

Abstract
Natural killer (NK) cells are cytotoxic lymphocytes that accumulate within the tumor microenvironment and are generally considered to be antitumorigenic. Using single-cell RNA sequencing and functional analysis of multiple triple-negative breast cancer (TNBC) and basal tumor samples, we observed a unique subcluster of Socs3highCD11b−CD27− immature NK cells that were present only in TNBC samples. These tumor-infiltrating NK cells expressed a reduced cytotoxic granzyme signature and, in mice, were responsible for activating cancer stem cells through Wnt signaling. NK cell–mediated activation of these cancer stem cells subsequently enhanced tumor progression in mice, whereas depletion of NK cells or Wnt ligand secretion from NK cells by LGK-974 decreased tumor progression. In addition, NK cell depletion or inhibition of their function improved anti–programmed cell death ligand 1 (PD-L1) antibody or chemotherapy response in mice with TNBC. Furthermore, tumor samples from patients with TNBC and non-TNBC revealed that increased numbers of CD56bright NK cells were present in TNBC tumors and were correlated to poor overall survival in patients with TNBC. Together, our findings identify a population of protumorigenic NK cells that may be exploited for both diagnostic and therapeutic strategies to improve outcomes for patients with TNBC.
INTRODUCTION
Breast cancer is the most frequently diagnosed cancer and remains the second leading cause of cancer-related death among women in the United States, with an estimated 276,480 new cases in 2020 (1). Triple-negative breast cancer (TNBC), one of the most aggressive molecular subtypes of breast tumors, is characterized by the lack of estrogen and progesterone receptors (ER/PR) as well as the absence of human epidermal growth factor receptor (HER2). TNBC accounts for 10 to 15% of all breast cancers, and its frequent recurrence and metastasis are associated with a poorer prognosis compared with other breast cancer subtypes (2). Unfortunately, the absence of ER, PR, and HER2 renders TNBC insensitive to commonly used breast cancer therapeutics. The wide degree of interpatient variability and molecular heterogeneity of TNBC tumors (3) further complicates development of “universal” targeted therapeutics for patients.
The tumor microenvironment (TME) of TNBC distinguishes it from other subtypes in that there are large numbers of tumor-infiltrating immune cells that may contribute to tumor progression through regulation of proliferation, angiogenesis, apoptosis, immune system suppression, and drug resistance (4). Phenotypic analysis of infiltrating immune cells in TNBC has prognostic utility (5, 6), suggesting that immunotherapeutic approaches targeting immune cells may have potential for improving clinical outcomes for patients with TNBC. Despite the success of immune checkpoint inhibitors in achieving durable clinical remission and prolonged survival in patients with many solid tumors, early-phase trials with monoclonal antibodies against programmed cell death ligand 1 (PD-L1) have shown very limited response in TNBC (7), possibly because only 20% of TNBC cells express PD-L1 (8). Moreover, contradictory findings from recent phase 3 clinical trials IMpassion130 and IMpassion131 (9–11) using an anti–PD-L1 antibody in combination with chemotherapy further demand the need to better understand the immune features and profiles of TNBC heterogeneity for future development of immune-modulatory strategies to stratify and target immune cells.
Natural killer (NK) cells are cytotoxic lymphocytes that can recognize and eliminate tumor cells and are therefore considered the early responders against tumors. Among various tumor-infiltrating immune cells, the presence of NK cells in the TME correlates with outcome in a variety of cancers (12–14). Although cytokine-stimulated NK cells can contribute to antitumor responses (15), the function of NK cells is affected by their activation and maturation status (16, 17). In several malignancies, intratumor NK cells differ phenotypically or functionally from peripheral NK cells (18–20). Notably, a specific subset of NK cells that do not express either CD27 or CD11b (CD11b−CD27− NK cells) has been reported to be associated with tumor progression in lung and hepatocellular cancers (21, 22); however, their function still remain elusive. In particular, the presence of specific NK cell subsets within TNBC tumors and their potential relationship to TNBC progression have not been fully characterized to date.
Single-cell RNA sequencing (scRNA-seq) has recently emerged as a powerful tool to define different cell phenotypes in the TME (23). In the present study, using scRNA-seq, we demonstrated that a distinct immature NK cell subset (CD11b−CD27−Socs3high) resides in the TME of TNBC and was associated with tumor progression and metastasis. Functionally, these tumor-associated NK cells had a greatly reduced granzyme-mediated cytolytic expression profile and increased Wnt16 ligand expression. Consequently, we found that these tumor-infiltrating NK cells supported the stemness of TNBC through up-regulation of Wnt signaling in tumor cells. Moreover, we demonstrated that combination therapy through NK cell depletion or inhibition of Wnt secretion, together with immunotherapy using anti–PD-L1 antibody or with the chemotherapy drug olaparib, reduced tumor progression. Data collected from analyzing patient samples showed a positive correlation between higher CD56 (a marker of NK cells in humans) expression and poorer overall survival (OS) of patients with TNBC. These data were further validated by immunohistochemistry (IHC) and flow cytometry analysis of human tissue specimens. Together, our preclinical murine studies along with patient samples identify a function for an immature NK cell subset in TNBC. Our data suggest that NK cell subsets may be used for patient stratification and could be harnessed for targeted immunotherapeutic approaches to improve patient outcomes.
RESULTS
scRNA-seq identified NK cell subpopulations in the microenvironment of murine TNBC tumors
TNBC cells are heterogeneous and highly immunogenic (3, 24, 25). Accumulating evidence suggests that infiltrating immune cells in the breast cancer TME can contribute to cancer progression and metastasis (26–29). To characterize the immune landscape of TNBC at a cellular level, we used a C3-T antigen (C3-T+)–based TNBC mouse model with a high rate of spontaneous metastasis (30). We carried out a scRNA-seq analysis to identify various tumor and immune cell subsets in aggressive and metastatic TNBC (C3-T+; Elf5+/−, hereafter referred to as C3-HET) and non-aggressive and low metastatic (C3-T+; Elf5+/+, hereafter referred to as C3-WT) primary tumors. Using a CD45 antibody to distinguish immune cells (CD45+) from tumor cells and fibroblasts (both CD45−), we identified 10 CD45− cell clusters (clusters 0 to 9) (Fig. 1A), including 6 epithelial clusters (clusters 0, 1, 2, 3, 4, and 9), defined by expression of Krt14, Krt18, Krt5, Krt8, and Vim, and 4 clusters (5, 6, 7, and 8) defined by expression of Sparc and Pdgfa, which we classified as fibroblasts and adipocytes, respectively (fig. S1A). Gene set enrichment analysis (GSEA) of genes expressed differentially between the epithelial clusters revealed enrichment of gene signatures associated with epithelial-mesenchymal transition (EMT), glycolysis, and hypoxia (table S1) in cell clusters 0, 1, 2, and 4, which appear increased in C3-HET tumors compared with cluster 9, which is increased in C3-WT (Fig. 1B). These data indicate an active metabolic state and propensity toward a potential EMT-like state for C3-HET tumors (31–33), supporting published data on TNBC (34–36). Expression of cancer stem cell and EMT markers such as Sox9, CD44, and Vimentin were significantly (P < 0.0001) higher in C3-HET tumor cell clusters as compared with C3-WT, suggesting stem-like properties of these tumor cells along with their aggressive, EMT-like state (Fig. 1, C to E). Furthermore, gene signatures from clusters higher in C3-HET tumors demonstrated enrichment for an immune-suppressive signature as reported in melanoma (37), compared with C3-WT cluster 9 (fig. S1B), thus suggesting an association between an immunosuppressive TME with TNBC tumors (36, 38, 39).
Fig. 1. Single-cell RNA sequencing (scRNA-seq) identifies the heterogeneity of TNBC tumors and the presence of a distinct NK cell subset.
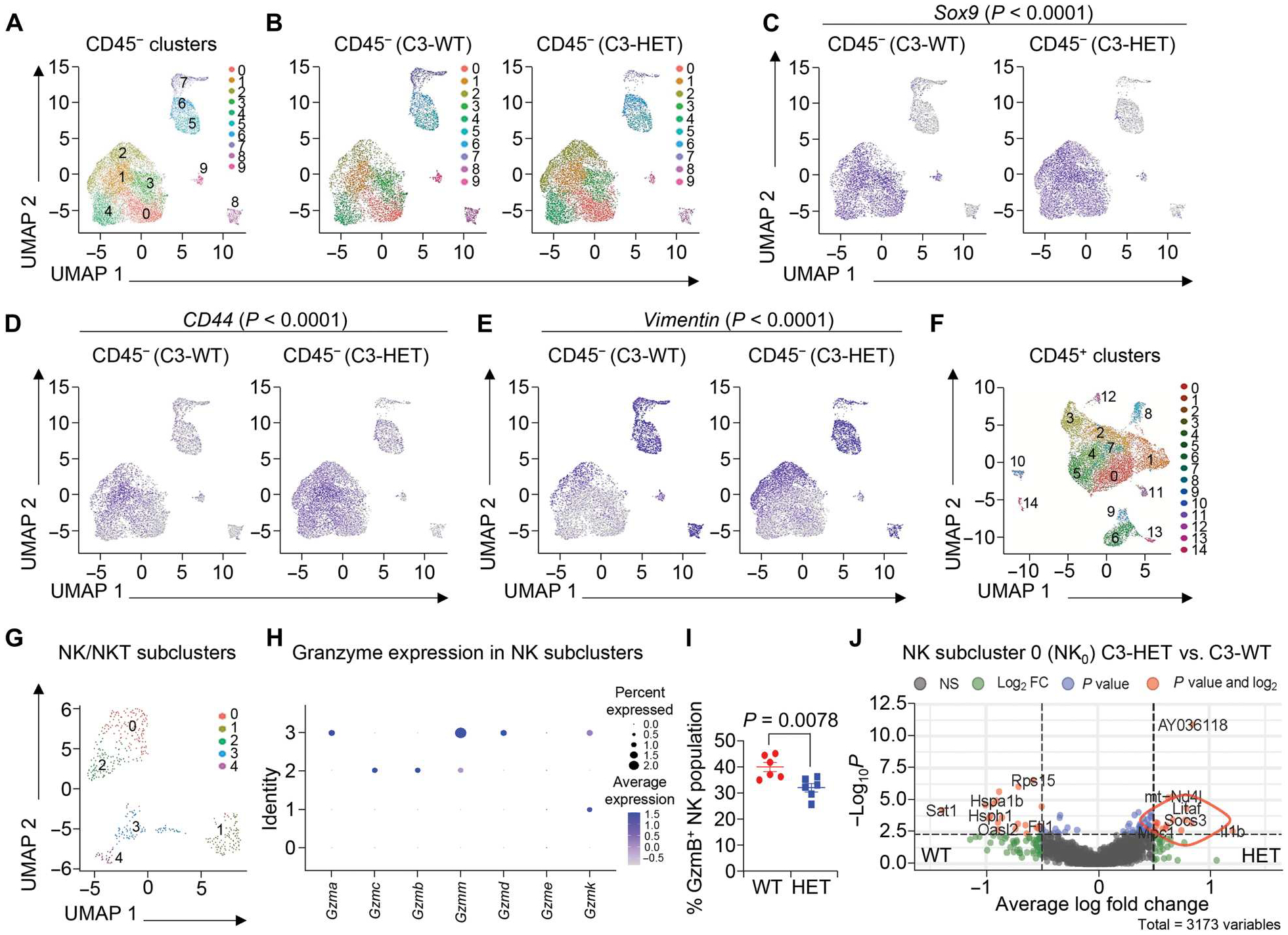
(A) Uniform manifold approximation and projection (UMAP) distribution of total CD45− cells from C3-WT (C3-T+ Elf5+/+) and C3-HET (C3-T+ Elf5+/−) tumors as 10 distinct clusters. (B) UMAPs depict the distribution and abundance of C3-WT and C3-HET CD45− cells among clusters. (C to E) Feature plots depicting the distribution and abundance of Sox9+ cells (C), CD44+ cells (D), and Vimentin+ cells (E) among C3-WT and C3-HET clusters. (F) UMAP distribution of total CD45+ immune cells from C3-WT and C3-HET tumors as 15 distinct clusters. (G) UMAP shows five distinct subclusters of Nkg7 expressing NK and NKT cells. (H) Dot plot of selected granzymes across subpopulations of NK cells. (I) Flow cytometry analysis shows granzyme B expression in C3-HET and C3-WT NK cells. Data are shown as a percentage of the total NK cell population. The scatterplot shows the number per group of samples from flow analysis, and the data are presented as means ± SEM. Statistical significance was determined by two-tailed Student’s t test. (J) Volcano plot of differentially expressed genes between C3-WT and C3-HET NK subcluster 0.
To further determine potential clinical correlations of our findings, we undertook in silico comparison of our data with published scRNA-seq data from patients with TNBC with poor survival (3, 40). This analysis revealed several similarities between a Claudin low-gene signature of human TNBC tumors and our C3-HET tumor cell signatures (fig. S1C and table S2). Another recent paper using scRNA-seq data in human TNBC demonstrated clusters of tumor cells that were highly enriched for genes encoding for glycosphingolipid, lysosome function, innate immune sensing, and inflammation (40), a signature that was also correlated with aggressive disease phenotypes such as metastasis and long-term poor outcome (40). This glycosphingolipid pathway gene signature was also enriched in C3-HET tumor cells compared with C3-WT (fig. S1D), explaining, in part, the reason behind the aggressive nature of these tumor cells. To further validate the impact of the glycosphingolipid pathway (41, 42) on tumor cells, we treated Epcam+ C3-HET tumor cells in vitro with 2 μM fingolimod (FTY720), an antagonist of sphingosine-1-phosphate receptor (S1PR1) (43). We observed a reduced number of tumorsphere formation upon FTY720 treatment, indicating a supportive role of glycosphingolipid metabolism in TNBC (fig. S1E). Collectively, our analysis revealed that CD45− tumor cell populations in the murine TNBC model recapitulated the heterogeneous and aggressive nature of human TNBC tumors, further validating this model for the study of the TME in TNBC.
In addition to its intratumoral heterogeneity, the TME of TNBC is characterized by its immune-rich microenvironment that likely contributes to tumor progression (25, 44). We found that the CD45+ immune cell population in C3-HET tumors is composed of 15 different clusters, containing myeloid cells, NK cells, dendritic cells, T cells, and macrophages (Fig. 1F and fig. S2, A and B). We observed altered myeloid cell populations in C3-HET tumors compared with less-aggressive and less-metastatic C3-WT tumors, supporting our earlier published work (26, 30); we further observed that Nkg7-expressing NK and natural killer T (NKT) cells had high numbers of differentially expressed genes between the two genotypes (fig. S2C), indicating the possibility of altered phenotypes for these cells between the two genotypes of mice. T cells showed no marked differences between the C3-HET and C3-WT tumors (fig. S2C). Furthermore, deep clustering of Nkg7-expressing cells identified five subclusters of NK and NKT cells, with subcluster 4 exclusively representing NKT-like cells based on their high expression of CD3e and CD3g (Fig. 1G and fig. S2D). These cells were excluded in further NK cell characterization. The percent distribution of cells within the NK and NKT subclusters showed that NK cells in subclusters 0, 2, and 3 were marginally enriched in C3-HET compared with C3-WT tumors, whereas the population of NKT cells in subcluster 4 was greatly reduced in C3-HET compared with C3-WT (Fig. 1G and fig. S2E). These data suggest that NK cells, and not NKT cells, were the predominant Nkg7-expressing cells in the TME of C3-HET tumors. Further analysis on the total NK cell population showed lower expression of genes responsible for cytotoxicity and NK cell activation, such as Hsp90ab1, Hsp90aa1, Ly6e, and Hsph1, in C3-HET tumor NK cells when compared with C3-WT tumor NK (fig. S3A) (45, 46). Moreover, subcluster analysis shows lower expression of various granzymes in NK subclusters 0 and 1 (Fig. 1H), suggesting that these two clusters comprise an altered NK cell population with less cytotoxicity. Low abundance of granzyme B was further confirmed in NK cells from C3-HET tumors by flow cytometry (Fig. 1I). An analysis of NK cell subcluster 0 further highlighted several genes that were enriched in C3-HET tumors, such as Socs3 (Fig. 1J), known to be associated with impaired differentiation of NK cells (47). Further flow cytometry analysis showed no difference in interferon-γ expression between C3-WT and C3-HET NK populations (fig. S3B); however, GSEA indicated that NK cells in C3-HET tumors had higher inflammatory responses (fig. S3C), indicating that they may have a proinflamma-tory role in the TME of TNBC tumors.
TNBC-associated NK cells are immature and are associated with increased PD-L1 expression, angiogenesis, and metastasis
Characterization of tumor-associated NK cells by flow cytometry showed overall greater numbers of CD45+CD3−DX5+ NK cells in C3-HET tumors as compared with C3-WT tumors (Fig. 2, A and B), which was further confirmed by IHC using anti-NK1.1 antibody (Fig. 2, C and D). C3-HET tumors also showed increased expression of CD34 and vascular endothelial growth factor (VEGF), markers for angiogenesis (48), further supporting their protumoral nature (fig. S4A). Moreover, PD-L1, which plays a major role in suppressing the adaptive arm of the immune response, was highly expressed in the C3-HET primary tumors (fig. S4B). Last, numerous K14+ metastatic cells were also observed in the lungs (K14+ cluster) and lymph nodes (K14+ single cells) of C3-HET mice (fig. S4, C and D) (30). An increased number of NK cells was also observed in lung metastatic sites of C3-HET tumor–bearing mice (fig. S4E), supporting an association between NK cells and TNBC metastasis.
Fig. 2. TNBC-associated NK cells are immature.
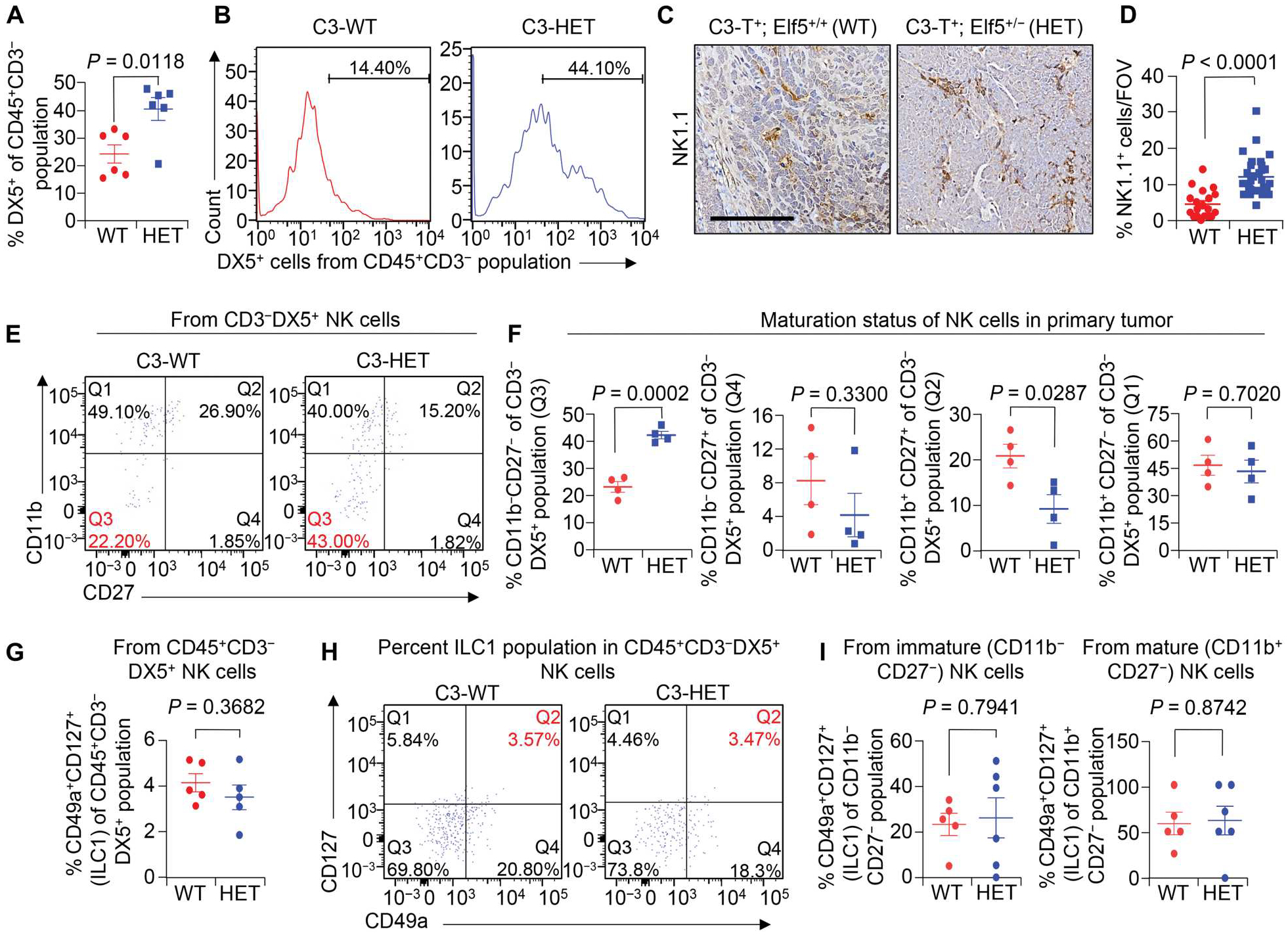
(A and B) Shown are the percentages of CD45+CD3−DX5+ NK cells of CD45+CD3− cells (A) and representative flow cytometry plots (B) in primary tumors of indicated groups. (C and D) Representative IHC staining (C) and quantification (D) of NK1.1+ cells in C3-HET and C3-WT primary tumors. n = 4 tumors for C3-WT and n = 5 tumors for C3-HET were used, with analysis of n = 19 random fields from C3-WT tumors and n = 29 random fields from C3-HET tumors for quantification. Scale bar, 100 μm. FOV, field of view. (E and F) Flow cytometry plots (E) and scatterplots (F) show comparison of NK cell subpopulations based on CD27 and CD11b expression in tumors from C3-HET and C3-WT mice. Percentages are of the total NK cell population. Q3 represents the immature NK cell population. (G) Flow cytometry analysis shows the percentage of ILC1 cells (CD45+CD3−DX5+CD49a+CD127+) between C3-WT and C3-HET tumors. (H) Representative flow cytometry plots show percentage of CD127+CD49a+ ILC1s gated from CD45+CD3−DX5+ NK cells of the indicated groups. Q2 represents the ILC1 gate. (I) Scatterplots show percentage of the CD127+CD49a+ ILC1 population gated either from CD11b−CD27− immature (left) or CD11b+CD27− mature (right) NK cells between C3-WT and C3-HET tumors. The scatterplots show the number per group of samples from flow analysis, and data are presented as means ± SEM. Statistical significance was determined by two-tailed Student’s t test.
Conventionally, mouse NK cells are divided into four functionally distinct subsets on the basis of surface expression of CD27 and CD11b, which reflects their maturation status. CD11b−CD27− NK cells are immune-regulatory and produce greater concentrations of cytokines, whereas CD11b+CD27− cells are considered mature NK cells and have a higher cytotoxic capacity (49, 50). In contrast to circulating NK cells that are primarily cytotoxic, we found increased numbers of the immature CD11b−CD27− NK cells in C3-HET primary tumors as compared with C3-WT (Fig. 2, E and F). Although these NK cells show some similarities with type 1 innate lymphocyte cells (ILC1s) (51, 52), the proportion of ILC1s did not differ between C3-WT and C3-HET in our flow cytometry and scRNA-seq data (Fig. 2, G to I, and fig. S5).
A high frequency of NK cells was also observed in another aggressive TNBC and basal enriched mouse model, Blg-Cre; p53KO BRCA1Floxed (hereafter termed as BRCA1cKO) (53); in contrast, their numbers were reduced in luminal mouse mammary tumor virus–polyoma middle tumor–antigen (MMTV-PyMT) tumors, which are not TNBC (fig. S6). These data suggest that the increased number of NK cells in the TME may be restricted to TNBC tumors as compared with luminal tumors. Overall, these findings indicate that NK cells present in the TME of TNBC are phenotypically distinct, are partly immature, and are associated with increased metastasis and angiogenesis.
Depletion of NK cells decreases tumor progression and metastasis
To test the functional importance of tumor-associated NK cells in the TME, we depleted NK cells in C3-HET mice at the hyperplasia stage (12 to 15 weeks) using anti-NK1.1 antibody (250 μg per mouse three times per week) (Fig. 3A and fig. S7A). Depletion of NK cells resulted in a significant (P = 0.0106) reduction in tumor incidence and progression (Fig. 3B), with a decreased mitotic index, lower necrosis score, and lower tumor margin score on average (fig. S7B) as well as delayed tumor progression (Fig. 3C). Delayed primary tumor progression was also associated with increased apoptosis of tumor cells, as confirmed by an increased number of cleaved caspase-3+ cells (Fig. 3D). NK cell depletion resulted in an increase in CD8+ cytotoxic T cells (Fig. 3E) and reduced PD-L1 expression in tumor cells (Fig. 3F), suggesting that this treatment affected immune suppression pathways. Moreover, K14+ (Fig. 3G) and Vimentin+ (Fig. 3H) single-cell metastases were reduced in the axillary lymph nodes of anti-NK1.1–injected mice as compared with controls. Together, these data strongly suggest a protumorigenic and prometastatic role for tumor-associated NK cells in TNBC.
Fig. 3. Spontaneous TNBC tumors have higher NK cell infiltration, and depletion of NK cells reduced tumor progression.
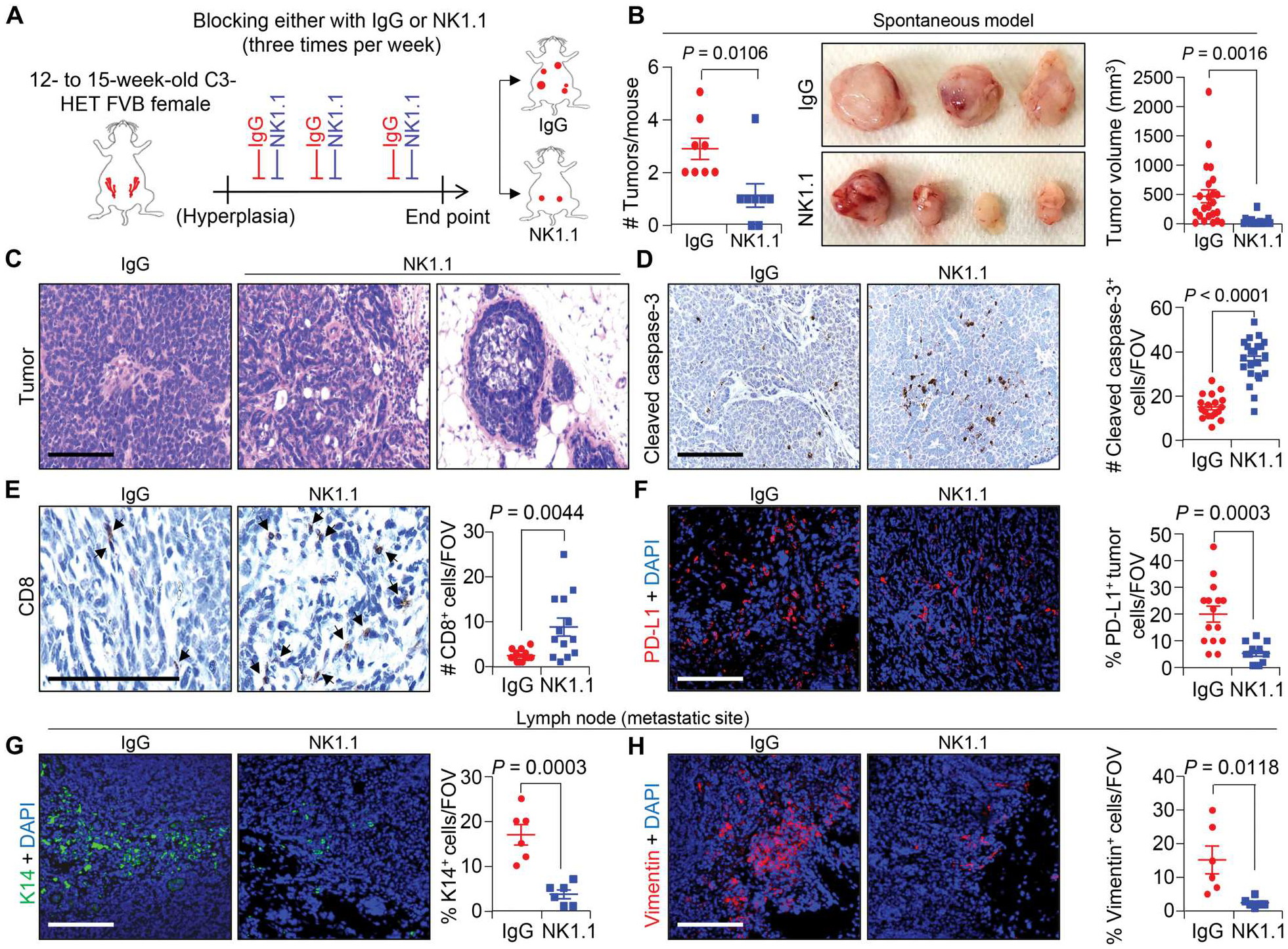
(A) The schematic shows the experimental approach for NK cell depletion in the spontaneous C3-HET murine model. Twelve- to 15-week-old female mice with hyperplasia of mammary gland received anti-NK1.1 or isotype control immunoglobulin G (IgG) antibodies three times per week, and tumor growth was measured until humane end point for analysis. (B) Tumor burden per mouse (left; n = 8 mice per group), representative tumor images (middle), and calculated tumor volume (right), V = ½ (length × width2) upon NK cell depletion in C3-HET mice. (C) Hematoxylin and eosin–stained images of primary tumors upon NK cell depletion compared with IgG control group. (D) Cleaved caspase-3 staining (left) and quantification (right) of apoptotic cells in primary tumors upon NK cell depletion (n = 3 individual tumors per group, with analysis of n = 18 random fields from IgG tumors and n = 22 random fields from NK1.1-depleted tumors for quantification). (E) Immunostaining of cytotoxic CD8+ T cells in tumors from NK cell–depleted mice (arrowheads indicate positive staining; left) with quantification on the right (n = 3 individual tumors per group and n = 13 random fields per group). (F) Representative immunofluorescence (IF) images show expression of PD-L1+ primary tumor cells (red) upon NK cell depletion. Scatterplots (right) represent quantification (n = 3 individual tumors per group, n = 15 random fields from IgG tumors, and n = 12 random fields from NK1.1-depleted tumor for quantification). (G and H) Representative IF images (left) and quantification (right) show K14+ tumor cells (green) (G) and Vimentin+ cells (red) (H) in the axillary lymph node upon NK cell depletion (n = 5 individual lymph nodes). Two-tailed Student’s t test was used to compute P values. Data are presented as the mean ± SEM. Scale bars, 100 μm. DAPI, 4′,6-diamidino-2-phenylindole.
To further understand the functional role of tumor-associated NK cells in tumor progression, we orthotopically implanted tumor cells into C3(1)/Tag-REAR (hereafter termed as REAR) mice, which bear all functional immune cells, including NK cells, but are tolerant to tumors carrying C3-T antigen (54) and in which size- and age-matched tumors can be generated faster than in the spontaneous model of TNBC. Injection of C3-HET or C3-WT tumor cells into this orthotopic REAR model resulted in tumors that were morphologically similar to primary spontaneous tumors (30), and C3-HET tumor cells grew faster than C3-WT tumor cells (fig. S7, C and D). NK cells were depleted with an anti-NK1.1 antibody when C3-HET tumors were either small (about 15 to 30 mm3; initiation setting) or established (about 65 to 150 mm3; preventative setting matching with clinical scenario of advanced TNBC) (Fig. 4, A to C, and fig. S7, E to G). In both cases, NK cell depletion (Fig. 4B) reduced tumor initiation and progression (Fig. 4C and fig. S7G). NK cell depletion also reduced tumor progression in the BRCA1cKO orthotopic TNBC model (Fig. 4, D and E) (53). No impact of NK cell depletion was observed on the tumor progression in established luminal breast cancer models [the MMTV-PyMT tumor–derived cell lines, MMTV-PyMT-B6 and MMTV-PyMT WT-B clone (WTB), were used for tumor growth] (fig. S7, H to K) (55), corroborating a protumorigenic function of NK cells in the TNBC/basal subset of breast cancer. We also observed an increased number of cleaved caspase-3+ apoptotic cells in NK cell–depleted C3-HET orthotopic tumors (Fig. 4F) along with a reduction in the number of angiogenic CD34+ vessels (Fig. 4G). These findings suggest that depletion of NK cells may affect cell death and angiogenesis in primary tumors. After NK cell depletion, there was also a reduction in the number of F4/80+ macrophages and CD4+ T cells, although no change was observed in the CD8+ T cell population (Fig. 4H and fig. S7, L and M). Depletion of NK cells did not alter the CD49a+−CD127+ ILC1 population in either the mature or immature subpopulation (fig. S7N). Increased cleaved caspase-3 was observed after NK cell depletion in BRCA1cKO TNBC models (fig. S8, A and B); CD8+ T cell proportions did not differ, consistent with the C3-HET model (fig. S8C). Reduction of Vimentin+ single-cell metastasis in axillary lymph nodes of NK cell–depleted C3-HET (fig. S8, D and E) and BRCA1cKO (fig. S8, F and G) mice further supported an association between NK cells and TNBC metastasis. Moreover, NK cell depletion caused a significant (P = 0.0008) reduction in PD-L1 expression in C3-HET tumor cells (Fig. 4I), indicating a role for NK cells in promoting PD-L1–related immune evasion in TNBC. Together, NK cell depletion is associated with a reduction in immune suppression that may be responsible for decreased tumor progression in TNBC.
Fig. 4. Pharmacological depletion of NK cells reduces tumor growth, progression, and metastasis in murine TNBC.
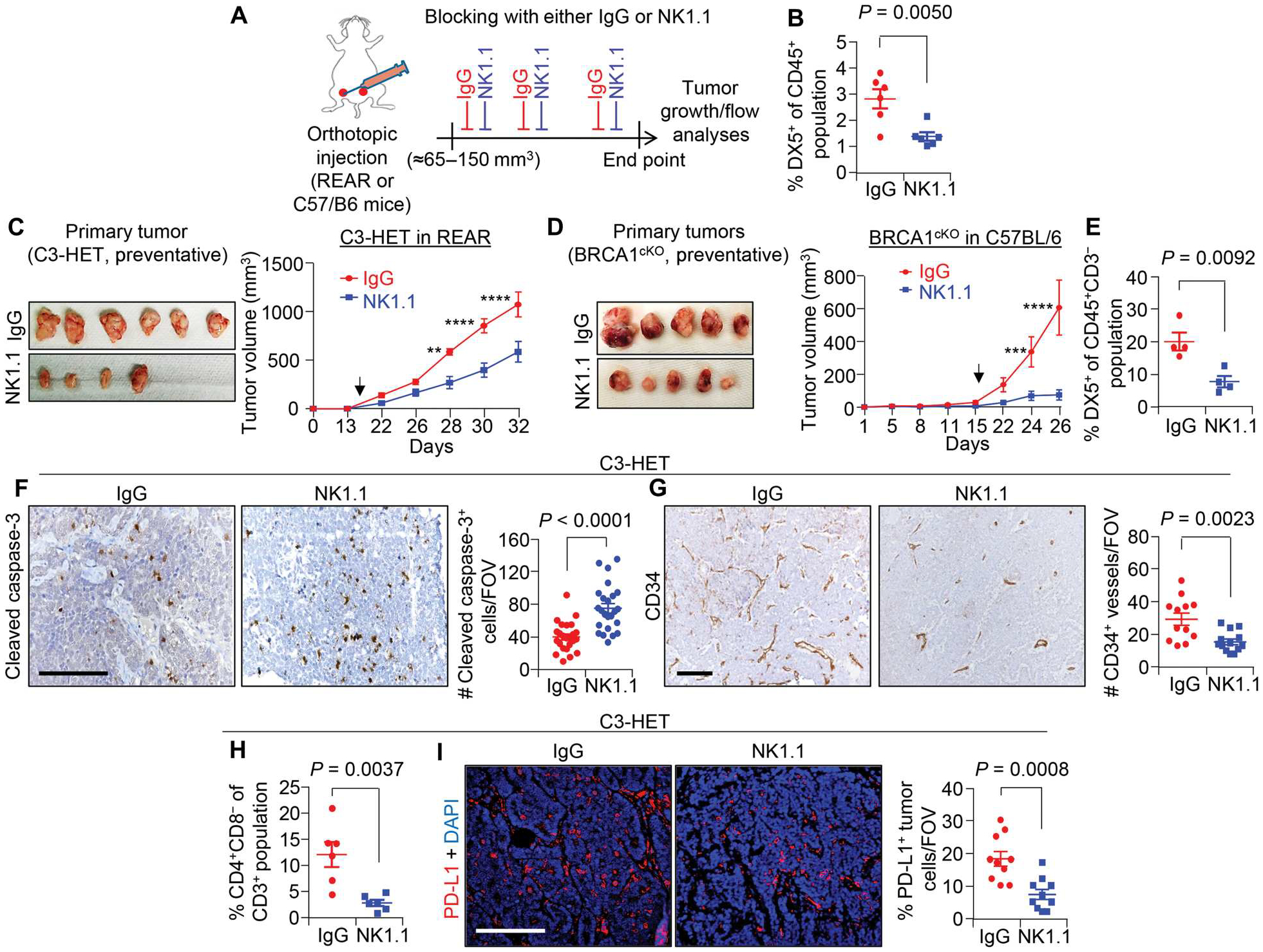
(A) Schematic shows experimental approach of NK cell depletion for preventative (large tumor) setting. A total of 140,000 primary tumor cells from C3-HET mice were injected into the mammary fat pads (MFPs) of REAR mice. (B) Percentage of NK cells (CD45+DX5+) of CD45+ cells in C3-HET primary tumors upon anti-NK1.1 treatment compared with control tumors. (C) Representative images of tumors (left) and tumor growth curve (right) upon NK cell depletion in the preventative setting (n = 8 tumors per group). (D) Representative tumor images (left) and tumor growth curve (right) showing tumor progression for BRCA1cKO tumor–bearing mice (n = 6 tumors for IgG and n = 8 tumors for NK1.1). A total of 125,000 primary tumor cells per MFP from BRCA1cKO mice were injected into C57BL/6 mice. Arrowheads in (C) and (D) indicate the start point for drug dose. (E) Percentage of NK cells (CD45+CD3−DX5+) of CD45+CD3− cells in primary BRCA1cKO tumors upon NK cell depletion compared with control tumors. (F) Cleaved caspase-3 staining (left) and quantification (right) show apoptotic cells in primary tumors upon NK cell depletion (n = 3 individual tumors per group and n = 24 random fields per group for quantification). (G) Representative IHC images (right) and quantification (left) show CD34+ blood vessels in NK cell–depleted primary tumors (n = 4 individual tumors for control and n = 3 for NK cell depletion, with analysis of n = 12 random fields from IgG tumors and n = 13 random fields from NK1.1-depleted tumors for quantification). (H) Flow cytometry analysis showing percentage of CD4+CD8− T cells of CD3+ T cells in primary C3-HET tumors upon NK cell depletion compared with control tumors. (I) IF staining (left) and quantification (right) of PD-L1 in primary tumors upon NK cell depletion (n = 4 individual tumors, with analysis of n = 10 random fields per group for quantification). The scatterplots show the number per group of samples from flow analysis. Two-tailed Student’s t test was used to compute P values for all scatterplots. Data are presented as the mean ± SEM. Statistical significance was determined by two-way ANOVA with Sidak’s multiple comparisons test in (C), (D), and (F). **P = 0.0011, ***P = 0.0003, and ****P < 0.0001. Scale bars, 100 μm.
TNBC-associated NK cells are noncytotoxic and support cancer stem cell properties through activation of Wnt signaling
To further evaluate the protumorigenic function of tumor-infiltrating NK cells, we assessed their potential effector function in coculture with TNBC and non-TNBC cell lines (fig. S9A). The viability of the 4T1 TNBC cell line was not changed, as measured by propidium iodide staining after 12 hours of coculture with sorted C3-HET NK cells (CD45+CD3−DX5+). In contrast, activated NK cells supplemented with interleukin-2 (IL-2) and IL-15 that were sorted from the spleens (positive control) of non–tumor-bearing mice retained their cytotoxic capability in coculture (Fig. 5, A and B), highlighting the reduced cytotoxic potential for C3-HET tumor-associated NK cells (P = 0.6755) compared with activated NK cells from the spleen (P < 0.0001).
Fig. 5. NK cells from C3-HET tumors are less cytotoxic.
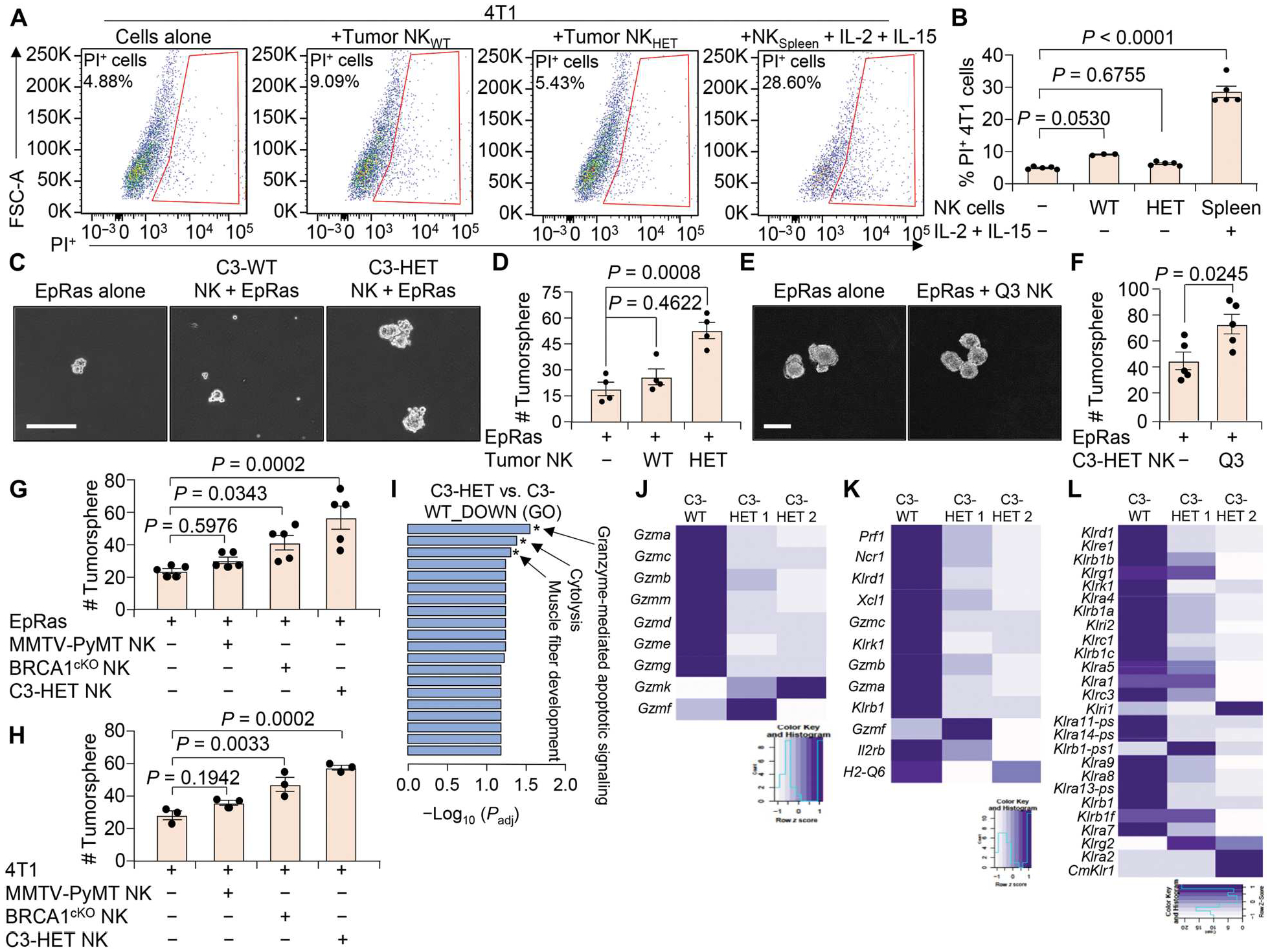
(A and B) Flow cytometry plots (A) and quantification (B) show the viability of 4T1 cells when cocultured with NK cells (CD45+CD3−DX5+) sorted from C3-WT and C3-HET tumors. Coculture with activated spleen NK cells (supplemented with IL-2 and IL-15) from non–tumor-bearing mice were used as control. FSC-A, forward scatter area; PI, propidium iodide. (C and D) Representative images (C) and number (D) of TNBC (EpRas)–derived tumorspheres are shown after coculture with tumor-associated NK cells isolated from the indicated groups. (E and F) Representative images (E) and quantification (F) of tumorspheres are shown for EpRas cells cultured for 3 days with or without sorted immature NK cells (Q3: DX5+CD11b−CD27−) from C3-HET tumors. (G and H) EpRas (G) and 4T1 (H) tumorsphere count is shown after coculture with NK cells isolated from indicated tumors. (I) Gene ontology (GO) enrichment analysis showing most significantly down-regulated pathways in NK cells sorted from C3-HET as compared with C3-WT tumors. Padj, adjusted P value. (J to L) Heatmap from bulk mRNA-seq showing expression of various granzymes (J), genes characteristic of cytotoxic NK cells (K), and killer cell lectin-like receptors (L) in NK cells sorted from C3-HET (n = 2 samples but pooled from several tumors) as compared with C3-WT (n = 1 sample but pooled from several tumors) tumors. Statistical significance was determined by one-way ANOVA with Dunnett’s multiple comparisons test in (B), (D), (G), and (H). Two-tailed Student’s t test was used to determine significance in (F). Data are presented as means ± SEM. Scale bars, 100 μm.
We found that NK cells from C3-HET tumors increased the number and size of TNBC tumorspheres formed in serum-free media in an in vitro surrogate stem cell assay as compared with tumor cells alone after 72 hours of coculture; however, NK cells from C3-WT tumors had no effect on tumorspheres (Fig. 5, C and D). Coculture of sorted immature NK cells [CD3−DX5+−CD11b−CD27− (defined as Q3)] from C3-HET tumors also led to increased number of TNBC tumorspheres as compared with TNBC cells alone (Fig. 5, E and F). These findings were confirmed by a similar set of coculture analysis with 4T1 cells as well (fig. S9, B and C). In contrast, coculture of similar Q3 NK cells (sorted from C3-HET tumors) with WTB cells (a luminal cell line established from transgenic mouse MMTV-PyMT tumors) (55, 56) had no significant (P = 0.0881) effect on WTB tumorsphere formation (fig. S9, D and E), suggesting a TNBC subtype–specific function of these tumor-associated NK cells. Last, both EpRas and 4T1 (TNBC cells) tumorsphere formations were increased after coculture with NK cells isolated from primary tumors of another aggressive TNBC tumor model, BRCA1cKO (53), but not by tumor-associated NK cells isolated from luminal tumors from MMTV-PyMT mice (Fig. 5, G and H). These findings indicated that NK cells derived from TNBC tumors are uniquely able to increase cancer stem cell properties in TNBC but not in non-TNBC luminal cells.
To identify how NK cells support cancer stem cell function, we performed unbiased bulk mRNA sequencing (mRNA-seq) on sorted NK (CD45+DX5+) cells from C3-WT and C3-HET tumors. Our data revealed that pathways related to granzyme-mediated apoptotic signaling and cytolysis were the most significantly down-regulated pathways in NK cells from C3-HET tumors (Fig. 5I), consistent with our scRNA-seq data (Fig. 1) and the observed lack of functional toxicity (Fig. 5, A and B). Reduced expression of genes for all major granzymes (Fig. 5J), genes characteristic of cytotoxic functioning of NK cells (Fig. 5K), and killer cell lectin-like receptors (Fig. 5L) were down-regulated in C3-HET tumor NK cells compared with C3-WT, further supporting earlier data.
GSEA of tumor cells from scRNA-seq revealed up-regulation of Wnt/β-catenin signaling (Fig. 6A) and high expression of Wnt target genes (Fig. 6B), such as the gene encoding β-catenin (Ctnnb1), Tcfs, and Lefs (57), in C3-HET tumors as compared with C3-WT tumor cells. Moreover, we found that expression of several Wnt ligands was up-regulated in NK cells from C3-HET compared with C3-WT TME-associated NK cells through mRNA-seq (Fig. 6C). Consequently, quantitative reverse transcription polymerase chain reaction (qRT-PCR) confirmed up-regulation of Wnt target genes such as Axin2, Ccnd1, and Ascl2 in tumor cells when cocultured with C3-HET NK cells compared with C3-WT (Fig. 6D). Wnt pathways classically govern molecular and cellular traits associated with stemness, and aberrant Wnt signaling mediates protumoral functions in several cancers, including TNBC (58–62). Thus, high expression of Wnt ligands in tumor-associated NK cells provides a potential mechanistic basis for the ability of NK cells to influence cancer stem cell properties. To test the functional relevance of Wnt signaling in TNBC cells, we used the pan-Wnt inhibitor JW-74 in a tumorsphere assay in which 4T1 TNBC cells were cocultured with sorted NK cells from C3-HET tumors. Addition of 10 μM JW-74 reverted the NK cell–mediated increase in TNBC tumorsphere count (Fig. 6E).
Fig. 6. NK cells in TME of aggressive TNBC secrete Wnt ligands and activate Wnt signaling in tumor cells.
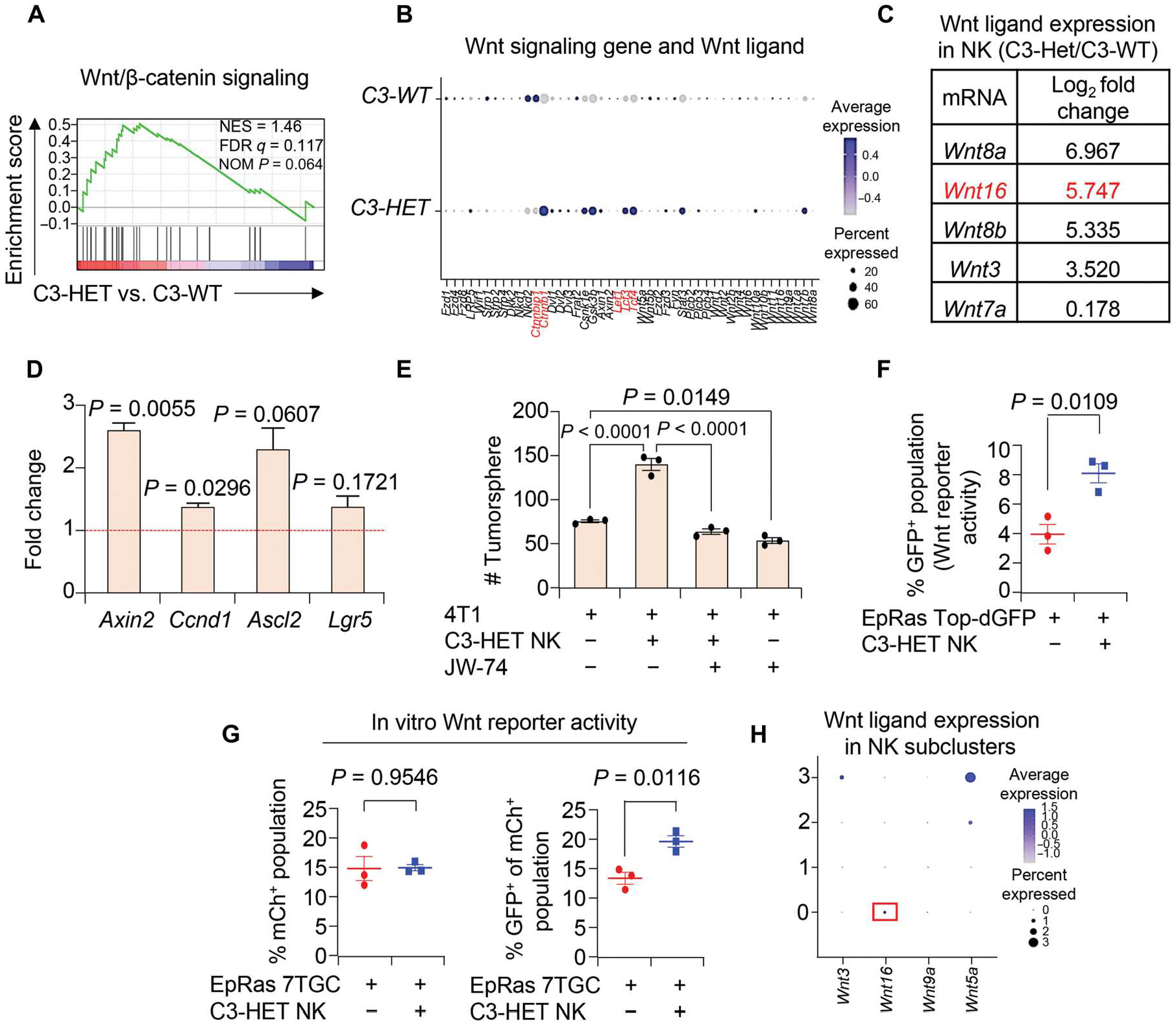
(A) Gene set enrichment analysis (GSEA) from scRNA-seq shows higher Wnt/β-catenin signaling in C3-HET as compared with C3-WT tumors (in CD45− cell population). NES, normalized enrichment score; FDR, false discovery rate; NOM, nominal. (B) Dot plot shows expression of key Wnt target genes across C3-WT and C3-HET tumor cells. Genes highlighted in red are Wnt signaling genes, including Wnt ligands. (C) List of Wnt ligands from bulk mRNA-seq (from NK cells) shows differential expression in NK cells sorted from C3-HET as compared with C3-WT tumors. (D) qRT-PCR analysis of Wnt target genes in primary tumor cells when cocultured with NK cells sorted from C3-HET as compared with C3-WT tumors in vitro. Relative expression is presented as fold change, and the dashed line represents the relative expression with respect to C3-WT. qRT-PCR values were normalized to Gapdh. Experiment was performed in technical duplicate. Each point represents a pool of five individual tumors. (E) The bar graph depicts the number of 4T1-derived tumorspheres after treatment with Wnt inhibitor (JW-74) and culture for 3 days with NK cells from C3-HET tumors. n = 2 independent experiments performed in technical triplicate. (F) Wnt reporter activity of EpRas-Top-dGFP (GFP+) upon coculture with NK cells from C3-HET tumors. (G) Wnt reporter activity of EpRas-7TGC (mCh+ GFP+) upon coculture with NK cells and EpRas tumor cells (right). mCh+ cells mark total infected cells with lentivirus (left). (H) Dot plot from scRNA-seq shows expression of selected Wnt ligands across four subpopulations of NK cells in C3-HET tumors. Statistical significance was determined by one-way ANOVA with Sidak’s multiple comparisons test in (E). All other data were analyzed using two-tailed Student’s t tests. The data are presented as means ± SEM.
To determine whether Wnt signaling in tumor cells is activated by Wnt ligands secreted by NK cells, we used lentiviral-based Wnt reporters, Top-dGFP (63) and 7TGC (64), to make EpRas cell lines stably expressing the reporters (EpRas-7TGC or EpRas-Top-dGFP). This allowed for measurement of Wnt signaling activation in the presence of NK cells. Coculture of NK cells sorted from C3-HET tumors with EpRas-Top-dGFP (Fig. 6F) and EpRas-7TGC (Fig. 6G) tumor cells showed activation of Wnt signaling [green fluorescent protein–positive (GFP+) cells] in these cells as compared with control, confirming NK cell–mediated activation of Wnt signaling in TNBC. To evaluate the effect of Wnt signaling on TNBC progression, we used LGK-974, a porcupine inhibitor that affects secretion of Wnt ligands. It is currently in a clinical trial for multiple solid malignancies that have progressed despite standard therapy or for which no effective standard therapy exists (ClinicalTrials.gov identifier: NCT01351103). Primary tumor cells from C3-HET tumors were injected in REAR mice and once tumors were established (about 65 to 150 mm3). Tumor-bearing mice were treated with a dose of LGK-974 (5 mg/kg of body weight) orally. Reduction in primary tumor growth was observed in the LGK-974–treated group when compared with control (fig. S9F). Down-regulation of Wnt signaling in primary tumors was confirmed by the lower expression of Wnt target genes Axin2, Ascl2, and Lgr5 (fig. S9G, left). Moreover, we observed down-regulation of stem cell genes such as Oct4, Nanog, and Sox2 in the tumor cells upon LGK-974 treatment (fig. S9G, right), supporting our finding that Wnt signaling in tumor cells activated by NK cells causes increased stemness of TNBC tumors. One of the up-regulated Wnt ligands, Wnt16, was found to be expressed relatively higher in the NK cell subcluster 0 population (Fig. 6H) that also has the lowest granzyme expression as indicated by scRNA-seq data previously (Fig. 1H), suggesting a Wnt16-mediated, non-immunological function of tumor-associated NK cells in inducing stemness in TNBC tumor cells. To further investigate this possibility, we stably knocked down Wnt16 in sorted C3-HET tumor NK cells (fig. S9H), followed by coculture with 4T1 cells. We observed a reduced number of 4T1-derived tumorspheres (fig. S9I) upon Wnt16 knockdown in NK cells, indicating the functional role of Wnt16 secreted by tumor-associated NK cells in inducing stem-like properties in tumor cells in some measure. Collectively, our data suggest that TNBC NK cells are protumorigenic and are responsible for activated Wnt signaling in TNBC cells.
Combinatorial targeting of NK cells along with immunotherapy or with chemotherapy improves therapeutic response in TNBC
Checkpoint blockade with antibodies specific for PD-L1 has proven its worth in the clinic, but the response to anti–PD-L1 immunotherapy in TNBC is limited, potentially because of the complex network of immune cells within these tumors (65). However, because our findings suggest that NK cells may promote PD-L1 expression in tumor cells, we next determined whether depletion of NK cells coupled with PD-L1 blocking antibodies could reduce tumor progression in TNBC. We found that the combination of anti-NK1.1 and anti–PD-L1 antibodies led to decreased tumor progression of C3-HET tumor cells (Fig. 7, A and B) and resulted in reduced K14+ single-cell metastasis in axillary lymph nodes (fig. S10, A and B) when compared with PD-L1 inhibition alone. Decreased numbers of NK cells in the tumor were confirmed through flow cytometry (Fig. 7C). Combined treatment of anti–PD-L1 and anti-NK1.1 antibodies resulted in increased apoptosis and reduced angiogenesis, as confirmed by increased cleaved caspase-3+ (fig. S10, C and D) and reduced CD34+ staining (fig. S10, E and F), respectively, in primary tumors as compared with control. In addition to immunotherapy, we also observed that depleting NK cells improves response to the chemotherapy drug olaparib (Fig. 7, D and E), which is approved by the U.S. Food and Drug Administration for adjuvant treatment of high-risk early breast cancer patients with germline BRCA (BReast CAncer gene) mutations (ClinicalTrials.gov identifier: NCT02032823). Increased evidence of apoptosis and CD8+ T cell infiltration was observed with combination treatment, supporting decreased immune suppression and reduced tumor growth in treated BRCA1cKO tumors (fig. S11, A and B).
Fig. 7. Targeting NK cells or Wnt signaling improves the efficacy of immunotherapy or chemotherapy in murine TNBC models.
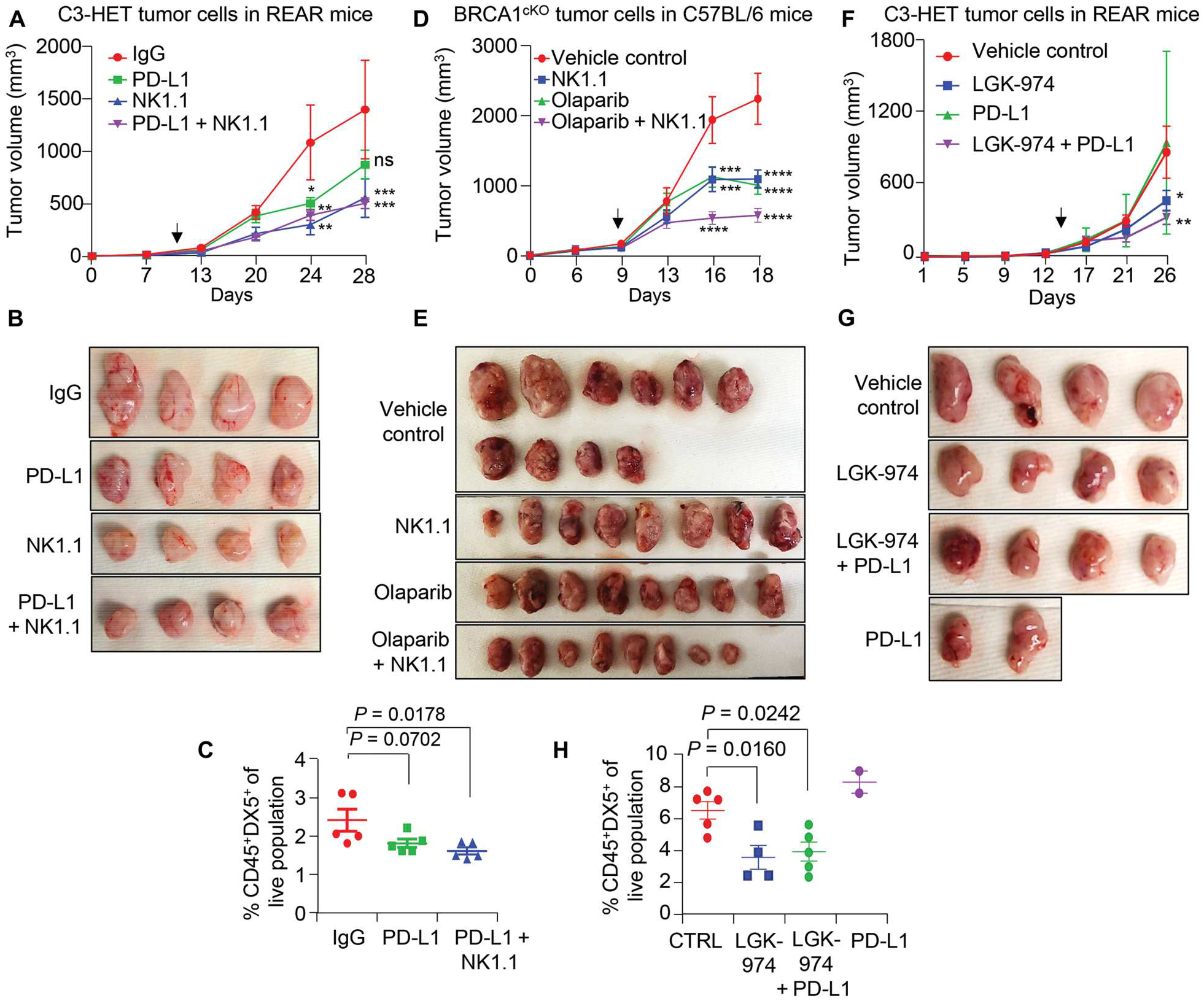
(A and B) Tumor growth curve (A) and representative tumor images (B) show tumor progression in anti-NK1.1 and anti-NK1.1 + anti–PD-L1 combination treatment groups (n = 8 tumors for IgG and n = 6 tumors for treated group). A total of 250,000 primary tumor cells per MFP from C3-HET tumors were injected into REAR mice. (C) Percentage of NK cell population (CD45+DX5+) of live cells in primary tumors of indicated groups. (D and E) Tumor growth curve (D) and representative tumor images (E) are shown for the indicated treatment group (n = 10 tumors for vehicle control and n = 8 tumors for treated group). A total of 150,000 primary tumor cells per MFP from BRCA1cKO tumors were injected into C57BL/6 mice. (F and G) Representative tumor images (F) and tumor growth curve (G) are shown for the indicated treatment group (n = 4 tumors for anti–PD-L1 and n = 6 tumors for the rest of the other groups). A total of 100,000 primary tumor cells per MFP from C3-HET mice were injected into REAR mice. Vehicle control and LGK-974 data points are shared with fig. S9F. (H) Flow cytometry analysis shows the NK cell frequency (CD45+DX5+) of live cells in C3-HET tumors. The scatterplots show the number per group of samples used for flow analysis. Statistical significance was determined by two-way ANOVA with Dunnett’s multiple comparisons test in (A), (D), and (F) and by one-way ANOVA with Dunnett’s multiple comparisons test in (C) and (H); *P < 0.05, **P < 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001. The data are presented as means ± SEM. Arrows indicate the start point for drug dose. Anti-IgG (250 μg per mouse), anti-NK1.1 (250 μg per mouse), and anti–PD-L1 (250 μg per mouse) antibodies were given three times per week. An oral dose of LGK-974 (5 mg/kg of body weight) was given every alternate day, and olaparib (50 mg/kg of body weight) was given daily by intraperitoneal injection. ns, not significant.
We next wanted to investigate the effect of blocking Wnt signaling using LGK-974, a Wnt ligand secretion inhibitor. We found that the combination of anti–PD-L1 and LGK-974 was more effective than anti–PD-L1 antibody alone, leading to slower tumor progression and reduced intratumoral NK cell abundance (Fig. 7, F to H). Together, these data suggested that combination therapy depleting NK cells or inhibiting Wnt ligand secretion along with immunotherapy using anti–PD-L1 antibody or chemotherapy may increase the efficacy of those therapies in TNBC tumors.
SOC3high immature CD56+ NK cells correlate with poor clinical outcome in patients with TNBC
To translate the data collected using experimental mouse models to TNBC clinical samples, we performed in silico analysis using large datasets such as the Cancer Genome Atlas in the KM plotter website (66). NK cells in human tumors express CD56 protein, which is encoded by the NCAM1 gene (fig. S12A). On the basis of proteinatlas.org datasets, NCAM1 is expressed predominantly in NK cells as compared with other immune cells (fig. S12A). We found a positive association between patients with ER-negative tumors with poor OS and high expression of NCAM1; in contrast, OS in patients with ER+ tumors were not correlated with NCAM1 expression (Fig. 8, A and B, and fig. S12B). High expression of NCAM1 was also correlated with poor OS and poor distant metastasis–free survival of patients with ER− tumors with lymph node metastasis (Fig. 8, C and D), patients with ER− tumors after chemotherapy (fig. S12, C and D), and patients with stage III ER− tumors (fig. S12E). Collectively, these findings indicate a positive correlation of high NCAM1 expression with poor clinical outcome in patients with TNBC.
Fig. 8. NCAM1 expression is associated with reduced survival of patients with TNBC.
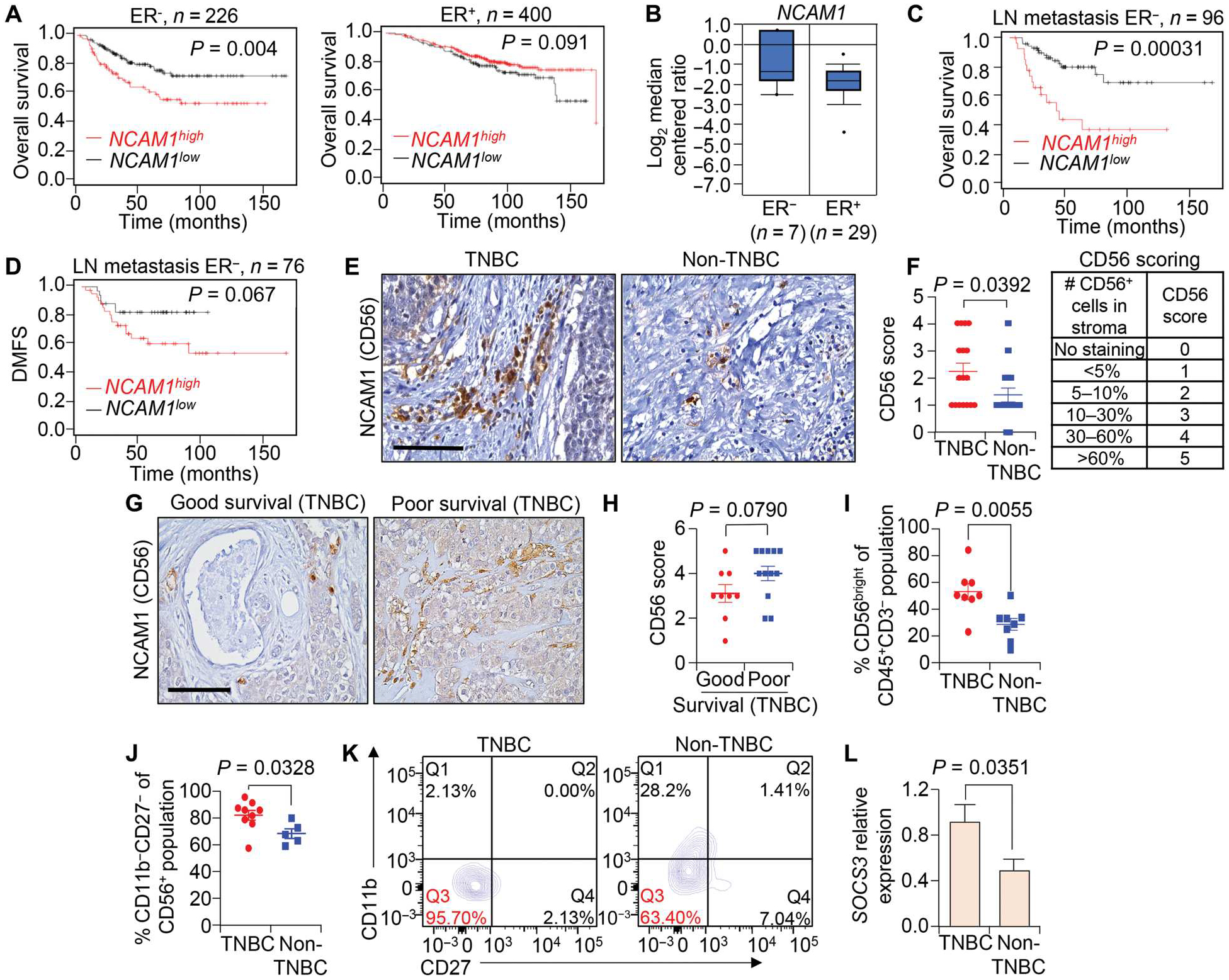
(A to D) The correlation between expression of NCAM1 (encoding CD56) and overall survival (OS) in patients with ER− (A) and ER+ tumors (B) as well as the correlation between expression of NCAM1 and overall survival (C) and distant metastasis–free survival (DMFS) (D) in patients with ER− tumors with lymph node metastasis. Data were analyzed using the KM plotter breast cancer database. (E and F) Representative IHC images (E) and quantification (F) are shown for CD56 expression in TNBC (n = 17) and non-TNBC (n = 16) tumor tissues. In all cases,“n” denotes independent biological samples. Quantification of CD56 scores was on a scale from 0 to 5 as indicated in (F). (G and H) Representative IHC images (G) and calculated CD56 scores (H) are shown for CD56bright cells in TNBC tissues stratified based on patient survival information (6 years cutoff value set to categorize poor versus good survival); n = 9 patient tumor tissues for good survival, and n = 12 for poor survival. (I) Scatterplot shows the percentage of the CD3−CD56bright NK cell population of the CD45+CD3− population in TNBC as compared with non-TNBC primary tumors. Fresh patient tumor tissues were used for analysis. (J and K) Scatterplot (J) and flow cytometry plots (K) show the percentage of CD56+CD11b−CD27− immature NK cells of the CD3−CD56+ population in TNBC as compared with non-TNBC primary tumors. Q3 represents immature NK cells. (L) qRT-PCR analysis shows relative mRNA expression of SOCS3 in NK cells from TNBC as compared with non-TNBC primary tumors. qRT-PCR values were normalized to Gapdh. The experiment was performed in technical duplicate (n = 3 individual tumors per group were used). Log-rank test was used for Kaplan-Meier plots to calculate P values in (A) to (D). Student’s t test was used to compute P values in (F), (I), (J), and (L), and Mann-Whitney U test was used for (H). The data are presented as means ± SEM. Scale bars, 100 μm.
To determine whether the correlation between CD56 (NCAM1) and patient survival is maintained at the protein level, we performed IHC analysis on TNBC and non-TNBC patient primary tumor sections. Our data from patients with non-TNBC (n = 16) and TNBC (n = 17) tumors showed presence of more CD56+ NK cells (P = 0.0392) in the TMEs of TNBC tumors as compared with tumors from patients with non-TNBC tumors (Fig. 8, E and F). Within TNBC, evaluation of tumor sections with patient survival information revealed that patients with poor survival (survival of less than 6 years) have relatively higher numbers of CD56+ NK cells than patients with longer survival (survival of ≥6 years), although this difference was not statistically significant (P = 0.0790; Fig. 8, G and H). An increase in the number of NK cells in TNBC was shown by flow cytometry analysis, where we observed a significantly (0.0055) higher percentage of CD3−CD56bright immature NK cells (67) in TNBC samples (Fig. 8I). Most of these tumor-associated NK cells were immature CD56+CD27−CD11b− in both TNBC and non-TNBC fresh primary tumor samples (Fig. 8,J and K). Furthermore, TNBC patient tumors showed higher expression of SOCS3 in sorted CD56+ NK cells from TNBC samples when compared with non–TNBC-associated NK cells, supporting our scRNA-seq data (Fig. 8L). These studies suggest that, in the TNBC TME, NK cells highly express SOCS3 and are CD56bright immature NK cells. Overall, our patient data support the findings from our murine experiments (fig. S13).
DISCUSSION
Functional interactions between tumor cells and the surrounding TME are critical for regulating signaling pathways that influence tumor growth and metastasis, promote angiogenesis, and maintain immune tolerance (68). The lack of expression of defined hormone receptors in TNBC prevents the use of conventional endocrine therapies that are effective in treating patients with other subtypes of breast cancers (3, 69), highlighting a critical need for alternative therapeutics to improve outcomes for patients with TNBC. scRNA-seq analysis of primary tumors from our preclinical spontaneous TNBC mouse model reveals that these tumors recapitulate the heterogeneity and diverse immune microenvironment of human TNBC, thus providing a model for understanding the TME in TNBC progression and assessing the efficacy of potential therapeutics.
Accumulating evidence highlights the role of tumor-infiltrating immune cells in dictating the fate of cancer cells (26–29). For example, NK cells within the TME can potentially control tumor growth by their cytotoxic activity (70). However, the function of NK cells highly depends on their maturation status and localization (71). In humans, terminally differentiated CD56dimCD16+ peripheral blood NK cells are cytotoxic in nature, whereas CD56bright CD16− NK cells that reside in secondary lymphoid tissues are considered to be immature, with reduced cytotoxic capability (72). Although there is some evidence that NK cells with reduced cytotoxicity profiles infiltrate solid tumors such as lung adenocarcinoma (73) and non–small-cell lung cancer (20, 74), the potential impact of NK cells on TNBC tumor progression is still poorly investigated.
In the present study, we showed that immature NK cells are increased in number in TNBC tumors using multiple TNBC models. Contrary to several findings supporting anticancer immunity of NK cells in cancer (75, 76), our data provide evidence that NK cells in the TNBC TME are protumorigenic in nature, because depletion of NK cells in our mouse model of TNBC decreased tumor progression while increasing apoptosis in primary tumor cells and decreasing single-cell metastasis. Depletion of NK cells affected tumor progression at early stages and tumor progression in established TNBC tumors at a later stage (C3-HET and BRCA1cKO), suggesting that it may be efficacious in patients with advanced-stage TNBC. Phenotypic characterization of tumor-associated NK cells further revealed that these NK cells differ from normal mature peripheral NK cells and, instead, have a more immature phenotype (CD3− DX5+CD11b−CD27−) as confirmed by flow cytometry. These immature NK cells also express Socs3, a gene that inhibits the JAKSTAT (Janus kinase–signal transducer and activator of transcription) pathway known to be important for NK cell maturation (77–79). Future studies will delineate how this pathway is crucial for NK cell function in the TNBC TME. Functionally, NK cells from these tumors promoted tumor cell proliferation and tumorsphere formation when cocultured in vitro with TNBC cell lines, a finding that was not observed with activated NK cells from the spleens of non–tumor-bearing mice. Tumor-associated NK cells isolated from TNBC tumors did not have a similar effect after coculture with luminal cell lines from MMTV-PyMT, suggesting a possible TNBC-specific role for tumor-associated NK cells. Our findings are in support with recent studies that demonstrate the correlation of CD11b−CD27− NK cells during tumor progression in lung and hepatocellular cancers (21, 22).
Comparative mRNA-seq and scRNA-seq analysis of NK cells from C3-HET and C3-WT tumors provided insight into the mechanistic basis for the protumorigenic effects of NK cells in the TNBC TME. Specifically, in addition to decreased expression of granzymes and NK cell killer lectin-like receptor, Wnt ligands such as Wnt16 were highly expressed in NK cells from TNBC tumors. Moreover, analysis using a Wnt inhibitor, Wnt16 KD, and Wnt reporters suggests that Wnt16 produced by NK cells may be, in part, responsible for Wnt signaling–dependent cancer stem cell function in TNBC tumor cells. However, a potential function of additional Wnt ligands cannot be ruled out and is still under investigation. In support of Wnt ligand secretion from NK cells playing a crucial function in TNBC TME, treatment of TNBC tumors with LGK-974, a well-characterized Wnt secretion inhibitor, decreased tumor progression in TNBC mouse models. The role of activated Wnt signaling in TNBC has recently been described (62, 80, 81), and the ability of Wnt/β-catenin signaling within the TME to support cancer cell survival and maintenance is well known (82). Moreover, Wnt signaling is positively correlated with PD-L1 expression and cancer stem cells in TNBC (83).
Immunotherapies using anti–PD-L1 antibodies are currently in clinical trials (84), but the current response rate to this treatment for patients with TNBC is very poor (85). Here, we show in our orthotopic transplant model that, when combined with NK cell depletion or Wnt secretion inhibition, the response of PD-L1 blocking antibody can be increased. This was further accompanied by reduced angiogenesis and increased apoptosis. Such a combined therapy may thus prove efficacious in improving outcomes for patients with aggressive TNBC cells who may not respond to immunotherapy alone. Similarly, combination targeting of NK cells along with olaparib, a chemotherapy drug used in BRCA1-associated TNBC, which is also known to not respond well, led to better response than olaparib alone in our model. Thus, our data highlight the effectiveness of targeting NK cells along with chemotherapy or immunotherapy in the context of TNBC.
Similar to our TNBC mouse models, a higher number of CD56+ cells was observed in samples from patients with TNBC compared with non-TNBC as seen by IHC. Flow cytometry data further confirmed that these CD56bright NK cells are mostly immature. However, future studies with larger numbers of patients with survival records are needed to validate the initial observation described here.
Our study has limitations. One of the limitations of the study is that we cannot exclude that other signaling pathway downstream of NK cells besides Wnt16 may have a cancer stem cell–supporting function in TNBC. Depletion of NK cells has no adverse effects in TNBC mouse models; however, depletion of immature NK cells needs to be cautiously tested in humans. Identification of additional immature NK cell features will help determine a specific marker for immature tumor–supporting NK cells in TNBC, allowing for better targeting in patients. We could not evaluate the correlation between immature NK cell abundance and patient response in a sufficiently large number of patients due to lack of samples, which needs to be addressed. Last, although we show that LGK-974 treatment reduced tumor growth in combination with anti–PD-L1 antibody, the effect should be further evaluated in combination with other standard care treatments such as chemotherapy and radiotherapy.
Together, our study provides insight into the paradoxical protumorigenic function of immature CD56brightCD11b−CD27− NK cells in TNBC. Our data further highlight a non-immunological function of NK cells on cancer stem cells in TNBC through activated Wnt signaling. Future studies with a large number of patients are needed to investigate the function of CD56bright immature NK cells in different subsets of patients with TNBC. Last, this study highlights the efficacy of combination therapy targeting NK cells along with anti–PD-L1 immunotherapy or chemotherapy.
MATERIALS AND METHODS
Study design
We used a minimum of three mice (six tumors as contralateral mammary fat pad injection was done) per group for in vivo experiments. Animals that experienced health issues or body score < 3 were excluded from analysis. We repeated our in vitro experiments at least two times in duplicate and to limit the use of animals for research; each in vivo experiment was performed twice, with sufficient power to detect effects. Mice were randomized on the basis of their age and body weight before the start of the experiment. No blinding was performed. All end points were prospectively determined by experimental design.
Statistical analysis
Raw data for experiments where n < 20 are presented in data file S1. The results were quantified using GraphPad Prism (version 5, 8, or 10). The significance of differences was calculated using two-tailed Student’s t test for normally distributed datasets. The tumor growth datasets were analyzed using the two-way analysis of variance (ANOVA) to compute statistical significance. For parametric data with multiple comparisons, a one-way ANOVA was used. For all the Kaplan-Meier plots, the KM plotter “auto select best cut-off” option was used for stratification. The differences between the survival groups were evaluated statistically using log-rank tests. The results are displayed as means ± SEM. The number of animals and the statistical tests to compute P value are reported in each of the corresponding figure legends.
Supplementary Material
Acknowledgments:
We thank L. King for critical reading of the manuscript and helpful discussions. We thank the Penn Vet Comparative Pathology Core and Cancer Modeling Shared Resource (CMSR) and the Sylvester Comprehensive Cancer Center at the University of Miami for assistance with embedding and sectioning of tissues. We also thank the members of the Flow Cytometry core at the University of Pennsylvania, Children’s Hospital of Philadelphia (CHOP) and the Flow Cytometry Shared Resource (FCSR) at the Sylvester Comprehensive Cancer Center of the University of Miami for all flow cytometry–based experiments. We thank the CHOP Next-Generation Sequencing Core for running scRNA-seq. We also thank Novogene Corporation Inc. for bulk mRNA-seq and analyses. We thank Y. Kang (Princeton University) for mouse EpRas, 4T1, WTB, and PYMT-B6 cell lines. We thank the Eastern Division of the Cooperative Human Tissue Network (CHTN), University of Pennsylvania and University of North Carolina, Chapel Hill for providing de-identified human breast cancer fixed tissues.
Funding:
This work was supported by grants from the American Cancer Society (RSG DDC-133604 to R.C.); the Emerson Collective Fund, University of Pennsylvania (20200315113131 to R.C.); the National Cancer Institute (R01CA237243 to R.C.); and the Rita Allen Scholar Award, the Pershing Square Sohn Prize for Cancer Research, NIH/NCI grant R01CA248158-01, and NIH/NIA grant R01 AG069727-01 (all to C.O.D.S.).
Footnotes
Supplementary Materials
This PDF file includes:
Other Supplementary Material for this manuscript includes the following:
Competing interests: The authors declare that they have no competing interests.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. scRNA-seq and bulk mRNA-seq datasets were deposited into the NCBI database (www.ncbi.nlm.nih.gov/), Sequence Read Archive PRJNA685201.
REFERENCES AND NOTES
- 1.Siegel RL, Miller KD, Jemal A, Cancer statistics, 2020. CA Cancer J. Clin 70, 7–30 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Garrido-Castro AC, Lin NU, Polyak K, Insights into molecular classifications of triple-negative breast cancer: Improving patient selection for treatment. Cancer Discov 9, 176–198 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA, Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest 121, 2750–2767 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deepak KGK, Vempati R, Nagaraju GP, Dasari VR, Nagini S., Rao DN, Malla RR, Tumor microenvironment: Challenges and opportunities in targeting metastasis of triple negative breast cancer. Pharmacol. Res 153, 104683 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Denkert C, von Minckwitz G, Darb-Esfahani S, Lederer B, Heppner BI, Weber KE, Budczies J, Huober J, Klauschen F, Furlanetto J, Schmitt WD, Blohmer JU, Karn T, Pfitzner BM, Kummel S, Engels K, Schneeweiss A, Hartmann A, Noske A, Fasching PA, Jackisch C, van Mackelenbergh M, Sinn P, Schem C, Hanusch C, Untch M, Loibl S, Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: A pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol 19, 40–50 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Denkert C, von Minckwitz G, Brase JC, Sinn BV, Gade S, Kronenwett R, Pfitzner BM, Salat C, Loi S, Schmitt WD, Schem C, Fisch K, Darb-Esfahani S, Mehta K, Sotiriou C, Wienert S, Klare P, Andre F, Klauschen F, Blohmer J-U, Krappmann K, Schmidt M, Tesch H, Kummel S, Sinn P, Jackisch C, Dietel M, Reimer T, Untch M, Loibl S, Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J. Clin. Oncol 33, 983–991 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Keenan TE, Tolaney SM, Role of immunotherapy in triple-negative breast cancer. J. Natl. Compr. Canc. Netw 18, 479–489 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Mittendorf EA, Philips AV, Meric-Bernstam F, Qiao N, Wu Y, Harrington S, Su X, Wang Y, Gonzalez-Angulo AM, Akcakanat A, Chawla A, Curran M, Hwu P, Sharma P, Litton JK, Molldrem JJ, Alatrash G, PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res 2, 361–370 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miles D, Gligorov J, Andre F, Cameron D, Schneeweiss A, Barrios C, Xu B, Wardley A, Kaen D, Andrade L, Semiglazov V, Reinisch M, Patel S, Patre M, Morales L, Patel SL, Kaul M, Barata T, O’Shaughnessy J; IMpassion131 investigators, Primary results from IMpassion131, a double-blind, placebo-controlled, randomised phase III trial of first-line paclitaxel with or without atezolizumab for unresectable locally advanced/metastatic triple-negative breast cancer. Ann. Oncol 32, 994–1004 (2021). [DOI] [PubMed] [Google Scholar]
- 10.Franzoi MA, de Azambuja E, Atezolizumab in metastatic triple-negative breast cancer: IMpassion130 and 131 trials—How to explain different results? ESMO Open 5, e001112 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Dieras V, Hegg R, Im SA, Shaw Wright G, Henschel V, Molinero L, Chui SY, Funke R, Husain A, Winer EP, Loi S, Emens LA; IMpassion130 Trial Investigators, Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N. Engl. J. Med 379, 2108–2121 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Villegas FR, Coca S, Villarrubia VG, Jiménez R, Chillón MJ, Jareño J, Zuil M, Callol L, Prognostic significance of tumor infiltrating natural killer cells subset CD57 in patients with squamous cell lung cancer. Lung Cancer 35, 23–28 (2002). [DOI] [PubMed] [Google Scholar]
- 13.Ishigami S, Natsugoe S, Tokuda K, Nakajo A, Che X, Iwashige H, Aridome K, Hokita S, Aikou T, Prognostic value of intratumoral natural killer cells in gastric carcinoma. Cancer 88, 577–583 (2000). [PubMed] [Google Scholar]
- 14.Coca S, Perez-Piqueras J, Martinez D, Colmenarejo A, Saez MA, Vallejo C, Martos JA, Moreno M, The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer 79, 2320–2328 (1997). [DOI] [PubMed] [Google Scholar]
- 15.Zwirner NW, Domaica CI, Cytokine regulation of natural killer cell effector functions. Biofactors 36, 274–288 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Marcenaro E, Notarangelo LD, Orange JS, Vivier E, Editorial: NK cell subsets in health and disease: New developments. Front. Immunol 8, 1363 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanna J, Mandelboim O, When killers become helpers. Trends Immunol 28, 201–206 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Costello RT, Sivori S, Marcenaro E, Lafage-Pochitaloff M, Mozziconacci M-J, Reviron D, Gastaut J-A, Pende D, Olive D, Moretta A, Defective expression and function of natural killer cell-triggering receptors in patients with acute myeloid leukemia. Blood 99, 3661–3667 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Bruno A, Focaccetti C, Pagani A, Imperatori AS, Spagnoletti M, Rotolo N, Cantelmo AR, Franzi F, Capella C, Ferlazzo G, Mortara L, Albini A, Noonan DM, The proangiogenic phenotype of natural killer cells in patients with non-small cell lung cancer. Neoplasia 15, 133–142 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carrega P, Morandi B, Costa R, Frumento G, Forte G, Altavilla G, Ratto GB, Mingari MC, Moretta L, Ferlazzo G, Natural killer cells infiltrating human nonsmall-cell lung cancer are enriched in CD56 bright CD16(−) cells and display an impaired capability to kill tumor cells. Cancer 112, 863–875 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Zhang Q-F, Yin W-W, Xia Y, Yi Y-Y, He Q-F, Wang X, Ren H, Zhang D-Z, Liver-infiltrating CD11b(−)CD27(−) NK subsets account for NK-cell dysfunction in patients with hepatocellular carcinoma and are associated with tumor progression. Cell. Mol. Immunol 14, 819–829 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jin J, Fu B, Mei X, Yue T, Sun R, Tian Z, Wei H, CD11b−CD27− NK cells are associated with the progression of lung carcinoma. PLOS ONE 8, e61024 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanay A, Regev A, Scaling single-cell genomics from phenomenology to mechanism. Nature 541, 331–338 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mills MN, Yang GQ, Oliver DE, Liveringhouse CL, Ahmed KA, Orman AG, Laronga C, Hoover SJ, Khakpour N, Costa RLB, Diaz R, Histologic heterogeneity of triple negative breast cancer: A National Cancer Centre Database analysis. Eur. J. Cancer 98, 48–58 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Liu Z, Li M, Jiang Z, Wang X, A comprehensive immunologic portrait of triple-negative breast cancer. Transl. Oncol 11, 311–329 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar S, Wilkes DW, Samuel N, Blanco MA, Nayak A, Alicea-Torres K, Gluck C, Sinha S, Gabrilovich D, Chakrabarti R, ΔNp63-driven recruitment of myeloid-derived suppressor cells promotes metastasis in triple-negative breast cancer. J. Clin. Invest 128, 5095–5109 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanahan D, Coussens LM, Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 21, 309–322 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Zhou J, Wang X-H, Zhao Y-X, Chen C, Xu X-Y, Sun Q, Wu H-Y, Chen M, Sang J-F, Su L, Tang X-Q, Shi X-B, Zhang Y, Yu Q, Yao Y-Z, Zhang W-J, Cancer-associated fibroblasts correlate with tumor-associated macrophages infiltration and lymphatic metastasis in triple negative breast cancer patients. J. Cancer 9, 4635–4641 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan Z-Y, Luo R-Z, Peng R-J, Wang S-S, Xue C, High infiltration of tumor-associated macrophages in triple-negative breast cancer is associated with a higher risk of distant metastasis. Onco. Targets. Ther 7, 1475–1480 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh S, Kumar S, Srivastava RK, Nandi A, Thacker G, Murali H, Kim S, Baldeon M, Tobias J, Blanco MA, Saffie R, Zaidi MR, Sinha S, Busino L, Fuchs SY, Chakrabarti R, Loss of ELF5-FBXW7 stabilizes IFNGR1 to promote the growth and metastasis of triple-negative breast cancer through interferon-γ signalling. Nat. Cell Biol 22, 591–602 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao H, Duan Q, Zhang Z, Li H, Wu H, Shen Q, Wang C, Yin T, Up-regulation of glycolysis promotes the stemness and EMT phenotypes in gemcitabine-resistant pancreatic cancer cells. J. Cell. Mol. Med 21, 2055–2067 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang R, Zong X, Aberrant cancer metabolism in epithelial-mesenchymal transition and cancer metastasis: Mechanisms in cancer progression. Crit. Rev. Oncol. Hematol 115, 13–22 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Georgakopoulos-Soares I, Chartoumpekis DV, Kyriazopoulou V, Zaravinos A, EMT factors and metabolic pathways in cancer. Front. Oncol 10, 499 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Z, Jiang Q, Dong C, Metabolic reprogramming in triple-negative breast cancer. Cancer Biol. Med 17, 44–59 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li C, Li X, Li G, Sun L, Zhang W, Jiang J, Ge Q, Identification of a prognosis-associated signature associated with energy metabolism in triple-negative breast cancer. Oncol. Rep 44, 819–837 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yuan J, Shi X, Chen C, He H, Liu L, Wu J, Yan H, High expression of CD47 in triple negative breast cancer is associated with epithelial-mesenchymal transition and poor prognosis. Oncol. Lett 18, 3249–3255 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cui C, Xu C, Yang W, Chi Z, Sheng X, Si L, Xie Y, Yu J, Wang S, Yu R, Guo J, Kong Y, Ratio of the interferon-γ signature to the immunosuppression signature predicts anti-PD-1 therapy response in melanoma. NPJ Genom. Med 6, 7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keren L, Bosse M, Marquez D, Angoshtari R, Jain S, Varma S, Yang S-R, Kurian A, Van Valen D, West R, Bendall SC, Angelo M, A structured tumor-immune microenvironment in triple negative breast cancer revealed by multiplexed ion beam imaging. Cell 174, 1373–1387.e19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xiao Y, Ma D, Zhao S, Suo C, Shi J, Xue MZ, Ruan M, Wang H, Zhao J, Li Q, Wang P, Shi L, Yang WT, Huang W, Hu X, Yu K-D, Huang S, Bertucci F, Jiang Y-Z, Shao ZM; AME Breast Cancer Collaborative Group, Multi-omics profiling reveals distinct microenvironment characterization and suggests immune escape mechanisms of triple-negative breast cancer. Clin. Cancer Res 25, 5002–5014 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Karaayvaz M, Cristea S, Gillespie SM, Patel AP, Mylvaganam R, Luo CC, Specht MC, Bernstein BE, Michor F, Ellisen LW, Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat. Commun 9, 3588 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guan F, Handa K, S.-i. Hakomori, Specific glycosphingolipids mediate epithelial-to-mesenchymal transition of human and mouse epithelial cell lines. Proc. Natl. Acad. Sci. U.S.A 106, 7461–7466 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liang YJ, Ding Y, Levery SB, Lobaton M, Handa K, S.-i. Hakomori, Differential expression profiles of glycosphingolipids in human breast cancer stem cells vs. cancer non-stem cells. Proc. Natl. Acad. Sci. U.S.A 110, 4968–4973 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huwiler A, Zangemeister-Wittke U, The sphingosine 1-phosphate receptor modulator fingolimod as a therapeutic agent: Recent findings and new perspectives. Pharmacol. Ther 185, 34–49 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Yu T, Di G, Role of tumor microenvironment in triple-negative breast cancer and its prognostic significance. Chin. J. Cancer Res 29, 237–252 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bae J, Munshi A, Li C, Samur M, Prabhala R, Mitsiades C, Anderson KC, Munshi NC, Heat shock protein 90 is critical for regulation of phenotype and functional activity of human T lymphocytes and NK cells. J. Immunol 190, 1360–1371 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.AlHossiny M, Luo L, Frazier WR, Steiner N, Gusev Y, Kallakury B, Glasgow E, Creswell K, Madhavan S, Kumar R, Upadhyay G, Ly6E/K signaling to TGFβ promotes breast cancer progression, immune escape, and drug resistance. Cancer Res 76, 3376–3386 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X, Sun R, Hao X, Lian ZX, Wei H, Tian Z, IL-17 constrains natural killer cell activity by restraining IL-15-driven cell maturation via SOCS3. Proc. Natl. Acad. Sci. U.S.A 116, 17409–17418 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Siemerink MJ, Klaassen I, Vogels IM, Griffioen AW, Van Noorden CJ, Schlingemann RO, CD34 marks angiogenic tip cells in human vascular endothelial cell cultures. Angiogenesis 15, 151–163 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fu B, Wang F, Sun R, Ling B, Tian Z, Wei H, CD11b and CD27 reflect distinct population and functional specialization in human natural killer cells. Immunology 133, 350–359 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fu B, Tian Z, Wei H, Subsets of human natural killer cells and their regulatory effects. Immunology 141, 483–489 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yuan X, Rasul F, Nashan B, Sun C, Innate lymphoid cells and cancer: Role in tumor progression and inhibition. Eur. J. Immunol 51, 2188–2205 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jacquelot N, Seillet C, Vivier E, Belz GT, Innate lymphoid cells and cancer. Nat. Immunol 23, 371–379 (2022). [DOI] [PubMed] [Google Scholar]
- 53.McCarthy A, Savage K, Gabriel A, Naceur C, Reis-Filho JS, Ashworth A, A mouse model of basal-like breast carcinoma with metaplastic elements. J. Pathol 211, 389–398 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Aprelikova O, Tomlinson CC, Hoenerhoff M, Hixon JA, Durum SK, Qiu T-H, He S, Burkett S, Liu Z-Y, Swanson SM, Green JE, Development and preclinical application of an immunocompetent transplant model of basal breast cancer with lung, liver and brain metastases. PLOS ONE 11, e0155262 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wan L, Lu X, Yuan S, Wei Y, Guo F, Shen M, Yuan M, Chakrabarti R, Hua Y, Smith HA, Blanco MA, Chekmareva M, Wu H, Bronson RT, Haffty BG, Xing Y, Kang Y, MTDH-SND1 interaction is crucial for expansion and activity of tumor-initiating cells in diverse oncogene- and carcinogen-induced mammary tumors. Cancer Cell 26, 92–105 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kumar S, Srivastav RK, Wilkes DW, Ross T, Kim S, Kowalski J, Chatla S, Zhang Q, Nayak A, Guha M, Fuchs SY, Thomas C, Chakrabarti R, Estrogen-dependent DLL1-mediated Notch signaling promotes luminal breast cancer. Oncogene 38, 2092–2107 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cadigan KM, Waterman ML, TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb. Perspect. Biol 4, a007906 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Spranger S, Gajewski TF, Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 18, 139–147 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhan T, Rindtorff N, Boutros M, Wnt signaling in cancer. Oncogene 36, 1461–1473 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lento W, Congdon K, Voermans C, Kritzik M, Reya T, Wnt signaling in normal and malignant hematopoiesis. Cold Spring Harb. Perspect. Biol 5, a008011 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khramtsov AI, Khramtsova GF, Tretiakova M, Huo D, Olopade OI, Goss KH, Wnt/beta-catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcome. Am. J. Pathol 176, 2911–2920 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chakrabarti R, Wei Y, Hwang J, Hang X, Andres Blanco M, Choudhury A, Tiede B, Romano RA, DeCoste C, Mercatali L, Ibrahim T, Amadori D, Kannan N, Eaves CJ, Sinha S, Kang Y, ΔNp63 promotes stem cell activity in mammary gland development and basal-like breast cancer by enhancing Fzd7 expression and Wnt signaling. Nat. Cell Biol 16, 1004–1013 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R, Weissman IL, A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 423, 409–414 (2003). [DOI] [PubMed] [Google Scholar]
- 64.Fuerer C, Nusse R, Lentiviral vectors to probe and manipulate the Wnt signaling pathway. PLOS ONE 5, e9370 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hayashi H, Nakagawa K, Combination therapy with PD-1 or PD-L1 inhibitors for cancer. Int. J. Clin. Oncol 25, 818–830 (2020). [DOI] [PubMed] [Google Scholar]
- 66.Györffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, Szallasi Z, An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat 123, 725–731 (2010). [DOI] [PubMed] [Google Scholar]
- 67.Poli A, Michel T, Theresine M, Andres E, Hentges F, Zimmer J, CD56bright natural killer (NK) cells: An important NK cell subset. Immunology 126, 458–465 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Quail DF, Joyce JA, Microenvironmental regulation of tumor progression and metastasis. Nat. Med 19, 1423–1437 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L, Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol 13, 674–690 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S, Functions of natural killer cells. Nat. Immunol 9, 503–510 (2008). [DOI] [PubMed] [Google Scholar]
- 71.Cooper MA, Fehniger TA, Caligiuri MA, The biology of human natural killer-cell subsets. Trends Immunol 22, 633–640 (2001). [DOI] [PubMed] [Google Scholar]
- 72.Moretta L, Dissecting CD56dim human NK cells. Blood 116, 3689–3691 (2010). [DOI] [PubMed] [Google Scholar]
- 73.Le Maux Chansac B, Moretta A, Vergnon I, Opolon P, Lecluse Y, Grunenwald D, Kubin M, Soria J-C, Chouaib S, Mami-Chouaib F, NK cells infiltrating a MHC class I-deficient lung adenocarcinoma display impaired cytotoxic activity toward autologous tumor cells associated with altered NK cell-triggering receptors. J. Immunol 175, 5790–5798 (2005). [DOI] [PubMed] [Google Scholar]
- 74.Platonova S, Cherfils-Vicini J, Damotte D, Crozet L, Vieillard V, Validire P, Andre P, Dieu-Nosjean MC, Alifano M, Regnard J-F, Fridman W-H, Sautes-Fridman C, Cremer I, Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res 71, 5412–5422 (2011). [DOI] [PubMed] [Google Scholar]
- 75.Woan KV, Miller JS, Harnessing natural killer cell antitumor immunity: From the bench to bedside. Cancer Immunol. Res 7, 1742–1747 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Laskowski TJ, Biederstadt A, Rezvani K, Natural killer cells in antitumour adoptive cell immunotherapy. Nat. Rev. Cancer 22, 557–575 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gotthardt D, Trifinopoulos J, Sexl V, Putz EM, JAK/STAT cytokine signaling at the crossroad of NK cell development and maturation. Front. Immunol 10, 2590 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kershaw NJ, Murphy JM, Liau NP, Varghese LN, Laktyushin A, Whitlock EL, Lucet IS, Nicola NA, Babon JJ, SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat. Struct. Mol. Biol 20, 469–476 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Babon JJ, Kershaw NJ, Murphy JM, Varghese LN, Laktyushin A, Young SN, Lucet IS, Norton RS, Nicola NA, Suppression of cytokine signaling by SOCS3: Characterization of the mode of inhibition and the basis of its specificity. Immunity 36, 239–250 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chakrabarti R, Celia-Terrassa T, Kumar S, Hang X, Wei Y, Choudhury A, Hwang J, Peng J, Nixon B, Grady JJ, DeCoste C, Gao J, van Es JH, Li MO, Aifantis I, Clevers H, Kang Y, Notch ligand Dll1 mediates cross-talk between mammary stem cells and the macrophageal niche. Science 360, eaan4153 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lindley LE, Curtis KM, Sanchez-Mejias A, Rieger ME, Robbins DJ, Briegel KJ, The WNT-controlled transcriptional regulator LBH is required for mammary stem cell expansion and maintenance of the basal lineage. Development 142, 893–904 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Patel S, Alam A, Pant R, Chattopadhyay S, Wnt signaling and its significance within the tumor microenvironment: Novel therapeutic insights. Front. Immunol 10, 2872 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Castagnoli L, Cancila V, Cordoba-Romero SL, Faraci S, Talarico G, Belmonte B, Iorio MV, Milani M, Volpari T, Chiodoni C, Hidalgo-Miranda A, Tagliabue E, Tripodo C, Sangaletti S, Di Nicola M, Pupa SM, WNT signaling modulates PD-L1 expression in the stem cell compartment of triple-negative breast cancer. Oncogene 38, 4047–4060 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vikas P, Borcherding N, Zhang W, The clinical promise of immunotherapy in triple-negative breast cancer. Cancer Manag. Res 10, 6823–6833 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Waks AG, Winer EP, Breast cancer treatment: A review. JAMA 321, 288–300 (2019). [DOI] [PubMed] [Google Scholar]
- 86.Shackleton M, Vaillant F, Simpson KJ, Stingl J, Smyth GK, Asselin-Labat M-L, Wu L, Lindeman GJ, Visvader JE, Generation of a functional mammary gland from a single stem cell. Nature 439, 84–88 (2006). [DOI] [PubMed] [Google Scholar]
- 87.Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, Gregory MT, Shuga J, Montesclaros L, Underwood JG, Masquelier DA, Nishimura SY, Schnall-Levin M, Wyatt PW,Hindson CM, Bharadwaj R, Wong A, Ness KD, Beppu LW, Deeg HJ, McFarland C,Loeb KR, Valente WJ, Ericson NG, Stevens EA, Radich JP, Mikkelsen TS, Hindson BJ, Bielas JH, Massively parallel digital transcriptional profiling of single cells. Nat. Commun 8, 14049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R, Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, Satija R, Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kiessling R, Klein E, Pross H, Wigzell H, “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur. J. Immunol 5, 117–121 (1975). [DOI] [PubMed] [Google Scholar]
- 91.Herberman RB, Nunn ME, Lavrin DH, Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic acid allogeneic tumors. I. Distribution of reactivity and specificity. Int. J. Cancer 16, 216–229 (1975). [DOI] [PubMed] [Google Scholar]
- 92.Godfrey DI, MacDonald HR, Kronenberg M, Smyth MJ, Van Kaer L, NKT cells: What’s in a name? Nat. Rev. Immunol 4, 231–237 (2004). [DOI] [PubMed] [Google Scholar]
- 93.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, Butte AJ, Bhattacharya M, Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol 20, 163–172 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Heng TS, Painter MW; Immunological Genome Project Consortium, The Immunological Genome Project: Networks of gene expression in immune cells. Nat. Immunol 9, 1091–1094 (2008). [DOI] [PubMed] [Google Scholar]
- 95.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Anders S, Huber W, Differential expression analysis for sequence count data. Genome Biol 11, R106 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chakrabarti R, Hwang J, Andres Blanco M, Wei Y, Lukacisin M, Romano R-A, Smalley K, Liu S, Yang Q, Ibrahim T, Mercatali L, Amadori D, Haffty BG, Sinha S, Kang Y, Elf5 inhibits the epithelial-mesenchymal transition in mammary gland development and breast cancer metastasis by transcriptionally repressing Snail2. Nat. Cell Biol 14, 1212–1222 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Choi YS, Chakrabarti R, Escamilla-Hernandez R, Sinha S, Elf5 conditional knockout mice reveal its role as a master regulator in mammary alveolar development: Failure of Stat5 activation and functional differentiation in the absence of Elf5. Dev. Biol 329, 227–241 (2009). [DOI] [PubMed] [Google Scholar]
- 99.Raj-Kumar P-K, Liu J, Hooke JA, Kovatich AJ, Kvecher L, Shriver CD, Hu H, PCA-PAM50 improves consistency between breast cancer intrinsic and clinical subtyping reclassifying a subset of luminal A tumors as luminal B. Sci. Rep 9, 7956 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. scRNA-seq and bulk mRNA-seq datasets were deposited into the NCBI database (www.ncbi.nlm.nih.gov/), Sequence Read Archive PRJNA685201.