

Abstract
Adenosine deaminases acting on RNA (ADARs) are endogenous enzymes catalyzing the deamination of adenosines to inosines, which are then read as guanosines during translation. This ability to recode makes ADAR an attractive therapeutic tool to edit genetic mutations and reprogram genetic information at the mRNA level. Using the endogenous ADARs and guiding them to a selected target has promising therapeutic potential. Indeed, different studies have reported several site-directed RNA-editing approaches for making targeted base changes in RNA molecules. The basic strategy has been to use guide RNAs (gRNAs) that hybridize and form a double-stranded RNA (dsRNA) structure with the desired RNA target because of ADAR activity in regions of dsRNA formation. Here we report on a novel pipeline for identifying disease-causing variants as candidates for RNA editing, using a yeast-based screening system to select efficient gRNAs for editing of nonsense mutations, and test them in a human cell line reporter system. We have used this pipeline to modify the sequence of transcripts carrying nonsense mutations that cause inherited retinal diseases in the FAM161A, KIZ, TRPM1, and USH2A genes. Our approach can serve as a basis for gene therapy intervention in knockin mouse models and ultimately in human patients.
Keywords: MT: RNA/DNA Editing, RNA editing, inherited retina diseases, ADAR, deaminization, yeast, pipeline, off-target
Graphical abstract
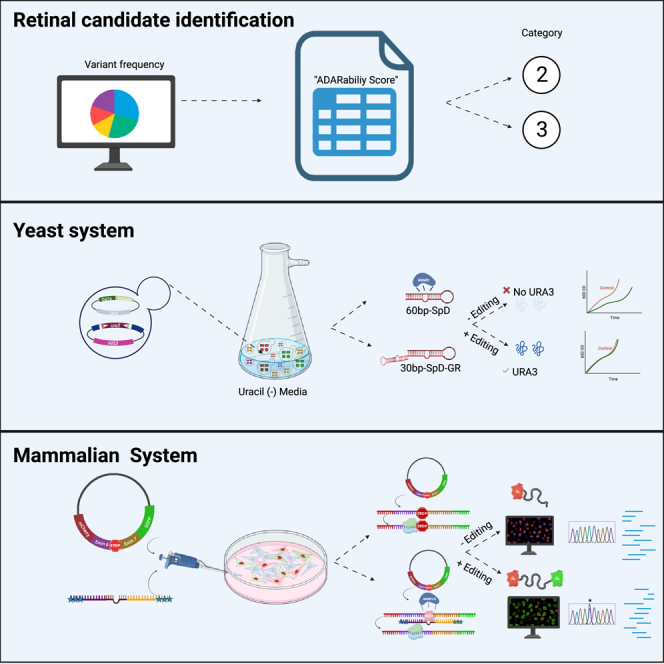
Ben-Aroya and colleagues introduce a novel pipeline that identifies inherited retinal disease mutations for ADAR-mediated editing, using a yeast-based screening system to select efficient gRNAs for editing nonsense mutations, and test them in a human cell line reporter system. This approach can be a basis for future gene therapy intervention.
Introduction
The most common type of RNA editing is the deamination of adenosine (A) to inosine (I), which is biochemically read as guanosine (G) by the translation machinery.1 The consequence is an A-to-G substitution that can lead to codon changes (recoding)2 and could rescue G-to-A mutations. A-to-I RNA editing is catalyzed by the highly conserved adenosine deaminase acting on RNA enzyme (ADAR) protein family, found in all metazoans.3,4,5,6 In mammalians, the ADAR family consists of two active enzymes, ADAR1 and ADAR2, which share the catalytical deaminase domain (DD),7 and double-stranded RNA (dsRNA) binding domains (RBDs). The RBDs bind not only to perfect dsRNA but also to imperfect double-stranded structures with mismatches at even higher affinity; accordingly, dsRNA is considered to be a prerequisite for editing by ADARs.3,8
In recent years, the prospect of using CRISPR-Cas9 for therapeutics has attracted much attention. This approach offers a solution to chronic conditions by introducing a lifelong permanent genetic modification.9,10 However, in some cases, RNA editing could be a more appropriate alternative, as it does not require the introduction of foreign immunogenic prokaryotic proteins and any off-target effects are impermanent.11 Thus, possible adverse effects would be reversible and tunable, making this approach safer.12
Advances made in understanding the mode of action of the ADAR proteins, enabled the development of several site-directed RNA-editing (SDRE) technologies as a therapeutic tool, one of which is by recruiting the endogenous ADAR, which is the method of choice in the present study. The ability of ADARs to recode makes them attractive therapeutic tools for correcting genetic mutations and reprogramming genetic information at the mRNA level. Several studies described pioneering approaches aiming to use ADAR-mediated editing as a therapeutic tool.13,14 Given that most adenosines are not likely to occur in a proper structure for editing, the main challenge is to design guide RNAs (gRNAs), which form an artificial dsRNA structure around a user-defined target and redirect human ADAR (hADAR) activity to this site.
Inherited retinal diseases (IRDs) are a clinically heterogeneous and complex group of visual impairment phenotypes caused by pathogenic variants in at least 277 nuclear and mitochondrial genes causing vision loss in individuals worldwide.15 It has been estimated that globally at least one in three individuals is a carrier of at least one recessive IRD-causing variant and that 1 in 1,380 individuals are affected by recessive IRDs.16,17 Taking into account that ∼41% of IRD-causing single-nucleotide variants (SNVs) can be candidates for ADAR G-to-A editing,15 the somewhat high editing activity in the retina,18 and the relative ease of gRNA delivery to the retina, SDRE appears to be a promising approach for genetic therapy of IRDs. There is currently no effective treatment for the vast majority of IRDs, but recent adeno-associated virus (AAV)-based gene therapies bring some hope. Although this is the case, many relatively prevalent IRD-causing genes (such as ABCA4 and USH2A) are too large for AAV vectors and/or have complex alternative splicing patterns yet to be well understood, and therefore new treatment modalities should be developed.
Here we developed a pipeline in which we selected common nonsense disease-causing mutations amenable for ADAR editing, used yeast screening systems that enable identification of efficient gRNAs that promote endogenous ADAR recruitment for SDRE, and verified their function in a human reporter cell line. Our study provides a proof of concept that RNA editing mediated by ADAR can be used as a novel approach for treating IRDs as well as other inherited diseases and forms the basis for better gRNA designs.
Results
Selecting “ADARable” candidate mutations
We selected four nonsense mutations (Table 1) to be edited in the present study on the basis of the following rationale: optimal ADAR editing targets for this study were identified by using the previously reported coding system,15 in which each SNV and therefore the corresponding mutated codon is assigned a score of 0–3, representing their amenability for undergoing ADAR editing. We assessed every disease-causing variant in a cohort of more than 2,100 Israeli and Palestinian families with IRDs as well as the international Global Retinal Inherited Disease (GRID) dataset.15 Within our scoring system, a score of 3 denotes that the wild-type (WT) amino acid can be reinstated by ADAR editing, 2 denotes a nonsense mutation in a codon that can be edited to a missense variant by either correcting the mutated nucleotide or a neighboring nucleotide, 1 denotes a mutated codon causing a missense variant that can be edited to a different missense variant, and 0 denotes an amino acid that is unable to undergo editing to another amino acid. In our cohort, 47.4% of solved families with IRD-causing missense or nonsense mutations have a score of 3 or 2. The distribution of the ADAR score 2 variants in our cohort is shown in Figure 1. We chose the three most common Israeli editable nonsense mutations (in the TRPM1, FAM161A, and KIZ genes) within our cohort as targets (Table 1), all of which are score 2 mutations. In addition, we also chose the most common worldwide nonsense mutation (c.11864G>A; p.W3955∗ in USH2A with a score of 3), identified in at least 49 cases on the basis of our previous analysis.15 All score 2 mutations had a neighboring adenosine whose subsequent deaminization would transform the premature stop codon into a tryptophan (W), while the original USH2A codon can be reinstated by ADAR. Aiming to verify that the missense variants generated by RNA editing in the KIZ, FAM161A, and TRPM1 genes are unlikely to affect protein function, we performed mutation and sequence analysis of these genes (Figure S1). Although these three particular amino acids are highly conserved, there are either no reported missense pathogenic variants (KIZ), a single “missense” mutation reported as pathogenic or likely pathogenic that is likely to affect splicing (FAM161A), or mainly null pathogenic variants (TRPM1) in these genes.
Table 1.
Four ADARable nonsense IRD-causing mutations selected for the present study
| Gene | NM | Inheritance pattern | gRNA | c. | p. | Number of patients in cohort | Original AA | Mutated AAa | ADAR edited AAb | % Editing ADAR1 (NGS) | % Editing ADAR2 (NGS) | % Editing ADAR1 (Sanger) | % Editing ADAR2 (Sanger) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TRPM1 | NM_002420.5 | AR | 60 bases | c.880A>T | p.K294∗ | 49 | AAG (K) | TAG (∗) | TGG (W) | 32.6% (n = 8) | 25.1% (n = 12) | 42.4% (n = 10) | 45.6% (n = 14) |
| FAM161A | NM_001201543 | AR | 60 bases | c.1567C>T | p.R523∗ | 34 | CGA (R) | TGA (∗) | TGG (W) | 2.5% (n = 2) | 4.7% (n = 2) | 1.7% (n = 2) | 5.2% (n = 2) |
| KIZ | NM_018474.4 | AR | 60 bases | c.226C>T | p.R76∗ | 21 | CGA (R) | TGA (∗) | TGG (W) | 7.4% (n = 6) | 7.8% (n = 6) | 3.3% (n = 2) | 9.9% (n = 3) |
| USH2A | NM_206933.2 | AR | 60 bases | c.11864G>A | p.W3955∗ | 49c | TGG (W) | TAG (∗) | TGG (W) | 31.2% (n = 5) | 20.5% (n = 4) | 30.0% (n = 3) | 29.5% (n = 5) |
| USH2A | NM_206933.2 | AR | 18 bases + 55 base GR | c.11864G>A | p.W3955∗ | 49c | TGG (W) | TAG (∗) | TGG (W) | 0.5% (n = 2) | 9% (n = 4) | 0% (n = 2) | 10.4% (n = 4) |
AA, amino acid; AR, autosomal recessive; NGS, next-generation sequencing.
The mutated nucleotide is in boldface type.
The edited nucleotide is in boldface type.
Based on a previous analysis of IRD variants worldwide.15
Figure 1.

Most prevalent “ADARable” IRD-causing mutations with ADAR scores of 2 in our cohort calculated by percentage of families
The 3 most prevalent variants are also shown (original codon > mutated codon > edited codon).
Creating a screening system in yeast for identifying the gRNA against IRD mutations
As a first step to identifying effective gRNAs for the aforementioned IRD-causing nonsense mutations, we created a yeast-based system to test candidate gRNAs (Figure 2A). The system is based on a strain carrying a LEU2-marked plasmid that can conditionally express hADAR1 or hADAR2 under a galactose-inducible promoter (GAL1p). This strain is a uracil auxotroph as a result of a deletion in the endogenous URA3 gene, and thus is unable to grow in a medium lacking uracil. A second HIS3-marked plasmid carries a copy of a URA3 gene that has been altered with a premature stop codon leading to the absence of the corresponding protein. This mutation was originated from 102 bp minigene fragments which were inserted in frame, immediately following the URA3 start codon, and contain the nonsense mutation in the selected target genes (termed TRPM1-p.K294∗-ura3, FAM161A-p.R523∗-ura3, KIZ-p.R76∗-ura3, and USH2A-p.W3955∗-ura3). As shown in Figures 2C and 2D we validated that such insertions minimally affected the function of the URA3 reporter gene. The 3′ end of the reporter gene is followed by a “tail” that can fold back and hybridize at the RNA level with the region flanking the nonsense mutation, thus creating a dsRNA structure that can promote ADAR-mediated editing of the UAG nonsense mutations, to UGG encoding tryptophan (Figure 2B). Thus, efficient hADAR-mediated editing of the IRD mutations should promote the expression of WT Ura3 and correlates with the strain’s growth in uracil-depleted medium.
Figure 2.

Yeast-based screening system for testing gRNAs that promote A-to-I editing
(A) Schematic representation of the yeast-based selection system. A uracil auxotroph yeast strain (deleted in the URA3 genes [Δura3]) harbors a LEU2-marked plasmid that enables the conditional expression of hADAR2 or hADAR2 under a galactose-inducible promoter (GAL1p-hADAR1/2) and a HIS3-marked plasmid carrying the gene URA3 as a reporter gene. The reporter gene expression was interrupted by the in-frame insertion of 102 bp minigene fragments containing the following nonsense mutations: TRPM1 K294∗, FAM161A R523∗, KIZ R76∗, and USH2A W3955∗ (denoted by the green arrow with the red bar). The 3′ end of each plasmid containing the mutated insert is followed by a reverse complement gRNA sequence (“tail”) of the region flanking the mutated insert (denoted a pink triangle in the HIS3-marked plasmid). When the “tail” folds back at the RNA level, it hybridizes with the corresponding mutated insert sequence and forms the dsRNA structure required for hADAR recruitment and editing, except from a single A-C mismatch at the target adenosine. If RNA editing is efficient, the translated wild-type Ura3 protein permits growth in a medium lacking uracil. (B) Sequences of the original amino acids, the specific changes leading to a nonsense mutation within the indicated mutations, and the result of ADAR-mediated editing. Nucleotides colored in red denote the specific mutation site. The target adenosines and the result of the ADAR-mediated editing are denoted in green. (C and D) Insertion of the corrected version of the minigene fragments minimally affect the function of the URA3 reporter gene. (C) Similar to (A), the corrected version of the mutated insert (∗>Trp) is denoted by the green bar. (D) Growth curves of the strains indicated in (C). Strains were grown to stationary phase in selective media supplemented with 2% raffinose (ADAR expression is off) and transferred to selective media lacking uracil, supplemented with 2% galactose (t-0) to induce the expression of hADAR2. Samples were brought to an optical density at a wavelength of 600 nm (O.D 600 nm) of 0.1, and the growth rate was assessed in a 96-well plate using a Tecan microplate reader by measuring O.D 600 nm every 30 min for 25 hours. Error bars represent the SD between three independent experiments. A plasmid carrying the URA3 gene without the in-frame insertion of the mutated insert fragment was used as a positive control.
Both hADAR1 and hADAR2 exhibit high expression levels in the retina, surpassing those found in most other tissues, including various brain regions where editing activity is known to be relatively high (Figures S2A and S2B). ADAR2 is particularly abundant in the retina (Figure S2A), and as it is responsible for most of the recoding activity,19,20,21 it is not surprising that the editing levels of the known recoding sites reach their peak in the retina (Figure S2C). Thus, we conducted experiments involving both hADAR1 and hADAR2.
To test whether the insertion of the minigene fragment affects the functionality of the URA3 reporter gene, a positive control strain was created as well, in which the stop codon within the 102 bp fragment was swapped with tryptophan (TGG), mimicking the outcome of perfect editing of the target adenosine (TRPM1-K294W-URA3, FAM161A-R523W-URA3, KIZ-R76W-URA3, and USH2A-W3955W-URA3) (Figures 2C and 2D). This would lead to a full-length Ura3 protein carrying an extra 34 amino acids. As shown in Figures 2C and 2D, we were able to validate that this addition had only a minor effect on the growth rates in the absence of uracil and revealed the expected maximum growth associated with each insertion.
Next, we identified and optimized the gRNA sequence for each IRD mutation. As mentioned above, the gRNAs used in previous studies to direct the endogenous ADARs are composed of two essential elements: the “specificity domain” and a 49 nt dsRNA structure originating from the GluR (GR) ion channel receptors used to recruit ADAR binding (the “recruitment element”). We decided to test the following gRNAs (Figure 3A) for each of the IRD mutations: (1) a 60 nt sequence representing the specificity domain, which fully complements the cDNA of the IRD sequence (termed 60nt-SpD), except for an A-C mismatch at the target adenosine, which is known to enhance editing efficiency,22 and (2) a shorter 30 nt specificity domain combined with GR as the recruiting element (termed 30nt-SpD-GR). These gRNAs were inserted in the above-mentioned 3′ end region. Next, we tested the growth of the indicated strains in the presence of galactose to induce hADAR2 or hADAR1 expression and under uracil depletion, for 25 hours. Growth was compared with the positive control carrying the corrected version of the IRD mutation (see above) and to the negative control carrying an empty LEU2-marked plasmid (without the hADAR) (Figure 3B). The results indicate that in the cases of TRPM1-p.K294∗ and USH2A-p.W3955∗, the 60nt-SpD induced growth similar to the WT, while the 30nt-SpD-GR gRNA induced mild growth only in the case of the USH2A variant. However, the 60nt-SpD gRNA induced only minimal or mild growth in the cases of KIZ-p.R76∗ and FAM161A p.R523∗, respectively (Figures 3C and 3D). No improvement was detected in the growth of these mutants even when we attempted to elongate the specificity domain to 60 nt and add GR as the recruiting element (60nt-SpD-GR) (Figure S3).
Figure 3.

hADAR2- and hADAR1-mediated editing of selected IRD mutations can be induced in the presence of gRNAs
(A) Schematic representation of the dsRNA structure formed when the “tail” containing the selected gRNA sequences folds back at the RNA level on the target to form the dsRNA structure for ADAR recruitment. (B) A table describing the contents of the strains (denoted as I–V) tested in (C)–(F). Strain I mimicked the outcome of perfect editing by ADAR and served as the positive control, while the negative controls (strains IV and V) carried an “hADAR1-mediated editing of selected empty” LEU2 plasmid without the ADAR enzyme. (C and D) Growth curves of the indicated strains were produced as described in two dimensions in the presence of hADAR2 (top) and hADAR1 (bottom). The numbers representing the editing levels at the target A’s are the average (n = 3) obtained on NGS analysis.
To validate the necessity of dsRNA formation in the vicinity of the target adenosine as an editing substrate for ADAR, we used a similar 60nt-SpD gRNA that in addition to the A-C mismatch, carried additional mismatches around the target adenosine. In comparison with the 60nt-SpD gRNA strain, which did not contain additional mismatches (termed “perfect tail”), we observed that the growth was largely impaired (Figure S4).
Overall, these results demonstrate that the combination of either hADAR2 or hADAR1, along with the hybridization of the dsRNA, can induce editing within the selected targets.
Next-generation sequencing confirms that yeast growth is associated with A-to-I editing
Next, we used next-generation sequencing (NGS) to validate that the yeast growth seen in our experimental system is a result of A-to-I editing carried out by hADAR2. To this end, we extracted RNA from the strains in which the presence of the selected IRD mutation, hADAR2, and either the gRNA 60nt-SpD (for variants in FAM161A, TRPM1, KIZ, and USH2A) or 30nt-SpD-GR (USH2A) induced yeast growth. cDNA of the region flanking the mutation was then amplified and subjected to NGS. The results indicate that in all cases, A-to-I editing at the target adenosine was detected. Strains carrying the FAM161A and KIZ variants exhibited the lowest editing levels of 5% and 1% (ADAR2), or 5% and 2.5% (ADAR1), which aligned with their limited growth, most probably because these mutants carry guanosines at the 5′ end of the target adenosine, which are largely depleted from the 5′ end of natively edited adenosines.23 The TRPM1 and USH2A variants showed a similar growth rate, with higher editing levels of 11% and 10% (ADAR2), or 40% and 30% (ADAR1), respectively, in the presence of the 60nt-SpD gRNA. Additionally, the presence of 30nt-SpD-GR induced editing levels of 0% and 7.3% (ADAR2), or 10.3% and 16% (ADAR1) (Figures 3C and 3D).
Overall, these results imply that the growth rate of the yeast strains in our screening system correlates with editing levels.
Expanding the yeast-based system for screening millions of candidate gRNAs
In naturally occurring editing sites, the dsRNA structure is imperfect in the vicinity of the targeted adenosine, and these mismatches are essential for efficient editing.24,25 Thus, although it is clear that perfect RNA duplexes make suboptimal structures for editing, the positions of the mismatches and bulges are difficult to predict. Hence, we aimed to investigate whether our yeast strains could be employed for their identification.
As a control, we initiated by introducing a specific G-G mismatch between the target and gRNA at the 5′ G of the adenosine to be edited in KIZ-p.R76∗ and FAM161A p.R523∗ mutants. It was recently demonstrated that such a mismatch can enhance editing efficiency in this context.26 Indeed, the results indicate a significant improvement in growth, which was correlated with increased editing efficiency 1%–38% (hADAR1) or 35% (hADAR2) in KIZ-p.R76∗ and 2.5%–10% (hADAR1), or 5%–8.6% (hADAR2), in FAM161A p.R523∗ (Figure 4). These findings underscore the potential of identifying specific mismatches that can enhance editing.
Figure 4.

Induction of editing with a G-G mismatch at the 5′ end of the target adenosine
Similar to Figure 3, but this time, the 60nt-SpD gRNA was compared with a gRNA containing a specific G-G mismatch between the target and gRNA at the 5′ G of the adenosine to be edited in KIZ-p.R76∗ and FAM161A p.R523∗ mutants (60nt-SpD G-G).
Next, we conducted an unbiased screening assay to identify the best performing gRNAs out of randomized pools. For this purpose, we focused on the KIZ-p.R76∗ mutant, which exhibited the lowest editing levels. We tested whether we could enhance the baseline editing levels obtained with the 60nt-SpD in the presence of hADAR2. To achieve this, we used the yeast strain described in Figure 2 to create a library of approximately 107 strains (Figure 5A). Each of these strains carries a distinct gRNA sequence that is mostly base-paired with the target, containing ≤6% mismatched positions, in addition to the A-C mismatch in the target adenosine. The 5′ and 3′ positions were left untouched, as we wanted to identify additional mismatches that could further contribute to the effect provided by the G:G mismatch at the 5′ G of the target as described above (see Figure 4). The library was then pooled and subjected to successive rounds of selection under uracil starvation (Figure 5B). Next, in order to identify the gRNA sequences responsible for enhancing editing and stimulating induced growth of cells in the pooled culture, we serially diluted a sample from the library and plated single cells on a medium that selects for the plasmid harboring the gRNAs and hADAR2. The sequences of the gRNAs that facilitated growth in the selected colonies were determined through Sanger sequencing of the PCR products, using universal primers flanking the insertion sites of the gRNA-encoding DNA. NGS analysis of RNA samples extracted from the cells containing the identified mismatched gRNAs (dubbed “B4,” “B12,” and “TT”) revealed that the minimal baseline editing level of 1% induced by the hADAR2 and 60nt-SpD was significantly increased to 4.5%–6% (Figure 5C).
Figure 5.

The high-throughput yeast-based screening system
(A) A library of approximately 107 yeast strains, described in Figure 2, was created by using in vivo homologous recombination to insert distinct gRNA sequences at the 5′ end of the reporter gene. (B) The process of selecting improved gRNA variants targeting the KIZ R67∗ mutation. The sample containing yeast strains with a gRNA that forms perfect dsRNA structures with the target mutation was used as a baseline growth control and compared with the pooled library. After iterative rounds of enrichment in a medium lacking uracil, we serially diluted a sample from the enriched library and plated single cells on a medium that selects for the HIS3 plasmid that harbors the gRNAs. Plasmids were prepared from the developing colonies, and the sequences of the gRNAs that facilitated growth in the selected colonies were determined through Sanger sequencing of the PCR products, using universal primers flanking the insertion sites of the gRNA-encoding DNA (blue arrows). (C) Sequence of the target adenosine and flanking sequence (green) and the complementary identified gRNAs (red) (termed “B4”, “B12”, and “TT”) compared with the control dsRNA containing a single A-C mismatch at the target adenosine. The editing levels of the control were determined using NGS as described above.
These results along with the validation in mammalian cells (see below), demonstrate that our screening system can be scaled up and customized for screening millions of candidate gRNAs to identify those that are particularly efficient in editing selected targets.
Confirming a functional reporter system
As the gRNAs identified as ADAR-recruiting candidates for the specific genes and mutations were tested in a yeast system, we chose to then test these in a mammalian cell line using a fluorescent reporter system. Validation of the fluorescent reporter system was done by cloning a 161 nt gene fragment of the GAPDH 3′ UTR into the pmCherry-EGFP reporter plasmid. Although this part of the gene is not translated, we cloned it into a reading frame in which one of the only two “stop codons” contains the target adenosine (supplemental methods), whose place in the sequence would cause termination of translation unless edited. We made a single base change (T>C) to the other “stop codon” in the 3′ UTR, 8 nt downstream to the target adenosine, to prevent termination of translation (TAG>CAG) unrelated to RNA editing. A chemically modified gRNA with an 18 nt long complementarity region and a 55 nt GR motif tail targeting the specific adenosine in the reporter plasmid, previously developed by the Stafforst group,27 was introduced to ADAR1 p.110 and ADAR2 (supplemental methods) overexpressing HeLa cells transiently transfected with the target plasmid, and all experimental wells showed both mCherry and EGFP fluorescence (Figure S5), while the negative control only showed mCherry fluorescence. Sanger sequencing showed that ADAR1 p.110-overexpressing and ADAR2-overexpressing HeLa cells underwent an average of 16.5% (n = 2) and 26.1% (n = 2) editing, respectively.
Editing levels are sequence dependent
Using our validated cellular reporter system in which a target nonsense mutation and surrounding exonic bases were inserted as a cassette between mCherry and EGFP in an expression plasmid and transfected into ADAR1 p.110-overexpressing and ADAR2-overexpressing HeLa cells, allowed us to measure the editing of our designed gRNAs (Figure 6A). The gRNAs used were those with the highest levels of editing and survival measured in the aforementioned yeast model. Introducing chemically modified 60-base oligomer gRNAs without a GR motif into ADAR1 p.110 or ADAR2-overexpressing stable HeLa cell lines induced varying levels of RNA editing.
Figure 6.

RNA editing in HeLa cells
Representative images from samples of ADAR1 p.110 (ADAR1)-overexpressing and ADAR2-overexpressing HeLa cells transfected with both the TRPM1 nonsense mutation reporter plasmid and 60-mer chemically modified gRNA at 96 h post-seeding. (A) Schematic of the in-vitro experiment. (B) Fluorescent microscopy. (C) Sanger sequence. (D) Next-generation sequencing reads in IGV viewer.
The highest levels of editing were observed in the samples transfected with a TRPM1 60-mer gRNA as measured by both Sanger sequencing and NGS. The minigene reporter construct expressed in the cell lines contained the TRPM1 mutation (c.880A>T, p.K294∗) and the target adenosine for editing was the 3′ neighboring base to the mutated A>T (Table 1). Both mCherry and EGFP expression were observed in all samples (Figure 6B). For samples of gRNA-treated ADAR1 p.110-overexpressing HeLa cells in which RNA editing was clearly observed, an average of 42.4% (n = 10) and 32.6% (n = 8) editing was observed in Sanger sequencing and NGS, respectively, compared with an average of 45.6% (n = 14) and 25.1% (n = 12) editing observed among HeLa cells expressing ADAR2 in Sanger sequencing and NGS with the maximum editing levels approaching 66% in Sanger sequencing (Table 1; Figures 6C and 6D).
Editing was also observed in cells transfected with a minigene reporter construct containing the nonsense USH2A mutation (c.11864G>A, p.W3955∗) and in this case the target adenosine for editing is the mutation itself, for a complete correction of the mutation (Table 1). Because of high levels of editing in the yeast model for both USH2A gRNAs with and without the GR motif, we tested each in the reporter system. Higher levels of editing were noticed in ADAR1 p.110-overexpressing and ADAR2-overexpressing HeLa cells transfected with a USH2A 60-mer gRNA without an GR motif compared with the levels observed in the samples transfected with the GR motif in which no editing was apparent in the ADAR1 p.110-overexpressing HeLa cells (Table 1; Figure S6). Both mCherry and EGFP expression were observed for all samples transfected with both types of gRNA.
Highest editing was seen in both ADAR1 p.110 and ADAR2 cells treated with the USH2A gRNA without the GR motif. For samples where RNA editing was observed, an average of 30.0% (n = 3) and 31.2% (n = 5) editing was observed in Sanger sequencing and NGS, respectively, for ADAR1 p.110-expressing cells compared with an average of 29.5% (n = 5) and 20.5% (n = 4) in ADAR2-expressing HeLa cells (Table 1). For samples transfected with USH2A gRNAs with an GR motif, the only levels of editing noted were in ADAR2-expressing HeLa cells with an average of 10.4% (n = 4) and 9% (n = 4) editing in Sanger sequencing and NGS, respectively (Table 1).
Both minigene reporter constructs containing nonsense mutations in the FAM161A gene (c.880A>T, p.K294∗) and KIZ gene (c.226C>T, p.R76∗) were less amenable to ADAR editing, probably because of a neighboring guanosine, 5′ to the target adenosine, previously described as being challenging to edit due hinderance of necessary base flipping for deaminization.28 Cell lines transfected with the mutant FAM161A gene fragment-containing reporter construct and treated with the 60-mer gRNA showed an average editing of 1.7% (n = 2) and 2.5% (n = 2) in Sanger sequencing and NGS, respectively, in ADAR1-overexpressing HeLa cells compared with 5.2% (n = 2) and 4.7% (n = 2) for ADAR2-overexpressing HeLa cells (Table 1). EGFP fluorescence was noted in some of the wells (Figure S4). Targeted editing of RNA transcribed from the KIZ reporter construct proved challenging as well. Among ADAR1-expressing HeLa cells treated with the KIZ gene fragment-containing reporter construct and 60-mer gRNA, where RNA editing was observed, an average of 3.3% (n = 2) and 7.4% (n = 6) of target adenosine editing was measured in Sanger sequencing and NGS, respectively, while an average of 9.9% (n = 3) and 7.8% (n = 6) editing was measured in ADAR2-expressing treated HeLa cells (Table 1). The same mammalian reporter system expressing ADAR2 was used to test the editing efficiency of the three library-identified gRNAs “B4,” “B12,” and “TT” (Figure 5C) complementary to the KIZ target with identified mismatches. One of the three tested gRNAs (“TT”) showed a 1.4× increase in editing compared with the original gRNA (Figure S7).
Off-target editing levels are also sequence dependent
A collateral consequence of using endogenous ADAR for RNA editing can be off-target editing affecting neighboring adenosines. Just as the editing levels of the RNA differed on the basis of gene and mutation, so too did the resulting off-target editing levels (Figure 7). TRPM1 has 16 potential off-target sites within the complementary region, 4 of them have a G 5′ nucleotide neighbor to the A and were unperturbed in both ADAR1 p.110-overexpressing and ADAR2-overexpressing samples, with only 2 sites and 5 sites showing levels of editing more than 2× background respectively, with an average of 1.1% and 2.9% for the adenosines that were edited (corrected for background). Each off-target adenosine that was edited would cause an amino acid change in that particular transcript. KIZ had the highest levels of off-target editing, while FAM161A and TRPM1 had almost none and minimal off-target editing, respectively. USH2A has 17 potential off-target sites; 7 are upstream to the target and 10 downstream. Only 3 sites show significant levels of editing with a maximum of 13%, 7 nt downstream to the target when the target averaged 29% editing. Moreover, when analyzing the off-target effects of the data obtained in yeast and comparing the two gRNAs used for the USH2A and TRPM1 variants (60nt-SpD or 30nt-SpD-GR), we observed that in the case of USH2A and hADAR1, no off-targets were detected. Interestingly, the total number of off-target sites was reduced from 4 to 2 when using 60nt-SpD and 30nt-SpD-GR respectively. Furthermore, no significant editing was detected in TRPM1 in the presence of these gRNAs and hADAR2. However, in cells expressing hADAR1, a similar effect was detected, resulting in a reduction in the total number of off-target sites from 4 to 0 (see Figure S8). These results imply that in addition to the previously described approaches,29,30,31,32 generally, shorter gRNAs can be used to reduce bystander editing. However, it may come with a cost, as in both cases, the RNA-editing efficiency of the target adenosine was negatively affected.
Figure 7.

Heatmap of average off-target and on-target editing rate for adenosines in complementarity region
Average editing level percentages are shown for TRPM1 (n = 8, n = 10), KIZ (n = 2, n = 2), FAM161A (n = 2, n = 2), and USH2A (without R/G) (n = 2, n = 2) in ADAR1 p.110-overexpressing and ADAR2-overexpressing HeLa cells, and USH2A (with GR) in ADAR2-overexpressing HeLa cells, corrected for NGS sequencing noise. Positions below the heatmap are relative to the target adenosine (0), bases in gray are non-adenosine bases, and adenosines with 0% editing are white, gradually increasing to deep green in correlation with increasing editing levels.
Discussion
IRDs are a set of devastating diseases that can cause blindness, with at least 5.5 million individuals affected worldwide.16 As the injured photoreceptors cannot regenerate, curing retinal degeneration is an extremely challenging task. Indeed, of the many therapeutic modalities developed and preclinically tested, there is currently only one US Food and Drug Administration (FDA)-approved treatment, namely, AAV-based gene therapy, which is suitable for but one of the 277 genes found to be mutated in the >50 IRD phenotypes. Here we propose a personalized approach for curing IRDs: applying RNA editing mediated by ADARs to target nonsense IRD mutations. Given that most adenosines are not likely to occur in a proper structure for editing, a main challenge is to design antisense gRNAs to artificially create an editable dsRNA structure around a user-defined target to redirect hADAR activity to this site. However, the rules for the construction of these elements are only partially known. To address this issue, we established for the first time a gRNA screening pipeline that is based on a powerful yeast-based initial screening system, followed by validation steps in a dual reporter system using HeLa cells which enables faster, easier, and cheaper approaches that can be tailored for the screening of a large number of candidate gRNAs to uncover those that are particularly more efficient in editing selected targets.
Yeast cells have been previously used as a model system to explore ADAR functionality and preferences. These studies have focused on hADARs and identified a novel set of editing substrates.33,34 Furthermore, the co-expression of hADARs with selected substrates has allowed monitoring the effects of mutations within the ADAR domains or the target dsRNA structure on the editing levels.35,36 These studies nicely demonstrated that ADAR enzymes can be expressed in yeast and carry out their biological function. Thus it was not surprising that the strains expressing the KIZ-p.R76∗ and FAM161A p.R523∗ mutations showed minimal growth and were difficult to edit. These mutants carry guanosines at the 5′ end of the target adenosine, which are largely depleted from the 5′ end of natively edited adenosines,23 most probably because of a steric clash between the 2-amino group of the 5′ guanosine and G489 of ADAR2 loop involved in stabilizing the flipped-out conformation required for the adenosine deamination reaction.7 This concept is supported experimentally by a recent study,26 as described in the control experiment outlined in Figure 4. By introducing a G:G mismatch at the 5′ end of the target adenosine, we observed a notable enhancement of editing, in line with the predictions made in this study that such mismatch should solve this steric problem.
This together with the clear correlation between the growth and editing levels in yeast and HeLa cells nicely demonstrate that the yeast-based screening system can be used as an efficient in-vitro platform for testing and optimizing selected “specificity domains” and recruitment elements. However, it is important to emphasize that the yeast system does not replicate the endogenous dsRNA structure surrounding the target adenosines. Furthermore, in addition to the ADARs’ dsRBDs, the human genome encodes for more than 1,000 RNA-binding proteins (RBPs), 16 of which contain dsRBDs that may directly compete with ADAR binding depending on the cellular context.37,38,39 Thus, it is crucial to highlight the significance of assessing and validating the results obtained in our yeast and HeLa cell pipeline in later stages, using model systems that mirror the endogenous dsRNA structure, and the dsRBP expression profile of the therapeutically relevant target cell.
Indeed, we have identified three gRNAs, termed “B4,” “B12,” and “TT,” that exhibited enhanced activity in yeast. Interestingly, this enhancement was replicated in mammalian cells only for the “TT” gRNA. Considering the aforementioned possibilities, it is plausible that only the bulge present in “TT,” a portion of which is also found in “B4” and “B12,” plays a crucial role in editing activity. Moreover, it is conceivable that the additional bulges in “B4” and “B12” might actually hinder their performance in mammalian cells. This specific hypothesis will be further explored in our forthcoming studies.
The pipeline we describe here is suitable mainly for nonsense mutations and in rare cases might be adapted for other type of mutations by shifting the frame of the target sequence to create a nonsense mutation around the target “A” if is surrounded by TAA, TGA, or TAG. Nonsense mutations are responsible for 18% of IRD-causing mutations11 and 11% of inherited diseases in humans,40 leaving a relatively large number of mainly missense and splice-site mutations that cannot be studied using this pipeline. Therefore additional screening systems need to be developed aiming to identify the most efficient gRNAs for each mutation using genetic libraries as gRNAs.41 Unlike the extremely large number of gRNAs that can be tested using the yeast screening tool described here, cloning-based libraries of gRNAs in human cells are limited by the number of studied inserts and therefore will allow only a more restricted analysis of missense and splice-site mutations.
The yeast model system described here is based on the dynamic range of the URA3 reporter gene, which leads to a WT-like growth of treated mutations even with a low editing level of about 12% editing. This feature will not enable us to distinguish between gRNAs with very high efficiency (more than 50%) to those in the lower range of 20%–25% editing. To overcome this issue, we are currently screening for other auxotrophic markers or fluorophores that will serve as alternative reporter genes.
Three out of the four nonsense mutations we studied are amenable for RNA editing by recoding nonsense to a missense variant, while only the USH2A nonsense mutation can be fully corrected by ADAR RNA editing. This distribution reflects nonsense mutations in general and therefore one needs to verify that the newly coded amino acid variant is not pathogenic. As functional assessment of protein isoforms is usually limited and is currently not available for most disease-causing proteins, other measures need to be taken to decrease the chances for such risk. We therefore selected genes (KIZ, FAM161A, and TRPM1) with either null reported mutations only or with a limited number of reported missense mutations. The encoded proteins are therefore likely to tolerate missense changes. This can be further validated by functional rescue of the retinal phenotype in an appropriate knockin mouse model. Currently only one such model is available42 for the FAM161A nonsense mutation. On the other hand, some of the major genes that cause retinal degeneration when mutated in humans do not cause a retinal phenotype in mouse models, therefore limiting the abilities of proving functional disease restoration.
One of the major concerns regarding nucleic acid manipulation as a therapeutic approach is the possibility of off-target effect and mainly non-targeted coding regions that might be edited by such manipulations.43 Although this concern is mainly valid for DNA editing using externally augmented proteins, such as CRISPR-Cas9, that might cause permanent DNA sequence abnormalities, ADAR-based RNA editing can only create missense changes,44,45 mainly in the vicinity of the targeted A, and not null mutations. In addition, RNA editing is temporary and can be tuned at any stage. Here we performed an off-target analysis of the gRNA region and show that once editing of the target “A” is efficient, off-targets are rare with extremely limited levels. On the other hand, if the target “A” shows limited editing levels, other “A”s in its vicinity might undergo relatively high editing. Our data also show that for both target and non-target “A”s, similar rules might apply and the editing of both might be low if the “A” is preceded by a “G” nucleotide. This is only one example of challenging sequences for editing and recent studies that are based on sequence libraries unravel potential sequence modifications that might overcome these challenges.41
The systems used in the present study use artificially expressed ADAR in both yeast and HeLa cell lines. Moving toward therapeutic options will require either recruiting endogenous ADAR or using AAV to deliver an exogenous ADAR to retinal cells. It is therefore difficult to predict how well the results obtained in the present study will be translated into editing levels in vivo. However, as the expression levels of ADAR2 in neurons generally, and in retinal cells particularly, are relatively high, one may hypothesize that RNA editing in photoreceptor cells will reach the required level to stop disease progression.
In summary, we describe here a pipeline for efficient design of gRNAs for RNA editing of IRD mutations. Together with previous preliminary studies,46,47 we provide here proof of concept that RNA editing might serve as an excellent therapeutic tool for genetic diseases. The next major challenge would be to obtain meaningful RNA editing levels in retinal cells using endogenous ADAR enzymes.
Materials and methods
Yeast strains
All the strains used in this study are isogenic to the diploid strain BY4743 (MATa/α ura3Δ0/ura3Δ0 leu2Δ0/leu2Δ0 his3Δ1/his3Δ1 lys2Δ0/LYS2 met15Δ0/MET15.48
The relevant genotypes are presented in Table S1.
Yeast plasmids
The relevant plasmids used in this study are presented in Table S1. The Gateway recombination cloning technology (Invitrogen, Waltham, MA) was used to clone the hADAR2 into the URA3 marked plasmid (pYES2-DEST52 Gateway destination vector [catalog #12286-019]; Thermo Fisher Scientific, Waltham, MA). The URA3 marker was later swapped with the LEU2 marker to create BSB641. This plasmid enables the conditional expression of the hADARs in yeast, under a galactose-inducible promoter (GAL1p-hADAR2).
The plasmid carrying the URA3 reporter gene (BSB656) was created using in vivo homologous recombination in yeast, by co-transforming the yeast URA3 gene (including its 5′ promoter region and 3′ UTR) into a linearized HIS3-marked plasmid (pRS31349) digested with XhoI/XbaI. In vivo homologous recombination was used to insert the target and reverse complement “tail” sequences (shown in Table S2) by linearizing BSB656 with either EcoRI (target insertion) or BamHI (“tail” insertion). Site-directed mutagenesis was used to insert the EcoRI (right before the URA3 start codon) and BamHI (right after the URA3 stop codon). The target and “tail” insertions were validated by PCR and Sanger sequence using OSB2366/OSB207 and OSB1202/OSB502, respectively (Table S3).
Yeast growth conditions
Yeast cells were grown in synthetic complete medium (SC; 0.17% yeast nitrogen base, 0.5% (NH4)2SO4, and amino acids), supplemented with either 2% glucose (SD), raffinose (SC+Raf), or galactose (SC+Gal), and 0.2% of either (-Histidine-Leucine) or (-Histidine-Leucine-Uracil) amino acid mix. Unless otherwise stated, cells were grown at 30°C. For logarithmic culture, cells were grown for 16–18 h and then back diluted 10× with fresh media and allowed to grow for the indicated time.
Yeast cell growth assays
To measure cells viability, exponentially growing cells were normalized to a density of 1 × 106 cells/mL into a 96-well plate containing selective medium supplemented with galactose to induce hADAR genes expression and incubated for 24 h at 30°C while shaking. Cell growth was then determined by measuring the absorbance at 600 nm in 30 min intervals using a Tecan Spark 10M multimode microplate reader. The area under the curve (AUC) was calculated using the Growthcurver package in R.
Yeast NGS
RNA samples were extracted from yeast cells using the MasterPure Yeast RNA purification Kit (Epicentre Technologies, Madison, WI). cDNA was synthesized using ProtoScript II Reverse transcriptase (OSB2798; NEB, Ipswich, MA). Then the IRD fragments were amplified using 15 PCR cycles, with specific primers for the relevant region (forward primers OSB2795-7 and OBS2840) and universal reverse primer (OSB2798). The primers included an overhang partial adapter for the UD indexes (sp p5 and sp p7). Adapters for Illumina NGS were added by 8 PCR cycles using the IDT for Illumina UD indexes. PCR products were then cleaned using KAPA pure beads in a 1:1 ratio, followed by DNA quantification and quality check using Qubit and tapestation, respectively. Finally, the samples were subjected to NGS by Illumina sequencing.
Library of gRNA construction and screening process
A library of random gRNA sequences was created using single-stranded DNA primers ordered from Integrated DNA Technologies (IDT). PCR amplification was performed using universal primers flanking the random sequences (OSB2874+2875). The resulting DNA fragments were co-transformed with a BamHI-digested plasmid containing the selected minigene into a strain carrying the hADAR2 plasmid. This step resulted in the integration of the PCR fragment at the 3′ UTR of the URA3 reporter gene through in vivo homologous recombination. The resulting yeast transformants, each carrying a unique gRNA, were pooled together and subjected to successive rounds of selection steps under uracil starvation in the presence of galactose (SC+GAL-HIS-LEU-URA). Next, to identify the gRNA sequences responsible for enhancing editing and stimulating induced growth of cells in the pooled culture, we serially diluted a sample from the library and plated single cells on a medium that selects for the plasmid harboring the gRNAs and hADAR2 (SD-HIS-LEU). Plasmids harboring the gRNAs that facilitated growth in the selected colonies were determined through Sanger sequencing of the PCR products, using universal primers flanking the insertion sites of the gRNA-encoding DNA (OSB502 and 1202).
Stable ADAR1 p.110-expressing and ADAR2-expressing HeLa cell line
HeLa cells conditionally overexpressing ADAR1 or ADAR2 were created using a CRISPR-CAS9-mediated system for the insertion of a doxycycline (Dox)-induced ADAR1 and ADAR2 cassette into the AAVS1 locus in HeLa cells.50
Reporter plasmids
An insert (ranging in size from 152 to 233 nt) harboring a cDNA fragment flanking the four studied mutations (TRPM1: c.880A>T, p.K294∗; FAM161A: c.1567C>T, p.R523∗; KIZ: c.226C>T, p.R76∗; USH2A: c.11864G>A, p.W3955∗) was cloned into the reporter plasmid expressing mCherry and EGFP (#86639; Addgene, Watertown, MA) in frame with the fluorescent proteins. Cloning was performed using restriction enzymes KpnI (#00914286; Thermo Fisher Scientific) and BamHI-HF (#R3136; New England Biolabs, Ipswich, MA) and subsequently ligated and transformed into DH5α-HIT-competent cells (#RH618; RBC, New Taipei City, Taiwan). Plasmids were purified using a plasmid purification kit (QIAprep Spin Miniprep Kit [#2714]; Qiagen, Hilden, Germany). Sequences of the cloned products were verified using Sanger sequencing.
gRNA generation
The chemically modified 73-mer gRNA (4 nmol) with an 18-nt-long complementarity region and a 55 nt GR motif tail targeting the specific adenosine in GAPDH cloned into the reporter plasmid was ordered from Biospring (Frankfurt, Germany) as previously described by Merkle et al.27 for the validation of the reporter system.
All chemically modified gRNAs used for the editing of IRD mutations were purchased as high-performance liquid chromatography (HPLC)-cleaned, 60-mer RNAs at a 4–6 nmol scale from Biospring. Both the 5′ and 3′ ends of the gRNA contained three 2′ O-methyl bases and two phosphorothioate bonds. All bases were complementary to the target sequence except for the thirtieth base, to be directly across from the target adenosine, which was a cytosine, creating a cytosine-adenosine mismatch (sequences are shown in Table S4).
In vitro editing in HeLa cells
Editing assays were performed with the stable ADAR-overexpressing HeLa cell line under transient expression controlled by the introduction of Dox, reporter plasmids, and chemically modified 60-mer gRNAs.
HeLa cells were cultured in DMEM + 10% fetal bovine serum (FBS) + 1% penicillin/streptomycin (P/S) (100 U/mL penicillin and 100 μg/mL streptomycin) + 1% L-glutamine. For editing, 4.0 × 104 of either ADAR1 p.110-expressing or ADAR2-expressing HeLa cells were seeded into 24-well plates in 500 μL DMEM + 10% FBS + 1% P/S (100 U/mL penicillin and 100 μg/mL streptomycin) + 1% L-glutamine + 1 ng/mL Dox. After 24 h, medium and Dox was changed and the reporter plasmid was transfected into each well; 50 μL Opti-MEM I Reduced-Serum Medium was mixed with 0.5 μg plasmid DNA and 1.0 μL TransfeX (AATC, Atlanta, GA) reagent, incubated at room temperature for fifteen minutes, and then distributed dropwise to each well. After 48 h, medium and Dox was changed, and the gRNA was transfected into each well; 1.5 μL Lipofectamine RNAiMAX (Thermo Fisher Scientific) and 25 pmol gRNA were each diluted in a total volume of 50 μL OptiMEM. After each solution was incubated for five minutes, the two solutions were combined, and after another 20 min incubation, the 100 μL transfection mix was evenly distributed into each well. After 72 h, the cells seeded on 10 mm coverslips were fixated using 4% paraformaldehyde (PFA) and mounted onto slides using DAPI mounting solution or harvested for RNA isolation using the Quick-RNA Miniprep Kit (Zymo Research, Irvine, CA) and Sanger sequencing as well as NGS.
Off-target analysis of NGS data
Binary alignment map (BAM) files were created by aligning the corresponding FASTQ files to a manually generated fasta file containing the insert sequence using bowtie2 (https://usegalaxy.org). Simultaneous analysis of NGS-generated samples was done using an RStudio code with BAM files used as input and nucleotide count at each reference sequence adenosine position as output. For each off-target site, the percentage of A-G in the negative control was deducted from the corresponding value in each sample, followed by heatmap generation.
Data and code availability
All data are available by request.
Acknowledgments
This work was supported by the Israel Science Foundation (grant 1778/20), the Foundation Fighting Blindness (grants TA-GT-0620-0790-HUJ and PPA-0923-0865-HUJ to S.B.-A., E.B., E.Y.L., and D.S.), and the STEP- GTP fellowship and the Ariane de Rothschild Women Doctoral Program (to N.S.). E.Y.L. was supported by grants from the Israeli Ministry of Science (grant 3-17916) and by the Israel Science Foundation (grant 2039/20 and 2637/23).
Author contributions
Conceptualization, N.S., R.S., A.B.D., J.V., E.B., E.Y.L., D.S., and S.B.-A.; data curation, N.S., R.S., A.B.D., and J.V.; formal analysis, N.S., R.S., A.B.D., and J.V.; funding acquisition, E.B., E.Y.L., D.S., and S.B.-A.; investigation, N.S., R.S., A.B.D., and J.V.; methodology, D.S. and S.B.-A.; software, J.V. and Z.R.; supervision, E.B., E.Y.L., D.S., and S.B.-A.; visualization, N.S., R.S., A.B.D., J.V., Z.R., D.S., and S.B.-A.; writing – original draft, N.S., R.S., S.B.-A., and D.S.; writing – review & editing, N.S., R.S., A.B.D., J.V., E.B., E.Y.L., D.S., and S.B.-A.
Declaration of interests
S.B.-A. and E.Y.L. are inventors of a filed patent based on the work published here: “Composition and Methods for Identifying Antisense Guide RNA for RNA Editing” (7632-PCT | PCT/IL2021/050800, Bar-Ilan University).
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2024.102130.
Contributor Information
Dror Sharon, Email: dror.sharon1@mail.huji.ac.il.
Shay Ben-Aroya, Email: Shay.Ben-Aroya@biu.ac.il.
Supplemental information
References
- 1.Bazak L., Haviv A., Barak M., Jacob-Hirsch J., Deng P., Zhang R., Isaacs F.J., Rechavi G., Li J.B., Eisenberg E., Levanon E.Y. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014;24:365–376. doi: 10.1101/gr.164749.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Licht K., Hartl M., Amman F., Anrather D., Janisiw M.P., Jantsch M.F. Inosine induces context-dependent recoding and translational stalling. Nucleic Acids Res. 2019;47:3–14. doi: 10.1093/nar/gky1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bass B.L. RNA Editing by Adenosine Deaminases That Act on RNA. Annu. Rev. Biochem. 2002;71:817–846. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eisenberg E., Levanon E.Y. A-to-I RNA editing - Immune protector and transcriptome diversifier. Nat. Rev. Genet. 2018;19:473–490. doi: 10.1038/s41576-018-0006-1. [DOI] [PubMed] [Google Scholar]
- 5.Porath H.T., Schaffer A.A., Kaniewska P., Alon S., Eisenberg E., Rosenthal J., Levanon E.Y., Levy O. A-to-I RNA Editing in the Earliest-Diverging Eumetazoan Phyla. Mol. Biol. Evol. 2017;34:1890–1901. doi: 10.1093/molbev/msx125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Savva Y.A., Rieder L.E., Reenan R.A. The ADAR protein family. Genome Biol. 2012;13:252. doi: 10.1186/gb-2012-13-12-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matthews M.M., Thomas J.M., Zheng Y., Tran K., Phelps K.J., Scott A.I., Havel J., Fisher A.J., Beal P.A. Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat. Struct. Mol. Biol. 2016;23:426–433. doi: 10.1038/nsmb.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Polson A.G., Bass B.L., Casey J.L. RNA editing of hepatitis delta virus antigenome by dsRNA-adenosine deaminase. Nature. 1996;380:454–456. doi: 10.1038/380454a0. [DOI] [PubMed] [Google Scholar]
- 9.Meisel R. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 2021;384:e91. doi: 10.1056/NEJMc2103481. [DOI] [PubMed] [Google Scholar]
- 10.Gillmore J.D., Maitland M.L., Lebwohl D. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med. 2021;385:1722–1723. doi: 10.1056/NEJMc2114592. [DOI] [PubMed] [Google Scholar]
- 11.Buchumenski I., Roth S.H., Kopel E., Katsman E., Feiglin A., Levanon E.Y., Eisenberg E. Global quantification exposes abundant low-level off-target activity by base editors. Genome Res. 2021;31:2354–2361. doi: 10.1101/gr.275770.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gold A., Levanon E.Y., Eisenberg E. The New RNA-Editing Era - Ethical Considerations. Trends Genet. 2021;37:685–687. doi: 10.1016/j.tig.2021.04.013. [DOI] [PubMed] [Google Scholar]
- 13.Xiang Y., Katrekar D., Mali P. Methods for recruiting endogenous and exogenous ADAR enzymes for site-specific RNA editing. Methods. 2022;205:158–166. doi: 10.1016/j.ymeth.2022.06.011. [DOI] [PubMed] [Google Scholar]
- 14.Pfeiffer L.S., Stafforst T. Precision RNA base editing with engineered and endogenous effectors. Nat. Biotechnol. 2023;41:1526–1542. doi: 10.1038/s41587-023-01927-0. [DOI] [PubMed] [Google Scholar]
- 15.Schneider N., Sundaresan Y., Gopalakrishnan P., Beryozkin A., Hanany M., Levanon E.Y., Banin E., Ben-Aroya S., Sharon D. Inherited retinal diseases: Linking genes, disease-causing variants, and relevant therapeutic modalities. Prog. Retin. Eye Res. 2022;89 doi: 10.1016/j.preteyeres.2021.101029. [DOI] [PubMed] [Google Scholar]
- 16.Hanany M., Rivolta C., Sharon D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc. Natl. Acad. Sci. USA. 2020;117:2710–2716. doi: 10.1073/pnas.1913179117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rivolta C., Sharon D., DeAngelis M.M., Dryja T.P. Retinitis pigmentosa and allied diseases: numerous diseases, genes, and inheritance patterns. Hum. Mol. Genet. 2002;11:1219–1227. doi: 10.1093/hmg/11.10.1219. [DOI] [PubMed] [Google Scholar]
- 18.Pinelli M., Carissimo A., Cutillo L., Lai C.-H., Mutarelli M., Moretti M.N., Singh M.V., Karali M., Carrella D., Pizzo M., et al. An atlas of gene expression and gene co-regulation in the human retina. Nucleic Acids Res. 2016;44:5773–5784. doi: 10.1093/nar/gkw486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hajji K., Sedmík J., Cherian A., Amoruso D., Keegan L.P., O’Connell M.A. ADAR2 enzymes: efficient site-specific RNA editors with gene therapy aspirations. RNA. 2022;28:1281–1297. doi: 10.1261/rna.079266.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chalk A.M., Taylor S., Heraud-Farlow J.E., Walkley C.R. The majority of A-to-I RNA editing is not required for mammalian homeostasis. Genome Biol. 2019;20:268. doi: 10.1186/s13059-019-1873-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tan M.H., Li Q., Shanmugam R., Piskol R., Kohler J., Young A.N., Liu K.I., Zhang R., Ramaswami G., Ariyoshi K., et al. Dynamic landscape and regulation of RNA editing in mammals. Nature. 2017;550:249–254. doi: 10.1038/nature24041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levanon E.Y., Eisenberg E., Yelin R., Nemzer S., Hallegger M., Shemesh R., Fligelman Z.Y., Shoshan A., Pollock S.R., Sztybel D., et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 2004;22:1001–1005. doi: 10.1038/NBT996. [DOI] [PubMed] [Google Scholar]
- 23.Eggington J.M., Greene T., Bass B.L. Predicting sites of ADAR editing in double-stranded RNA. Nat. Commun. 2011;2:319. doi: 10.1038/ncomms1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lehmann K.A., Bass B.L. The importance of internal loops within RNA substrates of ADAR1. J. Mol. Biol. 1999;291:1–13. doi: 10.1006/jmbi.1999.2914. [DOI] [PubMed] [Google Scholar]
- 25.Harvey S.C., Köster A., Yu H., Skolnick P., Baumbarger P., Nisenbaum E.S. AMPA receptor function is altered in GLUR2-deficient mice. J. Mol. Neurosci. 2001;17:35–43. doi: 10.1385/JMN:17:1:35. [DOI] [PubMed] [Google Scholar]
- 26.Doherty E.E., Karki A., Wilcox X.E., Mendoza H.G., Manjunath A., Matos V.J., Fisher A.J., Beal P.A. ADAR activation by inducing a syn conformation at guanosine adjacent to an editing site. Nucleic Acids Res. 2022;50:10857–10868. doi: 10.1093/nar/gkac897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Merkle T., Merz S., Reautschnig P., Blaha A., Li Q., Vogel P., Wettengel J., Li J.B., Stafforst T. Precise RNA editing by recruiting endogenous ADARs with antisense oligonucleotides. Nat. Biotechnol. 2019;37:133–138. doi: 10.1038/s41587-019-0013-6. [DOI] [PubMed] [Google Scholar]
- 28.Schneider M.F., Wettengel J., Hoffmann P.C., Stafforst T. Optimal guideRNAs for re-directing deaminase activity of hADAR1 and hADAR2 in trans. Nucleic Acids Res. 2014;42:e87. doi: 10.1093/nar/gku272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reautschnig P., Wahn N., Wettengel J., Schulz A.E., Latifi N., Vogel P., Kang T.-W., Pfeiffer L.S., Zarges C., Naumann U., et al. CLUSTER guide RNAs enable precise and efficient RNA editing with endogenous ADAR enzymes in vivo. Nat. Biotechnol. 2022;40:759–768. doi: 10.1038/s41587-021-01105-0. [DOI] [PubMed] [Google Scholar]
- 30.Katrekar D., Yen J., Xiang Y., Saha A., Meluzzi D., Savva Y., Mali P. Efficient in vitro and in vivo RNA editing via recruitment of endogenous ADARs using circular guide RNAs. Nat. Biotechnol. 2022;40:938–945. doi: 10.1038/s41587-021-01171-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qu L., Yi Z., Zhu S., Wang C., Cao Z., Zhou Z., Yuan P., Yu Y., Tian F., Liu Z., et al. Programmable RNA editing by recruiting endogenous ADAR using engineered RNAs. Nat. Biotechnol. 2019;37:1059–1069. doi: 10.1038/s41587-019-0178-z. [DOI] [PubMed] [Google Scholar]
- 32.Yi Z., Qu L., Tang H., Liu Z., Liu Y., Tian F., Wang C., Zhang X., Feng Z., Yu Y., et al. Engineered circular ADAR-recruiting RNAs increase the efficiency and fidelity of RNA editing in vitro and in vivo. Nat. Biotechnol. 2022;40:946–955. doi: 10.1038/s41587-021-01180-3. [DOI] [PubMed] [Google Scholar]
- 33.Eifler T., Pokharel S., Beal P.A. RNA-Seq analysis identifies a novel set of editing substrates for human ADAR2 present in Saccharomyces cerevisiae. Biochemistry. 2013;52:7857–7869. doi: 10.1021/bi4006539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Avram-Shperling A., Kopel E., Twersky I., Gabay O., Ben-David A., Karako-Lampert S., Rosenthal J.J.C., Levanon E.Y., Eisenberg E., Ben-Aroya S. Identification of exceptionally potent adenosine deaminases RNA editors from high body temperature organisms. PLoS Genet. 2023;19 doi: 10.1371/journal.pgen.1010661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y., Havel J., Beal P.A. A Phenotypic Screen for Functional Mutants of Human Adenosine Deaminase Acting on RNA 1. ACS Chem. Biol. 2015;10:2512–2519. doi: 10.1021/acschembio.5b00711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y., Beal P.A. Probing RNA recognition by human ADAR2 using a high-throughput mutagenesis method. Nucleic Acids Res. 2016;44:9872–9880. doi: 10.1093/nar/gkw799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pfaller C.K., Li Z., George C.X., Samuel C.E. Protein kinase PKR and RNA adenosine deaminase ADAR1: new roles for old players as modulators of the interferon response. Curr. Opin. Immunol. 2011;23:573–582. doi: 10.1016/j.coi.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Freund E.C., Sapiro A.L., Li Q., Linder S., Moresco J.J., Yates J.R., Li J.B. Unbiased Identification of trans Regulators of ADAR and A-to-I RNA Editing. Cell Rep. 2020;31 doi: 10.1016/j.celrep.2020.107656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Booth B.J., Nourreddine S., Katrekar D., Savva Y., Bose D., Long T.J., Huss D.J., Mali P. RNA editing: Expanding the potential of RNA therapeutics. Mol. Ther. 2023;31:1533–1549. doi: 10.1016/j.ymthe.2023.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mort M., Ivanov D., Cooper D.N., Chuzhanova N.A. A meta-analysis of nonsense mutations causing human genetic disease. Hum. Mutat. 2008;29:1037–1047. doi: 10.1002/humu.20763. [DOI] [PubMed] [Google Scholar]
- 41.Uzonyi A., Nir R., Shliefer O., Stern-Ginossar N., Antebi Y., Stelzer Y., Levanon E.Y., Schwartz S. Deciphering the principles of the RNA editing code via large-scale systematic probing. Mol. Cell. 2021;81:2374–2387.e3. doi: 10.1016/j.molcel.2021.03.024. [DOI] [PubMed] [Google Scholar]
- 42.Matsevich C., Gopalakrishnan P., Obolensky A., Banin E., Sharon D., Beryozkin A. Retinal Structure and Function in a Knock-in Mouse Model for the FAM161A-p.Arg523∗ Human Nonsense Pathogenic Variant. Ophthalmol. Sci. 2023;3 doi: 10.1016/j.xops.2022.100229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y., Wang M., Zheng T., Hou Y., Zhang P., Tang T., Wei J., Du Q. Specificity profiling of CRISPR system reveals greatly enhanced off-target gene editing. Sci. Rep. 2020;10:2269. doi: 10.1038/s41598-020-58627-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Song B., Shiromoto Y., Minakuchi M., Nishikura K. The role of RNA editing enzyme ADAR1 in human disease. Wiley Interdiscip. Rev. RNA. 2022;13:e1665. doi: 10.1002/wrna.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Merkle T., Stafforst T. New Frontiers for Site-Directed RNA Editing: Harnessing Endogenous ADARs. Methods Mol. Biol. 2021;2181:331–349. doi: 10.1007/978-1-0716-0787-9_19. [DOI] [PubMed] [Google Scholar]
- 46.Kumar S., Fry L.E., Wang J.H., Martin K.R., Hewitt A.W., Chen F.K., Liu G.S. RNA-targeting strategies as a platform for ocular gene therapy. Prog. Retin. Eye Res. 2023;92 doi: 10.1016/j.preteyeres.2022.101110. [DOI] [PubMed] [Google Scholar]
- 47.Fry L.E., Peddle C.F., Barnard A.R., McClements M.E., MacLaren R.E. RNA Editing as a Therapeutic Approach for Retinal Gene Therapy Requiring Long Coding Sequences. Int. J. Mol. Sci. 2020;21:777. doi: 10.3390/ijms21030777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brachmann C.B., Davies A., Cost G.J., Caputo E., Li J., Hieter P., Boeke J.D. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 49.Sikorski R.S., Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qian K., Huang C.T.-L., Chen H., Blackbourn L.W., Chen Y., Cao J., Yao L., Sauvey C., Du Z., Zhang S.-C. A simple and efficient system for regulating gene expression in human pluripotent stem cells and derivatives. Stem Cell. 2014;32:1230–1238. doi: 10.1002/stem.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available by request.