

Abstract
The apicomplexan parasite Toxoplasma gondii is a leading opportunistic pathogen associated with AIDS and congenital birth defects. Due to the need for identifying new parasite-specific treatments, the possibility of targeting sphingolipid biosynthesis in the parasite was investigated. Aureobasidin A, an inhibitor of the enzyme synthesizing the sphingolipid inositol phosphorylceramide, which is present in fungi, plants, and some protozoa but absent in mammalian cells, was found to block in vitro T. gondii replication without affecting host cell metabolism. Aureobasidin A treatment did not induce tachyzoite to bradyzoite stage conversion in T. gondii but resulted in a loss of intracellular structures and vacuolization within the parasite. In addition, aureobasidin A inhibited sphingolipid synthesis in T. gondii. Sphingolipid biosynthetic pathways may therefore be considered targets for the development of anti-T. gondii agents.
The apicomplexan parasite Toxoplasma gondii is an important agent of human disease. The broad host range of the parasite, including virtually all warm-blooded animals (10), and its high infection rates, which in humans average 10 to 30% but can reach 90% (6), contribute to the widespread distribution of T. gondii. In immunocompetent individuals, T. gondii induces chronic, normally asymptomatic infections following its conversion from the invasive tachyzoite stage to the slowly replicating bradyzoite stage. Bradyzoites reside in quiescent tissue cysts in the brain of the host organism and generally persist in this form throughout the host life span (10). However, serious cases of toxoplasmosis most commonly occur either following congenital transmission of the parasite to the fetus (often resulting in abortion or neurological disorders and blindness) or following immunosuppression in previously asymptomatic adults (commonly associated with organ transplantation or diseases such as AIDS), resulting in toxoplasmic encephalitis with mortality rates exceeding 30% (40). With the significantly increased number of immunocompromised individuals, T. gondii is now recognized as one of the most common opportunistic parasitic infections (8).
Despite efforts to develop effective anti-T. gondii vaccines, chemotherapy remains the only treatment for T. gondii infections. Drugs affecting nucleotide metabolism, such as sulfadiazine and pyrimethamine, are among the most effective therapies to date (26). However, toxicity, side effects, and the development of resistance to currently available compounds (1) highlight the need to improve our understanding of T. gondii biology in order to identify parasite-specific drug targets.
In a search for a key biosynthetic pathway that is present in T. gondii but absent from mammalian cells, we focused on sphingolipid biosynthesis.
Sphingolipids are ubiquitous components of plasma membranes. Their biosynthetic pathways are similar in all eukaryotic cells up to the formation of ceramide. At this point, the addition of the polar head group forms a branch in the pathway, separating animals on one side from fungi, plants, and kinetoplastid parasites on the other (Fig. 1) (21). The end points of sphingolipid synthesis in fungi are inositol phosphorylceramide (IPC) and its derivatives (25). For these organisms, the enzyme that catalyzes the formation of IPC, IPC synthase, is well characterized and plays a pivotal role in the regulation of intracellular levels of sphingolipids and ceramide (21). Moreover, IPC synthase is an essential enzyme in fungi, as its inhibition causes arrest in the cell cycle progression at G1, followed by a loss of viability and alterations in the cytoskeleton (7, 11, 15, 16, 34). The antibiotic aureobasidin A is a potent inhibitor of IPC synthase, causing lethality in a broad range of pathogenic fungi without affecting the mammalian synthesis of sphingolipids (11, 35).
FIG. 1.
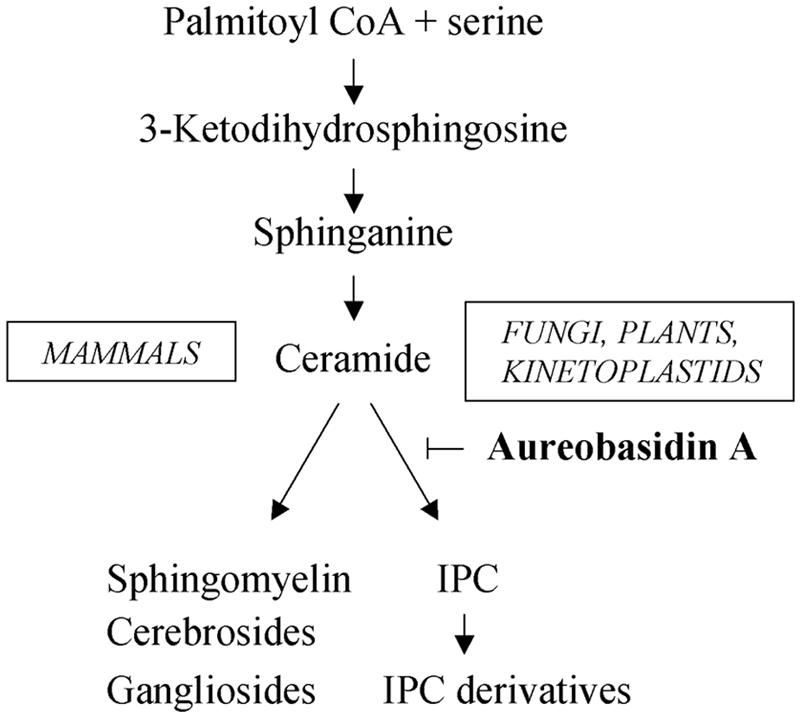
Sphingolipid synthesis in eukaryotic cells. Schematic representation of the synthetic pathways and site of action of the inhibitor aureobasidin A.
In kinetoplastid parasites, such as Leishmania and Trypanosoma, IPC has been shown to be a major component of the sphingolipid pool (25, 29). However, in T. gondii, which is known to synthesize mammal-like sphingolipids, such as sphingomyelin (2), neither the presence of IPC nor the effect of aureobasidin A has yet been characterized.
In the present study, we analyzed the effects of aureobasidin A after in vitro treatment of T. gondii-infected cells with regard to parasite survival, structural integrity, and lipid synthesis.
MATERIALS AND METHODS
Reagents and antibodies.
Unless otherwise stated, all reagents were purchased from Sigma. The antibiotic aureobasidin A was purchased from Takara Bio Inc. (Shiga, Japan). Stock solutions of aureobasidin A were prepared at a concentration of 2 mM in ethanol and freshly diluted to the concentrations required for each experiment. All cell culture reagents were supplied by Gibco-BRL. Radiolabeled reagents were purchased from Amersham Biosciences (Otelfingen, Switzerland). The primary antibodies used in this study were a rabbit polyclonal anti-T. gondii tachyzoite hyperimmune antiserum (13) and rabbit polyclonal anti-BAG5 antibodies (24). Fluorophore-conjugated secondary antibodies were purchased from Molecular Probes (Eugene, Oreg.).
Parasite and tissue culture.
T. gondii tachyzoites of the RH strain expressing Escherichia coli β-galactosidase (31) were used in this study. Parasites were maintained by serial passages in human foreskin fibroblasts (HFF). HFF were routinely cultured in RPMI 1640 medium supplemented with 10% fetal calf serum, 2 mM glutamine, 50 U of penicillin/ml, and 50 μg of streptomycin/ml at 37°C with 5% CO2 in tissue culture flasks. Cultures were trypsinized at least once a week. Parasites were harvested from infected host cells after cell trypsinization, repeatedly passed through a 25-gauge needle, and separated from host cell debris by separation in Sephadex-G25 columns (Amersham Biosciences, Otelfingen, Switzerland) as described previously (18). Prior to analyses, HFF were grown to confluence on glass slides or 96-well plates.
Bradyzoite differentiation was induced in vitro by alkaline treatment as described previously (33). Briefly, T. gondii tachyzoites were allowed to infect a host cell monolayer for 2 h, and then the medium was replaced with RPMI 1640 containing 1-g/liter NaHCO3 and 50 mM Tricine that had been adjusted to pH 8.1 with NaOH. Cultures were incubated at 37°C in ambient CO2 for 4 days.
Immunofluorescence analysis.
For analyses of intracellular T. gondii, infected HFF monolayers grown on glass slides were washed in phosphate-buffered saline (PBS), fixed in 4% paraformaldehyde in PBS, and permeabilized in 100% methanol. After blocking in 1.5% bovine serum albumin in PBS, parasites were visualized by staining with rabbit polyclonal sera against T. gondii tachyzoites at a 1:1,000 dilution, followed by a fluorescein-conjugated anti-rabbit secondary antibody. For analyses of parasite stage conversion to the bradyzoite form, infected HFF monolayers were fixed in 4% paraformaldehyde and permeabilized in 0.2% Triton X-100 in PBS, and parasites were detected by double fluorescence staining after blocking in 1.5% bovine serum albumin. Parasites were first stained with the bradyzoite-specific polyclonal antibody anti-BAG5 (1:250 dilution), followed by a Cy3-conjugated anti-rabbit secondary antibody. Parasites were then visualized by staining with rabbit polyclonal sera against T. gondii tachyzoites (1:1,000 dilution), followed by a fluorescein-conjugated anti-rabbit secondary antibody. Incubation with primary antibodies was performed for 30 min, followed by rinsing in PBS and incubation with the secondary antibodies for an additional 30 min. Parasite and host cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) for 2 min, rinsed in PBS, and mounted in Fluoroguard antifade reagent (Bio-Rad, Hercules, Calif.). Samples were analyzed under a Zeiss Axioplan 2 fluorescence microscope (Jena, Germany).
Replication assay.
HFF monolayers were infected for 2 h with T. gondii tachyzoites (multiplicity of infection, 0.1). HFF monolayers were then washed to remove extracellular parasites and were incubated with fresh medium containing different concentrations of aureobasidin A, as indicated in the individual figure legends. Parasite numbers were determined either after 24 h of drug treatment by direct counting of parasites after immunolabeling, as described above, or after 48 h of drug treatment by colorimetric detection of the β-galactosidase activity expressed by the parasites, as described previously (27). Colorimetric analysis was chosen to determine parasite numbers after 48 h of drug treatment because at this time point the numbers of T. gondii tachyzoites contained in single vacuoles do not allow precise visual counting of the parasites.
Host cell metabolic assay.
The metabolic activity of host cells was tested by using the AlamarBlue assay (BioSource International Inc., Camarillo, Calif.). Briefly, HFF were grown to confluence in 96-well plates, treated for 48 h with the test compounds indicated in the figure legends, and processed according to the instructions provided by the manufacturer.
Transmission electron microscopy.
HFF monolayers were grown in 75-cm2 flasks, infected with T. gondii tachyzoites at a multiplicity of infection of 1, and treated with 10 μg/ml of aureobasidin A or vehicle alone. After 24 h of drug treatment, the cells were fixed in 100 mM sodium cacodylate, pH 7.2, containing 2.5% glutaraldehyde for 2 h at room temperature, followed by several washes in 100 mM sodium cacodylate, pH 7.2. Monolayers were scraped from the surfaces of the tissue flasks by use of a rubber policeman. Postfixation in 2% OsO4 in cacodylate buffer was carried out for 2 h at 4°C. Subsequently, specimens were washed in water and then prestained with 1% uranyl acetate in water for 1 h at 4°C, followed by extensive washing. Specimens were then dehydrated in a graded series of ethanol (50, 70, 90, and 100% [three times]) and were embedded in Epon 820 resin. They were incubated at 40°C for 1 h, followed by a resin change, and this procedure was repeated twice before the resin was allowed to infiltrate for 2 days at room temperature. The resin was polymerized at 65°C over a period of 24 h. Ultrathin sections were cut on a Reichert and Jung ultramicrotome and were loaded onto 200- or 300-mesh copper grids (Plano GmbH, Marburg, Germany). Staining with uranyl acetate and lead citrate was performed as previously described (19). Finally, the grids were viewed with a Phillips 300 transmission electron microscope operating at 60 kV.
Labeling and analysis of sphingolipids.
For the labeling of lipids containing inositol, T. gondii-infected HFF were incubated with myo-[3H]inositol (10 μCi/ml) in the presence of 10 μg/ml of aureobasidin A or vehicle alone for 24 h in inositol-free RPMI 1640 medium (Sigma) supplemented with 10% calf serum. Cells were then extensively washed in PBS, and intracellular parasites were harvested from trypsinized host cells as described above. Lipids were extracted twice from 108 parasites in chloroform-methanol-water (10:10:3) and dried under a nitrogen stream, and aliquots were processed by mild alkaline hydrolysis to deacylate the lipids. The samples were then butanol extracted before being dried and applied to Silica Gel 60 HPTLC plates (20 by 20 cm; Merck, Darmstadt, Germany). For total sphingolipid labeling, 108 purified parasites or 107 host cells were incubated with [3H]palmitate (10 μCi/ml) in the presence of 10 μg/ml of aureobasidin A or vehicle alone for 7 h in RPMI 1640 medium supplemented with 10% delipidated calf serum (Sigma). Lipids were extracted according to the method of Bligh and Dyer (3), dried, and applied to Silica Gel 60 HPTLC plates. Plates were developed in chloroform-methanol-0.25% KCl (55:45:10) (solvent system A) for inositol-labeled lipid analysis or in chloroform-methanol-2 N NH4OH (80:15:2) (solvent system B) for total sphingolipid analysis. Radiolabeled spots were visualized by use of a tritium-sensitive screen (Perkin-Elmer, Boston, Mass.) and were quantified in a Storm 840 phosphorimager using ImageQuant software (Amersham, Otelfingen, Switzerland). Ten micrograms each of ceramide (Avanti Polar, Alabama) and sphingomyelin (Sigma) was used as standards, and their positions were visualized by the use of iodine vapors.
RESULTS
Aureobasidin A effect on T. gondii replication.
In order to analyze the sensitivity of T. gondii to the IPC synthase inhibitor aureobasidin A, we infected HFF monolayers with purified tachyzoites and subsequently treated them with different concentrations of the drug, as described in Materials and Methods. In untreated cells, parasites completed up to four replication cycles during the 24 h postinfection, with 60% of vacuoles containing 8 parasites and 10% containing 16 parasites after this period (Fig. 2A and B). In contrast, a 10-μg/ml aureobasidin A treatment severely inhibited parasite replication immediately following infection, with 60% of vacuoles containing only one parasite and no vacuoles containing more than four parasites. Moreover, immunofluorescence analysis of intracellular T. gondii treated with aureobasidin A showed the presence of abnormal parasites with an altered morphology (Fig. 2C).
FIG. 2.

Effect of aureobasidin A on T. gondii replication after 24 h of drug treatment. (A) HFF monolayers were infected for 2 h with T. gondii, washed to remove extracellular parasites, and subsequently treated with the indicated concentrations of aureobasidin A (AbA). After 24 h of drug treatment, cells were fixed and permeabilized, and intracellular parasites were quantified by staining with a polyclonal anti-T. gondii tachyzoite serum, followed by a fluorescein-conjugated secondary antibody. The distributions of the parasite numbers in single vacuoles are expressed as percentages of the total vacuoles examined (n > 30). (B) Immunofluorescence analysis of intracellular parasites treated with vehicle (control) or 10 μg/ml of aureobasidin A (AbA) by the use of anti-T. gondii tachyzoite serum (α-T. gondii), followed by fluorescein-conjugated secondary antibodies and DAPI staining. (C) Immunofluorescence analysis of intracellular parasites treated with 10 μg/ml of aureobasidin A (AbA) by the use of anti-T. gondii tachyzoite serum, followed by fluorescein-conjugated secondary antibodies. N, normal parasites; A, aberrant parasites. Bar = 5 μm.
To analyze whether T. gondii remained sensitive to aureobasidin A or could adapt to the drug after prolonged incubation, we quantified parasite replication at 48 h postinfection (Fig. 3A). T. gondii replication was potently inhibited by aureobasidin A at 48 h, and this inhibition was dose dependent, with a 50% inhibitory concentration of 0.3 μg/ml. Furthermore, we tested whether the blocking of parasite replication required the presence of aureobasidin A within the first 24 h of vacuole formation or whether it also occurred after the establishment of infection. As shown in Fig. 3B, an aureobasidin A treatment 24 h after T. gondii infection was still effective at inhibiting parasite replication, indicating that the initial formation of the parasite-containing vacuoles was not the crucial step inhibited by the drug.
FIG. 3.
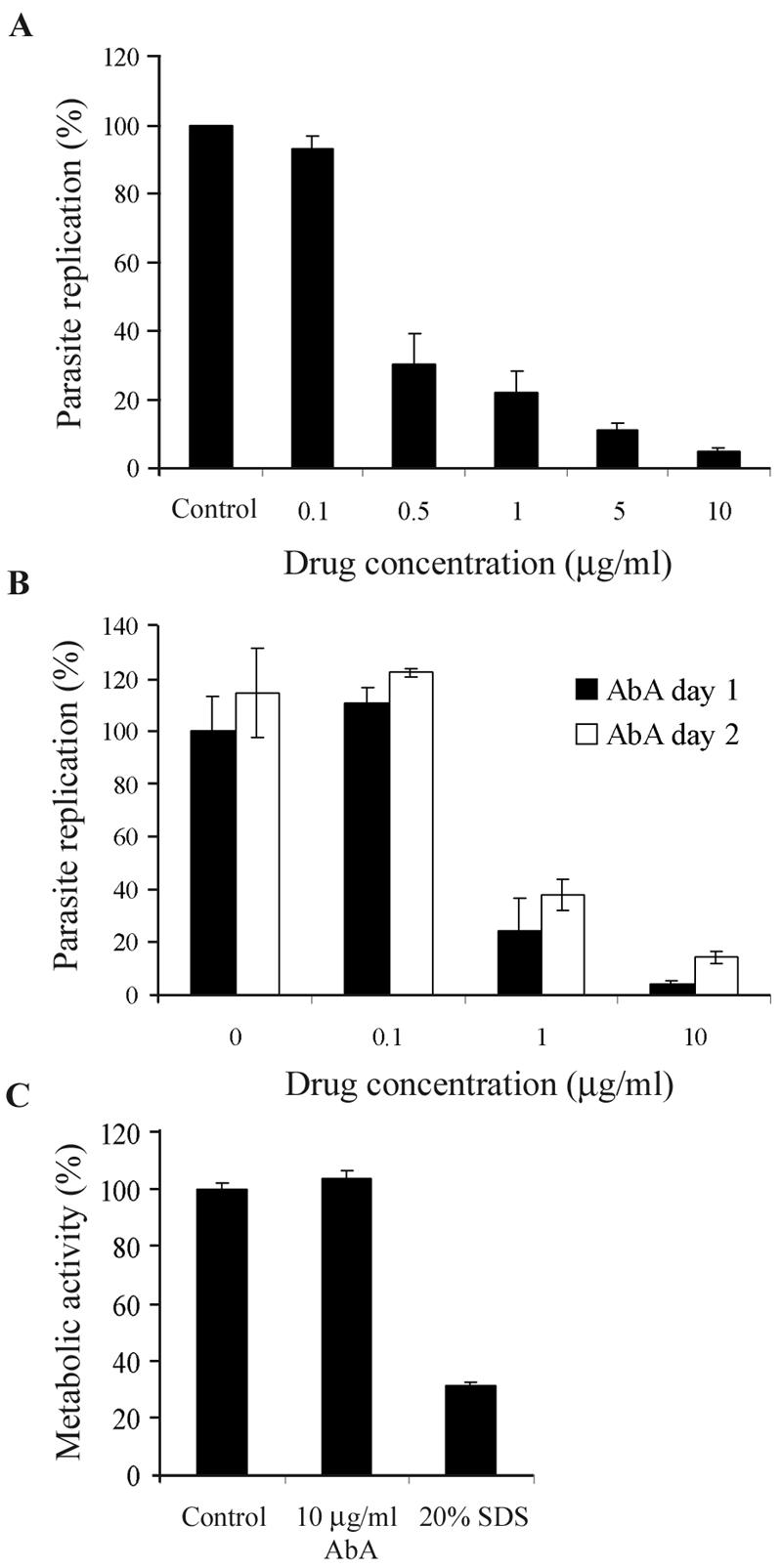
Effect of aureobasidin A on T. gondii replication after 48 h of drug treatment. (A) HFF monolayers were infected for 2 h with T. gondii, washed to remove extracellular parasites, and subsequently treated with the indicated concentrations of aureobasidin A (AbA). After 48 h of drug treatment, intracellular parasites were quantified by a colorimetric assay measuring the amount of parasite-expressed β-galactosidase. Results are presented as percentages of the control (vehicle-treated cells) values ± standard errors (n = 3). Results of a representative experiment (out of three) are shown. (B) HFF monolayers were infected for 2 h with T. gondii, washed to remove extracellular parasites, and treated with the indicated concentrations of aureobasidin A immediately after infection (AbA day 1) or 24 h postinfection (AbA day 2). Intracellular parasites were quantified at 48 h postinfection by a colorimetric assay measuring the parasite-expressed β-galactosidase, and data are expressed as described for panel A. (C) Metabolic activity of host cells treated for 48 h with vehicle (control), 10 μg/ml of aureobasidin A, or 20% sodium dodecyl sulfate (SDS), assessed by measuring AlamarBlue reduction (Biosource).
To analyze the effect of aureobasidin A on host cell functions, we tested the metabolic activity as a measure of cell viability. As shown in Fig. 3C, metabolic activities of host cells were comparable in the absence or presence of aureobasidin A at 10 μg/ml and impaired after cell lysis by a sodium dodecyl sulfate treatment, indicating that reduced host cell viability was not responsible for the inhibition of parasite replication.
Reversibility of aureobasidin A inhibitory effect.
The reversibility of aureobasidin A inhibition of parasite replication was tested by removing the drug from T. gondii-infected cells after 24 or 48 h of treatment, followed by a chase in the absence of the drug. Parasite replication was assessed by counting the vacuoles containing viable parasites, i.e., vacuoles with at least eight parasites after 24 h of drug removal (viable vacuoles). About 60% of the parasites treated with 10 μg/ml of aureobasidin A for 24 h formed viable vacuoles in the 24 h following drug removal, suggesting a recovered replication capability. However, in cells treated for 48 h, viable vacuoles were not observed even after an additional 48-h incubation in the absence of the drug (Fig. 4). These results indicate that a prolonged exposure to aureobasidin A caused an irreversible inhibition of parasite replication.
FIG. 4.
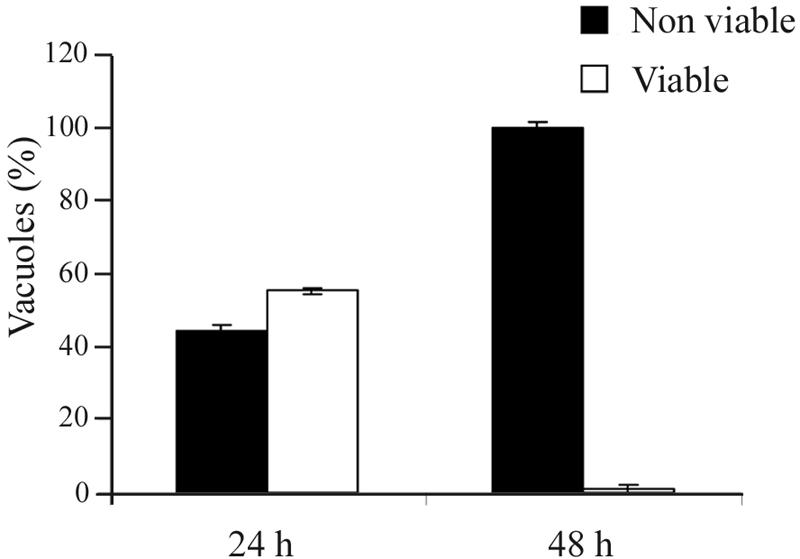
Reversibility of aureobasidin A-mediated inhibition of T. gondii replication. HFF monolayers were infected for 2 h with T. gondii, washed to remove extracellular parasites, and treated with 10 μg/ml of aureobasidin A for 24 or 48 h, followed by an additional 24- or 48-h incubation without the drug, respectively. After cell fixation and permeabilization, intracellular parasites were visualized with an antiserum against T. gondii tachyzoites followed by fluorescein-conjugated secondary antibodies. Parasites were considered viable when present in at least eight units per vacuole after 24 or 48 h of incubation in the absence of the drug. Data are expressed as percentages of vacuoles containing viable or nonviable parasites (total number of vacuoles examined, >50) ± standard errors of a representative from three experiments done in duplicate.
Analysis of bradyzoite stage conversion during aureobasidin A treatment.
To test whether the treatment with aureobasidin A arrested parasite replication by inducing tachyzoite to bradyzoite stage conversion instead of causing a lethal effect on tachyzoites, we performed immunofluorescence analysis with the bradyzoite-specific antibody BAG5. Intracellular parasites treated with the drug for up to 6 days did not reveal any appearance of the BAG5 antigen, whereas infected cells treated with alkaline medium and ambient CO2 to induce bradyzoite differentiation showed a positive signal with the BAG5 antibody, indicating that the arrest in parasite replication in the presence of aureobasidin A was not accompanied by stage conversion to the bradyzoite form (Fig. 5).
FIG. 5.

Analysis of bradyzoite stage conversion. HFF monolayers were infected for 2 h with T. gondii, washed to remove extracellular parasites, and treated either for 6 days with 10 μg/ml of aureobasidin A (AbA) or for 4 days with alkaline medium and ambient CO2 (pH 8.1). After cell fixation in 4% paraformaldehyde and permeabilization in 0.2% Triton X-100, intracellular parasites were visualized with an antiserum against T. gondii tachyzoites, followed by fluorescein-conjugated secondary antibodies (α-T. gondii), or with an antiserum raised against the bradyzoite-specific antigen BAG5, followed by Cy3-conjugated secondary antibodies (α-BAG5). Bar = 5 μm.
Aureobasidin A effect on parasite structure.
The aberrant parasite morphology observed in the immunofluorescence analysis, the irreversibility of the drug-mediated inhibition of parasite replication, and the lack of bradyzoite stage conversion suggest that the effect of aureobasidin A on T. gondii is parasiticidal rather than parasitistatic. To further assess the effect of aureobasidin A treatment on the parasite at the ultrastructural level, we performed electron microscopy analysis of T. gondii-infected HFF treated with 10 μg/ml of aureobasidin A for 24 h. As shown in Fig. 6, the presence of the drug caused severe alterations in the parasite-containing vacuoles, with an increase in T. gondii vacuolization and a loss of intracellular structures. In addition, the intravacuolar space between the damaged parasites showed the presence of amorphous material, whereas untreated cells showed a more defined intravacuolar space with a structured membranous tubular network. The morphological analysis suggests the occurrence of lysis of parasites and shedding of intracellular material in the intravacuolar space.
FIG. 6.

Analysis of T. gondii ultrastructure upon aureobasidin A treatment. HFF monolayers were infected for 2 h with T. gondii, washed to remove extracellular parasites, and treated for 24 h with a vehicle (control) or 10 μg/ml of aureobasidin A (AbA). Samples were fixed and processed for transmission electron microscopy analysis as described in Materials and Methods. Bar = 2 μm.
Aureobasidin A effect on sphingolipid biosynthetic pathways.
Aureobasidin A treatment is lethal to yeast cells by inhibiting the enzyme IPC synthase and the formation of the sphingolipid IPC (11). To analyze whether T. gondii contains, together with the sphingomyelin pathway, a sphingolipid pathway leading to the formation of IPC which can be inhibited by aureobasidin A, we radiolabeled the cells with inositol, a precursor of IPC and phosphatidylinositol (PI). PI and IPC can be biochemically distinguished by a mild alkaline treatment, which hydrolyzes only PI (alkaline sensitive) and not IPC (alkaline resistant) (12). An analysis of lipids from intracellular T. gondii incubated with [3H]inositol showed the presence of an inositol-labeled lipid that was resistant to alkaline treatment comigrating with alkaline-sensitive PI (Fig. 7A, asterisk). Aureobasidin A treatment inhibited the synthesis of this inositol-labeled lipid, with the concomitant appearance of a slower migrating band (arrowhead).
FIG. 7.

Analysis of sphingolipid synthesis upon aureobasidin A treatment. (A) Representative thin-layer chromatography (TLC) results for inositol-labeled lipids extracted from T. gondii. HFF monolayers were infected with T. gondii and labeled with myo-[3H]inositol in the presence of vehicle (control) or 10 μg/ml of aureobasidin A (AbA) for 24 h. After washing of the cells, T. gondii tachyzoites were harvested, lipids were extracted from 108 parasites, and aliquots were processed by mild alkaline hydrolysis (NaOH) and resolved by TLC analysis in solvent system A. (B) Representative TLC of palmitate-labeled lipids extracted from T. gondii or host cells. Purified parasites (108) or 107 host cells were labeled with [3H]palmitate in the presence of vehicle (Cntl) or 10 μg/ml of aureobasidin A (AbA) for 7 h. Lipids were extracted and resolved by TLC analysis in solvent system B. Cer, ceramide; SM, sphingomyelin. (C) Densitometric quantification of newly synthesized ceramide versus sphingomyelin ratio after labeling with [3H]palmitate. Data are expressed as percentages of newly synthesized ceramide/sphingomyelin in vehicle (Cntl)-treated parasites or host cells.
In yeast, the aureobasidin A-mediated inhibition of IPC formation is associated with increased levels of ceramide, the direct precursor of IPC (7). Therefore, ceramide levels in T. gondii were tested to assess possible variations in this lipid concentration due to drug treatment. T. gondii parasites were labeled with [3H]palmitate, the general precursor of sphingolipids, and the extracted lipids were separated in a suitable solvent system for resolving ceramide. [3H]palmitate labeling of purified extracellular parasites for 7 h effectively labeled T. gondii lipids, resulting in a labeling pattern comparable to the one obtained when parasites were labeled intracellularly for 24 h. In contrast, inositol labeling required a 24-h incubation time of intracellular parasites, possibly due to a different cell permeation characteristic of the lipid precursor. Aureobasidin A treatment increased the ratio of newly synthesized ceramide/sphingomyelin and concomitantly decreased the total synthesis of complex sphingolipids in T. gondii. An analysis of newly synthesized lipids in uninfected host cells did not reveal any alteration in sphingolipid levels, confirming that aureobasidin A does not target any sphingolipid biosynthetic enzyme in mammalian cells (Fig. 7B and C).
DISCUSSION
Sphingolipid synthesis is essential in a variety of fungi, thus providing an attractive target for antifungal agents (22, 34). Aureobasidin A, a potent antibiotic which is active against a variety of pathogenic fungi, has been shown to inhibit IPC synthase, the enzyme responsible for the synthesis of the sphingolipid IPC, which is present in fungi, plants, and some protozoa but absent from animal cells (21).
In the present work, we showed that aureobasidin A is a strong inhibitor of T. gondii's in vitro replication. The inhibition of parasite replication had occurred already during the first replication cycles, suggesting that the target of aureobasidin A is crucial for the immediate survival of T. gondii inside host cells.
An analysis of metabolic activity together with direct microscopic observations of host cells treated with aureobasidin A indicated that the presence of the drug does not cause host cell cytotoxicity and that the parasite inhibition observed is therefore most likely due to a direct effect of aureobasidin A on T. gondii. Together, these results suggest that aureobasidin A can be used to prevent T. gondii replication without adverse effects on host cell biology.
Stage conversion from the rapidly dividing tachyzoite to the quiescent bradyzoite form is the physiological parasitic response to stress conditions, among which are temperature (33), pH (33, 39), chemical stress (33), and nitric oxide (4). Stage conversion to the bradyzoite form has also been shown to occur upon treatment with some agents that are able to reduce the T. gondii replication rate (37, 38) and was detectable within 3 days of drug treatment. However, T. gondii treated with aureobasidin A did not show any stage conversion even after 6 days of drug exposure. This observation, together with the finding that the majority of parasites treated with the drug could no longer replicate after 48 h of drug treatment and showed severe damage to intracellular structures, suggests that aureobasidin A promotes the death of intracellular parasites.
The rapid action of the drug as well as the lethal effect observed mirrors the effect of aureobasidin A in yeast cells. Several studies investigating the mechanism of aureobasidin A action have revealed that the lethality observed in yeast following the inhibition of IPC synthase results from both a decrease in the level of the sphingolipid IPC and an increase in the level of ceramide, the substrate for IPC synthase (7). An analysis of lipid synthesis in T. gondii revealed the presence of a lipid that was resistant to alkaline treatment, a known property of IPC. In accordance with the effect of aureobasidin A on yeast IPC, the synthesis of this alkaline-resistant lipid was inhibited by aureobasidin A treatment. While these results support the hypothesis that IPC is present in T. gondii, further analyses are required to confirm the structure of this lipid and the presence of an IPC synthetic pathway together with mammal-like pathways in the parasite.
Increases in relative ceramide levels were also monitored in the presence of aureobasidin A. The drug treatment was shown to decrease the total synthesis of sphingolipids and to raise the ceramide/sphingomyelin ratio in T. gondii, indicating that the lipid synthetic pathway blocked by the drug contains ceramide as an intermediate step. Ceramide, beside being an intermediate element in the biosynthetic pathway of sphingolipids, is also an important second messenger with a pivotal role in sphingolipid-mediated signal transduction (23). In yeast, as well as in mammalian cells, increased levels of ceramide have been linked to cell cycle arrest and apoptosis (14). Therefore, the increased relative levels of cellular ceramide observed in T. gondii could induce cell death and account for the inhibition of total sphingolipid synthesis observed. Moreover, the inhibition of alkaline-resistant sphingolipid synthesis, such as that of IPC, may play an important role in the observed damage to the parasite's structure. Indeed, it has been reported that IPC synthesis is required for the proper organization of filamentous yeast cells (7). Collectively, these results indicate that the lethal phenotype observed could have been caused by both the accumulation of ceramide and/or the lack of a lipid inhibited by aureobasidin A.
In kinetoplastid parasites, such as Leishmania and Trypanosoma, IPC is a major component of the sphingolipid pool (25, 29). Interestingly, sphingolipids seem to have different functions in the biology of these parasites. In Leishmania major, de novo sphingolipid synthesis is necessary in the extracellular stage for proper trafficking, infectivity, and differentiation to the intracellular stage of the parasite (9). On the other hand, sphingolipid synthesis is down-regulated upon differentiation, indicating that this synthesis is not crucial for survival and proliferation inside the macrophages of the mammalian host. Conversely, in the case of Trypanosoma cruzi, production of the sphingolipid IPC and of glycoinositolphospholipids is up-regulated during differentiation to the intracellular stage. In T. cruzi, aureobasidin A treatment at concentrations of 5 to 20 μg/ml inhibits the synthesis of both IPC and glycoinositolphospholipids and leads to compromised differentiation and cellular toxicity (29). Thus, it is clear that in these parasites, sphingolipids, particularly IPC, play different yet interconnected roles which remain partly elusive. However, these studies reveal that sphingolipids are likely to have, also in protozoa, both structural and regulative roles, including membrane modification, signal transduction, and vesicular trafficking. In light of this knowledge and our findings, it would be interesting to further analyze whether T. gondii also exhibits a stage-specific regulation of sphingolipid composition during differentiation into the bradyzoite form as an adaptation mechanism for bradyzoite physiology.
A BLAST search for an IPC synthase in the T. gondii genome did not reveal IPC synthase candidates. Possible reasons for the absence of significant homology include species-specific variations in the enzyme sequence. A comparison of IPC sequences from a variety of fungi, including the human pathogens Candida, Cryptococcus, and Aspergillus, revealed that the C and N termini of the enzyme have little homology among the different species and that the only conserved regions are the internal 250 amino acids (17). Therefore, the sequence similarity may be too low to reveal a possible T. gondii IPC synthase by a homology search.
Aureobasidin A has been shown to be a substrate for the human MDR1,2 P-glycoproteins (Pgp) (20, 36), which are members of the ATP-binding cassette (ABC) transporter family and mediate the efflux of solutes across cell membranes. Some Pgp inhibitors, including cyclosporine A and its derivatives, were found to inhibit T. gondii replication, suggesting a possible role of Pgp in T. gondii physiology (32). In order to assess a role of Pgp in the inhibition of parasite replication induced by aureobasidin A, we measured the activity of Pgp in both host cells and isolated parasites in the presence of aureobasidin A and verapamil, a known efficient Pgp modulator. Both drugs inhibited the efflux of the dye rhodamine 123 in host cells as well as in extracellular parasites (data not shown), supporting the presence of an efflux system inhibited by Pgp modulators in T. gondii (30, 32). However, as no direct correlation between Pgp inhibition and parasite replication was observed, the contribution of a putative Pgp homologue to T. gondii survival within host cells remains unclear.
In summary, our studies indicate that the replication of T. gondii is severely and rapidly inhibited by treatment with the antibiotic aureobasidin A and that such a treatment causes an inhibition of parasite sphingolipid synthesis without affecting lipid synthesis and metabolic activity in the host cells. Future analyses will further characterize the nature of the T. gondii lipid which is modulated by aureobasidin A treatment and evaluate the efficacy of aureobasidin A both on T. gondii infection in vivo and on the bradyzoite form of the parasite, which is responsible for the latent form of infection.
A key factor in developing specific anti-T. gondii treatments is the identification of essential metabolic pathways that are restricted to the parasite and absent from mammalian cells, such as the recently described complete shikimate pathway (5, 28). Our results indicate that the characterization of sphingolipid biosynthetic pathways in T. gondii may lead to the development of new compounds specifically targeting sphingolipid synthesis in the parasite as promising candidates for anti-T. gondii treatments.
Acknowledgments
We thank John Boothroyd for kindly providing T. gondii tachyzoites of the βRH strain and Howard Riezman for invaluable advice and discussions.
This work was supported by grants obtained from Roche Research Foundation, Novartis Foundation, Swiss National Science Foundation, and Fondation Pierre Mercier pour la Science, Switzerland.
REFERENCES
- 1.Aspinall, T. V., D. H. Joynson, E. Guy, J. E. Hyde, and P. F. Sims. 2002. The molecular basis of sulfonamide resistance in Toxoplasma gondii and implications for the clinical management of toxoplasmosis. J. Infect. Dis. 185:1637-1643. [DOI] [PubMed] [Google Scholar]
- 2.Azzouz, N., B. Rauscher, P. Gerold, M. F. Cesbron-Delauw, J. F. Dubremetz, and R. T. Schwarz. 2002. Evidence for de novo sphingolipid biosynthesis in Toxoplasma gondii. Int. J. Parasitol. 32:677-684. [DOI] [PubMed] [Google Scholar]
- 3.Bligh, E. G., and W. J. Dyer. 1959. A rapid method of total lipid extraction and purification. Can. J. Med. Sci. 37:911-917. [DOI] [PubMed] [Google Scholar]
- 4.Bohne, W., J. Heesemann, and U. Gross. 1994. Reduced replication of Toxoplasma gondii is necessary for induction of bradyzoite-specific antigens: a possible role for nitric oxide in triggering stage conversion. Infect. Immun. 62:1761-1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campbell, S. A., T. A. Richards, E. J. Mui, B. U. Samuel, J. R. Coggins, R. McLeod, and C. W. Roberts. 2004. A complete shikimate pathway in Toxoplasma gondii: an ancient eukaryotic innovation. Int. J. Parasitol. 34:5-13. [DOI] [PubMed] [Google Scholar]
- 6.Carruthers, V. B. 2002. Host cell invasion by the opportunistic pathogen Toxoplasma gondii. Acta Trop. 81:111-122. [DOI] [PubMed] [Google Scholar]
- 7.Cheng, J., T. S. Park, A. S. Fischl, and X. S. Ye. 2001. Cell cycle progression and cell polarity require sphingolipid biosynthesis in Aspergillus nidulans. Mol. Cell. Biol. 21:6198-6209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dannemann, B. R., D. M. Israelski, G. S. Leoung, T. McGraw, J. Mills, and J. S. Remington. 1991. Toxoplasma serology, parasitemia and antigenemia in patients at risk for toxoplasmic encephalitis. AIDS 5:1363-1365. [DOI] [PubMed] [Google Scholar]
- 9.Denny, P. W., D. Goulding, M. A. Ferguson, and D. F. Smith. 2004. Sphingolipid-free Leishmania are defective in membrane trafficking, differentiation and infectivity. Mol. Microbiol. 52:313-327. [DOI] [PubMed] [Google Scholar]
- 10.Dubey, J. P. 1998. Advances in the life cycle of Toxoplasma gondii. Int. J. Parasitol. 28:1019-1024. [DOI] [PubMed] [Google Scholar]
- 11.Endo, M., K. Takesako, I. Kato, and H. Yamaguchi. 1997. Fungicidal action of aureobasidin A, a cyclic depsipeptide antifungal antibiotic, against Saccharomyces cerevisiae. Antimicrob. Agents Chemother. 41:672-676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fischl, A. S., Y. Liu, A. Browdy, and A. E. Cremesti. 2000. Inositolphosphoryl ceramide synthase from yeast. Methods Enzymol. 311:123-130. [DOI] [PubMed] [Google Scholar]
- 13.Fuchs, N., S. Sonda, B. Gottstein, and A. Hemphill. 1998. Differential expression of cell surface- and dense granule-associated Neospora caninum proteins in tachyzoites and bradyzoites. J. Parasitol. 84:753-758. [PubMed] [Google Scholar]
- 14.Hannun, Y. A. 1996. Functions of ceramide in coordinating cellular responses to stress. Science 274:1855-1859. [DOI] [PubMed] [Google Scholar]
- 15.Hashida-Okado, T., A. Ogawa, M. Endo, R. Yasumoto, K. Takesako, and I. Kato. 1996. AUR1, a novel gene conferring aureobasidin resistance on Saccharomyces cerevisiae: a study of defective morphologies in Aur1p-depleted cells. Mol. Gen. Genet. 251:236-244. [DOI] [PubMed] [Google Scholar]
- 16.Hashida-Okado, T., R. Yasumoto, M. Endo, K. Takesako, and I. Kato. 1998. Isolation and characterization of the aureobasidin A-resistant gene, aur1R, on Schizosaccharomyces pombe: roles of Aur1p+ in cell morphogenesis. Curr. Genet. 33:38-45. [DOI] [PubMed] [Google Scholar]
- 17.Heidler, S. A., and J. A. Radding. 2000. Inositol phosphoryl transferases from human pathogenic fungi. Biochim. Biophys. Acta 1500:147-152. [DOI] [PubMed] [Google Scholar]
- 18.Hemphill, A., B. Gottstein, and H. Kaufmann. 1996. Adhesion and invasion of bovine endothelial cells by Neospora caninum. Parasitology 112:183-197. [DOI] [PubMed] [Google Scholar]
- 19.Hemphill, A., and S. L. Croft. 1997. Electron microscopy in parasitology, p. 227-268. In M. Rogan (ed.), Analytical parasitology. Springer-Verlag, Heidelberg, Germany.
- 20.Kino, K., Y. Taguchi, K. Yamada, T. Komano, and K. Ueda. 1996. Aureobasidin A, an antifungal cyclic depsipeptide antibiotic, is a substrate for both human MDR1 and MDR2/P-glycoproteins. FEBS Lett. 399:29-32. [DOI] [PubMed] [Google Scholar]
- 21.Lester, R. L., and R. C. Dickson. 1993. Sphingolipids with inositolphosphate-containing head groups. Adv. Lipid Res. 26:253-274. [PubMed] [Google Scholar]
- 22.Mandala, S. M., R. A. Thornton, J. Milligan, M. Rosenbach, M. Garcia-Calvo, H. G. Bull, G. Harris, G. K. Abruzzo, A. M. Flattery, C. J. Gill, K. Bartizal, S. Dreikorn, and M. B. Kurtz. 1998. Rustmicin, a potent antifungal agent, inhibits sphingolipid synthesis at inositol phosphoceramide synthase. J. Biol. Chem. 273:14942-14949. [DOI] [PubMed] [Google Scholar]
- 23.Mathias, S., L. A. Pena, and R. N. Kolesnick. 1998. Signal transduction of stress via ceramide. Biochem. J. 335:465-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McAllister, M. M., S. F. Parmley, L. M. Weiss, V. J. Welch, and A. M. McGuire. 1996. An immunohistochemical method for detecting bradyzoite antigen (BAG5) in Toxoplasma gondii-infected tissues cross-reacts with a Neospora caninum bradyzoite antigen. J. Parasitol. 82:354-355. [PubMed] [Google Scholar]
- 25.McConville, M. J., and A. Bacic. 1989. A family of glycoinositol phospholipids from Leishmania major. Isolation, characterization, and antigenicity. J. Biol. Chem. 264:757-766. [PubMed] [Google Scholar]
- 26.McFadden, D. C., M. Camps, and J. C. Boothroyd. 2001. Resistance as a tool in the study of old and new drug targets in Toxoplasma. Drug Resist. Updat. 4:79-84. [DOI] [PubMed] [Google Scholar]
- 27.McFadden, D. C., F. Seeber, and J. C. Boothroyd. 1997. Use of Toxoplasma gondii expressing beta-galactosidase for colorimetric assessment of drug activity in vitro. Antimicrob. Agents Chemother. 41:1849-1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roberts, F., C. W. Roberts, J. J. Johnson, D. E. Kyle, T. Krell, J. R. Coggins, G. H. Coombs, W. K. Milhous, S. Tzipori, D. J. Ferguson, D. Chakrabarti, and R. McLeod. 1998. Evidence for the shikimate pathway in apicomplexan parasites. Nature 393:801-805. [DOI] [PubMed] [Google Scholar]
- 29.Salto, M. L., L. E. Bertello, M. Vieira, R. Docampo, S. N. Moreno, and R. M. de Lederkremer. 2003. Formation and remodeling of inositolphosphoceramide during differentiation of Trypanosoma cruzi from trypomastigote to amastigote. Eukaryot. Cell 2:756-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sauvage, V., D. Aubert, A. Bonhomme, J. M. Pinon, and J. M. Millot. 2004. P-glycoprotein inhibitors modulate accumulation and efflux of xenobiotics in extra and intracellular Toxoplasma gondii. Mol. Biochem. Parasitol. 134:89-95. [DOI] [PubMed] [Google Scholar]
- 31.Seeber, F., and J. C. Boothroyd. 1996. Escherichia coli beta-galactosidase as an in vitro and in vivo reporter enzyme and stable transfection marker in the intracellular protozoan parasite Toxoplasma gondii. Gene 169:39-45. [DOI] [PubMed] [Google Scholar]
- 32.Silverman, J. A., M. L. Hayes, B. J. Luft, and K. A. Joiner. 1997. Characterization of anti-Toxoplasma activity of SDZ 215-918, a cyclosporin derivative lacking immunosuppressive and peptidyl-prolyl-isomerase-inhibiting activity: possible role of a P glycoprotein in Toxoplasma physiology. Antimicrob. Agents Chemother. 41:1859-1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soete, M., D. Camus, and J. F. Dubremetz. 1994. Experimental induction of bradyzoite-specific antigen expression and cyst formation by the RH strain of Toxoplasma gondii in vitro. Exp. Parasitol. 78:361-370. [DOI] [PubMed] [Google Scholar]
- 34.Takesako, K., K. Ikai, F. Haruna, M. Endo, K. Shimanaka, E. Sono, T. Nakamura, I. Kato, and H. Yamaguchi. 1991. Aureobasidins, new antifungal antibiotics. Taxonomy, fermentation, isolation, and properties. J. Antibiot. (Tokyo) 44:919-924. [DOI] [PubMed] [Google Scholar]
- 35.Takesako, K., H. Kuroda, T. Inoue, F. Haruna, Y. Yoshikawa, I. Kato, K. Uchida, T. Hiratani, and H. Yamaguchi. 1993. Biological properties of aureobasidin A, a cyclic depsipeptide antifungal antibiotic. J. Antibiot. (Tokyo) 46:1414-1420. [DOI] [PubMed] [Google Scholar]
- 36.Tiberghien, F., T. Kurome, K. Takesako, A. Didier, T. Wenandy, and F. Loor. 2000. Aureobasidins: structure-activity relationships for the inhibition of the human MDR1 P-glycoprotein ABC-transporter. J. Med. Chem. 43:2547-2556. [DOI] [PubMed] [Google Scholar]
- 37.Wei, S., F. Marches, B. Daniel, S. Sonda, K. Heidenreich, and T. Curiel. 2002. Pyridinylimidazole p38 mitogen-activated protein kinase inhibitors block intracellular Toxoplasma gondii replication. Int. J. Parasitol. 32:969-977. [DOI] [PubMed] [Google Scholar]
- 38.Weiss, L. M., and K. Kim. 2000. The development and biology of bradyzoites of Toxoplasma gondii. Front. Biosci. 5:D391-D405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weiss, L. M., D. Laplace, P. M. Takvorian, H. B. Tanowitz, A. Cali, and M. Wittner. 1995. A cell culture system for study of the development of Toxoplasma gondii bradyzoites. J. Eukaryot. Microbiol. 42:150-157. [DOI] [PubMed] [Google Scholar]
- 40.Wong, S. Y., and J. S. Remington. 1993. Biology of Toxoplasma gondii. AIDS 7:299-316. [DOI] [PubMed] [Google Scholar]