcium in nigral and locus coeruleus neurons [27].
At a genetic level, polymorphisms in the SNCA promoter are associated with idiopathic PD. Gene dosage also plays an important factor in SNCA pathology, with gene duplications and triplications reported in PD and DLB—with the severity of disease displaying a dose dependency [10]. Pathological coding mutations in the SNCA gene have been reported in PD, including A30P, G51D, A53T and E46K, with variants increasing the propensity of α-synuclein to aggregate (box figure (b)). α-Synuclein abnormalities have been associated with mitochondrial dysfunction such as mitochondrial DNA fragmentation, decreased protein import, decreased ATP production, and increased reactive oxygen species (ROS) production, as well as endo-lysosomal dysfunction. This includes impairment of several steps in the endo-lysosomal machinery, thus impacting the autophagic pathway and lysosomal degradation, and disruption of synaptic vesicle transport. α-Synuclein dysfunction also leads to alteration in the retromer trafficking and toxic aggregates in the form of cytoplasmic inclusions through the endo-lysosomal pathway. One of the most prominent theories of α-synuclein aggregation is its spread from neuron to neuron in a prion-like propagation, supposedly through endosome or through tunnelling nanotube formation [20].
Prior to this, PD was considered a prototypic non-genetic form of neurological disorder—occurring sporadically or spontaneously [28–30]. Since then, substantial advances have been made with regards to the identification of monogenic forms of Parkinson's disease, spanning autosomal dominant, autosomal recessive and X-linked patterns of inheritance [28–30]. The monogenic forms of PD may not always follow classical Mendelian inheritance, and may present with varying ages of disease onset, depending on the gene studied. Monogenic loci can be further subtyped according to the clinical phenotypes such as tremor-dominant, akinetic-rigid, postural instability, gait-difficulties, mixed, or indeterminate subtype, depending on the primary symptomatic predispositions [13,31]. They may also be classified according to the progression of motor and non-motor symptoms, and it is apparent that auxiliary genetic or environmental factors may affect disease causation in monogenic PD. A summary of monogenic variants associated with Parkinson's disease is shown in table 1.
Table 1.
Monogenic Parkinson's disease genes. GWAS, genome-wide association studies; JOPD, juvenile-onset PD; YOPD, young-onset PD; MOPD, middle-onset PD; LOPD, late-onset PD; AD, autosomal-dominant; AR, autosomal-recessive; GOF, gain-of-function; LOF, loss-of-function; probable, higher likelihood/orthologue evidence; possible, high likelihood/associative evidence; ER, endoplasmic reticulum, EGFR, epidermal growth factor receptor. *Same PRKN locus.
| no. | gene symbol | gene name | age of onset | inheritance pattern | GWAS hit | confidence as PD gene | mechanism | proposed cellular functions |
|---|---|---|---|---|---|---|---|---|
| 1 | SNCA | synuclein-alpha | YOPD or MOPD | AD | yes | very high | LOF/GOF | synaptic vesicle trafficking, molecular chaperone activity [32,33] |
| 2 | LRRK2 | leucine-rich repeat kinase 2 | LOPD | AD | yes | very high | GOF | vesicle trafficking [34] |
| 3 | GBA1 | glucosylceramidase-beta 1 | YOPD | AR | yes | very high | LOF/GOF? | lysosomal ceramide metabolism [35] |
| 4 | VPS13C | vacuolar protein sorting 13 homologue C | YOPD | AR | yes | very high | LOF | lipid transfer from ER to lysosomes [36] |
| 5 | VPS35 | VPS35 retromer complex component | LOPD | AD | no | very high | LOF | endo-lysosomal sorting [37] |
| 6 | ATP13A2 | ATPase cation transporting 13A2 | YOPD | AR | no | very high | LOF | lysosomal polyamine transporter [38] |
| 7 | PRKN | Parkin RBR E3 ubiquitin-protein ligase | YOPD or JOPD | AR | no | very high | LOF | mitochondrial quality control [39] |
| 8 | PINK1 | PTEN-induced kinase 1 | YOPD | AR | no | very high | LOF | mitochondrial quality control [39] |
| 9 | DJ-1 | parkinsonism associated deglycase | YOPD | AR | no | very high | LOF | involved in oxidative stress, mitochondrial quality control [40] |
| 10 | PLA2G6 | phospholipase A2 group VI | YOPD | AR | no | very high | LOF | membrane lipid biosynthesis [41,42] |
| 11 | FBXO7 | F-box protein 7 | YOPD | AR | no | very high | LOF | E3 ubiquitin-protein ligase [43] |
| 12 | CHCHD2 | coiled-coil–helix–coiled-coil–helix domain-containing 2 | LOPD | AD | no | very high | both | hypoxia-responsive transcription factor; mitochondrial electron transport [44] |
| 13 | GCH1 | GTP cyclohydrolase 1 | YOPD | AD | yes | very high | LOF | involved in neurotransmitter synthesis, nitric oxide synthesis, GTP binding [45,46] |
| 19 | LRP10 | LDL receptor-related protein 10 | YOPD or MOPD | AD | no | very high | LOF | lipoprotein uptake and endosomal sorting [47,48] |
| 14 | DNAJC6 | DnaJ heat shock protein family (Hsp40) member C6 | YOPD | AR | no | high | LOF | uncoating of clathrin-coated vesicles [49] |
| 15 | SYNJ1 | synaptojanin 1 | YOPD | AR | no | high | LOF | synaptic vesicle phosphoinositide metabolism [50] |
| 16 | ATP10B | ATPase phospholipid-transporting 10B | YOPD | AR | no | high | LOF | lipid transfer from ER to lysosomes [51] |
| 17 | DCTN1 | dynactin subunit 1 | YOPD or MOPD | AD | no | high | LOF | transport along microtubules [52] |
| 18 | POLG | DNA polymerase-gamma, catalytic subunit | YOPD | AR and AR | no | high | LOF? | replication of mitochondrial DNA [53] |
| 20 | TMEM230* | transmembrane protein 230 | LOPD | AD | no | low | LOF | involved in trafficking and recycling of synaptic vesicles [54] |
| 21 | DNAJC13* | DnaJ heat shock protein family (Hsp40) member C13 | LOPD | AD | no | low | GOF | endosomal membrane tubulation and trafficking [55] |
| 22 | PSAP | prosaposin | MOPD or LOPD | AD | no | low | LOF? | lysosomal sphingolipid metabolism [56] |
| 23 | HTRA2 | HtrA serine peptidase 2 | YOPD or LOPD | AD | no | low | LOF | serine protease activity, response to oxidative stress, apoptosis [57] |
| 24 | EIF4G1 | eukaryotic translation initiation factor 4 gamma 1 | LOPD | AD | no | low | LOF | ER stress-dependent translation [58] |
| 25 | GIGYF2 | GRB10-interacting GYF protein 2 | LOPD | AD | no | low | undetermined | component of translation initiation repressor complex [59] |
| 26 | SCA2 | ataxin 2 | YOPD or MOPD | AD | no | undetermined | LOF?/GOF? | involved in EGFR trafficking, stress granule formation, regulation of translation [60] |
The identification of monogenic forms of PD has provided the foundations for functional characterization of the biological changes that result in neurodegeneration, studies that have revealed several common pathways. These include, but are not limited to, mitochondrial dysfunction, protein aggregation and disruption of the endo-lysosomal system, portrayed and summarized in figure 1 and table 1, and reviewed in the references cited therein and in [5,61].
Figure 1.

Endo-lysosomal pathways and monogenic Parkinson's disease. Parkin and Pink1 (PRKN and PINK1) play a central role in mitochondrial quality control. LRRK2 and α-synuclein (LRRK2 and SNCA) have been implicated in the regulation of macroautophagy, with LRRK2 also linked to vesicle trafficking at the trans-Golgi and lysosomal damage response. Glucocerebrosidase and ATP13a2 (GBA1 and ATP13a2) have roles in maintaining lysosomal function. Auxilin (DNAJC6) acts to uncoat clathrin-coated vesicles during clathrin-mediated endocytosis.
3. Common genetic risk for idiopathic Parkinson's disease
Monogenic PD represents a minority of cases—estimated to be about 1 in 10, with some variation depending on the population studied [62,63]. The overwhelming majority (>90%) of people living with Parkinson's do not have a single genetic cause of their disease, and the overlap between the functional biology of monogenic and idiopathic disease has only recently begun to emerge [12]. This has been driven by advances in genomic technology, reducing the cost of extensive genetic analysis of large numbers of individuals, and facilitating genome-wide association studies (GWAS) for idiopathic PD, in order to identify common genetic risk for the disease [64,65]. These analyses identify allelic variants that modulate the likelihood of a particular phenotype [66]. Several large-scale GWAS have now been carried out for risk of Parkinson's disease, with the most recent, published in 2019, presenting a meta-analysis of over 30 000 cases and 1 000 000 controls [67]. This identified dozens of loci across the human genome significantly associated with the risk of disease (frequently projected as a Manhattan plot, as shown in figure 2). With rapidly increasing numbers of participants, additional studies have been carried out moving beyond absolute risk of disease to assess genomic variation influencing age of onset, progression, motor subtypes, and risk of dementia, as well as providing insights into multi-ethnic heterogeneity in risk [69–73]. Together, these are beginning to map out how an individual's genetic background influences the likelihood of developing PD, and what happens following the onset of symptoms.
Figure 2.

A Manhattan plot for genome-wide associated variants in Parkinson's disease, showing –log10p for single nucleotide polymorphisms across the autosomes. The horizontal dotted lines indicate the corrected thresholds for achieving genome-wide significance, with example candidate genes for loci discussed in this review indicated [68].
4. Challenges of understanding genome-wide association at a molecular level
The data emerging from GWAS are complex and represent a starting point for translating genetic risk for PD into functional insights rather than being a functional end point in and of themselves. First, in the case of PD, the impact of each individual associated locus is relatively modest, with a small increase in overall risk—to the point where taken individually there is negligible predictive power. It is only when polygenic risk is considered, merging multiple common risk factors, that anything approaching clinical relevance can be arrived at [74,75]. Secondly, moving from the identification of associated loci to pinpointing the biological changes driving that association is a significant challenge [76]. The initial issue is the task of nominating a gene or genes as candidates underlying the association. In some cases, this might be relatively straightforward, with only one gene falling under the significantly associated single nucleotide polymorphisms and/or with a clear functionally impactful coding variant within the candidate gene. In many cases, however, there may be multiple genes falling under the association peak at a genome-wide significant locus. Of the loci identified in the most recent meta-analysis for genome-wide association in Parkinson's disease, only a small fraction of genes under the association peaks presented with conspicuous functional links to previously identified Mendelian forms of PD. For one example, in the SNCA gene on chromosome 4 (figure 3), the association is driven by non-coding variation regulating expression, distinct from the coding or copy number structural variants observed in the Mendelian form of the disease [78,79]. Notably, the majority of genes in the Nalls et al. study [67] were nominated based upon proximity to the sentinel single nucleotide polymorphism (that is, the polymorphism with the most significant association) for the locus. Identifying the gene driving association, and defining the mechanisms underlying association are, therefore, a non-trivial undertaking—and one that will require substantial efforts to complete [80]. An example of how to approach this is provided by Kia and co-workers, who applied analysis of expression, epigenetic modifications, and protein networks to triage and nominate likely candidates emerging from the 2019 GWAS meta-analysis across the genome in PD [81]. There are limitations to such an approach, for example not having accounted for epistasis or pleiotropy of the analysed genes, besides assumptions that the true causal variant is being triaged with certainty [81]. One possible way to overcome this challenge is by using the Mendelian randomization (MR) method, which can draw inferences on the effect of the genetic risk variants, thus possibly providing a better prediction of causation, based on stronger statistical powers [82,83].
Figure 3.

The SNCA locus on chromosome 4 and genome-wide associated single nucleotide polymorphisms. Data are derived from the Meta Five genome-wide association analysis, and accessed through the IPDGC genome browser [77].
Additionally, the majority of current GWAS studies have been based upon data derived from Western cohorts [30]. This issue is being mitigated by more inclusive studies and global initiatives like the Global Parkinson's Genetics Program, focusing on diverse cohorts. The novel hits derived from such studies underscore the heterogeneity of PD in different populations. For example, Rizig and coworkers identified a GBA1 risk variant in African and African-admixed PD cohorts that is distinct from previously identified GBA1 risk alleles [84], Foo and coworkers found SV2C and WBSCR17 as novel hits, along with GBA1, TMEM175, and LRRK2 etc., in East Asian cohorts [85], an Indian study found hits such as SNCA, TMEM175, GBA1, PRKN and BSN [86], and a multi-ethnic study including Latin and Latin-admixed populations identified SNCA, STXBP6 and RPS6KA2 as novel loci in their meta-analysis [87]. Interestingly, the effect of sample size is further demonstrated by Grenn and coworkers, who found GBA1 as a risk factor in European populations only after using larger sample size obtained from diverse datasets [77].
Furthermore, the prevalence of idiopathic PD has been found to be higher in males than in females, whereas there has been no gender bias observed in monogenic forms of PD, such as in LRRK2 G2019S mutation carriers. Recent GWAS studies have explored the causation behind gender differences in European cohorts. However, they have been unable to find any conclusive argument from the analyses of the autosomal and sex chromosomes [88–90]. A comprehensive meta-analysis of several diverse cohorts also did not offer any leads on gender differences [91]. Similar findings were observed in a recent study, where gender differences in GBA1 variants could not explain PD risk but predicted a stronger association of males with DLB [92]. In summary, there are several challenges associated with the molecular dissection of Parkinson's disease GWAS, which can only be resolved by larger and inclusive studies, with refined analyses. Until then, caution needs to be exercised before drawing definitive conclusions.
5. Pathways to parkinsonism—mitochondrial and endo-lysosomal dysfunction
Intriguingly, even with our incomplete knowledge of the genes underlying genome-wide association, there is evidence of convergence between the monogenic and common risk variants found in PD. There are a number of genes that are found in both aspects of genetic risk for Parkinson's disease—SNCA, as outlined above, but also LRRK2 on chromosome 12 and GBA1 on chromosome 1 (see table 1) [93,94]. This overlap implies shared biology between monogenic and idiopathic PD, and reinforces a role for specific cellular pathways in the pathogenesis of PD. Genes highlighted by GWAS and underlying monogenetic risk point to the endo-lysosomal system and mitochondrial quality control as two distinct, but overlapping, pathways leading to PD [95,96]. The endo-lysosomal system is formed by dynamic membrane-bound structures that are involved in processes such as endocytosis, phagocytosis and autophagy to carry out complex functions like macromolecule sorting, cellular signalling, proteostasis, organelle homeostasis and membrane organization. The terminal organelle, the lysosome, is a primary catabolic organelle that has the capacity to break down aggregated proteins or even entire organelles, such as mitochondria. It also serves as a signalling hub that communicates information about amino acid availability or proteotoxic stress to the rest of the cell. Mitochondria, in contrast, are the main producers of cellular energy but also ROS. As ROS can be damaging to the cell and mitochondria themselves, a functioning mitochondrial quality control system is vital in maintaining cellular health. The Parkinson's disease-associated cluster of proteins implicated in the endo-lysosomal system includes the lysosomal β-glucosylceramidase (encoded by GBA1), the lysosomal polyamine exporter ATP13A2 [97], the endoplasmic reticulum (ER)-to-lysosome lipid transfer protein VPS13C [98,99], the lysosomal proton channel TMEM175 [100] and VPS35, which is part of the retromer complex involved in endo-lysosomal sorting of proteins (table 1 and figure 1). Of note, some of these candidates, e.g. VPS13C, have been identified in monogenic forms of PD and via GWAS. Together, these proteins highlight deficits in lysosomal metabolic processes and sorting events along the endo-lysosomal pathway as disease-causing in PD. On the other hand, PINK1 and PRKN are key players in the mitochondrial quality control system by instigating the removal of damaged mitochondria via autophagy. Additionally, CHCHD2 and VPS13C can impact mitochondrial quality control in a PINK1/PRKN-dependent manner [101–103]. Notably, perturbations of genes influencing the endo-lysosomal system may also affect mitochondrial quality control and vice versa. Lysosomal dysfunction will impact mitochondrial quality control as fusion with the lysosome is the final degradative step in the autophagic removal of organelles, and organelle cross-talk, e.g. via lysosome–mitochondria contact sites, impacts the function of each organelle [104].
However, the task of defining the candidate genes at loci identified through studies for Parkinson's disease is still ongoing. To illustrate some of the challenges it presents, and how novel associations are expanding a role for the endo-lysosomal system and mitochondrial dysfunction in Parkinson's disease, two case studies are presented here—examining the loci at chromosomes 7p15.3 and 16q11.2/17q21.
6. Chromosome 7p15.3, GPNMB and lysosomal function
The association at chromosome 7p15.3 was first identified as a Parkinson's disease risk-associated allele through a two-stage meta-analysis as an extension of a previously conducted GWAS [105]. This association occurs as a non-coding A to G change at rs199347 on chromosome 7 [106], with three candidate genes (KLHL7, NUPL2 and GPNMB) in linkage disequilibrium falling under the association peak (figure 4). To discriminate between these three candidate genes, an expression quantitative trait locus analysis was carried out, indicating the risk allele is associated with increased brain expression of GPNMB, but not KLHL7 or NUPL2 [107,108]. Coincident with this, the sentinel single nucleotide polymorphism sits within the GPNMB locus. These data, therefore, support increased expression of GPNMB in brain tissue as being associated with heightened PD risk at this locus. It is worth noting, however, that this does not exclude a role of KLHL7 or NUPL2, as additional transcript-specific expression quantitative trait loci (eQTLs) at this locus are associated with differential expression of both genes in immune tissues [108]. This indicates a potentially complex function of this eQTL, and highlights the need to consider the roles that several differentially expressed genes can play in different cell types and tissues, and at different disease stages.
Figure 4.

The chromosome 7p15.3 locus and GPNMB. Significant single nucleotide polymorphism (SNPs cover a range of genes, including NUPL2, KLHL7 and GPNMB. The most significant SNP is located in a GPNMB intronic region. Data are derived from the Meta Five genome-wide association analysis, and accessed through the IPDGC genome browser [77].
GPNMB has been implicated in melanosome formation, autophagy, inflammation and various human diseases. Specifically, increased GPNMB expression has been linked to poorer prognosis in breast cancer patients [109], and mutations in the GPNMB gene have been named as causal of the skin pigmentation disorder amyloidosis cutis dyschromica (ACD) [110]. Since the discovery of the association between GPNMB and Parkinson's disease, several studies have observed GPNMB function in the context of neurodegeneration and models of PD-related pathology. Increased Gpnmb transcription has been observed in rodent models of PD [111], while post-mortem brain tissues of PD patients display increased GPNMB protein, specifically within the substantia nigra [112]. Moloney and co-workers observed raised GPNMB levels in the context of both α-synucleinopathy and lipidopathy. While elevated α-synuclein levels showed no effect on Gpnmb expression, inhibition of glucocerebrosidase activity (and therefore induction of lipidopathy) resulted in marked increases in GPNMB expression, implying a role of GPNMB in the context of lipid accumulation and lysosomal dysfunction. Additional work, motivated by the linkage of increased GPNMB expression and PD risk, hypothesized a protective effect of Gpnmb ablation in various mouse models of neurodegeneration, but found no effect of Gpnmb knockout when compared with wild-type mice [113]. By contrast, a recent study functionally implicates GPNMB in the cellular uptake of α-synuclein, giving rise to the possibility that GPNMB aids the spreading of α-synuclein throughout the brain in PD [107]. Intriguingly, there is evidence accruing to support a role for GPNMB in lysosomal integrity—although how these data relate to a putative role for GPNMB in PD is, to date, unclear [114,115]. Taken together, there is an emerging yet still incomplete picture of GPNMB's involvement in PD pathology, especially regarding the endo-lysosomal system. Indeed, most of GPNMB's proposed involvement in lysosomal function has been the result of observations made in lysosomal storage disorders [116–118]. Van der Lienden et al. [118] describe GPNMB as an emerging biomarker for lysosomal dysfunction, but its cellular and molecular role in disease mechanisms remains elusive [118]. While strides have been made in our understanding of GPNMB's role in PD, evidence suggests it performs a complex function in PD pathobiology and the endo-lysosomal system, necessitating further investigation.
7. Chromosomes 16q11.2/17q21, KAT8/KANSL1 and mitophagy
Mechanistic insight into the association at 16q11.2 emerged from an unbiased functional screening approach. Based on the hypothesis that idiopathic and Mendelian PD share common underlying disease pathomechanisms, Soutar and co-workers sought to investigate whether Parkinson's disease risk GWAS candidates regulate the PINK1/Parkin-dependent mitophagy process [119]. In this study, 36 PD risk GWAS candidate genes from significant risk loci reported in the 2017 PD GWAS were first prioritized through a triage process applying bioinformatic strategies [67,120,121]. Phenotypic screening and a primary validation of prioritized PD GWAS risk genes, revealed KAT8 to be a novel regulator of the PINK1/Parkin-dependent mitophagy process, with KAT8 knockdown leading to a reduction in phospho-ubiquitin (Ser65) deposition. The KAT8 gene is located within the 16q11.2 risk locus (figure 5), although, as shown by the association signal at this locus, there are many potential candidate genes. KAT8 is a lysine acetyltransferase that represents the catalytically active subunit of the non-specific lethal (NSL) epigenetic remodelling complex, which is responsible for the deposition of pro-transcriptional histone H4 acetylation modification [122]. The NSL complex has been shown to be associated with organism development, cellular homeostasis, and mitochondrial DNA transcription [123–125]. Specifically, KAT8 is involved in cerebral and neural stem cell development, and its variants have been linked to intellectual disability, epilepsy and autism [126]. Soutar et al. showed that knockdown of several other components of the NSL complex also lead to impairments in Ser65 phospho-ubiquitin deposition, including the NSL complex member KANSL1, which is itself another PD GWAS candidate risk gene [119,127]. KANSL1 is located within the 17q21 PD risk locus, which is in linkage disequilibrium with the commonly occurring MAPT H1 haplotype [119,127,128]. Besides this, genetic changes in KANSL1 play a causative role in Koolen–de Vries syndrome in children, which is characterized by intellectual disability, epilepsy, neuromotor phenotypes and other neurocognitive abnormalities [129]. KANSL1 and KAT8 KD were both shown to reduce the mitochondrial accumulation of PINK1 upon mitochondrial membrane depolarization, and consequently downstream PINK1-dependent steps of the mitophagy cascade. Given the canonical function of the NSL complex as a pro-transcriptional epigenetic remodelling complex, impairments in PINK1 gene expression represented a strong candidate mechanism accounting for the PINK1-deficits observed. In line with this hypothesis, the authors showed reduced PINK1 mRNA levels following both KANSL1 and KAT8 siRNA knockdown. Of relevance, eQTLs and allele-specific expression (ASE) analysis linked PD risk at these two loci with reduced KANSL1 and KAT8 expression [130,131], in line with the phenotypic data highlighting impaired PINK1-dependent mitophagy initiation. In addition to PINK1 mitophagy initiation, KANSL1 and KAT8 have also been shown to play an important role in regulating the expression of autophagy-related genes, autophagy and lysosomal function, further implicating the NSL complex in endo-lysosomal regulation, beyond PINK1 mitophagy alone [123,132,133].
Figure 5.

The chromosome 16q11.2 locus and KAT8. The signal at 16q11.2 covers multiple genes, including SETD1A, which was originally nominated as the lead single nucleotide polymorphism, and KAT8, which has been nominated through functional cell-based screening. Data are derived from the Meta Five genome-wide association analysis, and accessed through the IPDGC genome browser [77].
8. Constructing a genetic architecture for Parkinson's disease
These two examples are functionally congruent with the evidence from a number of other genome-wide associated candidate genes as contributing to dysregulation of endo-lysosomal function in idiopathic PD, mapping onto that observed in monogenic PD (figure 6).
Figure 6.

Endo-lysosomal pathways and genes implicated by genome-wide association studies for Parkinson's disease. KAT8 and KANSL1 (KAT8 and KANSL1) have been implicated in the regulation of mitochondrial quality control through mitophagy. As for monogenic associations, LRRK2 and α-synuclein (LRRK2 and SNCA) have been implicated in the regulation of macroautophagy, with LRRK2 and Rab29 (RAB29) also linked to vesicle trafficking at the trans-Golgi and lysosomal damage response. Glucocerebrosidase (GBA1), cathepsin B (CTSB), GPNMB (GPNMB) and TMEM 175 (TMEM175) are all linked to catabolic lysosomal function. Finally, GAK (GAK) plays an analogous role to DNAJC6 in uncoating clathrin-coated vesicles.
Building on the insights from GWAS, it is now possible to move beyond the dichotomous divide between monogenic and idiopathic (including polygenic) forms of the disorder and start to construct a comprehensive genetic architecture for PD. There is substantial evidence for a continuum of risk, quantified as altered odds ratio for disease, for several of the genes involved in monogenic Parkinson's disease, such as SNCA, LRRK2 and GBA1 (all with direct or close links to the endo-lysosomal system). These can be integrated into an increasing number of more common variants in strong candidate genes for increased risk of disease—as well as a smaller number of variants associated with lowered odds ratio and therefore decreased risk of disease (figure 7). This architecture has important implications for our understanding of PD. It provides evidence for shared aetiology between monogenic and idiopathic PD, while simultaneously highlighting the breadth of the disease pathways involved in the disorder. A key consequence of the former is that it increases the likelihood of drugs being developed to target monogenic PD, such as those linked to α-synuclein and LRRK2 forms, having relevance to the wider population of people living with the disease. Several of these, such as the antibody therapies aiming to remove α-synuclein from the brain, or small molecule kinase inhibitors of LRRK2, are currently undergoing clinical trials (NCT04777331, NCT05424276, NCT05670782, NCT03976349, NCT03710707, NCT04056689) [134–136]. Importantly, the identification of pathways linked to disease risk, beyond individual genes and proteins, also opens the door to targeting processes rather than individual genes—expanding the spectrum of drug discovery in Parkinson's.
Figure 7.

The emergent genetic architecture of Parkinson's disease, showing monogenic and genome-wide associated loci. Odds ratios and population frequencies are derived from the IPDGC genome browser [77].
9. Conclusion and future perspectives
Taken together, the past two decades have borne witness to substantial advances in our understanding of the causes of PD, with much of this deriving from enhanced comprehension of the molecular genetics of the disorder. However, much remains to be done. Most obviously, the functional changes underlying the majority of genome-wide associated loci are obscure—information that has the potential to provide even greater insight into the events that precede and drive neurodegeneration in the PD brain. The increasing application of multi-omic analysis, alongside detailed mechanistic characterization, provides hope that many more genome-wide associated loci will be clarified in the coming years. It is also clear that efforts to increase the diversity of genetic analyses of Parkinson's will yield important novel insights into the genetic architecture of the disease. Coupled to more sophisticated stratification of patient populations and moving beyond absolute risk to disease modifiers and progression, there will undoubtedly be more genetic information feeding into functional investigations—including those targeting mitochondrial quality control and the endolysosomal system. Although categorizing risk genes into functional categories can help to focus research efforts, assigning GWAS risk genes requires unbiased approaches and an open mind in order not to overlook novel avenues to understanding and treating PD. Crucially, insights deriving from these studies will aid in defining molecular sub-types of Parkinson's and support the development of novel drug targets for this disorder.
Acknowledgements
Image of a Lewy body in box 1 adapted from [137], with permission.
Contributor Information
Patrick A. Lewis, Email: plewis@rvc.ac.uk.
Susanne Herbst, Email: sherbst@rvc.ac.uk.
Data accessibility
This article has no additional data.
Declaration of AI use
We have not used AI-assisted technologies in creating this article.
Authors' contributions
N.B.: visualization, writing—original draft, writing—review and editing; E.C.B.: writing—original draft, writing—review and editing; B.O'C.: writing—original draft, writing—review and editing; H.P.-F.: writing—original draft, writing—review and editing; P.A.L.: conceptualization, visualization, writing—original draft, writing—review and editing; S.H.: conceptualization, visualization, writing—original draft, writing—review and editing.
All authors gave final approval for publication and agreed to be held accountable for the work performed herein.
Conflict of interest declaration
We declare we have no competing interests.
Funding
N.B., S.H. and P.A.L. are supported by MJFF grant no. 021184. This research was funded in whole or in part by Aligning Science Across Parkinson's (grant no. ASAP 000478) through the Michael J. Fox Foundation for Parkinson's Research (MJFF). For the purpose of open access, the author has applied a CC BY public copyright licence to all author accepted manuscripts arising from this submission.
References
- 1.Parkinson J. 2002. An essay on the shaking palsy. J. Neuropsych. Clin. 14, 223-236. ( 10.1176/jnp.14.2.223) [DOI] [PubMed] [Google Scholar]
- 2.Parkinson J. 1817. An essay on the shaking palsy. London, UK: Whittingham and Rowland for Sherwood, Neely, and Jones. [Google Scholar]
- 3.Seidel K, et al. 2015. The brainstem pathologies of Parkinson's disease and dementia with Lewy bodies. Brain Pathol. 25, 121-135. ( 10.1111/bpa.12168) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Repnik U, Cesen MH, Turk B. 2013. The endolysosomal system in cell death and survival. Cold Spring Harb. Perspect. Biol. 5, a008755. ( 10.1101/cshperspect.a008755) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vidyadhara DJ, Lee JE, Chandra SS. 2019. Role of the endolysosomal system in Parkinson's disease. J. Neurochem. 150, 487-506. ( 10.1111/jnc.14820) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poewe W. 2008. Non-motor symptoms in Parkinson's disease. Eur. J. Neurol. 15, 14-20. ( 10.1111/j.1468-1331.2008.02056.x) [DOI] [PubMed] [Google Scholar]
- 7.Guo YJ, Xiong H, Chen K, Zou JJ, Lei P. 2022. Brain regions susceptible to alpha-synuclein spreading. Mol. Psychiatr. 27, 758-770. ( 10.1038/s41380-021-01296-7) [DOI] [PubMed] [Google Scholar]
- 8.Braak H, Del Tredici K. 2017. Neuropathological staging of brain pathology in sporadic Parkinson's disease: separating the wheat from the chaff. J. Parkinson Dis. 7, S71-S85. ( 10.3233/JPD-179001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bloem BR, Okun MS, Klein C. 2021. Parkinson's disease. Lancet 397, 2284-2303. ( 10.1016/S0140-6736(21)00218-X) [DOI] [PubMed] [Google Scholar]
- 10.Greenland JC, Williams-Gray CH, Barker RA. 2019. The clinical heterogeneity of Parkinson's disease and its therapeutic implications. Eur. J. Neurosci. 49, 328-338. ( 10.1111/ejn.14094) [DOI] [PubMed] [Google Scholar]
- 11.Wüllner U, Borghammer P, Choe C, Csoti I, Falkenburger B, Gasser T, Lingor P, Riederer P. 2023. The heterogeneity of Parkinson's disease. J. Neural Transmiss. 130, 827-838. ( 10.1007/s00702-023-02635-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ben-Shlomo Y, Darweesh S, Llibre-Guerra J, Marras C, San Luciano M, Tanner C. 2024. The epidemiology of Parkinson's disease. Lancet 403, 283–292. ( 10.1016/S0140-6736(23)01419-8) [DOI] [PMC free article] [PubMed]
- 13.Dulski J, Uitti RJ, Ross OA, Wszolek ZK. 2022. Genetic architecture of Parkinson's disease subtypes – review of the literature. Front. Aging Neurosci. 14, 1023574. ( 10.3389/fnagi.2022.1023574) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mestre TA, et al. 2021. Parkinson's disease subtypes: critical appraisal and recommendations. J. Parkinson Dis. 11, 395-404. ( 10.3233/JPD-202472) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Polymeropoulos MH, et al. 1996. Mapping of a gene for Parkinson's disease to chromosome 4q21-q23. Science. 274, 1197-1199. ( 10.1126/science.274.5290.1197) [DOI] [PubMed] [Google Scholar]
- 16.Polymeropoulos MH, et al. 1997. Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science 276, 2045-2047. ( 10.1126/science.276.5321.2045) [DOI] [PubMed] [Google Scholar]
- 17.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. 1998. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605-608. ( 10.1038/33416) [DOI] [PubMed] [Google Scholar]
- 18.Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F. 2002. A new locus for Parkinson's disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann. Neurol. 51, 296-301. ( 10.1002/ana.10113) [DOI] [PubMed] [Google Scholar]
- 19.Singleton AB, et al. 2003. α-Synuclein locus triplication causes Parkinson's disease. Science 302, 841. ( 10.1126/science.1090278) [DOI] [PubMed] [Google Scholar]
- 20.Bernal-Conde LD, Ramos-Acevedo R, Reyes-Hernández MA, Balbuena-Olvera AJ, Morales-Moreno ID, Argüero-Sánchez R, Schüle B, Guerra-Crespo M. 2019. Alpha-synuclein physiology and pathology: a perspective on cellular structures and organelles. Front. Neurosci. 13, 1399. ( 10.3389/fnins.2019.01399) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. 1998. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc. Natl Acad. Sci. USA 95, 6469-6473. ( 10.1073/pnas.95.11.6469) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mahul-Mellier A-L, Burtscher J, Maharjan N, Weerens L, Croisier M, Kuttler F, Leleu M, Knott GW, Lashuel HA. 2020. The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc. Natl Acad. Sci. USA 117, 4971-4982. ( 10.1073/pnas.1913904117) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walker L, Stefanis L, Attems J. 2019. Clinical and neuropathological differences between Parkinson's disease, Parkinson's disease dementia and dementia with Lewy bodies – current issues and future directions. J. Neurochem. 150, 467-474. ( 10.1111/jnc.14698) [DOI] [PubMed] [Google Scholar]
- 24.Sulzer D, Edwards RH. 2019. The physiological role of α-synuclein and its relationship to Parkinson's disease. J. Neurochem. 150, 475-486. ( 10.1111/jnc.14810) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Surguchev AA, Surguchov A. 2017. Synucleins and gene expression: ramblers in a crowd or cops regulating traffic? Front. Mol. Neurosci. 10, 224. ( 10.3389/fnmol.2017.00224) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vicario M, Cieri D, Brini M, Cali T. 2018. The close encounter between alpha-synuclein and mitochondria. Front. Neurosci. 12, 388. ( 10.3389/fnins.2018.00388) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perez RG. 2020. Editorial: The protein alpha-synuclein: its normal role (in neurons) and its role in disease. Front. Neurosci. 14, 116. ( 10.3389/fnins.2020.00116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herbst S, Lewis PA, Morris HR. 2022. The emerging role of LRRK2 in tauopathies. Clin. Sci. 136, 1071-1079. ( 10.1042/CS20220067) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nishioka K, Imai Y, Yoshino H, Li Y, Funayama M, Hattori N. 2022. Clinical manifestations and molecular backgrounds of Parkinson's disease regarding genes identified from familial and population studies. Front. Neurol. 13, 764917. ( 10.3389/fneur.2022.764917) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bandres-Ciga S, Diez-Fairen M, Kim JJ, Singleton AB. 2020. Genetics of Parkinson's disease: an introspection of its journey towards precision medicine. Neurobiol. Dis. 137, 104782. ( 10.1016/j.nbd.2020.104782) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jia FZ, Fellner A, Kumar KR. 2022. Monogenic Parkinson's disease: genotype, phenotype, pathophysiology, and genetic testing. Genes 13, 471. ( 10.3390/genes13030471) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bellani S, Sousa VL, Ronzitti G, Valtorta F, Meldolesi J, Chieregatti E. 2010. The regulation of synaptic function by α-synuclein. Commun. Integr. Biol. 3, 106-109. ( 10.4161/cib.3.2.10964) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rekas A, Ahn KJ, Kim J, Carver JA. 2012. The chaperone activity of α-synuclein: utilizing deletion mutants to map its interaction with target proteins. Proteins 80, 1316-1325. ( 10.1002/prot.24028) [DOI] [PubMed] [Google Scholar]
- 34.Hur EM, Jang EH, Jeong GR, Lee BD. 2019. LRRK2 and membrane trafficking: nexus of Parkinson's disease. BMB Rep. 52, 533-539. ( 10.5483/BMBRep.2019.52.9.186) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paciotti S, Albi E, Parnetti L, Beccari T. 2020. Lysosomal ceramide metabolism disorders: implications in Parkinson's disease. J. Clin. Med. 9, 594. ( 10.3390/jcm9020594) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leonzino M, Reinisch KM, De Camilli P. 2021. Insights into VPS13 properties and function reveal a new mechanism of eukaryotic lipid transport. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids. 1866, 159003. ( 10.1016/j.bbalip.2021.159003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Williams ET, Chen X, Moore DJ. 2017. VPS35, the retromer complex and Parkinson's disease. J Parkinson Dis. 7, 219-233. ( 10.3233/JPD-161020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Veen S, et al. 2020. ATP13A2 deficiency disrupts lysosomal polyamine export. Nature 578, 419-424. ( 10.1038/s41586-020-1968-7) [DOI] [PubMed] [Google Scholar]
- 39.Ge P, Dawson VL, Dawson TM. 2020. PINK1 and Parkin mitochondrial quality control: a source of regional vulnerability in Parkinson's disease. Mol. Neurodegener. 15, 20. ( 10.1186/s13024-020-00367-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Strobbe D, Robinson AA, Harvey K, Rossi L, Ferraina C, De Biase V, Rodolfo C, Harvey RJ, Campanella M. 2018. Distinct mechanisms of pathogenic DJ-1 mutations in mitochondrial quality control. Front. Mol. Neurosci. 11, 68. ( 10.3389/fnmol.2018.00068) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen C, Lou M-M, Sun Y-M, Luo F, Liu F-T, Luo S-S, Wang W-Y, Wang J. 2022. Serum metabolomic characterization of PLA2G6-associated dystonia–parkinsonism: a case-control biomarker study. Front Neurosci. 16, 879548. ( 10.3389/fnins.2022.879548) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kinghorn KJ, et al. 2015. Loss of PLA2G6 leads to elevated mitochondrial lipid peroxidation and mitochondrial dysfunction. Brain 138, 1801-1816. ( 10.1093/brain/awv132) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Teixeira FR, Randle SJ, Patel SP, Mevissen TET, Zenkeviciute G, Koide T, Komander D, Laman H. 2016. Gsk3β and Tomm20 are substrates of the SCFFbxo7/PARK15 ubiquitin ligase associated with Parkinson's disease. Biochem. J. 473, 3563-3580. ( 10.1042/BCJ20160387) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lumibao J, et al. 2023. CHCHD2 mediates glioblastoma cell proliferation, mitochondrial metabolism, hypoxia-induced invasion and therapeutic resistance. Int. J. Oncol. 63, 117. ( 10.3892/ijo.2023.5565) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fanet H, Capuron L, Castanon N, Calon F, Vancassel S. 2021. Tetrahydrobioterin (BH4) pathway: from metabolism to neuropsychiatry. Curr. Neuropharmacol. 19, 591-609. ( 10.2174/1570159X18666200729103529) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hussein D, Starr A, Heikal L, Mcneill E, Channon KM, Brown PR, Sutton BJ, Mcdonnell JM, Nandi M. 2015. Validating the GTP-cyclohydrolase 1-feedback regulatory complex as a therapeutic target using biophysical and in vivo approaches. Br. J. Pharmacol. 172, 4146-4157. ( 10.1111/bph.13202) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sugiyama T, et al. 2000. A novel low-density lipoprotein receptor-related protein mediating cellular uptake of apolipoprotein E-enriched β-VLDL in vitro. Biochemistry 39, 15 817-15 825. ( 10.1021/bi001583s) [DOI] [PubMed] [Google Scholar]
- 48.Brodeur J, Larkin H, Boucher R, Theriault C, St-Louis SC, Gagnon H, Lavoie C. 2009. Calnuc binds to LRP9 and affects its endosomal sorting. Traffic 10, 1098-1114. ( 10.1111/j.1600-0854.2009.00933.x) [DOI] [PubMed] [Google Scholar]
- 49.Ungewickell E, Ungewickell H, Holstein SEH, Lindner R, Prasad K, Barouch W, Martini B, Greene LE, Eisenberg E. 1995. Role of auxilin in uncoating clathrin-coated vesicles. Nature 378, 632-635. ( 10.1038/378632a0) [DOI] [PubMed] [Google Scholar]
- 50.Ando K, et al. 2020. The lipid phosphatase Synaptojanin 1 undergoes a significant alteration in expression and solubility and is associated with brain lesions in Alzheimer's disease. Acta Neuropathol. Commun. 8, 79. ( 10.1186/s40478-020-00954-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martin S, et al. 2020. Mutated ATP10B increases Parkinson's disease risk by compromising lysosomal glucosylceramide export. Acta Neuropathol. 139, 1001-1024. ( 10.1007/s00401-020-02145-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Deshimaru M, et al. 2021. DCTN1 binds to TDP-43 and regulates TDP-43 aggregation. Int. J. Mol. Sci. 22, 3985. ( 10.3390/ijms22083985) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Copeland WC. 2010. The mitochondrial DNA polymerase in health and disease. Subcell. Biochem. 50, 211-222. ( 10.1007/978-90-481-3471-7_11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deng H-X, et al. 2016. Identification of TMEM230 mutations in familial Parkinson's disease. Nat. Genet. 48, 733-739. ( 10.1038/ng.3589) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Treutlein J, Kraemer B, Rex-Haffner M, Awasthi S, Ripke S, Binder EB, Gruber O. 2023. Gene set-based analysis of the endosomal sorting processes cargo selection and membrane tubulation with human reward system reactivity. bioRxiv, 549434. ( 10.1101/2023.07.18.549434) [DOI]
- 56.Moskot M, Bochenska K, Jakobkiewicz-Banecka J, Banecki B, Gabig-Ciminska M. 2018. Abnormal sphingolipid world in inflammation specific for lysosomal storage diseases and skin disorders. Int. J. Mol. Sci.. 19, 247. ( 10.3390/ijms19010247) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.You H, Jin Y, Kang J, Mao Y, Su J, Sun L, Wang L, Meng H. 2020. Mitochondrial serine protease Omi/HtrA2 accentuates brain ischemia/reperfusion injury in rats and oxidative stress injury in vitro by modulating mitochondrial stress proteins CHOP and ClpP and physically interacting with mitochondrial fusion protein OPA1. Bioengineered 11, 1058-1070. ( 10.1080/21655979.2020.1822105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jaud M, Philippe C, Di Bella D, Tang W, Pyronnet S, Laurell H, Mazzolini L, Rouault-Pierre K, Touriol C. 2020. Translational regulations in response to endoplasmic reticulum stress in cancers. Cells 9, 540. ( 10.3390/cells9030540) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hickey KL, et al. 2020. GIGYF2 and 4EHP Inhibit translation initiation of defective messenger RNAs to assist ribosome-associated quality control. Mol. Cell 79, 950-962. ( 10.1016/j.molcel.2020.07.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chu PY, Tai YL, Shen TL. 2019. Grb7, a critical mediator of EGFR/ErbB signaling, in cancer development and as a potential therapeutic target. Cells 8, 435. ( 10.3390/cells8050435) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wallings RL, Humble SW, Ward ME, Wade-Martins R. 2019. Lysosomal dysfunction at the centre of Parkinson's disease and frontotemporal dementia/amyotrophic lateral sclerosis. Trends Neurosci. 42, 899-912. ( 10.1016/j.tins.2019.10.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vollstedt EJ, et al. 2023. Embracing monogenic Parkinson's disease: the MJFF Global Genetic PD Cohort. Mov. Disord. 38, 286-303. ( 10.1002/mds.29288) [DOI] [PubMed] [Google Scholar]
- 63.Skrahina V, et al. 2021. The Rostock International Parkinson's Disease (ROPAD) Study: protocol and initial findings. Mov. Disord. 36, 1005-1010. ( 10.1002/mds.28416) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Blauwendraat C, Nalls MA, Singleton AB. 2020. The genetic architecture of Parkinson's disease. Lancet Neurol. 19, 170-178. ( 10.1016/S1474-4422(19)30287-X) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schilder BMM, Navarro E, Raj T. 2022. Multi-omic insights into Parkinson's disease: from genetic associations to functional mechanisms. Neurobiol. Dis. 163, 105580. ( 10.1016/j.nbd.2021.105580) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Manolio TA. 2010. Genomewide association studies and assessment of the risk of disease. N. Engl. J. Med. 363, 166-176. ( 10.1056/NEJMra0905980) [DOI] [PubMed] [Google Scholar]
- 67.Nalls MA, et al. 2019. Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Iwaki H, et al. 2021. Accelerating Medicines Partnership: Parkinson's disease. Genetic resource. Mov. Disord. 36, 1795-1804. ( 10.1002/mds.28549) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tan MMX, et al. 2021. Genome-wide association studies of cognitive and motor progression in Parkinson's disease. Mov. Disord. 36, 424-433. ( 10.1002/mds.28342) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Iwaki H, et al. 2019. Genomewide association study of Parkinson's disease clinical biomarkers in 12 longitudinal patients' cohorts. Mov. Disord. 34, 1839-1850. ( 10.1002/mds.27845) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Alfradique-Dunham I, et al. 2021. Genome-wide association study meta-analysis for Parkinson disease motor subtypes. Neurol. Genet. 7, e557. ( 10.1212/NXG.0000000000000557) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grover S, et al. 2022. Genome-wide association and meta-analysis of age at onset in Parkinson disease: evidence from the COURAGE-PD Consortium. Neurology 99, e698-e710. ( 10.1212/WNL.0000000000200699) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kim JJ, et al. 2022. Multi-ancestry genome-wide meta-analysis in Parkinson's disease. medRxiv, 22278432. ( 10.1101/2022.08.04.22278432) [DOI]
- 74.Paul KC, Schulz J, Bronstein JM, Lill CM, Ritz BR. 2018. Association of polygenic risk score With cognitive decline and motor progression in Parkinson disease. JAMA Neurol. 75, 360-366. ( 10.1001/jamaneurol.2017.4206) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pihlstrøm L, et al. 2022. Genetic stratification of age-dependent Parkinson's disease risk by polygenic hazard score. Mov. Disord. 37, 62-69. ( 10.1002/mds.28808) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tam V, Patel N, Turcotte M, Bosse Y, Pare G, Meyre D. 2019. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 20, 467-484. ( 10.1038/s41576-019-0127-1) [DOI] [PubMed] [Google Scholar]
- 77.Grenn FP, et al. 2020. The Parkinson's Disease Genome-Wide Association Study locus browser. Mov. Disord. 35, 2056-2067. ( 10.1002/mds.28197) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pihlstrøm L, et al. 2018. A comprehensive analysis of SNCA-related genetic risk in sporadic Parkinson disease. Ann. Neurol. 84, 117-129. ( 10.1002/ana.25274) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Soldner F, et al. 2016. Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 533, 95-99. ( 10.1038/nature17939) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pierce SE, Booms A, Prahl J, Van Der Schans EJC, Tyson T, Coetzee GA. 2020. Post-GWAS knowledge gap: the how, where, and when. NPJ Parkinsons Dis. 6, 23. ( 10.1038/s41531-020-00125-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kia DA, et al. 2021. Identification of candidate Parkinson disease genes by integrating genome-wide association study, expression, and epigenetic data sets. JAMA Neurol. 78, 464-472. ( 10.1001/jamaneurol.2020.5257) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Davies NM, Holmes MV, Davey Smith G. 2018. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ 362, k601. ( 10.1136/bmj.k601) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dang X, Zhang Z, Luo XJ. 2022. Mendelian randomization study using dopaminergic neuron-specific eQTL nominates potential causal genes for Parkinson's disease. Mov. Disord. 37, 2451-2456. ( 10.1002/mds.29239) [DOI] [PubMed] [Google Scholar]
- 84.Rizig M, et al. 2023. Genome-wide association identifies novel etiological insights associated with Parkinson's disease in African and African admixed populations. medRxiv, 023.05.05.23289529. ( ) [DOI]
- 85.Foo JN, et al. 2020. Identification of risk loci for Parkinson disease in Asians and comparison of risk between Asians and Europeans: a genome-wide association study. JAMA Neurol. 77, 746-754. ( 10.1001/jamaneurol.2020.0428) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Andrews SV, et al. In press.. The genetic drivers of juvenile, young, and early-onset Parkinson's disease in India. Mov. Disord. ( 10.1002/mds.29676) [DOI]
- 87.Loesch DP, et al. 2021. Characterizing the genetic architecture of Parkinson's disease in Latinos. Ann. Neurol. 90, 353-365. ( 10.1002/ana.26153) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Blauwendraat C, et al. 2021. Investigation of autosomal genetic sex differences in Parkinson's disease. Ann. Neurol. 90, 35-42. ( 10.1002/ana.26090) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Le Guen Y, et al. 2021. Common X-chromosome variants are associated with Parkinson disease risk. Ann. Neurol. 90, 22-34. ( 10.1002/ana.26051) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Grenn FP, Makarious MB, Bandres-Ciga S, Iwaki H, Singleton AB, Nalls MA, Blauwendraat C. 2022. Analysis of Y chromosome haplogroups in Parkinson's disease. Brain Commun. 4, fcac277. ( 10.1093/braincomms/fcac277) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zirra A, Rao SC, Bestwick J, Rajalingam R, Marras C, Blauwendraat C, Mata IF, Noyce AJ. 2023. Gender differences in the prevalence of Parkinson's disease. Mov. Disord. Clin. Pract. 10, 86-93. ( 10.1002/mdc3.13584) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Toffoli M, Schapira AHV, Proukakis C. 2023. Sex distribution of GBA1 variants Carriers with dementia with Lewy bodies and Parkinson's disease. Mov. Disord. 38, 2137-2139. ( 10.1002/mds.29609) [DOI] [PubMed] [Google Scholar]
- 93.Fernandez-Santiago R, Sharma M. 2022. What have we learned from genome-wide association studies (GWAS) in Parkinson's disease? Ageing Res. Rev. 79, 101648. ( 10.1016/j.arr.2022.101648) [DOI] [PubMed] [Google Scholar]
- 94.Ye H, Robak LA, Yu MG, Cykowski M, Shulman JM. 2023. Genetics and pathogenesis of Parkinson's syndrome. Annu. Rev. Pathol. Mech. 18, 95-121. ( 10.1146/annurev-pathmechdis-031521-034145) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Robak LA, et al. 2017. Excessive burden of lysosomal storage disorder gene variants in Parkinson's disease. Brain 140, 3191-3203. ( 10.1093/brain/awx285) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dehestani M, Liu H, Sreelatha AAK, Schulte C, Bansal V, Gasser T. 2022. Mitochondrial and autophagy-lysosomal pathway polygenic risk scores predict Parkinson's disease. Mol. Cell. Neurosci. 121, 103751. ( 10.1016/j.mcn.2022.103751) [DOI] [PubMed] [Google Scholar]
- 97.Jeunemaitre X, Kreft-Jais C, Chatellier G, Julien J, Degoulet P, Plouin PF, Ménard J, Corvol P. 1988. Long-term experience of spironolactone in essential hypertension. Kidney Int. Suppl. 26, S14-S17. [PubMed] [Google Scholar]
- 98.Glaves D. 1986. Intravascular death of disseminated cancer cells mediated by superoxide anion. Invasion Metastasis 6, 101-111. [PubMed] [Google Scholar]
- 99.Cai S, Wu Y, Guillen-Samander A, Hancock-Cerutti W, Liu J, De Camilli P. 2022. In situ architecture of the lipid transport protein VPS13C at ER–lysosome membrane contacts. Proc. Natl Acad. Sci. USA 119, e2203769119. ( 10.1073/pnas.2203769119) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hu M, et al. 2022. Parkinson's disease-risk protein TMEM175 is a proton-activated proton channel in lysosomes. Cell 185, 2292-2308. ( 10.1016/j.cell.2022.05.021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Narendra DP, Jin SM, Tanaka A, Suen D-F, Gautier CA, Shen J, Cookson MR, Youle RJ. 2010. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8, e1000298. ( 10.1371/journal.pbio.1000298) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Meng H, et al. 2017. Loss of Parkinson's disease-associated protein CHCHD2 affects mitochondrial crista structure and destabilizes cytochrome c. Nat. Commun. 8, 15500. ( 10.1038/ncomms15500) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lesage S, et al. 2016. Loss of VPS13C function in autosomal-recessive parkinsonism causes mitochondrial dysfunction and increases PINK1/Parkin-dependent mitophagy. Am. J. Hum. Genet. 98, 500-513. ( 10.1016/j.ajhg.2016.01.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cisneros J, Belton TB, Shum GC, Molakal CG, Wong YC. 2022. Mitochondria-lysosome contact site dynamics and misregulation in neurodegenerative diseases. Trends Neurosci. 45, 312-322. ( 10.1016/j.tins.2022.01.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.International Parkinson's Disease Genomics Consortium. 2011. A two-stage meta-analysis identifies several new loci for Parkinson's disease. PLoS Genet. 7, e1002142. ( 10.1371/journal.pgen.1002142) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nalls MA, et al. 2014. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat. Genet. 46, 989-993. ( 10.1038/ng.3043) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Diaz-Ortiz ME, et al. 2022. GPNMB confers risk for Parkinson's disease through interaction with α-synuclein. Science 377, eabk0637. ( 10.1126/science.abk0637) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Murthy MN, Blauwendraat C, Guelfi S, Hardy J, Lewis PA, Trabzuni D. 2017. Increased brain expression of GPNMB is associated with genome wide significant risk for Parkinson's disease on chromosome 7p15.3. Neurogenetics 18, 121-133. ( 10.1007/s10048-017-0514-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rose AAN, et al. 2010. Glycoprotein nonmetastatic B is an independent prognostic indicator of recurrence and a novel therapeutic target in breast cancer. Clin. Cancer Res. 16, 2147-2156. ( 10.1158/1078-0432.CCR-09-1611) [DOI] [PubMed] [Google Scholar]
- 110.Yang C-F, Lin S-P, Chiang C-P, Wu Y-H, H'Ng WS, Chang C-P, Chen Y-T, Wu J-Y. 2018. Loss of GPNMB causes autosomal-recessive amyloidosis cutis dyschromica in humans. Am. J. Hum. Genet. 102, 219-232. ( 10.1016/j.ajhg.2017.12.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kanaan NM, et al. 2015. The longitudinal transcriptomic response of the substantia nigra to intrastriatal 6-hydroxydopamine reveals significant upregulation of regeneration-associated genes. PLoS One 10, e0127768. ( 10.1371/journal.pone.0127768) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Moloney EB, Moskites A, Ferrari EJ, Isacson O, Hallett PJ. 2018. The glycoprotein GPNMB is selectively elevated in the substantia nigra of Parkinson's disease patients and increases after lysosomal stress. Neurobiol. Dis. 120, 1-11. ( 10.1016/j.nbd.2018.08.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Brendza R, et al. 2021. Genetic ablation of Gpnmb does not alter synuclein-related pathology. Neurobiol. Dis. 159, 105494. ( 10.1016/j.nbd.2021.105494) [DOI] [PubMed] [Google Scholar]
- 114.Suda M, et al. 2022. Glycoprotein nonmetastatic melanoma protein B regulates lysosomal integrity and lifespan of senescent cells. Scient. Rep. 12, 6522. ( 10.1038/s41598-022-10522-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Robinet P, Ritchey B, Lorkowski SW, Alzayed AM, Degeorgia S, Schodowski E, Traughber CA, Smith JD. 2021. Quantitative trait locus mapping identifies the Gpnmb gene as a modifier of mouse macrophage lysosome function. Scient. Rep. 11, 10249. ( 10.1038/s41598-021-89800-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kinghorn KJ, Asghari AM, Castillo-Quan JI. 2017. The emerging role of autophagic-lysosomal dysfunction in Gaucher disease and Parkinson's disease. Neur. Regen. Res. 12, 380-384. ( 10.4103/1673-5374.202934) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Saade M, De Souza GA, Scavone C, Kinoshita PF. 2021. The Role of GPNMB in inflammation. Front. Immunol. 12, 674739. ( 10.3389/fimmu.2021.674739) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Van der Lienden MJC, Gaspar P, Boot R, Aerts JMFG, Van Eijk M. 2019. Glycoprotein non-metastatic protein B: an emerging biomarker for lysosomal dysfunction in macrophages. Int. J. Mol. Sci. 20, 66. ( 10.3390/ijms20010066) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Soutar MPM, et al. 2022. Regulation of mitophagy by the NSL complex underlies genetic risk for Parkinson's disease at 16q11.2 and MAPT H1 loci. Brain 145, 4349-4367. ( 10.1093/brain/awac325) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. 2014. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 205, 143-153. ( 10.1083/jcb.201402104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ordureau A, et al. 2014. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell. 56, 360-375. ( 10.1016/j.molcel.2014.09.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sheikh BN, Guhathakurta S, Akhtar A. 2019. The non-specific lethal (NSL) complex at the crossroads of transcriptional control and cellular homeostasis. EMBO Rep. 20, e47630. ( 10.15252/embr.201847630) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li T, et al. 2022. Kansl1 haploinsufficiency impairs autophagosome-lysosome fusion and links autophagic dysfunction with Koolen-de Vries syndrome in mice. Nat. Commun. 13, 931. ( 10.1038/s41467-022-28613-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Moreno-Igoa M, Hernández-Charro B, Bengoa-Alonso A, Pérez-Juana-del-Casal A, Romero-Ibarra C, Nieva-Echebarria B, Ramos-Arroyo MA. 2015. KANSL1 gene disruption associated with the full clinical spectrum of 17q21.31 microdeletion syndrome. BMC Med. Genet. 16, 68. ( 10.1186/s12881-015-0211-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chatterjee A, et al. 2016. MOF acetyl transferase regulates transcription and respiration in mitochondria. Cell 167, 722. ( 10.1016/j.cell.2016.09.052) [DOI] [PubMed] [Google Scholar]
- 126.Li L, et al. 2020. Lysine acetyltransferase 8 is involved in cerebral development and syndromic intellectual disability. J. Clin. Invest. 130, 1431-1445. ( 10.1172/JCI131145) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chang D, et al. 2017. A meta-analysis of genome-wide association studies identifies 17 new Parkinson's disease risk loci. Nat. Genet. 49, 1511-1516. ( 10.1038/ng.3955) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pittman AM, et al. 2005. Linkage disequilibrium fine mapping and haplotype association analysis of the tau gene in progressive supranuclear palsy and corticobasal degeneration. J. Med. Genet. 42, 837-846. ( 10.1136/jmg.2005.031377) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Koolen DA, et al. 2016. The Koolen-de Vries syndrome: a phenotypic comparison of patients with a 17q21.31 microdeletion versus a KANSL1 sequence variant. Eur. J. Hum. Genet. 24, 652-659. ( 10.1038/ejhg.2015.178) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Koks S, Pfaff AL, Bubb VJ, Quinn JP. 2021. Transcript variants of genes involved in neurodegeneration are differentially regulated by the APOE and MAPT haplotypes. Genes 12, 423. ( 10.3390/genes12030423) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ferrari R, et al. 2017. Genetic architecture of sporadic frontotemporal dementia and overlap with Alzheimer's and Parkinson's diseases. J. Neurol. Neurosurg. Psychiatry 88, 152-164. ( 10.1136/jnnp-2016-314411) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Linda K, et al. 2022. Imbalanced autophagy causes synaptic deficits in a human model for neurodevelopmental disorders. Autophagy 18, 423-442. ( 10.1080/15548627.2021.1936777) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Füllgrabe J, et al. 2013. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature 500, 468-471. ( 10.1038/nature12313) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mcfarthing K, Simuni T. 2019. Clinical trial highlights: targetting alpha-synuclein. J. Parkinson Dis. 9, 5-16. ( 10.3233/JPD-189004) [DOI] [PubMed] [Google Scholar]
- 135.Hebron ML, Lonskaya I, Olopade P, Selby ST, Pagan F, Moussa CE. 2014. Tyrosine kinase inhibition regulates early systemic immune changes and modulates the neuroimmune response in α-synucleinopathy. J. Clin. Cell. Immunol. 5, 259. ( 10.4172/2155-9899.1000259) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Levin J, et al. 2022. Safety, tolerability and pharmacokinetics of the oligomer modulator anle138b with exposure levels sufficient for therapeutic efficacy in a murine Parkinson model: a randomised, double-blind, placebo-controlled phase 1a trial. eBioMedicine 80, 104021. ( 10.1016/j.ebiom.2022.104021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Forno 1996. Neuropathology of Parkinson's disease. J. Neuropathol. Exp. Neurol. 55, 259-272. ( 10.1097/00005072-199603000-00001) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This article has no additional data.