

Summary
CD3δ SCID is a devastating inborn error of immunity caused by mutations in CD3D, encoding the invariant CD3δ chain of the CD3/TCR complex necessary for normal thymopoiesis. We demonstrate an adenine base editing (ABE) strategy to restore CD3δ in autologous hematopoietic stem and progenitor cells (HSPCs). Delivery of mRNA encoding a laboratory-evolved ABE and guide RNA into a CD3δ SCID patient’s HSPCs resulted in 71.2±7.85% (n=3) correction of the pathogenic mutation. Edited HSPCs differentiated in artificial thymic organoids produced mature T cells exhibiting diverse TCR repertoires and TCR-dependent functions. Edited human HSPCs transplanted into immunodeficient mice showed 88% reversion of the CD3D defect in human CD34+ cells isolated from mouse bone marrow after 16 weeks, indicating correction of long-term repopulating HSCs. These findings demonstrate preclinical efficacy of ABE in HSPCs for the treatment of CD3δ SCID, providing a foundation for the development of a one-time treatment for CD3δ SCID patients.
Introduction
CD3δ severe combined immune deficiency (SCID) is a life-threatening inborn error of immunity (IEI) caused by biallelic mutations in the autosomal CD3D gene. During normal T cell development, T cell receptor (TCR) assembly begins in the endoplasmic reticulum as CD3 heterodimers associate with TCR chains for export to the Golgi apparatus, where interactions with the ζζ/CD2472 homodimer allow for transport to the cell surface.1 CD3δ is essential for the productive assembly of TCR complexes; thus, the absence of CD3δ chains results in the intracellular retention of defective TCR ensembles, leading to early arrest of thymopoiesis.1 A homozygous mutation in CD3D (c.202C>T), predominately found in a Mennonite population, results in a premature stop codon (p.R68X) and the complete absence of CD3δ protein and the CD3/TCR complex. CD3δ SCID patients present with a profound deficiency of circulating, mature αβ and γδ T cells, with present B and NK cells (T-B+NK+ SCID),2 often leading to infant mortality.
Allogeneic hematopoietic stem cell transplantation (HSCT) can be curative but may be complicated by the risk of potentially fatal graft-versus-host disease (GvHD), and treatment-related toxicities.3 In a multi-center study reported in 2011, survival of CD3δ SCID patients undergoing allogeneic HSCT was only 61.5% (n=13) with most patients experiencing acute GvHD and two patients developing chronic GvHD.3
Developing a strategy for autologous HSCT utilizing a patient’s own gene-corrected hematopoietic stem and progenitor cells (HSPCs) would abrogate many complications associated with allogeneic HSCT. Previous work has explored gene therapy for devastating monogenic IEIs, such as SCID-X1,4 adenosine deaminase (ADA)-SCID,5 Artemis-Deficient SCID,6 and RAG-1 SCID7 through ex vivo lentiviral vector (LV) gene addition. Although LV modification of HSCs to restore CD3δ expression is a feasible strategy, LVs can induce oncogenic insertional mutagenesis. Therefore, restoration of the endogenous temporal expression of CD3δ necessary for thymopoiesis via a T cell specific LV approach may prove difficult.8 CRISPR/Cas9 HDR-mediated correction of HSCs presents a promising therapeutic strategy for IEIs such as SCID-X1,9 however, this technology is yet to be used in clinical trials to correct SCID in humans. Furthermore, CRISPR/Cas9 HDR necessitates double-stranded breaks (DSBs) by Cas9 nuclease, is cell cycle dependent and therefore difficult to achieve with high efficiency in long-term HSCs, and carries risks associated with uncontrolled mixtures of indel byproducts, p53 activation, translocations, and loss or rearrangement of large chromosomal segments (chromothripsis).10 As an alternative approach, base editing (BE) can correct the pathogenic mutation without requiring donor DNA templates or DSBs and may overcome the limitations of LV gene addition or Cas9 nuclease-mediated HDR. Adenine base editors (ABEs) are comprised of a catalytically impaired Cas9 nickase (Cas9n) fused to a DNA-modifying deaminase enzyme, enabling direct conversion of A•T-to-G•C base pairs, without introducing DSBs and minimizing indel byproducts.11
Here, we describe the development of an ABE approach able to precisely revert the CD3D c.202C>T mutation in 1) a Jurkat T cell line disease model, 2) human CD34+ HSPCs from healthy donors transduced with an LV carrying a CD3D c.202C>T mutation target, and 3) CD34+ HSPCs from a CD3δ SCID patient. We demonstrate highly efficient and specific correction of the CD3D mutation in each cell type, with restoration of CD3δ protein expression and CD3/TCR complex signaling in response to antigenic stimuli. Edited human HSPCs persisted in humanized mouse models, maintaining 88% CD3D c.202C>T correction after 16 weeks.
We utilized the 3D artificial thymic organoid (ATO) system12 to determine restoration of CD3 and TCR surface expression in base-edited CD3δ SCID HSPCs undergoing in vitro T cell maturation. Previous ATO studies have demonstrated robust and unique recapitulation of thymocyte positive selection with remarkable fidelity to both mouse13 and human14,15 T cell differentiation in the thymus. ATOs have also been adopted to characterize and diagnose SCIDs that result in T cell lymphopenias like CD3δ SCID.12 Our results demonstrate that edited CD3δ SCID HSPCs produced functional T lymphocytes with diverse TCR repertoires in the ATO. These data suggest an ABE-mediated autologous gene therapy is a promising treatment strategy for CD3δ SCID.
Results
Adenine Base Editing Functionally Restores Wildtype Levels of CD3/TCR Expression and Signaling in a Jurkat T cell Disease Model
Cas9-mediated HDR and ABE therapies have recently been utilized to eliminate the point mutations causing monogenic diseases such as sickle cell disease and β-thalassemia.10,16–18 To determine whether ABE or Cas9 nuclease-mediated HDR gene correction could be suitable strategies for CD3δ SCID, we generated a clonal Jurkat T cell disease model (CD3D(C202T) Jurkat T cells) containing the pathogenic CD3D c.202C>T mutation in one CD3D allele with deleterious indels in the other three alleles of a pseudo-tetraploid Jurkat T cell line, rendering the latter three alleles non editable. (Materials and Methods; Fig. S1a). The disease-causing defect can be corrected by 1) evolved adenine base editors recognizing non-canonical (non-NGG) protospacer-adjacent motifs (PAM) (Fig. 1a) or by 2) Cas9 nuclease-mediated HDR utilizing a single-stranded oligodeoxynucleotide (ssODN) homologous donor and ribonucleoprotein (RNP) complex of rCas9 protein and a single-guide RNA (sgRNA). Electroporation of CD3D(C202T) Jurkat T cells with Cas9 nuclease RNP and an ssODN to mediate HDR resulted in 28 ± 4.6% (mean ± standard deviation) correction of the pathogenic mutation with 53 ± 5.2% indel byproducts (Fig. 1b). In contrast, electroporation of the same cells with plasmids encoding CD3D-targeting sgRNA and ABEmax-NRTH (GenScript codon optimized bis-bpNLS ABE7.1019,20; CGTT PAM), ABE8e-NRTH (CGTT PAM), ABE8e-NG (CG PAM), ABE8e-VRER (TGCG PAM), or ABE8e-xCas9(3.7) (CGT PAM) produced 93 ± 2.3%, 92 ± 3.1%, 86 ± 2.9%, 33 ± 4.8%, and 18 ± 4.7% correction of the CD3D c.202C>T mutation, respectively, with minimal indels (Fig. 1b). Analysis of edited CD3D(C202T) Jurkat T cells by flow cytometry revealed a positive correlation between CD3D c.202C>T correction and surface CD3 complex restoration, with rescued CD3 surface expression in up to 85 ± 2.1%, 79.4 ± 1.8%, 77.9% ± 1.9, and 29.4 ± 2.9% of cells manipulated with ABEmax-NRTH, ABE8e-NRTH, ABE8e-NG, or RNP + ssODN, respectively (Fig. 1c–d; Fig. S1b–c).
Figure 1. Adenine Base Editing Efficiently Rescues CD3/TCR Expression and Signaling in a T Cell Line Disease Model.
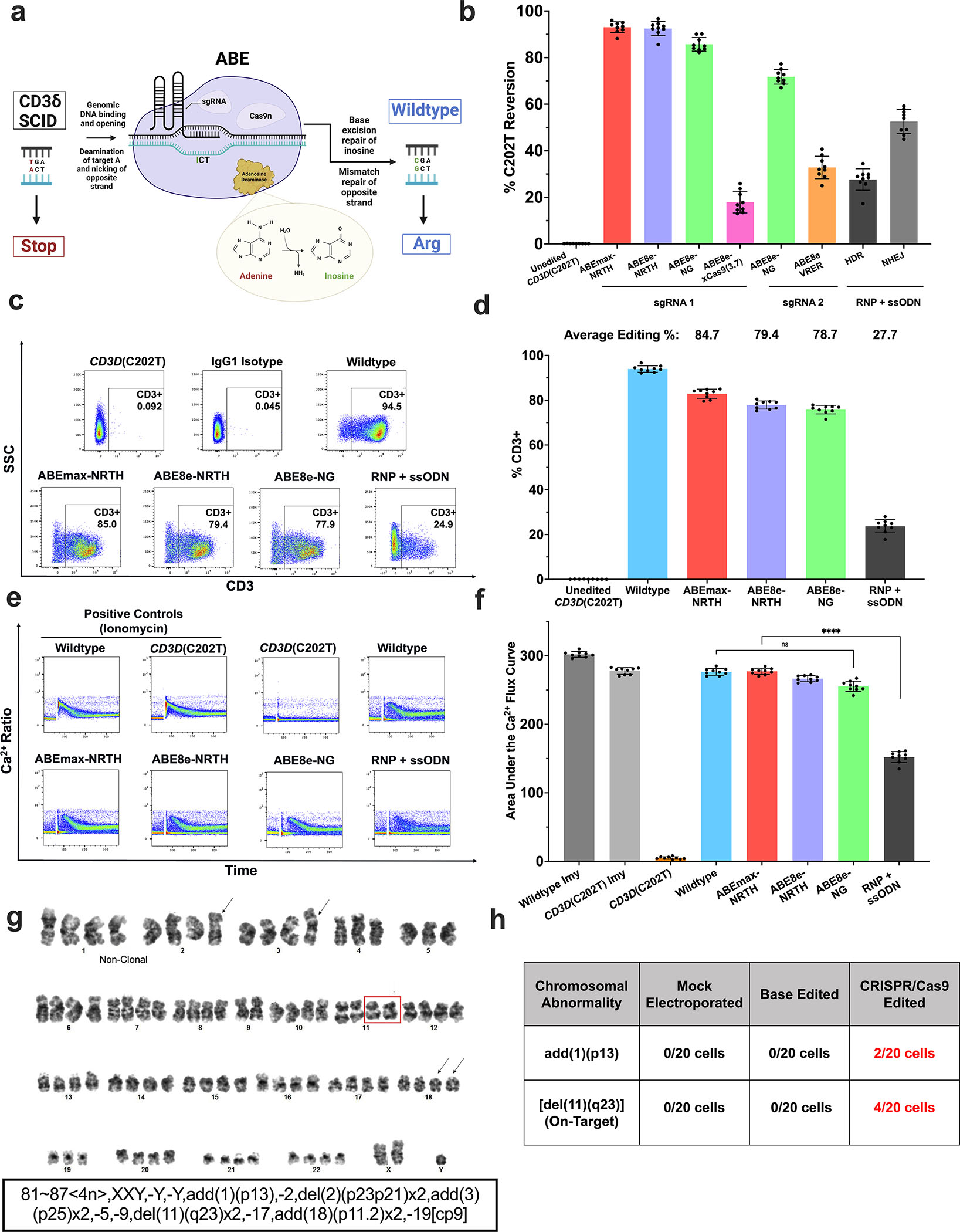
a) Schematic of ABE for CD3δ SCID. b) Plasmids encoding a CD3D-targeting sgRNA and either ABEmax-NRTH, ABE8e-NRTH, ABE8e-NG, ABE8e-xCas9(3.7), or ABE8e-VRER were transfected by electroporation into CD3D(C202T) Jurkat T cells. To assess restoration of CD3 by CRISPR/Cas9 HDR-mediated correction, sgRNA and rCas9 protein (RNP) and ssODN donor were co-electroporated into CD3D(C202T) Jurkat T cells. sgRNAs utilized for BE and HDR approaches were different and designed specifically for their respective use. c-d) Editing efficiencies were measured 5 days after electroporation by high-throughput sequencing (HTS) and restoration of CD3 expression was measured by flow cytometry with an anti-CD3 antibody. e-f) Calcium flux assay and quantified area under the calcium flux curve of treated and untreated CD3D(C202T) Jurkat T cells following stimulation with anti-CD3 and anti-CD28. g) CD3D(C202T) Jurkat T cells treated with RNP + ssODN (CRISPR/Cas9-edited), ABEmax-NRTH and sgRNA, or mock electroporated controls were harvested 24 hours after electroporation for G-banded karyotype analysis. Representative karyotype of one cell edited with Cas9 RNP and ssODN. Representative abnormalities described using the International System for Human Cytogenomic Nomenclature (ISCN). Clonal structural abnormalities inherent to the pseudo-tetraploid Jurkat T cell line (black arrows); “clonal” = at least two cells with the same chromosomal rearrangement. Clonal deletion of 11q23 distal to the on-target editing site (red box). h) Additional clonal structural abnormalities only observed in the CRISPR/Cas9-edited Jurkat T cells. b), d), f) Data shown as mean ± SD of nine replicates from 3 independent experiments. Statistical significance calculated using non-parametric t-test (****p<0.0001); ns, not significant.
During T cell activation, the engagement of a T cell with an antigen-presenting cell results in rapid cytoskeletal rearrangements and an increase of intracellular calcium concentration.21 Therefore, to assess functional rescue of CD3/TCR signaling, we performed a calcium flux assay with unedited and edited CD3D(C202T) Jurkat T cells, where a flux of intracellular calcium can be used as an indicator of TCR-dependent activation in response to an antigenic stimulus.21 Consistent with gene editing frequencies and CD3D rescue, adenine base editing with ABEmax-NRTH, ABE8e-NRTH, or ABE8e-NG restored CD3/TCR signaling in response to anti-CD3 and anti-CD28 to wildtype levels, while RNP + ssODN treatment restored calcium flux to only 58% of wildtype (Fig. 1e–f).
Previous studies have reported induction of large chromosomal rearrangements or deletions as on-target consequences of Cas9 nuclease-mediated DSBs.22 Importantly, chromosomal abnormalities involving the CD3D on-target site, 11q23, have frequently been associated with acute myeloid leukemia and poor prognosis for chronic myeloid leukemia patients.23,24 Therefore, to evaluate the effects of ABE and CRISPR/Cas9 manipulation on chromosomal integrity, we performed standard karyotype analysis of 20 metaphases each of mock electroporated (without cargo), ABE-treated, and RNP and ssODN-treated (CRISPR/Cas9) CD3D(C202T) Jurkat T cells. Four of 20 metaphase cells treated with Cas9 nuclease and ssODN for HDR demonstrated a large deletion distal to the chromosome 11q23 region [del(11)(q23)], with a subset of cells displaying rearrangements involving 11q23 (Fig. 1g karyotype; Fig. 1h (Table); Table S1; Fig. S1d–f). Unbalanced rearrangements involving chromosomal region 1p13 [add(1)(p13)] were also observed in CRISPR/Cas9-edited cells, consistent with off-target sites predicted by the in silico Cas-OFFinder tool for the CRISPR/Cas9 sgRNA (Table S2). Notably, no clonal structural abnormalities in ABE-treated cells were observed beyond those present in all pseudo-tetraploid Jurkat T cells. Thus, these findings suggested that ex vivo ABE manipulation can efficiently correct the pathogenic CD3δ SCID mutation without the deleterious byproducts associated with DSBs.
Evaluating Local Bystander and Genome-Wide Off-Target Editing in CD3D(C202T) Jurkat T cells and CD3δ SCID Patient CD34+ HSPCs
Recognizing local bystander editing, or base editing within or near the protospacer other than the target adenine, as a potential limitation of ABE,25 we sought to characterize the effects of detectable bystander editing on CD3/TCR signaling. High-throughput sequencing (HTS) analysis of CD3D(C202T) Jurkat T cells treated with plasmids encoding lead candidate base editors, ABEmax-NRTH, ABE8e-NRTH, or ABE8e-NG, revealed less than 1.35% indels, with the only detectable bystander edits occurring at positions A0 and A-2 (Fig. 2a–b). We noted significantly increased levels of bystander editing produced by the highly processive ABE8e variants (up to 50.4% and 13.9% at positions A0 and A-2, respectively) compared to ABEmax treatment (up to 1.4% at position A0), consistent with the increased deaminase activity characteristic of ABE8e-mediated editing26 (Fig 2b). These data suggested ABEmax-NRTH as the lead therapeutic candidate for safe and efficient correction of CD3D c.202C>T.
Figure 2. Characterization of Local Bystander and Genome-Wide Off-Target Editing in CD3D(C202T) Jurkat T cells and CD3δ SCID Patient CD34+ HSPCs.
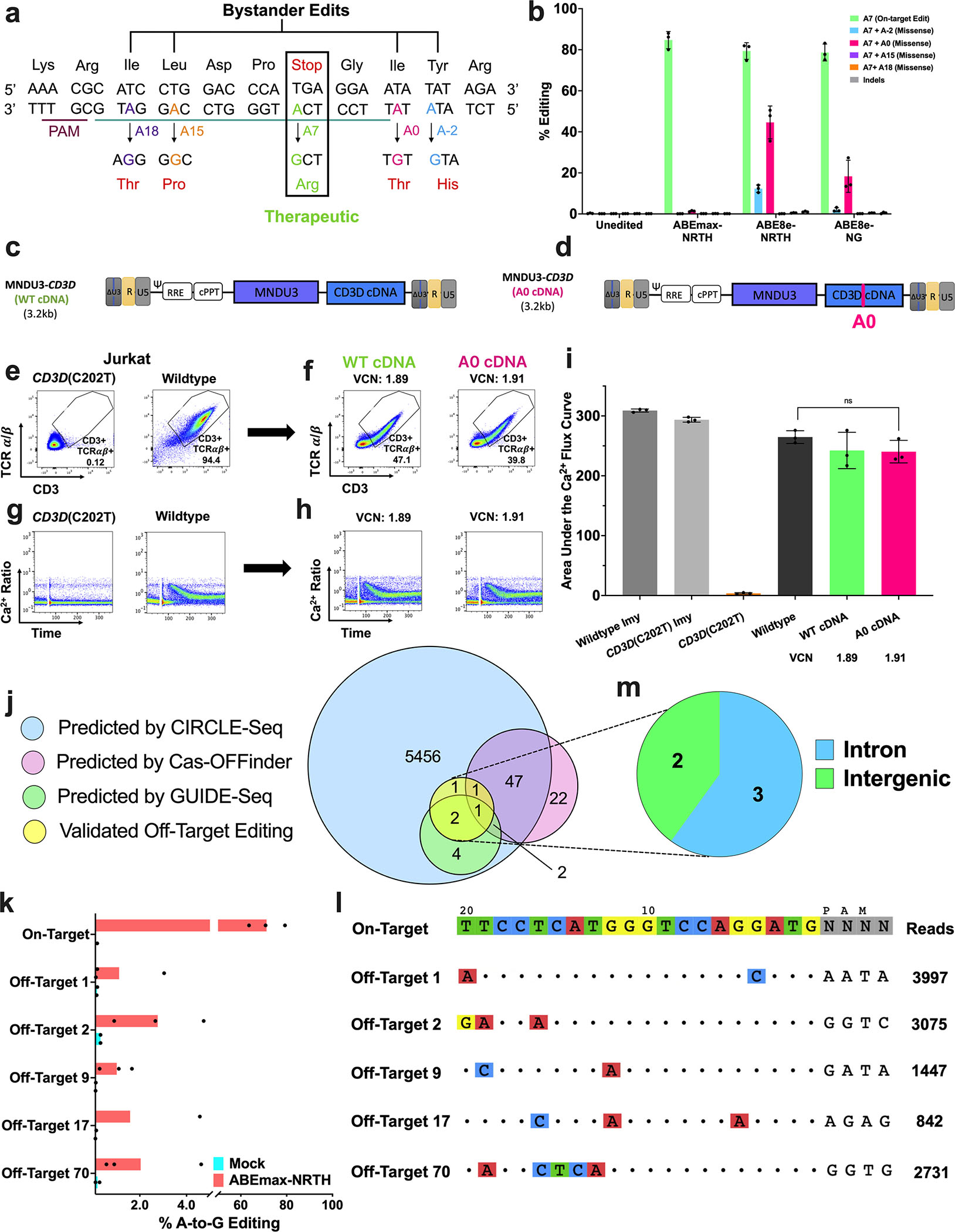
a) Schematic representation of the CD3D target with the on-target A at protospacer position A7 (green) and potential missense bystander edits A18 (purple), A15 (orange), A0 (pink), and A-2 (blue). Resulting amino acid substitutions (red text below). b) Plasmids encoding the CD3D-targeting sgRNA and either ABEmax-NRTH, ABE8e-NRTH, or ABE8e-NG were delivered by electroporation in CD3D(C202T) Jurkat T cells. Editing efficiencies were measured by HTS at on-target and bystander adenines five days after electroporation. c-d) Proviral maps of LVs used to characterize the effects of A0 bystander editing. MNDU3 (Myeloproliferative Sarcoma Virus, Negative Control region deleted Long Terminal Repeat promoter) is used to drive expression of the CD3D cDNA (with or without the A0 mutation). e-i) 14 days after transduction, a flow cytometry and a calcium flux assay were performed. LV vector copy number (VCN) was quantified by droplet digital PCR (ddPCR). j) Venn diagram of potential off-target sites assessed by multiplexed-targeted HTS nominated by CIRCLE-seq (blue), Cas-OFFinder (pink), GUIDE-seq (green), and predicted sites for which off-target editing was observed by HTS (yellow) in CD3δ SCID HSPCs electroporated with ABEmax-NRTH mRNA and sgRNA. k) Bar graphs demonstrate the percentage of sequencing reads containing A•T-to-G•C point mutations within protospacer positions 4–10 at on- and off-target sites in genomic DNA from treated and untreated CD3δ SCID HSPCs (n=3). l) CIRCLE-seq read counts and alignment to the on-target guide sequence for each validated off-target site. m) Genomic locations of validated off-target sites. b and i) Data shown as mean ± SD of 3 independent experiments. Statistical significance was calculated by non-parametric t-test; ns, not significant.
To further investigate the effect of the only detectable bystander edit (A0) induced by ABEmax-NRTH, we transduced CD3D(C202T) Jurkat T cells with one of two LVs expressing either: 1) a wildtype CD3D cDNA (MNDU3-CD3D WT cDNA) or 2) a CD3D cDNA containing the A0 bystander mutation (MNDU3-CD3D A0 cDNA) (Fig. 2c–f). Encouragingly, CD3D(C202T) Jurkat T cells transduced with MNDU3-CD3D A0 cDNA or MNDU3-CD3D WT cDNA demonstrated wildtype levels of CD3/TCR signaling in response to anti-CD3 and anti-CD28 stimulation (Fig. 2g–i; Fig. S2a). These findings suggested that low levels of bystander editing at position A0 would not interfere with rescue of healthy T cell function.
To identify and characterize genome-wide, Cas-dependent off-target editing resulting from ABEmax-NRTH mRNA and CD3D-directed sgRNA treatment, we utilized in vitro and in silico methods including, CIRCLE-seq,27 GUIDE-seq,28 and Cas-OFFinder.29 We performed CIRCLE-seq, a sensitive, in vitro off-target detection method, to identify nuclease-mediated cleavage sites induced by Cas9-NRTH and CD3D-localizing sgRNA in human genomic DNA. Recognizing the relaxed PAM consensus motif of the NRTH nuclease,30 we conducted CIRCLE-seq analysis to permit six mismatched nucleotides or fewer in aligned sequences, without specifying the PAM (NNNN), resulting in 5,514 candidate off-target sites (Table S2). To further validate off-target nominations, we performed GUIDE-seq, an unbiased detection method of off-target events, by electroporating CD3D(C202T) K562 cells with plasmids encoding Cas9-NRTH nuclease and sgRNA and a double-stranded DNA oligo for capture at DSBs. GUIDE-seq identified nine candidate sites, all of which overlapped with CIRCLE-seq nominations. The Cas-OFFinder in silico algorithm nominated 73 human genomic sites with ≤ 3 mismatches to the target protospacer, 51 of which were also nominated by CIRCLE-seq. Of the 5,514 sites predicted by CIRCLE-seq, the nine sites identified by GUIDE-seq, and the 73 sites nominated Cas-OFFinder, only three sites were shared between all off-target identification methods (Fig. 2j; Fig. S2b).
Next, we performed multiplex-targeted high-throughput sequencing in CD3δ SCID patient HSPCs treated with ABEmax-NRTH mRNA and sgRNA (edited patient HSPCs described in Fig. 4b) at the 57 off-target sites nominated by two or more prediction methods and the remaining top 143 sites nominated by CIRCLE-seq (n=200). Despite high levels of on-target CD3D c.202C>T editing (71.2 ± 7.85%), we observed A•T-to-G•C point mutations, characteristic of adenine base editing, at 2.5% (5/200) of the sequenced sites (Fig. 2k–l; Fig. S2c–d). All five validated sites were nominated by CIRCLE-seq, with three sites also identified by GUIDE-seq, and two sites predicted by Cas-OFFinder, demonstrating the importance of using experimental methods when investigating off-target sites. Of the five verified sites, three sites were found in introns greater than 100 bp away from any coding region and two sites occurred in intergenic regions (Fig. 2m). Indel frequencies were less than 0.54% at all sequenced sites after subtraction of mock control reads (Fig. S2e). Altogether, our assessment of local bystander editing and genome-wide off-target editing did not indicate clinically concerning off-target editing, despite high levels of on-target correction.
Figure 4. Base Editing of CD3δ SCID CD34+ HSPCs Rescues T cell Differentiation.
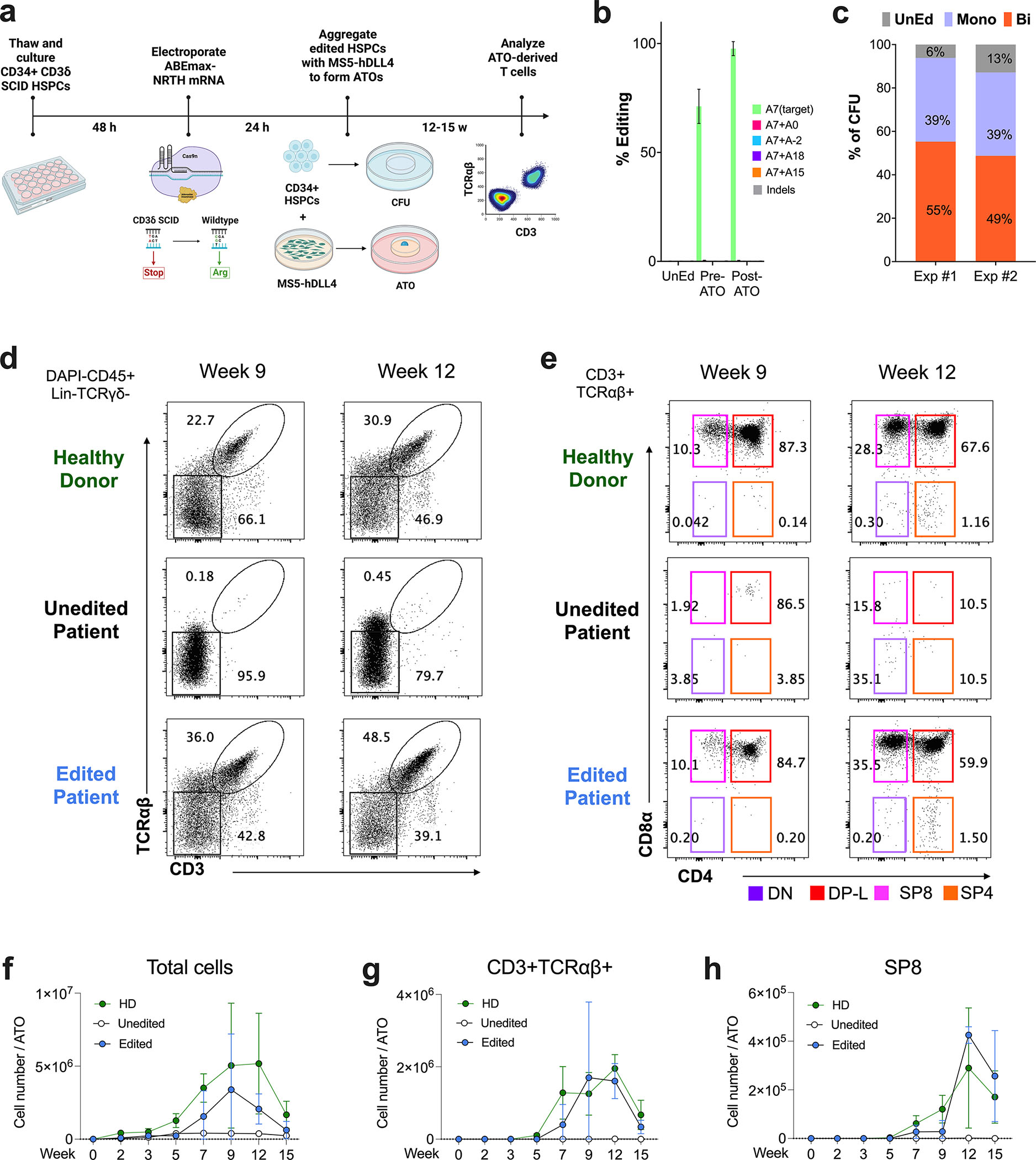
a) Workflow of T cell differentiation: HSPCs were isolated from bone marrow of a patient with CD3δ SCID and electroporated with ABEmax-NRTH mRNA and sgRNA localizing to the CD3D c.202C>T mutation. Treated cells were aggregated with MS5-hDLL4 stromal cells and installed on a cell culture insert for ATO differentiation. b) HTS editing efficiencies at target and bystander adenines (see Fig. 2a for descriptions of nomenclature) and indels after 5 days of in vitro culture post-electroporation (‘pre-ATO’) and 12–15 weeks after T cell differentiation (‘post-ATO’), UnEd, unedited. A portion of cells were plated in methylcellulose for a CFU assay. c) Clonal editing outcomes determined by HTS of the CD3D target by analysis of individual day 14 CFUs. Exp #1, n=100 CFUs and Exp #2, n=130 CFUs. Mono, monoallelic; Bi, biallelic. d-h) Kinetics of T cell differentiation in ATOs derived from CD34+ HSPC, d-e) Representative flow cytometry profiles of d) CD3+ and TCRαβ+ expression gated on DAPI-CD45+Lin-(CD56-CD14-)TCRγδ-, and CD4 and CD8α expression in e) CD3+TCRαβ+ cells gated on CD45+Lin-. HD (top), unedited patient (middle), and edited patient (bottom) ATOs (n=6–9 for each time point). Cell counts of f) total cell output, g) CD3+TCRαβ+, and h) SP8 T cells per ATO (n=6–12 per time point).
Long-Term Correction of Healthy Human HSPCs in a Humanized Mouse Model
We next explored the ability to base edit the pathogenic CD3D mutation in long-term, repopulating cells in a humanized xenograft model. Healthy human donor (HD) CD34+ HSPCs were transduced with an LV expressing a CD3D cDNA disease target containing the CD3D c.202C>T mutation under the control of the MNDU3 promoter (MNDU3-CD3D c.202C>T-cDNA) (Fig. 3a). Codon optimized N- and C- termini (20 bp) of the LV cDNA enabled differentiation of the corrected mutation from endogenous CD3D sequence (Fig. 3b) by PCR amplification. 24 after transduction HSPCs were electroporated with mRNA encoding ABEmax-NRTH and sgRNA. [The same approach was utilized to revert two other recurrent CD3δ SCID-causing mutations identified in Ecuador (CD3D c.274+5G>A)31 and Japan (CD3D c.275–2A>G)32, generating LVs carrying CD3D cDNA with either mutation for correction by base editing in HD CD34+ cells (Fig. S3)]. The day following electroporation, resulting CD3D c.202C>T LV-transduced and edited HSPCs along with transduced-only control cells were each transplanted into 4–10 NOD,B6.SCID IL2rg−/−KitW41/W41(NBSGW) immunodeficient mice.33 As a BE-only control, we separately electroporated HD HSPCs with mRNA encoding ABE8e-NG and sgRNA targeting an endogenous adenine base (adenine A0 described in Fig. 2a) because the CD3D c.202C>T-targeting sgRNA does not bind to healthy CD3D locus. 24 hours after electroporation, the resulting edited cells and untreated control cells were additionally transplanted into 2–7 NBSGW mice (Fig. S3).
Figure 3. Engrafted Healthy Human HSPCs Retain High Levels of Gene Correction in a Humanized Mouse Model.
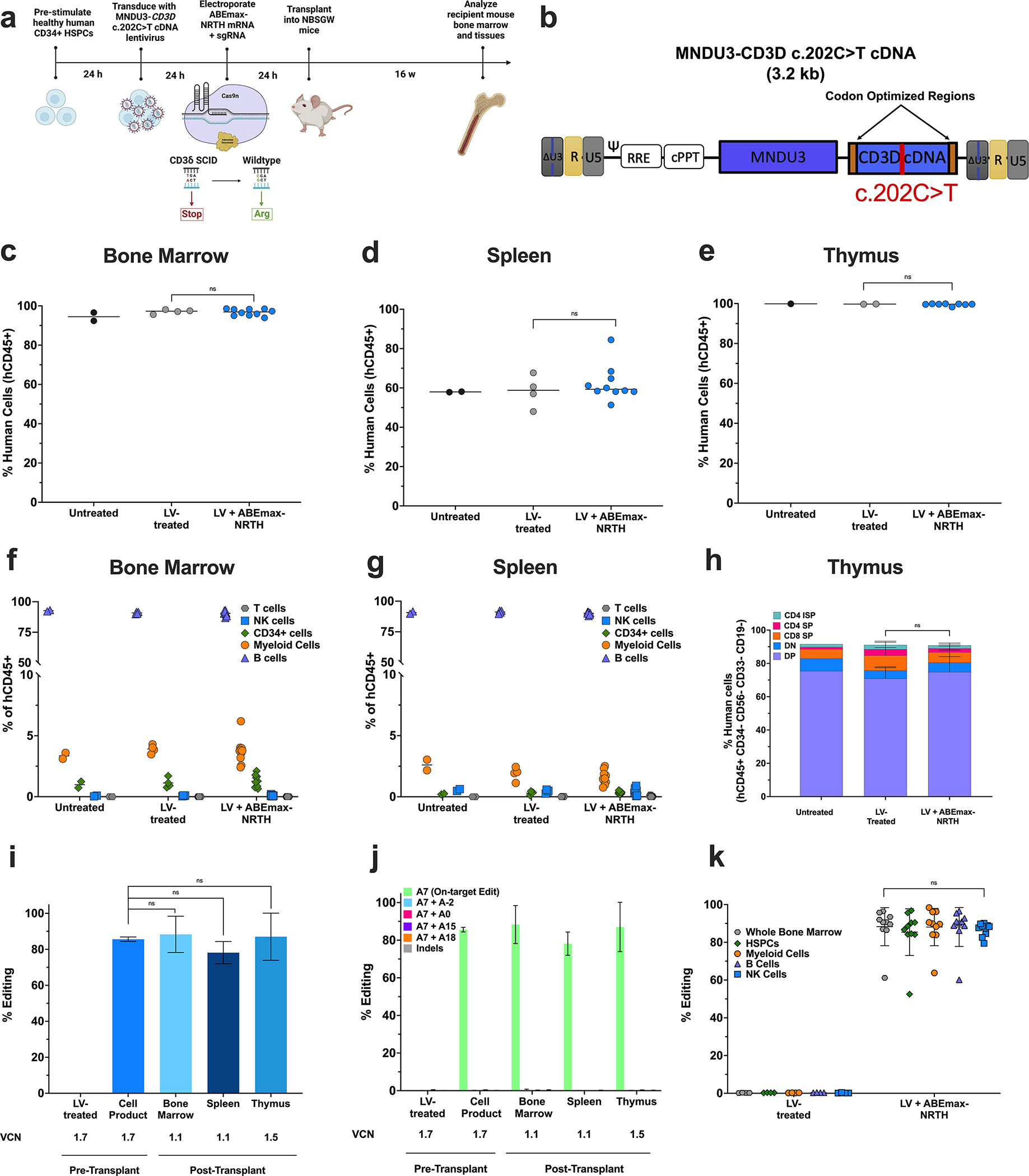
a) Experimental timeline for xenograft studies. b) Proviral map of lentiviral disease target for integration in healthy CD34+ HSPCs. Components of the LV are similar to those described in Fig. 2c–d, with the exception of 20 bp codon optimized regions on N- and C-termini (orange boxes) of the CD3D cDNA to allow for specific targeted DNA amplification of the CD3D cDNA (not the endogenous CD3D gene) for base editing analysis. 16 weeks after infusion, engraftment was measured by percentage of human CD45+ cells in recipient mice c) bone marrow, d) spleen, and e) thymus. Abundance of human CD19+ B cells, CD33+ myeloid, CD34+ HSPCs, CD56+ NK cells, and CD3+ T cells were measured as percentages of the hCD45+ population in transplant recipient f) bone marrow and g) spleen. h) Thymocytes as percentages of the hCD45+ population in recipient mouse thymus. i) CD3D c.202C>T editing efficiency and VCN determined by HTS and ddPCR, respectively, in cells cultured for 14 days after electroporation (pre-transplant) or in whole tissues 16 weeks after transplant. j) HTS of on-target and bystander adenines in the pre-transplant HSPC cell product and bulk tissues post-transplant. k) CD3D c.202C>T editing efficiency in human-derived hematopoietic lineages FACS sorted from mouse bone marrow. n=2 mice that received untreated cells, n=4 mice that received LV-transduced cells, and n=10 mice that received LV-transduced and edited cells. Data shown as mean ± SD; k) one-way ANOVA, c-j) non-parametric t-test; ns, not significant.
To assess the effects of base editing on engraftment and lineage maintenance, we extracted bone marrow (BM), spleen, and thymus from the recipient mice for analysis 16 weeks after transplant. Flow cytometry demonstrated 96.2 ± 1.45%, 58.3 ± 0.40%, and 99.8 ± 0.10% of hCD45+ human cells in all mice BM, spleen, and thymus, respectively. Furthermore, we did not observe statistically significant differences in engraftment between untreated, LV-treated, and LV + BE-treated human cells (p=0.63), indicating that engraftment was not altered by base editing (Fig. 3c–e). To determine if CD3D-targeted base editing influenced HSPC differentiation potential and lineage maintenance, we investigated the proportions of human CD19+ B cells, CD33+ myeloid, CD34+ HSPCs, CD56+ NK cells, and CD3+ T cells in engrafted mice (Fig. S3a). Relative abundances of hematopoietic lineages were equivalent across control and treatment arms in the BM and spleen, suggesting that base editing did not alter hematopoiesis (Fig. 3f–g). Although mature human T cells develop minimally from healthy stem cells engrafted in the adult NBSGW model due to thymic atrophy,34 analysis of reconstituted donor thymocytes demonstrated no changes in sub-population distribution, indicating that base editing did not disrupt thymocyte differentiation potential (p=0.97) (Fig. 3h; Fig. S3b, S3h).
Engraftment of gene-corrected, repopulating HSCs is a critical objective for sustained and effective hematopoiesis and survival following autologous HSCT.35 To investigate whether base editing can effectively correct the pathogenic mutation in long-term HSCs, we quantified CD3D c.202C>T editing efficiencies five days after electroporation (‘pre-transplant’, 85 ± 1.2%) and at the 16-week harvest from recipient mice. Notably, 16 weeks after infusion, editing frequencies measured from whole BM, spleen, and thymus of transplant recipients demonstrated durable base editing (84.5 ± 5.52%, 78.2 ± 6.18%, and 87 ± 13.1%, respectively), suggesting high levels of gene correction in repopulating HSCs (p=0.73, p=0.13, and p=0.89) (Fig. 3i–j).
Additionally, we explored if base editing could influence multipotency of repopulating HSCs. Different lineages of human donor-derived (hCD45+) mononuclear cells (hCD45+ whole bone marrow, CD34+ HSPCs, CD33+ myeloid, CD19+ B cells, and CD56+ NK cells) were isolated by fluorescence-activated cell sorting (FACS) from recipient mouse bone marrow. HTS of the CD3D disease target revealed no changes in base editing frequencies across all isolated populations (87.0 ± 1.15%; p=0.95); bystander edits were <1% (Fig. 3k).
Engraftment, differentiation potential, and multipotency were similarly unaffected in cells treated with ABE8e-NG mRNA and sgRNA targeting endogenous adenine A0 without LV transduction (Fig. S3c–h, S3j–k). Before transplantation, 78% editing was observed in the HSPC pool, and in repopulating HSCs that were engrafted, 54% editing was maintained (Fig. S3i–k). A pool level 50% editing efficiency could be therapeutic given the autosomal recessive nature of CD3δ SCID and that monoallelic CD3D correction restored T lymphopoiesis in patient cells (Fig. S6a–b). Altogether, these findings suggested that ABEmax-NRTH-treated CD34+ HSPCs can successfully repopulate the hematopoietic system and maintain therapeutic CD3D c.202C>T correction in all hematopoietic progeny.
Base Editing of CD3δ SCID HSPCs Rescues T cell development
To evaluate whether base editing of CD3δ SCID HSPCs can rescue CD3/TCR surface expression and normal T cell development, we employed an in vitro T cell differentiation assay (the artificial thymic organoid [ATO] model) that recapitulates normal human thymopoiesis from uncommitted HSPCs.12,14,36 CD34+ bone marrow-derived HSPCs from an infant with CD3δ SCID were electroporated with ABEmax-NRTH mRNA and sgRNA and tested for their capacity to generate mature T cells in ATOs compared to untreated patient HSPCs and healthy donor (HD) bone marrow CD34+ HSPC controls (Fig. 4a).
Electroporation of ABEmax-NRTH mRNA and sgRNA achieved 71.2 ± 7.85% correction of the CD3D c.202C>T mutant alleles in HSPCs by HTS prior to plating in ATOs, with minimal bystander editing or indels (Fig. 4b). One day after electroporation, an aliquot of cells from each arm was plated in methylcellulose for a colony forming unit (CFU) assay to assess base editing at the clonal myelo-erythroid progenitor level. Sequence analysis of individual CFUs demonstrated that 52 ± 4.24% of cells contained biallelic correction of the CD3D c.202C>T mutation, 39 ± 0.10% of cells showed monoallelic editing, and only 9.5 ± 4.95% of cells remained unedited (n=230) (Fig. 4c). Additionally, no impact of editing was observed on myelo-erythroid differentiation (Fig. S4a–b).
T cell development was evaluated by flow cytometry at 2, 3, 5, 7, 9, 12, and 15 weeks after electroporation. As expected, HD ATOs generated cells that co-expressed CD3 and TCRαβ at increasing percentages over time (Fig. 4d; Fig. S4c–e), with maturation to late double positive (“DP-L”, CD3+TCRαβ+CD4+CD8α+), single positive 8 T cells (“SP8”, CD3+TCRαβ+CD4−CD8α+CD8β+) and single positive 4 T cells (“SP4”, CD3+TCRαβ+CD4+CD8α-) (Fig. 4e; Fig. S4c, S4f–g). In contrast, cells from unedited patient ATOs had undetectable CD3 and TCRαβ surface expression across all time points (Fig. 4d; Fig. S4c–e). Because TCR expression was absent in unedited patient HSPCs, T cell differentiation was severely disrupted with an accumulation of unedited cells in the DN (CD4−CD8−) precursor stage and an inability to progress past the DP (CD4+CD8+) developmental stage into either SP8 T cells or SP4 T cells (Fig. 4e; Fig. S4c; Fig. S4e–g). Surface CD3 and TCRαβ co-expression was robustly rescued in edited patient ATOs (Fig. 4d; Fig. S4c–e), appearing first at the DP stage and persisting in SP8 and SP4 T cell populations (Fig. 4e; Fig. S4c–g). The cell number output of total, CD3+TCRαβ+, and SP8 T subsets per ATO (Fig. 4f–h; Fig. S4h–i) was similar between edited patient and HD ATO cultures across all time points and dramatically decreased in unedited cells.
Previous reports have described faulty development of TCRγδ+ T cells in patients with CD3δ SCID.2,37,38 Unedited patient ATOs recapitulated this clinical finding, demonstrating the absence of TCRγδ+ T cell production across all time points. In contrast, edited patient and HD ATOs supported the development of TCRγδ+ cells to similar extents (Fig. S4j–k).
Differentiation of Unedited CD3δ SCID HSPCs Cannot Proceed Past DP T cell Precursor Stage
A single prior report of an individual patient with CD3δ SCID characterized the block in thymopoiesis at the DN (CD3−TCRαβ-CD8α-CD4−) stage by western blot of a thymic biopsy.2 In contrast, the ATO system allowed us to interrogate thymopoiesis kinetics in distinct precursor stages. As previously reported, unedited patient ATOs demonstrated increased DN populations as compared to HD and edited patient ATOs (Fig. 5a–c). However, we identified maturation past the DN stage to the immature single positive (“ISP4”) and DP stages in unedited patient ATOs (Fig. 5a–d). While TCR-CD3-DP cells (DP-E) precursors could be detected in unedited patient ATOs, their absolute numbers were low (Fig. 5e).
Figure 5. T cell Differentiation from CD3δ SCID HSPCs is Blocked at the DP Stage.
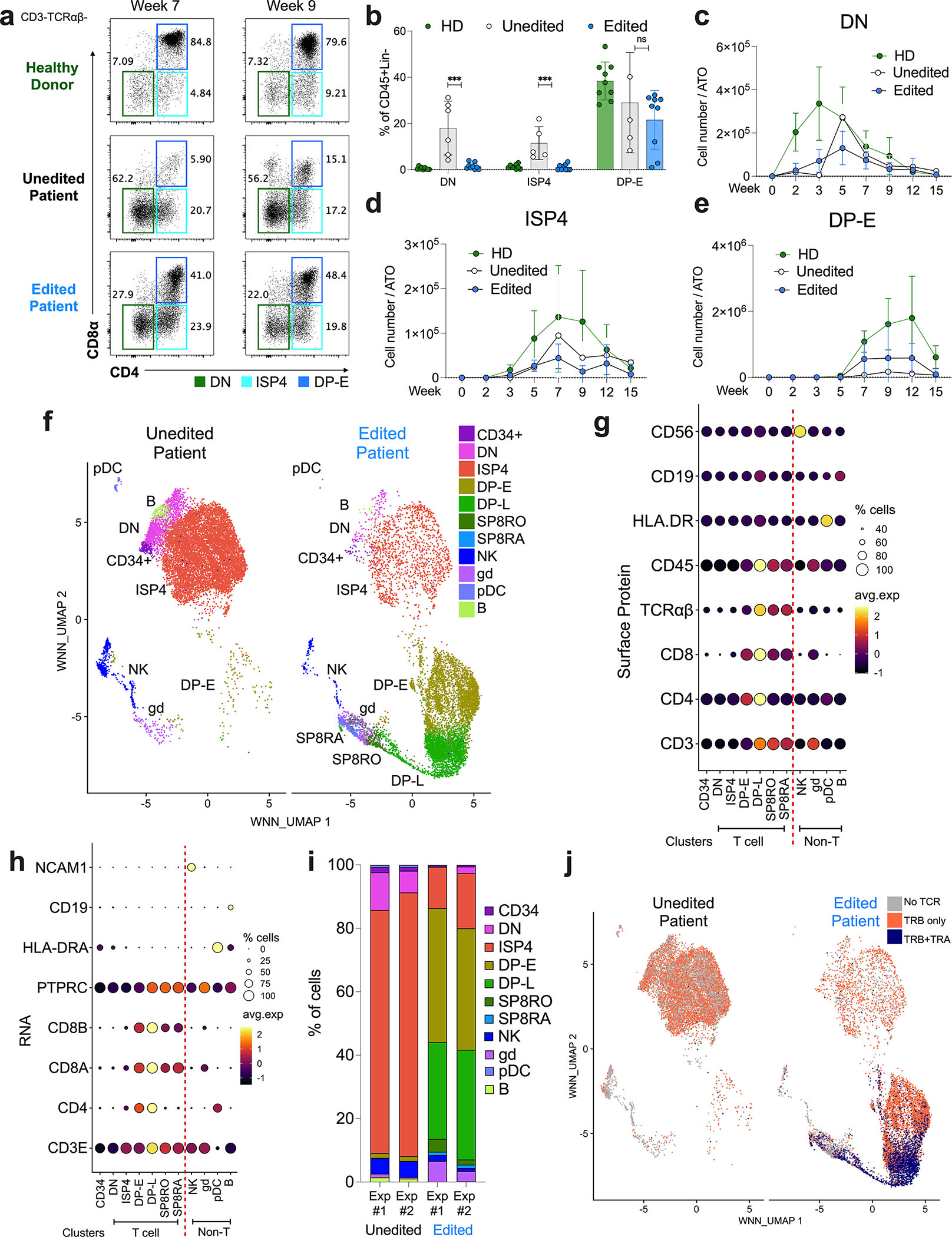
a-e) T cell differentiation of HD, unedited patient, and edited patient ATOs, n=6–12 from 4 independent experiments. a) Representative flow cytometry profiles depicting T cell differentiation of DN (green), ISP4 (aqua), and DP-E (blue) populations in cells gated on CD3-TCRαβ- cells at weeks 7 and 9. b) Frequency of DN, ISP4, and DP-E cells in CD45+Lin- cells at week 12. Data shown as mean ± SD. Statistical significance calculated by unpaired nonparametric t-test ***p<0.001. Cell counts of c) DN, d) ISP4, and e) DP-E cells per ATO. f-j) CITE-seq analysis of unedited and edited CD3δ SCID patient ATOs at week 8 (n=4). f) WNN_UMAP visualizations of annotated populations in unedited (left) and edited (right) patient ATOs. Expression of lineage defining g) surface proteins and h) RNA across clusters. i) Frequency of developing T cell (DN, ISP4, DP-E, DP-L, SP8RO, and SP8RA) and other immune cell (CD34+, NK, innate, pDC, γδ T cell, B cell) subsets in unedited (left) or edited (right) samples. j) WNN_UMAP visualization of no TRA or TRB (grey), TRB only (orange), and both TRA and TRB (purple) expression.
Single-cell RNA Sequencing Identifies Initial TRA Expression in DP-L Precursors
To provide a more detailed analysis of how base editing of CD3δ SCID affected T cell development, cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq)39 was utilized to integrate surface protein, transcriptional profile, and TCR clonotype expression at single cell resolution. Cells were isolated from unedited and edited CD3δ SCID ATOs, stained for 130 unique surface antigens, and sequencing libraries were generated using the 10X Chromium Single Cell Sequencing workflow. Individual samples were cleaned (Material and Methods) and −22,000 cells were identified after bioinformatic cleaning for downstream analysis (Fig. S5a).
Weighted Nearest Neighbor (WNN) Uniform Manifold Approximation and Projection (WNN_UMAP) using Seurat v4.2.040 (Fig. 5f) visualized individual cell subsets based on a combination of surface protein (Fig. 5g; Fig. S5b) and RNA gene expression (Fig. 5h; Fig. S5c). The following subsets were identified: CD34+ (CD34+CD4−CD8−TCRαβ-), DN (CD34-CD8−CD4−), ISP4 (CD3−TCRαβ-CD8−CD4+), DP Early (DP-E: CD3−TCRαβ-CD8+CD4+); late DP (DP-L, CD3+TCRαβ+CD8+CD4+), SP8 (CD3+TCRαβ+CD8+CD4−; further divided into SP8RO and SP8RA), NK cells (CD56+), γδ T cells (TCRαβ-TCR.Vδ2), pDC (CD4+RAG1-RAG2-HLADR+), and B cells (PAX5+CD19+).
WNN_UMAP visualization confirmed that unedited patient ATOs contained high proportions of DN and ISP4 subsets (Fig. 5f, 5i). While FACS analysis identified a higher proportion of DP-E precursors (Fig. 5a), than CITE-Seq analysis (Fig. S5a), a dead-cell removal kit was applied to ATOs prior to CITE-seq, likely depleting a proportion of rapidly apoptosing DP-E cells. As expected, CITE-seq analysis confirmed that populations defined by the co-expression of CD3 and TCRαβ (DP-L and SP8 subsets) were absent in unedited patient ATOs and restored in edited ATOs.
The TCR comprises two subunits: TRB and TRA, which must undergo rearrangement of germline variable (V), diversity (D), and joining (J) gene segments to generate a mature TCR.41 TRB rearrangement begins at the DN stage while TRA rearrangement begins at the DP stage.42 Because the development of unedited patient ATOs is blocked at the DP stage, we assessed TRB and TRA usage by single-cell TCR sequencing as described above by CITE-seq. Analysis of each TCR subunit found that single cells expressing both TRB and TRA belonged to cells with CD3/TCR surface expression (i.e. DP-L, SP8RO and SP8RA clusters, whereas single cells expressing only TRB were found in precursor populations that lacked CD3/TCR surface expression: DN, ISP4, and DP-E) (Fig. 5j). Unedited patient ATO-derived cells expressed TRB but not TRA and were unable to proceed to the DP-L stage when TRA is normally expressed.
T cells Derived from Edited CD3δ SCID HSPCs ATOs Show Mature, Naive Phenotype
Due to the autosomal recessive nature of CD3δ SCID, correction of a single CD3D allele is expected to rescue disease phenotype. Single-cell monoallelic and biallelic correction frequencies were measured by presence of RNA abundance in both unedited and edited patient ATOs. We observed nonsignificant differences in relative abundances of T cell precursors and in T cell maturation of patient-derived ATO cells containing a monoallelic or biallelic edit (p=0.99) (Fig. S6a–b).
SP8 T cells derived from edited patient ATOs expressed markers consistent with transition from an immature (CD45RO+CD45RA-CD27+CCR7−) to mature (CD45RO-CD45RA+CD27+CCR7+) thymocyte phenotype; both immature and mature subsets co-expressed CD62L and CD28 (Fig. 6a). Expression levels of maturation markers were similar between edited patient and HD ATOs by flow cytometry (Fig. 6a), and CITE-seq analysis of cells derived from edited patient ATOs confirmed protein expression of maturation markers (CD27, CD28, CD45RA, CD45RO, and TCRαβ), while lacking expression of activation markers CD25 and CD137 in SP8RO/RA cells (Fig. S6c).
Figure 6. Edited CD3δ SCID ATO-derived T cells Express Features of Maturation without Evidence of Exhaustion.
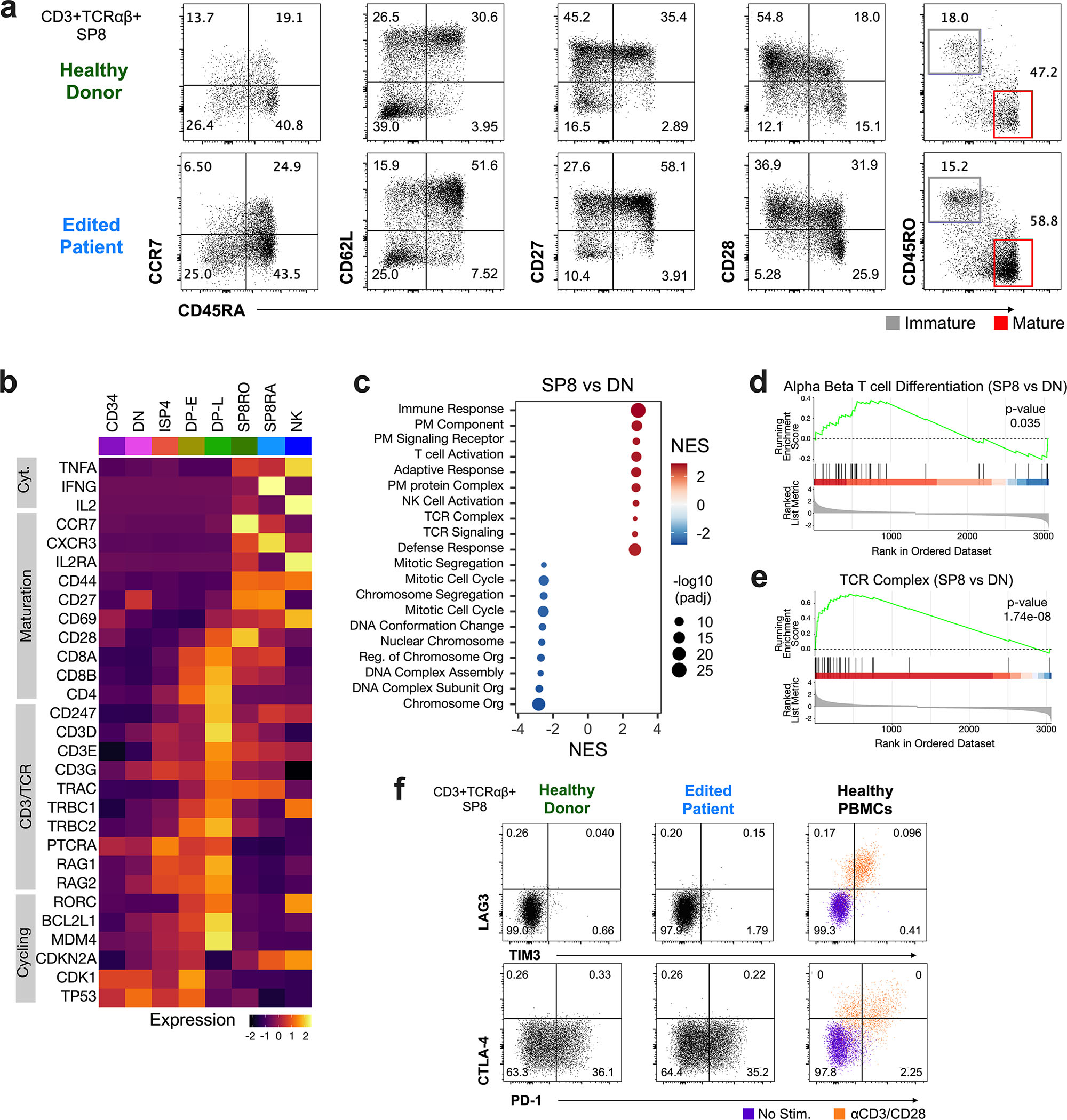
a) Representative flow cytometry profiles depicting maturation markers (CCR7, CD62L, CD27, CD28, CD45RO, and CD45RA) in cells gated on SP8 T cells - CD3+TCRαβ+CD8α+CD8β+, in week 12 ATOs (n=9, from four independent experiments). b) RNA expression of selected genes (y-axis) across clusters in edited patient ATOs by CITE-seq; Cyt., cytokine. c) Gene Set Enrichment Analysis (GSEA) of differentially expressed genes from GOBP (Gene Ontology Biological Process) and GOCC (Gene Ontology Cellular Compartment) between SP8 T and DN cells from edited ATOs. Dot size represents adjusted p-value (padj; two-sided permutation test). NES, normalized enrichment score; PM, plasma membrane. GSEA plots of representative gene sets d) alpha beta T cell differentiation (p=0.035), and e) TCR complex (p=1.74E-8) in SP8 vs DN T cells from edited ATOs. f) Representative flow cytometry profiles of exhaustion markers in SP8 T cells directly from week 15 HD (n=9) and edited patient ATOs (n=9), and compared to PBMCs (n=3); PBMC were stimulated with (orange) and without (purple) anti-CD3/28 beads and IL2 for 24 hours.
Single-cell transcriptomic analysis (Fig. 6b) demonstrated that mature SP8 T cells derived from edited patient ATOs expressed high levels of genes found in mature T cells (CXCR3, IL2RA, and CD44). DP-L cells derived from edited patient ATOs expressed high levels of genes found in CD3/TCR signaling (CD247, CD3D/E/G, and TRA/TRB), and cell cycling/proliferation (RORC, BCL2L1, and MDM4). Gene Set Enrichment Analysis (GSEA) was utilized to identify relevant biological processes and pathways that differed across developing thymocyte subsets in edited ATO-derived cells, where CD3/TCR expression was rescued (Fig. 6c). T cell activation, T cell differentiation, and TCR signaling were upregulated in SP8 (both SP8RO and SP8RA) relative to DN cells (Fig. 6c–e). Comparison of DP-L vs. DN cells identified upregulation of T cell differentiation and TCR signaling pathways in DP-L cells (Fig. S6d). Comparison of SP8 (both SP8RO and SP8RA) T vs. DP-L cells highlighted upregulation of ribosomal pathways required for protein translation in SP8 T cells (Fig. S6e).
Restoration of T cell development in base-edited ATOs resulted in normal production of SP8 T cells in culture. FACS analysis of SP8 T cells from late (15 week) edited patient and healthy donor ATO cultures lacked expression of exhaustion markers LAG3, TIM3, and CTLA-4.43–45 PD-1 expression was detected in both edited patient and HD ATOs at similar levels (Fig. 6f). SP4 T cells derived from edited patient ATOs demonstrated similar expression of maturation markers and lacked expression of exhaustion markers (Fig. S6f–g).
Base-Edited CD3δ SCID HSPCs Develop into Functional T cells with a Diverse TCR Repertoire
To evaluate the ability of base editing to produce T cells with functional TCRs, week 12–15 ATOs were harvested and calcium flux analysis was performed as a proxy for early CD3/TCR activation. Consistent with lack of CD3/TCR, unedited patient ATO cells displayed no calcium flux in response to stimulation with anti-CD3 and anti-CD28 antibodies. Base editing restored calcium flux to similar levels as HD ATO cells (381.0 ± 56.9 and 316 ± 24.1 AUC) (Fig. 7a–b). Mature SP8 T cells isolated from edited patient ATOs and HD ATOs demonstrated polyfunctional production of IFNγ, IL-2, and TNFα in response to stimulation with anti-CD3/CD28 beads with IL-2 for 24 hours (Fig. 7c–f; Fig. S7a). SP8 T cells upregulated CD25 and 4–1BB and proliferated in response to anti-CD3/CD28 beads and IL-2 for 5 days (Fig. 7g–h; Fig. S7b–c).
Figure 7. Base Editing of CD3δ SCID HSPCs Generates Functional T cells with TCR diversity.
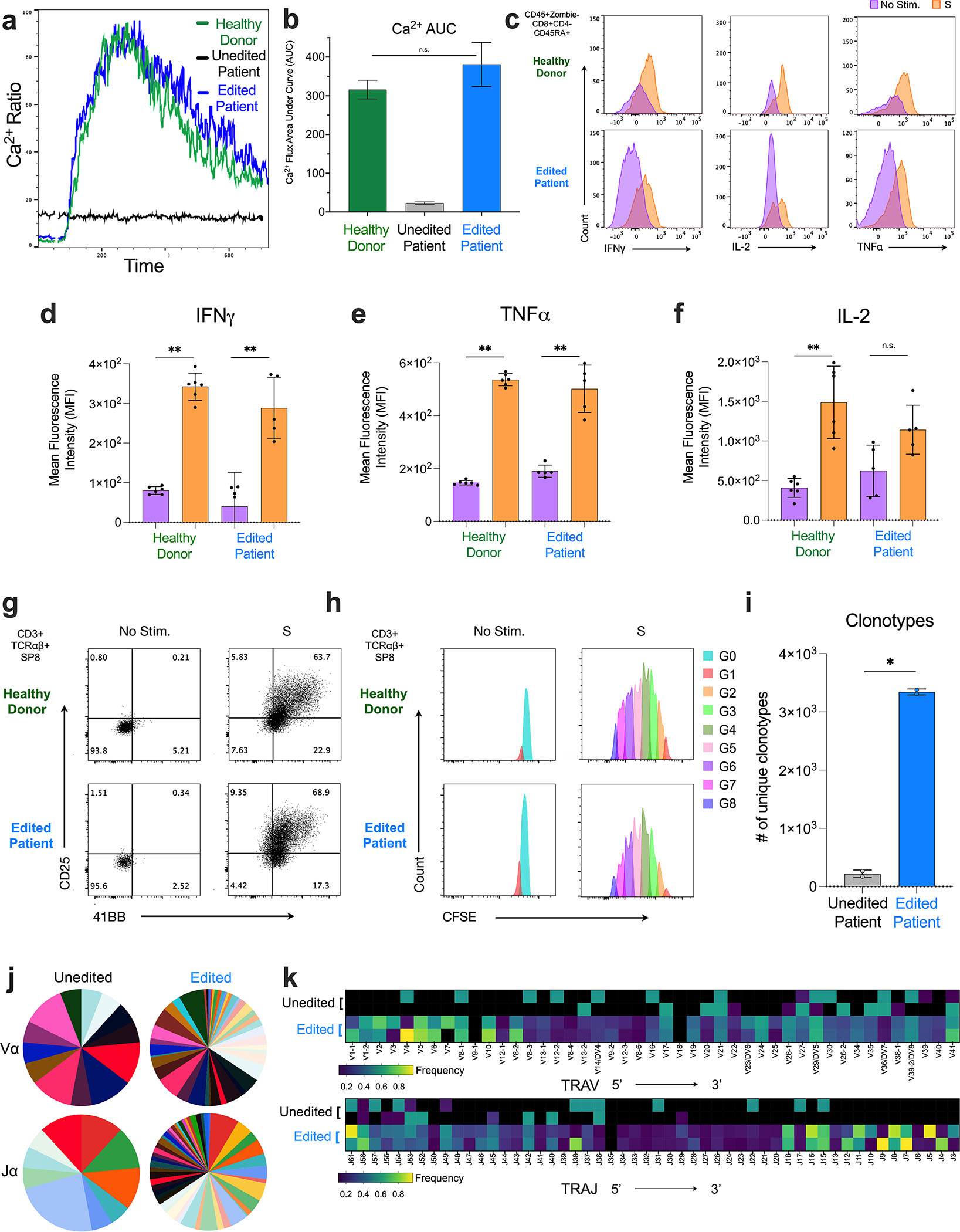
a) Calcium flux of cells isolated from HD (green), edited patient (blue), and unedited patient (black) ATOs stimulated with anti-CD3 and anti-CD28. b) Quantified area under the calcium flux curve of HD (green), edited patient (blue), and unedited patient (black) ATO cells. c-f) HD (green) and edited patient (blue) ATOs stimulated with and without anti-CD3/CD28 beads and IL2 for 24 hours (n=6). c) Representative flow cytometry histogram profiling and mean fluorescence intensity (MFI) of d) IFNγ, e) TNFα, and f) IL-2 production in mature SP8s (Zombie-CD45+CD8α+CD4−CD45RA+). Production of IFNγ and TNFα with and without stimulation was statistically significant (p<0.01). Production of IL-2 was not statistically significant (p=0.055). g) Activation (upregulation of CD25 and 4–1BB) and h) proliferation (CFSE dilution) of isolated HD and edited patient ATO-derived SP8 T cells after culture with anti-CD3/CD28 bead and IL-2 for 5 days. Data is representative of three independent experiments. i-k) Single-cell TCR sequencing by CITE-seq of unedited and edited patient ATOs harvested at week 8, n=2 for each arm. Data are representative of two independent experiments. i) Number of unique TCR clonotypes. j) Frequency of individual TRAV (top) and TRAJ (bottom) usage. k) Heatmap visualization of individual TRAV and TRAJ segments displayed in genomic order from 5’ distal -> 3’ proximal ends. Statistical significance was calculated by unpaired non-parametric t-test (**p < 0.01).
A diverse TCR repertoire is essential for an effective T cell immune response. Unedited CD3δ SCID ATOs demonstrated significantly fewer TCR clonotypes as compared to edited patient ATOs (217.5 ± 65.8, n=2 vs. 3344 ± 50.1, n=2, p<0.002) (Fig. 7i) as evidenced by decreased Chao 1 index46 (Fig. S7d). Chord diagrams of T cell populations from unedited and edited patient ATOs illustrate shared TCR clonotypes between developmentally neighboring subsets (Fig. S7e). In unedited patient ATOs, ISP4 precursors expressed the highest diversity of TCR clonotypes, and shared TCR clonotypes with DP-E precursors. In edited patient ATOs, DP-E precursors, yet to undergo positive selection, expressed the highest diversity of TCR clonotypes, and shared TCR clonotypes with DP-L precursors. In contrast, positively selected SP8RO and SP8RA T cells expressed fewer TCR clonotypes.
Further independent analysis of TRA and TRB usage revealed impaired 5’ distal TRAV and 3’ proximal TRAJ usage in unedited patient ATOs. These segments represent the regions of Vα and Jα that rearrange last during VDJ recombination. Base editing of CD3δ SCID HSPCs restored diverse TRAV and TRAJ usage in edited patient ATOs (Fig. 7j–k). No significant differences were found in TRBV or TRBJ usage between unedited and edited patient ATOs (Fig. S7f–g).
Taken together, these data demonstrate robust restoration of T cell development from CD3δ SCID HSPCs by ABE-mediated gene therapy. Extensive phenotyping of edited T cells in ATOs revealed rescue of mature T cell function and diverse TCR repertoire, indicating clinical promise for this approach.
Discussion
The ability to correct pathogenic point mutations that cause life-threatening monogenic diseases is becoming a clinical reality for precision medicine. One promising approach is base editing to efficiently and precisely correct disease-causing alleles.10,47,48 Base editing has advantages over approaches using HDR to correct mutations as it can be achieved without producing DSBs, generating uncontrolled mixtures of indel byproducts, requiring provision of donor DNA templates, or being limited to cells in certain phases of the cell cycle. Here, we describe an ABE-mediated approach to revert the mutation underlying most CD3δ SCID cases (CD3D c.202C>T) to wildtype sequence. This approach successfully reverted the premature stop codon in a Jurkat T cell line disease model, in healthy CD34+ HSPCs transduced with an LV carrying a CD3D c.202C>T target, and in CD34+ HSPCs isolated from a patient with CD3δ SCID. This base editing strategy was precise and efficient in all blood cell types analyzed (up to 85% in CD3D(C202T) Jurkat T cells, 96% in repopulating healthy HSPCs, and 79% in CD3δ SCID patient-derived HSPCs), with minimal bystander edits or indels.
The capacity to precisely position the ABE editing window at the target base may be limited by the availability of an appropriate protospacer adjacent motif (PAM) to direct localization of the base editor by a sgRNA. As demonstrated here, Cas9 variants with expanded targeting scope beyond the canonical NGG PAM of native SpCas9 can enable efficient and precise targeting of human pathogenic gene variants. Investigation of five ABE variants including three newly developed ABEs, ABE8e-xCas9(3.7), ABE8e-VRER, and ABE8e-NRTH, and two previously generated editors, ABE8e-NG and ABEmax-NRTH,20,26 resulted in robust correction of the c.202C>T mutation (18%, 33%, 92%, 86%, and 93%, respectively) whereas an HDR approach only achieved 28% correction to the wildtype sequence, accompanied by an excess of indel byproducts (53%).
Cas-nuclease mediated DSBs are well established to induce chromosomal abnormalities at on- or off-target sites.22 Indeed, we observed large deletions distal to the on-target CD3D locus (11q23) when CD3D(C202T) Jurkat T cells were treated with RNP + ssODN, but not when treated with ABEmax-NRTH. These deletions are clinically concerning since some chromosomal abnormalities at 11q23 have been associated with AML and poor prognosis for CML patients.49 Therefore, ABE may be a safer and more efficacious treatment for CD3δ SCID by circumventing the production of DSBs.
We observed infrequent (<1%) adenine editing at position A0 (counting position 1 as the PAM-distal end of the protospacer) in cells electroporated with ABEmax-NRTH; whereas ABE8e induced bystander edits at a much higher frequency (18–45%). The rare bystander editing at A0 by the lead candidate ABEmax-NRTH produced an isoleucine to threonine substitution that did not have a clear adverse effect on function of the CD3δ protein; expression of this variant corrected CD3/TCR signaling in CD3D(C202T) Jurkat T cells to levels equivalent in cells receiving a wildtype control. Thus, this low-level of bystander editing utilizing ABEmax-NRTH will not likely impair ABE efficacy for CD3D c.202C>T correction.
Furthermore, we examined the occurrence of genome-wide off-target base editing in primary CD3δ SCID patient HSPCs treated with ABEmax-NRTH mRNA and sgRNA. Of the 200 sites evaluated, HTS of ABEmax-treated CD3δ SCID patient cells verified only five sites containing point mutations consistent with ABE, despite high levels of on-target CD3D editing. Of these five validated off-target sites, three sites occurred in intronic regions and the remaining two sites were found in intergenic regions. Without the induction of DSBs necessary for CRISPR/Cas9-mediated editing and the apparent low frequency of off-target edits, base editing is less likely to induce genotoxicity.
Despite its prevalence in rural Mennonite communities of North America, comprising over 20% of SCID-causing genotypes in Alberta, Canada (unpublished data), CD3δ SCID is an ultra-rare disease, thus limiting access to patient-derived HSPCs in numbers sufficient for in vivo xenograft studies of long-term repopulating HSPCs. Therefore, we utilized HD CD34+ HSPCs transduced with a lentiviral vector carrying the CD3D mutation target and then base edited the cells for transplantation into immunodeficient mice as a surrogate model to test engraftment potential of edited repopulating HSCs. Gene correction in long-term HSCs able to repopulate the hematopoietic system is essential to generate a clinical benefit from autologous HSCT. Encouragingly, we did not observe changes in engraftment, multipotency or corrective base editing of human cells treated with ABEmax-NRTH compared to LV-treated controls after 16 weeks in mice. We posit that the modest, yet measurable, drop in editing efficiency seen when utilizing ABE8e-NG editor and adenine A0-targeting sgRNA (Fig. S3) reflects the higher editing efficiency of the ABEmax-NRTH/sgRNA combination compared to the ABE8e-NG/sgRNA combination at their respective target sites. Differences in editing efficiency of long-term HSCs could be an effect mediated by the Cas9n variant sgRNA affinity for the target site, or the relative stability or expression of the editor variant. Because long-term HSCs cycle slowly and restrict protein synthesis to reduce the biogenesis of defective translational products,50 uptake and translation of mRNAs in repopulating HSCs is likely less efficient. Therefore, a small difference in observed editing in an HSPC mixture cell product could become a significantly larger difference in repopulating cells after 16 weeks of engraftment.
Although xenografts provide a feasible surrogate assay for long-term HSPC activity, definitive evidence of gene modification in repopulating HSCs can only be determined by longer observations in large animal HSCT models such as canines or nonhuman primates, or in human studies. The precision of base editing does not provide a convenient clonal tag commonly used with randomly integrating LV-based therapies. Nevertheless, the presence of unchanged, high-frequency ABEmax-mediated base editing in unfractionated bone marrow and in four isolated hematopoietic lineages from bone marrow after 16 weeks (CD34+ HSPCs, CD33+ Myeloid, CD19+ B cells, and CD56+ NK cells) suggests engraftment of edited long-term HSCs.
Additionally, the method of using LV transduction of disease target mutations into HD CD34+ HSPCs facilitated proof-of-concept studies for correction of two additional pathogenic CD3D mutations reported to cause CD3δ SCID in Japan and Ecuador.31,51 These surrogate studies in HD HSPCs demonstrate a base editing pipeline capable of treating the most prevalent CD3δ SCID-causing mutations reported to date.
The ATO platform allows rigorous assessment of the effects of base editing on the CD3δ SCID disease phenotype due to its ability to support in vitro development of mature T cells from HSPCs. Edited ATO-derived mature T cells demonstrated rescue of CD3/TCR surface expression and TCR-dependent function: calcium flux, cytokine production, proliferation; and revealed a highly diverse TCR repertoire.
Prior characterization of the block in T cell development in CD3δ SCID was hindered by the extreme rarity of the disease and limited patient samples. A thymic biopsy on a single CD3δ SCID patient reported in 2003 showed reduced CD4 and CD8 protein expression by western blot and absent CD4 and CD8 protein expression by immunohistochemistry.2 These authors therefore posited a block in T cell development at the DN stage.2 Because the ATO system allows for robust in vitro recapitulation of each stage of thymopoiesis, we were able to interrogate this question at distinct stages of development. Our data revealed that unedited CD3δ SCID HSPCs developed past the DN stage to the ISP4 and DP-E stages. While the numbers of DP-E (CD3−TCR−) cells in unedited patient ATOs were lower as compared to edited patient and HD ATOs, a DP-E population is clearly present, in contrast to prior understanding. These data suggest inefficient development of unedited CD3δ SCID HSPCs to the DP-E stage, and a complete inability to proceed to the DP-L stage.
Prior groups have described the disparate role of CD3δ in surface expression of TCRγδ in mice versus humans.52 In mice with mutations in CD3δ, development of TCRαβ+ T cells is blocked, but TCRγδ T cells appear normal.52 Our data support the conclusion that in human T lymphopoiesis, CD3δ is critical for the development of both TCRαβ+ and TCRγδ+ T cells.12
Single-cell analysis of TCR usage in ATO-derived cells revealed that unedited patient T cells demonstrated normal TRB rearrangement (completed by the DN stage) but were defective in TRA rearrangement. We describe for time that lack of CD3D leads to reduced 5’ distal TRAV and 3’ proximal TRAJ usage. This spatiotemporal pattern corresponds to genomic positions that are rearranged later in VDJ rearrangement. RORC deficiency, also an IEI, results in a similar pattern of TRA usage,53 which is believed to result from absent downstream apoptosis regulator BCL2L1, which is highly expressed in DP cells.54,55 In the case of CD3δ SCID, our data from patient ATOs suggest that reduced 5’ TRAV and 3’ TRAJ usage likely results from the requirement for cells to express surface CD3/TCRαβ to survive and proceed through complete positive selection. Our data from edited patient ATOs further supports this hypothesis because base editing of CD3δ SCID HSPCs restored RORC RNA expression in DPs by CITE-seq. The inability for unedited CD3δ SCID HSPCs to efficiently mature past the DP-E stage is likely due to incomplete TRA rearrangement resulting in impaired surface expression of diverse TCRs. As such, base editing of CD3δ SCID HSPCs restores CD3/TCRαβ expression and allows for complete TRA rearrangement at the DP stage, leading to restored TCR diversity and positive selection.
Taken together, we demonstrate that highly efficient base editing to correct the CD3δ SCID mutation enabled robust rescue of T cell development and function. These results demonstrate a potential genome editing approach for autologous HSCT to successfully correct CD3δ SCID. Although this work is limited to a single IEI, translation to the clinic will have significant implications for numerous other rare, monogenic diseases, illuminating a potential translational pathway for the one-time treatment of these disorders.
Limitations of the Study
CD3δ SCID is an extremely rare disease and therefore, due to limited availability of patient HSPCs, xenograft experiments were performed using healthy donor HSPCs obtained from donors undergoing G-CSF mobilization. Restoration of T cell development from base edited patient HSPCs was tested via in vitro T cell differentiation in ATOs. The ATO system is biased towards the development of SP8 versus SP4 T cells due to MHC-I restriction; however, because base editing of CD3δ SCID HSPC restores CD3/TCR expression, we would expect restoration of SP4 T cell production in patients. Further in vivo studies utilizing base-edited CD3δ SCID patient HSPCs are vital to clarify these limitations as gene therapy advances towards the clinic.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to, and will be fulfilled by, the Lead Contact, Donald B. Kohn M.D. (dkohn1@mednet.ucla.edu).
Materials Availability
Plasmids generated in this study have been deposited to Addgene.
Data and Code Availability
Sequencing data have been deposited at NCBI Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-human CD3 | Biolegend | Clone UCHT1 |
| Anti-human CD3 (for in vivo studies) | Biolegend | Clone SK7 |
| Purified NA/LE Mouse Anti-Human CD3 (for calcium flux assay) | BD Biosciences | Clone HIT3a |
| Anti-human CD4 | Biolegend | Clone RPA-T4 |
| Anti-human CD5 | Biolegend | Clone UCHT2 |
| Anti-human CD7 | Biolegend | Clone M-T701 |
| Anti-human CD8a | Biolegend | Clone SK1 |
| Anti-human CD8b | Miltenyi Biotec | Clone REA-715 |
| Anti-human CD14 | Biolegend | Clone M5E2 |
| Anti-human CD19 | Biolegend | Clone HIB19 |
| Anti-human CD25 | Biolegend | Clone BC96 |
| Anti-human CD27 | Biolegend | Clone O323 |
| Anti-human CD28 | Biolegend | Clone CD28.2 |
| Purified NA/LE Mouse Anti-Human CD28 (for calcium flux assay) | BD Biosciences | Clone CD28.2 |
| Anti-human CD33 | Biolegend | Clone WM53 |
| Anti-human CD34 | Biolegend | Clone 561 |
| Anti-human CD45 | Biolegend | Clone HI30 |
| Anti-mouse CD45 | Biolegend | Clone 30-F11 |
| Anti-human CD45RA | Biolegend | Clone HI100 |
| Anti-human CD45RO | Biolegend | Clone UCHL1 |
| Anti-human CD56 | Biolegend | Clone HCD56 |
| Anti-human CD56 (for in vivo studies) | BD Biosciences | Clone B159 |
| Anti-human CD62L | Biolegend | Clone DREG-56 |
| Anti-human CCR7 | Biolegend | Clone G043H7 |
| Anti-human CTLA-4 | Biolegend | Clone BNI3 |
| Anti-human IFNγ | Biolegend | Clone 4S.B3 |
| Anti-human IL-2 | Biolegend | Clone MQ1-17H12 |
| Anti-human LAG-3 | Biolegend | Clone 11-C3C65 |
| Anti-human TIM-3 | Biolegend | Clone F38-2E2 |
| Anti-human TCRαβ | Biolegend | Clone IP26 |
| Anti-human TCRγδ | Biolegend | Clone B1 |
| Anti-human TNFα | Biolegend | Clone Mab11 |
| Anti-human CD137 (41-BB) | Biolegend | Clone 4B4-1) |
| TotalSeq-C Human Universal Cocktail, V1.0 | Biolegend | Cat. 399905 |
| Bacterial and virus strains | ||
| NEB Stable Competent E. coli (High Efficiency) | New England Biolabs | Cat. C3040I |
| LV pCCL-c-MNDU3-CD3D WT cDNA | This paper | Addgene |
| LV pCCL-c-MNDU3-CD3D A0 cDNA | This paper | Addgene |
| LV pCCL-c-MNDU3-CD3D c.202C>T-cDNA | This paper | Addgene |
| LV pCCL-c-MNDU3-CD3D c.274+5G>A | This paper | Addgene |
| LV pCCL-c-MNDU3-CD3Dc.275-2A>G | This paper | Addgene |
| Biological samples | ||
| Human CD34+ HSPCs | Charles River | Cat. BM34C |
| Human CD34+ HSPCs from CD3δ SCID Patient | Alberta Children’s Hospital | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| rhIL-2 | Miltenyi Biotec | Cat. 130-097-748 |
| rhFLT3L | Peprotech | Cat. 300-19 |
| rhIL-7 | Peprotech | Cat. 200-07 W |
| rhTPO | Peprotech | Cat. 300-18 |
| rhSCF | Peprotech | Cat. 300-07 |
| B27 supplement | Gibco | Cat. 17504-044 |
| TruStain FcX | Biolegend | Cat. 422302 |
| Purified Rat Anti-Mouse CD16/CD32 (Mouse BD Fc Block) | BD Biosciences | Cat. 553142 |
| DAPI | Life technologies | Cat. D1306 |
| AB serum | Gemini Bio-Products | Cat. 50-753-3011 |
| Ghost Dye Violet 510 | TONBO Biosciences | Cat. 13-0870-T100 |
| Indo-1 Dye, AM, cell permeant | ThermoFisher Scientific | Cat. I1203 |
| X-vivo-15 | Lonza | Cat. 04-744Q |
| RPMI | Lonza | Cat. 12-115Q |
| StemSpan SFEM | Stem Cell Technologies | Cat. 09650 |
| MethoCult H4435 Enriched | Stem Cell Technologies | Cat. 04445 |
| Q5 Hot Start High-Fidelity DNA Polymerase | New England Biolabs | Cat. M0493L |
| CleanCap Reagent AG | Trilink | Cat. N-7113 |
| N1-Methylpseudouridine-5’-Triphosphate | Trilink | Cat. N-1081 |
| LiCl Precipitation Solution | ThermoFisher Scientific | Cat. AM9480 |
| Poloxamer F108 (Kolliphor P 338) | BASF | Cat. 9003-11-6 |
| Recombinant Cas9-NRTH protein | QB3 Macrolab: UC Berkeley | N/A |
| Recombinant Hifi Cas9 protein | QB3 Macrolab: UC Berkeley | N/A |
| Ionomycin, Calcium Salt | ThermoFisher Scientific | Cat. I24222 |
| Critical commercial assays | ||
| CFSE proliferation assay | Biolegend | Cat. 423801 |
| antiCD3/CD28 beads | ThermoFisher Scientific | Cat. 11161D |
| Zombie UV™ Fixable Viability Kit | Biolegend | Cat. 423107 |
| Dead cell removal kit | Miltenyi Biotec | Cat. 130-090-101 |
| CD8+ T cell Isolation Kit | Miltenyi Biotec | Cat. 130-09-495 |
| Intracellular Fixation & Permeabilization Buffer Set | eBiosciences | Cat. 88-8824-00 |
| CD34 Microbead Kit, human | Miltenyi Biotec | Cat. 130-046-702 |
| ddPCR Supermix for Probes | Biorad | Cat. 186-3026 |
| NEBuilder HiFi DNA Assembly Cloning Kit | New England Biolabs | Cat. E5520S |
| Gibson Assembly Cloning Kit | New England Biolabs | Cat. E5510S |
| Purelink Genomic DNA Mini Kit | ThermoFisher Scientific | Cat. K182002 |
| HiScribe® T7 High Yield RNA Synthesis Kit | New England Biolabs | Cat. E2040S |
| QIAquick PCR Purification Kit | Qiagen | Cat. 28104 |
| rhAmpSeq CRISPR Library Kit | IDT | Cat.10007317 |
| Ficoll Pacque Plus | GE Healthcare | Cat. 17144003 |
| Q5 Site-Directed Mutagenesis Kit | New England Biolabs | Cat. E0554S |
| P3 Primary Cell 4D-Nucelofector X Kit S | Lonza | Cat. V4XP-3032 |
| SE Cell Line 4D-Nucleofector X Kit | Lonza | Cat. V4XC-1024 |
| SF Cell Line 4D-Nucleofector X Kit | Lonza | Cat. V4XC-2012 |
| AMPure XP Reagent | Beckman Coulter | Cat. A63881 |
| NucleoSpin Tissue XS, Micro kit for DNA from cells and tissue | Macherey Nagel | Cat. 740901.50 |
| Gentra Puregene Buccal Cell Kit | Qiagen | Cat. 158845 |
| KAPA HTP Library Preparation Kit | KAPA Biosystems | Cat. KR0426 |
| Deposited data | ||
| CITE-seq data | This paper | GSE220611 |
| rhAmpSeq | This paper | NCBI SRA Accession Number: PRJNA914907 |
| HTS Editing data | This paper | NCBI SRA Accession Number: PRJNA914907 |
| Experimental models: Cell lines | ||
| MS5-hDLL4 | Crooks Lab | N/A |
| Jurkat | ATCC | TIB-152 |
| K562 | ATCC | CCL-243 |
| HEK293T-CHEDAR | Kohn Lab | N/A |
| HT-29 | ATCC | HTB-38 |
| Experimental models: Organisms/strains | ||
| Mouse: NOD.Cg-KitW-41J Tyr + Prkdcscid Il2rgtm1Wjl/ThomJ | Jackson Laboratories | RRID:IMSR_JAX:026622 |
| Oligonucleotides | ||
| Gen_CD3D(C202T)_sgRNA: 5’-CGAGGAATATATAGGTGTAA-3’ | SYNTHEGO | N/A |
| Gen_CD3D(C202T)_ssODN: 5’-ACCCAAAGGGTTCAGGAAGCACGTACTTCGATAATGAACTTGCACGGTAGATTCTTTGTCCTTGTATATATCTGTCCCATTACATCTATATATTCCTCATGGGTCCAGGATGCGTTTTCCCAGGTC-3’ | IDT | N/A |
| CD3DF Forward Primer: 5’-CTTGGTGCAGATCAAAGAGC - 3’ | IDT | |
| CD3DR Reverse Primer: 5’-CTGGTGATGGGCTTGCCAC -3’ | IDT | N/A |
| Therapeutic_sgRNA1: 5’-TTCCTCGTGGGTCCAGGATG-3’ | SYNTHEGO | N/A |
| Therapeutic_sgRNA2: 5’-TATTCCTCGTGGGTCCAGGA-3’ | SYNTHEGO | N/A |
| Therapeutic HDR sgRNA: 5’-TTACACCTATATATTCCTCG-3’ | SYNTHEGO | N/A |
| Therapeutic_HDR_ssODN: 5’-TGCAATACCAGCATCACATGGGTAGAGGGAACGGTGGGAACACTGCTCTCAGACATTACAAGACTGGACCTGGGAAAACGCATCCTGGATCCACGAGGAATATATAGATGTAATGGGACAGATATA-3’ | IDT | N/A |
| CD3D_LibF_Forward Primer: 5’-ACACGACGCTCTTCCGATCTNNNNGAGGACAGAGTGTTTGTGAA -3’ | IDT | N/A |
| CD3D_LibR_Reverse Primer: 5’-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTCTCTAGCCAGAAAGTTCTCAC -3’ | IDT | N/A |
| ddPCR P5 Primer: 5’-AATGATACGGCGACCACCGA -3’ | IDT | N/A |
| ddPCR P7 Primer: 5’-CAAGCAGAAGACGGCATACG -3’ | IDT | N/A |
| Transcription Forward Primer: 5’-TCGAGCTCGGTACCTAATACGACTCACTATAAGG -3’ | IDT | N/A |
| Transcription Reverse Primer: 5’-TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTCTTCCTACTCAGGCTTTATTCAAAGACCA -3’ | IDT | N/A |
| GUIDE-seq_dsODN_Forward: 5’/Phos/GTTTAATTGAGTTGTCATATGTTAATAACGGT*A*T -3’ | IDT | N/A |
| GUIDE-seq_dsODN_Reverse: 5’/Phos/ATACCGTTATTAACATATGACAACTCAATTAA*A*C -3’ | IDT | N/A |
| Psi U5 Forward Primer: 5’ AAG TAG TGT GTG CCC GTC TG 3’ | IDT | N/A |
| Psi U5 Reverse Primer: 5’ CCT CTG GTT TCC CTT TCG CT 3’ | IDT | N/A |
| Psi Probe: 5′ FAM-CCC TCA GAC-ZEN*-CCT TTT AGT CAG TGT GGA AAA TCT CTA G-IBFQ** 3′; *ZEN = internal modification from IDT-Integrated DNA Technologies, **IBFQ = Iowa Black FQ | IDT | N/A |
| SDC4 Forward Primer: 5’ CAG GGT CTG GGA GCC AAG T 3’ | IDT | N/A |
| SDC4 Reverse Primer: 5’ GCA CAG TGC TGG ACA TTG ACA 3’ | IDT | N/A |
| SDC4 Probe: 5′ HEX-CCC ACC GAA-ZEN*-CCC AAG AAA CTA GAG GAG AAT-IBFQ** 3′; *ZEN = internal modification from IDT-Integrated DNA Technologies, **IBFQ = Iowa Black FQ | IDT | N/A |
| CD3D_LV_Forward Primer: 5’-GGAGCACAGCACCTTCCTG -3’ | IDT | N/A |
| CD3D_LV_Reverse Primer: 5’-CCCAGTTGCCGCCCAGG -3’ | IDT | N/A |
| Recombinant DNA | ||
| pCMV-ABE8e-NRTH | This paper | N/A |
| pCMV-ABE8e-VRER | This paper | N/A |
| pCMV-ABE8e-xCas9(3.7) | This paper | N/A |
| pCMV-ABE8e-NG | Addgene | Plasmid 138491 |
| pCMV-ABEmax-NRTH | Addgene | Plasmid 136922 |
| CCL vector backbone | Addgene | Plasmid 12251 |
| Software and algorithms | ||
| FlowJo | Tree Star Inc. | https://www.flowjo.com/solutions/flowjo |
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Cell Ranger v7.0.0 | 10X genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| Seurat 4.2.0 | Satija Lab | https://satijalab.org/seurat/ |
| cb_sniffer | sridnona | https://github.com/sridnona/cb_sniffer |
| MAST algorithm | RGLab | https://github.com/RGLab/MAST |
| fgsea v1.22.0 (GSEA) | Alexey Sergushichev | https://github.com/ctlab/fgsea/ |
| msigdbr v7.15.1 (MSigDB) | Igor Dolgalev | https://CRAN.R-project.org/package=msigdbr |
| enrichplot v1.16.2 (GSEA visualization) | Yu Lab | https://yulab-smu.top/biomedical-knowledge-mining-book/ |
| CasOFFinder.23 | BAE Lab | http://www.rgenome.net/cas-offinder/ |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
Jurkat (male) and K562 (female) cell lines were obtained from ATCC (Manassas, VA). Cells were maintained in R10 (RPMI [GIBCO]/10% FBS [GIBCO]/1x Penicillin/Streptomycin/Glutamine [PSG, Gemini Bio Products; Sacramento, CA]) at 37°C with 5% CO2 and were confirmed to be negative for mycoplasma by testing with MycoAlert (Lonza Biologics).
Animals
The NOD,B6.SCID IL2rg−/−KitW41/W41 (NBSGW) murine xenografts were purchased from Jackson Labs (Jackson Laboratory; Bar Harbor, ME) and colonies were maintained at UCLA under an approved protocol (2008–167) by the UCLA Animal Research Committee under the Division of Laboratory Medicine. NBSGW mice were housed in a specialized barrier facility designed for immunocompromised mice as previously described.56 Animals were handled in laminar flow hoods and housed in a pathogen-free colony in a biocontainment vivarium. At 5–7 weeks of age, female mice were randomly assigned to experimental groups and distributed evenly among cages.
Healthy Human Donors
Leukopaks from healthy donors were purchased from HemaCare (HemaCare BioResearch Products; Van Nuys, CA). Mobilized peripheral blood (mPB) was collected from normal, healthy donors on days 5 and 6 after 5 days of stimulation with granulocyte-colony stimulating factor (G-CSF) as described.57 Platelet depletion was performed from the centrifuged bags at each wash step using a plasma expressor extractor (Fenwal). CD34+ cell enrichment was performed using the CliniMACS Plus (Miltenyi; Bergish Gladbach, Germany). CD34+ cells were cryopreserved in CryoStor CS5 (StemCell Technologies; Vancouver, Canada) using a CryoMed controlled-rate freezer (Thermo Fisher Scientific). CD34+ HSPCs from healthy donors were cultured in X-VIVO 15 medium (Lonza; Basel, Switzerland) (50 ng/mL each of hSCF, hFLT3-L, and hTPO) 2 days prior to electroporation of BE and sgRNA at 37°C with 5% CO2.. Healthy donor samples were obtained from male and female donors 25–45 years of age.
CD3δ SCID Patient
Bone marrow cells were collected from a biologically male, 16-months old infant diagnosed with CD3δ SCID following local Research Ethics Board (REB) approval and informed parental consent (study ID# REB21-0375). Procedure was performed under general anesthetic at the same time as central line placement. Using sterile technique, 10 mL of bone marrow was aspirated from the right posterior superior iliac spine with a 16-gauge x 2.688 inch bone marrow aspirate needle (Argon medical Devices, Inc). Specimen was anticoagulated with preservative free heparin (100 units/mL). The use of bone marrow samples from CD3δ SCID patients was approved under UCLA IRB# 2010-001399.
METHOD DETAILS
Generating CD3D(C202T) Jurkat T Cell Line
Jurkat T cells were modified to contain the pathogenic CD3δ SCID allele by electroporation of SpCas9 recombinant protein (QB3 Macrolab, UC Berkeley; Berkeley, CA) complexed to sgRNA (5’-CGAGGAATATATAGGTGTAA-3’) (Synthego; Redwood City, CA) and ssODN homologous donor (5’- ACCCAAAGGGTTCAGGAAGCACGTACTTCGATAATGAACTTGCACGGTAGATTCTTTGTCCTTGTATATATCTGTCCCATTACATCTATATATTCCTCATGGGTCCAGGATGCGTTTTCCCAGGTC-3’) (Integrated DNA Technologies {IDT}; Coraville, IA) carrying the pathogenic mutation and FACS single-cell sorted and cultured in R20 (RPMI [GIBCO]/20% FBS [GIBCO]/1x Penicillin/Streptomycin/Glutamine [PSG, Gemini Bio Products]). Primers for amplification of the CD3D locus to confirm knock-in of the pathogenic mutation were CD3DF: 5’- CTTGGTGCAGATCAAAGAGC - 3’; CD3DR: 5’- CTGGTGATGGGCTTGCCAC −3’. A pseudo-tetraploid clonal cell line containing the CD3δ SCID mutation in 1/4 CD3D alleles and deleterious indels in 3/4 CD3D alleles (measured by HTS) was established (‘CD3D(C202T) Jurkat T cells’). Absence of CD3 surface expression was confirmed by flow cytometry (CD3-APC-Cy7, SK1, BioLegend; San Diego, CA ). Cells were cultured in R10 at 37°C with 5% CO2.
Generating CD3D(C202T) K562 Cell Line
K562 cells were modified to contain the pathogenic CD3δ SCID allele by electroporation of RNP and ssODN homologous donor (5’- ACCCAAAGGGTTCAGGAAGCACGTACTTCGATAATGAACTTGCACGGTAGATTCTTTGTCCTTGTATATATCTGTCCCATTACATCTATATATTCCTCATGGGTCCAGGATGCGTTTTCCCAGGTC −3’) carrying the pathogenic mutation were FACS single-cell sorted and cultured in R10. Primers for amplification of the CD3D locus to confirm knockin of the pathogenic mutation were CD3DF: 5’- CTTGGTGCAGATCAAAGAGC - 3’; CD3DR: 5’- CTGGTGATGGGCTTGCCAC −3’. A clonal cell line containing the CD3δ SCID mutation in all CD3D alleles (measured by HTS) was established (‘CD3D(C202T) K562 cells). Cells were cultured in R10 at 37C with 5% O2.
CD34+ HSPC Isolation from Patient Bone Marrow
CD34+ cells were isolated using microbeads conjugated to monoclonal mouse anti-human CD34 antibodies (Milteny Biotech CD34 MicroBead Kit. Cat# 130-046-702) according to manufacturer’s instructions. Briefly, mononuclear cells (MNC) obtained from patient bone marrow were isolated using Ficoll-Paque (Sigma) gradient centrifugation according to established methods. A total of 108 cells were collected, washed with sterile phosphate-buffered saline (PBS) to remove platelets and re-suspended in MACS buffer (PBS, pH 7.2, 0.5% bovine serum albumin [BSA], and 2 mM EDTA). To the cell pellet (108 cells), 100 ul of FcR blocking reagent and 100 ul of CD34 microbeads were added to the cell pellet, mixed well and incubated at 40°C for 30 minutes. Cells were then washed with 10 ml of MACS buffer by centrifugation at 300g for 10 minutes and re-suspended in 500 ul of the same buffer and loaded onto a prepared MACS column placed in a magnetic field. Flow through cell fraction (CD34 negative population) was collected. The column was then washed and removed from the magnet, placed on a collection tube and the bound cells were eluted using a plunger. The collected CD34+ cell fraction was then washed, viability checked and re-suspended in 1 ml of MACS buffer containing 10% DMSO and stored frozen in liquid nitrogen until processing. For transportation, cells in freezer vials were shipped by overnight courier in containers with excess dry ice. CD3δ SCID patient CD34+ HSPCs were cultured and electroporated with BE and sgRNA under the same conditions as CD34+ HSPCs isolated from healthy donors.
Cloning of Adenine Base Editor Variant Plasmids
pCMV-ABE8e-NG (Plasmid #138491) and pCMV-ABEmax-NRTH (Plasmid #136922) plasmids were obtained from AddGene (Watertown, MA). We generated all base editor variants derived from the same parental pCMV-ABE8e-NG backbone. Key substitutions were introduced to Cas9n genes to allow for alternative PAM recognition (other than canonical NGG). Substitutions were introduced by Q5 site-directed mutagenesis (New England Biolabs {NEB}, Ipswich, MA) and were as follows (relative to NGG-recognizing Cas9n): 1) ABE8e-VRER: D1135V, G1218R, R1335E, and T1337R, 2) ABE8e-xCas9(3.7) A262T, R324L, S409I, E480K, E543D, M694I, and E1219V. To generate plasmid encoding ABE8e-NRTH, we utilized Gibson Assembly (NEB) cloning to amplify and ligate the ABE8e deaminase gene and Cas9n-NRTH gene.
CD3D(C202T) Jurkat T cell Electroporation
CD3D(C202T) Jurkat T cells were electroporated at −85% confluency. Cells were counted on ViCell (Beckman Coulter; Brea, CA) and 5 × 105 cells per condition were centrifuged at 90 xg for 15 min at RT, resuspended in 20 uL of SE electroporation buffer (Lonza; Basel, Switzerland), and combined with 1 ug sgRNA (sgRNA1: 5’- TTCCTCGTGGGTCCAGGATG-3’; sgRNA2: 5’-TATTCCTCGTGGGTCCAGGA-3’) and 3 ug of BE expression plasmids. In the case of CRISPR/Cas9-HDR, 200 pmol of sgRNA (5’-TTACACCTATATATTCCTCG-3’) were combined with 100 pmol of rCas9 nuclease protein for 15 minutes at RT for RNP complex formation. Cells were resuspended in 20 uL of SE electroporation buffer and combined with RNP and 250 pmol of ssODN ultramer donor (5’-TGCAATACCAGCATCACATGGGTAGAGGGAACGGTGGGAACACTGCTCTCAGACATTACAAGACTGGACCTGGGAAAACGCATCCTGGATCCACGAGGAATATATAGATGTAATGGGACAGATATA-3’). The underlined base represents the target site. Cells were electroporated using the CL-120 setting on the Amaxa 4D Nucleofector X Unit (Lonza). As previously described,58 immediately after electroporation, cells were rested in 16-well electroporation strips (Lonza) for 10 min at RT and then recovered with 480 uL of R20 medium. In the case of CRISPR/Cas9-HDR, cells were recovered in 480 uL of R20 medium supplemented with 1.2 pmol of Alt-R HDR Enhancer and washed with phosphate-buffered saline (PBS) 24 hours later according to the manufacturer’s instructions (Integrated DNA Technologies {IDT}; Coraville, IA). Editing outcomes were measured by HTS, 5 days after electroporation from gDNA extracted using PureLink Genomic DNA Mini Kit (Invitrogen/ThermoFisher Scientific; Waltham MA).
Karyotype
24 hours post-electroporation, CD3D(C202T) Jurkat T cells treated with RNP + ssODN (CRISPR/Cas9-edited) or plasmids encoding ABEmax-NRTH and CD3D-localizing sgRNA were exposed to mitotic arresting agents to collect metaphases, and harvested for G-banded karyotype analysis adhering to standard cytogenetics procedures (UCLA Cytogenetics Laboratory, Los Angeles CA). Twenty cells were analyzed per experimental condition. Composite karyotype nomenclature (not all indicated abnormalities were identified in all abnormal cells analyzed) was used to describe the abnormal clones according to the International System for Human Cytogenomic Nomenclature (ISCN).
Illumina MiSeq Library Preparation for the CD3D locus in CD3D(C202T) Jurkat T cells and CD34+ CD3δ SCID HSPCs
DNA libraries for HTS were prepared as previously described.57,59 Five days after editing, an outer PCR was performed on genomic DNA to amplify 608 bp of the CD3D locus using CD3DF: 5’- CTTGGTGCAGATCAAAGAGC - 3’; CD3DR: 5’- CTGGTGATGGGCTTGCCAC −3’. A second PCR was performed to add a unique index to the PCR product of each sample; CD3D_LibF: 5’- ACACGACGCTCTTCCGATCTNNNNGAGGACAGAGTGTTTGTGAA −3’; CD3D_LibR 5’-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTCTCTAGCCAGAAAGTTCTCAC −3’. Underlined sequences represent Illumina adapter sequences. Following Illumina barcoding, PCR products were pooled at equal concentrations. The pooled library was purified twice using AMPure XP beads (Beckman Coulter; Brea, CA) and then quantified using ddPCR (QX 200; Bio-Rad Laboratories Inc.; Hercules, CA). HTS was performed at the UCLA Technology Center for Genomics & Bioinformatics (TCGB) using an Illumina MiSeq instrument 2 × 150 paired-end reads (Illumina; San Diego, CA). The sequences for all HSPC editing experiments were deposited to NCBI Sequence Read Archive.
Calcium Flux Assay
As previously described,60 cells were suspended at 106/mL in cell loading medium (CLM; RPMI, 2% BSA, 25mM HEPES (pH 7.4)). Cells were stained at a 1.5–5uM concentration with cell permeable Indo-1 acetoxymethyl ester (AM) (ThermoFisher Scientific; Waltham, MA). Cells were incubated for 50 min at 37°C in the dark and then washed 2x with CLM. Cells were gently resuspended by pipetting in CLM at 1 × 106/ mL and samples were protected from light until flow cytometric analysis. Individual samples were warmed at 37°C in the dark for 10 min prior to analysis. A baseline Ca2+ ratio was recorded for 60 seconds after which purified NA/LE mouse anti-human CD3 (HIT3a) and purified NA/LE mouse anti-human CD28 (CD28.2) antibodies were added to stimulate cells (10 μg and 30 μg of each antibody for stimulating Jurkat T cells and ATO-derived thymocytes, respectively) (BD Biosciences; Franklin Lakes NJ). Intracellular esterases cleave Indo-1 AM, producing non-cell permeable Indo-1, a high affinity calcium indicator. Once excited by UV light, the emission spectrum of Indo-1 changes from blue (510 nm) to violet (420 nm) when bound to calcium, allowing for ratiometric measurements of calcium flux.61 The stimulus was added 60 seconds after a baseline ratio was recorded.62 Ionomycin (Imy), a calcium ionophore which rapidly increases intracellular calcium concentration by releasing calcium from its intracellular stores and facilitating transport of calcium across the plasma membrane, was used as a positive control.63
ABE mRNA
ABE8e-NG and ABEmax-NRTH template plasmids were cloned via USER cloning to encode a dT7 promoter17 followed by a 5’ UTR, Kozak sequence, ORF, and 3’UTR. BE portions of the template plasmids were PCR amplified using Q5 Hot Start Mastermix (NEB) and PCR products were purified using QiaQuick PCR Purification Kit (Qiagen Inc., Valencia CA ). ABE8e-NG and ABEmax-NRTH mRNA were in vitro transcribed according to manufacturer’s guidelines from the purified PCR product using T7 HiScribe Kit (NEB) with full substitution of N1-methylpseudouridine for uridine and co-transcriptional 5’ capping using CleanCap AG analogue (TriLink Biotechnologies; San Diego, CA). Lastly, mRNA was purified according to manufacturer’s instructions using LiCl Precipitation Solution (Thermo Fisher). Resulting mRNA was run on the Agilent Bioanalyzer to confirm mRNA integrity and identity.
Colony-Forming Unit Assay
CFU assays were performed as previously described64 using Methocult H4435 Enriched Methylcellulose (StemCell Technologies; Vancouver, Canada. Cat. # 04445) according to the manufacturer’s instructions with minor modifications. Briefly, 100, 300, and 900 CD34+ PBSCs were plated in duplicates into 35 mm gridded cell culture dishes. After 14 days of culture at 5% CO2, 37°C and humidified atmosphere, mature colonies were counted and identified based on their specific morphology. CFUs were then plucked for genomic DNA isolation (NucleoSpin Tissue XS, Clontech Laboratories Inc.; Mountain View, CA).
CIRCLE-Seq Off-Target Editing Analysis
CIRCLE-Seq off-target editing analysis was performed as previously described.10 Genomic DNA from HEK293T cells was isolated using Gentra Puregene Kit (Qiagen; Hilden, Germany) according to the manufacturer’s instructions. Purified genomic DNA was sheared with a Covaris S2 instrument to an average length of 300 bp. The fragmented DNA was end repaired, A-tailed, and ligated to a uracil-containing stem-loop adaptor, using the KAPA HTP Library Preparation Kit, PCR Free (KAPA Biosystems; Wilmington MA). Adaptor-ligated DNA was treated with Lambda Exonuclease (NEB) and Escherichia coli Exonuclease I (NEB) and then with USER enzyme (NEB) and T4 poly- nucleotide kinase (NEB). Intramolecular circularization of the DNA was performed with T4 DNA ligase (NEB) and residual linear DNA was degraded by Plasmid-Safe ATP-dependent DNase (Lucigen; Middleton WI). In vitro cleavage reactions were performed with 250 ng Plasmid-Safe-treated circularized DNA, 90 nM Cas9-NRTH protein, Cas9 nuclease buffer (NEB) and 90 nM synthetic chemically modified sgRNA (Synthego; Redwood City, CA), in a 100-μl volume. Cleaved products were A-tailed, ligated with a hairpin adaptor (NEB), treated with USER enzyme (NEB) and amplified by PCR with barcoded universal primers (NEBNext Multiplex Oligos for Illumina (NEB), using Kapa HiFi Polymerase (KAPA Biosystems). Libraries were sequenced with 150-bp paired-end reads with an Illumina MiSeq instrument. CIRCLE-seq data analyses were performed using open-source CIRCLE-seq analysis software and default recommended parameters (https://github.com/tsailabSJ/circleseq).
GUIDE-Seq Off-Target Editing Analysis
CD3D(C202T) K562 cells were electroporated with plasmids encoding CD3D-targeting sgRNA and ABEmax-NRTH and a DS oligo for capture at DSBs. Two weeks after electroporation, cells were harvested and genomic DNA was extracted to prepare a library for Illumina HTS as previously described.65 In summary, genomic DNA was sonicated to an average size of 500 bp using a Bioruptor Pico Sonication Device (Diagenode; Liège, Belgium) and was 1x purified using AMPure XP beads (Beckman Coulter, Brea, CA). Purified product was then end-repaired and A-tailed (Fisher Scientific, Carlsbad, CA). Y-adapters were ligated using T4 DNA ligase (Fisher Scientific) according to manufacturer’s instructions. The ligated product was purified using 0.9x volumes of AMPure XP beads and the adapter ligated product was split into two PCR reactions for sense and antisense reactions. Site specific PCR1 was performed using Platinum Taq polymerase (Fisher Scientific,) and the product was purified using 1.2x volumes of AMPure XP beads. The purified product was utilized as a template for a second PCR (PCR2) to add P7 Illumina indexes for sequencing. PCR2 product was quantified by densitometry and pooled at equal concentrations. The pooled library was purified using 0.7x volumes of AMPure XP beads and then quantified using ddPCR (QX 200). HTS was performed at UCLA Technology Center for Genomics & Bioinformatics (TCGB) using an Illumina MiSeq instrument 2 × 150 paired-end reads. The sequences for all HSPC editing experiments were deposited to NCBI Sequence Read Archive.
CasOFFinder Off-Target Editing Analysis
Computational prediction of potential off-target sites with minimal mismatches relative to the intended target site (three or fewer mismatches overall, or two or fewer mismatches allowing G:U wobble base pairings with the guide RNA) was performed using CasOFFinder.29
Multiplex-Targeted Sequencing by rhAMPseq
On- and off-target sites identified by CIRCLE-seq, GUIDE-seq, and CasOFFinder were amplified from genomic DNA from ABEmax-NRTH edited CD34+ CD3δ SCID cells or unedited control CD3δ SCID cells using rhAMPSeq multiplexed library preparation (IDT), with amplification coordinates listed in Supplemental Table S2. Sequencing libraries were generated according to the manufacturer’s instructions and sequenced with 150-bp paired-end reads using an Illumina NextSeq instrument.
Quantification of Base Editing Efficiency at Off-Target Sites
The A•T-to-G•C editing frequency for each position in the protospacer was quantified as previously described10 using CRISPResso Pooled (v2.0.41) (https://github.com/pinellolab/CRISPResso2) with quantification_ window_size10, quantification_window_centre-10, base_editor_output, conversion_nuc_from A, conversion_nuc_to G. The genomic features of off-target sites were initially annotated using HOMER (v4.10) (http://homer.ucsd.edu/homer/). Confirmed off-target sites were inspected manually and annotated using the NCBI Genome Data Browser. The editing frequency for each site was calculated as the ratio between the number of reads containing the edited base in a window from position 4 to 10 of each protospacer and the total number of reads. To calculate the statistical significance of off-target editing for the ABEmax-NRTH mRNA treatment compared to control samples, we applied a χ2 test for each of three samples (one donor, with three replicates). The 2 × 2 contingency table was constructed using the number of edited reads and the number of unedited reads in treated and untreated groups and the false discovery rate (FDR) was calculated using the Benjamini–Hochberg method as previously described.10 The code used to conduct off-target quantification and statistical analysis was customized from Newby et al. 2020 (https://github.com/tsailabSJ/MKSR_off_targets), and will be available in our repository at date of publication.
Lentiviral Vector Packaging, Titering, and Transduction
LVs are pCCL HIV-derived LVs of self-inactivating (SIN) LTR configurations. Construction of pCCL-MND-GFP has been described66 and wild-type CD3D cDNA, CD3D cDNA containing the A0 bystander edit, and CD3D cDNA containing the c.202C>T mutation were cloned into the multi-cloning site of the vector. The CCL-MND-CD3D LV was packaged in a VSV-G pseudotype using a monoclonal HEK293T cell line, CRISPRed HEK293T to Disrupt Antiviral Response (CHEDAR)67, and titered as previously described.68
Determination of Vector Copy Number (VCN) per Cell
Genomic DNA was extracted using PureLink Genomic DNA Mini Kit (Invitrogen/ThermoFisher Scientific). Average VCN was measured using ddPCR with primers and probes specific to the HIV-1 Psi region and normalized using primers to the autosomal human gene SDC4 ddPCR as previously described.56
ABEmax-NRTH mRNA Electroporation in Human HSPCs
Cells were pre-stimulated for two days in X-VIVO 15 medium (50 ng/mL each of hSCF, hFLT3-L, and hTPO) with 2 × 105 cells per condition that were washed 2x and pelleted at 300 × g for 8 min at RT. Cells were resuspended in electroporation buffer (P3 buffer) (Lonza) (CD3δ SCID cells) or, in the case of HD HSPCs for in vivo studies, EP Buffer (Maxcyte, Gaithersburg, MD), and combined with 1 μg of sgRNA and 4.5 μg of BE mRNA. Cells were electroporated using programs DS-130 (Lonza) or HSC-3 (ATX MaxCyte). Electroporated cells were recovered in the same medium at 37°C, 5% CO2. 24 hours post-electroporation, samples of the cells were diluted 1:2 with trypan blue and counted manually using a hemocytometer to determine viability (number of live cells/number of total cells × 100). Cells were re-plated into 1 mL (or 5 mL, for 1 × 106 cells) of myeloid expansion medium (Iscove’s Modified Dulbecco’s Medium (IMDM, Thermo Fisher Scientific) + 20% FBS [HI FBS, Gibco/ThermoFisher) + 5 ng/mL Interleukin 3 (IL3), 10 ng/mL Interleukin 6 (IL6), 25 ng/mL SCF (Peprotech; Rocky Hill, NJ)], and cultured for 5 days prior to harvesting for genomic DNA (gDNA). gDNA was extracted using PureLink Genomic DNA Mini Kit (Invitrogen/ThermoFisher Scientific).
In Vivo Studies
Animals were handled in laminar flow hoods and housed in a pathogen-free colony in a biocontainment vivarium. Adult females (5–7 weeks old) were injected with 5 × 105 - 1 × 106 cells/mouse via retro-orbital injection of untreated, LV-treated, or LV and BE human CD34+ cells, and allowed to engraft over 12–16 weeks. After 12–16 weeks, mice were sacrificed by CO2 inhalation followed by cervical dislocation. Bone marrow, thymus, and spleen were harvested for subsequent analysis of chimerism and cell lineage composition. Lineage distribution was measured using cell-type specific antibodies on the Fortessa flow cytometer (BD Biosciences) and sorted using an Aria H cell sorter (BD Biosciences). The antibodies used were: anti-human CD45 (BD Biosciences, Cat. No. 560367), anti-mouse CD45 (Biolegend, Cat. No. 103107), anti-human CD34 (Biolegend, Cat. No. 343607), anti-human CD19 (Biolegend, Cat. No. 302215), anti-human CD56 (BD Biosciences, Cat. No. 555516), anti-human CD3 (Biolegend, Cat. No. 344817), anti-human CD33 (Biolegend, Cat. No. 303423), anti-human CD4 (Biolegend, Cat. No. 300501), and anti-human CD8 (Biolegend, Cat. No. 980902).
Bone Marrow Artificial Thymic Organoid (ATO) cultures
Bone Marrow ATOs were generated as previously described.36 MS5-hDLL4 cells were harvested by trypsinization and resuspended in serum free ATO culture medium (“RB27”) composed of RPMI 1640 (Corning, Manassas, VA), 4% B27 supplement (ThermoFisher Scientific, Grand Island, NY), 30 μM L-ascorbic acid 2-phosphate sesquimagnesium salt hydrate (Sigma-Aldrich, St. Louis, MO) reconstituted in PBS, 1% penicillin/streptomycin (Gemini Bio-Products, West Sacramento, CA), 2% Glutamax (ThermoFisher Scientific, Grand Island, NY), 5 ng/ml rhFLT3L and 2.5 ng/ml rhIL-7 (Peprotech, Rocky Hill, NJ). RB27 was made fresh weekly. 1.5×105 MS5-hDLL1 cells were combined with 1.5×103 CD34+ cells per ATO in 1.5 ml Eppendorf tubes and centrifuged at 300 g for 5 min at 4°C in a swinging bucket centrifuge. Supernatants were carefully removed, and the cell pellet was resuspended in 5 μl RB27 per ATO and mixed by brief vortexing. ATOs were plated on a 0.4 μm Millicell transwell insert (EMD Millipore, Billerica, MA; Cat. PICM0RG50) placed in a 6-well plate containing 1 ml RB27 per well. Medium was changed completely every 3–4 days by aspiration from around the cell insert followed by replacement with 1 ml with fresh RB27/cytokines. ATO cells were harvested by adding FACS buffer (PBS/0.5% bovine serum album/2mM EDTA) to each well and briefly disaggregating the ATO by pipetting with a 1 ml “P1000” pipet, followed by passage through a 50 μm nylon strainer.
T cell Cytokine Assays
ATOs were harvested at week 12 (as above) and resuspended in 48-well plates in 1 ml AIM V (ThermoFisher Scientific, Grand Island, NY) with 5% human AB serum (Gemini Bio-Products, West Sacramento, CA) at a concentration of 1×10E6 cells/ml. anti-CD3/CD28 beads (ThermoFisher Scientific, Grand Island, NY) in AIM V/5% human AB serum with 100 IU/mg rhIL-2 (Miltenyi), were added to cells for 24 hours. Because anti-CD3/CD28 bead stimulation is known to down-regulate surface CD3 and TCRαβ expression,69 mature SP8s T cells are defined as CD45+CD8+CD4−CD45RA+. Cells were stained for CD3, TCRαβ, CD45, CD4, CD8, CD45RA, and Zombie Aqua fixable viability dye (Biolegend, San Diego, CA) prior to fixation and permeabilization with an intracellular staining buffer kit (eBioscience, San Diego, CA) and intracellular staining with antibodies against IFNγ, TNFα, and IL-2 (Biolegend, San Diego, CA).
T cell Proliferation Assays
For CFSE proliferation assays, at least 100,000 ATO-derived CD8SP T cells were isolated by negative selection MACS using CD8+ T cell Isolation Kit, human (Miltenyi, Cat. 130-09-495) and labeled with 5 μM CFSE (Biolegend, San Diego, CA). Labeled cells were incubated with anti-CD3/CD28 beads (ThermoFisher Scientific, Grand Island, NY) in AIM V/5% human AB serum with 100 IU/mg rhIL-2 (Miltenyi), co-stained for CD25 and 4–1BB (Biolegend, San Diego, CA) and analyzed by flow cytometry on day 5.
Flow Cytometry and antibodies
All flow cytometry stains were performed in PBS/0.5% BSA/2 mM EDTA for 30 min on ice. FcX (Biolegend, San Diego, CA) was added to all samples during antibody staining. DAPI or Zombie Aqua fixable viability dye (Biolegend, San Diego, CA) was added to all samples prior to analysis. Analysis was performed on an LSRII Fortessa, and FACS on an ARIA or ARIA-H instrument (BD Biosciences, San Jose, CA) at the UCLA Broad Stem Cell Research Center Flow Cytometry Core. For all analyses DAPI+ or Zombie Aqua+ cells were gated out, and single cells were gated based on FSC-H vs. FSC-W.
scRNA-seq and CITE-seq library preparation and sequencing
ATOs were harvested at week 8 (as above) and subjected to MACs Dead Cell Removal Kit (Miltenyi, Cat. 130-090-101), and −5 × 105 cells were stained with TotalSeq-C Human Universal Cocktail, V1.0 (Biolegend, Cat. 399905) per the manufacturer’s protocol. Labeled cells were submitted to the UCLA Technology Center for Bioinformatics and Genomics for unique molecular identifier (UMI) tagging and generation of gene expression (GEX), human TCR repertoire (VDJ), and Feature Barcoding libraries using the 10X Chromium Next GEM Single Cell 5’ Kit v2 (10X Genomics, Pleasanton, CA). Fully constructed libraries for all samples were run in one S4 flowcell on the Illumina Novaseq platform.
scRNA-seq and CITE-seq data filtration and integration
Sequenced reads from each sample were aligned to the human reference genome GRCh38 and processed using the Cell Ranger v7.0.0 (10X Genomics) “multi” pipeline that generated count matrices from the GEX libraries, and assembled full TCR contigs from the VDJ libraries along with cell-surface protein expression from the Feature Barcoding libraries. On average, we achieved >70K mean reads per cell with >9000 mean UMIs per cell, and a median of >3,300 genes per cell. GEX (RNA) and Feature Barcoding (protein) count matrices from each sample were combined and loaded with Seurat v4.2.0 (Satija Lab), and barcoded cells were filtered for cells with outlier UMI counts <3000 (low quality cells) and >45000 (indicative of doublets), high mitochondrial gene expression (due to cellular stress or loss of cytoplasmic RNA), and low number of sequenced genes (<1200).
After initial data filtration for low-quality and outlier cells, the combined Seurat object was split by each modality, RNA and Protein, and then batch corrected for technical and biological variations using the Reciprocal Principal Component Analysis (RPCA) integration method in Seurat. Seurat utilized an unsupervised framework to learn cell-specific modality weights that allows integrated cell clustering based on both modalities. For integration of the combined RNA modality, molecular count data for each sample were individually normalized and variance stabilized using SCTransform, which bypasses the need for pseudocount addition and log-transformation, and then cell cycle phase scores were calculated for each individual sample based on the expression of canonical cell cycle genes within a specific barcoded cell. Following cell cycle scoring, raw counts were normalized and variance stabilized again using SCTransform with the additional step of regressing calculated cell cycle scores in order to mitigate the effects of cell cycle heterogeneity. In order to perform RPCA integration, highly variable genes (nfeatures = 3000) were then identified from each sample and then used to find integration anchors between datasets (k.anchor = 10). For integration of the protein modality, samples were individually normalized using centered log ratio transformation (CLR) prior to identification of highly variable features (nfeatures = 3000). Samples were then scaled and PCAs were calculated for log-normalized integration of datasets.
Weighted Nearest Neighbor multimodal analysis of scRNA-seq and CITE-seq data scRNA-seq clustering and visualization
Integrated Seurat objects of all samples from both modalities (RNA and surface protein) were combined and PCA were calculated for both modalities with the first 50 PCs taken for gene expression (RNA) and first 20 PCs for feature barcoding (surface protein) datasets. Visualization and clustering of both modalities was performed using Weighted Nearest Neighbor (WNN) multimodal analysis in Seurat v4.2.0, which utilizes an unsupervised framework to learn cell-specific modality weights that allow integrated cell clustering on both modalities (RNA and surface protein) at multiple resolutions (0.6, 0.8, and 1.0). Using the 1.0 resolution, clusters were labeled and collapsed into T cell developmental subsets (CD34, DN, ISP4, DP Early, DP Late, SP8+Tγδ, NK, pDC) based on expression of surface protein as well as RNA expression of key T cell developmental markers. Notably, two populations were removed from the dataset based on irregular gene expression: one population expressed both hCD45 and hDLL4, which could have been epithelial or stromal cells carried over from bone marrow aspirate collection of CD34+ cells used for generation of ATOs; and the other population stained for most antibodies, indicating the presence of a myeloid-lineage cell population.
Following initial labeling, specific subpopulations were subset out of the combined datasets and individually examined for key T cell developmental markers from surface protein and RNA expression profiles at high clustering resolutions in order to confirm cell identities, and correct for any grouping errors as a result of high order clustering of all cells from the combined datasets: the “CD34+” cluster was redefined, as only a specific subset expressed CD34 RNA within the cluster, with the remaining cells categorized as “DN”; a population of “B” cells were identified within the “DN” population, which expressed both CD19 transcriptionally and on the cell surface; and all DP populations (DP Early, DP Late) were redefined at higher resolution based on WNN_UMAP mapping coordinates (DP Early) and surface expression of TCRαβ and CD3 (DP Late).
To identify “SP8” T cells from the “SP8+Tγδ” population from high order clustering, fully reconstructed TCR contigs from VDJ sequencing libraries were added as metadata for their corresponding cell identities into the Seurat object using scRepertoire v1.7.2 (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7400693/). Based on cell surface expression of TCRαβ and metadata from full TCR contigs, “SP8” T cells were separated from “γδ” T cells, as sequencing of Tγδ TCRs was not performed. Further analysis of the “SP8” T cell population identified the “SP8RA” (CD45RA+CD45RO-) and “SP8RO” (CD45RA-CD45RA+) subsets.
Visualization and identification of gene-edited cells from scRNA-seq
Cellular barcodes from cleaned datasets were extracted from the integrated Seurat object and exported as individual lists for the identification of cells that were gene-corrected from scRNA-seq datasets. Cellular barcode lists were used by cb_sniffer70 (https://github.com/sridnona/cb_sniffer) to call mutant and edited RNA transcripts for CD3D (Chr 11:118340447-118340447, G [“Reference”] -> A [“Mutant”]) from BAM outputs from the Cell Ranger v7.0.0 (10X Genomics) “multi” pipeline alignment to the GRCh38 reference genome. Cells were assigned as “Biallelic” (Reference > 0, Mutant = 0), “Monoallelic” (Reference > 0, Mutant > 0), and “Uncorrected” (Reference = 0, Mutant > 0) based on the presence of reference and mutant CD3D RNA from BAM alignments. Cells that did not have read for CD3D RNA were labeled as “Dropout” due to dropouts that can occur stochastically from scRNA-sequencing. Cellular labels were added back into the Seurat object as metadata, and visualization was performed on the WNN_UMAP.
Visualization and identification of TCR rearrangements within scRNA-seq datasets
The integrated Seurat object including fully reconstructed TCRs in the metadata from VDJ sequencing was analyzed in order to visualize and identify cells that expressed no TRAV or TRBV, only TRBV, and both TRAV+TRBV. From the GEX sequencing data (RNA) in the integrated Seurat object, cells expressing no TRAV or TRBV, only TRBV, and both TRBV+TRAV were identified and labeled in a separate column of the metadata. As RNA sequencing of total genes could lead to dropouts, fully reconstructed TCRs from VDJ sequencing within the metadata of the Seurat object were also analyzed to determine cells that had no TRAV or TRBV, only TRBV, and both TRBV+TRAV in an additional column of the metadata. After identifying the intersections between both columns of the metadata (GEX and VDJ), visualization of TCR rearrangements within the datasets was performed on the WNN_UMAP. Chord diagrams were generated using the circlize v0.4.15 package71 using VDJ sequencing data embedded in the Seurat object with scRepetoire, as described above.
Differentially expressed gene analysis of scRNA-seq datasets
Differentially expressed genes (DEGs) were calculated using the “MAST” algorithm72 which is tailored to scRNA-seq data DEG analysis using a model that parameterizes both stochastic dropout and characteristic bimodal expression distributions, for the FindMarkers function of Seurat (min.pct = 0.1, logfc.threshold = 0.25). DEGs from FindMarkers were used to generate ranked gene lists ordered by log-fold change for Gene Set Enrichment Analysis (GSEA) using the fgsea v1.22.073 package and gene signatures were pulled from the Molecular Signatures Database (MSigDB) using msigdbr v7.15.174 (<https://CRAN.R-project.org/package=msigdbr>). Visualization of GSEA results was performed using the enrichplot v1.16.2 package75 (<https://yulab-smu.top/biomedical-knowledge-mining-book/>).
QUANTIFICATION AND STATISTICAL ANALYSIS
In all figures, n represents independent biological replicates and data are represented as mean ± standard deviation (SD). Statistical analysis was performed using GraphPad Prism software and p-values were calculated from the two-tailed unpaired t test or multiple t test, unless otherwise noted in figure legend. p-values of *p < 0.05; **p < 0.01; and ***p < 0.001, ****p<0.0001 were considered statistically significant, unless otherwise noted in figure legend.
Supplementary Material
Figure S1. Base Editing Efficiently Restores CD3 Expression without Inducing Chromothripsis, Related to Fig. 1.
a) Sanger sequencing traces confirm knockin of the CD3D c.202C>T pathogenic mutation in CD3D alleles with the remaining three alleles containing disruptive indels induced by CRISPR-mediated DSBs (rendering the latter three alleles non-editable, and therefore could be omitted from HTS analysis). Traces show the sense strand read 5’ to 3’. The black dashed vertical line represents the Cas9 cut site during CRISPR editing, with −150,000 total reads per population. b) Representative flow cytometry plots for CD3 surface expression. Top right: gating strategy to distinguish single cells. Bottom right: live cells. Bottom left: gating strategy to identify cells expressing CD3. SSC-A, side scatter area; FSC-A, forward scatter area; FSC-H, forward scatter height. c) Flow cytometry histogram profiling mean fluorescence intensity (MFI) of CD3 expression in CD3D(C202T) Jurkat T cells (orange), wildtype Jurkat T cells (blue), and cells treated with ABEmax-NRTH (red), ABE8e-NRTH (purple), ABE8e-NG (green), and RNP + ssODN-treated cells (black). G-banded karyotypes each representing clonal abnormalities observed in cells from d) mock electroporated, e) base-edited with plasmids encoding ABEmax-NRTH and sgRNA, or f) edited with Cas9 RNP and ssODN. Due to the complexity of the cell lines, the clonal abnormalities are described as composite karyotypes (not all indicated abnormalities were identified in all abnormal cells analyzed) using standard cytogenetic nomenclature (ISCN). Diploid, triploid, and tetraploid cells were observed. Black arrows indicate clonal structural abnormalities inherent to the Jurkat T cell line.
Figure S2. Base Editing of CD3δ SCID HSPCs Reveals Minimal Local Bystander and Genome-Wide Off-Target Editing and Indel Formation, Related to Fig. 2.
a) Representative flow cytometry plots to measure CD3/TCRαβ surface expression in CD3D(C202T) Jurkat T cells transduced with either LV expressing WT CD3D cDNA or CD3D cDNA containing the A0 bystander mutation. Top right: gating strategy to distinguish single cells. Bottom right: live cells. Bottom left: gating strategy to identify cells expressing CD3/TCRαβ. b) Venn diagram of candidate off-target sites predicted by CIRCLE-seq, GUIDE-seq, and Cas-OFFinder. Pie graph depicts the predicted genomic locations of the 57 candidate off-target sites nominated by two or more prediction tools. c-d) Bar graphs demonstrate the percentage of sequencing reads containing A•T-to-G•C point mutations consistent with adenine base editing and within protospacer positions 4–10 in gDNA from CD3δ SCID HSPCs treated with ABEmax-NRTH and sgRNA (peach) or untreated controls (teal). (n=3) e) Bar graphs demonstrate percent of sequencing reads containing indels across 200 genome-wide off-target sites. Sites were sequenced with HTS in gDNA from CD3δ SCID HSPCs treated with ABEmax-NRTH mRNA (peach) or untreated controls (teal). (n=3)
Figure S3. Base Editing is Durable in a Humanized Mouse Model and Can Rescue Additional CD3δ SCID-Causing Mutations in Human HSPCs, Related to Fig. 3.
a) Gating strategy to quantify relative abundances of hematopoietic lineages in recipient mouse bone marrow. b) Gating strategy to determine relative abundances of thymocytes across T cell development in recipient mouse thymi. 16 weeks after infusion of untreated or ABE8e-NG and adenine A0-targeting sgRNA-treated HD HSPCs, engraftment was measured by percentage of human CD45+ cells in recipient mouse c) bone marrow, d) spleen, and e) thymus for mice receiving untreated cells or ABE8e-NG treated cells. Abundance of human CD19+ B cells, CD33+ myeloid, CD34+ HSPCs, CD56+ NK cells, and CD3+ T cells were measured as percentages of the hCD45+ population in transplant recipient f) bone marrow and g) spleen. h) Human ISP4, SP4, SP8, DN, and DP T cells as percentages of the hCD45+ population in recipient mouse thymus. i) on-target adenine A0 editing efficiency and VCN determined by HTS and ddPCR, respectively, in cells cultured for 14 days after electroporation (pre-transplant) or in whole tissues 16 weeks after transplant. j) Gating strategy for FACS isolation of CD34+ HSPCs, CD33+ Myeloid, CD19+ B cells, and CD56+ NK cells from mouse bone marrow. k) Editing efficiency in human-derived hematopoietic lineages from mouse bone marrow. Populations were FACS sorted using hCD34+, hCD33+, hCD19+, and hCD56+ antibodies for HSPC, myeloid, B cell, and NK cell collection, respectively. n=2 mice that received untreated cells and n=7 mice that received ABE8e-NG edited cells. Data shown as mean ± SD; k) one-way ANOVA, non-parametric t-test elsewhere; ns, not significant. l) Schematic of the splicing mutation (identified in Ecuador) known to cause CD3δ SCID. A homozygous mutation in the splice donor site of intron 2 leads to abnormal splicing and exon 2 skipping (CD3D c.274+5G>A).31 m) Schematic of the splicing mutation, CD3D c.275–2A>G, (identified in Japan) known to cause CD3δ SCID.32 A homozygous mutation in the splice acceptor site of intron 2 leads to abnormal splicing and exon 3 skipping. n) HD CD34+ HSPCs were pre-stimulated for 24 hours and transduced with a lentiviral vector [containing either the CD3D c.274+5G>A mutation (MNDU3-CD3D-274+5G>A) or CD3D c.275–2A>G mutation (MNDU3-CD3D-275–2A>G) in intron 2]. Intron 2 was retained in the LV by positioning the internal expression cassette in reverse orientation. 24 hours after transduction, HSPCs were electroporated with ABE8e-NG, ABE8e-NGG, or BE4max-NG mRNA and sgRNA. Data shown as mean ± SD.
Figure S4. Base Editing Retains Clonogenic Potential and Rescues T cell Differentiation and Maturation, Related to Fig. 4.
a-b) Clonogenic potential. 24 hours following electroporation of CD3δ SCID CD34+ HSPCs, a CFU assay was performed to assess a) hematopoietic potential and b) hematopoietic lineage distribution of CD34+ HSPC HD, unedited, and edited patient cells prior to ATO formation (n=12). Statistics were calculated using a non-parametric t-test; ns, not significant. c-k) Rescue of T cell differentiation. c and d) FACS analysis of HD (green), unedited patient (black), and edited patient (blue) ATOs, n=6–9, from four independent experiments for all data. c) CD3 and TCRαβ co-expression (left), and developing T cell subsets (DN, DP-L, SP8, and SP4) (right) in ATOs at week 15. d) Early CD3 and TCRαβ co-expression at weeks 2, 3, 5, and 7. Frequency of e) CD3-TCRαβ-, DN, ISP4, DP, CD4−CD8α+, and CD3+TCRαβ+ cells from CD45+Lin- cells, and f) DP-L, CD8ɑ, CD8ɑβ (SP8 T cells), and CD8ɑɑ cells from CD45+Lin- cells at week 12. g) Representative flow cytometry profiles of CD8αα and CD8αβ cells in cells gated on CD3+TCRαβ+CD4− in HD, unedited patient, and edited patient ATOs at weeks 9, 12, and 15. Cell counts of h) CD3+ and i) TCRαβ+ cells per ATO over time. j) Representative flow cytometry profiles of TCRγδ expression in cells gated on CD45+Lin- at week 12. k) Cell counts of γδ T cells per ATO over time. Statistical significance was calculated using unpaired nonparametric T tests, **p < 0.01; and ***p < 0.001.
Figure S5. Identification of Developing T cell and Immune Cell Subsets in Unedited and Edited CD3δ SCID ATOs by CITE-seq, Related to Fig. 5.
a) Number of bioinformatically cleaned cells in indicated cell subsets in unedited and edited patient ATOs harvested at week 8 from two individual experiments. b) Surface protein and c) RNA gene expression of selected markers across cell subsets.
Figure S6. Monoallelic and Biallelic CD3D Correction Rescues SP8 and SP4 T cell Development, Related to Fig. 6.
a-b) Monoallelic and biallelic CD3D correction. a) WNN_UMAP visualization of unedited (grey), monoallelic (orange), or biallelic (blue) correction of patient ATOs. b) Bar graphs show relative T cell precursor abundances binned by the presence of unedited, monoallelic, or biallelic correction in single cells. Monoallelic vs. biallelic classification was determined by the presence of RNA strands with or without the CD3D c.202C>T edit. UnEd, unedited; Mono, monoallelic; Bi, biallelic. c-g) Edited CD3δ SCID ATO-derived SP8 T and SP4 T cells express features of maturation without evidence of exhaustion. c) Expression of indicated surface proteins (y-axis) across clusters in edited patient ATOs. d-e) Gene Set Enrichment Analysis (GSEA) of differentially expressed genes from GOBP (Gene Ontology Biological Process) and GOCC (Gene Ontology Cellular Compartment) between d) DP-L and DN, or e) SP8 T and DP-L cells. Dot size represents adjusted P-value (Padj; two-sided permutation test). NES, normalized enrichment score; PM, plasma membrane; RNP, ribonucleoprotein; LSU, large ribosomal subunit; SSU, small ribosomal subunit. f) Representative flow cytometry profiles depicting maturation markers (CCR7, CD62L, CD27, CD28, CD45RO, and CD45RA) in cells gated on SP4 T cells (CD3+TCRαβ+CD4+CD8α-), in week 12 ATOs (n=9, from four independent experiments). g) Representative flow cytometry profiles of exhaustion markers in SP4 T cells derived from week 15 HD (green) and edited patient (blue) ATOs, and Healthy PBMCs (right) stimulated with (orange) and without (purple) anti-CD3/28 beads + IL2 for 24 hours, n=9 for ATO groups and n=3 for PBMC controls.
Figure S7. Base editing of CD3δ SCID HSPCs recapitulates functional T cells with TCR diversity, Related to Fig. 7
a-c) Representative flow cytometry profiles of HD (green) and edited patient (blue) ATOs. a) IFNγ, IL-2, and TNFɑ production in SP8 T cells (CD3+TCRαβ+CD8α+CD4−CD45RA+) stimulated with or without anti-CD3/CD28 beads and IL2 for 24 hours, n=6. b) CD25 and c) 4–1BB expression vs proliferation (CFSE dilution) of MACs isolated ATO-derived SP8 cells after culture without stim, with IL2 alone, anti-CD3/CD28 bead alone, and anti-CD3/CD28 bead and IL-2 for 5 days. Data is representative of three independent experiments. d-g) TCR diversity by CITE-seq of unedited (black) and edited (blue) patient ATOs harvested at week 8, n=2 per arm, two independent experiments. d) TCR diversity measured by CHAO-1 index. Statistical significance was calculated by Hutchinson t-test (*p<5e10–8). e) Chord diagrams depicting interconnection of TCR clonotypes in developing cell subsets in unedited (left) and edited (right) patient ATOs. Area of each segment correlates to the relative abundance of TCR clonotypes in indicated cells subsets. Curved lines indicate TCR clonotypes shared between cell subsets. f) Frequency of individual TRBV (top) and TRBJ (bottom) usage. g) Heatmap visualization of individual TRBV and TRBJ segments displayed in genomic order from 5’ distal -> 3’ proximal ends.
Acknowledgements
These studies were supported by a Translational Research Grant from the Jeffrey Modell Foundation (D.B.K.); NIH T32HL086345 (P.C.C.), NIH U01AI142756 (D.R.L.), NIH RM1 HG009490 (D.R.L.), R35 GM118062 (D.R.L.), the Bill and Melinda Gates Foundation (D.R.L.), the Howard Hughes Medical Institute (D.R.L.), NIH K99 award HL163805 (G.A.N.); NIH K08CA235525 and V Foundation Scholar Award (C.S.S.); and G.Y. is the recipient of the A.P. Giannini Postdoctoral Research Fellowship and Leadership Award. N.W. is supported by the Barb Ibbotson Chair in Pediatric Hematology, Alberta Children’s Hospital Research Institute. Core services were supported by the UCLA Technology Center for Bioinformatics and Genomics. Karyotype studies were performed in the UCLA Cytogenetics Laboratory. We would like to thank the UCLA Broad Stem Cell Research Center (BSCRC) Flow Cytometry Core for assistance with FACS sorting.
Inclusion and Diversity
We support inclusive, diverse, and equitable conduct of research. We worked to ensure gender balance in the recruitment of human subjects. We worked to ensure diversity in experimental samples through the selection of the cell lines. We worked to ensure diversity in experimental samples through the selection of the genomic datasets. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research. One or more of the authors of this paper self-identifies as a member of the LGBTQIA+ community. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
Footnotes
Declaration of Interests
G.M.C. and C.S.S. are founders of Pluto Immunotherapeutics Inc and serve as consultants to this company. D.R.L. is a consultant and equity holder of Prime Medicine, Beam Therapeutics, Pairwise Plants, and Chroma Medicine, companies that use gene editing or genome engineering.
References
- 1.Garcillán B, Fuentes P, Marin A. v., Megino RF, Chacon-Arguedas D, Mazariegos MS, Jiménez-Reinoso A, Muñoz-Ruiz M, Laborda RG, Cárdenas PP, et al. (2021). CD3G or CD3D Knockdown in Mature, but Not Immature, T Lymphocytes Similarly Cripples the Human TCRαβ Complex. Front Cell Dev Biol 9. 10.3389/fcell.2021.608490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dadi HK, Simon AJ, and Roifman CM (2003). Effect of CD3 d Deficiency on Maturation of a / b and g / d T-Cell Lineages in Severe Combined Immunodeficiency. [DOI] [PubMed] [Google Scholar]
- 3.Marcus N, Takada H, Law J, Cowan MJ, Gil J, Regueiro JR, Plaza Lopez De Sabando D, Lopez-Granados E, Dalal J, Friedrich W, et al. (2011). Hematopoietic stem cell transplantation for CD3δ deficiency. Journal of Allergy and Clinical Immunology 128, 1050–1057. 10.1016/j.jaci.2011.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mamcarz E, Zhou S, Lockey T, Abdelsamed H, Cross SJ, Kang G, Ma Z, Condori J, Dowdy J, Triplett B, et al. (2019). Lentiviral Gene Therapy Combined with Low-Dose Busulfan in Infants with SCID-X1. New England Journal of Medicine 380, 1525–1534. 10.1056/nejmoa1815408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kohn DB, Booth C, Shaw KL, Xu-Bayford J, Garabedian E, Trevisan V, Carbonaro-Sarracino DA, Soni K, Terrazas D, Snell K, et al. (2021). Autologous Ex Vivo Lentiviral Gene Therapy for Adenosine Deaminase Deficiency. New England Journal of Medicine 384, 2002–2013. 10.1056/nejmoa2027675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cowan MJ, Yu J, Facchino J, Fraser-Browne C, Sanford U, Kawahara M, Dara J, Long-Boyle J, Oh J, Chan W, et al. (2022). Lentiviral Gene Therapy for Artemis-Deficient SCID. N Engl J Med 387, 2344–2355. 10.1056/NEJMoa2206575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garcia-Perez L, van Eggermond M, van Roon L, Vloemans SA, Cordes M, Schambach A, Rothe M, Berghuis D, Lagresle-Peyrou C, Cavazzana M, et al. (2020). Successful Preclinical Development of Gene Therapy for Recombinase-Activating Gene-1-Deficient SCID. Mol Ther Methods Clin Dev 17, 666–682. 10.1016/j.omtm.2020.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Romero Z, Torres S, Cobo M, Mũoz P, Unciti JD, Martín F, and Molina IJ (2011). A tissue-specific, activation-inducible, lentiviral vector regulated by human CD40L proximal promoter sequences. Gene Ther 18, 364–371. 10.1038/gt.2010.144. [DOI] [PubMed] [Google Scholar]
- 9.Pavel-Dinu M, Wiebking V, Dejene BT, Srifa W, Mantri S, Nicolas CE, Lee C, Bao G, Kildebeck EJ, Punjya N, et al. (2019). Gene correction for SCID-X1 in long-term hematopoietic stem cells. Nat Commun 10. 10.1038/s41467-019-09614-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newby GA, Yen JS, Woodard KJ, Mayuranathan T, Lazzarotto CR, Li Y, Sheppard-Tillman H, Porter SN, Yao Y, Mayberry K, et al. (2021). Base editing of haematopoietic stem cells rescues sickle cell disease in mice. Nature 595, 295–302. 10.1038/s41586-021-03609-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, et al. (2016). ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44, D862–D868. 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bosticardo M, Pala F, Calzoni E, Delmonte OM, Dobbs K, Gardner CL, Sacchetti N, Kawai T, Garabedian EK, Draper D, et al. (2020). Artificial thymic organoids represent a reliable tool to study T-cell differentiation in patients with severe T-cell lymphopenia. Blood Adv 4, 2611–2616. 10.1182/bloodadvances.2020001730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Montel-Hagen A, Sun V, Casero D, Tsai S, Zampieri A, Jackson N, Li S, Lopez S, Zhu Y, Chick B, et al. (2020). In Vitro Recapitulation of Murine Thymopoiesis from Single Hematopoietic Stem Cells. Cell Rep 33. 10.1016/j.celrep.2020.108320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Montel-Hagen A, Tsai S, Seet CS, and Crooks GM (2022). Generation of Artificial Thymic Organoids from Human and Murine Hematopoietic Stem and Progenitor Cells. Curr Protoc 2. 10.1002/cpz1.403. [DOI] [PubMed] [Google Scholar]
- 15.Montel-Hagen A, Seet CS, Li S, Chick B, Zhu Y, Chang P, Tsai S, Sun V, Lopez S, Chen HC, et al. (2019). Organoid-Induced Differentiation of Conventional T Cells from Human Pluripotent Stem Cells. Cell Stem Cell 24, 376–389.e8. 10.1016/j.stem.2018.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeWitt MA, Magis W, Bray NL, Wang T, Berman JR, Urbinati F, Heo SJ, Mitros T, Muñoz DP, Boffelli D, et al. (2016). Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci Transl Med 8. 10.1126/scitranslmed.aaf9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chu SH, Packer M, Rees H, Lam D, Yu Y, Marshall J, Cheng LI, Lam D, Olins J, Ran FA, et al. (2021). Rationally Designed Base Editors for Precise Editing of the Sickle Cell Disease Mutation. CRISPR Journal 4, 169–177. 10.1089/crispr.2020.0144. [DOI] [PubMed] [Google Scholar]
- 18.Gehrke JM, Cervantes O, Clement MK, Wu Y, Zeng J, Bauer DE, Pinello L, and Joung JK (2018). An apobec3a-cas9 base editor with minimized bystander and off-target activities. Nat Biotechnol 36, 977. 10.1038/nbt.4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koblan LW, Doman JL, Wilson C, Levy JM, Tay T, Newby GA, Maianti JP, Raguram A, and Liu DR (2018). Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat Biotechnol 36, 843–848. 10.1038/nbt.4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller SM, Wang T, Randolph PB, Arbab M, Shen MW, Huang TP, Matuszek Z, Newby GA, Rees HA, and Liu DR (2020). Continuous evolution of SpCas9 variants compatible with non-G PAMs. Nat Biotechnol 38, 471–481. 10.1038/s41587-020-0412-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joseph N, Reicher B, and Barda-Saad M (2014). The calcium feedback loop and T cell activation: How cytoskeleton networks control intracellular calcium flux. Biochim Biophys Acta Biomembr 1838, 557–568. 10.1016/j.bbamem.2013.07.009. [DOI] [PubMed] [Google Scholar]
- 22.Leibowitz ML, Papathanasiou S, Doerfler PA, Blaine LJ, Sun L, Yao Y, Zhang CZ, Weiss MJ, and Pellman D (2021). Chromothripsis as an on-target consequence of CRISPR–Cas9 genome editing. Nat Genet 53, 895–905. 10.1038/s41588-021-00838-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang W, Tang G, Cortes JE, Liu H, Ai D, Yin CC, Li S, Khoury JD, Bueso-Ramos C, Medeiros LJ, et al. (2015). Chromosomal rearrangement involving 11q23 locus in chronic myelogenous leukemia: A rare phenomenon frequently associated with disease progression and poor prognosis. J Hematol Oncol 8. 10.1186/s13045-015-0128-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baer MR, Stewart CC, Lawrence D, Arthur DC, Mró Zek M, Strout MP, Davey FR, and Bloomfield CD (1998). Acute myeloid leukemia with 11q23 translocations: myelomonocytic immunophenotype by multiparameter flow cytometry. [DOI] [PubMed] [Google Scholar]
- 25.Rees HA, and Liu DR (2018). Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet 19, 770–788. 10.1038/s41576-018-0059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richter MF, Zhao KT, Eton E, Lapinaite A, Newby GA, Thuronyi BW, Wilson C, Koblan LW, Zeng J, Bauer DE, et al. (2020). Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat Biotechnol 38, 883–891. 10.1038/s41587-020-0453-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsai SQ, Nguyen NT, Malagon-Lopez J, Topkar V. v., Aryee MJ, and Joung JK (2017). CIRCLE-seq: A highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat Methods 14, 607–614. 10.1038/nmeth.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar V. v., Thapar V, Wyvekens N, Khayter C, Iafrate AJ, Le LP, et al. (2015). GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 33, 187–198. 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bae S, Park J, and Kim JS (2014). Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475. 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Collias D, and Beisel CL (2021). CRISPR technologies and the search for the PAM-free nuclease. Nat Commun 12. 10.1038/s41467-020-20633-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garcillán BM, Mazariegos MS MS, Fisch P, Res PC, Muñoz-Ruiz MM, Gil J, López-Granados E, Fernández-Malavé E, and Regueiro JR (2014). Enrichment of the rare CD4+ γδ T-cell subset in patients with atypical CD3δ deficiency. 10.1016/j.jaci.2013.09. [DOI] [PubMed] [Google Scholar]
- 32.Takada H, Nomura A, Roifman CM, and Hara T (2005). Severe combined immunodeficiency caused by a splicing abnormality of the CD3δ gene. Eur J Pediatr 164, 311–314. 10.1007/s00431-005-1639-6. [DOI] [PubMed] [Google Scholar]
- 33.McIntosh BE, Brown ME, Duffin BM, Maufort JP, Vereide DT, Slukvin II, and Thomson JA (2015). Nonirradiated NOD,B6.SCID Il2rγ−/− kitW41/W41 (NBSGW) mice support multilineage engraftment of human hematopoietic cells. Stem Cell Reports 4, 171–180. 10.1016/j.stemcr.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hess NJ, Lindner PN, Vazquez J, Grindel S, Hudson AW, Stanic AK, Ikeda A, Hematti P, and Gumperz JE (2020). Different Human Immune Lineage Compositions Are Generated in Non-Conditioned NBSGW Mice Depending on HSPC Source. Front Immunol 11. 10.3389/fimmu.2020.573406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hutt D (2018). Engraftment, Graft Failure, and Rejection. In The European Blood and Marrow Transplantation Textbook for Nurses (Springer International Publishing; ), pp. 259–270. 10.1007/978-3-319-50026-3_13. [DOI] [Google Scholar]
- 36.Seet CS, He C, Bethune MT, Li S, Chick B, Gschweng EH, Zhu Y, Kim K, Kohn DB, Baltimore D, et al. (2017). Generation of mature T cells from human hematopoietic stem and progenitor cells in artificial thymic organoids. Nat Methods 14, 521–530. 10.1038/nmeth.4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Saint Basile G, Geissmann F, Flori E, Uring-Lambert B, Soudais C, Cavazzana-Calvo M, Durandy A, Jabado N, Fischer A, and le Deist F (2004). Severe combined immunodeficiency caused by deficiency in either the δ or the ε subunit of CD3. Journal of Clinical Investigation 114, 1512–1517. 10.1172/JCI200422588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Recio MJ, Moreno-Pelayo MA, Kiliç SS, Guardo AC, Sanal O, Allende LM, Pérez-Flores V, Mencía A, Modamio-Høybjør S, Seoane E, et al. (2007). Differential Biological Role of CD3 Chains Revealed by Human Immunodeficiencies. The Journal of Immunology 178, 2556–2564. 10.4049/jimmunol.178.4.2556. [DOI] [PubMed] [Google Scholar]
- 39.Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, and Smibert P (2017). Simultaneous epitope and transcriptome measurement in single cells. Nat Methods 14, 865–868. 10.1038/nmeth.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davis MM, and Bjorkman PJ (1988). T-cell antigen receptor genes and T-cell recognition. Nature 334. [DOI] [PubMed] [Google Scholar]
- 42.Taghon T, and Rothenberg E. v. (2008). Molecular mechanisms that control mouse and human TCR-αβ and TCR-γδ T cell development. Semin Immunopathol 30, 383–398. 10.1007/s00281-008-0134-3. [DOI] [PubMed] [Google Scholar]
- 43.Anderson AC, Joller N, and Kuchroo VK (2016). Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 44, 989–1004. 10.1016/j.immuni.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, and Anderson AC (2010). Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. Journal of Experimental Medicine 207, 2187–2194. 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mclane LM, Abdel-Hakeem MS, and Wherry EJ (2019). CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. 10.1146/annurev-immunol-041015. [DOI] [PubMed] [Google Scholar]
- 46.Chao A (1984). Board of the Foundation of the Scandinavian Journal of Statistics Nonparametric Estimation of the Number of Classes in a Population Nonparametric Estimation of the Number of Classes in a Population. [Google Scholar]
- 47.Siegner SM, Ugalde L, Clemens A, Garcia-Garcia L, Bueren JA, Rio P, Karasu ME, and Corn JE (2022). Adenine base editing efficiently restores the function of Fanconi anemia hematopoietic stem and progenitor cells. Nat Commun 13, 6900. 10.1038/s41467-022-34479-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koblan LW, Erdos MR, Wilson C, Cabral WA, Levy JM, Xiong ZM, Tavarez UL, Davison LM, Gete YG, Mao X, et al. (2021). In vivo base editing rescues Hutchinson–Gilford progeria syndrome in mice. Nature 589, 608–614. 10.1038/s41586-020-03086-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bejar R, Levine R, and Ebert BL (2011). Unraveling the molecular pathophysiology of myelodysplastic syndromes. Journal of Clinical Oncology 29, 504–515. 10.1200/JCO.2010.31.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chua BA, and Signer RAJ (2020). Hematopoietic stem cell regulation by the proteostasis network. Curr Opin Hematol 27, 254–263. 10.1097/MOH.0000000000000591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gil J, Busto EM, Garcillán B, Chean C, García-Rodríguez MC, Díaz-Alderete A, Navarro J, Reiné J, Mencía A, Gurbindo D, et al. (2011). A leaky mutation in CD3D differentially affects αβ and γδ T cells and leads to a Tαβ- Tγδ +B +NK + human SCID. Journal of Clinical Investigation 121, 3872–3876. 10.1172/JCI44254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dave VP, Cao Z, Browne C, Alarcon B, Fernandez-Miguel G, Lafaille J, de la Hera A, Tonegawa S, and Kappes DJ (1997). CD3δ deficiency arrests development of the αβ but not the γδ T cell lineage. EMBO J Vol.16, 1360–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Okada S, Markle JG, Deenick EK, Mele F, Averbuch D, Lagos M, Alzahrani M, Al-Muhsen S, Halwani R, Ma CS, et al. (2015). Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science (1979) 349, 606–613. 10.1126/science.aaa4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun Z, Unutmaz D, Zou Y-R, Sunshine MJ, Pierani A, Brenner-Morton S, Mebius RE, and Littman DR Requirement for ROR in Thymocyte Survival and Lymphoid Organ Development. [DOI] [PubMed] [Google Scholar]
- 55.Ligons DL, Hwang SJ, Waickman AT, Park JY, Luckey MA, and Park JH (2018). RORt limits the amount of the cytokine receptor c through the prosurvival factor Bcl-xL in developing thymocytes. Sci Signal 11. 10.1126/scisignal.aam8939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Masiuk KE, Laborada J, Roncarolo MG, Hollis RP, and Kohn DB (2019). Lentiviral Gene Therapy in HSCs Restores Lineage-Specific Foxp3 Expression and Suppresses Autoimmunity in a Mouse Model of IPEX Syndrome. Cell Stem Cell 24, 309–317.e7. 10.1016/j.stem.2018.12.003. [DOI] [PubMed] [Google Scholar]
- 57.Lomova A, Clark DN, Campo-Fernandez B, Flores-Bjurström C, Kaufman ML, Fitz-Gibbon S, Wang X, Miyahira EY, Brown D, DeWitt MA, et al. (2019). Improving Gene Editing Outcomes in Human Hematopoietic Stem and Progenitor Cells by Temporal Control of DNA Repair. Stem Cells 37, 284–294. 10.1002/stem.2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Benitez EK, Lomova Kaufman A, Cervantes L, Clark DN, Ayoub PG, Senadheera S, Osborne K, Sanchez JM, Crisostomo RV, Wang X, et al. (2020). Global and Local Manipulation of DNA Repair Mechanisms to Alter Site-Specific Gene Editing Outcomes in Hematopoietic Stem Cells. Front Genome Ed 2. 10.3389/fgeed.2020.601541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hoban MD, Lumaquin D, Kuo CY, Romero Z, Long J, Ho M, Young CS, Mojadidi M, Fitz-Gibbon S, Cooper AR, et al. (2016). CRISPR/Cas9-mediated correction of the sickle mutation in human CD34+ cells. Molecular Therapy 24, 1561–1569. 10.1038/mt.2016.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Camelia Botnar Laboratories, F.C.C.F. Calcium Flux on LSRII Background. [Google Scholar]
- 61.Grynkiewicz G, Poenie M, and Tsien RY (1985). A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry 260, 3440–3450. 10.1016/s0021-9258(19)83641-4. [DOI] [PubMed] [Google Scholar]
- 62.Bannwarth M, Corrêa IR, Sztretye M, Pouvreau S, Fella C, AnninaAebischer, Royer L, Ríos E, and Johnsson K (2009). Indo-1 derivatives for local calcium sensing. ACS Chem Biol 4, 179–190. 10.1021/cb800258g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morgan AJ, and Jacob R (1994). lonomycin enhances Ca2+ influx by stimulating store-regulated cation entry and not by a direct action at the plasma membrane. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kuo CY, Long JD, Campo-Fernandez B, de Oliveira S, Cooper AR, Romero Z, Hoban MD, Joglekar A. v., Lill GR, Kaufman ML, et al. (2018). Site-Specific Gene Editing of Human Hematopoietic Stem Cells for X-Linked Hyper-IgM Syndrome. Cell Rep 23, 2606–2616. 10.1016/j.celrep.2018.04.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Malinin NL, Lee GH, Lazzarotto CR, Li Y, Zheng Z, Nguyen NT, Liebers M, Topkar V. v., Iafrate AJ, Le LP, et al. (2021). Defining genome-wide CRISPR–Cas genome-editing nuclease activity with GUIDE-seq. Nat Protoc 16, 5592–5615. 10.1038/s41596-021-00626-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Logan AC, Nightingale SJ, Haas DL, Cho GJ, Pepper KA, and Kohn DB (2004). Factors influencing the titer and infectivity of lentiviral vectors. Hum Gene Ther 15, 976–988. 10.1089/hum.2004.15.976. [DOI] [PubMed] [Google Scholar]
- 67.Han J, Tam K, Tam C, Hollis RP, and Kohn DB (2021). Improved lentiviral vector titers from a multi-gene knockout packaging line. Mol Ther Oncolytics 23, 582–592. 10.1016/j.omto.2021.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cooper AR, Patel S, Senadheera S, Plath K, Kohn DB, and Hollis RP (2011). Highly efficient large-scale lentiviral vector concentration by tandem tangential flow filtration. J Virol Methods 177, 1–9. 10.1016/j.jviromet.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.San José E, Borroto A, Niedergang F, Alcover A, and Alarcó B (2000). Triggering the TCR Complex Causes the Downregulation of Nonengaged Receptors by a Signal Transduction-Dependent Mechanism. [DOI] [PubMed] [Google Scholar]
- 70.Petti AA, Williams SR, Miller CA, Fiddes IT, Srivatsan SN, Chen DY, Fronick CC, Fulton RS, Church DM, and Ley TJ (2019). A general approach for detecting expressed mutations in AML cells using single cell RNA-sequencing. Nat Commun 10. 10.1038/s41467-019-11591-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gu Z, Gu L, Eils R, Schlesner M, and Brors B (2014). Circlize implements and enhances circular visualization in R. Bioinformatics 30, 2811–2812. 10.1093/bioinformatics/btu393. [DOI] [PubMed] [Google Scholar]
- 72.Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, et al. (2015). MAST: A flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol 16. 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, and Sergushichev A Fast gene set enrichment analysis. 10.1101/060012. [DOI] [Google Scholar]
- 74.Dolgalev I (2005). MSiMSigDB Gene Sets for Multiple Organisms in a Tidy Data FormatgDB Gene Sets for Multiple Organisms in a Tidy Data Format. Proc Natl Acad Sci U S A 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yu G (2022). Visualization of Functional Enrichment Result_. R package version 1.16.2 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Base Editing Efficiently Restores CD3 Expression without Inducing Chromothripsis, Related to Fig. 1.
a) Sanger sequencing traces confirm knockin of the CD3D c.202C>T pathogenic mutation in CD3D alleles with the remaining three alleles containing disruptive indels induced by CRISPR-mediated DSBs (rendering the latter three alleles non-editable, and therefore could be omitted from HTS analysis). Traces show the sense strand read 5’ to 3’. The black dashed vertical line represents the Cas9 cut site during CRISPR editing, with −150,000 total reads per population. b) Representative flow cytometry plots for CD3 surface expression. Top right: gating strategy to distinguish single cells. Bottom right: live cells. Bottom left: gating strategy to identify cells expressing CD3. SSC-A, side scatter area; FSC-A, forward scatter area; FSC-H, forward scatter height. c) Flow cytometry histogram profiling mean fluorescence intensity (MFI) of CD3 expression in CD3D(C202T) Jurkat T cells (orange), wildtype Jurkat T cells (blue), and cells treated with ABEmax-NRTH (red), ABE8e-NRTH (purple), ABE8e-NG (green), and RNP + ssODN-treated cells (black). G-banded karyotypes each representing clonal abnormalities observed in cells from d) mock electroporated, e) base-edited with plasmids encoding ABEmax-NRTH and sgRNA, or f) edited with Cas9 RNP and ssODN. Due to the complexity of the cell lines, the clonal abnormalities are described as composite karyotypes (not all indicated abnormalities were identified in all abnormal cells analyzed) using standard cytogenetic nomenclature (ISCN). Diploid, triploid, and tetraploid cells were observed. Black arrows indicate clonal structural abnormalities inherent to the Jurkat T cell line.
Figure S2. Base Editing of CD3δ SCID HSPCs Reveals Minimal Local Bystander and Genome-Wide Off-Target Editing and Indel Formation, Related to Fig. 2.
a) Representative flow cytometry plots to measure CD3/TCRαβ surface expression in CD3D(C202T) Jurkat T cells transduced with either LV expressing WT CD3D cDNA or CD3D cDNA containing the A0 bystander mutation. Top right: gating strategy to distinguish single cells. Bottom right: live cells. Bottom left: gating strategy to identify cells expressing CD3/TCRαβ. b) Venn diagram of candidate off-target sites predicted by CIRCLE-seq, GUIDE-seq, and Cas-OFFinder. Pie graph depicts the predicted genomic locations of the 57 candidate off-target sites nominated by two or more prediction tools. c-d) Bar graphs demonstrate the percentage of sequencing reads containing A•T-to-G•C point mutations consistent with adenine base editing and within protospacer positions 4–10 in gDNA from CD3δ SCID HSPCs treated with ABEmax-NRTH and sgRNA (peach) or untreated controls (teal). (n=3) e) Bar graphs demonstrate percent of sequencing reads containing indels across 200 genome-wide off-target sites. Sites were sequenced with HTS in gDNA from CD3δ SCID HSPCs treated with ABEmax-NRTH mRNA (peach) or untreated controls (teal). (n=3)
Figure S3. Base Editing is Durable in a Humanized Mouse Model and Can Rescue Additional CD3δ SCID-Causing Mutations in Human HSPCs, Related to Fig. 3.
a) Gating strategy to quantify relative abundances of hematopoietic lineages in recipient mouse bone marrow. b) Gating strategy to determine relative abundances of thymocytes across T cell development in recipient mouse thymi. 16 weeks after infusion of untreated or ABE8e-NG and adenine A0-targeting sgRNA-treated HD HSPCs, engraftment was measured by percentage of human CD45+ cells in recipient mouse c) bone marrow, d) spleen, and e) thymus for mice receiving untreated cells or ABE8e-NG treated cells. Abundance of human CD19+ B cells, CD33+ myeloid, CD34+ HSPCs, CD56+ NK cells, and CD3+ T cells were measured as percentages of the hCD45+ population in transplant recipient f) bone marrow and g) spleen. h) Human ISP4, SP4, SP8, DN, and DP T cells as percentages of the hCD45+ population in recipient mouse thymus. i) on-target adenine A0 editing efficiency and VCN determined by HTS and ddPCR, respectively, in cells cultured for 14 days after electroporation (pre-transplant) or in whole tissues 16 weeks after transplant. j) Gating strategy for FACS isolation of CD34+ HSPCs, CD33+ Myeloid, CD19+ B cells, and CD56+ NK cells from mouse bone marrow. k) Editing efficiency in human-derived hematopoietic lineages from mouse bone marrow. Populations were FACS sorted using hCD34+, hCD33+, hCD19+, and hCD56+ antibodies for HSPC, myeloid, B cell, and NK cell collection, respectively. n=2 mice that received untreated cells and n=7 mice that received ABE8e-NG edited cells. Data shown as mean ± SD; k) one-way ANOVA, non-parametric t-test elsewhere; ns, not significant. l) Schematic of the splicing mutation (identified in Ecuador) known to cause CD3δ SCID. A homozygous mutation in the splice donor site of intron 2 leads to abnormal splicing and exon 2 skipping (CD3D c.274+5G>A).31 m) Schematic of the splicing mutation, CD3D c.275–2A>G, (identified in Japan) known to cause CD3δ SCID.32 A homozygous mutation in the splice acceptor site of intron 2 leads to abnormal splicing and exon 3 skipping. n) HD CD34+ HSPCs were pre-stimulated for 24 hours and transduced with a lentiviral vector [containing either the CD3D c.274+5G>A mutation (MNDU3-CD3D-274+5G>A) or CD3D c.275–2A>G mutation (MNDU3-CD3D-275–2A>G) in intron 2]. Intron 2 was retained in the LV by positioning the internal expression cassette in reverse orientation. 24 hours after transduction, HSPCs were electroporated with ABE8e-NG, ABE8e-NGG, or BE4max-NG mRNA and sgRNA. Data shown as mean ± SD.
Figure S4. Base Editing Retains Clonogenic Potential and Rescues T cell Differentiation and Maturation, Related to Fig. 4.
a-b) Clonogenic potential. 24 hours following electroporation of CD3δ SCID CD34+ HSPCs, a CFU assay was performed to assess a) hematopoietic potential and b) hematopoietic lineage distribution of CD34+ HSPC HD, unedited, and edited patient cells prior to ATO formation (n=12). Statistics were calculated using a non-parametric t-test; ns, not significant. c-k) Rescue of T cell differentiation. c and d) FACS analysis of HD (green), unedited patient (black), and edited patient (blue) ATOs, n=6–9, from four independent experiments for all data. c) CD3 and TCRαβ co-expression (left), and developing T cell subsets (DN, DP-L, SP8, and SP4) (right) in ATOs at week 15. d) Early CD3 and TCRαβ co-expression at weeks 2, 3, 5, and 7. Frequency of e) CD3-TCRαβ-, DN, ISP4, DP, CD4−CD8α+, and CD3+TCRαβ+ cells from CD45+Lin- cells, and f) DP-L, CD8ɑ, CD8ɑβ (SP8 T cells), and CD8ɑɑ cells from CD45+Lin- cells at week 12. g) Representative flow cytometry profiles of CD8αα and CD8αβ cells in cells gated on CD3+TCRαβ+CD4− in HD, unedited patient, and edited patient ATOs at weeks 9, 12, and 15. Cell counts of h) CD3+ and i) TCRαβ+ cells per ATO over time. j) Representative flow cytometry profiles of TCRγδ expression in cells gated on CD45+Lin- at week 12. k) Cell counts of γδ T cells per ATO over time. Statistical significance was calculated using unpaired nonparametric T tests, **p < 0.01; and ***p < 0.001.
Figure S5. Identification of Developing T cell and Immune Cell Subsets in Unedited and Edited CD3δ SCID ATOs by CITE-seq, Related to Fig. 5.
a) Number of bioinformatically cleaned cells in indicated cell subsets in unedited and edited patient ATOs harvested at week 8 from two individual experiments. b) Surface protein and c) RNA gene expression of selected markers across cell subsets.
Figure S6. Monoallelic and Biallelic CD3D Correction Rescues SP8 and SP4 T cell Development, Related to Fig. 6.
a-b) Monoallelic and biallelic CD3D correction. a) WNN_UMAP visualization of unedited (grey), monoallelic (orange), or biallelic (blue) correction of patient ATOs. b) Bar graphs show relative T cell precursor abundances binned by the presence of unedited, monoallelic, or biallelic correction in single cells. Monoallelic vs. biallelic classification was determined by the presence of RNA strands with or without the CD3D c.202C>T edit. UnEd, unedited; Mono, monoallelic; Bi, biallelic. c-g) Edited CD3δ SCID ATO-derived SP8 T and SP4 T cells express features of maturation without evidence of exhaustion. c) Expression of indicated surface proteins (y-axis) across clusters in edited patient ATOs. d-e) Gene Set Enrichment Analysis (GSEA) of differentially expressed genes from GOBP (Gene Ontology Biological Process) and GOCC (Gene Ontology Cellular Compartment) between d) DP-L and DN, or e) SP8 T and DP-L cells. Dot size represents adjusted P-value (Padj; two-sided permutation test). NES, normalized enrichment score; PM, plasma membrane; RNP, ribonucleoprotein; LSU, large ribosomal subunit; SSU, small ribosomal subunit. f) Representative flow cytometry profiles depicting maturation markers (CCR7, CD62L, CD27, CD28, CD45RO, and CD45RA) in cells gated on SP4 T cells (CD3+TCRαβ+CD4+CD8α-), in week 12 ATOs (n=9, from four independent experiments). g) Representative flow cytometry profiles of exhaustion markers in SP4 T cells derived from week 15 HD (green) and edited patient (blue) ATOs, and Healthy PBMCs (right) stimulated with (orange) and without (purple) anti-CD3/28 beads + IL2 for 24 hours, n=9 for ATO groups and n=3 for PBMC controls.
Figure S7. Base editing of CD3δ SCID HSPCs recapitulates functional T cells with TCR diversity, Related to Fig. 7
a-c) Representative flow cytometry profiles of HD (green) and edited patient (blue) ATOs. a) IFNγ, IL-2, and TNFɑ production in SP8 T cells (CD3+TCRαβ+CD8α+CD4−CD45RA+) stimulated with or without anti-CD3/CD28 beads and IL2 for 24 hours, n=6. b) CD25 and c) 4–1BB expression vs proliferation (CFSE dilution) of MACs isolated ATO-derived SP8 cells after culture without stim, with IL2 alone, anti-CD3/CD28 bead alone, and anti-CD3/CD28 bead and IL-2 for 5 days. Data is representative of three independent experiments. d-g) TCR diversity by CITE-seq of unedited (black) and edited (blue) patient ATOs harvested at week 8, n=2 per arm, two independent experiments. d) TCR diversity measured by CHAO-1 index. Statistical significance was calculated by Hutchinson t-test (*p<5e10–8). e) Chord diagrams depicting interconnection of TCR clonotypes in developing cell subsets in unedited (left) and edited (right) patient ATOs. Area of each segment correlates to the relative abundance of TCR clonotypes in indicated cells subsets. Curved lines indicate TCR clonotypes shared between cell subsets. f) Frequency of individual TRBV (top) and TRBJ (bottom) usage. g) Heatmap visualization of individual TRBV and TRBJ segments displayed in genomic order from 5’ distal -> 3’ proximal ends.
Data Availability Statement
Sequencing data have been deposited at NCBI Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-human CD3 | Biolegend | Clone UCHT1 |
| Anti-human CD3 (for in vivo studies) | Biolegend | Clone SK7 |
| Purified NA/LE Mouse Anti-Human CD3 (for calcium flux assay) | BD Biosciences | Clone HIT3a |
| Anti-human CD4 | Biolegend | Clone RPA-T4 |
| Anti-human CD5 | Biolegend | Clone UCHT2 |
| Anti-human CD7 | Biolegend | Clone M-T701 |
| Anti-human CD8a | Biolegend | Clone SK1 |
| Anti-human CD8b | Miltenyi Biotec | Clone REA-715 |
| Anti-human CD14 | Biolegend | Clone M5E2 |
| Anti-human CD19 | Biolegend | Clone HIB19 |
| Anti-human CD25 | Biolegend | Clone BC96 |
| Anti-human CD27 | Biolegend | Clone O323 |
| Anti-human CD28 | Biolegend | Clone CD28.2 |
| Purified NA/LE Mouse Anti-Human CD28 (for calcium flux assay) | BD Biosciences | Clone CD28.2 |
| Anti-human CD33 | Biolegend | Clone WM53 |
| Anti-human CD34 | Biolegend | Clone 561 |
| Anti-human CD45 | Biolegend | Clone HI30 |
| Anti-mouse CD45 | Biolegend | Clone 30-F11 |
| Anti-human CD45RA | Biolegend | Clone HI100 |
| Anti-human CD45RO | Biolegend | Clone UCHL1 |
| Anti-human CD56 | Biolegend | Clone HCD56 |
| Anti-human CD56 (for in vivo studies) | BD Biosciences | Clone B159 |
| Anti-human CD62L | Biolegend | Clone DREG-56 |
| Anti-human CCR7 | Biolegend | Clone G043H7 |
| Anti-human CTLA-4 | Biolegend | Clone BNI3 |
| Anti-human IFNγ | Biolegend | Clone 4S.B3 |
| Anti-human IL-2 | Biolegend | Clone MQ1-17H12 |
| Anti-human LAG-3 | Biolegend | Clone 11-C3C65 |
| Anti-human TIM-3 | Biolegend | Clone F38-2E2 |
| Anti-human TCRαβ | Biolegend | Clone IP26 |
| Anti-human TCRγδ | Biolegend | Clone B1 |
| Anti-human TNFα | Biolegend | Clone Mab11 |
| Anti-human CD137 (41-BB) | Biolegend | Clone 4B4-1) |
| TotalSeq-C Human Universal Cocktail, V1.0 | Biolegend | Cat. 399905 |
| Bacterial and virus strains | ||
| NEB Stable Competent E. coli (High Efficiency) | New England Biolabs | Cat. C3040I |
| LV pCCL-c-MNDU3-CD3D WT cDNA | This paper | Addgene |
| LV pCCL-c-MNDU3-CD3D A0 cDNA | This paper | Addgene |
| LV pCCL-c-MNDU3-CD3D c.202C>T-cDNA | This paper | Addgene |
| LV pCCL-c-MNDU3-CD3D c.274+5G>A | This paper | Addgene |
| LV pCCL-c-MNDU3-CD3Dc.275-2A>G | This paper | Addgene |
| Biological samples | ||
| Human CD34+ HSPCs | Charles River | Cat. BM34C |
| Human CD34+ HSPCs from CD3δ SCID Patient | Alberta Children’s Hospital | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| rhIL-2 | Miltenyi Biotec | Cat. 130-097-748 |
| rhFLT3L | Peprotech | Cat. 300-19 |
| rhIL-7 | Peprotech | Cat. 200-07 W |
| rhTPO | Peprotech | Cat. 300-18 |
| rhSCF | Peprotech | Cat. 300-07 |
| B27 supplement | Gibco | Cat. 17504-044 |
| TruStain FcX | Biolegend | Cat. 422302 |
| Purified Rat Anti-Mouse CD16/CD32 (Mouse BD Fc Block) | BD Biosciences | Cat. 553142 |
| DAPI | Life technologies | Cat. D1306 |
| AB serum | Gemini Bio-Products | Cat. 50-753-3011 |
| Ghost Dye Violet 510 | TONBO Biosciences | Cat. 13-0870-T100 |
| Indo-1 Dye, AM, cell permeant | ThermoFisher Scientific | Cat. I1203 |
| X-vivo-15 | Lonza | Cat. 04-744Q |
| RPMI | Lonza | Cat. 12-115Q |
| StemSpan SFEM | Stem Cell Technologies | Cat. 09650 |
| MethoCult H4435 Enriched | Stem Cell Technologies | Cat. 04445 |
| Q5 Hot Start High-Fidelity DNA Polymerase | New England Biolabs | Cat. M0493L |
| CleanCap Reagent AG | Trilink | Cat. N-7113 |
| N1-Methylpseudouridine-5’-Triphosphate | Trilink | Cat. N-1081 |
| LiCl Precipitation Solution | ThermoFisher Scientific | Cat. AM9480 |
| Poloxamer F108 (Kolliphor P 338) | BASF | Cat. 9003-11-6 |
| Recombinant Cas9-NRTH protein | QB3 Macrolab: UC Berkeley | N/A |
| Recombinant Hifi Cas9 protein | QB3 Macrolab: UC Berkeley | N/A |
| Ionomycin, Calcium Salt | ThermoFisher Scientific | Cat. I24222 |
| Critical commercial assays | ||
| CFSE proliferation assay | Biolegend | Cat. 423801 |
| antiCD3/CD28 beads | ThermoFisher Scientific | Cat. 11161D |
| Zombie UV™ Fixable Viability Kit | Biolegend | Cat. 423107 |
| Dead cell removal kit | Miltenyi Biotec | Cat. 130-090-101 |
| CD8+ T cell Isolation Kit | Miltenyi Biotec | Cat. 130-09-495 |
| Intracellular Fixation & Permeabilization Buffer Set | eBiosciences | Cat. 88-8824-00 |
| CD34 Microbead Kit, human | Miltenyi Biotec | Cat. 130-046-702 |
| ddPCR Supermix for Probes | Biorad | Cat. 186-3026 |
| NEBuilder HiFi DNA Assembly Cloning Kit | New England Biolabs | Cat. E5520S |
| Gibson Assembly Cloning Kit | New England Biolabs | Cat. E5510S |
| Purelink Genomic DNA Mini Kit | ThermoFisher Scientific | Cat. K182002 |
| HiScribe® T7 High Yield RNA Synthesis Kit | New England Biolabs | Cat. E2040S |
| QIAquick PCR Purification Kit | Qiagen | Cat. 28104 |
| rhAmpSeq CRISPR Library Kit | IDT | Cat.10007317 |
| Ficoll Pacque Plus | GE Healthcare | Cat. 17144003 |
| Q5 Site-Directed Mutagenesis Kit | New England Biolabs | Cat. E0554S |
| P3 Primary Cell 4D-Nucelofector X Kit S | Lonza | Cat. V4XP-3032 |
| SE Cell Line 4D-Nucleofector X Kit | Lonza | Cat. V4XC-1024 |
| SF Cell Line 4D-Nucleofector X Kit | Lonza | Cat. V4XC-2012 |
| AMPure XP Reagent | Beckman Coulter | Cat. A63881 |
| NucleoSpin Tissue XS, Micro kit for DNA from cells and tissue | Macherey Nagel | Cat. 740901.50 |
| Gentra Puregene Buccal Cell Kit | Qiagen | Cat. 158845 |
| KAPA HTP Library Preparation Kit | KAPA Biosystems | Cat. KR0426 |
| Deposited data | ||
| CITE-seq data | This paper | GSE220611 |
| rhAmpSeq | This paper | NCBI SRA Accession Number: PRJNA914907 |
| HTS Editing data | This paper | NCBI SRA Accession Number: PRJNA914907 |
| Experimental models: Cell lines | ||
| MS5-hDLL4 | Crooks Lab | N/A |
| Jurkat | ATCC | TIB-152 |
| K562 | ATCC | CCL-243 |
| HEK293T-CHEDAR | Kohn Lab | N/A |
| HT-29 | ATCC | HTB-38 |
| Experimental models: Organisms/strains | ||
| Mouse: NOD.Cg-KitW-41J Tyr + Prkdcscid Il2rgtm1Wjl/ThomJ | Jackson Laboratories | RRID:IMSR_JAX:026622 |
| Oligonucleotides | ||
| Gen_CD3D(C202T)_sgRNA: 5’-CGAGGAATATATAGGTGTAA-3’ | SYNTHEGO | N/A |
| Gen_CD3D(C202T)_ssODN: 5’-ACCCAAAGGGTTCAGGAAGCACGTACTTCGATAATGAACTTGCACGGTAGATTCTTTGTCCTTGTATATATCTGTCCCATTACATCTATATATTCCTCATGGGTCCAGGATGCGTTTTCCCAGGTC-3’ | IDT | N/A |
| CD3DF Forward Primer: 5’-CTTGGTGCAGATCAAAGAGC - 3’ | IDT | |
| CD3DR Reverse Primer: 5’-CTGGTGATGGGCTTGCCAC -3’ | IDT | N/A |
| Therapeutic_sgRNA1: 5’-TTCCTCGTGGGTCCAGGATG-3’ | SYNTHEGO | N/A |
| Therapeutic_sgRNA2: 5’-TATTCCTCGTGGGTCCAGGA-3’ | SYNTHEGO | N/A |
| Therapeutic HDR sgRNA: 5’-TTACACCTATATATTCCTCG-3’ | SYNTHEGO | N/A |
| Therapeutic_HDR_ssODN: 5’-TGCAATACCAGCATCACATGGGTAGAGGGAACGGTGGGAACACTGCTCTCAGACATTACAAGACTGGACCTGGGAAAACGCATCCTGGATCCACGAGGAATATATAGATGTAATGGGACAGATATA-3’ | IDT | N/A |
| CD3D_LibF_Forward Primer: 5’-ACACGACGCTCTTCCGATCTNNNNGAGGACAGAGTGTTTGTGAA -3’ | IDT | N/A |
| CD3D_LibR_Reverse Primer: 5’-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTCTCTAGCCAGAAAGTTCTCAC -3’ | IDT | N/A |
| ddPCR P5 Primer: 5’-AATGATACGGCGACCACCGA -3’ | IDT | N/A |
| ddPCR P7 Primer: 5’-CAAGCAGAAGACGGCATACG -3’ | IDT | N/A |
| Transcription Forward Primer: 5’-TCGAGCTCGGTACCTAATACGACTCACTATAAGG -3’ | IDT | N/A |
| Transcription Reverse Primer: 5’-TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTCTTCCTACTCAGGCTTTATTCAAAGACCA -3’ | IDT | N/A |
| GUIDE-seq_dsODN_Forward: 5’/Phos/GTTTAATTGAGTTGTCATATGTTAATAACGGT*A*T -3’ | IDT | N/A |
| GUIDE-seq_dsODN_Reverse: 5’/Phos/ATACCGTTATTAACATATGACAACTCAATTAA*A*C -3’ | IDT | N/A |
| Psi U5 Forward Primer: 5’ AAG TAG TGT GTG CCC GTC TG 3’ | IDT | N/A |
| Psi U5 Reverse Primer: 5’ CCT CTG GTT TCC CTT TCG CT 3’ | IDT | N/A |
| Psi Probe: 5′ FAM-CCC TCA GAC-ZEN*-CCT TTT AGT CAG TGT GGA AAA TCT CTA G-IBFQ** 3′; *ZEN = internal modification from IDT-Integrated DNA Technologies, **IBFQ = Iowa Black FQ | IDT | N/A |
| SDC4 Forward Primer: 5’ CAG GGT CTG GGA GCC AAG T 3’ | IDT | N/A |
| SDC4 Reverse Primer: 5’ GCA CAG TGC TGG ACA TTG ACA 3’ | IDT | N/A |
| SDC4 Probe: 5′ HEX-CCC ACC GAA-ZEN*-CCC AAG AAA CTA GAG GAG AAT-IBFQ** 3′; *ZEN = internal modification from IDT-Integrated DNA Technologies, **IBFQ = Iowa Black FQ | IDT | N/A |
| CD3D_LV_Forward Primer: 5’-GGAGCACAGCACCTTCCTG -3’ | IDT | N/A |
| CD3D_LV_Reverse Primer: 5’-CCCAGTTGCCGCCCAGG -3’ | IDT | N/A |
| Recombinant DNA | ||
| pCMV-ABE8e-NRTH | This paper | N/A |
| pCMV-ABE8e-VRER | This paper | N/A |
| pCMV-ABE8e-xCas9(3.7) | This paper | N/A |
| pCMV-ABE8e-NG | Addgene | Plasmid 138491 |
| pCMV-ABEmax-NRTH | Addgene | Plasmid 136922 |
| CCL vector backbone | Addgene | Plasmid 12251 |
| Software and algorithms | ||
| FlowJo | Tree Star Inc. | https://www.flowjo.com/solutions/flowjo |
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Cell Ranger v7.0.0 | 10X genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| Seurat 4.2.0 | Satija Lab | https://satijalab.org/seurat/ |
| cb_sniffer | sridnona | https://github.com/sridnona/cb_sniffer |
| MAST algorithm | RGLab | https://github.com/RGLab/MAST |
| fgsea v1.22.0 (GSEA) | Alexey Sergushichev | https://github.com/ctlab/fgsea/ |
| msigdbr v7.15.1 (MSigDB) | Igor Dolgalev | https://CRAN.R-project.org/package=msigdbr |
| enrichplot v1.16.2 (GSEA visualization) | Yu Lab | https://yulab-smu.top/biomedical-knowledge-mining-book/ |
| CasOFFinder.23 | BAE Lab | http://www.rgenome.net/cas-offinder/ |