

Abstract
Many bacteria, particularly actinomycetes, are known to produce secondary metabolites synthesized by polyketide synthases (PKS). Bacterial polyketides are a particularly rich source of bioactive molecules, many of which are of potential pharmaceutical relevance. To directly access PKS gene diversity from soil, we developed degenerate PCR primers for actinomycete type II KSα (ketosynthase) genes. Twenty-one soil samples were collected from diverse sources in New Jersey, and their bacterial communities were compared by terminal restriction fragment length polymorphism (TRFLP) analysis of PCR products generated using bacterial 16S rRNA gene primers (27F and 1525R) as well as an actinomycete-specific forward primer. The distribution of actinomycetes was highly variable but correlated with the overall bacterial species composition as determined by TRFLP. Two samples were identified to contain a particularly rich and unique actinomycete community based on their TRFLP patterns. The same samples also contained the greatest diversity of KSα genes as determined by TRFLP analysis of KSα PCR products. KSα PCR products from these and three additional samples with interesting TRFLP pattern were cloned, and seven novel clades of KSα genes were identified. Greatest sequence diversity was observed in a sample containing a moderate number of peaks in its KSα TRFLP. The nucleotide sequences were between 74 and 81% identical to known sequences in GenBank. One cluster of sequences was most similar to the KSα involved in ardacin (glycopeptide antibiotic) production by Kibdelosporangium aridum. The remaining sequences showed greatest similarity to the KSα genes in pathways producing the angucycline-derived antibiotics simocyclinone, pradimicin, and jasomycin.
Prokaryotic microorganisms are the largest reservoir of genetic diversity on earth, yet to date only a small fraction (maybe 0.1 to 1%) of all microbes have been cultured and rigorously described. Over 17,000 antibiotics and other biologically active molecules have been found in prokaryotic microbes (2, 7, 25). By the logic that only a small fraction of microbes have been described, it can be expected that the vast majority of pharmaceutically and industrially important molecules of microbial origin remain to be discovered.
A large number of biologically active molecules are synthesized by polyketide synthase (PKS) pathways. Polyketides are structurally diverse secondary metabolites that have already found widespread application as pharmaceuticals (20), in particular as antibiotics. Industrially important polyketides include rapamycin (immunosuppressant), erythromycin (antibiotic), lovostatin (anticholesterol drug), and epothilone B (anticancer drug). Molecules are synthesized from simple two-, three-, and four-carbon building blocks such as acetyl-coenzyme A, propionyl-coenzyme A, and butyryl-coenzyme A (14) by means of a stepwise, decarboxylative condensation pathway analogous to fatty acid synthesis. Unlike in fatty acid synthesis, however, polyketide maturation is not limited to a rigid sequence of ketoreduction, dehydration, and enoyl reduction (14). Instead, modification may include some, all, or none of these steps as well as other modifications such as cyclization and aromatization (among others). Because different starter and chain elongation units can be used and multiple stereo-centers are formed during synthesis, a tremendous structural diversity of PKS products can be achieved.
Several classes of PKS genes are commonly recognized (12-14, 31). Type I PKS, also known as modular PKS, are large, multidomain enzymes which carry a series of functional sites for stepwise polyketide synthesis. Type II PKS are composed of three or more mono- or bi-functional enzymes, which act in an iterative manner during synthesis. A core of three enzymes referred to as the minimal polyketide synthase is shared by all type II pathways: a ketoacyl-synthase (KSα), a chain length factor (KSβ) and an acyl-carrier protein. This study focuses on the actinomycete type II minimal PKS.
Many actinomycetes contain PKS gene clusters and this group of bacteria has long been recognized as an important source of bioactive molecules. Screening of actinomycete cultures has for decades yielded novel industrially important products and pharmaceuticals. However, the rate of discovery has not kept pace with resource expenditure and advances in technology and has even declined in recent years (19, 34). This unfortunate circumstance arises because previously described strains and molecules are being rediscovered and redescribed (19, 33). Better methods are clearly needed for the more efficient identification and screening of actinomycete cultures and soils containing potentially interesting secondary metabolites. Most importantly, it is hoped that culture-independent methods may prove worthwhile. Such methods may not only prevent reinvestigation of previously described bioactive molecules, but also alleviate some of the biases introduced by the use of standard cultivation techniques.
In this context Metsä-Ketelä (20) obtained the partial sequences of ketosynthase genes from six known and 29 unidentified actinomycete soil isolates using degenerate PCR primers. Pigment- and antibiotic-producing PKS gene clusters could be clearly distinguished based on their KSα sequences, indicating that functional information may be derived from PKS sequence information. A later study (19) found that the phylogenies of 16S rRNA and PKS genes in actinomycete soil isolates were not congruent, indicating that the phylogenetic grouping of actinomycetes is an inadequate predictor for the type of secondary metabolites these strains produce.
The purpose of this study was to develop a culture-independent method to identify genes encoding novel and unique PKS pathways. Specifically, we hypothesized that soil containing a large diversity of actinomycetes, as indicated by terminal restriction fragment length polymorphism (TRFLP), also contains a large diversity of PKS gene sequences. In addition, we hypothesized that soil with unique TRFLP patterns for both the actinomycete 16S rRNA and PKS genes would yield particularly novel PKS gene sequences.
MATERIALS AND METHODS
Sampling sites.
Soil was collected in four separate times (1S, 2S, 3S, and 4S) from several sites throughout the state of New Jersey at the beginning of March of 2003. The 1S samples originated from the Greenwood Forest Wildlife Management Area in the New Jersey Pinelands, Ocean County. Samples (n = 6) were collected roughly one meter apart across a sand dune. Vegetation varied from grass to small shrubs. The 2S samples (n = 6) were collected along N.J. State Road 72 at 0.5-mile intervals. The first sample was taken in the Greenwood Forest area, while the last sample originated from soil in the Lebanon State Forest area. There was a significant difference in elevation between the first and the last sample. Soil contained little organic material and was dry at the higher elevation. The forest floor was water saturated and organic material dominated the soil samples at the lower elevation. Vegetation changed from stunted pines in the Greenwood Forest area to a mix of large pines and oak trees in the Lebanon State Forest.
The 3S samples (n = 6) were taken near Pakim Pond in Lebanon State Forest (N.J. Ocean County). Samples were collected roughly 10 m apart. The first sample was taken from Pakim Pond sediment. The remaining samples were collected at progressively greater distance from the pond. Soil was mostly composed of leaf litter and roots, with virtually no mineral (sand or clay) content. The 4S samples (n = 5) were collected from several sites in the Picatinny Arsenal (Morris County). Samples were frozen at −80°C upon return to the laboratory (ca. 2 h) and thawed for DNA extraction. Sample labels throughout the text are shown as NSX, where NS indicates the sampling collection site (1S, 2S, 3S, and 4S) and X indicates the sample number.
DNA extraction and PCR.
DNA was extracted using the Ultraclean Soil DNA kit (MO BIO Laboratories, Carlsbad, CA). This method was first compared to several other extraction protocols. The MO BIO kit produced better DNA yields and copurified less humic contaminants than a freeze-thaw approach, an SDS boiling lysis method, and the DNeasy tissue kit (QIAGEN, Valencia CA) (data not shown). Soil samples (0.2 g) were extracted as recommended by the manufacturer. DNA was analyzed on a 1% agarose gel and quantified using a Kodak EDAS 290 Gel Imaging system by comparing band intensities to known quantities of lambda HindIII markers. Five ng of DNA was then added to 50-μl PCRs.
The total bacterial community was analyzed using the following 16S rRNA gene PCR primers: 27F, 5′-AGAGTTTGATCMTGGCTCAG-3′ and 1525R, 5′-AAGGAGGTGATCCAGCC-3′. The 27F primer was carboxyfluorescein (FAM) labeled for TRFLP analysis. The actinomycete community was analyzed using a FAM-labeled specific 16S rRNA gene forward primer, 243F (9): 5′-GGATGAGCCCGCGGCCTA-3′. Neither 1525R nor 1492R were suitable as reverse primers for TRFLP analysis, since multiple and unspecific banding was observed in combination with 243F. 1401R (5′-CGGTGTGTACAAGACCC-3′) was instead utilized, since it produced no unspecific amplification. PCRs contained 200 nM of each PCR primer, 2.5 mM MgCl, 5 U Taq DNA polymerase (Promega, Madison WI), and 0.4 μl 10 mg ml−1 acetylated bovine serum albumin (Promega). PCR conditions were as follows: (for 27F/1525R) 5 min at 95°C followed by 30 cycles of 1 min at 95°C, 1 min at 55°C, and 1.5 min at 72°C. The annealing temperature for the 243F/1401R primer pair was optimized to 60°C (the highest temperature where no significant loss of PCR product formation was observed in an ethidium bromide stained gel), while the remaining parameters remained identical. A 15-min extension step at 72°C was applied at the end of the PCR program.
PKS cloning and library generation.
The sequences of 69 actinomycete type II KSα genes were obtained from GenBank and aligned. Conserved regions were located and primers designed for a 554 bp fragment located in the 5′ portion of the gene. The primer sequences are 540F, 5′-GGITGCACSTCIGGIMTSGAC-3′, and 1100R, 5′-CCGATSGCICCSAGIGAGTG-3′. PCRs contained 5 ng DNA template, 1 μM primer each, 2.5 mM MgCl, 5 U Taq DNA polymerase (Promega) and 0.4 μl of 10 mg ml−1 BSA. PCR conditions were as follows: 5 min at 95°C followed by 40 cycles of 1 min at 95°C, 1 min at 64°C and 1.5 min at 72°C followed by a 15 min extension step at 72°C. PCR products were gel purified using a gel extraction kit (QIAGEN) and cloned into pCR4-TOPO vector using a TOPO-TA cloning kit (Invitrogen, Carlsbad CA). Forty white colonies from each transformation were grown in LB medium containing 50 μg ml−1 kanamycin and ampicillin and plasmids extracted using QIAprep spin miniprep columns (QIAGEN). Plasmids were screened for inserts by digestion with the restriction enzyme EcoRI and insert containing plasmids were sequenced. To obtain TRFLP patterns of the KSα PCR products, 5 ng of template DNA were first amplified for 30 cycles as indicated above using unlabeled primers. Five μl of this reaction was then transferred to a new PCR containing the FAM-labeled forward (540F) primer and amplified for an additional 10 cycles.
TRFLP and data analysis.
FAM-labeled PCR products were diluted to 5 ng μl−1 and 30 ng were independently digested for 4 h using 2 units of MnlI or AluI (NEB, Beverly, MA). Digested DNA was precipitated with glycogen as a carrier, resuspended in 20 μl of deionized formamide and denatured at 95°C for 5 min. Samples were then analyzed using an ABI 310 automated sequencer (Applied Biosystems, Foster City, CA) to obtain the TRFLP patterns. For the soil samples in this study (n = 23) this procedure generated 16S TRFLP patterns with average maximum peak heights of 4.36 × 103 fluorescence units (FU) and average cumulative peak areas of 3.92 × 105 FU2.
TRFLP data were exported to Matlab (Natick, MA) and analyzed by considering the presence/absence of peaks only. Data were not binned, because two peaks that are only a fraction of a base pair apart in two different TRFLP patterns, can sometimes be binned into different nucleotide length, even though they may represent the same organism (e.g., 120.48 is rounded to 120 and 120.52 is rounded to 121). This can bias and skew similarities calculated from TRFLP data (15). Instead, a search window of ± 0.5 bp was applied for analysis and each peak was individually compared to all peaks of all other TRFLP pattern in the data set. The minimum peak height was set at 3% of the highest peak in each individual TRFLP. In addition, only those peaks, which cumulatively accounted for < 97% of the total peak area were considered. Pairwise similarities were calculated as previously described (28) using the Sorenson index: Cs = 2Nab/(Na + Nb), where Nab = number of shared peaks and Na and Nb are the number of peaks in each sample (18, 21). Distance data for both restriction enzymes was averaged. This should help reduce biases introduced by particular enzymes (uneven distribution of cut sites). Data were then exported as 1 − Cs into MEGA (Molecular Evolutionary Genetics Analysis Software, version 2.1) (16) to generate neighbor-joining dendrograms.
Phylogenetic analysis.
DNA sequences obtained from the PCR product cloning experiments were trimmed by removing all vector and primer sequences from the ends. They were then aligned with their closest match in GenBank (obtained by translated and untranslated BLAST searches) as well as sequences used to design the PCR primers. Neighbor-Joining trees were generated using MEGA by using the Tajima-Nei distance model. One thousand bootstrap replicates were performed and consensus trees are shown.
Nucleotide accession numbers.
DNA sequences obtained in this study were deposited in GenBank under the accession numbers AY684435 to AY684585.
RESULTS AND DISCUSSION
Bacterial community composition of soils collected for this study was analyzed for their 16S bacterial rRNA gene composition using TRFLP (Fig. 1A). Overall, the geographic origin of samples was reflected by their TRFLP patterns (for sampling labeling scheme see Materials and Methods). Samples from the 1S collection were most similar to one another with similarities among TRFLP patterns ranging between 55% and 81% (average = 68%). This was not surprising, as samples were collected from within a very small area (<6 m apart). Samples from the 2S and 3S collections also formed clusters indicating similarities among their bacterial communities. Pairwise similarities among all samples ranged between 12% and 81% (average = 41%). Samples from the 4S collection did not group in cluster analysis and were in average only 32% and 35% similar to each other and the remaining samples, respectively.
FIG. 1.
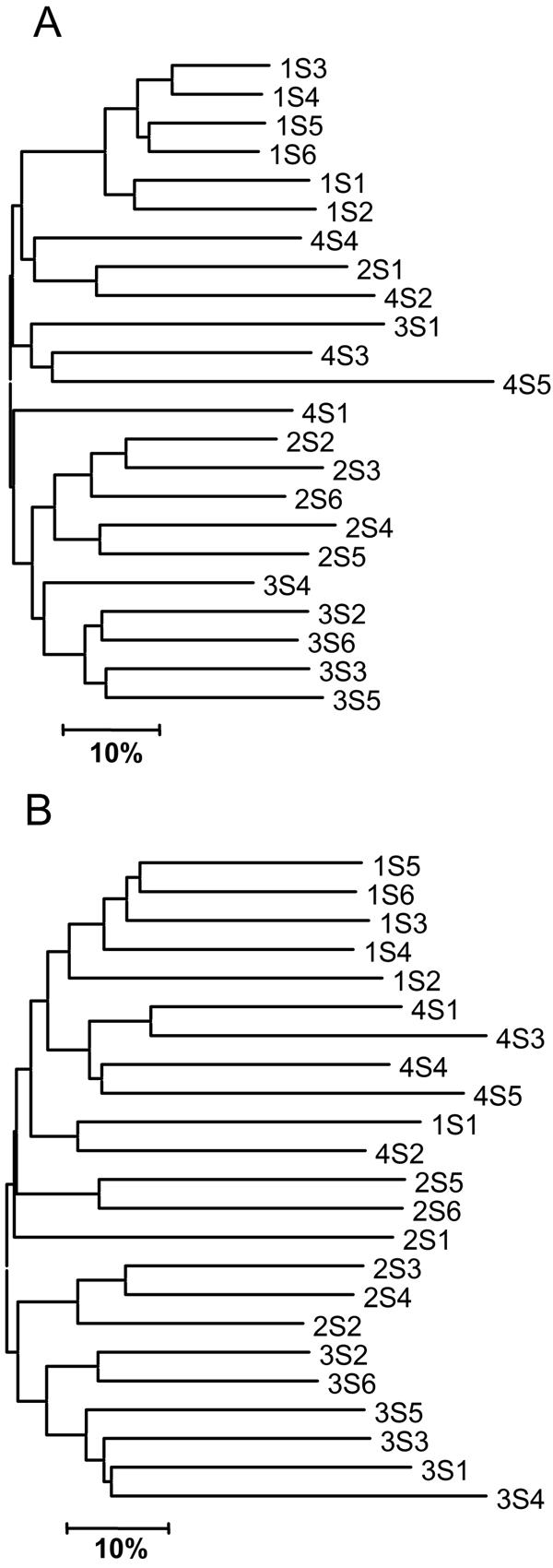
TRFLP dendrograms. Labels indicate the identity of soil samples (NSX) where X is the sample number and NS (1S, 2S, 3S, and 4S) indicates the sampling collection site (see Materials and Methods). (A) Bacterial 16S rRNA genes (27F/1525R). (B) Actinomycete 16S rRNA genes (243F/1401R). A distance of 10% (scale bar) indicates a dissimilarity of 10% between two samples as calculated by the Sorenson index.
With the exception of samples 1S1 and 4S4, the clustering pattern obtained for the actinomycete-specific rRNA gene TRFLP data (Fig. 1B) was highly similar to the clustering of samples based on the total bacterial community composition (Fig. 1A). Similarities between actinomycete TRFLPs were on average significantly lower than those observed for the total bacterial community TRFLPs (paired t test: P < 0.001). This indicates actinomycetes were similarly distributed as the overall bacterial community in our soil samples, but that actinomycete species composition showed greater sample to sample variability than the total bacterial community. Some of these differences may be due to the fact that the specificity of the actinomycete group-specific primer F243 is slightly suboptimal, because it does not match the 16S sequences of all actinomycetes and does match a few nonactinomycetes (9). The inability of F243 to amplify all actinomycete 16S genes was nonetheless not seen as a problem for this study. Only a small fraction of actinomycetes (10 out of 364 sequences that were analyzed in the original reference) are excluded. Similarly, the bias introduced by unspecific amplification of nonactinomycetes is almost certainly small. In the original study only thirteen out of the 2,270 nonactinomycete 16S rRNA gene sequences were homologous at positions 239 to 243 (the 3′ terminus, which determines-specificity).
Table 1 shows the total number of peaks observed in the actinomycete-specific 16S TRFLP as well as the number of unique peaks (not observed in any other TRFLP in the data set). A significantly larger number (Student's t test: P < 0.001) of peaks was observed in the TRFLP of samples from collection 2S (average = 47) than in the remaining samples (average = 17), indicating that these samples contained particularly rich actinomycete communities. Samples 2S4 and 2S6 produced no amplification product using our KSα primers, eliminating them from further analysis. The TRFLP of sample 2S2 contained a small number of peaks (n = 21) similar to samples from other collections (S1, S3, and S4). Sample 2S3 only contained one unique peak. Samples 2S1 and 2S5 were thus selected for PCR clone library generation and further analysis because of the high level of diversity observed.
TABLE 1.
Actinomycete and KSα TRFLP dataa
| Sample | No. of actinomycete TRFLP peaks
|
No. of KSα TRFLP peaks
|
||
|---|---|---|---|---|
| Total | Unique | Total | Unique | |
| 1S1 | 14 | 4 | ||
| 1S2 ▴ | 14 | 1 | 19 | 3 |
| 1S3 | 28 | 1 | ||
| 1S4 | 39 | 1 | ||
| 1S5 | 20 | 0 | 15 | 3 |
| 1S6 | 23 | 2 | 25 | 3 |
| 2S1 ▪ | 45 | 7 | 33 | 14 |
| 2S2 | 21 | 1 | 10 | 4 |
| 2S3 | 21 | 0 | 14 | 4 |
| 2S4 | 54 | 1 | ||
| 2S5 ▪ | 58 | 8 | 38 | 14 |
| 2S6 | 51 | 8 | ||
| 3S1 | 19 | 1 | ||
| 3S2 | 17 | 0 | 11 | 3 |
| 3S3 | 14 | 0 | 5 | 0 |
| 3S4 | 17 | 1 | ||
| 3S5 | 15 | 0 | 6 | 0 |
| 3S6 ▴ | 21 | 1 | 13 | 4 |
| 4S1 | 26 | 3 | 9 | 0 |
| 4S2 | 2 | 0 | ||
| 4S3 | 7 | 1 | 8 | 0 |
| 4S4 ▴ | 16 | 2 | 16 | 1 |
| 4S5 | 4 | 0 | 16 | 1 |
Shown are the number of total and unique peaks in the actinomycete-specific 16S ribosomal RNA and KSα gene TRFLP patterns. ▪, samples chosen for cloning because of high actinomycete diversity; ▴, samples chosen for cloning because of high KSα diversity or number of total/unique peaks.
DNA from all samples was PCR amplified using PKS primers developed for this study and PCR products were analyzed using TRFLP (Table 1). Eight samples produced no KSα PCR product. These samples amplified well using 16S rRNA gene primers, suggesting that PCR inhibitor was not preventing amplification of KSα sequences. Two interpretations of these data are possible. Either no PKS gene sequences were present or our PCR primers did not match the ones that were. For the remaining samples, however, a relatively good correlation between the number of peaks in the actinomycete and KSα TRFLPs was observed (R2 = 0.71). Samples 2S1 and 2S5, which contained the most diverse actinomycete community, contained the largest number of total and unique peaks in their PKS TRFLP patterns. 2S1 and 2S5 were thus targeted for further analysis and PCR library generation. Three additional samples, one from each of the remaining three collections (1S, 3S, and 4S), were also chosen. 3S6 and 4S4 contained the largest number of total and unique peaks among samples from their collections (3S and 4S). Sample 1S2 was chosen at random among the three samples from collection 1S.
Despite the large number of peaks in the actinomycete and KSα TRFLP patterns of samples 2S1 and 2S5, only limited sequence diversity was detected in the KSα PCR clone libraries generated from both samples (Fig. 2). All sequences originating from 2S1 and 2S5 formed tightly linked monophyletic clades. With the exception of clone 2S1-1′ all sequences from sample 2S1 were > 90% identical, with the majority of percent identities ranging between 97% and 99%. Only one pair of clones contained identical sequences (2S1-3′ and 2S1-16′). Percent identity of sequences obtained from sample 2S5 ranged between 88 and 99% (the majority was >95%). PCR and sequencing error are likely to have accounted for less than 1% of the sequence heterogeneity we observed, suggesting that there was considerable microdiversity within the PKS bearing actinomycete population in these samples.
FIG. 2.

Phylogenetic analysis of DNA sequences obtained by cloning of KSα PCR products from samples 2S1 and 2S5, which were chosen because they contained a large number of peaks in their actinomycete 16S TRFLPs. Species names indicate sequences obtained from GenBank. A subset of GenBank sequences used to design the PCR primer set as well as GenBank sequences most similar to cloned sequences (Actinomadura hibisca for 2S5 and Streptomyces antibioticus for 2S1 clones) was used for analysis. Numbers indicate bootstrap percentages. Only bootstraps >65% are shown. A star (*) after a sequence identifier indicates that this sequence and its identity to other sequences are shown in Table 2.
Microdiversity has been defined as high bacterial diversity below the species and subspecies level (30) and is thought to arise from mechanisms such as random mutation, chromosomal rearrangements (6), gene duplication (32), or horizontal gene transfer (17). At the nucleotide level microdiversity manifests itself as a large number of slightly dissimilar (<10%) DNA sequences of functional genes found in the environment. Microdiversity has for example been observed for the nosZ (27), nifH (40), and rbcL (37) loci, suggesting that it may be a salient feature of microbial populations. A satisfying hypothesis explaining microdiversity has thus far not been elaborated. It has been speculated that it may indicate a fast growing and rapidly evolving population of cells accumulating high frequencies of mutations in the absence of strong selection and an equalizing mechanism, such as sexual reproduction and genetic recombination (37). Antibiotic-producing actinomycetes should then, however, not be prone to exhibiting the microdiversity, because their competitiveness depends on the efficient production of the antibiotic. The responsible PKS genes should be under strong selection and exhibit less variability in a natural population.
The DNA sequences within the 2S1 and 2S5 groups of sequences are highly similar to each other, yet they are no more than 75% and 80% identical (Table 2) to sequences found in GenBank (as of July 2004). Sequences from sample 2S1 are most similar to the simocyclinone D8 biosynthetic gene cluster of Streptomyces antibioticus Tü6040 (GenBank accession no. AF324838). Simocyclinone D8 is composed of four structural elements, an angucyclinone (polyketide) core, a deoxysugar, an octatetraene dicarboxylate, and aminocoumarin moiety (10). The molecule has been shown to have antibiotic activity against gram-positive bacteria and to exhibit cytostatic activity against human tumor cells (29, 36).
TABLE 2.
Percent identity of KSα PCR product nucleotide sequences and best GenBank BlastN search resulta
| % Identity
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sequences from Fig. 2
|
Sequences from Fig. 3
|
||||||||||
| 2S1-1 | 2S1-18 | 2S1-27 | 2S5-8 | 2S5-27 | 1S2-11 | 1S2-20 | 1S2-25 | 3S6-15 | 4S4-1 | 4S4-30 | |
| BLAST | 73C | 74C | 75C | 80A | 78A | 79A | 80D | 79B | 80A | 80A | 79A |
| 2S1 1 | 78 | 78 | 72 | 70 | 72 | 71 | 74 | 71 | 72 | 71 | |
| 2S1 18 | 97 | 72 | 71 | 73 | 70 | 72 | 73 | 73 | 72 | ||
| 2S1 27 | 73 | 71 | 73 | 70 | 73 | 73 | 73 | 73 | |||
| 2S5 8 (B) | 86 | 77 | 73 | 76 | 91 | 89 | 88 | ||||
| 2S5 27 (A) | 75 | 70 | 74 | 96 | 90 | 90 | |||||
| 1S2 11 (A) | 77 | 76 | 77 | 77 | 76 | ||||||
| 1S2 20 (C) | 79 | 73 | 72 | 71 | |||||||
| 1S2 25 (B) | 76 | 75 | 75 | ||||||||
| 3S6 15 | 92 | 92 | |||||||||
| 4S4 1 | 97 | ||||||||||
Sequences from sample 2S5 are most similar to the pradimicin biosynthetic gene cluster of Actinomadura hibisca (GenBank accession no. D87924). Pradimicin antibiotics are antifungal compounds requiring a calcium cofactor and are composed of a dihydrobenzo[α]naphthacenequinone aglycone core as well as a d-alanine and sugar moieties (11). These data suggest that sequences recovered from samples 2S1 and 2S5 likely originated from PKS pathways involved in antibiotic synthesis. The products of the 2S1 and 2S5 pathways can however not be predicted using the data at hand. Simocyclinone and pradimicin are greatly dissimilar molecules and their KSα genes in the region of our PCR product are about as similar to one another (ca. 76% identical) as they are similar to the 2S1 and 2S5 clusters (Table 2), suggesting that products of the 2S1 and 2S5 pathways will be chemically unique and quite different from simocyclinone and pradimicin.
It is troubling that in silico digestion of sequences obtained from sample 2S1 revealed only 3 (out of 33, Table 1) fragments corresponding to peaks detected by TRFLP analysis of PCR products. This suggests that sequencing 40 clones at random vastly under-samples clone libraries generated from PCR products composed of a large number of different sequences. How to overcome this cloning bias, which has also been encountered in other studies (4, 22), is unfortunately not known. Sequencing a large enough number of clones to obtain all PCR product components may not be feasible and screening clones by other methods for selective sequencing may not be viable as sequencing cost likely approximates the time and effort spent in screening. Better methods are clearly needed for cloning and/or identifying rare PCR product components.
Five additional clusters of sequences were recovered from samples 1S2, 3S6 and 4S4, which were chosen based on their KSα TRFLP patterns (Fig. 3). Sequences from samples 3S6 and 4S4 formed two nonexclusive clades most similar to the KSα of the pradimicin biosynthetic gene cluster of Actinomadura hibisca (ca. 79 to 80% identical, Table 2). However, the 3S6 and 4S4 sequence clusters (Fig. 3) are only 86 to 96% and 79 to 90%, respectively, identical to sequences from the 2S5 clade (Table 2) and there was good bootstrap support to indicate that these sequences are phylogenetically distinct from the sequences of the 2S5 cluster (Fig. 3).
FIG. 3.

Phylogenetic analysis of DNA sequences obtained by cloning of KSα PCR products from samples 1S2, 3S6, and 4S4, which were chosen because they contained a large number of peaks in their KSα TRFLPs. Only the most divergent subset (n = 35) of the sequences we obtained (n = 97) for these three samples are shown. Also shown is a subset of sequences from the 2S1 and 2S5 samples. Species names indicate sequences obtained from GenBank. A subset of GenBank sequences used to design the PCR primer set as well as GenBank sequences most similar to cloned sequences (Actinomadura hibisca for 3S6 and 4S4, Streptomyces venezuelae for 1S2 A, and Kibdelosporangium ardium for 1S2 B and C clones) was used for analysis. Numbers indicate bootstrap percentages. Only bootstraps >65% are shown. A star (*) after a sequence identifier indicates that this sequence and its % identity to other sequences are shown in Table 2.
The most diverse set of KSα sequences was recovered from sample 1S2, a sample with a moderate number of peaks in its KSα TRFLP pattern (Fig. 1). One sequence originating from 2S1 (2S1-11 A, Fig. 3) was also most similar to the KS from Actinomadura hibisca (79% identical, Table 2), but was less than 78% identical to any other sequence in our data set indicating its phylogenetic distinctness. The remaining 1S2 sequences formed two clades (1S2 B and 1S2 C, Fig. 3) most closely associated with the KSα of the jadomycin and ardicin pathways in Streptomyces venezuelae (GenBank accession no. AF126429) and Kibdelosporangium aridum, respectively (GenBank accession no. L24518.1). Jadomycin B is a angucycline-derived benzoxazolophenanthridine antibiotic containing a l-digitoxose moiety (5, 24). Jadomycins have a wide spectrum of biological activity and are effective against gram-positive and gram-negative bacteria and even yeasts. Ardacin is a glycopeptide antibiotic shown to be effective against gram-positive bacteria (23, 41). As with the 2S1 and 2S5 clades of sequences, their percent identity to known KSα genes was less than 80%, indicating that the organisms containing these genes have yet to be isolated or have not been analyzed for their PKS gene pathways.
The emergence of antibiotic-resistant pathogenic bacteria over the past decades has become an important public health threat, and options for confident chemotherapy in the clinical setting are becoming limited (1, 38). The spread of drug-resistant bacteria has been exacerbated by inappropriate antibiotic use such as improper prescription, inconsistent patient use and feed amendment in domestic livestock (3, 8, 35, 39). To compound the problem, the rate of discovery of new antibiotics is slowing (26) as traditional methods for antibiotics discovery rely on standard isolation techniques and are thus limited to the small fraction of bacteria that can be cultured on standard media. Novel means for the identification and isolation of antibiotic producing microbes are clearly needed. Culture-independent methods will allow direct access to the genetic material, i.e., the metabolic capabilities of hitherto uncultured microbes.
The purpose of this study was to develop a method for identifying soil samples most likely to contain novel and unique PKS pathways and to ascertain whether KSα gene diversity is reflected by actinomycete 16S rRNA gene diversity. We made the assumption that it is most sensible to select for soils containing dissimilar bacterial communities. This should allow a greater diversity of bacteria to be screened with fewer samples. We hypothesized that samples containing more diverse and unique actinomycete communities are most likely to contain novel and unique PKS genes. Our data provides only partial support for this hypothesis. Among the samples that were amplified using our KSα primers, the two samples with the richest and most unique actinomycete community in our sample set (2S1 and 2S5) also contained the largest diversity of KSα sequences as indicated by TRFLP.
Statistical support for the linkage between actinomycete species and KSα sequence diversity based on TRFLP data was weak, mainly because eight samples produced no KSα PCR product using the primers developed in this study. Also, the largest sequence diversity obtained by cloning PCR products came from a sample with only an average number of peaks in it's KSα TRFLP. Overall, KSα sequences obtained in this study belong to seven novel clades, none of which were >81% identical at the DNA level to sequences already in GenBank (as of August 2004). In particular there are six clades with high similarity to the KSα genes in pathways for the production of angucycline-derived antibiotics. These data indicate that group-specific TRFLP analysis of bacterial communities and functional genes can be a practical tool for prescreening environmental samples for diverse and unique gene sequences, but cannot be used to predict which sample will yield the most diverse sequence data in cloning experiments. Prescreening methods, such as the one described here, would be of very large benefit to projects aiming to identify novel bioactive molecules, because they increase throughput and efficiency, reduce cost, and reduce the probability of reinvestigating previously described targets.
Acknowledgments
This work was supported by the Fogarty International Center of the NIH under U01 TW006674 for the International Cooperative Biodiversity Groups.
We thank Laurie Seliger for DNA sequencing assistance.
REFERENCES
- 1.Allsop, A., and R. Illingworth. 2002. The impact of genomics and related technologies on the search for new antibiotics. J. Appl. Microbiol. 92:7-12. [DOI] [PubMed] [Google Scholar]
- 2.Borders, D. B. 1999. Isolation and identification of small molecules, p. 257-265. In A. L. Demain, and J. E. Davies (ed.), Manual of industrial microbiology and biotechnology, 2nd ed. ASM Press, Washington, D.C.
- 3.Chen, S., S. Zhao, D. G. White, C. M. Schroeder, R. Lu, H. Yang, P. F. McDermott, S. Ayers, and J. Meng. 2004. Characterization of multiple-antimicrobial-resistant salmonella serovars isolated from retail meats. Appl. Environ. Microbiol. 70:1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cottrell, M. T., and D. L. Kirchman. 2000. Community composition of marine bacterioplankton determined by 16S rRNA gene clone libraries and fluorescence in situ hybridization. Appl. Environ. Microbiol. 66:5116-5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doull, J. L., A. K. Singh, M. Hoare, and S. W. Ayer. 1994. Conditions for the production of jadomycin B by Streptomyces venezuelae ISP5230: effects of heat shock, ethanol treatment and phage infection. J. Indust. Microbiol. 13:120-125. [DOI] [PubMed] [Google Scholar]
- 6.Ginard, M., J. Lalucat, B. Tummler, and U. Romling. 1997. Genome organization of Pseudomonas stutzeri and resulting taxonomic and evolutionary considerations. Int. J. Syst. Bacteriol. 47:132-143. [DOI] [PubMed] [Google Scholar]
- 7.Glasby, J. S. 1992. Dictionary of antibiotic-producing organisms. Prentice Hall Professional Technical Reference, New York.
- 8.Gomez-Lus, R. 1998. Evolution of bacterial resistance to antibiotics during the last three decades. Int. Microbiol. 1:279-284. [PubMed] [Google Scholar]
- 9.Heuer, H., M. Krsek, P. Baker, K. Smalla, and E. M. Wellington. 1997. Analysis of actinomycete communities by specific amplification of genes encoding 16S rRNA and gel-electrophoretic separation in denaturing gradients. Appl. Environ. Microbiol. 63:3233-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holzenkampfer, M., M. Walker, A. Zeeck, J. Schimana, and H. P. Fiedler. 2002. Simocyclinones, novel cytostatic angucyclinone antibiotics produced by Streptomyces antibioticus Tu 6040 II. Structure elucidation and biosynthesis. J. Antibiot. 55:301-307. [DOI] [PubMed] [Google Scholar]
- 11.Hu, M., Y. Ishizuka, Y. Igarashi, T. Oki, and H. Nakanishi. 2000. Interaction of three pradimicin derivatives with divalent cations in aqueous solution. Spectrochim. Acta A Mol. Biomol. Spectr. 56:1233-1243. [DOI] [PubMed] [Google Scholar]
- 12.Hutchinson, C. R. 1997. Antibiotics from genetically engineered microorganisms, p. 659-682. In W. R. Strohl (ed.), Biotechnology of industrial antibiotics. Marcel Dekker, Baltimore, Md.
- 13.Katz, L., and S. Donadio. 1993. Polyketide synthesis: prospects for hybrid antibiotics. Annu. Rev. Microbiol. 47:875-912. [DOI] [PubMed] [Google Scholar]
- 14.Khosla, C., R. S. Gokhale, J. R. Jacobsen, and D. E. Cane. 1999. Tolerance and specificity of polyketide synthases. Annu. Rev. Biochem. 68:219-253. [DOI] [PubMed] [Google Scholar]
- 15.Kitts, C. L. 2001. Terminal restriction fragment patterns: a tool for comparing microbial communities and assessing community dynamics. Curr. Issues Intestinal Microbiol. 2:17-25. [PubMed] [Google Scholar]
- 16.Kumar, S., K. Tamura, I. B. Jakobsen, and M. Nei. 2001. MEGA2: molecular evolutionary genetics analysis software. Bioinformatics. 17:1244-1245. [DOI] [PubMed] [Google Scholar]
- 17.Lorenz, M. G., and W. Wackernagel. 1994. Bacterial gene transfer by natural genetic transformation in the environment. Microbiol. Rev. 58:563-602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Magurran, A. E. 1998. Ecological diversity and its measurement. Princeton University Press, Princeton, N.J.
- 19.Metsä-Ketelä, M., L. Halo, E. Munukka, J. Hakala, P. Mäntsälä, and K. Ylihonko. 2002. Molecular evolution of aromatic polyketides and comparative sequence analysis of polyketide ketosynthase and 16S ribosomal DNA genes from various Streptomyces species. Appl. Environ. Microbiol. 68:4472-4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Metsä-Ketelä, M., V. Salo, L. Halo, A. Hautala, J. Hakala, P. Mäntsälä, and K. Ylihonko. 1999. An efficient approach for screening minimal PKS genes from Streptomyces. FEMS Microbiol. Lett. 180:1-6. [DOI] [PubMed] [Google Scholar]
- 21.Murray, A. E., C. M. Preston, R. Massana, L. T. Taylor, A. Blakis, K. Wu, and E. F. DeLong. 1998. Seasonal and spatial variability of bacterial and archaeal assemblages in the coastal waters near Anvers Island, Antarctica. Appl. Environ. Microbiol. 64:2585-2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Phelps, C. D., L. Kerkhof, and L. Y. Young. 1998. Molecular characteriztion of a sulfate-reducing consortium which mineralizes benzene. FEMS Microbiol. Ecol. 27:269-279. [Google Scholar]
- 23.Piecq, M., P. Dehottay, A. Biot, and J. Dusart. 1994. Cloning and nucleotide sequence of a region of the Kibdelosporangium aridum genome homologous to polyketide biosynthetic genes. DNA Sequence. 4:219-229. [DOI] [PubMed] [Google Scholar]
- 24.Rix, U., J. Zheng, L. L. Remsing Rix, L. Greenwell, K. Yang, and J. Rohr. 2004. The dynamic structure of jadomycin B and the amino acid incorporation step of its biosynthesis. J. Am. Chem. Soc. 126:4496-4497. [DOI] [PubMed] [Google Scholar]
- 25.Roberts, A. D., and A. A. Higton. 1988. Dictionary of antibiotics and related substances. Chapman and Hall, London, England.
- 26.Russell, A. D. 2002. Antibiotic and biocide resistance in bacteria: introduction. J. Appl. Microbiol. 92(Suppl.):1S-3S. [PubMed] [Google Scholar]
- 27.Scala, D. J., and L. J. Kerkhof. 1999. Diversity of nitrous oxide reductase (nosZ) genes in continental shelf sediments. Appl. Environ. Microbiol. 65:1681-1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scala, D. J., and L. J. Kerkhof. 2000. Horizontal heterogeneity of denitrifying bacterial communities in marine sediments by terminal restriction fragment length polymorphism analysis. Appl. Environ. Microbiol. 66:1980-1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schimana, J., H. P. Fiedler, I. Groth, R. Sussmuth, W. Beil, M. Walker, and A. Zeeck. 2000. Simocyclinones, novel cytostatic angucyclinone antibiotics produced by Streptomyces antibioticus Tu 6040. I. Taxonomy, fermentation, isolation and biological activities. J. Antibiot. 53:779-787. [DOI] [PubMed] [Google Scholar]
- 30.Schloter, M., M. Lebuhn, T. Heulin, and A. Hartmann. 2000. Ecology and evolution of bacterial microdiversity. FEMS Microbiol. Rev. 24:647-660. [DOI] [PubMed] [Google Scholar]
- 31.Seow, K. T., G. Meurer, M. Gerlitz, E. Wendt-Pienkowski, C. R. Hutchinson, and J. Davies. 1997. A study of iterative type II polyketide synthases, using bacterial genes cloned from soil DNA: a means to access and use genes from uncultured microorganisms. J. Bacteriol. 179:7360-7368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith, J. M., N. H. Smith, M. O'Rourke, and B. G. Spratt. 1993. How clonal are bacteria? Proc. Natl. Acad. Sci. USA 90:4384-4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strohl, W. R. 1997. Industrial antibiotics: today and the future. In W. R. Strohl (ed.), Biotechnology of antibiotics, 2nd ed. Marcel Dekker, New York, N.Y.
- 34.Strohl, W. R. 2000. The role of natural products in modern drug discovery. Drug Discovery Today. 5:39-41. [DOI] [PubMed] [Google Scholar]
- 35.Tollefson, L., S. F. Altekruse, and M. E. Potter. 1997. Therapeutic antibiotics in animal feeds and antibiotic resistance. Rev. Sci. Technol. 16:709-715. [DOI] [PubMed] [Google Scholar]
- 36.Trefzer, A., S. Pelzer, J. Schimana, S. Stockert, C. Bihlmaier, H. P. Fiedler, K. Welzel, A. Vente, and A. Bechthold. 2002. Biosynthetic gene cluster of simocyclinone, a natural multihybrid antibiotic. Antimicrob. Agents Chemother. 46:1174-1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wawrik, B., J. H. Paul, L. Campbell, D. Griffin, L. Houchin, A. Fuentes-Ortega, and F. Muller-Karger. 2003. Vertical structure of the phytoplankton community associated with a coastal plume in the Gulf of Mexico. Mar. Ecol. Prog. Ser. 251:87-101. [Google Scholar]
- 38.Witte, W. 1998. Medical consequences of antibiotic use in agriculture. Science 279:996-997. [DOI] [PubMed] [Google Scholar]
- 39.World Health Organization. 1996. The World Health Report. W.H.O. Publications. World Health Organization, Geneva, Switzerland.
- 40.Zehr, J. P., M. T. Mellon, and S. Zani. 1998. New nitrogen-fixing microorganisms detected in oligotrophic oceans by amplification of Nitrogenase (nifH) genes. Appl. Environ. Microbiol. 64:3444-3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zinn, R. A., M. K. Song, and T. O. Lindsey. 1991. Influence of ardacin supplementation on feedlot performance and digestive function of cattle. J. Anim. Sci. 69:1389-1396. [DOI] [PubMed] [Google Scholar]