

Abstract
In contrast to previous steady-state analyses of the O2-responsive transcriptome, here we examined the dynamics of the response to short-term anaerobiosis (2 generations) in both catabolite-repressed (glucose) and derepressed (galactose) cells, assessed the specific role that Msn2 and Msn4 play in mediating the response, and identified gene networks using a novel clustering approach. Upon shifting cells to anaerobic conditions in galactose medium, there was an acute (∼10 min) yet transient (<45 min) induction of Msn2- and/or Msn4-regulated genes associated with the remodeling of reserve energy and catabolic pathways during the switch from mixed respiro-fermentative to strictly fermentative growth. Concomitantly, MCB- and SCB-regulated networks associated with the G1/S transition of the cell cycle were transiently down-regulated along with rRNA processing genes containing PAC and RRPE motifs. Remarkably, none of these gene networks were differentially expressed when cells were shifted in glucose, suggesting that a metabolically derived signal arising from the abrupt cessation of respiration, rather than O2 deprivation per se, elicits this “stress response.” By ∼0.2 generation of anaerobiosis in both media, more chronic, heme-dependent effects were observed, including the down-regulation of Hap1-regulated networks, derepression of Rox1-regulated networks, and activation of Upc2-regulated ones. Changes in these networks result in the functional remodeling of the cell wall, sterol and sphingolipid metabolism, and dissimilatory pathways required for long-term anaerobiosis. Overall, this study reveals that the acute withdrawal of oxygen can invoke a metabolic state-dependent “stress response” but that acclimatization to oxygen deprivation is a relatively slow process involving complex changes primarily in heme-regulated gene networks.
In response to environmentally stressful conditions, all organisms mount defensive strategies essential for survival. The first line of defense often consists of low-molecular-weight compounds (e.g., trehalose) and proteins (e.g., chaperones), which afford immediate protection against a variety of environmental insults. Activation of stress signaling pathways, in turn, leads to the transcription of genes that serve additional protective roles. These can include both stress-specific genes (e.g., oxidative stress genes) and a common set involved in a more general response to stress. Genomic studies in Saccharomyces cerevisiae have revealed a large, overlapping set of genes that are differentially expressed in response to variety of environmental insults, including temperature shock, osmotic stress, oxidative stress, low pH, glucose or nitrogen starvation, and DNA-damaging agents (12, 28, 63, 70). These studies identified Msn2 and Msn4 as key regulators of both the environmental stress response (28) and common environmental response (12). Here, we examined the remodeling of the transcriptome during acclimatization to short-term anaerobiosis under different metabolic states (catabolite repressed or derepressed) and assessed the specific role these factors play in mediating the response.
Msn2 and Msn4 are Cys2His2 zinc finger proteins that activate the expression of a number of stress-inducible genes (60, 73). Although often considered to be functionally redundant in part because they activate gene expression through a common site, the stress response element (STRE), they are differentially regulated and may play distinct roles under different environmental conditions (26, 29). Their activity is regulated by their subcellular localization, residing in the cytosol under standard growth conditions and translocating to the nucleus under stressful conditions (30). This translocation is thought to be controlled by their phosphorylation state, which may dictate their interactions with cytosolic anchoring proteins (Bmh2 and Bmh1) (9).
Several condition-specific signaling pathways are thought to influence the activity of Msn2 and/or Msn4 (hereafter referred to as Msn2/4 for simplicity) and other factors responsible for the environmental stress response (reviewed in reference 29). These include the target of rapamycin (TOR) pathway (9), the protein kinase C-mitogen-activated protein (MAP) kinase pathway (34, 44, 55, 65), the high-osmolarity glycerol-MAP kinase cascade (70), the Snf1 protein kinase pathway (61), and the protein kinase A-MAP kinase pathway (30, 79, 80). Additional signaling pathways that respond to changes in other environmental parameters may also feed in to the regulation of Msn2/4 and the general stress response. Although seemingly complex, such a multiplex of signaling cascades is likely required for dictating specificity in the cellular response to an environment in which changes in a multitude of different parameters (e.g., temperature, osmolarity, pH, O2 availability, etc.) can occur simultaneously.
Previous studies of the oxygen-responsive transcriptome in yeast have focused on genes that are differentially expressed between steady-state aerobic and anaerobic conditions (8, 51, 68, 77) and the gene networks controlled by key regulators, such as Rox1 (8, 27, 51, 78), Upc2 (87), Hap1 (8, 78), and others (8). Although these studies have helped to identify O2-responsive genes and the role these factors play in controlling expression, the dynamics of remodeling activity elicited by acute changes in oxygen availability remain largely unexplored. In this study, we examined dynamical changes in the transcriptome associated with the acute withdrawal of oxygen, compared the response under different metabolic states (respiro-fermentative and fermentative), and identified the gene networks involved using a novel clustering approach.
In regard to Msn2/4, although our previous analyses indicated little overlap between genes that are differentially expressed in response to O2 deprivation and those activated by other environmental insults, a substantive fraction of anaerobically induced genes are annotated under “cell stress” and little is known about their regulation. This poor overlap could simply reflect differences in the time courses examined (e.g., acute versus chronic) or, alternatively, O2 deprivation may activate entirely different gene networks than those activated by other environmentally stressful conditions. To distinguish between these possibilities and explore the dynamics of the response, we compared genomewide expression profiles obtained with 70-mer oligonucleotide microarrays between an msn2/4 strain and its isogenic parent during short-term acclimatization to anaerobiosis. To aid in the identification of gene networks, we present a novel method for determining a clustering approach (algorithm and distance metric) and number of clustering divisions that results in the most nonrandom configuration of transcription factor motifs (TFMs) among gene clusters.
MATERIALS AND METHODS
Yeast strains, media, and growth conditions.
The following yeast strains were used: JM43 (MATα leu2-3,112 his4-580 trp1-289 ura3-52 [rho+]) (wild type) and the isogenic strain KKY8, which contains msn2::LEU2 and msn4::KanMX disruptions. KKY8 was constructed by transforming JM43 with a BglII fragment from pmsn2::LEU2 and then transforming the resulting strain with a BamHI-EcoRI fragment from pmsn4::KanMX (plasmids were a gift from R. S. Zitomer). Gene disruptions were confirmed by Southern blot analysis.
Cultures were grown in a semisynthetic galactose or glucose medium containing Tween 80, ergosterol, and silicon antifoam (SSG-TEA and SSD-TEA, respectively) (10). Amino acids and nucleotides were added, as appropriate, at a concentration of 40 mg/liter. Liquid precultures were grown at 28°C on a shaker (200 rpm) and kept in early to mid-exponential growth phase (<100 Klett units, optical density at 600 nm <1.0) for 3 to 4 days prior to inoculating a New Brunswick BioFlo III fermentor (3.5-liter working volume) (51). The fermentor was inoculated with an appropriate volume of preculture so as to yield an initial cell density of ∼0.2 Klett unit. The cultures were sparged with air (1.2 vol/vol of medium per min) for six generations. A sample (aerobic control) was then harvested before switching the gas to 2.5% CO2 in O2-free N2 for 2 generations of anaerobic growth. This procedure ensured the cell density upon final harvesting was ∼60 Klett units. The dissolved O2 concentration was calculated from the output current of a 12-mm Ingold polarographic O2 sensor, which was calibrated for 0% O2 saturation with Ingold sensor-checking gel and for 20.94% O2 with air-saturated medium. Corrections were made for temperature and barometric pressure (for SSG-TEA medium at 101 kPa and 28°C, 20.94% O2 = 238 μM O2).
To compare the response of the wild-type and msn2/4 strains, samples were harvested at the same time points (0, 10, 20, 45, and 480 min) after the shift to anaerobiosis as well as after the same relative number of cell mass doublings (0, 0.04, 0.08, 0.19, and 2 generations) as assessed by turbidity measurements (Klett meter). As discussed in Results, only the latter comparison provides good alignment of the temporal signatures between the strains. Thus, this same “generation-specific” sampling regimen (0, 0.04, 0.08, 0.19, and 2 generations) was used for comparing the effects of carbon source (galactose or glucose) in the wild-type strain. For sampling, cells were harvested using a vacuum filtration apparatus (11) onto AcetatePlus membranes (GE Osmonics, Minnetonka, MN). The filtered cells were washed with either sterile oxygenated or deoxygenated water, as appropriate, flash-frozen in liquid N2 within one minute of initiating the sampling, and stored at −80°C for later RNA isolation (11). At least three independent fermentor experiments were completed for each strain and growth condition examined.
RNA extraction, cDNA synthesis, and microarray hybridization.
Total RNA was extracted from the filtered cells using hot phenol as described previously (11). Thirty micrograms of total RNA was used for first-strand cDNA synthesis and microarray target preparation following previously described methods (32) with the following modifications. For cDNA synthesis, Superscript III reverse transcriptase (Invitrogen, Carlsbad, CA) was used with a 4:1 ratio of amino-allyl-dUTP to dTTP so as to yield about 1 dye molecule per 15 to 20 nucleotides. QIAquick mini-PCR purification columns (QIAGEN, Valencia, CA) were used for cDNA purification and unincorporated dye removal. For the latter, an additional first wash step with 750 μl of 35% guanidine hydrochloride was included for more efficient free-dye removal. The amount of cDNA obtained and dye (Cy3 or Cy5) incorporated was quantified using wavelength-scanning (750 to 200 nm) spectrophotometry with a 50-μl microcuvette.
Fluorescent cDNA targets were dried under vacuum (SpeedVac) and resuspended in an appropriate volume of hybridization buffer (50% formamide, 5× SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate], 0.1% sodium dodecyl sulfate, and 0.5 μg/μl of tRNA) so as to yield equal amounts of Cy3 (query cDNA) and Cy5 (reference cDNA) dyes. A reference design was used for all microarray hybridizations. The reference consisted of a pool of equal masses of RNA from each time point sampled. The 80-μl samples (2.5 μl per cm2 of coverslip) were denatured at 95°C for 2 min, centrifuged (16,000 × g for 1 min), and immediately applied to the microarrays. The arrays were sealed in a humidified aluminum chamber and placed in a 42°C water bath for 16 h.
After hybridization, the arrays were washed individually with mild agitation in 50 ml each of 2× SSC and 0.1% SDS at 42°C for 5 min, 0.2× SSC at room temperature for 1 min, and 0.1× SSC at room temperature for 1 min. The slides were dried by centrifugation (1,000 rpm for 5 min) and scanned within 2 h of processing using a GenePix 4000B confocal laser scanner (Axon, Union City, CA). The custom microarrays consisted of QIAGEN/Operon's yeast genomic 70-mer oligonucleotide set (QIAGEN, Valencia, CA; version 1.1) spotted in duplicate at a concentration of 20 μM in 150 mM sodium phosphate (pH 8.5), Arabidopsis oligonucleotide spike controls (Stratagene SpotReport, La Jolla, CA) spotted in quadruplicate, and 10 human and 10 yeast oligonucleotide negative controls spotted in duplicate. The oligonucleotides were printed on Codelink slides (Amersham, Piscataway, NJ) by Microarrays, Inc. (Nashville, TN). Postprint processing was conducted according to the manufacturer's recommendations.
Microarray and statistical analyses.
GenePix Pro software (v4.1) was used for spot identification and fluorescence intensity quantification. Spots with aberrant measurements due to array artifacts or of poor quality were manually flagged and removed from analyses. Local background fluorescence was subtracted from the median Cy3 and median Cy5 fluorescence intensity values. Any resulting negative intensity values, which accounted for <0.5% of the total observations here, were set to zero, and a constant of one fluorescent unit was then added to all intensity values. Outliers were identified using SAS (SAS Institute, Cary, NC) as those observations whose fluorescence intensity deviated significantly (P < 0.01) from the average of all six observations (duplicate spots on each of three independent replicate slides per treatment).
After removing outliers, the log2 Cy3 intensity (query cDNA) for all observations on a slide was normalized against that of the log2 Cy5 intensity (reference cDNA) using locally weighted linear regression (Loess) in SAS. The linearity of the resulting Cy3 and Cy5 intensities across each slide was checked against the Arabidopsis spike controls (fluorescence intensity versus spike mRNA amount [ranging from 0.02 to 2 ng]) and a slope adjustment was made if the latter deviated significantly (P < 0.01) from a value of 1 (zero occurrences in this study). The log2 (Cy3/Cy5) ratio for each spot was calculated, and the mean log2 (Cy3/Cy5) ratio across all observations on a slide was normalized to a value of zero. The mean of the normalized log2 (Cy3/Cy5) ratio for each gene was then calculated by averaging the duplicate observations on each slide and pooling replicate slides by strain, medium, and sampling time.
Statistical analyses were performed as a three-factor analysis of variance (ANOVA) using the SAS MIXED procedure with repeated measures. The factors were medium (galactose or glucose), strain (wild-type or msn2/4), and time (0, 0.04, 0.08, 0.19 or 2 generations after the switch to anaerobiosis). A step-down Bonferroni post hoc P value adjustment was used to minimize false positives. Unless otherwise noted, all P values reported were adjusted using this procedure. Postmodel analyses included promoter searches (−1 to −800 bp excluding upstream ORFs) using regulatory sequence analysis (RSA) tools (83) or other freely available, web-based bioinformatic tools. Note that the full data set has been deposited at GEO with accession number GSE1879 (http://www.ncbi.nlm.nih.gov/projects/geo/query/acc.cgi?acc=GSE1879), and the results of all statistical comparisons (see items S1 to S5 in the supplemental material) are available.
Data clustering and gene network discovery.
To aid in the identification of the gene networks, we developed a method that uses two metrics to assess the quality of clustering results obtained with different algorithms and distance metrics and determine an optimal number of clusters (K) based on the distribution of transcription factor motifs (TFMs): consensus share (CS), the percentage of genes that are consistently grouped together over multiple runs of a clustering algorithm, and the motif configuration statistic (MCS), a novel metric that determines which clustering approach and cluster number (K) results in the most nonrandom configuration of transcription factor motifs (TFMs) among gene clusters.
To begin, the temporal profiles in gene expression were clustered ten times with different algorithms (K-means, K-medoids, or self-organizing map [SOM]) and distance metrics (Euclidean, Manhattan, Sup, or correlation) using a range of K values, in this case 2 to 50. These metrics were then calculated for each value of K and clustering approach (algorithm and distance metric) examined. Rather than using mean expression values from the microarrays, or models for the inclusion of the variance estimate (38, 89), individual replicates (n ≥ 3) were used as features in clustering. Genes that were not consistently clustered together over replicate runs of a given clustering approach were placed into a separate group and excluded from MCS calculations. The configuration of 1,813 transcription-factor consensus binding sequences (see Table S1 in the supplemental material), taken from both experimental and comparative phylogenic studies (15, 46, 54, 69, 76, 91), was assessed among the gene clusters by calculating the MCS for each.
The MCS metric is presented using the following definitions. Let n = (n1… ni) be a vector of cluster sizes and m = (m1… mk) be a vector of motif counts per cluster (motif configuration). The vector of relative cluster sizes is defined as q = (q1… qk), where
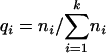 |
For the null hypothesis we assume that m came from the multinomial distribution M(q), i.e., larger clusters have higher motif counts. In this case the value of the probability mass function for each motif configuration m can be calculated as:
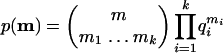 |
(1) |
and the MCS is then given by the probability P(m) = P[p(r) ≤ p(m)], where r is a sample drawn from M(q). Because it is not feasible to compute the statistic exactly, we use the following algorithm to approximate it. Starting with counter c = 0, we repeat the following steps 106 times: distribute the total motif count uniformly among genes; calculate the vector of motif counts per cluster r = (r1… rk) by grouping gene counts per cluster; compute p(r) according to equation 1; if p(r) ≤ p(m), then increase counter c. The approximation of MCS P value is given by c/N. An average MCS value is calculated for all motifs in the list. By comparing the values of MCS and CS for different clustering approaches (both algorithms and distance metrics), we can determine which approach consistently uncovers the most structure from the expression profiles (highest CS) and which value of K yields the most nonrandom configuration of TFMs (lowest MCS) among gene clusters.
RESULTS
Dynamics of the response to short-term anaerobiosis in galactose medium.
In contrast to previous steady-state analyses of the O2-responsive transcriptome, here we examined dynamical changes elicited by the acute withdrawal of O2 in sparged batch cultures of yeast. To minimize the impact of changing medium components in batch, cultures were maintained at low cell densities (<60 Klett units) and harvested in early log-phase growth. Inoculation volumes were adjusted to achieve identical cell densities among treatments (strains or media) for all samples compared. Finally, cells were allowed to acclimate to fermentor conditions for six generations before harvesting the aerobic control and switching the sparge gas from air to O2-free N2.
By design these experiments examined the dynamics of the genomic response to a rapid change in O2 availability during the transition from pseudo-steady-state aerobiosis to anaerobiosis, and, out of necessity, cultures were grown in batch because of the rapid sampling regimen and quantity of cells required at each time point using our 3.5-liter fermentation apparatus. As shown in Fig. 1, even with relatively high gas sparge rates (1.2 vol gas/vol of medium per minute) several minutes are required to purge O2 from the medium after switching the gas from air to 2.5% CO2 in O2-free N2. Previous studies of the O2 dependency for transcription of aerobic and hypoxic genes suggest 1 μM O2 is a critical threshold (50), with maximal expression of aerobic genes above this value and that of hypoxic genes below it. Thus, the first sampling in this study (10 min) roughly corresponds to the time at which this oxygen concentration is achieved.
FIG. 1.

Change in O2 concentration over the first 10 min of the shift to anaerobiosis. The change in dissolved O2 concentration (μM) in the fermentor is plotted as a function of time over the first ten minutes after switching the sparge gas from air to 2.5% CO2 in O2-free N2. The O2 concentration was calculated from the dissolved oxygen level measured with a 12-mm Ingold polarographic O2 sensor and is based upon the solubility of O2 in the media at 28°C and ambient barometric pressure.
Statistical analyses (mixed ANOVA with repeated measures) of the genomic response revealed 938 genes whose transcript levels deviated significantly (P < 0.01, step-down Bonferroni, n ≥ 3) from that of the aerobic control (time zero) after shifting the wild-type strain to anaerobic conditions in galactose medium (see item S1 in the supplemental material). The step-down Bonferroni P value adjustment was used to minimize the false discovery rate and is a stringent cut of the data, with a value of 0.01 roughly equivalent to a raw P value of 10−8. Of the 938 genes identified, 387 were significantly up-regulated at one or more time points after the shift to anaerobiosis (0, 10, 20, 45, and 480 min), 533 were down-regulated, and 18 genes were both up- and down-regulated at different time points after the shift. Figure 2 shows the dynamics of the anaerobic response presented as the number of genes that were differentially expressed (P < 0.01) for the first time at each time point (black bars) and the total number of differentially up- and down-regulated genes in each sample (combined height of black and gray bars). From this figure it is clear that the response to anaerobiosis in galactose medium is biphasic, consisting of a large set of genes (599 of 938) that show an acute (10 to 20 min) yet transient (≤45 min) response followed by a smaller set of genes (277 of 938) that exhibit a delayed (>45 min), more chronic (2 generations) response. Thus, the majority of genes that are differentially expressed during the shift to anaerobiosis exhibit a transient response over the first ∼45 min.
FIG. 2.

Dynamics of oxygen-responsive gene induction and repression during short-term anaerobiosis in galactose medium. The number of genes that responded significantly (P < 0.01) to the shift in O2 concentration in the wild-type strain (JM43) grown in galactose medium (SSG-TEA) is plotted as a function of both time and relative number of doublings in cell mass (generations) after the shift to anaerobiosis. Genes are divided into those that were significantly up-regulated and those that were significantly down-regulated. Black bars indicate the number of genes that were identified for the first time at that time point to exhibit a significant change in expression from that of the aerobic control (time zero). Gray bars indicate the number of genes that were differentially expressed relative to the aerobic control yet had already been identified at an earlier time point to have responded significantly to the shift in O2 concentration. The combined height of the black and gray bars indicates the total number of genes at each time point that showed a significant change in expression relative to the aerobic control.
Gene network identification.
To aid in the identification of the transcriptional networks, we developed a method that uses quality assessment metrics to choose an appropriate clustering approach (algorithm and distance metric) and number of clustering divisions (K) for analyses. The first metric is CS, which is the percentage of genes that are consistently grouped together over multiple runs of a clustering algorithm. It provides a measure of both the robustness and extent of structure that can be recovered from unbiased clustering of the gene expression profiles with a given approach and number of clustering divisions. The second metric is a novel one we call the MCS. It estimates the probability that the observed configuration of a given transcription factor motif (TFM) among gene clusters arose from a multinomial distribution by chance alone. The lower the MCS P value, the more nonrandom the distribution of a TFM among gene clusters. By calculating the average value of this metric using a comprehensive list of TFMs (see Table S1 in the supplemental material), we can determine what clustering approach and K value results in the most nonrandom configuration of TFMs among gene clusters (lowest MCS value).
Figure 3 compares CS (upper panel) and MCS (lower panel) as a function of cluster number for three different algorithms: SOM with one-dimensional (1D) ring topology (solid line), K-means (dashed line), and K-medoids (dotted line). The results were obtained from unbiased clustering of the temporal profiles of the 938 differentially expressed genes (P < 0.01) in the wild-type strain grown in galactose media (SSG-TEA) using Pearson correlation as the distance metric and replicates as features. From this figure it is clear that the SOM algorithm finds far more structure in the gene expression profiles than either K-means or K-medoids as evidenced by higher CS values for all K > 2. Moreover, the SOM algorithm partitions the gene expression profiles in a manner that results in a more nonrandom configuration of TFMs among clusters (lower MCS) for nearly all values of K examined (2 to 50). We interpret the inferior performance of both K-means and K-medoids to be due, in part, to the fact these algorithms do not use topological information.
FIG. 3.

Comparison of the performance of different clustering algorithms in terms of consensus share and the motif configuration statistic as a function of cluster number (K). Three clustering algorithms were evaluated in terms of the resulting gene-to-cluster consensus (consensus share) and the motif configuration statistic (MCS) as a function of cluster numbers (K = 2 to 50) using Pearson correlation as the distance metric: SOM (Kohonen map) algorithm with 1D ring topology (solid line), K-means (dashed line), and K-medoids (dotted line). Consensus share (upper panel) is the percentage of genes that were consistently grouped together over ten runs of the algorithm. An average MCS P value (lower panel) for 1,813 transcription factor consensus binding motifs (TFMs) was calculated using a compiled list of both experimentally verified and putative motifs (see Table S1 in the supplemental material) as described in Materials and Methods. Lower MCS values indicate more nonrandom configurations of TFMs among gene clusters. The clustered data are individual replicate expression levels of all genes that significantly (P < 0.01) responded to the shift to anaerobiosis in SSG-TEA (galactose) medium.
Topology defines the neighborhood relation between clusters, which depends on dimensionality and configuration of the space in which clusters are located. Most clustering methods, including K-means and K-medoids, use zero-dimensional or discrete topologies in which different clusters are unrelated to each other. SOM is one of few algorithms that allow for topologies of higher dimensions. Although the true topology of the transcriptional network is not known, the fact that 1D ring results in much higher CS and lower MCS values than other topologies we examined, including 1D string, 2D surface, and the more commonly used 2D torus (data not shown), suggests this topology more correctly approximates the underlying structure.
In interpreting the results presented in Fig. 3, it is important to realize that genes were clustered based solely on temporal changes in their expression (unbiased clustering) and these quality metrics (CS and MCS) were calculated from the results obtained. As presented, the MCS is a global metric that examines the configuration of all TFMs provided among gene clusters to determine which clustering approach and K value results in the most nonrandom configuration of TFMs among gene clusters (lowest MCS). Although MCS can be approximated by other, computationally more tractable, methods (e.g., Chi-square fitness test), we developed the generally more applicable Monte-Carlo sampling approach given that our motif analyses frequently did not meet the central limit theorem (CLT) criterion for normal approximation, i.e., that each cluster contain at least 5 expected occurrences of each motif provided in the TFM list. The resolution of this approach depends on the performance of the algorithm in terms of its ability to partition genes into coregulated groups based upon expression profiles alone and, second, on the use of a TFM list that includes all motifs that are responsible for the observed expression differences.
In regard to the latter, we use a comprehensive list of 1,813 consensus sequences (see Table S1 in the supplemental material) taken from both experimental and comparative phylogenic studies (15, 46, 54, 69, 76, 91). Although the list includes experimentally unverified and thus perhaps dubious motifs, their inclusion should have little effect on the choice of an appropriate clustering approach and K value; such motifs or ones that are “inactive” under the experimental conditions examined are expected to have a random distribution among network defined clusters, and, thus, their inclusion should have little influence on the average value of MCS. The lower the average MCS value for all TFMs the more biased the distribution of those that are most likely to be responsible for the observed differences in expression profiles.
From the clustering quality assessment shown in Fig. 3, we chose to further analyze the gene networks recovered with the SOM algorithm for K = 17 as this yields the lowest MCS P value (0.37) while retaining high consensus share (94%). Panel D in Fig. 4 shows the clustered expression profiles as heat maps for the 938 genes that responded significantly (P < 0.01) to the shift to anaerobiosis in the wild-type strain grown in galactose medium. Because of the 1D ring topology, expression profiles of genes in adjacent clusters are most similar to each other, with cluster 17 to cluster 1 serving to close the ring. The 54 genes shown in cluster 0 are those that were not consistently grouped together over ten runs of the SOM algorithm and were omitted from the MCS calculations. From this figure it is clear that the algorithm nicely divides the temporal signatures into those that are primarily down-regulated (clusters 1 to 9) from those that are up-regulated (clusters 10 to 17), with further partitioning based upon differences in the timing of the response. Comparison of these temporal signatures to those obtained in glucose medium (Fig. 4A to C) and to an msn2/4 strain (F to H) will be discussed in separate sections below.
FIG. 4.

Heat maps and statistical comparisons of oxygen-responsive genes in a wild-type strain grown in both galactose and glucose media and in an msn2/4 strain grown in galactose. The temporal profiles of genes whose transcript levels responded significantly (P < 0.01) to the shift to anaerobiosis (wild-type strain, SSG-TEA) were clustered using an SOM algorithm with 1D ring topology (K = 17). Panel D is a heat map of the temporal changes in gene expression relative to the aerobic control (time zero) plotted as a function of the number of generations of anaerobiosis (0, 0.04 0.08, 0.19, and 2). Green indicates down-regulated expression and red indicates up-regulated expression. Genes are sorted into 17 primary clusters as indicated in panel E. Cluster 0 contains genes that exhibited unstable cluster membership. Panel B shows the response of the same genes shown in panel D but in glucose (SSD-TEA) as opposed to galactose medium (wild-type strain). The black bars in panel A indicate genes that responded significantly (P < 0.01) to the shift in O2 concentration in glucose medium. Black bars in panel C indicate genes that showed a differential response (P < 0.01) in the two media (galactose versus glucose) as assessed by ANOVA. The purple bars in panel F identify putative Msn2/4-regulated genes, that is, genes that were anaerobically induced (P < 0.01) in the wild type but expressed at significantly (P < 0.01) lower levels in the msn2/4 strain and contained one or more STRE sites. Panel G is an expansion of the heat map shown in panel D for Msn2/4-regulated genes in the wild-type strain. Panel H is the expression profiles of the Msn2/4-regulated genes in the mutant strain, with yellow boxes indicating samples for which the transcript level was significantly (P < 0.01) lower in the msn2/4 strain compared to its wild-type parent.
Table 1 is a partial list of TFMs and functional categories (Munich Information Center for Protein Sequences [MIPS]) that were significantly [−log10 (p) ≥ 2] enriched in each of the gene clusters shown in panel D of Fig. 4 (see Table S2 in the supplemental material for gene cluster membership and a full listing of enriched TFMs, 6-mer oligonucleotides, and MIPS functional categories). Compared to other clustering methods we examined, this approach results in remarkable enrichment for TFMs and associated functional categories in clusters of genes in which a large fraction is known to be regulated by such factors.
TABLE 1.
Enrichment of transcription factor motifs (TFMs) and MIPS functional categories in clusters of genes that were differentially expressed in response to anaerobiosis in galactose medium (wild-type strain, SOM clustering with 1D ring topology)
| Cluster no. | na | TFM | Reference(s) | Percentage share |
P
|
MIPS functional category | Pd (genomic) | |
|---|---|---|---|---|---|---|---|---|
| In setb | Genomicc | |||||||
| 1 | 55 | MCB | 58, 91 | 65 | 8.9 | 7.9 | Cell cycle and DNA processing | 7.6 |
| SCB | 91 | 47 | 4.9 | 4.5 | DNA synthesis and replication | 7.0 | ||
| MBP1 | 46, 54, 76 | 45 | 10.4 | ≥38 | Nucleus | 4.8 | ||
| SWI4 | 46, 54, 76 | 45 | 2.7 | 3.3 | DNA recombination and DNA repair | 2.7 | ||
| SWI6 | 46, 54 | 45 | 8.4 | 11.1 | ||||
| 2 | 70 | RRPE | 46 | 37 | 4.9 | 8.8 | Translation | 2.3 |
| rRNA transcription | 2.1 | |||||||
| 3 | 78 | PAC | 46 | 74 | 11.7 | 19.1 | rRNA processing/transcription | ≥5.0 |
| RRPE | 46 | 41 | 7.2 | ≥38 | tRNA synthesis | 2.5 | ||
| ABF1 | 46, 54, 76, 91 | 46 | 2.1 | 2.7 | Polynucleotide degradation | 2.4 | ||
| 4 | 56 | PAC | 46 | 79 | 10.3 | 16.1 | Pyrimidine ribonucleotide metabolism | 2.7 |
| RRPE | 46 | 54 | 10.3 | 15.3 | rRNA transcription | 2.6 | ||
| ABF1 | 91 | 52 | 2.7 | 3.3 | ||||
| 5 | 51 | PAC | 46 | 57 | 2.7 | 5.8 | Nucleotide metabolism | 2.2 |
| RRPE | 46 | 31 | 2.3 | 4.5 | ||||
| 6 | 18 | |||||||
| 7 | 43 | HAP1 | 46, 76, 85, 91 | 30 | 3.6 | 5.0 | Mitochondrion/respiration/energy | ≥4.6 |
| INO2 | 46 | 30 | 2.8 | 2.7 | ||||
| 8 | 48 | MAC1 | 46 | 40 | 2.8 | 3.6 | Mitochondrion/respiration/energy | ≥5.7 |
| HAP2/3/4/5 | 46 | 33 | 2.3 | 3.4 | Mitochondrial transport | 5.4 | ||
| Homeostasis of cations | 4.0 | |||||||
| Cellular transport and transport mechanisms | 2.4 | |||||||
| 9 | 76 | Ribosome biogenesis | 11.3 | |||||
| Protein synthesis | 10.0 | |||||||
| Mitochondrion/respiration/energy | ≥3.7 | |||||||
| 10 | 30 | |||||||
| 11 | 74 | MSN2/4 | 91 | 68 | 2.9 | 4.4 | C-compound, carbohydrate transport/metabolism | ≥4.7 |
| MIG1 | 46 | 28 | 3.6 | 4.0 | Metabolism of energy reserves (glycogen, trehalose) | 3.1 | ||
| 12 | 91 | MSN2/4 | 91 | 64 | 2.4 | 3.9 | C-compound, carbohydrate transport/metabolism | ≥5.9 |
| Proteolytic degradation | 5.6 | |||||||
| Metabolism of energy reserves (glycogen, trehalose) | 4.9 | |||||||
| C-compound & carbohydrate utilization | 2.4 | |||||||
| 13 | 47 | INO2 | 76 | 87 | 2.1 | NSe | C-compound & carbohydrate metabolism | 3.1 |
| MIG1 | 46 | 19 | 2.1 | 2.7 | Metabolism of energy reserves (glycogen, trehalose) | 2.7 | ||
| Cell rescue, defense and virulence | 2.6 | |||||||
| 14 | 32 | GCR1 | 76 | 50 | 2.1 | 2.3 | Nitrogen & sulfur metabolism | 6.1 |
| PHO4 | 46, 76, 91 | 50 | 2.4 | 3.0 | Amino acid metabolism | 4.2 | ||
| ACE2 | 54, 91 | 44 | 2.5 | 2.9 | Lipid, fatty acid & isoprenoid metabolism activities | 2.7 | ||
| C-compound & carbohydrate utilization | 2.0 | |||||||
| 15 | 29 | MAC1 | 46 | 45 | 2.6 | 3.2 | Lipid, fatty acid & isoprenoid metabolism | 2.6 |
| UPC2 | 16 | 21 | 2.3 | 3.8 | Tricarboxylic acid pathway | 2.2 | ||
| Phosphate metabolism | 2.0 | |||||||
| 16 | 71 | ROX1 | 17 | 89 | 3.9 | 3.5 | Cell rescue, defense & virulence | 8.1 |
| UPC2 | 16 | 70 | 18.8 | 21.4 | Cell wall | 5.5 | ||
| ABC transporters | 2.4 | |||||||
| 17 | 15 | HAP2/3/4/5 | 54 | 33 | 2.2 | 2.7 | Lipid, fatty acid & isoprenoid metabolism | 8.1 |
| UPC2 | 16 | 27 | 2.1 | 3.1 | Endoplasmic reticulum | 4.6 | ||
| 0 | 54 | HAP2/3/4/5 | 46 | 11 | 2.4 | 3.7 | rRNA transcription | 2.6 |
| Mitochondrion/respiration/energy | ≥2.3 | |||||||
| Vitamin, cofactor & prosthetic group biosynthesis/metabolism | ≥2.2 | |||||||
n = number of genes in cluster.
Hypergeometric P [−log10(p)] for TFM enrichment relative to clustered genes (938).
Hypergeometric P [−log10(p)] for TFM enrichment relative to genome.
Hypergeometric P [−log10(p)] for MIPS functional category enrichment.NS, not significant.
NS, not significant.
Moreover, in over half of the clusters, 50% or more of the genes contain binding sites for the same transcription factor. Not all gene clusters were enriched for specific TFMs and, in several cases, the same TFM was significantly enriched in adjacent clusters (e.g., RRPE in clusters 2 to 5, PAC in 3 to 5, MSN2/4 in 11 to 12, and UPC2 in 15 to 17), which could occur if the number of divisions allowed were greater than that required to partition all true sets of coregulated genes. In all of these cases, however, additional TFMs were differentially enriched in adjacent clusters (see Table S2 in the supplemental material), accounting for the lower MCS P value that is afforded by additional clustering divisions. Based upon this differential enrichment, the slight differences in temporal signatures are predicted to be due to differential regulation.
An example where this is apparent is the partitioning of genes between clusters 1 and 2, where there is only a slight difference in the timing of the response (Fig. 4D) yet no overlap in the TFMs enriched in each. In general, this clustering approach can be used to screen for the predominate TFMs that are most likely to be responsible for the observed expression patterns. However, caution must be exercised in interpreting the results, as one cannot deduce from the mere presence of an enriched motif that changes in the activity of its associated transcription factor are directly responsible. Motifs that are “inactive” under the experimental conditions examined may also be enriched, especially in clusters that contain functional regulons that are controlled by a multiplex of transcriptional networks. Thus, the clustering results are best interpreted in the context of any additional knowledge of regulation and/or function that may be available.
In terms of the specific genes networks identified here, several TFMs and functional categories were predictably enriched based upon previous experimental studies (reviewed in references 50 and 92), including steady-state analyses of the O2-responsive transcriptome (51, 68, 77). These include HAP1 and HAP2/3/4/5 in clusters 7 and 8, respectively, which were significantly enriched for genes involved in respiration and energy metabolism; UPC2 in clusters 15 to 17, which were enriched for lipid, fatty acid, and isoprenoid metabolism, and cell wall genes; and ROX1 in cluster 16, which was enriched for cell wall and cell rescue, defense and virulence genes. Moreover, there was a temporal delay (≥45 min) in the response of these gene clusters, a predicted result based upon studies of their regulation by heme (reviewed in references 50 and 92). In addition to these motifs, others not previously associated with the anaerobic response were also enriched, notably in clusters of transiently responding genes (clusters 1 to 5 and 12 to 13). These included MCB, SCB, MBP1, SWI4, and SWI6 in cluster 1, which was significantly enriched for DNA synthesis/replication and the cell cycle genes; PAC and RRPE in clusters 2 to 5, which were enriched for rRNA processing genes; and MSN2/4 in clusters 12 to 14, which were enriched for carbohydrate and reserve energy metabolism genes.
Interestingly, many of the same motifs have been found in sets of genes that transiently respond to other environmental challenges (12, 28, 63, 66, 67, 70, 90). Indeed, a comparison of these transiently responding gene networks with those identified in the environmental stress response (12, 28) reveals substantive overlap: 51% (173 of 340) of those that were transiently down-regulated are also down-regulated in the environmental stress response (585 total) and 23% (59 of 259) of those that were transiently up-regulated are also up-regulated in the environmental stress response (283 total). In contrast, there is very little overlap between the environmental stress response and the set of genes that were more chronically down-regulated (1%) or up-regulated (2%) here. From these analyses it is clear that there is a bifunctional response to the shift in oxygen: one that consists of acutely yet transiently responding gene networks that appear to function in a general stress response and a more delayed and chronic one comprised of heme-responsive gene networks that are associated more directly with acclimatization to oxygen deprivation. In the following sections, we explore functional attributes of these transiently responding gene networks.
DNA synthesis and repair and the cell cycle.
Some of the first genes to respond to the O2 shift were transiently down-regulated (cluster 1 in Fig. 4D) and are associated with DNA synthesis/repair and the cell cycle (Table 1). Overall, there is remarkable enrichment for TFMs whose factors, i.e., Mbp1p-Swi6p (MBF) and Swi4p-Swi6p (SBF), are known to coregulate genes required for progression through the G1/S transition of the cell cycle (36, 39, 42). Genes associated with such functions include several for chromosomal replication initiation (CDC45, CDC54, TAH11, and YLR003C), DNA replication (POL1, -12, and -30, DBP2, and RNH201), concomitant repair (LRP1, RFA2, and RDH54), checkpoint function (RFA2 and MEC3), and chromosomal structure (IRR1, MCD1, ASF1, and PDS5). DNA synthesis and chromosome maintenance are closely linked and monitored at the replication fork and, thus many of the genes affected are associated with telomeres and their regulation (e.g., ESC8, DOT1, RIF1, and TBF1) or more than one process (e.g., CDC9, CTF18, HO, MSH2, RAD27, SMC5, TOF1, TOP1, TOS4, and TRF5). Genes that supply nucleotides for DNA synthesis were also transiently down-regulated (ADE1, -4, -12 and -17, GUA1, PRS3, RNR1, TRZ1, and URA2 and -7), as were salvage pathway genes (DCD1, HPT1 and URK1) and the uridine transporter (FUI1). Finally, genes associated with cytokinesis (e.g., CBF2, NKP1, SLK19, GIN4, and KCC4) (6) and bud site selection (STE20, RHO4, BEM3, PWP2, and SKG6) were also transiently down-regulated. Given the function of these genes, it would appear that the abrupt cessation in respiration during the shift to anaerobiosis in galactose medium results in a transient arrest in the cell cycle, predictably at the G1/S transition, i.e., before the cells commit to another round of DNA replication.
rRNA processing genes.
In conjunction with cell cycle-related genes, a large number involved in rRNA processing were also transiently down-regulated (clusters 2 to 5, Table 1). Remarkably, 96% of these have one or more PAC sites in their promoter and about half have RRPE sites (37, 69). Studies of environmental stress responses (12, 28) have observed a similar down-regulation of such genes, followed by a Rap1-mediated down-regulation of ribosomal protein genes. However, unlike the environmental stress response, few genes encoding structural components of the ribosomes (only RPL4A, RPP2B, and RPS2 at P < 0.01) were significantly affected here. Notably, these rRNA processing genes include the majority that encode the U3 snoRNP complex (small subunit processome; BUD21, DIP2, ECM16, IMP3 and -4, MMP10, NOC4, NOP14, PWP2, RRP9, SAS10, and UTP4, -5, -7, -8, -14, -18, -20, and -21), which functions in the earliest steps of ribosome biogenesis and is essential for pre-18S rRNA processing (20). In addition, several involved in 20S pre-rRNA processing (EMG1, ENP1, NOP7 and -14, RIO2, UTP22, and YGR272C), 35S primary transcript processing (DBP3, -6, -8, -9 and -10, DRS1, FAL1, MRD1, MTR3 and 4, PXR1, RNT1, and RRP3), large ribosomal subunit biogenesis, processing, and assembly (ARX1, IPI3, MAK16, NMD3, NOC2, NOG2, NOP16, NSA2, RLP7 and -24, RPF1 and -2, RRB1, SQT1, SSF1 and -2, TIF6, and YTM1), and a large group involved in general rRNA processing and modification (CGR1, EBP2, ENP2, ERB1, IFH1, IMP3 and -4, IPI1, KRR1, LCP5, MAK5, MPP10, MRT4, NHP2, NOC3, NOP6 and -12, NSR1, NUG1, PNO1, POP1, RCL1, REX4, RRP1, -8 and -9, RRS1, SPB1, TSR1, and YJL010C) were also similarly affected. Genes for tRNA synthesis and/or processing (ALA1, DUS1, FRS2, GCD14, KRS1, MES1, PUS7, TRM1, TRM10, TRM2, TRM82, and YKR079C) as well as subunits of RNA polymerases I (RPA12, -14, -43, and -49), II (R-3), and III (RPC19, -40, and -53 and RPO31) were also down-regulated, similar to that observed during the environmental stress response.
From a functional viewpoint, rather than chronically down-regulating the capacity for translation under anoxia, it would appear there is a brief interruption in the processing and de novo synthesis of the cytoplasmic translational apparatus. Such a transcriptional program makes functional sense given the acute interruption in the steady-state rate of energy production during the switch from mixed respirofermentative to strictly fermentative growth in galactose. Although no factors have been identified that directly bind to PAC or RRPE, Sfp1, and/or Sch9 are likely involved given their role in regulating nucleolar genes for ribosomal biogenesis and translation at START (22, 43, 59).
In contrast to the cytoplasmic ribosomal complexes, the majority of genes encoding or associated with the mitochondrial 15S complex (MRP10, -13, -20 and -51, MRPS17, -18 and -35, RSM7, -18, -24, -26 and -28, and NAM9) and 21S complex (MRPL1, -3, -7, -8, -15, -19, -20, -22, -23, -33, -35, -36 and -51, MRP20, IMG1 and -2, RML2, and YML6) were chronically down-regulated after a substantial delay. Their chronic down-regulation is predictable as there would be little apparent benefit in supporting the mitochondrial translational apparatus to full aerobic capacity when mitochondrial function is restricted due to O2 lack. Although these genes appear to be coregulated given their tight clustering pattern (primarily cluster 9), no TFMs were significantly enriched and directed promoter searches failed to reveal any overrepresented sequences. Hap2/3/4/5 may regulate some of them (31), which could account for the delay in their transcriptional down-regulation.
Carbohydrate utilization and reserve energy metabolism.
Concomitant with the transient down-regulation of rRNA processing and cell cycle genes was the transient up-regulation of genes associated with carbohydrate and reserve energy metabolism (clusters 11 to 14, Table 1). Their up-regulation here suggests a program designed for preserving cellular energy status during the metabolic transition from mixed respiro-fermentative to fermentation growth. This is evidenced in part by the transient induction of genes for the import and catalysis of hexose sugars (GAL2 and HXT3, -4, -6, -7, -9, -11, -13, -15, -16, and -17), as well as the exploitation of alternative carbon sources such as maltose (MAL11, -12, -31, -32, and -33) and xylose (XKS1). Genes encoding key regulators in hexose dissimilation were also transiently up-regulated, including those for glucokinase (GLK1), hexokinase (HXK1), glucose-6-phosphate isomerase (PGI1), phosphofructokinase (PFK2), and 6-phosphofructo-2-kinase (PFK26 and -27).
Evidence for a drop in the cellular energy state includes the transient induction of negative regulators of gluconeogenesis (GID7, FYV10, VID24, -28 and -30, and UBC8) and genes that accumulate under glucose-limiting conditions (DCS1, DCS2, GPX1, SIP2, and the regulator SNF3). Genes encoding catalytic and regulatory subunits of trehalose-6-phosphate synthase (TPS1, TPS2, TPS3, and TSL1) were also transiently induced, similar to that observed during the environmental stress response. Trehalose is not only an important reserve energy store but is also involved in controlling glycolytic flux (23, 81), facilitating the transition from respiratory to fermentative growth (64), preserving protein structure during anoxia (14), and maintaining membrane integrity in the face of increasing ethanol concentrations (2). Unlike the environmental stress response, genes involved in its degradation (ATH1 and NTH1 and -2) were not differentially expressed here. Glycogen is also an important reserve energy source and stress protectant, especially during restricted growth (23, 67), and several genes encoding key regulators (MRK1, PCL6 and -10, PSK1, and GLC8) or enzymes involved in its elongation (GSY2) or branching (GLC3) were significantly induced. The cyclins PCL6 and PCL10 for Pho85 kinase, which may be responsible for the accumulation of glycogen and trehalose in G1 for later use in the cell cycle (23), were also transiently up-regulated.
Overall, the transient induction of genes for trehalose and glycogen processing in conjunction with those for the dissimilation of sugars suggests a transcriptional program designed for ensuring adequate energy supplies during the transition from mixed respiro-fermentative to strictly fermentative growth. Indeed, when all transiently responding gene networks are viewed together, it is clear they are involved in balancing energetic supply and demand during this transition. Although this response is phenotypically similar to what has been referred to as the “environmental stress response” (28) or “common environmental response” (12), here it is clear that the “stress” encountered is one associated with the acute cessation of respiration and associated changes in energy metabolism during this metabolic transition. Given the commonalities in these responses to environmental change, we sought to define the specific role that the stress-inducible factors Msn2 and Msn4 play in mediating acclimatization to anaerobiosis.
Role of Msn2/4 in mediating the anaerobic response.
Msn2/4 appear to play a ubiquitous role in regulating the response to environmentally stressful conditions. Thus, it was not surprising to find significant enrichment for Msn2/4 binding sites in groups of genes that were transiently induced in response to anaerobiosis (clusters 12 to 14, Table 1). To further define the role these factors play, we compared the temporal profiles of an msn2/4 double mutant strain to that of its isogenic wild-type parent. For these studies, cells were grown on the nonrepressing substrate galactose (SSG-TEA medium) to both facilitate comparisons to our previous steady-state analyses of the O2-responsive transcriptome (51) and circumvent confounding effects of carbon source regulation on specific subsets of O2-responsive genes (e.g., Hap2/3/4/5-regulated genes). However, as has been reported for other msn2/4 strains (21), the mutant grew significantly (P < 0.001) more slowly than its parent, especially under anaerobic conditions (Table 2).
TABLE 2.
Comparison of aerobic and anaerobic growth rates between wild-type (JM43) and msn2/4 (KKY8) strains in galactose (SSG-TEA) and glucose (SSD-TEA) media
| Strain | Medium | Mean doubling time (h) ± SD (no. of samples)
|
||||
|---|---|---|---|---|---|---|
| Air | Air + anti-mycin A (1 μM) | N2 | ||||
| JM43 | SSG-TEA | 2.4 ± 0.5 (6) | 3.7 ± 0.3 (3) | 3.9 ± 0.3 (4) | ||
| SSD-TEA | 2.1 ± 0.2 (3) | N/Aa | 1.9 ± 0.2 (3) | |||
| KKY8 | SSG-TEA | 4.3 ± 0.4 (7) | 10.7 ± 0.6 (3) | 11.5 ± 2.0 (4) | ||
N/A, not available.
To determine if this phenotype is simply due to slow rates of galactose utilization, as opposed to the failed induction of Msn2/4-regulated genes required for efficient anaerobic growth that could complicate analysis, we measured growth rates supported solely by galactose fermentation by poisoning the respiratory chain under aerobiosis. Aerobic growth rates in the presence of 1 μM anti-mycin A were not significantly (P > 0.05) different from their corresponding anaerobic growth rates (Table 2), suggesting that this slow anaerobic growth phenotype simply results from slow rates of galactose fermentation. Although the specific nature of the defect is not known, phenotypically it is similar to the Kluyver effect where low rates of sugar import have a pronounced effect on fermentative growth but relatively little effect on respiratory growth (reviewed in reference 24).
Compared to the wild-type strain, preliminary genomic analysis of the msn2/4 strain revealed a substantial delay (∼3×) in its response to the shift in O2 availability, corresponding well to its three times slower anaerobic growth rate (Table 2). Given this difference, we harvested anaerobic samples from each strain after the same number of doublings in cell mass (generations), and at the same time points for comparison, and then determined how best to align the temporal profiles for statistical comparisons. A time-warping algorithm (1) provided poor alignment (data not shown), presumably due to the limited number of time points sampled. However, comparison of the temporal profiles on a generation-specific versus time-specific scale (ANOVA) revealed the former provides good alignment between the strains, with far fewer genes (5% versus 14%) exhibiting a significant (P < 0.01) strain-time interaction. These results are perhaps not surprising given that we are assaying the end products (mRNAs) of a highly energy-dependent process (transcription) and the primary defect in the mutant is one associated with limiting rates of galactose fermentation. This same generation-specific time scale was used for comparing the effects of carbon source (galactose versus glucose) on the anaerobic response of the wild-type strain, as described in a separate section below.
Statistical comparison (mixed ANOVA with repeated measures) of the msn2/4 and wild-type strains revealed a total of 93 putative Msn2/4-regulated genes, i.e., genes that were anaerobically induced (P < 0.01) in the wild type but expressed at significantly (P < 0.01) lower levels in the mutant (see items S2 and S3 in the supplemental material for full results). Promoter searches (−1 to −800 bp) revealed that 65% (60 genes) contain one or more STRE sites (AGGGG), corresponding to a binomial enrichment P value of 4.8 × 10−10. Predictably, most of these genes are found in clusters 11 and 12 (Fig. 4D), i.e., within clusters of genes that were transiently induced and significantly enriched for Msn2/4-binding sites (Table 1).
The expression profiles of these 60 genes are shown in panels G and H of Fig. 4 for the wild type and mutant, respectively. The yellow boxes in panel H indicate samples for which the transcript level in the mutant was significantly (P < 0.01) lower than that of its parent. From this figure it is clear that Msn2/4-regulated genes begin to respond by 0.04 generations (10 min in the wild type) after the shift to anaerobiosis, are maximally induced at ∼0.08 generation, and their transcript levels diminish thereafter. This time course is similar to that observed for the transient induction of Msn2/4-regulated genes in response to other environmental challenges (12, 28) and fits well with the kinetics of Msn2 nuclear import and export determined in previous studies (40). From this comparison it is also clear that factors in addition to Msn2/4 likely regulate these genes given that nearly half (25 of 60) are anaerobically induced in the mutant but to a significantly (P < 0.01) lower extent than in the wild type. Comparison of the genes identified here with those from studies of heat shock (28), H2O2 addition (28), or acidic conditions (12) reveals only moderate overlap (22 genes of 60), indicating modularity in the network of genes these factors control depending on the specific nature of the stress encountered.
In regard to the function of these genes, most are annotated under categories of energy metabolism (carbohydrate and reserve energy metabolism) or general cell stress. They include genes for import and utilization of hexose sugars (HXT13, -15, -16, and -17) or maltose (MAL31 and -32), reduction of pentose sugars (YJR096W and GRE3) or aldose (YDL124W), and for processing of mannitol (YEL070W) and fructose-2,6-bisphosphate (YLR345W, DSC2, and GPX1). Several are also involved in the processing of trehalose (TPS1, -2 and -3, TSL1) and glycogen (GDB1, GLC3, GSY2, and MRK1). Finally, others are associated with the mitochondrial function, including ubiquinone synthesis (COQ1 and -6), chaperone activity (TCM62 and MBA1), division (FIS1), aldehyde processing (ALD5), and other functions (SFT2 and CBP6). The function of the remaining Msn2/4-regulated genes is not known and, thus, this study should help with their annotation. Overall, given the function of the genes affected here, it would appear that Msn2/4 are involved in the retooling of catabolic pathways and energy reserves, presumably to ensure adequate energy supplies during the metabolic transition from mixed respiro-fermentative metabolism to the slower growth rates supported by fermentative metabolism alone. In the following section, we further explore this hypothesis.
Response to anaerobiosis in glucose medium.
As noted above, the acute response to anaerobiosis in galactose medium is phenotypically similar to that invoked by a number of other environmental insults. This includes the transient down-regulation of genes for rRNA processing genes (PAC and RRPE) and DNA transcription and repair (MCB and SCB), as well as the transient up-regulation of Msn2/4-regulated genes involved in carbohydrate and reserve energy metabolism (Table 1). Although a number of explanations have been proposed for such “stress-induced” changes (12, 28), from a functional standpoint it would appear to be simply part of an energy balancing measure. In this study, this would be required as diminishing O2 availability limits respiration, resulting in a shift to slower growth rates supported by galactose fermentation alone (Table 2). A similar response might also be elicited by any stress-induced decrease in energy production, whether chronic in nature, as here, or transient. Thus, rather than directly mitigating the environmental stress per se, Msn2/4 appear to orchestrate acute changes in catabolism (18, 49). To further explore this hypothesis, we asked if a similar genomic response would be elicited during the shift to anaerobiosis when growth rate, and the corresponding cellular energy status, is unaltered during the shift (see Table 2 for O2- and medium-dependent growth rate comparisons). To examine this, we shifted cells to anaerobic conditions in glucose medium and repeated the microarray analyses.
ANOVA revealed that far fewer genes responded to the shift in O2 availability in glucose (352 genes) than in galactose medium (938 genes) (see items S4 and S5 in the supplemental material for full results). For direct comparison, panel B of Fig. 4 shows the glucose-dependent response of those genes that responded significantly to the shift in galactose (panel D). Note that only those genes indicated by a bar in panel A were differentially (P < 0.01) expressed with respect to O2 in glucose. Bars in panel C indicate genes that showed a differential response (P < 0.01) in the two media. From this figure it is clear that the majority of genes that transiently respond to the O2 shift in galactose medium (clusters 1 to 5 and 10 to 15) fail to do so in glucose. Indeed, the biphasic nature of the response observed in galactose (Fig. 1) is compressed to a single, delayed response in glucose, as shown in Fig. 5. From Fig. 4C it is also clear that the majority of genes that respond similarly in the two media exhibit a delayed response to the O2 shift. These comparisons reveal substantive carbon-source-dependent differences that are specific to the acute phase of the anaerobic response. This suggests that glucose either represses the expression of the transiently responding networks observed in galactose or that a signal other than the change in O2 availability is responsible for eliciting such a response.
FIG. 5.

Dynamics of oxygen-responsive gene induction and repression during short-term anaerobiosis in glucose medium. The number of genes that responded significantly (P < 0.01) to the change in O2 concentration in the wild-type strain (JM43) grown in glucose medium (SSD-TEA) is plotted as a function of the relative number of cell doublings (generations) after the shift to anaerobiosis. Genes are divided into those that were significantly up-regulated and those that were significantly down-regulated. Black bars indicate the number of genes that were identified for the first time at that time point to exhibit a significant change in expression from that of the aerobic control (time zero). Gray bars indicate the number of genes that were differentially expressed relative to the aerobic control yet had already been identified at an earlier time point to have responded significantly to the shift in O2 concentration. The combined height of the black and gray bars indicates the total number of genes at each time point that showed a significant change in expression relative to the aerobic control.
In regard to the possibility of catabolite repression, statistical comparison of the aerobic controls reveals 78 genes whose transcript levels were significantly (P < 0.01) lower in glucose compared to galactose. A search of their promoters reveals significant (P = 3.3 × 10−4) enrichment for a single motif (TCCCCG) with similarity to a Mig1-binding site (TA/TCCCA/C) (46). However, few of these genes were transiently expressed in galactose. Indeed, 30 of the 43 that are shown in panel D of Fig. 4 are in clusters of genes (7 to 9) that exhibit a delayed, chronic down-regulation in galactose and are associated with mitochondrial function (Table 1). Moreover, aerobic transcript levels of only four (MRK1, HXT15, HXT16, and YOL155C) of the 60 Msn2/4-regulated genes identified here were significantly (P < 0.01) lower in glucose. Finally, none of the Msn2/4-regulated genes that were anaerobically induced in glucose (EMI2, COQ6, ECM3, YOL155C, YLR177W, and YPL109C) exhibit a transient response. Taken together, it is clear that the gene networks that transiently respond to the O2 shift in galactose are not repressed by glucose and, thus, changes in the O2 concentration per se cannot be directly responsible for the change in their expression.
To further explore these carbon source-specific differences in the networks that respond to the shift in O2 availability, we clustered the temporal profiles of the 352 genes that were differentially (P < 0.01) expressed in glucose. From the clustering quality assessment shown in Fig. 6 (panel A), we chose to analyze the results obtained with K = 11 as it results in the most nonrandom configuration of TFMs among clusters (lowest MCS P value) while retaining high consensus share (CS). As shown in the heat map of Fig. 6 (panel B), the SOM algorithm nicely partitions the transcript profiles into temporally shifted clusters of up- and down-regulated genes. Bars to the left of the heat map indicate genes that failed to respond (P > 0.01) to the O2 shift in galactose whereas bars to the right indicate genes that were differentially (P < 0.01) expressed in the two media. Overall, this figure further illustrates similarities in the genes that exhibit a delayed response to the O2 shift in the two media (clusters 4 to 6 and 8 to 10) yet substantive differences for those that more acutely respond (clusters 1, 2, and 11).
FIG. 6.

Clustering quality assessment (A) and heat maps (B) for genes that significantly responded to the shift to anaerobiosis in glucose medium. The temporal profiles of genes whose expression responded significantly (P < 0.01) to the shift to anaerobiosis in glucose medium (SSD-TEA) were clustered using an SOM algorithm with 1D ring topology. Panel A shows the motif configuration statistic (solid line, left ordinate) and consensus share (dotted line, right ordinate) as a function of cluster number. Panel B is the heat map of the temporal changes in gene expression relative to the aerobic control (time zero) partitioned with 11 clusters. Green indicates down-regulated expression and red indicates up-regulated expression. Bars to the left of the heat map indicate genes that failed to respond (P > 0.01) to the O2 shift in galactose medium, and bars to the right indicate genes that exhibited a differential response (P < 0.01) in glucose versus galactose medium.
Comparison of the TFMs enriched from clustering each of these responses (Table 3 for glucose and Table 1 for galactose) reveals that the glucose set is largely a subset of the galactose one, with the conspicuous absence of TFMs (e.g., PAC, RRPE, MCB, SCB, MBP1, SWI4, and SWI6) associated with genes that transiently respond in galactose (see Table S3 in the supplemental material for a full listing of enriched, TFMs, 6-mer oligonucleotides, and functional categories in glucose). Interestingly, although MSN2/4 binding sites were significantly enriched in glucose clusters 1 to 2 (Table 3), they are found in genes that were down-regulated as opposed to transiently up-regulated as in galactose. These include 19 (13 with STRE sites) that show differential expression in the msn2/4 strain, including key regulators for the metabolism of trehalose (TPS2 and TSL1), glycogen (MRK1, GLC3, and GSY2) and carbohydrates (YLR345W and DSC2). In addition to STRE sites, many of these have MIG1 sites, which may account for their down-regulation in glucose.
TABLE 3.
Enrichment of transcription factor motifs (TFMs) and MIPS functional categories in clusters of genes that were differentially expressed in response to anaerobiosis in glucose medium (wild-type strain, SOM clustering with 1D ring topology)a
| Cluster no. | n | TFM | Reference(s) | Percentage share |
P
|
MIPS functional category | P (genomic) | |
|---|---|---|---|---|---|---|---|---|
| In set | Genomic | |||||||
| 1 | 20 | MSN2/4 | 46 | 95 | 5.3 | 8.0 | Metabolism of energy reserves (glycogen, trehalose) | 11.0 |
| MIG1 | 46 | 50 | 3.7 | 4.5 | C-compound and carbohydrate metabolism | 5.7 | ||
| 2 | 16 | MSN2/4 | 46 | 88 | 3.1 | 5.0 | Cell rescue, defense, and virulence | 4.4 |
| 3 | 16 | ABF1 | 91 | 63 | 2.0 | 2.1 | Respiration/mitochondrion/energy | ≥3.5 |
| Transport ATPases | 3.8 | |||||||
| 4 | 34 | HAP2/3/4/5 | 46 | 44 | 3.0 | 4.8 | Respiration/mitochondrion/energy | ≥8.7 |
| HAP1 | 48 | 26 | 2.3 | 4.9 | ||||
| 5 | 40 | PDR1 | 54 | 13 | 2.4 | 3.9 | Respiration/mitochondrion | ≥2.7 |
| Homeostasis of cations | 2.2 | |||||||
| 6 | 75 | Mitochondrion | 14.0 | |||||
| Ribosome biogenesis | 8.5 | |||||||
| Phosphate metabolism | 2.2 | |||||||
| 7 | 10 | ACE2 | 54, 91 | 70 | 3.0 | 3.2 | C-compound and carbohydrate metabolism | 3.6 |
| NDD1 | 54 | 50 | 2.8 | 3.4 | ||||
| SWI4 | 54 | 50 | 2.6 | 3.0 | ||||
| 8 | 42 | ROX1 | 17 | 95 | 2.7 | 4.0 | Vitamin, cofactor, and prosthetic group biosynthesis | 2.1 |
| HAP2/3/4/5 | 54 | 62 | 2.6 | 2.8 | ||||
| 9 | 34 | UPC2 | 16 | 76 | 7.9 | 12.7 | ABC transporters | 3.4 |
| Cell rescue, defense, and virulence | 3.2 | |||||||
| 10 | 38 | MCM1 | 46, 76, 91 | 82 | 3.6 | 5.2 | Cell wall | 5.6 |
| GLN3 | 91 | 71 | 2.1 | 2.3 | Cell rescue, defense, and virulence | 3.7 | ||
| UPC2 | 16 | 63 | 4.8 | 9.0 | C-compound and carbohydrate utilization | 2.4 | ||
| 11 | 20 | |||||||
| 0 | 7 | Mitochondrion | 2.2 | |||||
See Table 1, footnotes a to d.
Of the remaining genes that were down-regulated (clusters 3 to 6), the majority exhibit a similar response in the two media (Fig. 6) and are primarily involved in mitochondrial function (Table 3). Although HAP2/3/4/5 and HAP1 sites were found in a large fraction of genes in cluster 4, the transcriptional networks that control the majority of these down-regulated genes (e.g., cluster 6) are not clearly evident from these analyses. Given their function, it is not surprising that most are chronically down-regulated for the duration of anaerobiosis as revealed by comparisons to steady-state analyses of the anaerobic response (51, 77). Finally, in regard to those that were up-regulated, the majority show a similar response in the two media (Fig. 4 and 6) and chronic expression patterns (51, 77). Most contain binding sites for heme-responsive transcription factors, notably Rox1 (Glu-cluster 8 and Gal-cluster 16) and Upc2 (Glu-clusters 9 to 10 and Gal-clusters 15 to 17). These genes are involved in diverse cellular processes required for acclimatization to anaerobiosis, and include genes for lipid, fatty acid, and isoprenoid metabolism, cell wall structure and modification, redox balance, and carbohydrate metabolism (Tables 1 and 3), as will be discussed further in the sections below.
Delayed yet chronically expressed gene networks.
In contrast to the transiently responding gene networks, there is very little overlap between the environmental stress response and the gene networks that were more chronically down-regulated (1%) or up-regulated (2%) in galactose, or indeed any of the genes that were either down-regulated (1%) or up-regulated (4%) in glucose. Changes in these networks include the chronic down-regulation of Hap1- and Hap2/3/4/5-regulated ones involved in mitochondrial function and the up-regulation of Rox1- and Upc2-regulated ones required for more long-term acclimatization to anaerobiosis. Given that we have only a limited view of the dynamical changes in these gene networks here, we limit our functional analyses to a few groups below.
Transition metal transport and utilization.
Transition metals, such as iron, copper, zinc, cobalt, and manganese, are essential to all organisms for participation in a variety of redox reactions, and their transport and processing are closely monitored because the metals themselves can be toxic. The bulk of proteins that utilize these metals are associated with the processing of oxygen or its by-products and, thus, both the transport of these ions and synthesis of proteins that require them is affected by oxygen availability.
Here we observed the down-regulation of number of genes for the import of metals, including those encoding high-affinity iron transporters (FRE1, UTR1, FTR1, and FET3), yet up-regulation of the relatively nonspecific (Fe, Cu, Mn, or Zn) low-affinity transporter encoded by FET4, which is regulated by Rox1, Aft1, and Zap1 (41, 86). Fet4 may thus assume a pivotal role in regulating general metal-ion homeostasis during anaerobic conditions. However, increased Fet4 activity can lead to transition metal sensitivity (56), which may account for the simultaneous up-regulation of the metallothionine Cup1 and the copper chaperone for Sod1 (LYS7) here. A number of other genes for transporting or processing metals were chronically down-regulated (e.g., ARN1, CCC2, CCH1, COX17, HMX1, ISU2, OCT1, and SMF1). The up-regulation of IZH4, which is a plasma membrane protein induced by high Zn (57) or hypoxia (45), may serve a critical role in coordinating both sterol and zinc metabolism under anoxia (57).
Lipids and sterols.
Although it has long been known that an exogenous source of unsaturated fatty acid and sterol is essential for long-term anaerobic growth in S. cerevisiae (3, 4), a more complete picture of the anoxia-induced remodeling in these pathways is revealed here. Notably, this remodeling is fairly specific for sterol and sphingolipid pathways, with very few genes for phospholipid or fatty acid synthesis (save for AAC1 and OLE1) showing changes in expression. For sterol, genes in the early portion of the pathway (for isoprenoid synthesis) exhibited complex expression patterns, with some showing transient (ERG10) or chronic up-regulation (IDI1 and HMG2) and others showing transient (ERG8 and MVD1) or chronic down-regulation (ERG13 and -20 and HMG1). However, nearly all of the genes (ARE1, ERG1, -2, -3, -6, -11, -24, -25, -26 and -28, and NCP1) in the later portion of the pathway were chronically up-regulated, as were genes involved in transport (PDR11 and AUS1) and their primary regulator (UPC2).
Apparently exogenous ergosterol is imported and cycles between membranes and lipid droplets but does not affect control points in the endoplasmic reticulum for its synthesis (5, 75), given that ergosterol synthesis is not possible without oxygen. In terms of their regulation, Upc2 and Rox1 control many of them (51, 84, 87), and binding sites for these factors were significantly enriched in clusters containing these genes (Tables 1 and 3). Finally, for sphingolipid synthesis, a number genes encoding the middle portion of the pathway, linking dihydrosphingosine to ceramide, and the putative signaling molecules (dihydrosphingosine-1-phosphate and phytosphingosine-1-phosphate) were affected here (e.g., SUR2, YSR3, LCB4, and YDC1). The net effect of these changes may be to increase phytosphingosine during the transition to slower anaerobic growth rates.
Cell wall, vesicle transport, and secretory networks.
Similar to previous steady-state analyses of the anaerobic transcriptome (51, 77), here we see substantive evidence for remodeling of the plasma membrane and cell wall during the shift to anaerobiosis. This is reflected in the delayed but chronic up-regulation of genes for cell wall structure, modifying enzymes, protein secretion, vesicle trafficking, as well as lipid and sterol metabolism. These networks are largely controlled by Upc2 and/or Rox1, whose binding sites were significantly enriched in Gal-clusters 15 to 17 and Glu-clusters 8 to 10. Notably, this includes the up-regulation of nearly all of the seripauperin and TIP1 gene family (DAN1, -2, -3 and -4; TIR1, -2, -3 and -4; PAU1, -2, -3, -4, -5, -6 and -7; and 10 other open reading frames) as well as other genes for biogenesis or modification of the cell wall (e.g., CSR1, GSC2, and YOL155C).
Although the expression patterns of secretory genes were more complex, and probably reflect the slowing of growth, a number of such genes were up-regulated here (e.g., EUG1, CPR4, SRO77, AKR2, YOL075C, and PLB2). Genes for modifying transported proteins were also up-regulated, including several for glycosylation (PMT3, PMT5, KRE9, UGP1, and KTR4), prenylation (BET2), and GPI anchors (GPI12). Finally, genes involved in endocytosis (SRO77 and AKR2) were also up-regulated, including several (e.g., IZH4, CLC1, AKR1, RVS161, AFR1, and YBR108W) that were induced much earlier in the time course. Overall, such remodeling activity can be expected to alter cell wall porosity to accommodate the transport and processing of essential nutrients that are required during prolonged periods of anaerobiosis.
DISCUSSION
In this study, we have found that the dynamics of the genomic response to acute oxygen deprivation consists of two distinct phases when cells are shifted from aerobiosis to anaerobiosis in galactose medium: (i) an acute, transitory one in which genes associated with the retooling of metabolism (respiro-fermentative to strictly fermentative and reserve energy) and slowing of growth rate are differentially expressed; and (ii) a delayed, chronic phase in which genes associated with long-term acclimatization to anaerobiosis are differentially expressed. The acute, transitory phase is absent when cells are shifted to anaerobiosis in glucose medium, conditions in which little catabolic remodeling is required and no change in the growth rate is observed.
Using a novel metric that we call the motif configuration statistic, which determines what clustering approach and number of clustering divisions results in the most nonrandom distribution of transcription factor motifs (TFMs) among gene clusters, we recover a transcriptional network from SOM clustering that shows that these phases are distinct from one another, both in terms of the gene networks that are involved and the signaling pathways that elicit these responses. This clustering approach identifies several gene networks, notably MBF, SBF, PAC/RRPE, and MSN2/4 networks, that were not previously known to be involved in the anaerobic response while simultaneously redefining those controlled by heme-responsive transcription factors, such as Hap1, Hap2/3/4/5, Rox1, and Upc2. Examination of an msn2/4 strain shows these stress factors (i.e., Msn2 and Msn4) are involved in the metabolic remodeling that occurs in galactose medium but do not directly respond to O2 deprivation per se, as evidenced by their conspicuous absence in playing a role in the O2-dependent remodeling of the transcriptome under glucose-repressed conditions.
Physiological acclimatization to anaerobiosis.
In comparison to other environmentally stressful conditions examined in S. cerevisiae (12, 28, 49, 66, 67, 70, 90), acclimatization to anaerobic conditions occurs more slowly, over multiple generations. Perhaps this is not surprising, given that O2 deprivation per se does not pose an immediate threat to cell survival or directly damage cellular components, unlike the case for oxidative, osmotic, or acid stresses, temperature shock, glucose and nitrogen starvation, or DNA-damaging agents. The major challenges facing a yeast cell growing anaerobically on a fermentable carbon source are apparently those associated with (i) the time-dependent depletion of essential cellular components that require molecular O2 for their biosynthesis, (ii) maintenance of cellular redox balance, and (iii) mitigation of damage associated with the accumulation of anaerobic end products.
In regard to the first, molecular oxygen is required for the activity of various hydroxylases, desaturases, and oxygenases involved in the de novo biosynthesis of membrane components such as sterols and unsaturated fatty acids (reviewed in references 50 and 92). Although cells growing on rich medium can typically undergo several (two to three) anaerobic divisions without supplementation, an exogenous source of unsaturated fatty acids and sterol is essential for long-term anaerobic growth (3, 4). One of the most striking patterns in gene expression observed during anaerobic growth is the remodeling activity associated with the cell wall and plasma membrane. This remodeling is required, in part, for the efficient import and processing of these supplements in order to combat the compromised ability to regulate membrane fluidity (51). However, these changes are slow to occur and, as will be described elsewhere (L.-C. Lai, P. V. Burke, and K. E. Kwast, unpublished data), take several generations (>4) for completion.
Molecular oxygen is also required for the biosynthesis of nicotinic acid (33), ubiquinone (62), and some enzymes (e.g., hemoproteins), as well as for the activity of oxidases involved in Fe3+ uptake and the reduction of free radicals (e.g., ascorbate) (reviewed in reference 71). However, these processes are primarily associated with aerobiosis, and the expression of genes involved in such processes is predictably down-regulated under anaerobiosis. Given the lack of O2 for oxidative phosphorylation and β-oxidation, predictable changes are also observed in the expression of genes involved in dissimilatory metabolism and mitochondrial function, but again they are slow to occur. Some molecular defenses associated with maintenance of redox balance (e.g., glycerol and fumarate accumulation) and mitigating the effects of anaerobic end product accumulation (e.g., trehalose production instep with ethanol accumulation) are activated more quickly, but still after a considerable delay (>0.2 generation).
Thus, in this study it was surprising to find that such a large number of genes (nearly a tenth of the genome) acutely (<15 min) yet transiently (∼45 min) respond to the shift to anaerobiosis on galactose medium. Comparison of these gene networks to those that respond in glucose revealed that they are unique to galactose and reflect the remodeling activity that is necessitated by the metabolic switch from mixed respiro-fermentative to strictly fermentative growth and the slowing of growth to a new steady state supported by galactose fermentation alone. Indeed, genes that are identifiably associated O2-dependent phenomena respond with similar kinetics in the two media and are differentially expressed only towards the latter part of the time series (2 generations) examined here.
Remodeling of the transcriptome in response to anaerobiosis.
Using a novel clustering approach, we recover a more detailed view of the genomic remodeling responsible for physiological acclimatization to anaerobiosis. Within 0.04 generation (10 min) after the shift in O2 availability in galactose, MBF and SBF networks are the first to be negatively affected, resulting in down-regulation of genes involved in DNA replication/repair and other processes associated with the G1/S transition of the cell cycle. This likely results in a delay in its progression as mass and energy levels are assessed before committing to another round of DNA replication; a similar, transient cell cycle arrest is thought to occur in response to other environmental insults (reviewed in reference 29). Concomitantly, or shortly thereafter, PAC/RRPE-associated gene networks that are involved in cytoplasmic rRNA processing are negatively affected, while Msn2/4-regulated networks involved in both the import and utilization of primary and alternative carbon sources and reserve energy metabolism are activated. This remodeling activity suggests simultaneous exploration and utilization of available carbon sources and sparing of energetic demand by an arrest in the de novo synthesis of the cytoplasmic translational machinery. The response is predictably transitory, during the transition in metabolic state alone, with maximal changes at 0.08 generation (20 min), a diminishing response by 0.2 generation, and no further evidence of such a response by 2 generations. Although similar to that observed during nutrient limitation or a switch to lower-quality carbon sources (66, 67), it is unique in affecting a subset of genes associated with the balancing of energetic supply and demand before initiating another round of the cell cycle. Such changes were not observed during the transition to anaerobiosis in glucose medium, yet those that were initiated after a considerable delay were.
Given that nutrient availability, save for O2, was constant during the transition to anaerobiosis, these carbon source-dependent differences are most easily explained by the differential contribution of respiration to growth in the two media. Whereas O2 availability has little or no effect on growth rate in catabolite (glucose)-repressed cells due to the Crabtree effect, respiration contributes substantially to growth under derepressed conditions (e.g., in galactose). Here we find a 40% reduction in growth rate when respiration is inhibited by either antimycin A or anoxia in cells grown in galactose medium (Table 2). The abrupt cessation of respiration will lead to a rapid, albeit transient change in the phosphorylation state of the purine nucleotide pool before compensatory mechanisms are activated (7, 19). A rapid but more sustained increase in cellular redox potential is also expected (82) yet should be similar, although not identical, in the two media.
Obviously, inhibition of respiration will result in a number of other metabolic signals, including changes in carbon flux rates (glycolysis, tricarboxylic acid), the proton motive force, et cetera. However, little is currently known about “small-molecule sensors” in yeast that may monitor the energetic or redox state of the cell. Candidate sensors of the energy status include the PAS kinase Psk2, which has been shown to coordinate both sugar storage and protein synthesis with respect to carbohydrate availability (72), and Snf1, which may respond to changes in adenylates, in particular AMP, although this hypothesis remains somewhat controversial. Signaling could occur by potentially most of the nutrient-regulated protein kinase pathways, including Ras/protein kinase A, TOR, the Snf1-kinase complex, and the cyclin-dependent kinase Pho85 (reviewed in references 25 and 88). Changes in the expression of some of the components in these pathways were observed, including the transient up-regulation of TCO89 and TIP41 for TOR and the Pho85 cyclins PCL6 and PCL10.
Inhibition of Ras/protein kinase A and/or TOR could lead to Msn2/4 activation and down-regulation of the PAC/RRPE and MBF/SBF networks. In addition to Ras/protein kinase A and TOR, Sfp1 and Sch9 are likely involved in coordinating the expression of these two networks, given their role in dictating critical cell size at the G1/S transition and coordinating the expression of genes for ribosomal biogenesis (22, 43, 59). Although few of the genes encoding structural components of the ribosomes were significantly (P < 0.01) affected here, many (∼80) do show a kinetically similar trend of down-regulation but with lower confidence (P > 0.01). Overall, with such a branched and overlapping network of signaling pathways and no clear indication of the sensing event, it is difficult to identify the specific signaling pathways involved without further investigation.
By ∼0.2 generation of anaerobiosis in both media, we begin to see evidence of more chronic, heme-dependent effects, including the down-regulation of Hap1- and possibly Hap2/3/4/5-regulated networks, derepression of Rox1-regulated networks, and, following a slight delay, activation of Upc2-regulated networks. Oxygen levels continue to decline for some time after the shift, reaching 1 μM O2 by ∼10 min, 0.5 μM O2 by ∼20 min, and < 0.1 μM O2 within 30 min (Fig. 1). A large body of evidence indicates that the effect of O2 on the activity of these factors is mediated by heme (35, 50, 53, 92). Given the low Km for O2 (<0.1 μM O2) of enzymes (Hem13 and Hem14) involved in its synthesis (52), the delay in the response of these heme-regulated gene networks is predictable as diminishing O2 inhibits heme biosynthesis and regulatory pools within the nucleus are diluted with time.
In addition to changes in their activity, the expression of several of these factors is directly affected. Here we observed the down-regulation of ROX1 and MOT3, whose expression is regulated by Hap1 and heme (17, 47, 74), and up-regulation of UPC2, whose expression is repressed by Rox1 (51). Interestingly, HAP1, which serves an essential function under anaerobiosis (13), was chronically up-regulated in both media.
Because we have primarily focused on the acute response to O2 deprivation in this study, we have only a limited view of the dynamical changes that occur in these heme-regulated networks, as many show differential expression only at the terminal time point examined in this study (2 generations). Thus, we alert interested readers to a recently completed study (Lai, Burke, and Kwast, unpublished data) that examines the dynamics of the response over six generations of anaerobiosis. In terms of the gene networks identified, there is significant overlap with those identified in previous steady-state analyses of the O2-responsive transcriptome (8, 51, 68, 77). They are involved in diverse cellular processes and include both Rox1- and Upc2-regulated networks involved in lipid, fatty acid, and isoprenoid metabolism, cell wall structure and modification, and carbohydrate metabolism, and Hap1- and Hap2/3/4/5-regulated networks involved in respiration and mitochondrial function. Changes in these gene networks appear to be uniquely required for physiological acclimatization to anaerobiosis (51).
Supplementary Material
Acknowledgments
We thank R. Zitomer for providing the plasmids for disruption. We are indebted to Joel Peek at Microarrays, Inc., for technical guidance in designing and printing the custom 70-mer oligonucleotide microarrays.
This work was supported by National Institutes of Health grant RO1-GM59826 to K.E.K.
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Aach, J., and G. M. Church. 2001. Aligning gene expression time series with time warping algorithms. Bioinformatics 17:495-508. [DOI] [PubMed] [Google Scholar]
- 2.Alexandre, H., V. Ansanay-Galeote, S. Dequin, and B. Blondin. 2001. Global gene expression during short-term ethanol stress in Saccharomyces cerevisiae. FEBS Lett. 498:98-103. [DOI] [PubMed] [Google Scholar]
- 3.Andreasen, A., and T. Stier. 1953. Anaerobic nutrition of Saccharomyces cerevisiae. I. Ergosterol requirement for growth in a defined medium. J. Cell. Comp. Physiol. 41:23-36. [DOI] [PubMed] [Google Scholar]
- 4.Andreasen, A., and T. Stier. 1954. Anaerobic nutrition of Saccharomyces cerevisiae. II. Unsaturated fatty acid requirement for growth in defined medium. J. Cell. Comp. Physiol. 43:271-281. [DOI] [PubMed] [Google Scholar]
- 5.Athenstaedt, K., D. Zweytick, A. Jandrositz, S. D. Kohlwein, and G. Daum. 1999. Identification and characterization of major lipid particle proteins of the yeast Saccharomyces cerevisiae. J. Bacteriol. 181:6441-6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barral, Y., M. Parra, S. Bidlingmaier, and M. Snyder. 1999. Nim1-related kinases coordinate cell cycle progression with the organization of the peripheral cytoskeleton in yeast. Genes Dev. 13:176-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beauvoit, B., M. Rigoulet, O. Bunoust, G. Raffard, P. Canioni, and B. Guerin. 1993. Interactions between glucose metabolism and oxidative phosphorylations on respiratory-competent Saccharomyces cerevisiae cells. Eur. J. Biochem. 214:163-172. [DOI] [PubMed] [Google Scholar]
- 8.Becerra, M., L. J. Lombardia-Ferreira, N. C. Hauser, J. D. Hoheisel, B. Tizon, and M. E. Cerdan. 2002. The yeast transcriptome in aerobic and hypoxic conditions: effects of hap1, rox1, rox3 and srb10 deletions. Mol. Microbiol. 43:545-555. [DOI] [PubMed] [Google Scholar]
- 9.Beck, T., and M. N. Hall. 1999. The TOR signaling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature 402:689-692. [DOI] [PubMed] [Google Scholar]
- 10.Burke, P. V., K. E. Kwast, F. Everts, and R. O. Poyton. 1998. A fermentor system for regulating oxygen at low concentrations in cultures of Saccharomyces cerevisiae. Appl. Environ. Microbiol. 64:1040-1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burke, P. V., L. C. Lai, and K. E. Kwast. 2004. A rapid filtration apparatus for harvesting cells under controlled conditions for use in genome-wide temporal profiling studies. Anal. Biochem. 328:29-34. [DOI] [PubMed] [Google Scholar]
- 12.Causton, H. C., B. Ren, S. S. Koh, C. T. Harbison, E. Kanin, E. G. Jennings, T. I. Lee, H. L. True, E. S. Lander, and R. A. Young. 2001. Remodeling of yeast genome expression in response to environmental changes. Mol. Biol. Cell. 12:323-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chantrel, Y., M. Gaisne, C. Lions, and J. Verdiere. 1998. The transcriptional regulator Hap1p (Cyp1p) is essential for anaerobic or heme-deficient growth of Saccharomyces cerevisiae: Genetic and molecular characterization of an extragenic suppressor that encodes a WD repeat protein. Genetics 148:559-569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen, Q., and G. G. Haddad. 2004. Role of trehalose phosphate synthase and trehalose during hypoxia: from flies to mammals. J. Exp. Biol. 207:3125-3129. [DOI] [PubMed] [Google Scholar]
- 15.Cliften, P., P. Sudarsanam, A. Desikan, L. Fulton, B. Fulton, J. Majors, R. Waterston, B. A. Cohen, and M. Johnston. 2003. Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science 301:71-76. [DOI] [PubMed] [Google Scholar]
- 16.Cohen, B. D., O. Sertil, N. E. Abramova, K. J. Davies, and C. V. Lowry. 2001. Induction and repression of DAN1 and the family of anaerobic mannoprotein genes in Saccharomyces cerevisiae occurs through a complex array of regulatory sites. Nucleic Acids Res. 29:799-808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deckert, J., A. M. Torres, S. M. Hwang, A. J. Kastaniotis, and R. S. Zitomer. 1998. The anatomy of a hypoxic operator in Saccharomyces cerevisiae. Genetics. 150:1429-1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DeRisi, J. L., V. R. Iyer, and P. O. Brown. 1997. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 278:680-686. [DOI] [PubMed] [Google Scholar]
- 19.Ditzelmuller, G., W. Wohrer, C. P. Kubicek, and M. Rohr. 1983. Nucleotide pools of growing, synchronized and stressed cultures of Saccharomyces cerevisiae. Arch. Microbiol. 135:63-67. [DOI] [PubMed] [Google Scholar]
- 20.Dragon, F., J. E. Gallagher, P. A. Compagnone-Post, B. M. Mitchell, K. A. Porwancher, K. A. Wehner, S. Wormsley, R. E. Settlage, J. Shabanowitz, Y. Osheim, A. L. Beyer, D. F. Hunt, and S. J. Baserga. 2002. A large nucleolar U3 ribonucleoprotein required for 18S ribosomal RNA biogenesis. Nature 417:967-970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Estruch, F., and M. Carlson. 1993. Two homologous zinc finger genes identified by multicopy suppression in a SNF1 protein kinase mutant of Saccharomyces cerevisiae. Mol. Cell. Biol. 13:3872-3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fingerman, I., V. Nagaraj, D. Norris, and A. K. Vershon. 2003. Sfp1 plays a key role in yeast ribosome biogenesis. Eukaryot. Cell 2:1061-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Francois, J., and J. L. Parrou. 2001. Reserve carbohydrates metabolism in the yeast Saccharomyces cerevisiae. FEMS Microbiol. Rev. 25:125-145. [DOI] [PubMed] [Google Scholar]
- 24.Fukuhara, H. 2003. The Kluyver effect revisited. FEMS Yeast Res. 3:327-331. [DOI] [PubMed] [Google Scholar]
- 25.Gagiano, M., F. F. Bauer, and I. S. Pretorius. 2002. The sensing of nutritional status and the relationship to filamentous growth in Saccharomyces cerevisiae. FEMS Yeast Res. 2:433-470. [DOI] [PubMed] [Google Scholar]
- 26.Garreau, H., R. N. Hasan, G. Renault, F. Estruch, E. Boy-Marcotte, and M. Jacquet. 2000. Hyperphosphorylation of Msn2p and Msn4p in response to heat shock and the diauxic shift is inhibited by cAMP in Saccharomyces cerevisiae. Microbiology 146:2113-2120. [DOI] [PubMed] [Google Scholar]
- 27.Gasch, A. P., M. Huang, S. Metzner, D. Botstein, S. J. Elledge, and P. O. Brown. 2001. Genomic expression responses to DNA-damaging agents and the regulatory role of the yeast ATR homolog Mec1p. Mol. Biol. Cell 12:2987-3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gasch, A. P., P. T. Spellman, C. M. Kao, O. Carmel-Harel, M. B. Eisen, G. Storz, D. Botstein, and P. O. Brown. 2000. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell 11:4241-4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gasch, A. P., and M. Werner-Washburne. 2002. The genomics of yeast responses to environmental stress and starvation. Funct. Integr. Genomics. 2:181-192. [DOI] [PubMed] [Google Scholar]
- 30.Gorner, W., E. Durchschlag, M. T. Martinez-Pastor, F. Estruch, G. Ammerer, B. Hamilton, H. Ruis, and C. Schuller. 1998. Nuclear localization of the C2H2 zinc finger protein Msn2p is regulated by stress and protein kinase A activity. Genes Dev. 12:586-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Graack, H. R., and B. Wittmann-Liebold. 1998. Mitochondrial ribosomal proteins (MRPs) of yeast. Biochem. J. 329:433-448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31a.Guldener U., M. Munsterkotter, G. Kastenmuller, N. Strack, J. van Helden, C. Lemer, J. Richelles, S. J. Wodak, J. Garcia-Martinez, J. E. Perez-Ortin, H. Michael, A. Kaps, E. Talla, B. Dujon, B. Andre, J. L. Souciet, J. De Montigny, E. Bon, C. Gaillardin, and H. W. Mewes. 2005. CYGD: the comprehensive yeast genome database. Nucleic Acids Res. 33:D364-D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hegde, P., R. Qi, K. Abernathy, C. Gay, S. Dharap, R. Gaspard, J. E. Hughes, E. Snesrud, N. Lee, and J. Quackenbush. 2000. A concise guide to cDNA microarray analysis. BioTechniques 29:548-. 4: 556. [DOI] [PubMed] [Google Scholar]
- 33.Heilmann, H. D., and F. Lingens. 1968. On the regulation of nicotinic acid biosynthesis in Saccharomyces cerevisiae. Hoppe Seylers Z. Physiol. Chem. 349:231-236. [PubMed] [Google Scholar]
- 34.Heinisch, J. J., A. Lorberg, H. P. Schmitz, and J. J. Jacoby. 1999. The protein kinase C-mediated MAP kinase pathway involved in the maintenance of cellular integrity in Saccharomyces cerevisiae. Mol. Microbiol. 32:671-680. [DOI] [PubMed] [Google Scholar]
- 35.Hon, T., A. Dodd, R. Dirmeier, N. Gorman, P. R. Sinclair, L. Zhang, and R. O. Poyton. 2003. A mechanism of oxygen sensing in yeast. Multiple oxygen-responsive steps in the heme biosynthetic pathway affect Hap1 activity. J. Biol. Chem. 278:50771-50780. [DOI] [PubMed] [Google Scholar]
- 36.Horak, C. E., and M. Snyder. 2002. ChIP-chip: a genomic approach for identifying transcription factor binding sites. Methods Enzymol. 350:469-483. [DOI] [PubMed] [Google Scholar]
- 37.Hughes, J. D., P. W. Estep, S. Tavazoie, and G. M. Church. 2000. Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J. Mol. Biol. 296:1205-1214. [DOI] [PubMed] [Google Scholar]
- 38.Hughes, T. R., M. J. Marton, A. R. Jones, C. J. Roberts, R. Stoughton, C. D. Armour, H. A. Bennett, E. Coffey, H. Dai, Y. D. He, M. J. Kidd, A. M. King, M. R. Meyer, D. Slade, P. Y. Lum, S. B. Stepaniants, D. D. Shoemaker, D. Gachotte, K. Chakraburtty, J. Simon, M. Bard, and S. H. Friend. 2000. Functional discovery via a compendium of expression profiles. Cell 102:109-126. [DOI] [PubMed] [Google Scholar]
- 39.Iyer, V. R., C. E. Horak, C. S. Scafe, D. Botstein, M. Snyder, and P. O. Brown. 2001. Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature 409:533-538. [DOI] [PubMed] [Google Scholar]
- 40.Jacquet, M., G. Renault, S. Lallet, J. De Mey, and A. Goldbeter. 2003. Oscillatory nucleocytoplasmic shuttling of the general stress response transcriptional activators Msn2 and Msn4 in Saccharomyces cerevisiae. J. Cell Biol. 161:497-505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jensen, L. T., and V. C. Culotta. 2002. Regulation of Saccharomyces cerevisiae FET4 by oxygen and iron. J. Mol. Biol. 318:251-260. [DOI] [PubMed] [Google Scholar]
- 42.Jorgensen, P., J. L. Nishikawa, B. J. Breitkreutz, and M. Tyers. 2002. Systematic identification of pathways that couple cell growth and division in yeast. Science 297:395-400. [DOI] [PubMed] [Google Scholar]
- 43.Jorgensen, P., I. Rupes, J. R. Sharom, L. Schneper, J. R. Broach, and M. Tyers. 2004. A dynamic transcriptional network communicates growth potential to ribosome synthesis and critical cell size. Genes Dev. 18:2491-2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kamada, Y., U. S. Jung, J. Piotrowski, and D. E. Levin. 1995. The protein kinase C-activated MAP kinase pathway of Saccharomyces cerevisiae mediates a novel aspect of the heat shock response. Genes Dev. 9:1559-1571. [DOI] [PubMed] [Google Scholar]
- 45.Karpichev, I. V., L. Cornivelli, and G. M. Small. 2002. Multiple regulatory roles of a novel Saccharomyces cerevisiae protein, encoded by YOL002c, in lipid and phosphate metabolism. J. Biol. Chem. 277:19609-19617. [DOI] [PubMed] [Google Scholar]
- 46.Kellis, M., N. Patterson, M. Endrizzi, B. Birren, and E. S. Lander. 2003. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 423:241-254. [DOI] [PubMed] [Google Scholar]
- 47.Keng, T. 1992. HAP1 and ROX1 form a regulatory pathway in the repression of HEM13 transcription in Saccharomyces cerevisiae. Mol. Cell. Biol. 12:2616-2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.King, D. A., L. Zhang, L. Guarente, and R. Marmorstein. 1999. Structure of HAP1-18-DNA implicates direct allosteric effect of protein-DNA interactions on transcriptional activation. Nat. Struct. Biol. 6:22-27. [DOI] [PubMed] [Google Scholar]
- 49.Koerkamp, M. G., M. Rep, H. J. Bussemaker, G. P. Hardy, A. Mul, K. Piekarska, C. A. Szigyarto, J. M. De Mattos, and H. F. Tabak. 2002. Dissection of transient oxidative stress response in Saccharomyces cerevisiae by using DNA microarrays. Mol. Biol. Cell 13:2783-2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kwast, K. E., P. V. Burke, and R. O. Poyton. 1998. Oxygen sensing and the transcriptional regulation of oxygen-responsive genes in yeast. J. Exp. Biol. 201:1177-1195. [DOI] [PubMed] [Google Scholar]
- 51.Kwast, K. E., L.-C. Lai, N. Menda, D. T. James III, S. Aref, and P. V. Burke. 2002. Genomic analyses of anaerobically induced genes in Saccharomyces cerevisiae: Functional role of Rox1 and other factors in mediating the anoxic response. J. Bacteriol. 184:250-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Labbe-Bois, R., and P. Labbe. 1990. Tetrapyrrole and heme biosynthesis in the yeast Saccharomyces cerevisiae, p. 235-285. In H. A. Dailey (ed.), Biosynthesis of heme and chlorophylls. McGraw-Hill, New York, N.Y.
- 53.Lee, H. C., T. Hon, C. Lan, and L. Zhang. 2003. Structural environment dictates the biological significance of heme-responsive motifs and the role of Hsp90 in the activation of the heme activator protein Hap1. Mol. Cell. Biol. 23:5857-5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee, T. I., N. J. Rinaldi, F. Robert, D. T. Odom, Z. Bar-Joseph, G. K. Gerber, N. M. Hannett, C. T. Harbison, C. M. Thompson, I. Simon, J. Zeitlinger, E. G. Jennings, H. L. Murray, D. B. Gordon, B. Ren, J. J. Wyrick, J. B. Tagne, T. L. Volkert, E. Fraenkel, D. K. Gifford, and R. A. Young. 2002. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 298:799-804. [DOI] [PubMed] [Google Scholar]
- 55.Levin, D. E., and E. Bartlett-Heubusch. 1992. Mutants in the S. cerevisiae PKC1 gene display a cell cycle-specific osmotic stability defect. J. Cell Biol. 116:1221-1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li, L., and J. Kaplan. 1998. Defects in the yeast high affinity iron transport system result in increased metal sensitivity because of the increased expression of transporters with a broad transition metal specificity. J. Biol. Chem. 273:22181-22187. [DOI] [PubMed] [Google Scholar]
- 57.Lyons, T. J., N. Y. Villa, L. M. Regalla, B. R. Kupchak, A. Vagstad, and D. J. Eide. 2004. Metalloregulation of yeast membrane steroid receptor homologs. Proc. Natl. Acad. Sci. USA 101:5506-5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Machado, A. K., B. A. Morgan, and G. F. Merrill. 1997. Thioredoxin reductase-dependent inhibition of MCB cell cycle box activity in Saccharomyces cerevisiae. J. Biol. Chem. 272:17045-17054. [DOI] [PubMed] [Google Scholar]
- 59.Marion, R. M., A. Regev, E. Segal, Y. Barash, D. Koller, N. Friedman, and E. K. O'Shea. 2004. Sfp1 is a stress- and nutrient-sensitive regulator of ribosomal protein gene expression. Proc. Natl. Acad. Sci. USA 101:14315-14322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martinez-Pastor, M. T., G. Marchler, C. Schuller, A. Marchler-Bauer, H. Ruis, and F. Estruch. 1996. The Saccharomyces cerevisiae zinc finger proteins Msn2p and Msn4p are required for transcriptional induction through the stress response element (STRE). EMBO J. 15:2227-2235. [PMC free article] [PubMed] [Google Scholar]
- 61.Mayordomo, I., F. Estruch, and P. Sanz. 2002. Convergence of the target of rapamycin and the Snf1 protein kinase pathways in the regulation of the subcellular localization of Msn2, a transcriptional activator of STRE (Stress Response Element)-regulated genes. J. Biol. Chem. 277:35650-35656. [DOI] [PubMed] [Google Scholar]
- 62.Meganathan, R. 2001. Ubiquinone biosynthesis in microorganisms. FEMS Microbiol. Lett. 203:131-139. [DOI] [PubMed] [Google Scholar]
- 63.Moskvina, E., C. Schuller, C. T. Maurer, W. H. Mager, and H. Ruis. 1998. A search in the genome of Saccharomyces cerevisiae for genes regulated via stress response elements. Yeast 14:1041-1050. [DOI] [PubMed] [Google Scholar]
- 64.Neves, M. J., S. Hohmann, W. Bell, F. Dumortier, K. Luyten, J. Ramos, P. Cobbaert, W. de Koning, Z. Kaneva, and J. M. Thevelein. 1995. Control of glucose influx into glycolysis and pleiotropic effects studied in different isogenic sets of Saccharomyces cerevisiae mutants in trehalose biosynthesis. Curr. Genet. 27:110-122. [DOI] [PubMed] [Google Scholar]
- 65.Nierras, C. R., and J. R. Warner. 1999. Protein kinase C enables the regulatory circuit that connects membrane synthesis to ribosome synthesis in Saccharomyces cerevisiae. J. Biol. Chem. 274:13235-13241. [DOI] [PubMed] [Google Scholar]
- 66.Parrou, J. L., B. Enjalbert, L. Plourde, A. Bauche, B. Gonzalez, and J. Francois. 1999. Dynamic responses of reserve carbohydrate metabolism under carbon and nitrogen limitations in Saccharomyces cerevisiae. Yeast 15:191-203. [DOI] [PubMed] [Google Scholar]
- 67.Parrou, J. L., M. A. Teste, and J. Francois. 1997. Effects of various types of stress on the metabolism of reserve carbohydrates in Saccharomyces cerevisiae: Genetic evidence for a stress-induced recycling of glycogen and trehalose. Microbiology 143:1891-1900. [DOI] [PubMed] [Google Scholar]
- 68.Piper, M. D., P. Daran-Lapujade, C. Bro, B. Regenberg, S. Knudsen, J. Nielsen, and J. T. Pronk. 2002. Reproducibility of oligonucleotide microarray transcriptome analyses. An interlaboratory comparison using chemostat cultures of Saccharomyces cerevisiae. J. Biol. Chem. 277:37001-37008. [DOI] [PubMed] [Google Scholar]
- 69.Pritsker, M., Y. C. Liu, M. A. Beer, and S. Tavazoie. 2004. Whole-genome discovery of transcription factor binding sites by network-level conservation. Genome Res. 14:99-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rep, M., M. Krantz, J. M. Thevelein, and S. Hohmann. 2000. The transcriptional response of Saccharomyces cerevisiae to osmotic shock. Hot1p and Msn2p/Msn4p are required for the induction of subsets of high osmolarity glycerol pathway-dependent genes. J. Biol. Chem. 275:8290-8300. [DOI] [PubMed] [Google Scholar]
- 71.Rosenfeld, E., B. Beauvoit, M. Rigoulet, and J. M. Salmon. 2002. Non-respiratory oxygen consumption pathways in anaerobically grown Saccharomyces cerevisiae: evidence and partial characterization. Yeast 19:1299-1321. [DOI] [PubMed] [Google Scholar]
- 72.Rutter, J., B. L. Probst, and S. L. McKnight. 2002. Coordinate regulation of sugar flux and translation by PAS kinase. Cell 111:17-28. [DOI] [PubMed] [Google Scholar]
- 73.Schmitt, A. P., and K. McEntee. 1996. Msn2p, a zinc finger DNA-binding protein, is the transcriptional activator of the multistress response in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 93:5777-5782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sertil, O., R. Kapoor, B. D. Cohen, N. Abramova, and C. V. Lowry. 2003. Synergistic repression of anaerobic genes by Mot3 and Rox1 in Saccharomyces cerevisiae. Nucleic Acids Res. 31:5831-5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Soustre, I., P. H. Dupuy, S. Silve, F. Karst, and G. Loison. 2000. Sterol metabolism and ERG2 gene regulation in the yeast Saccharomyces cerevisiae. FEBS Lett. 470:102-106. [DOI] [PubMed] [Google Scholar]
- 76.Svetlov, V. V., and T. G. Cooper. 1995. Compilation and characteristics of dedicated transcription factors in Saccharomyces cerevisiae. Yeast 11:1439-1484. [DOI] [PubMed] [Google Scholar]
- 77.ter Linde, J. J., H. Liang, R. W. Davis, H. Y. Steensma, J. P. van Dijken, and J. T. Pronk. 1999. Genome-wide transcriptional analysis of aerobic and anaerobic chemostat cultures of Saccharomyces cerevisiae. J. Bacteriol. 181:7409-7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.ter Linde, J. J., and H. Y. Steensma. 2002. A microarray-assisted screen for potential Hap1 and Rox1 target genes in Saccharomyces cerevisiae. Yeast 19:825-840. [DOI] [PubMed] [Google Scholar]
- 79.Thevelein, J. M., L. Cauwenberg, S. Colombo, J. H. de Winde, M. Donation, F. Dumortier, L. Kraakman, K. Lemaire, P. Ma, D. Nauwelaers, F. Rolland, A. Teunissen, P. Van Dijck, M. Versele, S. Wera, and J. Winderickx. 2000. Nutrient-induced signal transduction through the protein kinase A pathway and its role in the control of metabolism, stress resistance, and growth in yeast. Enzyme Microb. Technol. 26:819-825. [DOI] [PubMed] [Google Scholar]
- 80.Thevelein, J. M., and J. H. de Winde. 1999. Novel sensing mechanisms and targets for the cAMP-protein kinase A pathway in the yeast Saccharomyces cerevisiae. Mol. Microbiol. 33:904-918. [DOI] [PubMed] [Google Scholar]
- 81.Thevelein, J. M., and S. Hohmann. 1995. Trehalose synthase: guard to the gate of glycolysis in yeast? Trends Biochem. Sci. 20:3-10. [DOI] [PubMed] [Google Scholar]
- 82.Unkefer, C. J., and R. E. London. 1984. In vivo studies of pyridine nucleotide metabolism in Escherichia coli and Saccharomyces cerevisiae by carbon-13 NMR spectroscopy. J. Biol. Chem. 259:2311-2320. [PubMed] [Google Scholar]
- 83.van Helden, J., B. Andre, and J. Collado-Vides. 2000. A web site for the computational analysis of yeast regulatory sequences. Yeast 16:177-187. [DOI] [PubMed] [Google Scholar]
- 84.Vik, A., and J. Rine. 2001. Upc2p and Ecm22p, dual regulators of sterol biosynthesis in Saccharomyces cerevisiae. Mol. Cell. Biol. 21:6395-6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vuidepot, A. L., F. Bontems, M. Gervais, B. Guiard, E. Shechter, and J. Y. Lallemand. 1997. NMR analysis of CYP1(HAP1) DNA binding domain-CYC1 upstream activation sequence interactions: recognition of a CGG trinucleotide and of an additional thymine 5 bp downstream by the zinc cluster and the N-terminal extremity of the protein. Nucleic Acids Res. 25:3042-3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Waters, B. M., and D. J. Eide. 2002. Combinatorial control of yeast FET4 gene expression by iron, zinc, and oxygen. J. Biol. Chem. 277:33749-33757. [DOI] [PubMed] [Google Scholar]
- 87.Wilcox, L. J., D. A. Balderes, B. Wharton, A. H. Tinkelenberg, G. Rao, and S. L. Sturley. 2002. Transcriptional profiling identifies two members of the ATP-binding cassette transporter superfamily required for sterol uptake in yeast. J. Biol.Chem. 277:32466-32472. [DOI] [PubMed] [Google Scholar]
- 88.Wilson, W. A., and P. J. Roach. 2002. Nutrient-regulated protein kinases in budding yeast. Cell 111:155-158. [DOI] [PubMed] [Google Scholar]
- 89.Yeung, K. Y., M. Medvedovic, and R. E. Bumgarner. 2003. Clustering gene-expression data with repeated measurements. Genome Biol. 4:R34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zahringer, H., J. M. Thevelein, and S. Nwaka. 2000. Induction of neutral trehalase Nth1 by heat and osmotic stress is controlled by STRE elements and Msn2/Msn4 transcription factors: variations of PKA effect during stress and growth. Mol. Microbiol. 35:397-406. [DOI] [PubMed] [Google Scholar]
- 91.Zhu, J., and M. Q. Zhang. 1999. SCPD: a promoter database of the yeast Saccharomyces cerevisiae. Bioinformatics 15:607-611. [DOI] [PubMed] [Google Scholar]
- 92.Zitomer, R. S., and C. V. Lowry. 1992. Regulation of gene expression by oxygen in Saccharomyces cerevisiae. Microbiol. Rev. 56:1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.