

Abstract
Salmonella enterica serotype Typhi and nontyphoidal Salmonella remain major causes of morbidity and mortality worldwide. Ampicillin, trimethoprim-sulfamethoxazole, and chloramphenicol no longer provide reliable coverage of Salmonella, and fluoroquinoloes have emerged as first-line treatment options. Due to mounting evidence of decreased in vitro susceptibility and diminished clinical response to fluoroquinolone therapy, it has been suggested that the NCCLS breakpoints for the salmonellae be reevaluated. We utilized an in vitro infection model to determine which pharmacokinetic-pharmacodynamic (PK-PD) measure was most closely linked to fluoroquinolone activity against salmonellae and the magnitude that was predictive of efficacy. Monte Carlo simulation was utilized to determine the probability of attaining potential susceptibility breakpoints for three fluoroquinolones. The free-drug area under the concentration-time curve from 0 to 24 h/MIC ratio was the PK-PD measure most predictive of efficacy, and a ratio of 105 corresponded to 90% of maximal activity. Simulation results suggested susceptible breakpoints of 0.12 μg/ml for ciprofloxacin and gatifloxacin and 0.25 μg/ml for levofloxacin. These proposed breakpoints correspond to the MIC separating the wild-type susceptible organism population from those strains possessing single-step mutations in the quinolone resistance-determining region. These results that integrate PK-PD measures and fluoroquinolone MIC distributions in the genetic context of examined Salmonella isolates clearly demonstrate that the prudent use of a lower susceptibility breakpoint minimizes the probability of clinical failure or delayed response in fluoroquinolone-treated patients.
Over the last decade, fluoroquinolones have emerged as the mainstay of therapy for invasive infection associated with Typhi and nontyphoidal Salmonella enterica serotypes. At the same time, the increasing incidence of infection with salmonellae resistant to nalidixic acid, which usually also display decreased susceptibility to fluoroquinolones, has raised considerable global concern (1, 10). The incidence of infection with nalidixic acid-resistant S. enterica serotype Typhi has been reported to be as high as 23.2% by the National Antimicrobial Resistance Monitoring System of the Centers for Disease Control and Prevention (2). The vast majority of nalidixic acid-resistant strains remain within the current susceptible range for ciprofloxacin (≤1 μg/ml) as recommended by the National Committee for Clinical Laboratory Standards (NCCLS). However, the probability of clinical response to fluoroquinolone therapy in patients with invasive Salmonella infection is lower in those with nalidixic acid-resistant than with -susceptible strains (7, 20, 21, 26, 32, 33). These data suggest that the current susceptibility breakpoints for fluoroquinolones published by the NCCLS may need to be altered to better predict clinical efficacy. Moreover, changing susceptibility breakpoints for fluoroquinolones will clearly negate the need for use of the nalidixic acid screening test, which is recommended by the NCCLS (25).
Nonclinical infection models, such as animal or in vitro models, are fundamental tools in the evaluation of the therapeutic efficacies of antimicrobial agents. From these models, one can gain important information about the time course of antimicrobial effect, which can be used to construct exposure-response relationships and to determine pharmacokinetic-pharmacodynamic (PK-PD) target measures that are predictive of clinical efficacy in humans (9). Over the past 5 years, Monte Carlo simulation has been used to integrate human pharmacokinetic data and PK-PD targets derived from nonclinical or clinical data in order to provide decision support for determination of in vitro susceptibility breakpoints (12, 13; P. G. Ambrose, Abstr. 42nd Intersci. Conf. Antimicrob. Agents Chemother., abstr. 1020, 2002; P. G. Ambrose, W. A. Craig, S. M. Bhavnani, and M. N. Dudley, Abstr. 42nd Intersci. Conf. Antimicrob. Agents Chemother., abstr. A-635, 2002; M. N. Dudley and P. G. Ambrose, Abstr. 42nd Intersci. Conf. Antimicrob. Agents Chemother., abstr. A-1263, 2002). This approach to evaluating antimicrobial regimens may be especially valuable in the circumstance when formal exposure-ranging clinical trials are unlikely, which may be the case in the study of invasive salmonellae infection, uncommon pathogens, unique patient populations, or agents of bioterrorism.
The objectives of these studies were fourfold: (i) to identify the PK-PD measure (i.e., free-drug [f] area under the concentration-time curve from 0 to 24 h [AUC0-24]/MIC, f Cmax/MIC, or the duration of time free drug concentrations remain above the MIC [T>MIC]) that best predicts efficacy; (ii) to determine the magnitude of the PK-PD measure required for 90, 95, and 99% maximal efficacy; (iii) to utilize Monte Carlo simulation to integrate human pharmacokinetic data and PK-PD magnitude targets in an effort to determine MIC susceptibility breakpoints of ciprofloxacin, gatifloxacin, and levofloxacin when testing salmonellae; and (iv) to correlate these measures with contemporary fluoroquinolone MIC population statistics.
MATERIALS AND METHODS
Bacteria, media, and susceptibility testing.
Two serotype Typhi isolates from the JONES Group/JMI Laboratories were selected for these experiments. These strains were from patients with bloodstream infections hospitalized in Italy and India. Isolate 85-1416G had an elevated MIC of fluoroquinolones of 0.25 to 0.5 μg/ml (gatifloxacin, 0.5 μg/ml), and strain G6/9 had a nonsusceptible-level MIC of fluoroquinolones (gatifloxacin, 4 μg/ml). The MICs were determined by replicate tests in cation-adjusted Mueller-Hinton (MH) broth by methods outlined by the NCCLS (24) and interpreted by M100-S14 (25). American Type Culture Collection control strain Escherichia coli ATCC 25922 was used for quality assurance of susceptibility testing. The quinolone resistance-determining regions (QRDR) for genes parC, parE, gyrA, and gyrB were amplified by PCR using primers and cycling conditions previously described (30). Sequencing was performed in both strands by the dideoxy chain termination method with a Perkin-Elmer Biosystems 377 DNA sequencer. The sequences were analyzed using the Lasergene software package (DNASTAR, Madison, WI).
The in vitro pharmacodynamic model also used cation-adjusted MH broth (Difco Laboratories, Detroit, MI), and quantitation of isolates was performed on MH agar plates. Gatifloxacin powder was provided by the Bristol-Myers Squibb Company (lot 0175650; Wallingford, CT). Immediately prior to each experiment, gatifloxacin powder was dissolved in 0.2 ml of 0.1 M NaOH, diluted to a final volume with distilled water, and sterilized by passage through a 0.2-μm syringe filter membrane.
MIC population distributions were generated by the SENTRY Antimicrobial Surveillance Program using reference, validated broth microdilution methods for the following fluoroquinolones: ciprofloxacin, gatifloxacin, and levofloxacin (30).
In vitro model and sample processing.
The one-compartment in vitro pharmacodynamic model used in these studies has been described previously (17). The model consists of a central infection compartment containing bacteria, bacterial growth medium, and magnetic stir bars, which ensure sufficient content mixture, all of which are placed into a temperature-controlled (37°C) circulating water bath. Drug-free bacterial growth medium is delivered into the central infection compartment by using a high-precision computer-programmable peristaltic pump; simultaneously, bacterial growth medium is removed from the central compartment via an exit port and contained in a waste flask. The test antimicrobial is introduced into the central infection compartment aseptically through a port sealed with a rubber septum, and the peristaltic pump rate is set in a manner that allows for the simulation of human drug clearance. At various times, the central infection compartment is aseptically sampled using a sterile syringe and needle for bacteria CFU (CFU/ml) and/or drug concentration determinations through a rubber septum-sealed port.
In these experiments, an initial inoculum of 107 CFU/ml of the test strain was prepared from an overnight culture. Following inoculation of the central infection compartment with the test strain, growth was permitted for 1 h to ensure logarithmic-phase growth prior to the introduction of gatifloxacin. Bacteria were then exposed to changing gatifloxacin concentrations simulating a gatifloxacin half-life of 8 h, similar to that observed in humans (19). Once-daily regimens were simulated to deliver steady-state fAUC/MIC ratios ranging from 6 to 185. Each experiment was run with appropriate growth control studies. Results from the once-daily dose-ranging studies guided further fractionated studies. The doses that resulted in exposures on the steep part of the exposure-response curve were subsequently given either on an every-12-hour schedule or as a continuous infusion. To confirm the simulation of human pharmacokinetic parameters, samples were collected throughout each model experiment and stored at −70°C until they were assayed for drug concentration, as described below. All studies were conducted over a period of 24 h.
Quantitative cultures were assessed at −1, 0, 0.25, 0.5, 1, 2, 3, 4, 6, 8, 12, and 24 h. For twice-daily regimens, quantitative cultures were also assessed at 13 and 14 h. All studies were performed in duplicate. Bacterial counts were determined by serially diluting 100-μl aliquots in cold 0.9% sodium chloride, with 10- and 100-μl aliquots of each dilution plated on MH agar plates. Plated samples were then incubated at 37°C for 24 h. The mean change in log10 CFU/ml was calculated for each duplicate study, and time-kill curves were constructed by plotting log10 CFU/ml versus time. The lower limit of detection was 102 CFU/ml.
Fluoroquinolone concentration determinations.
Aliquots from each sample obtained from each of the treatment models were placed in cryovials and stored at −70°C until assayed for gatifloxacin concentration. Gatifloxacin concentrations were determined by a previously validated ion-paired high-performance liquid chromatography method assay (19). The assay of gatifloxacin was linear over the range from 0.1 to 4 μg/ml. The intraday coefficients of variation for the low and high concentrations were 1.3 and 2.8, respectively. The interday coefficients of variation for the low and high concentrations were 3.6 and 1.0, respectively. The lower limit of quantification of the assay was 0.03 mg/liter.
Dose-ranging studies for establishing 25, 50, and 90% Emax.
Initial dose-ranging experiments, consisting of once-daily administration of gatifloxacin, were conducted to characterize the dose-response relationship versus salmonellae and to further guide dose selection for dose fractionation studies. The chosen dosing regimens yielded fCmax gatifloxacin concentrations ranging from 0.5 to 64 μg/ml, resulting in fCmax/MIC and fAUC0-24/MIC ratios ranging from 1 to 128 and 6 to 185, respectively. Administration of doses resulting in fCmax concentrations higher than those typically observed in humans were required to fully delineate the sigmoidal maximal effect (Emax) dose-response relationship of gatifloxacin against salmonellae.
PK-PD analyses and Monte Carlo simulation.
Noncompartmental methods (WINNONLIN version 2.1; Pharsight Corp, Lexington, KY) and actual drug concentration data from each experiment were used to determine the following pharmacokinetic parameters: fCmax, elimination rate constant, half-life, and fAUC0-24. The fAUC0-24 was calculated using the trapezoidal method. Experimental pharmacokinetic and baseline gatifloxacin MIC data were used to determine the following PK-PD measures for each experiment: fAUC0-24/MIC ratio, fCmax/MIC ratio, and percent T>MIC.
To accommodate all available data generated for each regimen tested and avoid biased conclusions based on single time points, an integrated PK-PD area measure was applied to all CFU data. For each regimen tested, the area under the CFU versus time curve (AUCFU) from 0 to 24 h was calculated via the trapezoidal rule for both growth control and drug-containing regimens. Drug effect was quantified by normalizing the 24-h drug AUCFU by the growth control area and taking the logarithm of the ratio, as in equation 1.
 |
(1) |
Log ratio values of zero indicate no drug effect, with larger negative values indicative of increasing drug effect. For example, a −1 log ratio implies 90% less area in the drug-containing experiment over 24 h compared to that with the growth control. Using nonlinear regression (version 10; Systat Software Inc., Richmond, CA), a Hill-type model was fit to the log ratio of the AUCFU to obtain estimates of Emax (fitted maximum log10 reduction in bacteria), fAUC0-24/MIC (the ratio required to achieve 50% of Emax), and H (Hill's constant, which accommodates sigmoidicity) (equation 2).
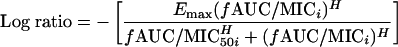 |
(2) |
Based on parameter estimates of Emax, fAUC0-24/MIC, and Hill's constant, the dose required to provide 25, 50, and 90% of Emax was calculated and a series of dose fractionation studies were conducted to determine whether the fAUC0-24/MIC ratio, fCmax/MIC ratio, or the T>MIC best characterized the effect of gatifloxacin on salmonellae. Each regimen, either twice-daily dosing or continuous infusion, delivered the same overall 24-hour drug exposure, providing a constant fAUC0-24/MIC ratio but differing fCmax/MIC or T>MIC values. Significance of differences in gatifloxacin activity between regimens that received the same total exposure (fAUC0-24) as once, twice, or continuous infusion dosing were evaluated by general linear modeling using backwards stepping, with the log10 ratio (AUCFUdrug/AUCFUgrowth control) as the dependent variable and the fAUC0-24/MIC ratio, fCmax/MIC ratio, and the T>MIC as the independent variables.
PK-PD target attainment analyses were carried out using Monte Carlo simulation. Dosing regimens modeled included gatifloxacin (400 mg once daily), ciprofloxacin (500 mg twice daily), and levofloxacin (500 mg once daily). Five thousand patient simulations were carried out using Crystal Ball, 2000.1 by Decisioneering, Inc. (Denver, Colo.) and using equation 3.
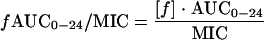 |
(3) |
Pharmacokinetics from healthy subjects were obtained from Food and Drug Administration-approved product labels (7, 14, 18, 27). The means (± standard deviations) AUC0-24 for gatifloxacin, ciprofloxacin, and levofloxacin was 34.4 (5.7), 28.2 (5.4), and 47.5 (6.7), respectively. The fraction of total drug bound to serum proteins was assumed to be 0.20, 0.30, and 0.31 for gatifloxacin, ciprofloxacin, and levofloxacin, respectively. The probability of attaining 90% Emax was estimated for each agent.
RESULTS
Susceptibility testing, organism growth in the model, and MIC distributions for Salmonella spp.
Based upon current NCCLS susceptibility breakpoints (29), one serovar Typhi strain (85-1416G) was susceptible to gatifloxacin with a MIC of 0.5 μg/ml, in contrast to strain G6/9, which had a gatifloxacin MIC of 4 μg/ml (intermediate). Sequencing of the QRDR revealed that strain 85-1416G had a single gyrA mutation at Asp87→Asn and strain G6/9 had four mutations as follows: gyrA, Ser83→Tyr and Asp87→Gly; gyrB, none; parC, Thr57→Ser and Ser80Ile; and parE, none. Both strains had a screening nalidixic acid MIC result of ≥32 μg/ml and ciprofloxacin MICs at >0.12 μg/ml (29). These organisms exhibited logarithmic growth prior to initiation of antimicrobial therapy in the model. Bacterial numbers at the start of therapy ranged from 7.5 to 8.1 log10 CFU/ml. Each isolate grew from 7.4 to 9.3 log10 CFU/ml by 24 h in the untreated control models.
The SENTRY Program MIC population distributions generally exhibited three peaks for the monitored fluoroquinolones (see Fig. 4, below). These peaks were characterized by MICs at ≤0.06 μg/ml, 0.12 to 0.5 μg/ml, and ≥8 μg/ml. Previous studies with these organisms exhibited elevated nalidixic acid MIC results (≥32 μg/ml) for all isolates having ciprofloxacin, gatifloxacin, or levofloxacin MICs of ≥0.25 μg/ml, each possessing a single gyrA target mutation (30), and greater numbers of mutations for Salmonella isolates having MICs in the current resistant range (29).
FIG. 4.

Fractional probability of PK-PD target (fAUC/MIC0-24 ≥ 105) attainment for ciprofloxacin, gatifloxacin, and levofloxacin and MIC histogram of ciprofloxacin, gatifloxacin, and levofloxacin against Salmonella spp. The MIC distribution was provided courtesy of the SENTRY Antimicrobial Surveillance Program (n = 2,805) (data from reference 30). Grey bars, ciprofloxacin; black bars, gatifloxacin; white bars, levofloxacin.
Pharmacokinetics.
The gatifloxacin pharmacokinetic profiles observed in the models were similar to intended target profiles. The mean (± standard deviation) simulated half-life observed in the model was 7.91 (1.12) hours. In each model experiment, the observed fAUC0-24 was within 15% of the targeted value, while the fCmax was within 10% of the targeted value.
In vitro efficacy.
The mean changes in log10 CFU/ml over time for gatifloxacin administered once daily against gatifloxacin-susceptible (85-1416G) and intermediately susceptible (G6/9) serovar Typhi strains are presented in Fig. 1 and 2, respectively. The pattern of bactericidal activity was similar for both strains. When fAUC0-24 ratios were 92 or greater, viable bacterial counts decreased 4 to 5.5 log units 3 to 4 h after dosing and no significant regrowth occurred by 24 h. Lesser exposures (fAUC0-24 ratios of ≤46) resulted in either no substantial bactericidal activity or an initial 3.8- to 4.6-log unit decrease in viable bacterial count followed by extensive regrowth. When the total drug exposure was delivered as a single dose, gatifloxacin was more rapidly bactericidal against both strains compared to fractionated or continuous infusion regimens (data not shown).
FIG. 1.

Mean PK-PD time-kill curves of gatifloxacin administered once daily against a gatifloxacin-susceptible serovar Typhi strain (85-1416G) (MIC, 0.5 μg/ml) in an in vitro infection model. The graph for the experiment with an AUC/MIC ratio of 185 was truncated following sterilization of the model (CFU/ml below limit of quantification).
FIG. 2.

Mean PK-PD time-kill curves of gatifloxacin administered once daily against a gatifloxacin-resistant serovar Typhi strain (G6/9) (MIC, 4 μg/ml) in an in vitro infection model. Gatifloxacin exposure was initiated at time = 1 h.
Correlation between PK-PD measures and in vitro efficacy.
As shown in Fig. 3, there was a strong correlation between bacterial killing and the fAUC0-24/MIC (R2 = 0.96) and the fCmax/MIC (R2 = 0.93) ratios for both gatifloxacin-susceptible and intermediately susceptible serovar Typhi strains. Testing for which PK-PD measure, either fAUC0-24/MIC or fCmax/MIC, best correlated with in vitro efficacy could not be done, since both parameters were colinear given our experimental design. The correlation between bacterial killing and the duration of time that free-drug gatifloxacin concentrations remained above the MIC was poor by comparison (R2 = 0.68).
FIG. 3.

Relationships between gatifloxacin fAUC0-24/MIC ratio (left), fCmax/MIC ratio (middle), and T>MIC (right) for two strains of serovar Typhi with differing MICs and changes in bacterial density. The square symbols represent a strain with a gatifloxacin MIC of 0.5 μg/ml, while the circles represent a strain with a gatifloxacin MIC of 4 μg/ml. GC, growth control.
Model-fitted parameter estimates (Table 1) did not differ significantly by strain (data not shown) and were therefore pooled for analysis. The fAUC0-24/MIC and fCmax/MIC ratios associated with 50% maximal gatifloxacin activity were 34.7 and 2.61, respectively. The fAUC0-24/MIC ratios associated with 90, 95, and 99% Emax were 105, 152, and 348, respectively. Similarly, the fCmax/MIC ratios associated with 90, 95, and 99% Emax were 12.7, 21.7, and 71.2, respectively (Table 1). The AUC/MIC ratios required for stasis, 1-, and 2-log10 reductions in bacteria at 24 h were approximately 33, 41, and 51, respectively.
TABLE 1.
Model-fitted parameter estimates and magnitude of the fAUC0-24/MIC and fCmax/MIC ratios producing 90, 95, and 99% Emax
| Model-fitted parameter | fAUC0-24/MICa | fCmax/MICa |
|---|---|---|
| Emax | −3.58 (−4.1, −3.0) | −3.80 (−4.5, −2.8) |
| Hill's constant | 2.04 (1.2, 2.8) | 1.39 (0.7, 2.4) |
| IC50b | 34.7 (27.0, 43.1) | 2.61 (1.6, 3.9) |
| 90% Emax | 105 | 12.7 |
| 95% Emax | 152 | 21.7 |
| 99% Emax | 348 | 71.2 |
Data are reported as maximum likelihood parameter estimates (Wald 95% confidence intervals are shown in parentheses).
IC50, 50% inhibitory concentration.
Monte Carlo simulation.
The fractional probability of PK-PD target attainment for an fAUC0-24/MIC ratio of 105 is presented in Fig. 4. An fAUC0-24/MIC ratio of 105, an exposure associated with 90% of maximal drug effect, was chosen as the PK-PD target. fAUC0-24/MIC ratios greater than 105 appeared to provide relatively little additional antibacterial effect. The probability of attaining a PK-PD target fAUC0-24/MIC ratio of 105 was high (>0.90) for all fluoroquinolones evaluated up to and including MICs of 0.12 μg/ml. Differences in target attainment among the fluoroquinolones were observed for MICs of 0.25 μg/ml. The probabilities of gatifloxacin and ciprofloxacin target attainment were 61.2 and 5.4%, whereas for levofloxacin the probability of PK-PD target attainment was 92.1%. The probability of attaining an fAUC0-24/MIC ratio of at least 105 approached zero for all three fluoroquinolones for MICs of 0.5 μg/ml and greater.
DISCUSSION
Over the last several years there has been growing controversy regarding the appropriateness of current NCCLS fluoroquinolone breakpoints for Salmonella (1, 10). Mounting clinical evidence suggests that infections due to serovar Typhi and nontyphoidal strains resistant to nalidixic acid but susceptible to ciprofloxacin by NCCLS criteria may not respond to fluoroquinolone therapy (8, 20, 21, 24, 31-33). While the reasons for treatment failure are multifactorial, PK-PD remains an important predictor of response to antimicrobial therapy, and failure to achieve adequate PK-PD targets may be a likely contributor to poor clinical outcomes. Based on our results, the current clinical breakpoints (1 to 2 μg/ml) are too high for fluoroquinolones against Salmonella, as standard dosing regimens provide poor PK-PD target attainment for MICs higher than 0.25 μg/ml, regardless of the fluoroquinolone considered. As serovar Typhi and nontyphoidal salmonellae are major causes of morbidity and mortality in many areas of the world and orally administered fluoroquinolones have emerged as first-line therapy, the reassessment of fluoroquinolone susceptibility breakpoints is imperative.
Nonclinical infection models, such as in vitro or animal models, have proven useful in predicting effective dosing regimens in humans and in establishing susceptibility breakpoints (4-6, 9, 12, 13, 15, 16, 29). These tools may be especially useful when formal exposure-ranging clinical trials are unlikely, which appears to be the case in the study of invasive, extraintestinal Salmonella infections. Further, the exposure-response relationships of fluoroquinolones determined using in vitro infection models appear to be generally concordant with those derived from animal infection models. Moreover, a number of clinical studies involving fluoroquinolones have demonstrated similar exposure-response relationships as those derived from animal and in vitro infection models, for both gram-positive and -negative organisms. Findings from in vitro and animals models are in agreement with clinical data, where fAUC0-24/MIC ratios of 125 are associated with optimal fluoroquinolone activity for gram-negative bacilli (3, 5, 6, 9, 11, 16, 22, 28).
In this analysis, we utilized an in vitro infection model to identify the fAUC0-24/MIC ratio as the PK-PD measure most closely associated with fluoroquinolone efficacy against Salmonella, and we found that an fAUC0-24/MIC ratio of 105 is associated with a 90% maximal drug effect. A target fAUC0-24/MIC ratio of 105 identified by our in vitro model is consistent with previous animal and human studies involving the optimization of fluoroquinolones for treatment of other gram-negative infections. Forrest et al. identified a ciprofloxacin AUC0-24/MIC ratio of 125 to be a significant breakpoint for both clinical and microbiological cures in seriously ill patients (16). Additionally, Andes and Craig identified similar PK-PD targets for gatifloxacin against enteric gram-negative bacilli and Pseudomonas aeruginosa in murine thigh and lung infection models (5).
There are few published animal studies providing adequate information to evaluate the PK-PD of fluoroquinolones against Salmonella and no human clinical trials. Magallanes et al. reported the results of a standard formulation of ciprofloxacin treatment with that of a liposomal preparation in a murine model of salmonellosis (23). Mice infected with a strain of S. enterica serovar Dublin (ciprofloxacin MIC of 0.03 μg/ml) by oral gavage were treated with a limited number of ciprofloxacin dosage regimens. Without treatment, mortality in this model was 100%. The 50% effective dose of the standard ciprofloxacin formulation was reported to be 5 mg/kg of body weight, corresponding to a total drug AUC0-24/MIC ratio of approximately 82. A total drug AUC0-24/MIC ratio of approximately 130 was effective in curing three of five animals studied. These observations are generally consistent with the results obtained in our in vitro infection model. Interestingly, the liposomal preparation of ciprofloxacin was more effective at similar plasma concentrations, due to an increased uptake of the drug into infected tissues compared to the standard ciprofloxacin formulation.
We utilized our model-identified fAUC0-24/MIC ratio target of 105 and Monte Carlo simulation to determine PK-PD-based susceptibility breakpoints for ciprofloxacin, gatifloxacin, and levofloxacin for Salmonella. PK-PD-based susceptibility breakpoints for fluoroquinolones in humans may be estimated as the highest MIC for which the fractional PK-PD target attainment is approximately 0.9 or higher. Based on standard oral dosing regimens for ciprofloxacin (500 mg twice daily), gatifloxacin (400 mg once daily), and levofloxacin (500 mg once daily), the PK-PD-derived susceptibility breakpoints would be 0.12, 0.12, and 0.25 mg/liter, respectively. Clearly, the existing breakpoints for these fluoroquinolones of ≤1 to ≤2 μg/ml would not detect those isolates which might be predicted to result in poor clinical outcomes.
There are a number of important issues to consider when evaluating the clinical applicability of these data. The in vitro infection model utilized in these experiments does not completely replicate the pathophysiology of all salmonellae-associated diseases in humans. In the case of typhoid fever, salmonellae may reside intracellularly, which is a space that is well penetrated by fluoroquinolones. However, some of the failures observed clinically may be attributable to the nature of the intracellular milieu rather than underdosing of the antimicrobial agent. It should be noted that healthy volunteer pharmacokinetic data were utilized for a Monte Carlo simulation, which may differ from those for patients with mesenteric lymphadenitis. Also, the MIC distributions could dictate a 1-log2 correction to better discriminate between organism populations with or without topoisomerase target mutations. In this case, a common susceptible breakpoint of ≤0.06 μg/ml without an intermediate category separates the organisms with highest accuracy. One could argue that salmonellae, and indeed any Enterobacteriaceae organism, with MICs of 0.12 to 0.5 μg/ml could be categorized as intermediate, thus indicating the need for maximal doses or combination antibiotic regimens.
In conclusion, our studies suggest that, based on PK-PD principles, the current NCCLS-approved fluoroquinolone breakpoints for Salmonella isolates are likely too high and may place patients at risk for treatment failure. These data may be useful as decision support in the breakpoint reevaluation process for fluoroquinolones and Salmonella isolates. Additional in vitro, animal, and human PK-PD studies are needed to better characterize the pharmacological effects of antibiotics in the treatment of Salmonella infection.
Acknowledgments
This study was supported in part by a grant from the Bristol-Myers Squibb Company.
REFERENCES
- 1.Aarestrup, F. M., C. Wiuff, K. Molbak, and E. J. Threlfall. 2003. Is it time to change fluoroquinolone breakpoints for Salmonella spp. Antimicrob. Agents Chemother. 47:827-829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ackers, M. L., N. D. Puhr, R. V. Tauxe, and E. D. Mintz. 2000. Laboratory-based surveillance of Salmonella serotype Typhi infections in the United States: antimicrobial resistance on the rise. JAMA 283:2668-2673. [DOI] [PubMed] [Google Scholar]
- 3.Ambrose, P. G., S. M. Bhavnani, and R. C. Owens, Jr. 2003. Clinical pharmacodynamics of quinolones. Infect. Dis. Clin. North Am. 17:529-543. [DOI] [PubMed] [Google Scholar]
- 4.Ambrose, P. G., D. M. Grasela, T. H. Grasela, J. Passarell, H. B. Mayer, and P. F. Pierce. 2001. Pharmacodynamics of fluoroquinolones against Streptococcus pneumoniae in patients with community-acquired respiratory tract infections. Antimicrob. Agents Chemother. 45:2793-2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andes, D., and W. A. Craig. 2002. Pharmacodynamics of the new fluoroquinolone gatifloxacin in murine thigh and lung infection models. Antimicrob. Agents Chemother. 46:1665-1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andes, D. R., and W. A. Craig. 1998. Pharmacodynamics of fluoroquinolones in experimental models of endocarditis. Clin. Infect. Dis. 27:47-50. [DOI] [PubMed] [Google Scholar]
- 7.Bristol-Myers Squibb Company. 2003. Gatifloxacin prescribing information. Bristol-Myers Squibb, Wallingford, Conn.
- 8.Chandel, D. S., and R. Chaudhry. 2001. Enteric fever treatment failures: a global concern. Emerg. Infect. Dis. 7:762-763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Craig, W. A. 1998. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin. Infect. Dis. 26:1-10. [DOI] [PubMed] [Google Scholar]
- 10.Crump, J. A., T. J. Barrett, J. T. Nelson, and F. J. Angulo. 2003. Reevaluating fluoroquinolone breakpoints for Salmonella enterica serotype Typhi and for non-Typhi salmonellae. Clin. Infect. Dis. 37:75-81. [DOI] [PubMed] [Google Scholar]
- 11.Drusano, G., M. T. Labro, O. Cars, P. Mendes, P. Shah, F. Sorgel, and W. Weber. 1998. Pharmacokinetics and pharmacodynamics of fluoroquinolones. Clin. Microbiol. Infect. 4(Suppl. 2):S27-S41. [PubMed] [Google Scholar]
- 12.Drusano, G. L., S. L. Preston, C. Hardalo, R. Hare, C. Banfield, D. Andes, O. Vesga, and W. A. Craig. 2001. Use of preclinical data for selection of a phase II/III dose for evernimicin and identification of a preclinical MIC breakpoint. Antimicrob. Agents Chemother. 45:13-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dudley, M. N., and P. G. Ambrose. 2000. Pharmacodynamics in the study of drug resistance and establishing in vitro susceptibility breakpoints: ready for prime time. Curr. Opin. Microbiol. 3:515-521. [DOI] [PubMed] [Google Scholar]
- 14.Flor, S. C., M. C. Rogge, and A. T. Chow. 1993. Bioequivalence of oral and intravenous ofloxacin after multiple-dose administration to healthy male volunteers. Antimicrob. Agents Chemother. 37:1468-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forrest, A., S. Chodosh, M. A. Amantea, D. A. Collins, and J. J. Schentag. 1997. Pharmacokinetics and pharmacodynamics of oral grepafloxacin in patients with acute bacterial exacerbations of chronic bronchitis. J. Antimicrob. Chemother. 40(Suppl. A):45-57. [DOI] [PubMed] [Google Scholar]
- 16.Forrest, A., D. E. Nix, C. H. Ballow, T. F. Goss, M. C. Birmingham, and J. J. Schentag. 1993. Pharmacodynamics of intravenous ciprofloxacin in seriously ill patients. Antimicrob. Agents Chemother. 37:1073-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garrison, M. W., K. Vance-Bryan, T. A. Larson, J. P. Toscano, and J. C. Rotschafer. 1990. Assessment of effects of protein binding on daptomycin and vancomycin killing of Staphylococcus aureus by using an in vitro pharmacodynamic model. Antimicrob. Agents Chemother. 34:1925-1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Israel, D., J. G. Gillum, M. Turik, K. Harvey, J. Ford, H. Dalton, M. Towle, R. Echols, A. H. Heller, and R. Polk. 1993. Pharmacokinetics and serum bactericidal titers of ciprofloxacin and ofloxacin following multiple oral doses in healthy volunteers. Antimicrob. Agents Chemother. 37:2193-2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.LaCreta, F. P., G. D. Kollia, G. Duncan, D. Behr, and D. M. Grasela. 2000. Age and gender effects on the pharmacokinetics of gatifloxacin. Pharmacotherapy 20:67S-75S. [DOI] [PubMed] [Google Scholar]
- 20.Launay, O., J. C. Nguyen Van, A. Buu-Hoi, and J. F. Acar. 1997. Typhoid fever due to a Salmonella typhi strain of reduced susceptibility to fluoroquinolones. Clin. Microbiol. Infect. 3:541-544. [DOI] [PubMed] [Google Scholar]
- 21.Le Lostec, Z., S. Fegueux, P. Jouve, M. Cheron, P. Mornet, and A. Boisivon. 1997. Reduced susceptibility to quinolones in Salmonella typhi acquired in Europe: a clinical failure of treatment. Clin. Microbiol. Infect. 3:576-577. [DOI] [PubMed] [Google Scholar]
- 22.Madaras-Kelly, K. J., A. J. Larsson, and J. C. Rotschafer. 1996. A pharmacodynamic evaluation of ciprofloxacin and ofloxacin against two strains of Pseudomonas aeruginosa. J. Antimicrob. Chemother. 37:703-710. [DOI] [PubMed] [Google Scholar]
- 23.Magallanes, M., J. Dijkstra, and J. Fierer. 1993. Liposome-incorporated ciprofloxacin in treatment of murine salmonellosis. Antimicrob. Agents Chemother. 37:2293-2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.National Committee for Clinical Laboratory Standards. 2004. Performance standards for antimicrobial susceptibility testing, document M7-A6. National Committee for Clinical Laboratory Standards, Wayne, Pa.
- 25.National Committee for Clinical Laboratory Standards. 2004. Performance standards for antimicrobial susceptibility testing, document M100-S14. National Committee for Clinical Laboratory Standards, Wayne, Pa.
- 26.Nguyen, J. C., and F. W. Goldstein. 2000. Low-level resistance to fluoroquinolones among Salmonella and Shigella. Clin. Microbiol. Infect. 6:231. [DOI] [PubMed] [Google Scholar]
- 27.Ortho-McNeil Pharmaceutical. 2003. Levofloxacin prescribing information. Ortho-McNeil Pharmaceutical Inc., Raritan, N.J.
- 28.Preston, S. L., G. L. Drusano, A. L. Berman, C. L. Fowler, A. T. Chow, B. Dornseif, V. Reichl, J. Natarajan, and M. Corrado. 1998. Pharmacodynamics of levofloxacin: a new paradigm for early clinical trials. JAMA 279:125-129. [DOI] [PubMed] [Google Scholar]
- 29.Smith, P. F., C. H. Ballow, B. M. Booker, A. Forrest, and J. J. Schentag. 2001. Pharmacokinetics and pharmacodynamics of aztreonam and tobramycin in hospitalized patients. Clin. Ther. 23:1231-1244. [DOI] [PubMed] [Google Scholar]
- 30.Stephen, J. M., M. A. Toleman, T. R. Walsh, and R. N. Jones. 2003. Salmonella bloodstream infections: report from the SENTRY Antimicrobial Surveillance Program (1997-2001). Int. J. Antimicrob. Agents 22:395-405. [DOI] [PubMed] [Google Scholar]
- 31.Threlfall, E. J., and L. R. Ward. 2001. Decreased susceptibility to ciprofloxacin in Salmonella enterica serotype Typhi, United Kingdom. Emerg. Infect. Dis. 7:448-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Threlfall, E. J., L. R. Ward, J. A. Skinner, H. R. Smith, and S. Lacey. 1999. Ciprofloxacin-resistant Salmonella typhi and treatment failure. Lancet 353:1590-1591. [DOI] [PubMed] [Google Scholar]
- 33.Wain, J., N. T. Hoa, N. T. Chinh, H. Vinh, M. J. Everett, T. S. Diep, N. P. Day, T. Solomon, N. J. White, L. J. Piddock, and C. M. Parry. 1997. Quinolone-resistant Salmonella typhi in Vietnam: molecular basis of resistance and clinical response to treatment. Clin. Infect. Dis. 25:1404-1410. [DOI] [PubMed] [Google Scholar]