

Abstract
Alkene difunctionalizations enable the synthesis of structurally elaborated products from simple and ubiquitous starting materials in a single chemical step. Carbohydroxylations of olefins represent a family of reactivity that furnishes structurally complex alcohols. While examples of this type of three-component coupling have been reported, catalytic asymmetric examples remain elusive. Here, we report an enzyme-catalyzed asymmetric carbohydroxylation of alkenes catalyzed by flavin-dependent ‘ene’-reductases to produce enantioenriched tertiary alcohols. Seven rounds of protein engineering reshapes the enzyme’s active site to increase activity and enantioselectivity. Mechanistic studies suggest that C–O bond formation occurs via a 5-endo-trig cyclization with the pendant ketone to afford an 𝛼-oxy radical which is oxidized and hydrolyzed to form the product. This work demonstrates photoenzymatic reactions involving ‘ene’-reductases can terminate radicals via mechanisms other than hydrogen atom transfer, expanding their utility in chemical synthesis.
Graphical Abstract

The tertiary alcohol motif is increasingly common in pharmaceutical and agrochemical compounds as it imbues molecules with a metabolically stable fully substituted carbon while maintaining the ability to form hydrogen bonding interactions which are beneficial for binding and specificity. 1 While nucleophilic additions to ketones represent a common strategy for preparing this motif, 2,3 the steric degeneracy of the two substituents limits the generality of these approaches. Consequently, the most selective methods transfer chirality from more easily formed enantioenriched motifs or kinetic resolutions. 4–7 Inspired by the convergence of a three-component coupling reaction, we sought to develop a catalytic asymmetric method for preparing tertiary alcohols from simple alkenes in a carbohydroxylation reaction. While there are various reports of transition metal, 8 electrochemical, 9 and photoredox methods to catalyze this type of reaction, there are no known asymmetric methods (Figure 1A). 10,11
Figure 1.
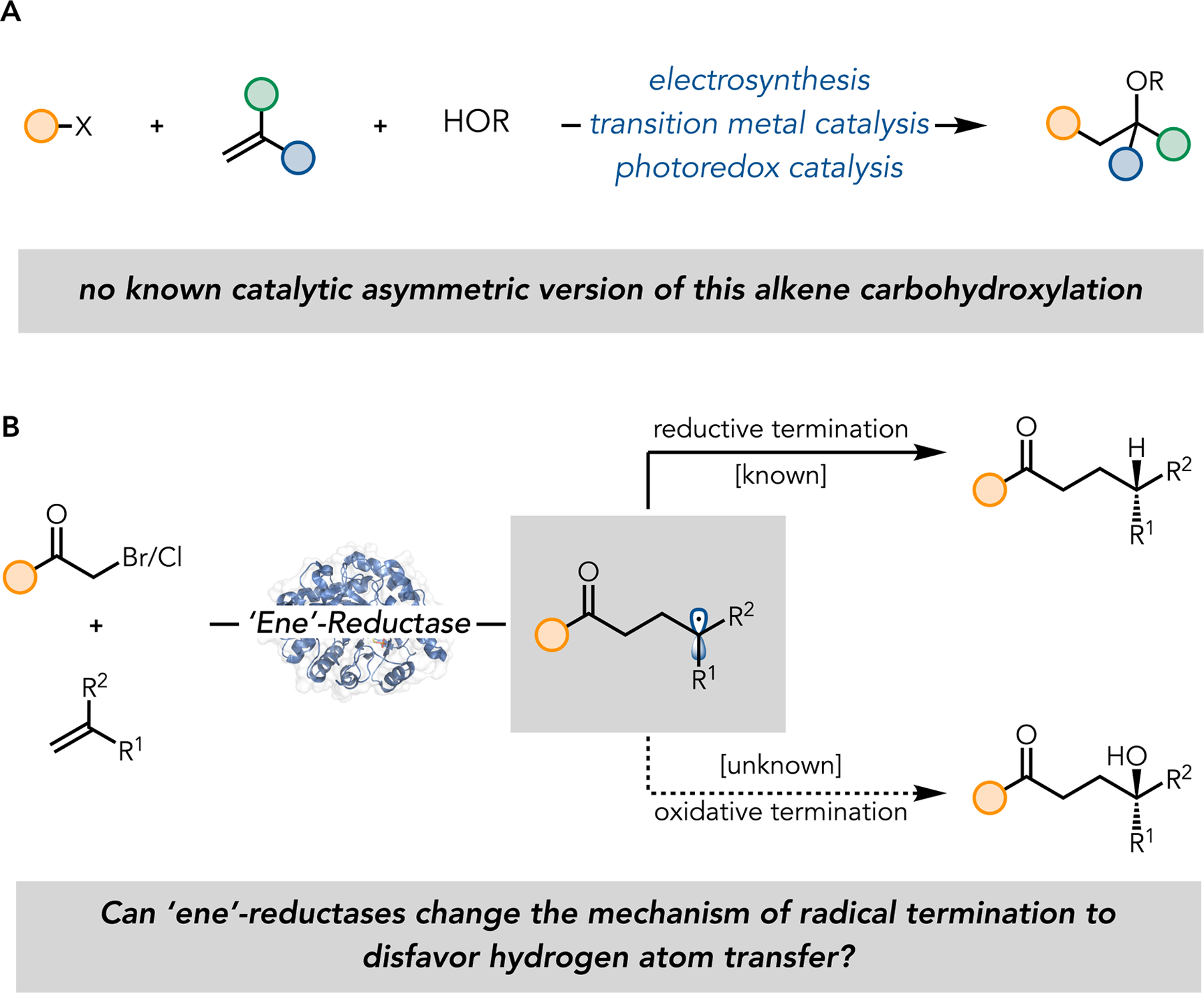
Tertiary Alcohol Synthesis and Mechanisms of Radical Termination in Photoenzymatic Catalysis. (A) Alkene difunctionalization by electrosynthesis, transition metal catalysis, and photoredox catalysis to prepare tertiary alcohols. (B) Changing the mechanism of radical termination in photoenzymatic reactions catalyzed by ‘ene’-reductases.
Enzymes are attractive catalysts for asymmetric synthesis because the protein scaffold can be evolved to tune the chemical environment for a reaction to achieve high levels of enantioselectivity. As enzymes typically catalyze only a single chemical reaction, efforts over the past decade have focused on expanding their catalytic abilities. Toward this end, our group has focused on developing new reaction mechanisms for flavin-dependent ‘ene’-reductases (EREDs). These enzymes catalyze the reduction of activated alkenes through a hydride transfer mechanism. 12–14 Our group found that the flavin cofactor in its hydroquinone (FMNhq) and semiquinone (FMNsq) oxidation state can initiate radical formation for C–C bond-forming reactions. Our group and others have exploited this initiation mode for various asymmetric reductive hydroalkylation and hydrosulfonylation reactions. 15–19 In these examples, 20–22radical termination is the enantiodetermining step and occurs via hydrogen atom transfer from FMNsq to the organic radical. 23 This pathway is favored because of the weak N–H bond strength of FMNsq (58 kcal/mol) and has limited EREDs to hydrofunctionalizations of alkenes. 24–27
To address this limitation, we questioned whether an ERED could be evolved to utilize a different mechanism of radical termination. To outcompete hydrogen atom transfer, the proteins’ structure would need to be modified to alter the kinetic profile to favor trapping via a different bond-forming event (Figure 1B). With the goal of synthesizing enantioenriched tertiary alcohols, we sought to develop an enzyme that could stereoselectively trap the radical with a nucleophilic oxygen species (Figure 1B). While we initially envisioned a radical/polar crossover to form cation which could be trapped with water, we recognized that alternative mechanisms could form the same product.
We began by looking at a carbohydroxylation of α-methylstyrene using α-bromoacetophenone as a radical precursor under anaerobic conditions based on our previous studies using flavin protein. In a previous study, we found that the EREDs from Gluconabacter (GluER) and Zymomonas mobilis (NCR) could reductively couple these compounds in good yield with excellent enantioselectivity either when irradiated with light or in the dark. 16 When reexamining these reactions under modified reaction conditions, we found small amounts of the carbohydroxylation product were formed (Table S1). After examining a broader set of EREDs under cyan light irradiation, we found that most favored the reductively coupled product while morphinone reductase from Pseudomonas putida (MorB) gave the best yield of 45% and 75:25 enantiomeric ratio, with only 10% of reductive coupling byproduct (Figure 2B and Table S1). To improve the yield and enantioselectivity, we subjected MorB to iterative saturation mutagenesis (ISM). 28,29 We began by targeting 21 residues lining the active site of MorB (Tables S3–S16). Over the first four rounds of ISM, we found a series of mutations located in the proximal binding site that increased the yield to 83% with 87:13 er. In the next round, we reinvestigated previously mutated positions and found that mutation of Y72C to threonine (C72T) led to the product in 94% yield with 92:8 er. Two additional rounds of mutagenesis led to MorB-Y72T-C191G-T240E-R110L-V73D-S34Q (MorB-B3), affording the hydroxylation product with 96% yield and 95:5 er. Notably, the hydrogen atom transfer pathway was suppressed through protein engineering, rendering the reductive coupling byproduct negligible.
Figure 2.
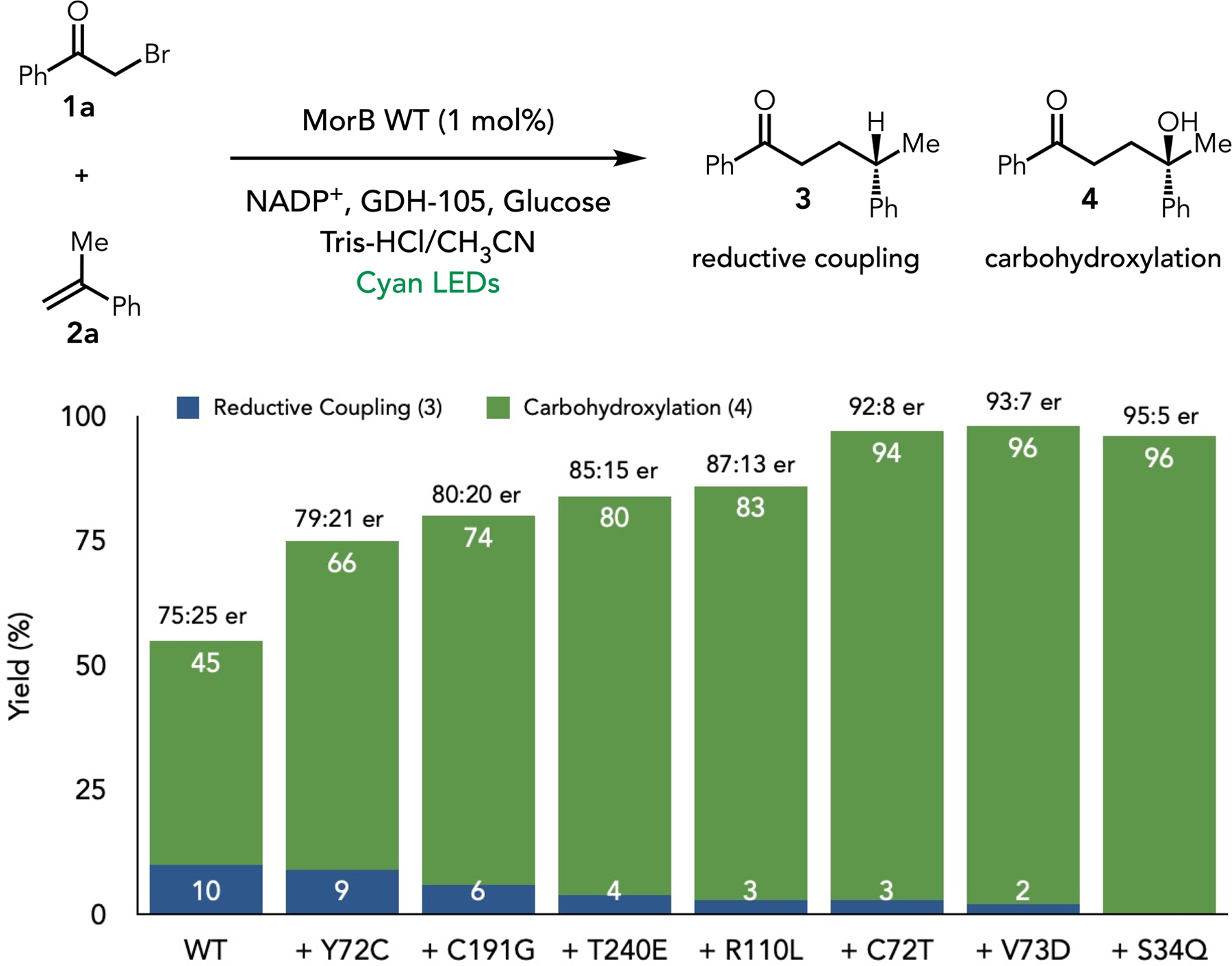
Optimization of the Photoenzymatic Carbohydroxylation. Reaction conditions: α-methylstyrene (0.65 uL, 0.01 mmol, 1.0 eq, 16.6 mM), α-bromoketone (5.94 mg, 0.03 mmol, 3 eq), GDH-105 (0.12 mg), NADP+ (0.4 mg), glucose (1.8 mg) and purified ‘ene’-reductases (1 mol% based on α-methylstyrene) in 100 mM Tris-HCl buffer pH 8.0, with 10% CH3CN (v/v) as cosolvent, final total volume is 660 μL. Reaction mixtures were stirred under anaerobic conditions and irradiated with cyan LED at room temperature for 24 h. Yield determined via LCMS relative to an internal standard (TBB). Enantiomeric ratio (er) determined by HPLC on a chiral stationary phase.
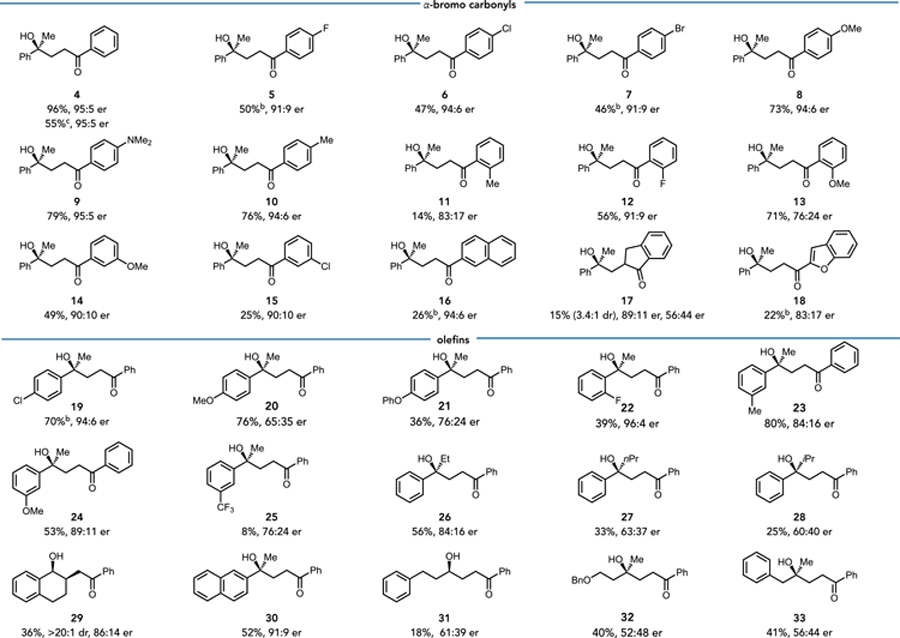
With the optimal condition in hand, we began to explore the scope and limitations of the reaction (Table 1). This enzyme is tolerant to substituents at the para- and meta- positions, albeit in slightly diminished yields by comparison to the model substrate. In general, electron-donating substituents furnished higher yields than electron-withdrawing ones. The enzyme is also tolerant of small ortho-substituents, such as fluoro- and methoxy-groups (12, 13). Even small increases in the steric bulk of the substituent lead to lower yields. The secondary bromoacetophenone form product in modest yield with 3.4:1 diastereoselectivity (17). Finally, larger aromatic groups, such as naphthyl groups and benzofurans (16, 18), result in lower yields.
Table 1.
Scope for the Photoenzymatic Carbohydroxylationa
|
Reaction conditions: α-methylstyrene 0.01 mmol, 1.0 eq, 16.6 mM), α-bromoketone (5.94 mg, 0.03 mmol, 3 eq), GDH-105 (0.12 mg), NADP+ (0.4 mg), glucose (1.8 mg) and purified ‘ene’-reductases (1 mol% based on α-methylstyrene) in 100 mM Tris-HCl buffer pH 8.0, with 10% CH3CN (v/v) as cosolvent, final total volume is 660 μL. Reaction mixtures were stirred under anaerobic conditions and irradiated with cyan LED at room temperature for 24 h. Yield determined via LCMS relative to an internal standard (TBB). Enantiomeric ratio (er) determined by HPLC on a chiral stationary phase.
1.5 mol% MorB-B3 was used.
1.0 mmol scale-up, isolated yield.
We next tested the scope of alkenes. Alkenes with various electronic substituents effectively produced the target products with moderate to excellent yields and good selectivity (19–24, 30). However, the meta electron-withdrawing groups, such as CF3, were less effective, resulting in poor yield and enantioselectivity (25). Regarding the larger group at α-position, Et, nPr, and iPr were well accommodated, although with modest yields and selectivity (26–28). Besides, the trisubstituted alkene was reactive and exhibited excellent diastereoselectivity favoring the cis isomer (29). Notably, non-styrene alkenes were also efficient, affording the hydroxylation product with moderate yields and selectivity (31–33). While this enzyme was not optimized for these interesting substrates, protein engineering can be used to improve the activity and selectivity with these substrates. The remaining mass balance in these reactions are cyclized coproducts where the nucleophilic benzylic radical underwent the radical addition to the phenyl ring of ketone to form the benzocyclohexanones (Figure S3). These products could be derivatized to other useful motifs. For instance, the ketone in the model substrate could be reduced to the corresponding 1,4-diol in a 1.2:1 diastereomeric ratio with 93:7 er (Figure S2). The chiral lactone 35 could also be accessed via Baeyer-Villiger oxidation in good yield and er (Figure S2).
Next, we conducted a series of experiments to elucidate the mechanism of this transformation. We began by investigating the mechanism of radical initiation. In the reductive coupling of alkyl halides with alkenes, we found that radical initiation occurred via electron transfer from FMNhq via an enzyme-templated charge transfer complex (CT complex). Unfortunately, this complex could not be observed via UV-Vis because FMNhq is oxidized in the presence of substrate without irradiation. Indeed, MorB-B3 can catalyze the carbohydroxylation without irradiation in 14% yield with no change in enantioselectivity (95:5 er) (Figure 3A). This observation suggests that FMNhq is responsible for initiating the reaction, with photoexcitation providing a stronger driving force for radical initiation.
Figure 3. Mechanistic Studies and Possible Mechanisms.
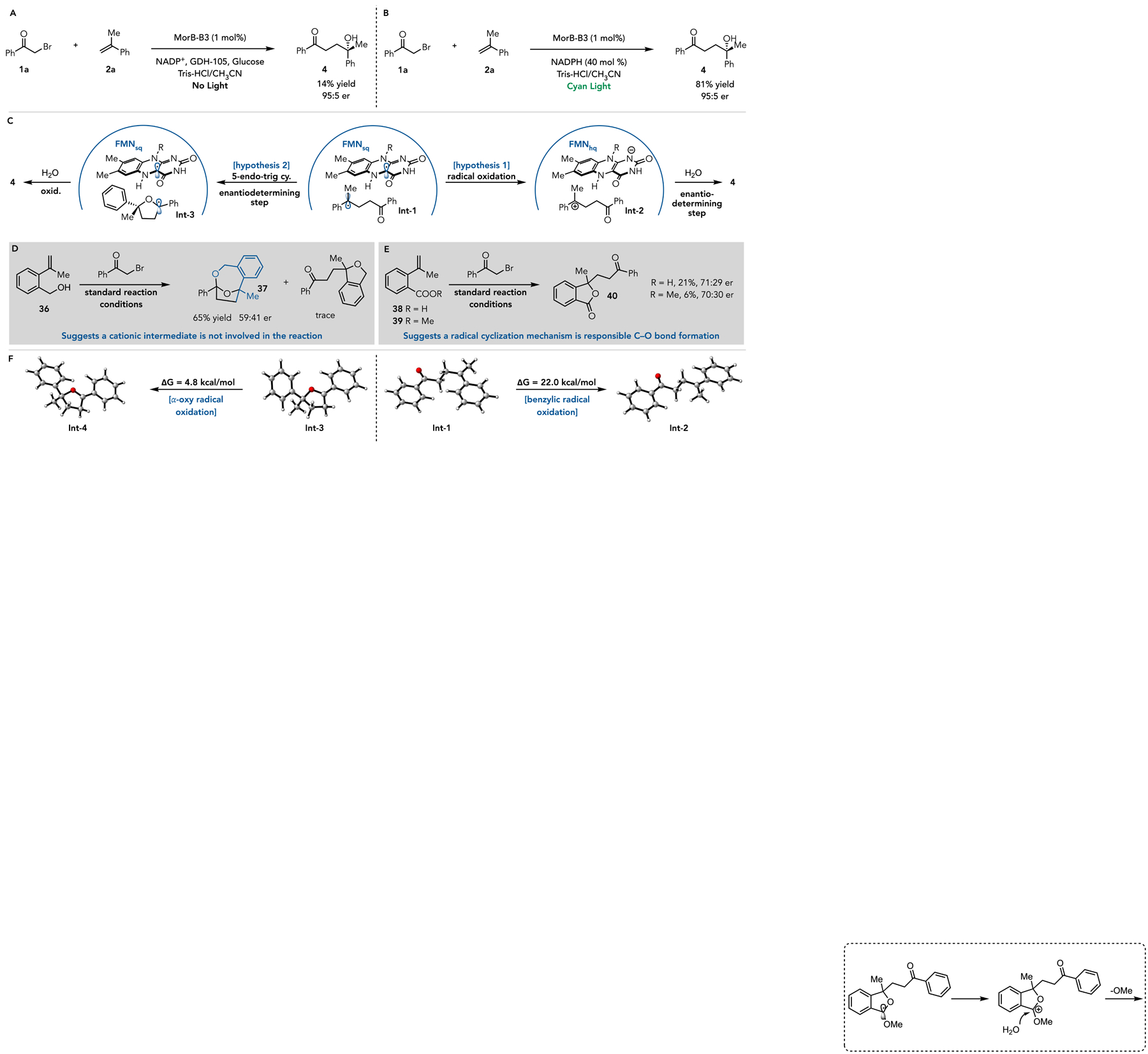
(A) Carbohydroxylation in the dark indicating a ground state electron transfer mechanism. (B) Reaction with 2 mol% NADPH suggesting that the overall reaction is redox-neutral. (C) Two possible mechanistic pathways. (D) Reactivity with an alkene bearing an alcohol suggests that radical termination does not occur via a radical/polar crossover. (E) Reactions with alkenes bearing a carboxylic acid/ester moiety suggest radical termination occurs via 5-endo-trig cyclization and oxidation of the resulting 𝛼-oxy radical. (F) Free-energy profile of possible intermediates.
Next, we turned our attention to understanding the radical termination event. Two possible pathways were considered: i) the benzylic radical Int-1 is oxidized by FMNsq to form the acyclic cation Int-2 which is trapped by water to form product, or ii) the radical engages in a 5-endo-trig cyclization with ketone carbonyl to form an 𝛼-oxy radical Int-3 which is oxidized by FMNsq to form an oxocarbenium ion which is hydrolyzed to form the product (Figure 3C). In both cases, the reaction is redox-neutral and thus only requires a catalytic amount of reductant. Indeed, when the cofactor turnover system is replaced with 40 mol % NADPH, the product is formed in 81% yield with 95:5 er (Figure 3B). To distinguish between the two mechanisms, we prepared an alkene 36 containing a pendant alcohol. We hypothesized that if a cation were formed, competitive cyclic ether formation would be observed. However, when 36 was subjected to the reaction conditions, the cyclic ether was not observed. Instead, a [4.2.1] bicyclic acetal 37, derived from the product of carbohydroxylation, was generated in 65% yield with 59:41 er (Figure 3D). This result suggests that a radical/polar crossover mechanism is not responsible for radical termination. Next, we prepared substrates with either a carboxylic acid 38 or ester 39 at the ortho-position of the arene. The hypothesis is that 𝜋-electrons in the carbonyl could react with the radical, leading to an 𝛼-oxy radical which, upon oxidation, would form a lactone. Indeed, when using the carboxylic acid, the lactone is formed in 21% yield with 71:29 er. The ester was less effective, providing the same lactone in only a 6% yield with 70:30 er (Figure 3E). Formation of the lactone product suggests that 𝜋-electrons are required for cyclization, indicating that a cation is not a likely intermediate in this reaction. As further support for this mechanism, we performed DFT calculations to compare the differences in Gibbs free energy for oxidizing the benzylic radical Int-2 versus the 𝛼-oxy radical Int-3. We found that oxidation of benzylic radical was uphill byΔG = 22.0 kcal/mol (Figure 3 and Figures S5–S9), while oxidation of the 𝛼-oxy radical Int-3 to the corresponding oxycarbenium ion Int-4 was only uphill by byΔG = 6.6 kcal/mol, which indicated the radical oxidation could be assisted by 𝜋-system of carbonyl. Collectively, these results are most consistent with a mechanism of radical termination where the carbonyl serves as a 𝜋-system for a 5-endo-trig cyclization.
We have developed an asymmetric synthesis of tertiary alcohols via a photoenzymatic alkene carbohydroxylation. While this reaction and the previously reported alkene hydroalkylation involve the same radical initiation step and benzylic radical intermediate, this evolved enzyme favors C–O bond formation over hydrogen atom transfer for radical termination, highlighting the ability of proteins to precisely control the kinetic profile of reactions. Moreover, this unique approach for alkene carbohydroxylation highlights the opportunity for enzymes to unlock new mechanisms for previously developed transformations.
Supplementary Material
ACKNOWLEDGMENT
The research reported here was supported by the National Institutes of Health National Institute of General Medical Sciences (R01 GM127703). Yao Ouyang acknowledges the SIOC Scholarship from Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences for financial support. This work made use of the Cornell University NMR Facility, which is supported, in part, by the NSF though MRI Award CHE-1531632. The authors thank Dr. Felix C. Raps for mechanism discussion.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Experimental procedures, characterization data, NMR spectra, and HPLC traces. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interests.
REFERENCES
- (1).Cramer J; Sager CP; Ernst B Hydroxyl Groups in Synthetic and Natural-Product-Derived Therapeutics: A Perspective on a Common Functional Group. J. Med. Chem 2019, 62, 8915–8930. [DOI] [PubMed] [Google Scholar]
- (2).Liu YL; Lin X-T Recent Advances in Catalytic Asymmetric Synthesis of Tertiary Alcohols via Nucleophilic Addition to Ketones. Adv. Synth. Catal 2019, 361, 876–918. [Google Scholar]
- (3).Ruan L-X; Sun B; Liu J-M; Shi S-L Dynamic Kinetic Asymmetric Arylation and Alkenylation of Ketones. Science 2023, 379, 662–670. [DOI] [PubMed] [Google Scholar]
- (4).Stymiest JL; Bagutski V; French RM; Aggarwal VK Enantiodivergent Conversion of Chiral Secondary Alcohols into Tertiary Alcohols. Nature 2008, 456, 778–783. [DOI] [PubMed] [Google Scholar]
- (5).Seliger J; Dong X; Oestreich M Kinetic Resolution of Tertiary Propargylic Alcohols by Enantioselective Cu−H-Catalyzed Si−O Coupling. Angew. Chem. Int. Ed 2019, 58, 1970–1974. [DOI] [PubMed] [Google Scholar]
- (6).Liao K; Gong Y; Zhu RY; Wang C; Zhou F; Zhou J Highly Enantioselective CuAAC of Functional Tertiary Alcohols Featuring an Ethynyl Group and Their Kinetic Resolution. Angew. Chem. Int. Ed 2021, 60, 8488–8493. [DOI] [PubMed] [Google Scholar]
- (7).Wang G; Li L; Jiang Y; Zhao X; Ban X; Shao T; Yin Y; Jiang Z Kinetic Resolution of Azaarylethynyl Tertiary Alcohols by Chiral Brønsted Acid Catalysed Phosphine-Mediated Deoxygenation. Angew. Chem. Int. Ed 2023, 62, e20221483. [DOI] [PubMed] [Google Scholar]
- (8).Wang Q; Shi P; Zeng R Copper(I) Reagent-Promoted Hydroxytrifluoromethylation of Enamides: Flexible Synthesis of Substituted-3-Hydroxy-2-Aryl-3-(2,2,2-Trifluoro-1-Arylethyl)Isoindolin-1-One. RSC Adv 2018, 8, 25961–25965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Xiong P; Long H; Song J; Wang Y; Li JF; Xu HC Electrochemically Enabled Carbohydroxylation of Alkenes with H 2 O and Organotrifluoroborates. J. Am. Chem. Soc 2018, 140, 16387–16391. [DOI] [PubMed] [Google Scholar]
- (10).Yan D; Xu S; Qian H; Gao P; Bi M; Xiao W; Chen J Photoredox-Catalyzed and Copper(II) Salt-Assisted Radical Addition/Hydroxylation Reaction of Alkenes, Sulfur Ylides, and Water. ACS Catal 2022, 12, 3279–3285. [Google Scholar]
- (11).Altmann LM; Zantop V; Wenisch P; Diesendorf N; Heinrich MR Visible Light Promoted, Catalyst-Free Radical Carbohydroxylation and Carboetherification under Mild Biomimetic Conditions. Chem. - A Eur. J 2021, 27, 2452–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Winkler CK; Tasnádi G; Clay D; Hall M; Faber K Asymmetric Bioreduction of Activated Alkenes to Industrially Relevant Optically Active Compounds. J. Biotechnol 2012, 162, 381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Winkler CK; Faber K; Hall M Biocatalytic Reduction of Activated C[Dbnd]C-Bonds and beyond: Emerging Trends. Curr. Opin. Chem. Biol 2018, 43, 97–105. [DOI] [PubMed] [Google Scholar]
- (14).Durchschein K; Hall M; Faber K Unusual Reactions Mediated by FMN-Dependent Ene- and Nitro-Reductases. Green Chem 2013, 15, 1764–1772. [Google Scholar]
- (15).Page CG; Cooper SJ; Dehovitz JS; Oblinsky DG; Biegasiewicz KF; Antropow AH; Armbrust KW; Ellis JM; Hamann LG; Horn EJ; Oberg KM; Scholes GD; Hyster TK Quaternary Charge-Transfer Complex Enables Photoenzymatic Intermolecular Hydroalkylation of Olefins. J. Am. Chem. Soc 2021, 143, 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Fu H; Lam H; Emmanuel MA; Kim JH; Sandoval BA; Hyster TK Ground-State Electron Transfer as an Initiation Mechanism for Biocatalytic C-C Bond Forming Reactions. J. Am. Chem. Soc 2021, 143, 9622–9629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Chen X; Zheng D; Jiang L; Wang Z; Duan X; Cui D; Liu S; Zhang Y; Yu X; Ge J; Xu J Photoenzymatic Hydrosulfonylation for the Stereoselective Synthesis of Chiral Sulfones. Angew. Chem. Int. Ed 2023, 62, e202218140. [DOI] [PubMed] [Google Scholar]
- (18).Biegasiewicz KF; Cooper SJ; Gao X; Oblinsky DG; Kim JH; Garfinkle SE; Joyce LA; Sandoval BA; Scholes GD; Hyster TK Photoexcitation of Flavoenzymes Enables a Stereoselective Radical Cyclization. Science 2019, 364, 1166–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Emmanuel MA; Bender SG; Bilodeau C; Carceller JM; DeHovitz JS; Fu H; Liu Y; Nicholls BT; Ouyang Y; Page CG; Qiao T; Raps FC; Sorigué DR; Sun S; Turek-Herman J; Ye Y; Rivas-Souchet A; Cao J; Hyster TK Photobiocatalytic Strategies for Organic Synthesis. Chem. Rev 2023, 123, 5459–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Huang X; Wang B; Wang Y; Jiang G; Feng J; Zhao H Photoenzymatic Enantioselective Intermolecular Radical Hydroalkylation. Nature 2020, 584, 69–74. [DOI] [PubMed] [Google Scholar]
- (21).Fu H; Cao J; Qiao T; Qi Y; Charnock SJ; Garfinkle S; Hyster TK An Asymmetric Sp3–Sp3 Cross-Electrophile Coupling Using ‘Ene’-Reductases. Nature 2022, 610, 302–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ye Y; Cao J; Oblinsky DG; Verma D; Prier CK; Scholes GD; Hyster TK Using Enzymes to Tame Nitrogen-Centred Radicals for Enantioselective Hydroamination. Nat. Chem 2022, 15, 206–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Warren JJ; Tronic TA; Mayer JM Thermochemistry of Proton-Coupled Electron Transfer Reagents and Its Implications. Chem. Rev 2010, 110, 6961–7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Li Z-L; Fang G-C; Gu Q-S; Liu X-Y Recent Advances in Copper-Catalysed Radical-Involved Asymmetric 1,2-Difunctionalization of Alkenes. Chem. Soc. Rev 2020, 49, 32–48. [DOI] [PubMed] [Google Scholar]
- (25).Zhou Q; Chin M; Fu Y; Liu P; Yang Y Stereodivergent Atom-Transfer Radical Cyclization by Engineered Cytochromes P450. Science 2021, 374, 1612–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Rui J; Zhao Q; Huls AJ; Soler J; Paris JC; Chen Z; Reshetnikov V; Yang Y; Guo Y; Garcia-Borràs M; Huang X Directed Evolution of Nonheme Iron Enzymes to Access Abiological Radical-Relay C(Sp3)−H Azidation. Science 2022, 376 , 869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Giofrè S; Molteni L; Beccalli EM Asymmetric Pd(II)‐Catalyzed C−O, C−N, C−C Bond Formation Using Alkenes as Substrates: Insight into Recent Enantioselective Developments. European J. Org. Chem 2023, 26, e2022009. [Google Scholar]
- (28).Reetz MT; Carballeira JD Iterative Saturation Mutagenesis (ISM) for Rapid Directed Evolution of Functional Enzymes. Nat. Protoc 2007, 2, 891–903. [DOI] [PubMed] [Google Scholar]
- (29).Reetz MT Laboratory Evolution of Stereoselective Enzymes: A Prolific Source of Catalysts for Asymmetric Reactions. Angew. Chem. Int. Ed 2011, 50, 138–174. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.