

Abstract
The Zyxin/Ajuba family of cytosolic LIM domain-containing proteins has the potential to shuttle from sites of cell adhesion into the nucleus and thus can be candidate transducers of environmental signals. To understand Ajuba's role in signal transduction pathways, we performed a yeast two-hybrid screen with the LIM domain region of Ajuba. We identified the atypical protein kinase C (aPKC) scaffold protein p62 as an Ajuba binding partner. A prominent function of p62 is the regulation of NF-κB activation in response to interleukin-1 (IL-1) and tumor necrosis factor signaling through the formation of an aPKC/p62/TRAF6 multiprotein signaling complex. In addition to p62, we found that Ajuba also interacted with tumor necrosis factor receptor-associated factor 6 (TRAF6) and PKCζ. Ajuba recruits TRAF6 to p62 and in vitro activates PKCζ activity and is a substrate of PKCζ. Ajuba null mouse embryonic fibroblasts (MEFs) and lungs were defective in NF-κB activation following IL-1 stimulation, and in lung IKK activity was inhibited. Overexpression of Ajuba in primary MEFs enhances NF-κB activity following IL-1 stimulation. We propose that Ajuba is a new cytoso lic component of the IL-1 signaling pathway modulating IL-1-induced NF-κB activation by influencing the assembly and activity of the aPKC/p62/TRAF6 multiprotein signaling complex.
The LIM domain is a tandem zinc finger structure mediating protein interactions. The targets that LIM domains interact with are multiple and quite varied in structure and function. Proteins containing LIM domains (LIM proteins) are present in the nucleus and cytoplasm, and some can shuttle between the cytoplasm and nucleus. LIM proteins have been classified based on sequence homology between LIM domains and their overall protein organization (2). Some nuclear LIM proteins contain homeodomains, while others are comprised of only LIM domains (LIM-only proteins or LMO proteins). In the nucleus, these proteins regulate transcription complex formation and function and thus cell lineage fate determination and organ development (31). LIM-only proteins are also present in the cytosol, but in addition, there are LIM domain-containing protein kinases and complex type LIM domain-containing proteins (2). In the cytosol, these proteins contribute to the regulation of cytoskeletal organization, adhesion of cells to the extracellular matrix and to other cells, and cell migration.
Of the complex cytosolic LIM proteins, the Zyxin/Ajuba family is characterized by the presence of three homologous LIM domains at their C terminus (LIM region) and distinct N-terminal PreLIM region. Mammalian members include the following: Ajuba (12), LIMD1 (20), LPP (33), Trip6 (46), and Zyxin (5). While these proteins associate with cytoskeletal elements and are components of cell adhesive complexes, they also shuttle in and out of the nucleus (19, 30, 32) and thus have the potential for transducing signals from environmental cues. The LIM region and the PreLIM region have unique, nonoverlapping binding partners and as such have the potential to link distinct proteins and/or functions. For example, Ajuba is recruited to cell-cell junctions in epithelial cells by virtue of the LIM region interacting with α-catenin bound to the cytoplasmic tail of E-cadherin (27). The PreLIM region associates directly with F-actin (27). As such, Ajuba is thought to contribute to the formation or stabilization of adherens junctions by linking adhesive receptor to the actin cytoskeleton. Primary skin keratinocytes from Ajuba null mice exhibit defects in cell-cell adhesion ex vivo, and Ajuba null mice have defects in skin wound healing (27).
Zyxin/Ajuba LIM proteins have also been implicated in the regulation of other signaling pathways and cellular responses. The PreLIM regions are proline rich and include putative SH3 recognition motifs, and the SH3 domains of Vav and Grb2 have been shown to interact with Zyxin and Ajuba, respectively (12, 17). The functional significance of the Zyxin-Vav interaction is not clear, but the Ajuba-Grb2 interaction augments serum-stimulated extracellular signal-regulated kinase (ERK) activation in a Ras-dependent manner. Ajuba's effect on ERK can result in enhanced fibroblast proliferation and meiotic maturation of Xenopus oocytes (12). Zyxin, LPP, and Trip6 but not Ajuba and LIMD1 contain multiple FPPPP binding motifs for EVH1 domains present in Ena/VASP proteins and may serve to recruit these proteins to the leading edge and thereby influence local actin assembly (36). Ajuba was recently found to regulate Rac activity in migrating cells by influencing the assembly of the p130Cas/Dock180 Rac guanine nucleotide exchange factor at sites of focal adhesions (34). As a result, fibroblasts and keratinocytes from Ajuba null mice exhibit decreased cell motility (34). Accumulation of Ajuba in the nucleus of P19 multipotent embryonal carcinoma cells results in spontaneous endodermal differentiation through a mechanism that is yet to be determined (19). Finally, Zyxin and Ajuba have been implicated as contributing to mitotic cell cycle regulation. Zyxin was found to associate with the tumor suppressor h-warts/LATS1 (16), while Ajuba interacts with and activates the mitotic kinase Aurora A (15).
To further understand Ajuba's role in environmental signal transduction, we performed a yeast two-hybrid screen with the LIM region of Ajuba. We identified the atypical protein kinase C (aPKC) scaffold protein, p62, as an Ajuba binding partner. A prominent function of p62 is the regulation of NF-κB activation in response to interleukin-1 (IL-1), nerve growth factor (NGF), tumor necrosis factor (TNF), and receptor activator of NF-κB ligand (RANKL) signaling through the formation of an aPKC/p62/TRAF6 multiprotein signaling complex. We present evidence indicating that Ajuba is a new cytosolic component of the IL-1 signaling pathway modulating IL-1-induced NF-κB activation by influencing the assembly and activity of the aPKC/p62/TRAF6 multiprotein signaling complex.
MATERIALS AND METHODS
Cells.
Mouse embryonic fibroblasts (MEFs) were isolated as described previously (34) and maintained in Dulbecco's minimal essential medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamate, and antibiotics. HEK293 and HepG2 cells were maintained in DMEM-10% FCS medium. HepG2.tet cell lines were maintained in DMEM medium containing 10% tetracycline-free FBS, 2 mM l-glutamate, and 5 μg/ml blasticidin. MC3T3 osteoblast cell line clone E14 was maintained in αMEM supplemented with 10% FBS, antibiotics, and 2 mM l-glutamate. Bone marrow-derived macrophages (BMDM) were maintained in αMEM-10% FCS plus 100 ng/ml macrophage colony-stimulating factor.
Antibodies and purified proteins.
Ajuba antiserum has been described previously (19, 27). Zyxin polyclonal antibody was kindly provided by M. Beckerle (University of Utah). Mouse monoclonal antibodies against p62 and rabbit polyclonal antibodies against the p50 and p65 subunits of NF-κB were from BD Transduction Laboratories. TRAF6 and PKCζ rabbit polyclonal antibodies were from Santa Cruz. M2AG (mouse monoclonal anti-Flag antibody immobilized on agarose) and horseradish peroxidase (HRP)-conjugated monoclonal anti-Flag, HRP-conjugated monoclonal anti-myc, and HRP-conjugated monoclonal antihemagglutinin (anti-HA) antibodies were all from Sigma (St. Louis, MO). Phospho-IκΒ, IκBα, phospho-ERK, ERK1, phospho-JNK, JNK1, phospho-p38, and p38 polyclonal antibodies were all from Cell Signaling. Mouse IL-1β, human IL-1β, and mouse TNF-α were from PerproTECH. Escherichia coli purified lipopolysaccharide (LPS) was from Sigma. Purified human PKCζ was from Calbiochem. Ajuba, the PreLIM region of Ajuba, and the LIM region of Ajuba were expressed in baculovirus and purified as described previously (27). Purified proteins were dialyzed against immunoprecipitation (IP) buffer without NP-40 and concentrated with centrifugal filter units (Millipore). Protein aliquots were stored at −80°C until use.
Yeast two-hybrid screen.
Competent yeast cells (strain AH109) containing the plasmid pAS2-human Ajuba LIM region were transformed with human keratinocytes Matchmaker cDNA library (Clontech). Transformants (5 × 106) were analyzed according to the manufacturer's instructions. Positive colonies were cured of the bait plasmid by segregation and the library plasmid rescued following transformation of XL1-Blue bacteria. Retransformation of yeast strain AH109 with appropriate specificity controls was carried out to confirm all positives. Library plasmid cDNA inserts were sequenced.
Plasmids.
Human Ajuba, PKCζ, and p62 cDNAs were PCR amplified from a human keratinocyte cDNA library and subcloned into pCMV14 containing various N-terminal epitope tags: Flag, HA, or Myc. TRAF6 cDNA was kindly provided by R. Arch (Washington University). The PreLIM and LIM regions of Ajuba were PCR amplified and likewise subcloned. Human Ajuba, containing a C-terminal Flag epitope, was subcloned into the tetracycline-inducible plasmid pcDNA4 (drug resistance was changed from Zeocin to G418; Invitrogen). TRAF6, p62, and PKC mutants were generated by PCR and subcloned. All plasmids were verified by DNA sequencing and Western blot analysis of cell extracts from HEK293 cells transfected with the respective plasmid. For plasmid transfection of all cell lines, Transit LT-1 (Mirus) was used.
Tetracycline-inducible HepG2 cell lines.
To generate the HepG2.tet repressor cell line, cells were transfected with pcDNA6 containing the tetracycline (tet) repressor (Invitrogen) and selected with 5 μg/ml blasticidin, and clones were isolated and confirmed by transfection of a tet-inducible green fluorescent protein plasmid. Control HepG2.tet- and HepG2.tet-Ajuba-F were obtained by transfection of HepG2.tet with pcDNA4 or pcDNA4-Ajuba-Flag plasmids, respectively, and selected with 1.5 mg/mg G418 and 5 μg/ml blasticidin; single clones were obtained and tested for tet induction followed by Western blotting for the presence of Ajuba-Flag.
IL-1, TNF, and LPS stimulation and cell fractionation.
MEFs or BMDM (2 × 106) were seeded in 100-mm dishes for 24 h and then washed and cultured in serum-free DMEM for 2 h before addition of mouse IL-1β (10 ng/ml), mouse TNF-α (10 ng/ml), or LPS (100 μg/ml) for the times indicated. After stimulation, cells were washed with ice-cold water and lysed with 200 μl of hypotonic buffer (20 mM HEPES [pH 7.5], 5 mM NaF, 1 mM sodium orthovanadate, 0.5 mM EDTA, 1 mM dithiothreitol [DTT], and protease inhibitor cocktail from Sigma). Samples were collected, NP-40 was added to 0.1%, and samples were vortexed and centrifuged at 2,000 rpm for 5 min. The nuclear pellet was then extracted with 20 μl of hypertonic buffer (hypotonic buffer containing 400 mM NaCl), and after centrifugation at 15,000 × g for 15 min, the supernatants were kept as nuclear extract. Total cell lysates were prepared by lysing stimulated cells directly with hypertonic buffer without fractionation and adding sodium dodecyl sulfate (SDS) sample buffer. The protein concentration was determined with a Bio-Rad protein assay kit. HepG2.tet cells were serum starved overnight in the presence or absence of tetracycline (2 ng/ml) and then stimulated with IL-1β, lysed, and fractionated.
For in vivo experiments, 6- to 10-week-old gender-matched wild-type and Ajuba null mice were injected intraperitoneally with 5 ng/g of body weight IL-1β. After various times following injection, mice were sacrificed and lungs were isolated, rinsed with cold phosphate-buffered saline, and frozen immediately on dry ice. Fractionation of lung was performed by adding 0.5 ml of hypotonic buffer per 0.1 g of frozen lung tissue; tissue was homogenized and NP-40 was then added to a 0.5% final concentration, and tissue was kept on ice for 5 min with vortexing every minute. The sample was spun at 5,000 rpm for 10 min and the pellets were washed with hypotonic buffer and then extracted with 200 μl of hypertonic buffer to obtain the nuclear extract. The remaining supernatant was centrifuged at 12,000 rpm for 10 min to obtain the cytosolic extract. The final concentration of NaCl in each sample was made 120 mM for further analysis. Total RNA was prepared from lung using Trizol reagent (Invitrogen) following the manufacturer's instructions.
Semiquantitative RT-PCR.
Reverse transcription (RT) was performed using the SuperScript III first-strand synthesis system (Invitrogen); 2 μg of total RNA was used for each reaction. Reverse transcribed products (1 μl) were used in PCRs. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used as internal control. The following PCR primers were used: MIP-2 (CXCL2) forward, 5′-CAGTGAGCTGCGCTGTCCAATG; reverse, 5′-CAGTTAGCCTTGCCTTTGTTCAG; GAPDH forward, 5′-GCCACCCAGAAGACTGTGGAT; reverse, 5′-TGGTCCAGGGTTTCTTACTCC.
Immunoprecipitation and Western blots.
For immunoprecipitation of endogenous Ajuba, HepG2 cells were harvested, washed with cold phosphate-buffered saline, and lysed with IP buffer (20 mM HEPES [pH 7.5], 120 mM NaCl, 5 mM NaF, 1 mM sodium orthovanadate, 0.5 mM EDTA, 1 mM DTT, 5% glycerol, 0.1% NP-40, and protease inhibitor cocktail from Sigma). Extracts were clarified by centrifugation at 15,000 × g for 15 min. For each IP, 2.5 mg of cell extract protein was mixed with 5 μg of affinity-purified Ajuba antibody or preimmune serum on ice for 2 h and then incubated with 25 μl of protein A-protein G slurry (1:1 [vol/vol]) overnight with gentle rotation at 4°C. The immunoprecipitates were washed five times with IP buffer and boiled in SDS loading buffer. After SDS-polyacrylamide gel electrophoresis under reducing conditions, products were transferred to a nitrocellulose membrane and subjected to p62 Western blot analysis with ECL reagent (Amersham).
NF-κB EMSA.
Oligonucleotides for NF-κB binding sites were labeled at the 5′ end with biotin during synthesis. The following oligonucleotides were used: 5′ biotin-AAGTTGAGGGGACTTTCCCAGGCT 3′ and 5′ biotin-AGCCTGGGAAAGTCCCCTCAACTT 3′. Annealed DNA was purified from a 15% polyacrylamide gel. Two micrograms of nuclear extract protein for each sample was mixed with electrophoretic mobility shift assay (EMSA) binding buffer [10 mM HEPES (pH 7.5), 1.5 mM MgCl2, 50 mM KCl, 2.5% glycerol, 1 mM DTT, 0.5 mM EDTA, 0.2 μg of poly(dI:dC), 0.2 nM purified biotin-labeled NF-κB], and the final volume was adjusted to 20 μl and kept at room temperature for 30 min. The mixture was subjected to 6% Tris-borate-EDTA-polyacrylamide gel electrophoresis. The NF-κB complex signal was detected using HRP-coupled streptavidin as instructed by the manufacturer (Pierce).
IKK kinase assay.
Lung cytosolic extracts containing 500 μg of total protein were incubated with 5 μl of IKKγ antibody for 2 h, followed by overnight incubation with 25 μl of protein A-protein G (1:1). Beads were washed with IP buffer, and then the IKKγ kinase assay was performed as described by Lallena et al. (23), using purified glutathione S-transferase (GST)-IκBα(1-54) as an exogenous substrate.
PKCζ kinase assay.
Purified PKCζ was mixed with baculovirus-produced and purified Ajuba, PreLIM peptide, and LIM peptide on ice for 20 min in kinase buffer (sonicated 20 mM MOPS [morpholinepropanesulfonic acid], pH 7.2, 25 mM β-glycerolphosphate, 1 mM sodium orthovanadate, 1 mM DTT, 1 mM CaCl2, 0.1 mg/ml phosphatidylserine). ATP (50 μM and containing 250 μCi/ml [γ-32P]ATP) and MgCl2 (1.5 mM) were added. Kinase reactions were started by mixing 40 μl of kinase buffer with enzyme mixture and incubated at 30°C for 10 min.
RESULTS
The LIM protein Ajuba associates with the aPKC-interacting protein p62.
To further understand the potential function(s) of Ajuba in intracellular signaling pathways, we sought to identify cellular proteins interacting with Ajuba. Towards this aim, a yeast two-hybrid protein-protein interaction screen was performed using the C-terminal LIM region (three LIM domains) of human Ajuba as bait (Fig. 1A). Since Ajuba is abundantly expressed in skin, a human keratinocyte library was screened. Two of the five true positive interacting clones coded for p62, an aPKC-interacting protein (35, 40), or phosphotyrosine-independent p56lck SH2-interacting protein (18). Both clones contained a partial cDNA encoding the N-terminal portion (amino acids [aa] 1 to 266 and aa 1 to 333) of p62.
FIG. 1.

The LIM protein Ajuba associates with the aPKC-interacting protein p62. (A) Stick figure representation of Ajuba. The gray boxes represent the three LIM domains present in the C-terminal LIM region. The PreLIM region is N terminal. (B) Ajuba and p62 associate in human HepG2 cells. Cell extracts were immunoprecipitated with preimmune serum (PI, lane 1) or Ajuba antiserum (lane 2). Bound products were Western blotted for the presence of p62. In lane 3, 0.5% of the amount of cell extract used for immunoprecipitation (input) was run. (C) Ajuba and p62 colocalize in cells. Fibroblasts were transfected with Flag-tagged p62 alone (panel i), myc-tagged Ajuba alone (panel ii), or both p62-Flagand myc-Ajuba (panels iii to v) and immunostained for p62 (anti-Flag, red, panels i and iii) or Ajuba (anti-myc, green, panels ii and iv). Panel v is a merged image of panels iii and iv. Arrows identify vesicular, endosomal structures. Arrowheads identify focal adhesion sites. (D) Ajuba and LIMD1 but not Zyxin or LPP associate with p62. HEK293 cells were transfected with p62 (Flag-tagged) and myc-tagged LIM proteins, as indicated. p62 was immunoprecipitated (anti-Flag), and bound products were Western blotted for the presence of LIM protein (anti-myc, upper panels) and p62 (anti-Flag, lower panels). For each set, input controls were run (5% of amount used for immunoprecipitation). (E) Mapping of the region of Ajuba that interacts with p62. HEK293 cells were transfected with myc-tagged p62 and Flag-tagged isoforms of Ajuba, as indicated. Ajuba was immunoprecipitated (anti-Flag), and bound products were Western blotted for the presence of p62 (anti-myc, left upper panel) and Ajuba isoforms (anti-Flag, left lower panel). Five percent of the cell extract used for immunoprecipitation was run as an input control (right panels).
To determine whether Ajuba and p62 interact with one another in cells expressing endogenous levels of each protein, Ajuba was immunoprecipitated from the human hepatocyte cell line, HepG2, and bound p62 was detected by Western blotting. A significant proportion of cellular p62 was present in Ajuba immunoprecipitates, while preimmune serum did not immunoprecipitate p62 protein (Fig. 1B). Next, we asked whether Ajuba and p62 colocalized in cells. To do so, MC3T3 cells were transfected with myc-tagged Ajuba and Flag-tagged p62 plasmids, either alone or together, and indirect immunofluorescence was performed on fixed cells. Consistent with previous reports, when expressed alone, p62 localized to punctate, vesicular structures (endosomes) (Fig. 1Ci, arrows) (40), while Ajuba was diffusely distributed throughout the cytoplasm and present at focal adhesion sites (Fig. 1Cii, arrowheads) (34). When p62 and Ajuba were coexpressed, Ajuba colocalized with p62 at vesicular structures (Fig. 1Ciii, iv, and v, arrows), and p62 colocalized with Ajuba at some, but not all, focal adhesion sites (Fig. 1Ciii, iv, and v, arrowheads). Together, these two results indicated that Ajuba interacted with p62 in cells.
In mammals there are five related Ajuba family members: Ajuba, LIMD1, Zyxin, LPP, and Trip6. The homology between these proteins is in the three C-terminal LIM domains. Phylogenetic analysis, protein sequence analysis, and overall protein organization suggest that Ajuba and LIMD1 may form a subfamily, while Zyxin, LPP, and Trip6 are more closely related to one another than Ajuba or LIMD1. To determine whether there was specificity among Ajuba/Zyxin family proteins for interacting with p62, HEK293 cells were cotransfected with myc-tagged LIM proteins and Flag-tagged p62, p62 was immunoprecipitated (anti-Flag), and then bound products were Western blotted for the presence of LIM protein (anti-Myc). Only Ajuba and related LIMD1 associated with p62 (Fig. 1D). Zyxin and LPP did not associate with p62 (Fig. 1D). Mapping studies were then performed to determine the region of Ajuba directing its association with p62. Full-length Ajuba and the LIM region of Ajuba associated with p62, while the PreLIM region of Ajuba did not (Fig. 1E). Similar results were obtained from LIMD1 mapping studies (data not shown). These results indicated that there was specificity in the LIM protein family members interacting with p62 and that the LIM region of Ajuba directed its interaction with p62.
Ajuba is required for efficient IL-1- and TNF-induced activation of NF-κB by affecting IKK activation.
p62 associates with the atypical PKCs (ζ and λ) (35, 40), receptor-interacting protein (RIP) (42), and TRAF6 (41) and as such is thought to function as a scaffolding protein influencing IL-1, TNF, NGF, and RANKL signaling to activate NF-κB (8, 41, 42, 45). Since the LIM protein Ajuba interacted with p62, we asked whether Ajuba influenced NF-κB activation in response to IL-1 or TNF stimulation. To test this possibility, primary cells from Ajuba null mice and in vivo stimulation of Ajuba null mice were both evaluated.
First, Ajuba−/− primary MEFs were compared to wild-type (wt) (Ajuba+/+), littermate-matched cells (loss of Ajuba model). MEFs were stimulated with IL-1β (two different litter-matched sets) or TNF-α. NF-κB activation was determined by EMSAs of nuclear extracts. In two different litter-matched sets of MEFs, NF-κB activation was reduced in Ajuba null MEFs in response to both IL-1 and TNF stimulation (Fig. 2A). The observed EMSA was specific for NF-κB, as the addition of antibodies to the nuclear p50 subunit of NF-κB to the EMSA reaction completely shifted the band, while control antibodies did not (see Fig. 5B). IL-1-induced IκB phosphorylation, which was required for the release of cytoplasmic NF-κB complexes and subsequent translocation into the nucleus, was also reduced in Ajuba−/− MEFs compared to wt controls (Fig. 2B, upper panel), suggesting that Ajuba contributed to the cytosolic activation of NF-κB. Consistent with decreased IκΒ phosphorylation, IκΒα protein was more stable in Ajuba null cells (Fig. 2B, lower panel). Finally, the transcription of IκΒα, an NF-κB-regulated gene, was decreased in Ajuba null cells subsequent to IL-1 stimulation (Fig. 2B, lower panel, 60-min time point).
FIG. 2.

Ajuba is required for efficient activation of NF-κB by IL-1 and TNF. (A) Two sets of litter-matched Ajuba−/− and control wt MEFs were stimulated with IL-1β (10 ng/ml) or TNF-α (10 ng/ml) for the indicated times. Nuclei were isolated, and EMSA with NF-κB-specific oligonucleotides was performed, as described in the text. Equal amounts of nuclear extracts were used in each lane. The percent NF-κB activity in Ajuba null cells compared to control wt cells is indicated under each lane. (B) Ajuba−/− and control wt MEFs were treated as in panel A, and cytosolic fractions from the same experimental cells were Western blotted for phospho-IκΒ (upper panel) and total IκΒα protein (lower panel). The percent NF-κB activity in Ajuba null cells compared to control wt cells is indicated under each lane. (C) Ajuba−/− (left lane) and Ajuba null MEFs stably expressing exogenous Ajuba (right lane) were treated as described for panel A, and nuclear EMSA for NF-κB activity was performed. The percent NF-κB activity in Ajuba-rescued cells compared to Ajuba null cells is indicated under each lane. The lower panel is an anti-Ajuba Western blot comparing the level of Ajuba protein in wt (+/+), Ajuba null (−/−), and Flag-tagged Ajuba-rescued Ajuba null MEFs (−/−, +A). Equal amounts of protein were loaded in each lane. (D) Zyxin−/− and control wt MEFs were stimulated with IL-1β (10 ng/ml) for the indicated times. Nuclei were isolated, and EMSA with NF-κB-specific oligonucleotides was performed, as in panel A. Equal amounts of nuclear extracts were used in each lane. The percent NF-κB activity in Zyxin null cells compared to control wt cells is indicated under each lane. (E) Ajuba−/− and control wt MEFs were treated as in panel A, and the same cytosolic fractions were Western blotted for phospho-JNK, total JNK-1, phospho-ERK, total ERK1, phospho-p38, and total p38 protein. Equal amounts of protein were loaded in each lane.
FIG. 5.

Ajuba recruits TRAF6 into the p62/PKCζ signaling complex. (A) HepG2 cells containing tet-regulated Ajuba (Flag-tagged) were stimulated with differing concentrations of tetracycline, and Ajuba (anti-Flag) Western blotting was performed. (B) NF-κB supershift assay. HepG2 cells containing tet-regulated Ajuba (Flag-tagged) were induced with tetracycline (2 ng/ml) and then stimulated with IL-1β (10 ng/ml). Nuclei were isolated, and NF-κB EMSA was performed in the absence (lane 2) or presence of antibodies to the p50 subunit of NF-κB (lane 3), Flag (lane 4), or Ajuba (lane 5). NS indicates nonspecific bands. (C and D) HepG2 cells containing tet-regulated Ajuba (Flag-tagged) were induced with tetracycline (2 ng/ml) and then stimulated with IL-1β (10 ng/ml) for the indicated times. Ajuba (anti-Flag) (C) or endogenous p62 (D) were immunoprecipitated, and bound products were Western blotted for the presence of endogenous p62, TRAF6, PKCζ, and p38 as a negative control. Right panels show input controls. (E) HEK293 cells were cotransfected with fixed amounts of TRAF6 (myc-tagged), either p62 (Flag-tagged) (left panels) or p62.Δ225-255 (right panels), and increasing amounts of HA-tagged Ajuba. p62 was immunoprecipitated (anti-Flag), and bound products were Western blotted for the presence of TRAF6 (anti-myc), Ajuba (anti-HA), and p62 (anti-Flag) (left panels). Right panels show input controls.
When Ajuba was stably reintroduced into Ajuba−/− MEFs, NF-κB activation, in response to IL-1β and TNF-α, returned to levels observed in wt MEFs or greater (Fig. 2C), indicating that the absence of Ajuba was directly related to decreased NF-κB activation. Furthermore, the level of exogenous Ajuba protein present in Ajuba-rescued Ajuba−/− MEFs was greater than that present in wt MEFs (fivefold) (Fig. 2C, lower panel) and NF-κB activity in Ajuba-rescued Ajuba−/− MEFs was greater than that observed in wt MEFs; this suggested that when Ajuba was overexpressed in primary MEFs, activation of NF-κB by IL-1 was enhanced and was consistent with the decrease in NF-κB activation observed in Ajuba null MEFs (Fig. 2A).
Since the Ajuba-related LIM protein Zyxin did not interact with p62 (Fig. 1D), we asked whether IL-1 signaling was influenced by loss of Zyxin. IL-1 stimulation of Zyxin−/− MEFs was compared to wt (Zyxin+/+) control MEFs. In contrast to Ajuba−/− MEFs, we observed no diminution of NF-κB activation (Fig. 2D) or significant alteration in any of the mitogen-activated protein kinase (MAPK) activation profiles in Zyxin−/− MEFs in response to IL-1 (see Fig. S1B in the supplemental material), suggesting that the interaction between Ajuba and p62 was important for Ajuba's effect upon NF-κB activation following IL-1 stimulation.
IL-1 signals also activate the MAPKs ERK, JNK, and p38, yet precisely how these kinases are activated is less clear (7). We determined whether the loss of Ajuba influenced IL-1-induced activation of JNK, ERK, and p38 by analyzing Ajuba−/− MEFs versus control wt MEFs. Following stimulation with IL-1, Ajuba−/− MEFs exhibited a slight reduction in p38 activation and a persistence of JNK activation relative to Ajuba+/+ MEFs (Fig. 2C). There were no significant changes in ERK activation between wt and Ajuba null MEFs, however (Fig. 2E).
The levels of p62, TRAF6, PKCζ, and other Ajuba family LIM proteins were determined in IL-1-stimulated Ajuba−/− and Ajuba+/+ MEFs (see Fig. S1A in the supplemental material). In the absence of Ajuba, the level of related LIM protein family members was not altered. IL-1 stimulation did not alter the protein level of any of the adapter proteins (p62 and TRAF6) or kinase (PKCζ) analyzed. A slight increase in Ajuba protein level was apparent after 60 min of IL-1 stimulation of wt MEFs.
These analyses in primary MEFs revealed that Ajuba was required for IL-1 and TNF signaling to efficiently activate NF-κB. This effect was specific to the Ajuba LIM protein, as loss of the related LIM protein Zyxin, which does not interact with p62, did not affect NF-κB activation following IL-1 stimulation, suggesting that Ajuba's interaction with p62 was important for its regulation of NF-κB activity.
The level of Ajuba protein in MEFs is relatively low compared to other tissues. Epithelial cells contain much higher levels of Ajuba (27). PKCζ, which also interacts with p62, was also shown to only modestly affect NF-κB activity in MEFs (low level of PKCζ); however, when the lung (high level of PKCζ) from PKCζ null mice was examined following IL-1 stimulation in vivo, a much greater inhibition of NF-κB was observed (24). Since Ajuba levels in the lung are much higher than in MEFs (26), NF-κB activation in the lungs of Ajuba null mice was determined following intraperitoneal injection of IL-1. The extent of NF-κB inhibition in the lungs of Ajuba null mice was found to be much greater than observed in MEFs (Fig. 3A). Moreover, as observed in PKCζ knockout mice, nuclear transport of the p65 subunit of NF-κB was also inhibited in Ajuba null lungs (Fig. 3A) (24). In addition, transcription of the NF-κB-regulated genes CXCL2 or MIP-2 (44) was decreased in Ajuba null lung in response to IL-1 (Fig. 3B).
FIG. 3.

Ajuba is required for IL-1-induced NF-κB activation in vivo through activation of IKK activity. (A) Ajuba−/− and control wt mice were injected with IL-1β for the indicated times. Nuclear extracts from the lungs of these mice were analyzed by NF-κB EMSA assays (upper panel), nuclear p65 subunit of NF-κB Western blotting (middle panel), and nucleophosmin Western blotting of nuclear extracts as a loading control (lower panel). (B) Semiquantitative RT-PCR analysis of lungs from IL-1β-injected mice for the level of the NF-κB-responsive gene CXCL2 (MIP-2) (44) (upper panel). Loading control is GAPDH (lower panel). (C) IKK in vitro kinase assay of cytosolic extracts from the lungs of the mice described for panel A. GST-IκΒα(1-54) was used as exogenous substrate.
PKCζ modulates NF-κB activity through an effect upon IKK activity in lung (24); therefore, we asked whether, in the absence of Ajuba, lung IKK activity was also inhibited following IL-1 stimulation. IKKγ was immunoprecipitated from IL-1-stimulated lung extracts, and in vitro kinase assays using GST-IκΒα as substrate were performed. IKK activity was inhibited in Ajuba null lungs, compared to wt controls, following IL-1 stimulation (Fig. 3C). Thus, when tissues (e.g., lung) expressing high levels of Ajuba were analyzed, a much greater inhibition of IL-1-stimulated NF-κB activity was observed. In lung, it appeared that Ajuba, like PKCζ, affected NF-κB activation by regulating IKK activity.
A unique domain in p62, not overlapping with aPKC or TRAF6 binding sites, directs its interaction with Ajuba.
p62 interacts with signaling proteins important for the activation of NF-κB by IL-1 (reviewed in reference 10). Specifically, the aPKCs interact with the aPKC-interacting domain (AID) near the N terminus (35), and TRAF6 interacts with a domain (T6) present at aa 225 to 251 of p62 (41). In addition, the TNF signaling intermediate RIP associates with p62 at the zinc finger domain (ZZ) (42). To determine the region of p62 mediating its interaction with Ajuba, a series of Flag-tagged p62 truncation and deletion mutants (Fig. 4D) was cotransfected with myc-tagged Ajuba into HEK293 cells. p62 mutants were immunoprecipitated using anti-Flag antibody and bound products Western blotted for the presence of Ajuba (anti-myc). Analysis of a panel of p62 truncation mutants indicated that the N-terminal 1 to 220 amino acids were critical for the interaction with Ajuba (Fig. 4A). Further deletion of aa 170 to 220 resulted in the loss of Ajuba binding to p62 (Fig. 4A, lane 6), indicating that this region of p62 was important to mediate the association between p62 and Ajuba.
FIG. 4.

A unique domain in p62, not overlapping with aPKC or TRAF6 binding sites, directs its interaction with Ajuba. HEK293 cells were cotransfected with myc-tagged Ajuba and Flag-tagged p62. p62 (anti-Flag) was immunoprecipitated, and bound products were Western blotted for the presence of Ajuba (anti-myc) and p62 (anti-Flag) (left panels). Right panels show input controls. (A) Mapping of the p62 truncation mutants. (B and C) Mapping of p62 deletion mutants. (D) Stick figure representation of p62 mutants tested and those that associate with Ajuba. AID is the atypical PKC-interacting domain, ZZ is the zinc finger RIP-interacting domain, LB is the novel LIM protein binding domain, and T6 is the TRAF6 binding domain.
To confirm that this region was indeed required to direct p62's association with Ajuba, a panel of p62 deletion mutants was tested. Deletion of aa 170 to 220, in the context of full-length p62, resulted in the loss of association with Ajuba (Fig. 4B, lane 3), while deletion of AID (aa 66 to 82), the ZZ domain (RIP-interacting domain, aa 128 to 163), and the TRAF6 (T6) binding domain (aa 225 to 255) had no effect upon p62's association with Ajuba (Fig. 4C, lanes 3, 4, and 5, respectively).
We have named the domain between aa 170 and 220 of p62 a new LIM protein-binding region (LB). Importantly, the LB domain of p62 did not overlap with the binding sites for other IL-1 or TNF signaling intermediates, such as aPKC, TRAF6, and RIP.
Ajuba levels affected the amount of TRAF6 present in the p62/PKCζ signaling complex.
To determine biochemically whether Ajuba regulates the p62 signaling complex and how, we turned to an Ajuba inducible cell line. We generated HepG2 cell lines containing a tetracycline-regulated, Flag-tagged Ajuba plasmid. HepG2 cells were used, as they are human and available antibodies to p62 only immunoprecipitate human p62, precluding a similar analysis in mouse cells. HepG2 cells express endogenous Ajuba, and this level was not affected by IL-1 stimulation (data not shown). Due to leakiness of the promoter, a small amount of Flag-Ajuba was expressed in the absence of tetracycline, but following the addition of increasing amounts of tetracycline (2 to 250 ng/ml) Flag-Ajuba expression progressively increased (Fig. 5A). To ensure that the observed NF-κB activity in IL-1-stimulated HepG2.tetAjuba cells was indeed NF-κB, cells were induced with tetracycline, and EMSA assays were performed in the presence of antibodies to the p50 subunit of NF-κB or control Ajuba or Flag antibodies. Anti-p50 antibodies completely shifted the NF-κB complex (Fig. 5B), while Ajuba and Flag antibodies had no effect on the mobility of the NF-κB complex (Fig. 5B). This result also indicated that Ajuba was not present in a nuclear complex containing the p50 protein of the NF-κB complex.
TRAF6 is an important adapter protein in IL-1-induced NF-κB activation. TRAF6 null MEFs are defective in NF-κB activation following IL-1 stimulation (25). TRAF6 is thought to activate NF-κB by interacting with transforming growth factor-activated kinase TAK1 and its regulator TAB-1 to activate IKK (29, 39). TRAF6 forms a complex with p62 and PKCζ in an IL-1-dependent manner, and this complex influences IL-1-induced NF-κB activity (41). Since Ajuba interacts with a unique domain in p62 and Ajuba affects IL-1-induced NF-κB activation, Ajuba may influence IL-1 signaling by affecting the assembly of the p62/PKCζ/TRAF6 multiprotein signaling complex. To test this hypothesis, we induced overexpression of Ajuba in HepG2 cells and then treated them with IL-1 for increasing periods of time (0 to 30 min). Exogenous Ajuba (Fig. 5C) or endogenous p62 (Fig. 5D) was immunoprecipitated, and bound products were Western blotted for endogenous TRAF6, PKCζ, and p62. In the absence of IL-1 Ajuba, p62, and PKCζ but not TRAF6 were present in a multiprotein complex coimmunoprecipitating with Ajuba (Fig. 5C, lane 2). Following IL-1 addition, the amount of p62 present in Ajuba immunoprecipitates increased slightly, while the levels of PKCζ did not appreciably change (Fig. 5C, lanes 3 to 5). Interestingly, however, following IL-1 stimulation TRAF6 was recruited into the Ajuba/p62/PKCζ complex (Fig. 5C, lanes 3 to 5). Similar results were observed when endogenous p62 was immunoprecipitated, and bound products were Western blotted for Ajuba (Flag), endogenous TRAF6, and endogenous PKCζ (Fig. 5D). These results suggested that Ajuba could increase the recruitment of TRAF6 to a p62/PKCζ complex following IL-1 stimulation.
To further test this hypothesis, HEK293 cells were transfected with fixed amounts of Flag-tagged p62 and myc-tagged TRAF6 and increasing amounts of HA-tagged Ajuba. p62 was immunoprecipitated, and bound products were Western blotted for TRAF6 and Ajuba. Overexpressed p62 and TRAF6 associated in HEK293 cells in the absence of exogenous Ajuba. Following transfection with increasing amounts of Ajuba, substantially more TRAF6 was now present in the p62 immunoprecipitates (Fig. 5E, left panel, lanes 3 and 4), suggesting that Ajuba levels influence the amount of TRAF6 recruited to p62.
To address the molecular mechanism of this process, we transfected cells with fixed amounts of a mutant form of p62 that no longer binds TRAF6 (p62.Δ225-255) (Fig. 4A), TRAF6, and increasing amounts of Ajuba. p62 was immunoprecipitated, and bound products were Western blotted for TRAF6 and Ajuba. In the absence of exogenous Ajuba, very little TRAF6 associated with p62.Δ225-255, as anticipated (Fig. 5E, right panel, lane 2); however, following transfection with increasing amounts of Ajuba, TRAF6 was still recruited to p62.Δ225-255 (Fig. 5E, right panel, lanes 3 and 4), albeit at levels less than detected in cells transfected with wild-type p62 (Fig. 5E, left panel). Therefore, although a p62 protein lacking its TRAF6 binding domain does not efficiently interact with TRAF6, Ajuba can promote the recruitment of TRAF6 to p62, suggesting that Ajuba may itself bind TRAF6, thereby further increasing TRAF6 recruitment into the p62/PKC signaling complex.
Ajuba interacts with TRAF6.
To test whether Ajuba does interact with TRAF6, HEK293 cells were transfected with Ajuba and various truncation mutants of TRAF6 (Fig. 6C) or TRAF2 as a negative control (not part of the IL-1 signaling pathway and does not interact with p62 [41]). TRAF6 associated with Ajuba in cells (Fig. 6A, lane 2), while TRAF2 did not (data not shown). TRAF6 mapping studies demonstrated that its interaction with Ajuba was largely directed by the TRAF-N region (aa 270 to 352) within the C-terminal TRAF domain (Fig. 6A, lane 6). Deletion of the TRAF-C region of the TRAF domain also had a small effect on the amount of Ajuba bound to TRAF6 (Fig. 6A, lane 5). Ajuba mapping studies demonstrated that the LIM region of Ajuba directed its association with TRAF6, and the PreLIM region did not interact with TRAF6 (Fig. 6B). Therefore, in addition to interacting with p62, Ajuba also interacts with TRAF6. Likewise, TRAF6 interacts with p62 and Ajuba.
FIG. 6.

Ajuba interacts with TRAF6. (A) Mapping of the TRAF6 domains required for an interaction with Ajuba. HEK293 cells were cotransfected with myc-tagged Ajuba and Flag-tagged TRAF6 isoforms. TRAF6 (anti-Flag) was immunoprecipitated, and bound products were Western blotted for the presence of Ajuba (anti-myc) and TRAF6 (anti-Flag) (left panels). Right panels show input controls. (B) Mapping of the Ajuba regions required for an interaction with TRAF6. HEK293 cells were cotransfected with Flag-tagged Ajuba isoforms and myc-tagged TRAF6. Ajuba isoforms (anti-Flag) were immunoprecipitated, and bound products were Western blotted for the presence of TRAF6 (anti-myc) and Ajuba (anti-Flag) (left panels). Right panels show input controls. (C) Stick figure representation of TRAF6 mutants and their association with Ajuba. The light gray box is the RING finger domain, the black box is the five zinc fingers, the dark gray box is the N-terminal region of the TRAF domain, and the hatched box is the C-terminal region of the TRAF domain.
Ajuba interacts with PKCζ, is a substrate for PKCζ, and activates PKCζ activity.
In addition to TRAF6, PKCζ also activates IKK, leading to activation of NF-κB following IL-1 stimulation (14, 24). TRAF6, through its interaction with p62, may also influence PKCζ activity (41). Since Ajuba interacts with p62 and TRAF6, increasing the recruitment of TRAF6 to p62, and affects IL-1-induced NF-κB activity, it begs the question: does Ajuba influence IL-1-induced NF-κB activity by interacting with PKCζ or affecting its enzyme activity?
When Ajuba was immunoprecipitated from HepG2 cells, p62 and PKCζ were present in the immunoprecipitate (Fig. 5C). PKCζ could be present in this Ajuba protein complex by virtue of its interaction with p62, through an association with Ajuba independent of p62, or both. Therefore, to determine whether PKCζ interacted with Ajuba, HEK293 cells were cotransfected with various Flag-tagged truncation mutants of PKCζ (Fig. 7C) and myc-tagged Ajuba. PKCζ was immunoprecipitated and bound products Western blotted for the presence of Ajuba. PKCζ did indeed associate with Ajuba, and this interaction mapped to the N-terminal 1 to 244 aa of PKCζ (Fig. 7A). Ajuba did not interact with the C-terminal kinase domain of PKCζ (Fig. 7A). Ajuba mapping studies demonstrated that the LIM region of Ajuba directed its association with PKCζ and that the PreLIM region did not interact (Fig. 7B).
FIG. 7.

Ajuba interacts with PKCζ, activates PKCζ, and is a substrate of PKCζ. (A) Mapping of the PKCζ region required to interact with Ajuba. HEK293 cells were cotransfected with myc-tagged Ajuba and Flag-tagged PKCζ mutants. PKCζ isoforms were immunoprecipitated (anti-Flag), and bound products were Western blotted for the presence of Ajuba (anti-myc) (left panels). Right panels show input controls. (B) Mapping of the Ajuba region required to interact with PKCζ, as described for panel A. (C) Stick figure representation of PKCζ mutants and their interaction with Ajuba. (D) Coomassie-stained gel of baculovirus-produced and purified Ajuba, PreLIM peptide of Ajuba, and LIM peptide of Ajuba. Molecular weight markers in thousands are on the left. (E) In vitro PKCζ kinase assays. A fixed amount of purified PKCζ (20 ng) was mixed with increasing amounts of Ajuba protein (upper panel) or PreLIM and LIM peptides (lower panel). Arrows identify the location of PKCζ and Ajuba peptides. (F) Purified PKCζ (20 ng) was mixed with PreLIM peptide (400 ng). Increasing amounts of Ajuba LIM peptide were added, and an in vitro kinase reaction was performed. In this assay, more nonradioactive ATP was present than in panel E. The arrow identifies a phosphorylated PreLIM peptide. The increase (n-fold) in PreLIM phosphorylation relative to that observed in the absence of LIM peptide (set at 1) is shown below each lane.
Ajuba is a phosphoprotein (12, 15). Therefore, we next asked whether Ajuba could be a substrate for PKCζ and whether Ajuba affected PKCζ activity. Full-length Ajuba, the PreLIM region of Ajuba, and the LIM region of Ajuba were expressed in baculovirus and purified (Fig. 7D). A fixed amount of pure PKCζ was mixed with increasing amounts of purified Ajuba protein, and kinase reaction in the presence of [32P]ATP was performed. Both full-length Ajuba and the PreLIM region were phosphorylated by PKCζ, while the LIM region was not (Fig. 7E). In addition, autophosphorylation of PKCζ was enhanced by the presence of full-length Ajuba and the LIM region of Ajuba (both interact with PKCζ), and the extent of PKCζ autophosphorylation increased with increasing amounts of added Ajuba or LIM region (Fig. 7E). The LIM region alone appeared to activate PKCζ to the greatest extent.
Next, we mixed PKCζ and Ajuba PreLIM peptide (“substrate”) and then added increasing amounts of LIM region peptide (“activator”) to the reaction. In the presence of increasing amounts of LIM region, more PreLIM peptide was phosphorylated (Fig. 7F). The intensity of phosphorylation was less than that observed in Fig. 7E because larger amounts of nonradioactive ATP, in excess of [32P]ATP, were added to the kinase reaction. These in vitro data suggested that Ajuba (specifically the PreLIM region) could be a substrate of PKCζ and that binding of Ajuba (particularly the LIM region) activated PKCζ enzyme activity.
DISCUSSION
Herein we demonstrate that the LIM protein Ajuba is a new component of the proinflammatory cytokines IL-1 and TNF signal transduction cascades regulating NF-κB activation.
IL-1 can activate NF-κB by interrelated signaling pathways (reviewed in references 7 and 10). First, IL-1 interacts with a specific cell surface receptor (IL-1R) on target cells. Receptor activation leads to the recruitment of the myeloid differentiation factor (MyD88) to the receptor complex. MyD88 serves to further recruit members of the IL-1R kinase family (IRAK) to the receptor complex. IRAK plays a critical role in the activation of NF-κB through its interaction with TRAF6. In response to IL-1, TRAF-6 becomes ubiquitinated, oligomerizes, and interacts with TAB-2, the transforming growth factor-activated kinase 1 (TAK1), and TAB-1. Activated TAK1 can then activate the IκΒ kinase complex (IKK), leading to phosphorylation of IκΒ and its subsequent degradation with release of NF-κB proteins and their translocation into the nucleus, where inflammatory, apoptotic, or antiapoptotic pathways are regulated, depending upon cell type (3, 4).
Alternatively, IL-1 can activate NF-κB through a cytosolic pathway involving aPKC (13, 38), p62 (41), and TRAF6 (41). p62 has been proposed to function as a scaffold protein, in that it interacts with both PKCζ and TRAF6 in an IL-1-dependent manner. Dominant negative aPKC blocks IL-1-induced and TRAF6-induced NF-κB activation (41), suggesting that TRAF6, when bound to p62, may activate aPKC. Furthermore, the aPKC/p62/TRAF6 complex appears to function downstream of IRAK (41). Cells and organs from mice deficient in p62 (41), TRAF6 (25, 28), or PKCζ (24) all exhibit defective NF-κB activation in response to proinflammatory cytokines of the IL-1 family.
Primary MEFs from Ajuba null mice and the lungs from Ajuba null mice injected with IL-1 have diminished NF-κB activation following IL-1 and TNF stimulation. This defect in NF-κB activation is biologically relevant, as Ajuba null MEFs are more sensitive to TNF-induced apoptosis (data not shown), and reintroduction of Ajuba into Ajuba null MEFs rescues the IL-1- and TNF-induced NF-κB signaling defect and prevents TNF-induced cell death. Likewise, overexpression of Ajuba in primary MEFs increased NF-κB activity following IL-1 stimulation.
We propose that Ajuba influences IL-1-induced NF-κΒ activation by affecting the assembly of the PKCζ/p62/TRAF6 multiprotein complex in cells and thus IKK activity. Using purified proteins in vitro, we also found that Ajuba could be a PKCζ substrate and enhance PKCζ activity. The aPKC-interacting protein p62 was identified as interacting with Ajuba in a two-hybrid screen, and endogenous p62 coimmunoprecipitates and colocalizes with endogenous Ajuba in cells. Within the Ajuba-related LIM protein family, there was specificity in interacting with p62. Importantly, p62-noninteracting Zyxin null MEFs have no defect in IL-1-induced NF-κB activity, suggesting that Ajuba's interaction with p62 was crucial for its regulation of NF-κB activity.
In addition to interacting with p62, Ajuba also interacted with TRAF6. The interaction with TRAF6 was also specific, as Ajuba did not interact with related TRAF2. Mapping studies identified the TRAF domain of TRAF6 as mediating its interaction with Ajuba. Within the TRAF family, the TRAF domain in TRAF6 is most divergent (1). Other proteins interacting with this domain are p62 (41) and ECSIT (22), and each, like Ajuba, is specific for TRAF6. The TRAF domain is responsible for oligomerization of TRAF6, which is thought to be critical for signaling by the N-terminal region (3). Progressive overexpression of Ajuba results in increased recruitment of TRAF6 to p62 and enhanced NF-κB activity. So it is possible that Ajuba and p62 serve to “activate” TRAF6, bringing its effector domain into close proximity to PKCζ bound to p62 or its other downstream kinase, TAK1 (Fig. 8).
FIG. 8.
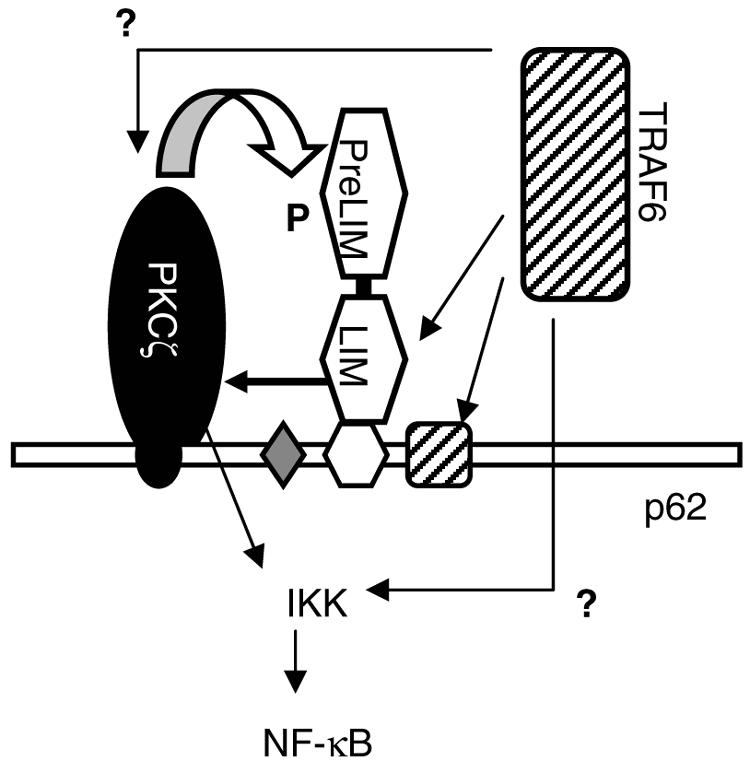
Working model for how Ajuba influences IL-1-induced NF-κB activation. PKCζ and the PKCζ binding site in p62 (AID) are in black. Ajuba and the LIM protein binding site in p62 (LB) are in white. TRAF6 and the TRAF6 binding site in p62 (T6) are hatched. The LIM region of Ajuba interacts with both TRAF6 and PKC. In vitro, the LIM region of Ajuba activates PKC, and the PreLIM region of Ajuba can be phosphorylated by PKC. Whether phosphorylation of Ajuba regulates either the assembly of the multiprotein complex or the capacity of the complex to activate IKK is not known.
Mapping studies identified a unique domain (LB domain) in p62 that directs its association with Ajuba. Importantly, the LB domain does not overlap with the aPKC, RIP, or TRAF6 binding domains previously identified in p62 (41, 42). Interestingly, the LB domain of p62 lies just N-terminal to the TRAF6 binding domain. Taken together, these observations suggest the following possibilities for how Ajuba could influence the recruitment of TRAF6 to p62: (i) following Ajuba binding to p62, TRAF6 could be recruited to p62 through an interaction with Ajuba or p62 (i.e., independent binding sites); (ii) a preformed Ajuba-TRAF6 protein complex could be recruited to p62; (iii) Ajuba could stabilize the association of TRAF6 with p62; (iv) Ajuba could also influence the activation (i.e., oligomerization and ubiquitination) of TRAF6 bound to p62 (6, 43); or (v) some combination of the above mechanisms. Future biochemical studies with purified protein components and genetic studies of mutant proteins expressed in cells from knockout mice should begin to address these possibilities.
We also found that Ajuba could interact with PKCζ independently of p62. In vitro, Ajuba activated PKCζ enzyme activity, and Ajuba could be a PKCζ substrate. Interestingly, the LIM region of Ajuba activates PKCζ but is not phosphorylated, whereas the PreLIM is readily phosphorylated by PKCζ but does not significantly activate PKCζ activity. That components of Ajuba are more efficient as activator and substrate than the full-length protein suggests the possibility that Ajuba may exist in an inhibited conformation, possibly with the PreLIM region folded back on the LIM regions, as has been suggested for Vinculin, WASP, and another LIM protein, Tes (9, 11), or may be inaccessible due to an interaction with another cellular protein. Phosphorylation of the PreLIM region of Ajuba could then possibly open up the structure or release it from a cellular complex, now allowing for the LIM region to interact with p62, TRAF6, and PKCζ. An interaction with PKCζ could further activate PKCζ activity, thereby influencing NF-κB activation.
In addition to regulating NF-κB activity, IL-1 signals also modulate the activity of the MAPKs, particularly JNK and p38. While Ajuba appeared to have a mild effect upon p38 activity, there was sustained activation of JNK in Ajuba null MEFs. JNK can be activated by TRAF6-activated TAK1, which can also activate NF-κB (3). Whether TRAF6-regulated TAK1 activity is altered in Ajuba null cells is unknown. Alternatively, sustained JNK activity in Ajuba null cells may simply result from the inhibition of NF-κB (37).
When one compares the activation of NF-κB by the IL-1 family of proinflammatory cytokines in primary cells from knockout mice, Ajuba's action overlaps with that observed for TRAF6−/− (21, 25, 28) and PKCζ−/− (24) mice. p62 knockout mice have not been fully analyzed (8). In vivo, TRAF6−/− and p62−/− mice both have defects in bone metabolism through effects upon IL-1, TNF, and RANKL signaling during osteoclast differentiation. Ajuba is present at low levels in BMDM and doesn't change during RANKL differentiation into osteoclasts (Y. Feng and G. Longmore, unpublished). Thus, we saw no change in LPS-induced NF-κB activity in Ajuba−/− BMDM (data not shown), and bone development in Ajuba null mice was not abnormal (data not shown). Ajuba-related LIMD1 also interacts with p62. LIMD1 was originally identified as an open reading frame present on a segment of chromosome 3 deleted in some cervical cancers (20). Little else is known about LIMD1 cell biology or function. Interestingly, LIMD1 expression, like p62, is upregulated during RANKL-induced osteoclast differentiation of BMDM (8; Y. Feng and G. Longmore, unpublished). Therefore, it will be interesting to determine whether genetic ablation of LIMD1 affects bone metabolism through modulation of IL-1, TNF, and RANKL signaling pathways. Finally, residual NF-κB activity in Ajuba null MEFs and lungs may reflect the presence of LIMD1 in these cells and/or tissues.
Supplementary Material
Acknowledgments
This work was supported by grant CA75315 from the NIH and a grant from the Washington University/Pfizer biomedical research program to G.D.L.
We thank Robert Arch, P. Ross, and S. Teitelbaum for comments and suggestions, Mary Beckerle for Zyxin null mice, J. Moscat and T. Diaz-Meco for plasmids and p62 antiserum, and J. Weber for nucleophosmin antiserum.
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Arch, R. H., R. W. Gedrich, and C. B. Thompson. 1998. Tumor necrosis factor receptor-associated factors (TRAFs)—a family of adapter proteins that regulates life and death. Genes Dev. 12:2821-2830. [DOI] [PubMed] [Google Scholar]
- 2.Bach, I. 2000. The LIM domain: regulation by association. Mech. Dev. 91:5-17. [DOI] [PubMed] [Google Scholar]
- 3.Baud, V., Z. G. Liu, B. Bennett, N. Suzuki, Y. Xia, and M. Karin. 1999. Signaling by proinflammatory cytokines: oligomerization of TRAF2 and TRAF6 is sufficient for JNK and IKK activation and target gene induction via an amino-terminal effector domain. Genes Dev. 13:1297-1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burstein, E., and C. S. Duckett. 2003. Dying for NF-κB? Control of cell death by transcriptional regulation of the apoptotic machinery. Curr. Opin. Cell Biol. 15:732-737. [DOI] [PubMed] [Google Scholar]
- 5.Crawford, A. W., and M. C. Beckerle. 1991. Purification and characterization of zyxin, an 82,000-dalton component of adherens junctions. J. Biol. Chem. 266:5847-5853. [PubMed] [Google Scholar]
- 6.Deng, L., C. Wang, E. Spencer, L. Yang, A. Braun, J. You, C. Slaughter, C. Pickert, and Z. Y. Chen. 2000. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103:351-361. [DOI] [PubMed] [Google Scholar]
- 7.Dunne, A., and L. A. O'Neill. 25 February 2003, posting date. The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Science STKE [Online.] 10.1126/stke.2003.171.re3. [DOI] [PubMed]
- 8.Duran, A., M. Serrano, M. Leiyges, J. M. Flores, S. Picard, J. P. Brown, and M. T. Diaz-Meco. 2004. The atypical PKC-interacting protein p62 is an important mediator of RANK-activated osteoclastogenesis. Dev. Cell 6:303-309. [DOI] [PubMed] [Google Scholar]
- 9.Garvalov, B. K., T. E. Higgins, J. D. Sutherland, M. Zettl, N. Scaplehorn, T. Kocher, E. Piddini, G. Griffiths, and M. Way. 2003. The conformational state of Tes regulates its zyxin-dependent recruitment to focal adhesions. J. Cell Biol. 161:33-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geetha, T., and M. W. Wooten. 2002. Structure and functional properties of the ubiquitin binding protein p62. FEBS Lett. 512:19-24. [DOI] [PubMed] [Google Scholar]
- 11.Gilmore, A. P., and K. Burridge. 1996. Regulation of vinculin binding to talin and actin by phosphatidyl-inositol-4-5-bisphosphate. Nature 381:531-535. [DOI] [PubMed] [Google Scholar]
- 12.Goyal, R. K., P. Lin, J. Kanungo, A. S. Payne, A. J. Muslin, and G. D. Longmore. 1999. Ajuba, a novel LIM protein, interacts with Grb2, augments mitogen-activated protein kinase activity in fibroblasts, and promotes meiotic maturation of Xenopus oocytes in a Grb2- and Ras-dependent manner. Mol. Cell. Biol. 19:4379-4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herrera-Velit, P., K. L. Knutson, and N. E. Reiner. 1997. Phosphatidyl 3-kinase-dependent activation of protein kinase C ζ in bacterial lipopolysaccharide-treated human monocytes. J. Biol. Chem. 272:16445-16452. [DOI] [PubMed] [Google Scholar]
- 14.Hirai, T., and K. Chida. 2003. Protein kinase Czeta (PKCzeta): activation mechanisms and cellular functions. J. Biochem. (Tokyo) 133:1-7. [DOI] [PubMed] [Google Scholar]
- 15.Hirota, T., N. Kunitoku, T. Sasayama, T. Marumoto, D. Zhang, M. Nitta, K. Hatakeyama, and H. Saya. 2003. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell 114:585-598. [DOI] [PubMed] [Google Scholar]
- 16.Hirota, T., T. Morisaki, Y. Nishiyama, T. Marumoto, K. Tada, T. Hara, N. Masuko, M. Inagaki, K. Hatakeyama, and H. Saya. 2000. Zyxin, a regulator of actin filament assembly, targets the mitotic apparatus by interacting with h-warts/LATS1 tumor suppressor. J. Cell Biol. 149:1073-1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hobert, O., J. W. Schilling, M. C. Beckerle, A. Ullrich, and B. Jallal. 1996. SH3 domain-dependent interaction of the proto-oncogene product Vav with the focal contact protein zyxin. Oncogene 12:1577-1581. [PubMed] [Google Scholar]
- 18.Juong, I., J. L. Strominger, and J. Shin. 1996. Molecular cloning of a phosphotyrosine-independent ligand of the p56lckSH2 domain. Proc. Natl. Acad. Sci. USA 93:5991-5995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanungo, J., S. J. Pratt, H. Marie, and G. D. Longmore. 2000. Ajuba, a cytosolic LIM protein, shuttles into the nucleus and affects embryonal cell proliferation and fate decisions. Mol. Biol. Cell 11:3299-3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kiss, H., D. Kedra, Y. Yang, M. Kost-Alimova, C. Kiss, K. P. O'Brien, I. Fransson, G. Klein, S. Imreh, and J. P. Dumanski. 1999. A novel gene containing LIM domains (LIMD1) is located within the common eliminated region 1 (C3CER1) in 3p21.3. Hum. Genet. 105:552-559. [DOI] [PubMed] [Google Scholar]
- 21.Kobayashi, N., Y. Kadono, A. Naito, K. Matsumoto, T. Yamamoto, S. Tanaka, and J. Inoue. 2001. Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis. EMBO J. 20:1271-1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kopp, E., R. Medzhitov, J. Carothers, C. Xiao, I. Douglas, C. A. Janeway, and S. Ghosh. 1999. ECSIT is an evolutionarily conserved intermediate in the Toll/IL-1 signal transduction pathway. Genes Dev. 13:2059-2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lallena, M.-J., M. T. Diaz-Meco, G. Bren, C. V. Payá, and J. Moscat. 1999. Activation of IκB kinase β by protein kinase C isoforms. Mol. Cell. Biol. 19:2180-2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leitges, M., L. Sanz, P. Martin, A. Duran, U. Braun, J. F. Garcia, F. Camacho, M. T. Diaz-Meco, P. D. Rennert, and J. Moscat. 2001. Targeted disruption of the zetaPKC gene results in the impairment of the NF-κB pathway. Mol. Cell 8:771-780. [DOI] [PubMed] [Google Scholar]
- 25.Lomaga, M. A., W. Yeh, I. Sarosi, G. S. Duncan, C. Furlonger, A. Ho, S. Morony, C. Capparelli, G. Van, S. Kaufman, A. van der Heiden, A. Itie, A. Wakeham, W. Khoo, T. Sasaki, Z. Cao, J. Penninger, C. Paige, D. Lacey, C. Dunstan, W. Boyle, D. V. Goeddel, and T. W. Mak. 1999. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 13:1015-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marie, H., D. Billups, F. K. Bedford, A. Dumoulin, R. K. Goyal, G. D. Longmore, S. J. Moss, and D. Attwell. 2002. The amino terminus of the glial glutamate transporter GLT-1 interacts with the LIM protein Ajuba. Mol. Cell. Neurosci. 19:152-164. [DOI] [PubMed] [Google Scholar]
- 27.Marie, H., S. J. Pratt, M. Betson, H. Epple, J. T. Kittler, L. Meek, S. J. Moss, S. Troyanovsky, D. Attwell, G. D. Longmore, and V. M. Braga. 2003. The LIM protein Ajuba is recruited to cadherin-dependent cell junctions through an association with alpha-catenin. J. Biol. Chem. 278:1220-1228. [DOI] [PubMed] [Google Scholar]
- 28.Naito, A., S. Azuma, S. Tanaka, T. Miyazaki, S. Takaki, K. Takatsu, K. Nakao, K. Nakamura, M. Katsuki, T. Yamamoto, and J. Inoue. 1999. Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells 4:353-362. [DOI] [PubMed] [Google Scholar]
- 29.Ninomiya-Tsuji, J., K. Kishimoto, A. Hiyama, J. Inoue, Z. Cao, and K. Matsumoto. 1999. The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature 398:252-256. [DOI] [PubMed] [Google Scholar]
- 30.Nix, D. A., and M. C. Beckerle. 1997. Nuclear-cytoplasmic shuttling of the focal contact protein, Zyxin: a potential mechanism for communication between sites of cell adhesion and the nucleus. J. Cell Biol. 138:1139-1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ostendorff, H. P., R. I. Peirano, M. A. Peters, A. Schluter, M. Bossenz, M. Scheffner, and I. Bach. 2002. Ubiquitination-dependent cofactor exchange on LIM homeodomain transcription factors. Nature 416:99-103. [DOI] [PubMed] [Google Scholar]
- 32.Petit, M. M., J. Fradelizi, R. M. Golsteyn, T. A. Ayoubi, B. Menichi, D. Louvard, W. J. Van de Ven, and E. Friederich. 2000. LPP, an actin cytoskeleton protein related to zyxin, harbors a nuclear export signal and transcriptional activation capacity. Mol. Biol. Cell 11:117-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petit, M. M., R. Mols, E. F. Schoenmakers, N. Mandahl, and W. J. Van de Ven. 1996. LPP, the preferred fusion partner gene of HMGIC in lipomas, is a novel member of the LIM protein gene family. Genomics 36:118-129. [DOI] [PubMed] [Google Scholar]
- 34.Pratt, S. J., H. Epple, M. Ward, Y. Feng, V. M. Braga, and G. D. Longmore. 2005. The LIM protein Ajuba modulates cell migration through regulation of Rac1 activity. J. Cell Biol. 168:813-824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Puls, A., S. Schmidt, F. Grawe, and S. Stabel. 1997. Interaction of protein kinase C zeta with ZIP, a novel protein kinase C binding protein. Proc. Natl. Acad. Sci. USA 94:6191-6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Renfranz, P. J., and M. C. Beckerle. 2002. Doing the (F/L)PPPPs: EVH1 domains and their proline-rich partners in cell polarity and migration. Curr. Opin. Cell Biol. 14:88-103. [DOI] [PubMed] [Google Scholar]
- 37.Reuther-Madrid, J. Y., D. Kashatus, S. Chen, X. Li, J. Westwick, R. J. Davis, H. S. Earp, C. Y. Wang, and A. S. Baldwin, Jr. 2002. The p65/RelA subunit of NF-κB suppresses the sustained, antiapoptotic activity of Jun kinase induced by tumor necrosis factor. Mol. Cell. Biol. 22:8175-8183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rzymkiewicz, D. M., T. Tetsuka, D. Daphna-Iken, S. Srivastava, and A. R. Morrison. 1996. Interleukin-1β activates protein kinase C ζ in renal mesangial cells. Potential role in prostaglandin E2 up-regulation. J. Biol. Chem. 271:17241-17246. [DOI] [PubMed] [Google Scholar]
- 39.Sakurai, H., H. Miyoshi, W. Toriumi, and T. Sugita. 1999. Functional interactions of transforming growth factor beta-activated kinase 1 with IkappaB kinases to stimulate NF-kappaB activation. J. Biol. Chem. 274:10641-10648. [DOI] [PubMed] [Google Scholar]
- 40.Sanchez, P., G. De Carcer, I. V. Sandoval, J. Moscat, and M. T. Diaz-Meco. 1998. Localization of atypical protein kinase C isoforms into lysosome-targeted endosomes through interaction with p62. Mol. Cell. Biol. 18:3069-3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sanz, L., M. T. Diaz-Meco, H. Nakano, and J. Moscat. 2000. The atypical PKC-interacting protein p62 channels NF-kB activation by the IL-1-Traf6 pathway. EMBO J. 19:1576-1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanz, L., P. Sanchez, M.-J. Lallena, M. T. Diaz-Meco, and J. Moscat. 1999. The interaction of p62 with RIP links the atypical PKCs to NF-kB activation. EMBO J. 18:3044-3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun, L., L. Deng, C. K. Ea, Z. P. Xia, and Z. J. Chen. 2004. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol. Cell 14:289-301. [DOI] [PubMed] [Google Scholar]
- 44.Wald, D., J. Qin, Z. Zhao, Y. Qian, M. Naramura, L. Tian, J. Towne, J. E. Sims, G. R. Stark, and X. Li. 2003. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat. Immunol. 4:920-927. [DOI] [PubMed] [Google Scholar]
- 45.Wooten, M. W., M. L. Seibenhener, V. Mamidipudi, M. T. Diaz-Meco, P. A. Barker, and J. Moscat. 2001. The atypical protein kinase C-interacting protein p62 is a scaffold for NF-kB activation by nerve growth factor. J. Biol. Chem. 276:7709-7712. [DOI] [PubMed] [Google Scholar]
- 46.Yi, J., and M. C. Beckerle. 1998. The human TRIP6 gene encodes a LIM domain protein and maps to chromosome 7q22, a region associated with tumorigenesis. Genomics 49:314-316. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.