

Summary
Abl family kinases are evolutionarily conserved regulators of cell migration and morphogenesis. Genetic experiments in Drosophila suggest that Abl family kinases interact functionally with microtubules to regulate axon guidance and neuronal morphogenesis. Vertebrate Abl2 binds to microtubules and promotes their plus-end elongation both in vitro and in cells, but the molecular mechanisms by which Abl2 regulates microtubule (MT) dynamics were unclear. We report here that Abl2 regulates MT assembly via condensation and direct interactions with both the MT lattice and tubulin dimers. We find that Abl2 promotes MT nucleation, which is further facilitated by the ability of the Abl2 C-terminal half to undergo liquid-liquid phase separation (LLPS) and form co-condensates with tubulin. Abl2 binds to regions adjacent to MT damage and facilitates their repair via fresh tubulin recruitment and increases MT rescue frequency and lifetime. Cryo-EM analyses strongly support a model in which Abl2 engages tubulin C-terminal tails along an extended MT lattice conformation at damage sites to facilitate repair via fresh tubulin recruitment. Abl2Δ688-790, which closely mimics a naturally occurring splice isoform, retains binding to the MT lattice but does not bind tubulin, promote MT nucleation, or increase rescue frequency. In COS-7 cells, MT reassembly after nocodazole treatment is greatly slowed in Abl2 knockout COS-7 cells compared to wild type cells, and these defects are rescued by re-expression of Abl2, but not Abl2Δ688-790. We propose that Abl2 locally concentrates tubulin to promote MT nucleation and recruits it to defects in the MT lattice to enable repair and rescue.
In Brief:
Genetic studies suggested that Abl family kinases interact functionally with microtubules (MT) to promote cell morphogenesis, but the underlying mechanisms were unknown. Duan and Lyu et al. show that Abl2 recruits tubulin dimers to sites of MT lattice expansion or damage to promote MT nucleation and rescue in vitro and MT reassembly in cells.
Introduction
Microtubules (MTs) play crucial roles in intracellular trafficking, cell morphogenesis, migration, and division1–4. MTs are mostly nucleated from the MT organizing center (MTOC), which mediates MT assembly during interphase and spindle formation during mitosis5,6. Once nucleated, MT plus ends undergo dynamic instability, switching from growth to shrinkage (catastrophe) or from shrinkage to growth (rescue)7. Microtubule-binding proteins (MBPs) regulate the dynamic behavior of MT plus ends8, enabling them to sample their environment, provide transport of MT-based cargoes, and regulate signaling pathways. MT lattice defects induced by mechanical stress, severing enzymes, or fast polymerization impact MT dynamics by triggering shrinkage or precluding rescue from catastrophe9–14. These defects are detected by tubulin itself and/or by MBPs such as SSNA115; and are repaired via incorporation of new tubulin dimers, which is stimulated by MBPs such as CSPP116 and CLASP217,18. MT dynamics can be facilitated by Liquid-Liquid Phase Separation (LLPS) of MT effector proteins, which concentrates them at sites of action19,20. For example, LLPS of EB1 promotes recruitment of +TIP binding proteins to regulate MT growth and catastrophe21–25.
Abl family nonreceptor tyrosine kinases, Abl1 and Abl2 in vertebrates, play essential roles in metazoan development26–28. Cell surface receptors signal through Abl kinases to phosphorylate key mediators that coordinate changes in actin cytoskeletal structure and adhesion dynamics29–33. However, the ability to regulate adhesion and actin dynamics does not fully explain the roles that Abl kinases play in neuronal development. Genetic studies in Drosophila suggest that functional interactions between Abl and the CLASP2 ortholog orbit are required for proper axonal growth cone guidance which is mediated by dynamic MT reorganization34,35. At the mitotic spindle, CLASP2 enhances MT nucleation and lattice repair by recruiting tubulin to MT tips and MT damage sites17,18,36,37. CLASPs bind MT ends and stabilize them by maintaining an intermediate state between phases of shrinkage and growth36,37. Abl2 directly binds to MTs and promotes MT elongation both in vitro and in cells33,38. Abl1 also binds to and phosphorylates γ-tubulin to promote assembly of the microtubule-nucleating γ-tubulin ring complex in cells39. These findings collectively led us to explore whether and how Abl2 impacts MT nucleation and repair.
We provide evidence here that Abl2 binding to the MT lattice and tubulin dimers promotes nucleation and repair. Abl2 binds with high affinity to tubulin dimers and forms condensates that recruit tubulin and facilitate MT nucleation. Abl2Δ688-790, which closely mimics a naturally occurring splice isoform, retains binding to the MT lattice but does not bind tubulin or promote MT nucleation. Biochemical and cryo-EM analyses reveal that Abl2 engages individual protofilaments within GTP-bound extended regions of the MT lattice to bridge adjacent tubulin dimers and recruit additional dimers. Specific recognition of the damaged MT segments and recruitment of tubulin by Abl2 mediate lattice repair and increase the rescue frequency and lifetime of dynamic MTs in vitro. Consistent with these findings, loss of Abl2 in cells greatly slows MT recovery after nocodazole-induced disassembly, which is rescued by re-expression of Abl2, but not Abl2Δ688-790. Collectively, our data suggest that Abl2 promotes MT nucleation, repair, and rescue by binding and concentrating tubulin dimers and facilitating their assembly and incorporation into the MT lattice.
Results
Abl2 binds to free tubulin dimers via the C-terminal half.
Abl2 promotes MT plus-end elongation in vitro and in cells, and the C-terminal half, 557-C, is both necessary and sufficient for this activity38. To test whether Abl2 binds tubulin dimers in vitro, purified 557-C and porcine tubulin dimers were applied, separately or after mixing, to a size exclusion column, and their elution profiles were monitored. Purified 557-C (68 kDa) eluted with an estimated Stokes radius (Rs) = 68 Å, corresponding to a globular protein of 350 kDa (Figure 1A, S1A). Size exclusion chromatography coupled with multi-angle light scattering (SEC-MALS) analysis revealed an estimated molecular weight of 73 kDa for the peak, corresponding to a monomer of 557-C (Figure S1D). Similarly, SEC-MALS revealed Abl2 to be a monomer of ~127 kDa, with Rs = 78 Å, corresponding to a globular protein of 480 kDa (Figure S1A, B) while Abl2Δ688-790, which closely mimics the alternatively spliced Abl2Δ688-791 isoform found in both humans and mice, was a monomer of ~104 kDa (Figure S1C). Peak elution fractions of Abl2, Abl2Δ688-790, and 557-C had A260nm/A280nm ratios ≈ 0.6, indicating a lack of nucleic acid contamination (Figure S1E–G). Together, these data indicate that Abl2 and its C-terminal half have an extended monomer conformation in solution. Tubulin dimers (105 kDa) eluted at Rs = 42 Å (108 kDa for globular proteins). Upon mixing 557-C and tubulin, the elution profile revealed a peak of Rs = 68 Å, and a less prominent new peak with a larger Rs = 101 Å (Figure 1A). SDS-PAGE analysis revealed that 557-C:tubulin complexes co-eluted in both peaks (Figure 1B).
Figure 1. Abl2 binds tubulin via two regions in the C-terminal half.
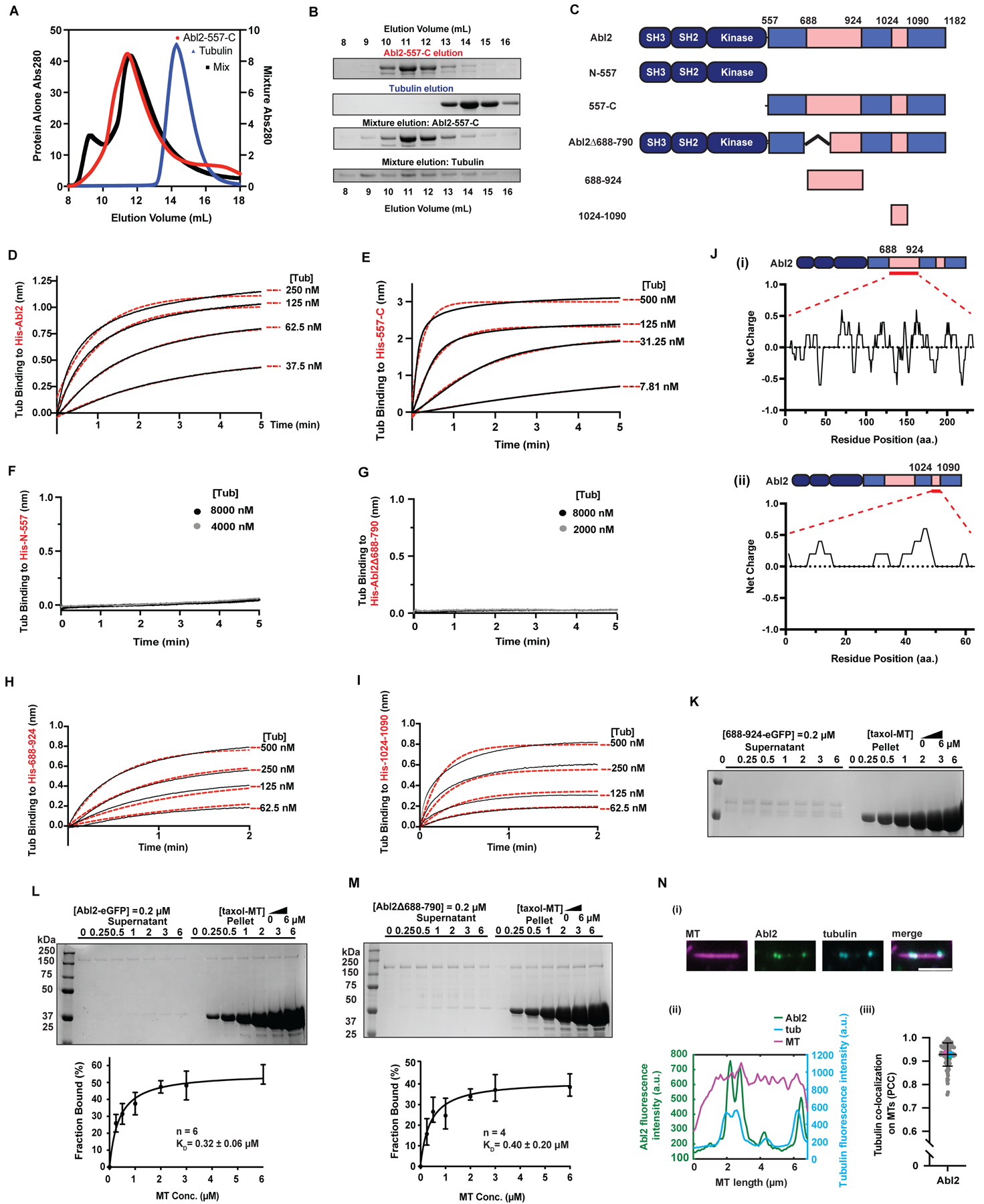
(A) SEC analysis of 557-C alone (left Y axis, red line), tubulin alone (left Y axis, blue line), and the mixture (right Y axis, black line) with (B) showing the corresponding SDS-PAGE analysis of the elution fractions. Tubulin alone was eluted at fractions 13–15 ml and was shifted to fractions 8–15 ml in the presence of 557-C. (C) Domain architecture of Abl2, Abl2-74-557 (N-557), Abl2-557-1182 (557-C), Abl2Δ688-790, Abl2-688-924, and Abl2-1024-1090. (D) 6XHis-Abl2, (E) 6XHis-557-C, (F) 6XHis-N-557, (G) 6XHis-Abl2Δ688-790, (H) 6XHis-688-924, or (I) 6XHis-1024-1090 were immobilized on a Ni-NTA biosensor and the association of tubulin at different concentrations was measured. Representative traces are shown, with data shown in black and global exponential fits in red dotted lines. Full concentration series (4 tubulin concentrations) were performed three independent times and used to calculate KD, as summarized in Figure S1K. (J) Net charges of every 5-residue block within Abl2-688-924 (i) and Abl2-1024-1090 (ii). The residue charge distribution was calculated with EMBOSS to indicate positively-charged clusters. (K-M) Representative gels of the MT co-sedimentation assays performed with 688-924-eGFP (K), Abl2-eGFP (L), and Abl2-Δ688-790-eGFP in (M) are shown. Plots of percentages of Abl2-eGFP or Abl2Δ688-790 bound versus MT concentration are shown in the bottom panels with best fit KD values shown. The number of experimental replicates is indicated by n. Data are mean ± SD. See also Figure S1J, S1L. (N) Abl2 co-localizes with tubulin dimers on GMPCPP-MTs. (i) Representative Abl2- and tubulin-bound MT. (ii) Quantification of fluorescence intensities of Abl2-eGFP and Alexa647-tubulin on the rhodamine-labeled GMPCPP-MT shown in (i). (iii) Pearson-correlation coefficient (PCC) analysis reveals that Abl2 and tubulin co-localize on a MT. Magenta, cyan, and green points indicate the mean PCC for each technical replicate. n = 94 filaments across 3 technical replicates. See also Figure S1.
We measured Abl2 binding affinity for tubulin dimers using biolayer interferometry. His-tagged Abl2 constructs tested in this assay are shown in Figure 1C, while purified His-tagged Abl2, porcine brain tubulin, and untagged Abl2 fragments are shown in Figure S1H, i–viii; S1I, ii–vii, respectively. Tubulin bound biosensor-immobilized His-Abl2 with a KD = 42 ± 13 nM (Figure 1D). His-557-C was sufficient to bind tubulin with high affinity (KD = 17 ± 8 nM) (Figure 1E), while the N-terminal half, His-N-557, did not bind tubulin (Figure 1F). Abl2Δ688-790 also did not bind tubulin dimers (Figure 1G). We discovered two distinct Abl2 fragments within the C-terminal half, comprised of amino acids 688–924 (site I) and 1024–1090 (site II), that bound independently to tubulin dimers, but with significantly reduced affinity (KD, His-688-924 = 118 ± 39 nM; KD, His-1024-1090 = 252 ± 123 nM; Figure 1H, I; S1K) relative to His-Abl2 or His-557-C. These two binding sites are not homologous to tubulin-binding TOG and CAP-Gly domains40, but are enriched in positively charged residues, with calculated pIs of site I = 9.05 (Figure 1J, i) and site II = 10.53 (Figure 1J, ii). To measure MT binding affinities, fixed concentrations of Abl2 proteins were mixed with increasing concentrations of taxol-stabilized MTs and the bound fraction at each concentration was measured after MT co-sedimentation38. Strikingly, Abl2Δ688-790, which lacks part of the higher-affinity site I, bound MTs with a similar affinity as Abl2-eGFP (KD, Abl2-eGFP = 0.32 ± 0.06 μM, KD, Abl2Δ688-790 = 0.40 ± 0.20 μM; Figure 1L, M; S1L), even though it did not bind tubulin. In contrast, neither tubulin binding fragment 688-924-eGFP or 1024-1090-eGFP, nor the region between sites I and II (924-1024-eGFP) bound detectably to MTs (Figure 1K, Figure S1J, i–iii), indicating that the MT- and tubulin-binding regions of Abl2 do not overlap completely. Abl2-eGFP also colocalized prominently with Alexa647-tubulin (Pearson coefficient = 0.93 ± 0.05) on rhodamine GMPCPP-MTs (Figure 1N).
Abl2 undergoes phase separation and co-condenses with tubulin.
A growing list of MT-binding proteins regulate MT dynamics through LLPS, which is associated with regions of disorder19,22–24,41–43. PONDR-FIT and Diso-Pred3 software predicted the Abl2 C-terminal half to be disordered (Figure 2A). Indeed, deconvolution of circular dichroism (CD) spectra of 557-C indicated 10–30% disordered secondary structure content (Figure 2B).
Figure 2. Abl2 undergoes phase separation and co-condenses with tubulin.
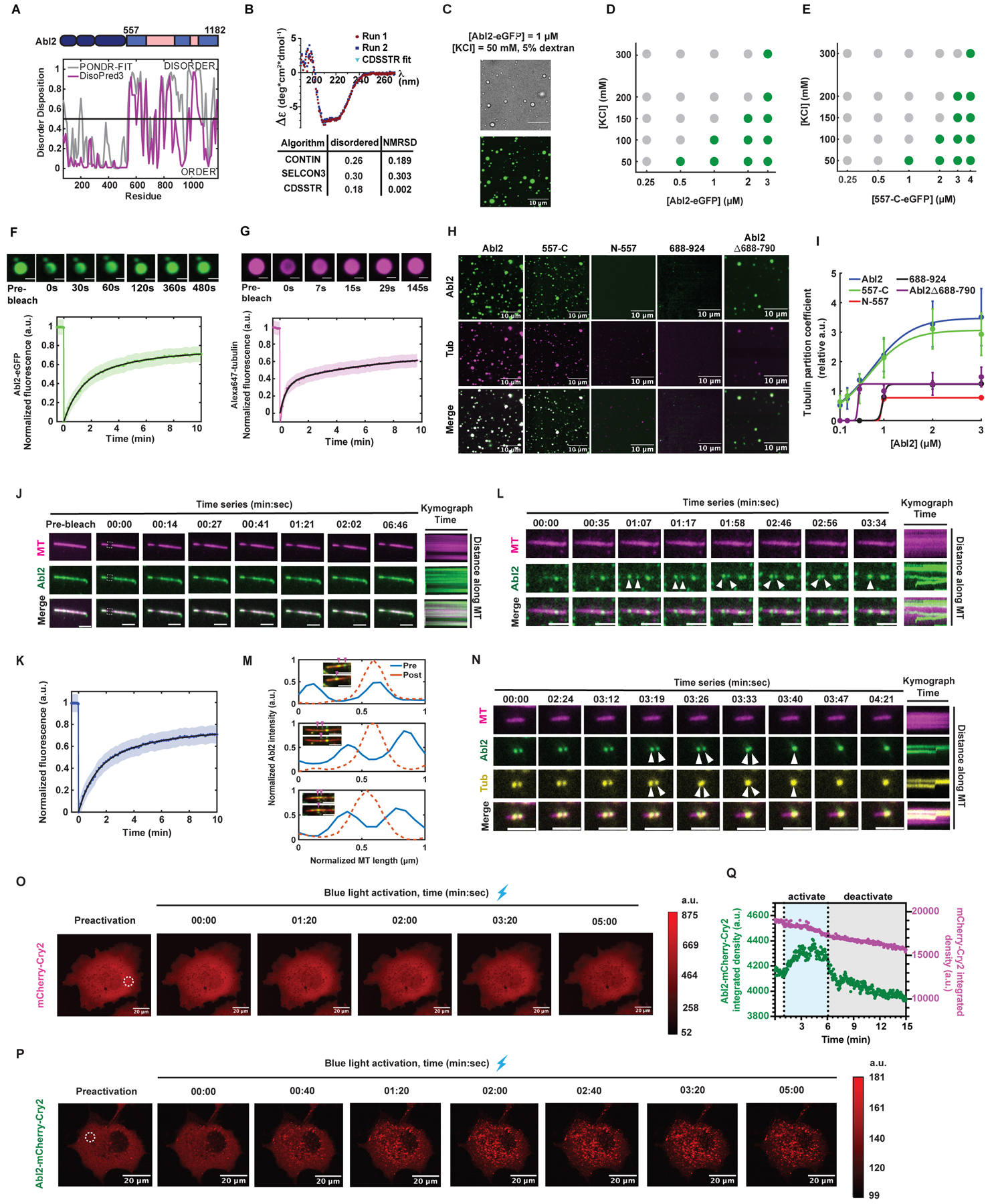
(A) Disorder prediction from primary sequence of murine Abl2 using PONDR-FIT and DisoPred3 algorithms. Disorder disposition of 0.5 was used as the threshold. (B) Buffer-subtracted CD spectra of 6XHis-tag free 557-C collected at 4°C. CD spectra deconvolution algorithms CONTIN, SELCON3, CDSSTR analyses reveal the disordered content of the 557-C. n = 2. (C) 1 μM Abl2-eGFP forms condensates in 5% dextran, 50 mM KCl, and BRB80. Condensates were imaged under brightfield (top) and fluorescence (bottom). Scale bar, 10 μm. Phase separation diagrams of the [Abl2-eGFP] vs. [KCl] (D) and [557-C-eGFP] vs. [KCl] (E) relationships. Partition coefficient (PC) ≥ 4 was the threshold for phase separation, shown as green dots, PC < 4 is defined as not phase separated, shown as gray dots. X-axis shown on log10 scale. 17–230 condensates were analyzed per condition. (F,G) FRAP recovery of Abl2-eGFP in condensates (F) and tubulin in Abl2-eGFP:Alexa647-tubulin co-condensates (G) in solution. Double-exponential fits shown in solid black with SEM shown as green. n ≥ 11 condensates. Scale bar, 2 μm in (F); 1 μm in (G). (H) 1 μM Abl2, 557-C, N-557, 688–924, and Abl2Δ688-790 were incubated with equimolar concentrations of Alexa647-tubulin in BRB80, 5% dextran, 50 mM KCl to test for co-condensation. (I) PC analysis of co-condensed tubulin in Abl2 condensates at increasing equimolar concentrations of Abl2-eGFP and tubulin at 50 mM KCl. At least 90 tubulin co-condensates were scored for Abl2, 557-C, and Abl2Δ688-790. N557: n2μM = 37; n3μM = 89; 688–924: n2μM = 53; n3μM = 147. Sigmoidal fits shown in solid lines. Data are mean ± SD. (J, K) Representative time series of FRAP on Abl2 puncta on GMPCPP-stabilized MTs (J). Bleached ROI shown in dashed white line. Scale bar, 3 μm. FRAP recovery curve reveals that Abl2 condensates are undergoing dynamic exchange with other molecules on the MT and/or from solution (K). Double-exponential fit in solid black with SEM shown as blue. n = 15 filaments. (L, M) Representative time series of fusion of Abl2 condensates (L, scale bar = 3 μm) or Abl2-tubulin co-condensates (M, scale bar = 5 μm) on a rhodamine GMPCPP-MT (pseudo-colored magenta). Kymographs are shown on right. (N) Fluorescence intensity analysis of fusion events of Abl2-eGFP puncta on stabilized MTs, as shown in (M). Post-fusion condensates yield higher mean fluorescence intensities (dashed orange line) relative to their pre-fusion condensates of various sizes (solid blue lines). (O,P) Representative time series of mCherry-Cry2 (O) and Abl2-mCherry-Cry2 (P) in transfected COS-7 cells, shown pre- and post-blue light activation (λ = 488 nm). Raw fluorescence intensity color bars shown on right. (Q) Quantification of mCherry intensity (integrated densities, a.u.) within the ROIs shown in (O, magenta) and (P, green) indicated by dashed white circles. Blue shaded region depicts 5 min blue light activation period. Gray shaded region depicts 10 min following blue light exposure. See also Figure S2.
To investigate whether Abl2 could undergo LLPS, we observed solutions containing Abl2-eGFP and 5% dextran, a non-physiological agent that mimics intracellular molecular crowding44,45. Abl2e-GFP puncta appeared spherical in both brightfield and fluorescence imaging (Figure 2C). We used a partition coefficient (PC), a ratio of the mean condensate intensity relative to background, ≥ 4 as a cutoff for LLPS19. LLPS increased with Abl2-eGFP concentration, but was attenuated by increasing salt concentration, a known disruptor of phase separation44,46,47 (Figure 2D). PC analysis revealed that 557-C-eGFP also phase separates (Figure 2E), consistent with its propensity to disorder. We utilized a sedimentation assay to measure the fraction of Abl2-eGFP entering the dense and dilute phases as a function of concentration. The proportion of Abl2-eGFP in the dense phase increased strikingly when the Abl2-eGFP concentration was raised from 0.5 to 1 μM in 50 mM KCl (Figure S2A), consistent with the phase transition observed in epifluorescence. This phase transition occurs below the concentration of 1.4 μM Abl2 previously measured in COS-7 cells38. Fluorescence recovery after photobleaching (FRAP) of a confocal plane within Abl2-eGFP condensates revealed a recovery half time (t1/2) ≈ 1.7 min (Figure 2F).
We next tested whether Abl2-eGFP formed coacervates with tubulin. When incubated under conditions that promote Abl2-eGFP LLPS, Alexa647-tubulin dimers partitioned into Abl2-eGFP condensates (Figure 2G, H). FRAP analysis revealed that tubulin dimers diffused into an internally bleached region within Abl2:tubulin coacervates (Figure 2G). The Abl2 C-terminal half was necessary and sufficient for coacervation with tubulin, as 557-C-eGFP both underwent phase separation and recruited tubulin dimers into the condensate, while N-557-eGFP and the tubulin binding site I, 688-924-eGFP, did not undergo phase separation (Figure 2H, I). While it underwent LLPS, tubulin-binding defective Abl2Δ688-790-eGFP condensates had significantly reduced ability to partition tubulin into the condensates (Figure 2H, I). Abl2-eGFP condensates recruited tubulin to reach partition coefficients over 3, while Abl2Δ688-790-eGFP condensates only reached tubulin partition coefficients just over the background of 1.
We also examined how Abl2-eGFP interacted with MTs, either by itself or if soluble tubulin was included. FRAP analysis revealed that 1 μM Abl2-eGFP bound to GMPCPP-stabilized MTs could undergo slow exchange with Abl2-eGFP molecules in solution, in absence of dextran or free tubulin (Figure 2J, K). Under these same conditions, Abl2-eGFP coacervates were also observed to form, move, and undergo fusion (Figure 2L), with a relative increase in fluorescence intensity proportional to the mean intensities of pre-fusion condensates (Figure 2M). This suggests MTs may serve as a platform to recruit Abl2 and enhance LLPS. We also found that at equimolar 0.5 μM concentrations, Abl2:tubulin co-condensates diffused along GMPCPP-MT templates and could undergo fusion throughout time (Figure 2N).
To determine whether Abl2 exhibits self-association in cells, we expressed Abl2-mCherry-Cry2 or control mCherry-Cry2 in COS-7 cells48. We used purified 6XHis-mCherry (Figure S1H, x) to generate a standard curve relating concentration to mean fluorescence intensity to cellular concentration (Figure S2B, i). Using the initial pre-activation images, we estimated intracellular concentrations of ~0.5–1.2 μM mCherry-Cry2 and ~0.4–0.6 μM Abl2-mCherry-Cry2, respectively (Figure S2B, ii). While Abl2-mCherry-Cry2 and mCherry-Cry2 were diffuse within the cytoplasm under pre-activation conditions, blue light induced rapid cytoplasmic clustering of Abl2-mCherry-Cry2, but not mCherry-Cry2 (Figure 2O–Q). Our data reveal that Abl2-mCherry-Cry2 forms puncta upon blue light exposure at concentrations below the endogenous concentrations of Abl2 – which is 1.4 μM in COS7 cells70 – while mCherry-Cry2 does not (Figure S2B–D).
Abl2 promotes MT nucleation.
The finding that Abl2 binds both the MT lattice and tubulin dimers led us to investigate whether Abl2 impacts MT nucleation. We measured changes in turbidity (OD350) over time, which increases upon MT polymerization, in the absence or presence of Abl2 (Figure 3A). We previously showed that 0–2 μM MBP-tagged 557-C increases MT assembly in a concentration-dependent manner38. Addition of 1 μM Abl2 to 18 μM tubulin increased the plateau of turbidity from ΔOD350,tub = 0.093 ± 0.000 to ΔOD350,tub+Abl2 = 0.210 ± 0.173 (mean ± SD), a 2.3-fold increase over tubulin alone, Figure 3A). The increased turbidity reflected greater MT polymerization, as revealed by sedimenting polymerized MTs and analyzing them by SDS-PAGE (Figure S3A). The tubulin critical concentration decreased 11-fold from 5.5 ± 1.1 μM in the absence of Abl2 to 0.5 ± 1.8 μM in the presence of Abl2, respectively (Figure 3B). Addition of 1 μM Abl2 also lowered nucleation lag time, when turbidity reaches one-tenth of its maximum71–73, 1.8-fold from 16 ± 2 min to 9 ± 3 min (Figure 3C). The ability of Abl2 to promote MT nucleation was also affirmed by visualizing reaction products at the initial phase of the reaction via negative-stain electron microscopy (EM), which revealed significantly more MTs when Abl2 was included (Figure 3D). Abl2Δ688-790 – which does not bind tubulin – had no effect on critical concentration or lag time (Figure 3C). To test if co-condensation of Abl2 and tubulin facilitates MT nucleation, we measured non-templated MT nucleation using low concentrations (8 μM) of Alexa647-tubulin, 1 μM Abl2-eGFP and 3% dextran (Figure 3E). Abl2:tubulin formed co-condensates from which numerous MTs grew. Control eGFP did not form condensates or promote MT formation under these conditions (Figure 3E). In the absence of 3% dextran, Abl2-eGFP remained diffuse in solution and few MTs were observed after a 1 hr reaction.
Figure 3. Abl2 promotes MT nucleation via interactions with tubulin and MTs.
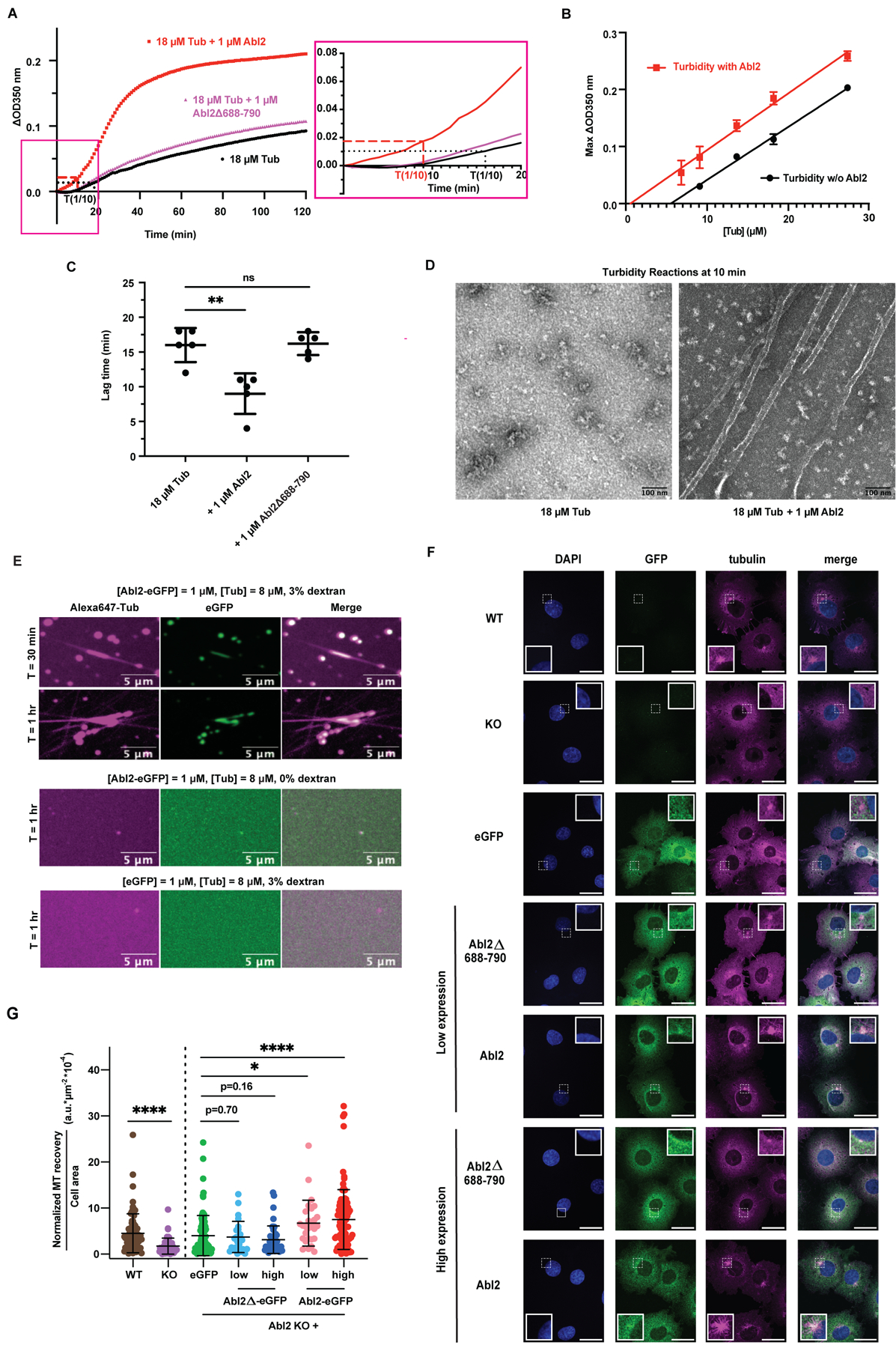
(A) MT assembly was monitored by measuring the increase in turbidity (ΔOD350). 18 μM tubulin and 2 mM GTP were incubated alone (black) or with 1 μM Abl2 (red) or Abl2Δ688-790 (magenta). Representative time series of OD350 measurements are shown. Curves in the blue dotted window were expanded in the right panel. Lag time of reactions with (red) and without Abl2 (black) are indicated with the dotted line. (B) Turbidity assays with different tubulin concentrations were performed and the maximal ΔOD350 was plotted against initial tubulin concentration to determine the critical concentration of tubulin polymerization. n = 3 replicates at each concentration for each of these experiments. (C) The lag time until ΔOD350 reaches 1/10 of the maximal ΔOD350 was measured. The inclusion of Abl2 significantly decreased the lag time for MT nucleation. Mann-Whitney test. n = 5. **, p < 0.001. (D) Representative samples taken 10 min after turbidity reactions were initiated were visualized under negative-stain EM. More polymerized MT segments were observed in the presence of Abl2. (E) Representative confocal images of the MT nucleation from the Abl2-eGFP:Alexa647-tubulin co-condensates under 3% dextran. Control reactions containing Abl2-eGFP and Alexa647-tubulin without dextran, or mixing eGFP and Alexa647-tubulin with 3% dextran did not show observable MTs growing after 1 hr. (F) WT COS-7, Abl2 KO COS-7, and eGFP-, Abl2Δ688-790-eGFP-, and Abl2-eGFP-expressing Abl2 KO cells were treated with 10 μM nocodazole for 1 hr, at which time nocodazole was removed and replaced with complete medium. Immunofluorescence images of cells upon 5 min after washout for quantification of MT reassembly from the microtubule organizing center (white dashed inlets with expanded views in solid white borders) shown. Scale bar, 25 μm. (G) The integrated fluorescence intensity of recovered MTs was measured and normalized to the total cell area. MT recovery values for each condition was normalized to the average MT recovery of WT cells per experimental replicate. Welch’s t-test. n ≥ 25 cells analyzed across 2–3 experimental replicates. Data are mean ± SD. *, p < 0.05; ****, p < 0.0001. See also Figure S3.
We then examined how Abl2 impacts MT reassembly following nocodazole treatment in WT and Abl2 KO COS-7 cells (Figures 3F, S3B, S3I). Cells were treated with 10 μM nocodazole for 1 hr and MT recovery was measured 5 minutes after washout. MT recovery from centrosomes was significantly higher (2.6-fold) in WT COS-7 cells relative to Abl2 KO COS-7 cells (MTRWT = 4.52 ± 4.23 a.u.*μm−2*10−4, MTRKO = 1.72 ± 1.74 a.u.*μm−2*10−4, p < 0.0001, Figure 3G). To assess whether Abl2 rescued this MT recovery deficit, we generated Abl2 KO cells stably expressing low or high Abl2-eGFP or Abl2Δ688-790-eGFP levels (Figure S3C, S3E–H). Cells selected with Zeocin for lentiviral infection yielded consistently higher MT recovery rates over parental cells and thus we used eGFP-expressing Abl2 KO cells as a control group (Figure S3C, D). Expression of Abl2-eGFP at low and high levels significantly increased MT recovery by 1.7- and 1.9-fold respectively (MTRlow Abl2-eGFP = 6.72 ± 4.98 a.u.*μm−2*10–4; MTRhigh Abl2-eGFP = 7.51 ± 6.50 a.u.*μm−2*10−4, p < 0.05, p < 0.0001 respectively; Figure 3F, G) over controls (MTReGFP = 4.02 ± 4.37 a.u.*μm−2*10−4). In contrast, even though Abl2Δ688-790-eGFP and Abl2-eGFP were both similarly enriched at MTOCs in both low- and high-expressing cells (Figure S3D, ii), tubulin binding-deficient Abl2Δ688-790-eGFP (Figure S3C–H) did not promote increased MT recovery at either expression level (MTRlow Abl2Δ688-790-eGFP = 3.71 ± 3.40 a.u.*μm-2*10–4; MTRhigh Abl2Δ688-790-eGFP = 3.12 ± 2.98 a.u.*μm−2*10−4; Figure 3F, G). Together, these data suggest that Abl2 promotes nucleation by binding and recruiting cytoplasmic tubulin and that this correlates with faster MT recovery in cells.
Abl2 preferentially binds expanded over compacted MT lattices.
MBPs bind MT lattices with distinct conformational states with different affinities41,49–51. Cryo-EM studies show that the GMPCPP-MT lattice is relatively expanded compared to the GDP-MT lattice52,53. Given that Abl2 promotes nucleation which generates tubulin oligomers or immature MT tubes, we measured the nucleotide state preference (NSP) of Abl2-eGFP and control proteins using MTs of GDP-polymerized tubulin, capped with GTP-like GMPCPP-containing segments to prevent catastrophe. The +TIP tracker EB1-eGFP bound GMPCPP-rich regions preferentially with NSPEB1 = 0.38 ± 0.37, where NSP = log10(fluorescence intensity on GMPCPP versus GDP segments) (Figure 4A)54, while tau-2N4R-eGFP preferentially bound GDP-rich regions (NSPtau = −0.67 ± 0.26; Figure 4A, purified EB1-eGFP and tau-eGFP are shown in Figure S1I, viii–ix), as expected41. Abl2-eGFP and its cytoskeletal-binding half 557-C-eGFP strongly preferred GMPCPP-MT segments over GDP-MT segments (NSPAbl2 = 0.35 ± 0.33, p < 0.0001 relative to NSPtau; NSP557-C = 0.41 ± 0.28, p < 0.0001 relative to NSPtau, Figure 4A).
Figure 4. Abl2 preferentially localizes onto extended MT lattices through multiple binding patterns.
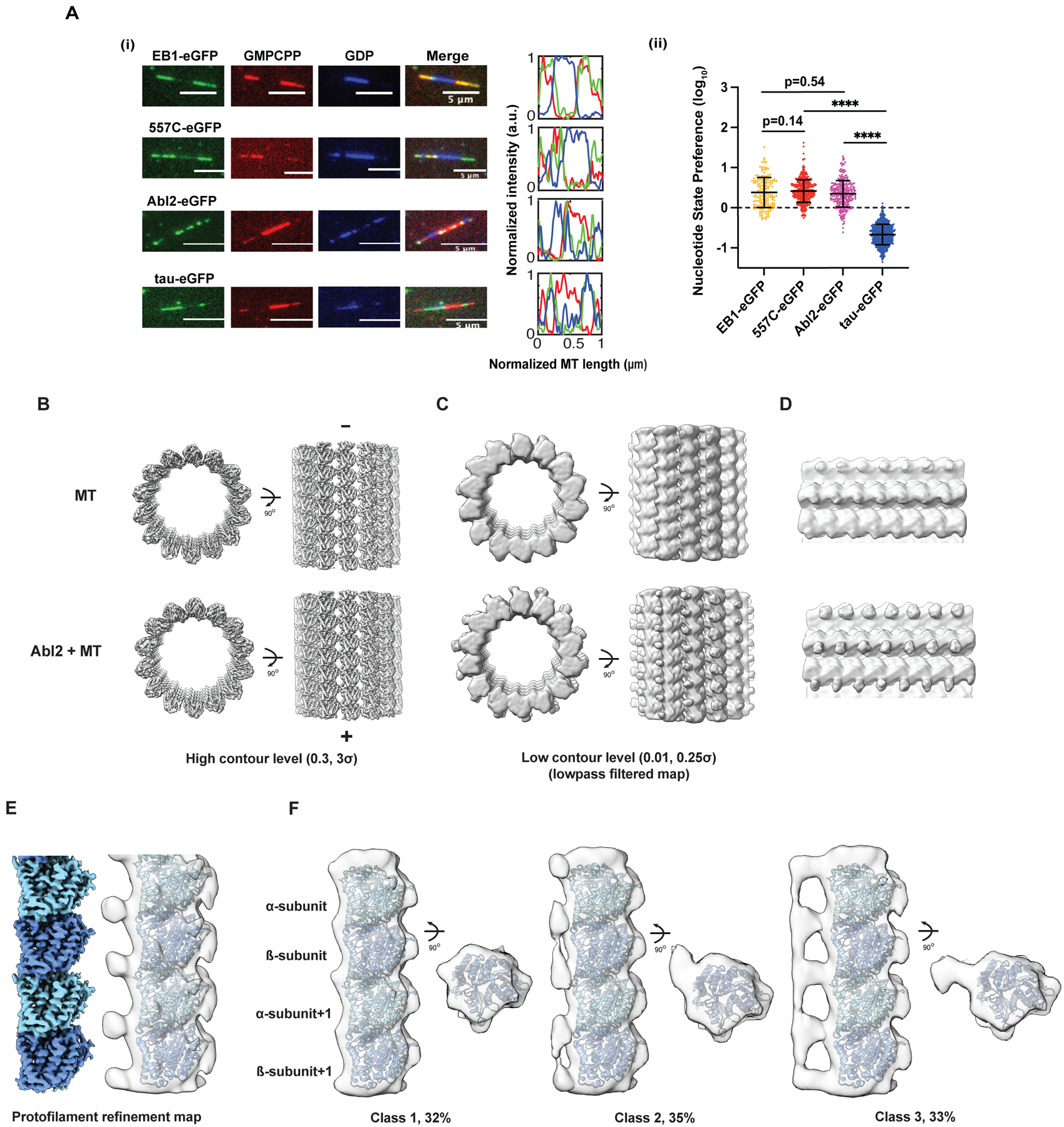
(A) (i) Representative images of 0.8 μM EB1-eGFP, 0.5 μM 557-C-eGFP, 0.5 μM Abl2-eGFP, and 25 nM tau 2N4R-eGFP on segmented end-stabilized MTs. eGFP-tagged MBP shown in green; GMPCPP segments in red; GDP segments in blue. 3 pixel-wide line scans were drawn lengthwise across the microtubule. Normalized lengths and corresponding fluorescence intensities shown on right. (ii) Nucleotide-state preference (NSP) of EB1-eGFP, tau-eGFP, 557-C-eGFP, and Abl2-eGFP shown in log10 scale. Data are mean ± SD. Mann-Whitney test. ****, p < 0.0001. (B, C) Cryo-EM reconstructions of GMPCPP MT lattice alone (top) or with 6XHis-tagged Abl2-557-1090 (bottom) visualized at high (left) and low (right) contour levels. For better comparison of cryo-EM maps at low contour levels, maps were filtered via convolution with a Gaussian filter (one standard deviation of 3 Å). (D) Zoomed-in view of maps shown in (C). (E) Protofilament refinement of the Abl2-MT cryo-EM dataset improves the map resolution and quality. The map after filtering (in transparency) contains extra density around tubulin C-terminal tails. (F) 3D classification reveals that Abl2 bridges multiple tubulins within a protofilament. Class 1 does not show clear extra density near tubulin C-terminal tails. Classes 2 and 3 show clear densities near tubulin C-terminal tails. The densities in class 2 show inter-dimer connectivity while the densities in class 3 show both inter- and intra-dimer connectivity. See also Figure S4, Table S1.
We performed cryo-EM analysis of MTs alone or incubated with 6XHis-tagged Abl2 C-terminal fragment (His-Abl2-557-1090) to understand the structural basis for Abl2 interactions with the MT lattice (Figure S4A, Table S1). Abl2-557-1090-eGFP binding to MTs was documented via TIRF microscopy (Figure S1J, iv). Only a portion of the tubulin C-terminal tail was observed via low-pass filtering the cryo-EM map and by setting it at low contour levels, likely due to its high flexibility55,56 (Figure 4B, C). We observed extra density near the tubulin C-terminal tail on the MT lattice decorated with Abl2-557-1090 (Figure 4C, D).
Abl2 binds MTs via both electrostatic interactions with the tubulin acidic C-terminal tails and via additional interfaces38. To identify all possible interfaces and capture stable complexes for atomic model building, we used a protofilament refinement method55 to merge different protofilaments into one single protofilament volume, greatly improving EM map resolution and quality (Figure 4E). Due to tubulin tail flexibility and non-uniform Abl2-557-1090 decoration, no additional density was observed at high contour levels (Figure 4B). However, we created a mask covering four tubulin monomers and their surfaces and performed a focused 3D classification (Figure S4B). Even after extensive 3D classification, complexes between the Abl2-557-1090 and the protofilament stable enough to enable high-resolution reconstructions were not observed, suggesting that Abl2-557-1090 interacts with the MT lattice in a flexible manner. We did observe, however, three classes with different binding patterns of Abl2-557-1090 (Figure 4F). The first class (32%) did not have extra density around tubulin C-terminal tails, no Abl2-557-1090 binding in this class. In the second class (35%), a connection of extra density was observed between the tubulin β-subunit and the tubulin α-subunit+1 of the adjacent tubulin dimer in the protofilament. In the third class (33%), the extra density was further connected along protofilaments. Together, the cryo-EM analysis suggests that Abl2-557-1090 flexibly and non-uniformly decorates the MT lattice to bridge multiple dimers within a protofilament.
Abl2 mediates repair in damaged MT lattices.
Given that Abl2:tubulin coacervates interact with the expanded MT lattice, we next asked if Abl2 impacts damaged MT lattice repair. MT lattice defects were induced by incubating rhodamine-labeled MTs overnight with taxol at 23°C and 37°C12, and visualized as regions of reduced rhodamine intensity along the MT (Figure 5A). Visualization by negative stain EM confirmed that taxol-stabilized MTs stored at 23°C overnight had more defects than those stored at 37°C (Figure S5A, B). We monitored repair via the incorporation of new Alexa647-tubulin at damaged sites, tracking both the number of incorporation events and the fraction of total MT length labeled with new Alexa647-tubulin, termed the reporter fraction (RF). In agreement with previous reports of greater damage to the MTs stored at 23°C versus 37°C12,57, we observed a higher RF in MT shafts stored at 23°C overnight relative to those stored at 37°C overnight (RF23°C,tub = 0.17 ± 0.13; RF37°C,tub = 0.12 ± 0.10; Figure 5D, S5C). Though Abl2-eGFP binds uniformly on fresh taxol-stabilized MTs (Figure S5E), Abl2-eGFP bound at 1.3-fold higher density to the more damaged 23°C-stored MTs than 37°C-stored MTs, with mean normalized fluorescence intensities of 1.02 ± 0.03 a.u.*μm−1 and 0.82 ± 0.03 a.u.*μm−1, respectively (Figure S5D).
Figure 5. Abl2 promotes damaged lattice repair and increases MT lifetime.
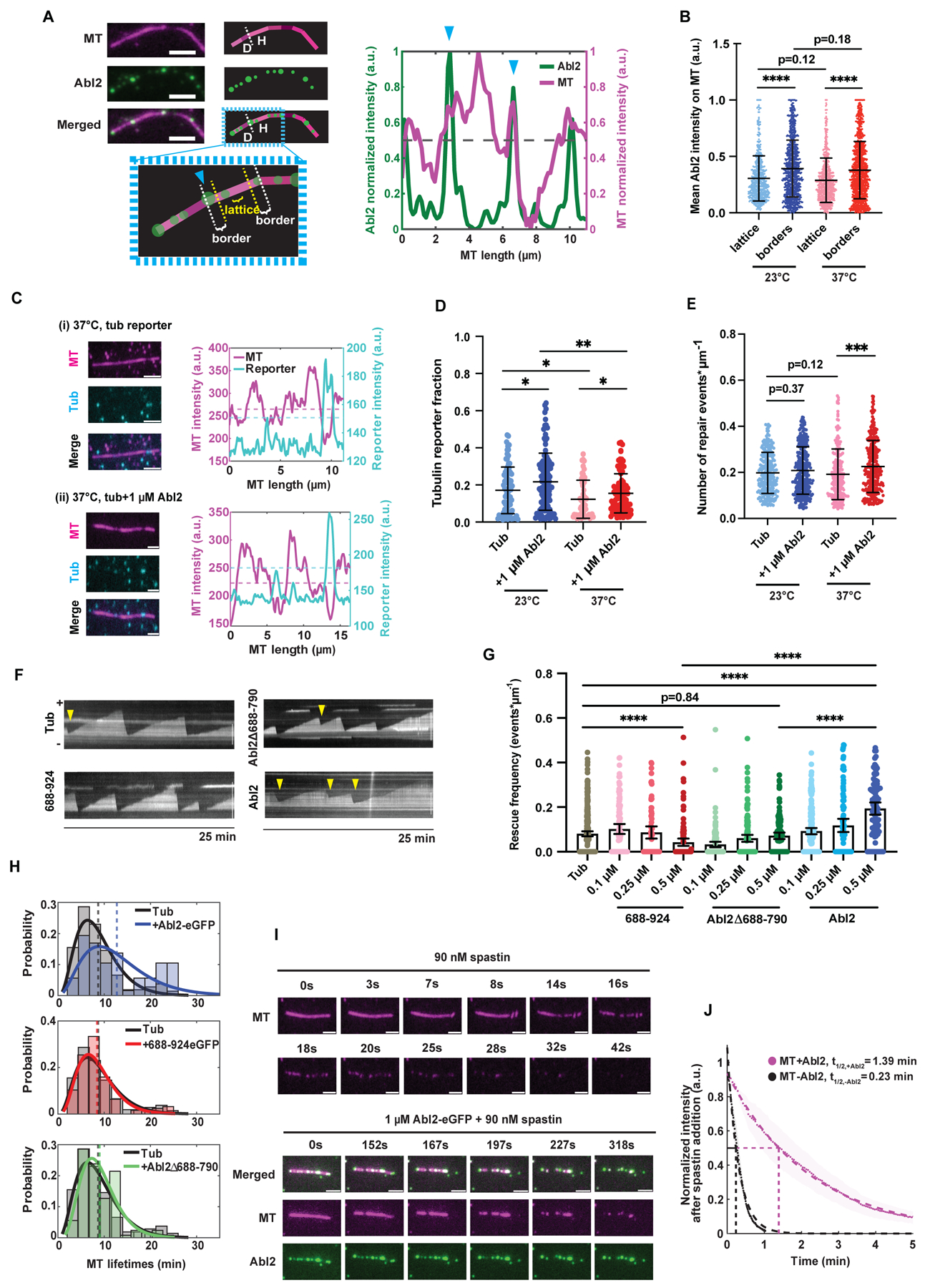
(A) Left: Representative Abl2-eGFP localization on a 37°C-overnight stored taxol-stabilized MT. Borders were defined as boundaries on ‘healthy’ (“H”) structurally intact MT segments adjacent to ‘damaged’ (“D”) segments, demarcated with a blue arrow. Lattices were stretches that are ≥ 1/5 of the total MT segment length away from either terminus. Scale bar, 3 μm. Right: Quantification of MT and Abl2-eGFP intensities along the taxol-stabilized MT shown on left. Dotted black line denotes the mean normalized MT fluorescence intensity, which was used as the threshold for scoring a segment as “H” or “D”. Normalized fluorescence intensity scale bar shown on left. (B) Mean intensities of 1 μM Abl2-eGFP at borders and lattices of healthy MT segments along filaments stored at 23°C and 37°C overnight. Means shown as solid horizontal black lines, 25–75% quartiles shown as box plots. n ≥ 600 healthy segments analyzed per condition. Wilcoxon rank sum test. ****, p < 0.0001. (C) Representative taxol-stabilized rhodamine-MTs stored at 37°C overnight (magenta) and repaired tubulin reporter (cyan). MT mixture was supplemented with 2 μM Alexa647-tubulin (cyan), 10 mM GTP, and 10 μM taxol were allowed to incorporate at damaged sites for 3 hr at 37°C alone (i) or with (ii) 1 μM Abl2-eGFP. Fluorescence intensities of MT and 647-tubulin were plotted. Dashed magenta line denotes the MT intensity threshold (mean normalized fluorescence intensity) to distinguish healthy from damaged segments. Dashed cyan line denotes the reporter intensity threshold to score whether 647-tubulin reporter is present or not. Scale bar, 3 μm. (D) Total tubulin reporter fraction per MT was quantified per storage condition in the presence or absence of 1 μM Abl2-eGFP. Data are mean ± SEM. n ≥ 50 filaments analyzed per condition. Wilcoxon rank sum test. *, p < 0.05; **, p < 0.01. (E) Total number of repair/incorporation events per MT was quantified per storage condition in the presence or absence of 1 μM Abl2-eGFP. n ≥ 200 incorporation events analyzed per condition. Data are mean ± SEM. Wilcoxon rank sum test. ***, p < 0.001. (F) Representative kymographs of dynamic MT filaments in presence of 10.5 μM rhodamine tubulin alone, or with 0.5 μM 688-924-eGFP, Abl2Δ688-790, or Abl2-eGFP. Yellow arrow denotes a rescue event. Scale bar, 8 μm. (G) Rescue frequencies (fres) were quantified for 10.5 μM rhodamine tubulin alone, or supplemented with 0.1, 0.25, and 0.5 μM 688-924-eGFP, 0.5 μM Abl2Δ688-790, or 0.5 μM Abl2-eGFP. Data are means with 95% CI as black lines. Wilcoxon rank sum test. ****, p < 0.0001. (H) MT lifetime distributions of filaments grown in presence of 0.5 μM Abl2-eGFP (blue), 0.5 μM 688-924-eGFP (red), and 0.5 μM Abl2Δ688-790 (green) relative to tubulin alone (black). Gamma distributions were fit to MT lifetime histograms, shown in solid blue, red, and green curves respectively, relative to tubulin control (solid black). Mean MT lifetimes are denoted as dashed lines. (I) Severing assays performed in TIRF chambers containing biotinylated, rhodamine GMPCPP-stabilized MTs (pseudo-colored magenta) with or without 1 μM Abl2-eGFP supplemented with 90 nM spastin and 2 mM ATP. Representative time series of MTs are shown. Intensity decay curves of severed MTs quantified in (J). Scale bar, 3 μm. (J) Mean intensity decays of MTs in absence (black) and presence of 1 μM Abl2-eGFP (magenta) shown in scatter dots, and SEMs shown as wider shadowing, respectively. Single exponential decay curves were fit to the data, shown as dashed lines. n ≥ 200 filaments analyzed per condition. See also Figure S5.
Abl2-eGFP did not uniformly decorate the damaged MTs. To measure whether Abl2 has preference for healthy versus damaged regions, we made line scans of MTs and defined segments of mean normalized fluorescence intensities equal to or above 0.5 as ‘healthy’ and those below as ‘damaged’. This threshold was used across experimental replicates as it best differentiated higher versus lower fluorescent intensities throughout a given MT. ‘Borders’ were defined as segments directly adjacent to damaged ones, with lengths one-fifth of the total healthy segment length away from damaged sites and ‘lattices’ defined as the remaining three-fifths middle portion of the healthy segment (Figure 5A). Abl2-eGFP bound preferentially to borders, with mean intensities of 0.39 ± 0.01 a.u. and 0.38 ± 0.01 a.u. on 23°C- and 37°C-stored MT damaged borders, respectively; and mean intensities of 0.31 ± 0.01 a.u. and 0.28 ± 0.01 a.u. on 23°C- and 37°C-stored MT lattices, respectively (mean ± SEM, Figure 5B).
To measure if Abl2-eGFP impacted tubulin incorporation at damage sites, we incubated 23°C- and 37°C-stored damaged MTs with and without 1 μM Abl2-eGFP (Figure 5C; representative EM micrographs for tubulin bound 23°C-stored MTs in Figure S5H). Including Abl2-eGFP increased the fraction of total MTs containing Alexa647-tubulin 1.27- and 1.26-fold to RF23°C, +Abl2 = 0.22 ± 0.15 and RF37°C, +Abl2 = 0.15 ± 0.11, respectively (Figure 5D, S5I). Abl2-eGFP also increased the frequency of new tubulin incorporation segments into existing shafts in the 37°C storage condition 1.21-fold, from fincorp, tub, 37°C = 0.19 ± 0.11 events*μm−1 of MT length to fincorp, +Abl2, 37°C = 0.23 ± 0.11 events*μm−1 in (p < 0.001; Figure 5E, S5G). Although the number of tubulin incorporation events did not change significantly for MTs stored at 23°C upon inclusion of 1 μM Abl2-eGFP (fincorp, tub, 23°C = 0.20 ± 0.09 events*μm−1 of MT length to fincorp, +Abl2, 23°C = 0.21 ± 0.10 events*μm−1), the relative length of newly incorporated tubulin (RL) per MT increased 1.34-fold in the presence of Abl2 (RLtub alone, 23°C = 2.40 ± 2.04 μm; RLtub+Abl2, 23°C = 3.24 ± 2.72 μm) (Figure S5F). Together, these data indicate that Abl2-eGFP can promote repair of MT lattice damage.
Abl2 increases rescue frequency to prolong MT lifetimes.
Repair of MT damage leads to the formation of GTP-tubulin enriched islands within MT segments58,59, which promotes rescue11,13,14,21,60. We tested how Abl2-eGFP impacts rescue frequency (fres) of MTs elongated from GMPCPP-MT seeds. Consistent with previous work38, Abl2 decreased the MT depolymerization rate 0.6-fold, from 13.49 μm*min−1 to 8.49 μm*min−1 (Figure S5J). Inclusion of Abl2-eGFP increased rescue frequency in a concentration-dependent manner (Figure 5F, G; S5K), such that inclusion of 0.5 μM Abl2-eGFP induced a 2.4-fold increase in rescue frequency, from fres, tub = 0.080 ± 0.006 events*μm−1 to fres, 0.5 μM Abl2 = 0.194 ± 0.014 events*μm−1 (mean ± SEM). However, 0.5 μM of tubulin-binding 688-924-eGFP fragment decreased rescue frequency relative to control (fres, 0.5 μM 688-924 = 0.043 ± 0.001 events*μm−1, p < 0.0001), whereas 0.5 μM of MT-binding Abl2Δ688-790 did not impact rescue (fres, 0.5 μM Abl2Δ688-790 = 0.071 ± 0.007 events*μm−1; mean ± SEM). We speculate that without the ability to bind MT lattices (Abl2-688-924) or tubulin dimers (Abl2Δ688-790), Abl2 cannot recruit tubulin dimers to the MT lattice. CLASPs and CSPP1 induce pauses prior to MT rescue16, but Abl2 did not impact the pausing frequency prior to growth and shrinkage phases, nor the time spent pausing until either phase (Figure S5L–N). We also measured the net effects of Abl2 and Abl2 fragments on MT lifetime distribution (MTLD). The addition of 0.5 μM Abl2 significantly extended the MTLD 1.5-fold to 12.7 ± 7.0 mins from 8.7 ± 4.5 mins for MTs alone. Neither 0.5 μM of Abl2-688-924 nor Abl2Δ688-790 altered MTLD relative to control (MTLDAbl2-688-924 = 8.4 ± 4.0 min; MTLDAbl2Δ688-790 = 9.0 ± 3.9 min; Figure 5H).
Considering its ability to maintain MT lattice integrity, we asked whether Abl2 could protect against MT lattice damage mediated by the ATP-dependent MT severing enzyme spastin (Figure 5I, protein purity shown in Figure S1H, ix). Spastin promoted rapid disassembly of MTs with t1/2 = 0.23 min, while pre-incubation of MTs with 1 μM Abl2-eGFP greatly slowed the spastin-mediated disassembly rate 6-fold to t1/2, Abl2 = 1.39 min (Figure 5J), suggesting that it protects MT lattices from active damage.
Discussion
We demonstrate here for the first time that Abl2 undergoes phase separation, forms coacervates with tubulin, and promotes MT nucleation. Abl2 also engages an extended and damaged MT lattice, spanning over single MT protofilaments to promote lattice repair and increase rescue and lifetime. Our results are consistent with a model in which Abl2 promotes the recruitment and addition of tubulin to MTs in different functional scenarios.
Abl2 binds tubulin using two distinct C-terminal regions.
We identified two tubulin-binding regions in Abl2, contained within amino acids 688–924 (site I) and 1024–1090 (site II), each sufficient to bind tubulin dimers itself, albeit with weaker affinity than Abl2 fragments containing both sites. Curiously, Abl2Δ688-790 did not bind detectably to tubulin even though it retains site II (Figure 1; S1K). We propose that Abl2 initially interacts with tubulin via site I (Figure 6A, i), which has a higher affinity to tubulin. Binding of tubulin to site I may be required to expose site II to an additional tubulin dimer and promote their interactions in a head-to-tail fashion (Figure 6A, ii–iii). This model would explain why Abl2Δ688-790, which lacks all or part of site I, fails to bind tubulin and promote MT nucleation. The two tubulin-binding sites in Abl2 do not resemble known tubulin-binding domains but are enriched in positively charged residues. Interestingly, Abl2 binds to tubulin with lower affinity than 557-C, suggesting that in addition to substrate phosphorylation, the N-terminal half sterically hinders tubulin binding. Most soluble Abl2 molecules undergo phase separation and co-condense with tubulin, and we believe this underlies the patchy appearance of Abl2:tubulin co-condensate binding to assembled MTs (Figure 1N).
Figure 6. Model for Abl2 in regulating MT dynamics.
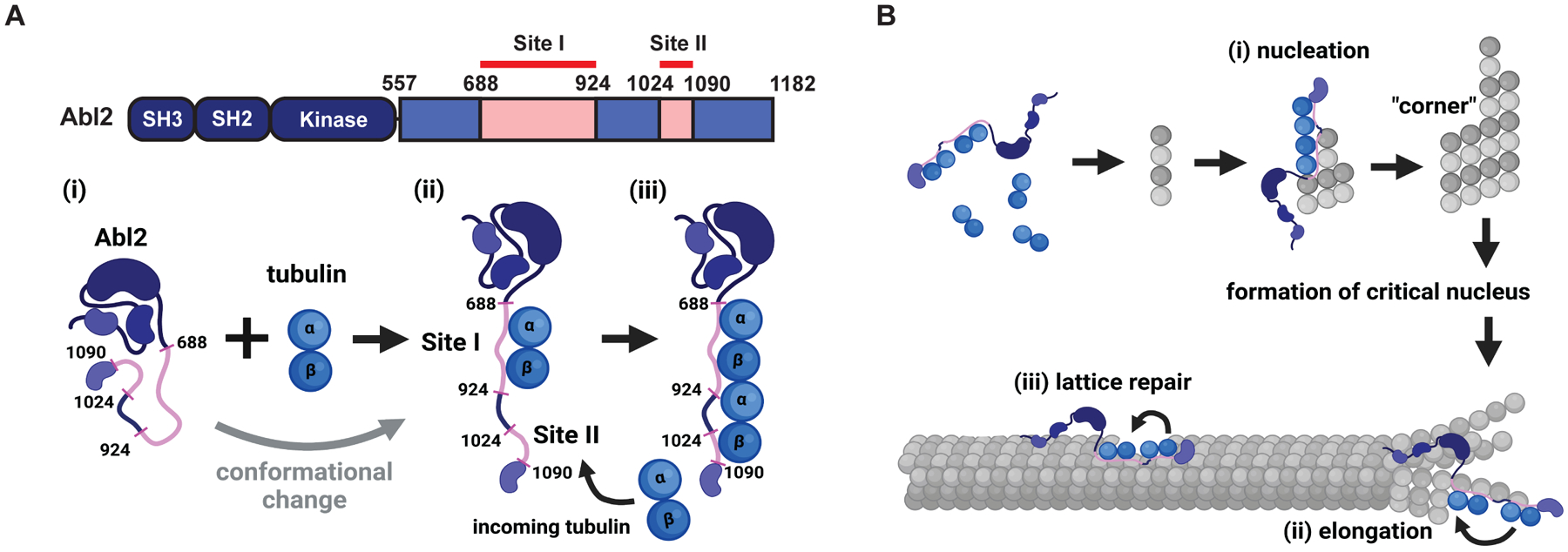
(A) Schematic of full length Abl2 regulating MT growth, nucleation, and lattice repair. (i) Abl2 forms a conformational change when it binds a tubulin dimer. (ii) Abl2 directly interacts with tubulin dimers using its solvent-exposed 688–924 (site I). The 1024–1090 region (site II) is less accessible in the native conformation. (iii) Binding to a single tubulin dimer via site I may induce a conformational change within 557-C, leading to solvent exposure of site II, which can further recruit an additional dimer. (B) Abl2 can dock onto: (i) tubulin oligomers to facilitate the incorporation of new tubulin dimers for nucleation; (ii) on growing MT ends to promote elongation; and (iii) on regions along MTs that harbor lattice defects to promote rescue and repair.
The tubulin-binding Abl2 splice isoform is more predominant in human tissues than in mice.
Murine Abl2, used in this study, is 95% identical to human Abl2, with near identical sequence of the alternatively spliced exon encoding amino acids 688–791, which sits entirely within a larger final coding exon in Abl2. Analysis of murine and human Abl2 across tissues indicated that the relative abundance of transcripts containing this alternatively spliced exon differed between species (Figure S6A, B; abbreviations found in Tables S2, S3, respectively). Namely, transcripts including the amino acids 688–791 predominate over those lacking it across human tissues, while the Δ688-791 isoform is predominant in mouse tissues (Figure S6A)61. Genomic engineering of mice that preferentially express Abl2 isoforms including amino acids 688–791 should reveal how this exon impacts microtubule dynamics and organismal development in vivo.
Abl2 interacts with MTs and tubulin to promote MT nucleation, growth, and lattice repair.
Abl2 reduces the critical concentration for spontaneous tubulin polymerization and shortens the MT nucleation lag time. Tubulin binding-deficient Abl2Δ688-790 did not impact nucleation, indicating that tubulin binding is critical to promote nucleation (Figure 3). Classical models for MT nucleation proposed that new MTs form via a nucleation-elongation mechanism. Recent studies demonstrated that ‘critical nuclei’ are first formed as 2D layers of a growing lattice prior to its maturation into a cylindrical tube62,63. Enlargement of 2D lattices is energetically favorable but kinetically impeded by the difficulty in adding new protofilament layers. Given our structural data that Abl2 binds to extended lattices and potentially bridges multiple dimers along a single protofilament (Figure 4), we propose that Abl2 may facilitate nucleation by binding a nascent protofilament and recruiting tubulin dimers to promote layer formation. In addition, Abl2 may then bind the lattice and facilitate tubulin addition at corners of 2D lattices, which may be structurally akin to the corners at growing MT ends and damaged holes (Figure 6B, i). While growing MT ends contain a heterogeneous mixture of curled multi-protofilament sheets, and straight, flared, or ragged ends64,65, damaged sites are likely to contain a heterogeneous mixture of these structures. We propose that Abl2 recognizes one or more of these structures within the damaged region, recruiting tubulin for incorporation (Figure 6B, ii–iii). It is also possible that Abl2 binding along the protofilament recruits new tubulin dimers for incorporation at protofilament ends. In doing so, Abl2 may stabilize inter-protofilament interactions to promote MT nucleation, growth, and lattice repair. Tools developed here should facilitate future studies to probe how Abl2 promotes MT assembly.
Abl2 localizes to expanded lattices via multiple binding patterns.
Our cryo-EM analysis reveals that Abl2-557-1090 primarily binds to the tails of polymerized tubulin (Figure 4B), a binding mode that differs from other conventional MBPs (e.g. tau, MAP7, and EB3), which bind multiple interfaces along a polymerized MT or protofilament56,66,67. Abl2-557-1090 did not form a highly stable complex with MTs, as we did not observe extra density surrounding the tails (Figure 4B). Our sequence and biophysical analyses (Figure 2A, B) and Alphafold2 (Figure S4C, D) indicate that Abl2-557-1090 is disordered, lacking a well-ordered microtubule-binding domain, which likely underlies the difficulty in obtaining highly ordered structures. Cryo-EM analysis did reveal that Abl2-557-1090 can bridge multiple tubulin dimers, which is supported by our observations of Abl2 diffusion along stabilized MT lattices (Figure 2L). We propose that Abl2 can dynamically “slide” across the negatively charged lattice surface with its net positively charged tubulin-binding fragment, which can simultaneously bind and potentially locally concentrate soluble dimers at damage sites. The ability of MT-bound Abl2 to move on a bed of negatively-charged tubulin tails may serve as an economic means to transport along the lattice for damage recognition and repair; or toward ends for recruiting dimers for growth68,69. A similar mode of diffusion along the MT lattice is employed by tau66,70.
Phase separation contributes to Abl2 function in regulating MT nucleation.
A growing number of MT plus-end regulators undergo phase separation22–25,71, but less is known about how LLPS contributes to microtubule nucleation. Under conditions of molecular crowding in solution or in the presence of stabilized MT lattices, Abl2 underwent phase separation and recruited tubulin into the dense phase. FRAP recovery times reported for other GFP-fusion protein condensates within highly compartmentalized structures in cells have timescales of seconds72,73. In contrast, FRAP recovery times of Abl2-eGFP and tubulin in condensates within minutes were consistent with measurements of other MT regulators in in vitro biomolecular condensates19,74. Taylor et al. have developed an infinite 2D model that accurately captures the relatively slower kinetics of extracellular molecular condensates which depend critically on factors such as bleach spot size and droplet size. As such, we appropriately implemented their model here to quantify the t1/2 values75.
Protein condensation is often driven by intrinsically disordered regions and multivalent binding regions43,44,46,47,76. Phase separation is a common feature of diverse MT regulators77 41,42,49. Consistent with this, we found that the Abl2 C-terminal half is significantly disordered and has two distinct sites for tubulin binding (Figure 1H, I; 2A, B). We showed that new MTs nucleated from Abl2-eGFP:tubulin co-condensates. Under the same conditions, eGFP alone was not able to phase separate or interact with tubulin to promote MT assembly (Figure 3E). Our data suggest that the ability of Abl2 to co-condense with tubulin dimers may help concentrate tubulin onto nascent or damaged protofilaments to facilitate nucleation and promote lattice repair.
In summary, we demonstrate that co-condensates of Abl2 and tubulin act as compartmentalized reactors which may contribute to (but not required for): 1) nucleate MTs; and 2) promote the incorporation of tubulin dimers at damaged lattice sites for repair and rescue. Our study provides a mechanistic model to probe how Abl2 regulates MT assembly and repair and provides tools to explore how these mechanisms regulate cell morphogenesis and migration.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Anthony J. Koleske (anthony.koleske@yale.edu).
Materials availability
All unique reagents used in this study are available from the lead contact.
Data and code availability
All data is available in the main text or supplemental information.
Custom-written MATLAB scripts for lattice damage analysis are available at https://github.com/koleskelab/lattice_damage.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture and stable cell line establishment
Mycoplasma-free COS-7 cell lines (ATCC) and the derived stable cell lines were grown in DMEM supplemented with 10% FBS, 100 units*ml−1 penicillin, 100 μg*ml−1 streptomycin, and 2 mM L-glutamine. Abl2 KO COS-7 cells were generated using CRISPR/Cas9, as described previously78. Resistant Abl2-eGFP, Abl2Δ688-790-eGFP, and eGFP were cloned into FUGW vector (Addgene #14883) and then transfected together with pLP1, pLP2, and pLP/VSVG into HEK293T cells to generate lentivirus. Abl2 KO COS-7 cells were infected with the lentivirus and selected with 400 μg*ml−1 Zeocin (Thermo Fisher Scientific) for 7 days.
Abl2-eGFP, Abl2Δ688-790-eGFP, and eGFP expressing cells were obtained by fluorescence-activated cell sorting using Abl2 KO COS-7 cells as a negative reference. Cell lysates were collected to determine the expression levels of Abl2 or Abl2 variants via immunoblotting. Cells were lysed with 1x LSB buffer (8% SDS, 20% glycerol, 100 mM Tris pH 6.8, 8% 2-mercaptoethanol) at 100°C. Lysates were run on SDS-PAGE and transferred to 0.45 μm nitrocellulose, blocked using 5% milk and immunoblotted with anti-GFP antibody (Novus Biologicals) or with Ar11 antibody, which specifically recognizes the Abl2 C-terminal half (residues 766–1182; gift from Peter Davies, Albert Einstein Medical College, Bronx, NY).
METHOD DETAILS
Tubulin purification and labeling
Porcine brain tubulin was prepared as previously described in Castoldi & Popov (2003)79. 2 ml 14 mg*ml−1 porcine brain tubulin was labeled with TAMRA as described in Peloquin et al. (2005)80. Biotinylated tubulin was obtained from Cytoskeleton.
Molecular cloning and Abl2 purification
Murine Abl2 (residues 74–1182), Abl2-eGFP, N terminal half (N-557), N-557-eGFP, C terminal half (557-C), 557-C-eGFP, and fragments contain amino acids 688–790, 688–924, 688-924-eGFP, 1024–1090 were cloned with an N-terminal 6XHis tag into the pFastBac1 vector (Invitrogen) for insect cell expression. Abl2Δ688-790 was generated using PCR-based mutagenesis and confirmed by DNA sequencing. Abl2 and Abl2Δ688-790 were cloned into pN1-EGFP expression vector for mammalian cell expression. Recombinant baculoviruses expressing Abl2 or Abl2 fragments in pFastBac were generated using the Bac-to-Bac expression system in Sf9 insect cells according to the manufacturer’s instructions (ThermoFisher, Waltham, MA). After 36–48 hr infection with baculoviruses, High Five cells were collected and centrifuged for 3,000 rpm for 5 min at 4°C. Cells were lysed using buffer containing 20 mM HEPES pH 7.25, 5% glycerol, 500 mM KCl, 20 mM imidazole, 1 mM DTT, 1 mM PMSF, 1X protease inhibitor cocktail, 1% Triton-X100 and left to incubate at 4°C on a rotisserie stand for 5–10 min. Lysates were ultracentrifuged using Ti70.1 rotor for 45 min, 40K rpm at 4°C. After collecting and 0.45 μm filtering the supernatant, proteins were passed through disposable columns with Ni-NTA resin beads via gravity flow three times. Resin was washed in order with the following buffers: wash A (20 mM HEPES pH 7.25, 5% glycerol, 500 mM KCl, 20 mM imidazole, 1 mM DTT, 1 mM PMSF, 1X protease inhibitor cocktail); B (20 mM HEPES pH 7.25, 5% glycerol, 1 M KCl, 20 mM imidazole, 1 mM DTT, 1 mM PMSF, 1X protease inhibitor cocktail), A, and C (20 mM HEPES pH 7.25, 5% glycerol, 300 mM KCl, 20 mM imidazole, 1 mM DTT, 1 mM PMSF, 1X protease inhibitor cocktail). Bound proteins were step eluted off the Ni-NTA resin (Invitrogen) using wash C buffer containing 300 mM imidazole in 5X1 ml fractions. 6XHis-Abl2 and 6XHis-Abl2 fragments for biolayer interferometry assays were exchanged into storage buffer containing 20 mM HEPES pH7.25, 5% glycerol, 100 mM KCl, 1 mM DTT using either Superdex 75 increase 10/300 GL column for proteins with MW < 75kDa, or Superdex 200 increase 10/300 GL column for proteins with MW ≥ 75 kDa. His tags were removed by adding TEV protease into purified Abl2 constructs, and were incubated on a rotisserie stand at 4°C for 2.5–4 hrs. His-tag cleaved proteins were exchanged into the storage buffer containing 20 mM HEPES, 5% glycerol, 300 mM KCl, and 1 mM DTT using either Superdex 75 increase 10/300 GL column or Superdex 200 increase 10/300 GL column. Peak fractions were collected, snap frozen, and stored at −80°C until use.
Tau 2N4R-eGFP purification
FUGW-tau 2N4R plasmid was a kind gift from the Liz Rhoades Lab. Human full-length tau 2N4R was cloned into pFastBac1 vector (Invitrogen) with an N-terminal 6XHis tag for insect cell expression. Baculovirus generation and Hi5 insect cell expression and purification follow that of His-cleaved Abl2 constructs, as described above. 6XHis-cleaved tau 2N4R-eGFP was buffer exchanged into 20 mM HEPES, 5% glycerol, 300 mM KCl, 1mM DTT using Superdex 200 increase 10/300 GL column. Each peak fraction was snap frozen and stored at −80°C until use.
EB1-eGFP purification
pETM-11-EB1-eGFP plasmid was a kind gift from the Joe Howard Lab. pETM-11-EB1-eGFP was transformed and expressed into E. coli BL21 cells. 4 l expression cultures were grown in 2XYT at 37°C, 200 rpm until A600 reached 0.6, followed by induction with 1 mM IPTG at 18°C for 24 hrs. Cells were harvested by centrifugation at 4K rpm in SLC6000 rotor 15 min at 4°C. Cell pellets were resuspended in lysis buffer (20 mM HEPES, 500 mM KCl, 5% glycerol, 1 mM DTT, 0.1% Triton X-100, 1X protease inhibitor cocktail) and lysed with the QSonica Q55 Compact Sonicator for 5 minutes on ice at amplitude 30 (on-off 10s pulses on the thumb switch setting). Cell lysates were ultracentrifuged using Ti70.1 rotor for 45 min, 40K rpm at 4°C. Purification of 6XHis-EB1-eGFP to generate 6XHis-cleaved EB1-eGFP follows that of His-cleaved Abl2 constructs as described above. 6XHis-cleaved EB1-eGFP was buffer exchanged into 20 mM HEPES, 5% glycerol, 300 mM KCl, 1mM DTT using Superdex 75 increase 10/300 GL column. Each peak fraction was snap frozen with the addition of 1X protease inhibitor cocktail and stored at −80°C until use.
Spastin purification
pGEX-PP-Spastin(87–616)DeltaExon4 was purchased from Addgene (item ID #128794). Spastin was transformed and expressed into E. coli BL21 cells. 4l expression cultures were grown in 2XYT media at 37°C, 200 rpm until A600 reached 0.6, which was then followed by induction with 0.5 mM IPTG. Cultures were grown 16°C for 16 hrs. Cells were harvested by centrifugation at 4K rpm in SLC6000 rotor 15 min at 4°C. Cell pellets were resuspended in lysis buffer (50 mM Tris-HCl pH 8.0, 5% glycerol, 0.1% Triton X-100, 5 mM MgCl2, 1 mM DTT, 1X PMSF, 1X protease inhibitor cocktail) and lysed with the QSonica Q55 Compact Sonicator for 5 minutes on ice at amplitude 30 (on-off 10s pulses on the thumb switch setting). Lysates were clarified for 20 min in SA600 rotor at 15K rpm at 4°C, and 0.45 μm filtered. 2 ml of glutathione agarose resin was equilibrated using lysis buffer without added detergent and pipetted into a disposable gravity flow column. Lysates were passed through column 3X. Resin was washed with buffers in the following order: A (50 mM Tris-HCl pH 8.0, 300 mM KCl, 5 mM MgCl2; 5% glycerol, 1X DTT, 1X PMSF); B (50 mM Tris-HCl pH 8.0, 1 M KCl, 5 mM MgCl2; 5% glycerol, 1X DTT, 1X PMSF); A; and C (50 mM Tris-HCl pH 8.0, 150 mM KCl, 5 mM MgCl2; 5% glycerol, 1X DTT, 1X PMSF). 5 ml of Wash C supplemented with 1 mg PreScission protease was added into the resin and left to incubate on the column overnight at 4°C. After ~16 hrs, GST-cleaved spastin was removed from PreScission protease by passing through Superdex 200 increase 10/300 GL column and stored in 20 mM HEPES, 5% glycerol, 300 mM KCl, 5 mM MgCl2, 1 mM DTT. The first three peak fractions were concentrated using 0.5 ml centrifugal filter units (EMD Millipore #UFC501096) that were pre-equilibrated with storage buffer and stored at −80°C until use.
eGFP purification
eGFP from pN1-eGFP plasmid was cloned C-terminus of GST into the pGEX-6p1-GST vector. pGEX-6p1-GST-eGFP was transformed into E. coli BL21 cells. 2l expression cultures were grown in 2XYT media at 37°C, 200 rpm until A600 reached 0.6, which was then followed by induction with 0.5 mM IPTG. Cultures were grown 16°C for 16 hr. Cells were harvested by centrifugation at 4K rpm in SLC6000 rotor 15 min at 4°C. Cell pellets were resuspended in lysis buffer (20 mM HEPES pH 7.25, 500 mM KCl, 5% glycerol, 0.1% Triton X-100, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1 mM PMSF, 1X protease inhibitor cocktail). Lysates were clarified for 20 min in SA600 rotor at 15K rpm at 4°C, and 0.45 μm filtered. 2 ml of glutathione agarose resin was equilibrated using lysis buffer without added detergent and pipetted into a disposable gravity flow column. Lysates were passed through column 3X. Resin was washed with buffers in the following order: A (20 mM HEPES pH 7.25, 500 mM KCl, 5% glycerol, 1 mM EDTA, 1mM EGTA, 1 mM DTT, 1 mM PMSF); B (20 mM HEPES pH 7.25, 1M KCl, 5% glycerol, 1 mM EDTA, 1mM EGTA, 1 mM DTT, 1 mM PMSF); A; and C (20 mM HEPES pH 7.25, 300 mM KCl, 5% glycerol, 1 mM EDTA, 1mM EGTA, 1 mM DTT, 1 mM PMSF). 5 ml of Wash C supplemented with 1 mg PreScission protease was added into the resin and left to incubate on the column overnight at 4°C. After ~16 hr, GST-cleaved eGFP was removed from PreScission protease by passing through Superdex 75 increase 10/300 GL column and stored in 20 mM HEPES, 5% glycerol, 300 mM KCl, 1 mM DTT, 1X protease inhibitor cocktail. Peak fractions were stored in −80°C until use.
mCherry purification
mCherry from pN1-mCherry-Cry2 was cloned in frame with the N-terminal 6XHis tag into pET28b via 5’ NheI and 3’ BamHI sites. The plasmid was transformed into BL21(DE3) cells and grown to OD600 ≈ 0.6 at 37°C in 2l of 2XYT, then induced with 0.5 mM IPTG at 16°C for 14 hrs. Cells were pelleted and resuspended in lysis buffer containing 20 mM HEPES pH 7.25, 5% glycerol, 500 mM KCl, 20 mM imidazole, 5 mM β-mercaptoethanol, 1 mM PMSF, 1X protease inhibitor cocktail, and lysed by sonication at 60% power for 10 minutes in repeated cycles of 30 s on, 30 s off. 1% Triton-X100 was added and left to incubate at 4°C for 5–10 min. Lysates were clarified in a SA600 rotor for 20 min at 15K rpm at 4°C in. Supernatants were further clarified by 0.45 μm filtration and passed through disposable columns with Ni-NTA resin (Invitrogen) beads via gravity flow three times. Resin was washed in order with the following buffers: wash A (20 mM HEPES pH 7.25, 5% glycerol, 500 mM KCl, 20 mM imidazole, 1 mM DTT, 1 mM PMSF, 1X protease inhibitor cocktail); B (20 mM HEPES pH 7.25, 5% glycerol, 1 M KCl, 20 mM imidazole, 1 mM DTT, 1 mM PMSF, 1X protease inhibitor cocktail). Bound proteins were step eluted off the Ni-NTA resin using wash C buffer containing 100 mM imidazole in 10X1 ml fractions.
TEV protease purification
The plasmid pRK793 TEV S219V was obtained as a gift from the Boggon Lab and transformed into BL21 E. coli for purification. The protein was expressed in 2 l of BL21 E. coli culture followed by 0.2 mM isopropylβ-D-thiogalactopyranoside (IPTG) induction at 37°C for 3 hrs. Cells were sonicated in buffer containing 20 mM Tris pH 8.0, 5 mM βME, 500 mM NaCl, 4 mM imidazole, 1 mM DTT, 1 mM PMSF, 1X protease inhibitor cocktail. Lysates were ultracentrifuged using Ti70.1 rotor for 45 min, 40K rpm at 4°C. After collecting and 0.45 μm filtering the supernatant, proteins were passed through disposable columns with Ni-NTA resin beads via gravity flow 3X. Resin was washed with the following buffers in the following order: wash A (20 mM Tris pH 8.0, 5 mM βME, 500 mM NaCl, 4 mM imidazole, 1 mM DTT, 1 mM PMSF), B (20 mM Tris pH 8.0, 5 mM βME, 250 mM NaCl, 40 mM imidazole, 1 mM DTT, 1 mM PMSF), and C (20 mM Tris pH 8.0, 5 mM βME, 40 mM Imidazole, 100 mM NaCl, 1 mM DTT, 1 mM PMSF). Bound proteins were eluted off the Ni-NTA resin using elution buffer (20 mM Tris pH 8.0, 5 mM βME, 400 mM Imidazole, 100 mM NaCl, 1 mM DTT, 1 mM PMSF, and 1X protease inhibitor cocktail). The eluted fractions were then applied to a mono S cation exchange column for clean up using buffer A (20 mM HEPES pH 7.25, 5% glycerol, 1 mM DTT) and buffer B (20 mM HEPES pH 7.25, 5% glycerol, 1 M KCl, 1 mM DTT). The eluted fractions containing TEV were pooled, flash-frozen in liquid nitrogen, and stored at 80°C.
Microtubule co-sedimentation assays and quantification
Co-sedimentation assays were performed as previously described33,81. Double-cycled GMPCPP (Jena Bioscience, Thuringia, Germany) stabilized MTs were grown as described82. Taxol-MTs were polymerized at a final concentration of 60 μM at 37°C in polymerization buffer [80 mM PIPES, pH 6.8, 1 mM MgCl2, 1 mM EGTA, 1 mM GTP, and 15 nM paclitaxel (taxol)]. The taxol-stabilized MTs and GMPCPP-stabilized MTs were set aside for co-sedimentation. For MT co-sedimentation assays, 0.2 μM Abl2 or Abl2 fragments were mixed with increasing concentration of MTs (0 to 6 μM) at 37°C for 20 minutes in binding buffer [80 mM PIPES, pH 6.8, 70 mM KCl, 1 mM GTP, 5 nM taxol (100 μL reaction volume)]. Mixtures were pelleted by high-speed centrifugation at 120,000 × g for 20 minutes at 37°C. Pellet and supernatant fractions were recovered and separated by SDS-PAGE, stained with Coomassie Blue G-250 (Bio-Rad Laboratories, Hercules, CA) then destained in water. The SDS-PAGE gels were then scanned with Bio-Rad ChemiDoc™ Touch Imaging System and quantified by densitometry using ImageJ software. Binding affinity was quantified either as the percentage of Abl2/Abl2 fragments bound to MTs over total amount of Abl2/Abl2 fragments in the reaction for each concentration or as the amount of Abl2 bound to MTs for each concentration of Abl2. Experiments were repeated at least 4 times for each experimental condition (n ≥ 4). Equation (Equation 1) was used to fit the curve83, where y is specific binding, x is the ligand concentration, Bmax is the maximal binding (same units as y), and KD is the binding affinity (same units as x). Binding curves, affinities (KD), and R2 values for curve fitting were calculated using GraphPad Prism 9 GraphPad.
Tubulin binding analysis with size-exclusion chromatography
Size-exclusion chromatography used a Superdex 200 increase 10/300 GL column equilibrated in 20 mM HEPES pH 7.25, 5% glycerol, 100 mM KCl, 1 mM DTT. The column was calibrated with standard proteins of known Stokes radii (Sigma-Aldrich, St. Louis, MO). Abl2 or 557-C and tubulin were mixed with tubulin at a ratio of 1:4 and incubated for 30 min on ice, and then injected onto the column. Control experiments were performed with each protein alone. The collected fractions were analyzed by SDS-PAGE, stained with Coomassie brilliant blue G250 (Sigma-Aldrich, St. Louis, MO), and scanned.
Tubulin binding affinity measurements using biolayer interferometry
The biolayer interferometry technique using the BLItz system (ForteBio) was used to measure binding kinetics for the tubulin interaction with 6XHis-Abl2 and 6XHis-Abl2 fragments. His tag-binding Ni-NTA biosensors were hydrated in binding buffer (20 mM HEPES pH 7.25, 5% glycerol, 100 mM KCl, 1 mM DTT, supplemented with 0.02% Tween to reduce noise) for 10 min. For each tubulin concentration (ranging from 7 nM to 2000 nM), the following procedure was performed. An initial baseline was collected by immersing the biosensor in binding buffer for 1 min, and then 4 μl of fixed concentrations of 6XHis-Abl2 or 6XHis-Abl2 fragments (0.3 μM or 1 μM) were loaded to the biosensor for 5 min. The Abl2-loaded biosensor was returned to binding buffer for collection of a second baseline for 1 min and then placed in 4 μl of tubulin for a 5-min association step. For each data point, the background binding was also measured. For each tubulin concentration, the difference in the signal (in nm) just prior to the association step and that at the end of the association step was subtracted from the difference in signal for background binding. The binding curves were plotted fit to the one-phase exponential curves assuming a shared koff using the “association kinetics (two or more ligand concentrations)” model with GraphPad Prism to obtain a dissociation constant.
Turbidity assay and preparation of Abl2-MT sample grids for EM imaging
18 μM porcine brain tubulin in BRB80 was incubated with the MT polymerization buffer (2 mM of GTP, 1 mM DTT, 15% glycerol) alone or in the presence of Abl2 and Abl2 fragments at 37°C. Tubulin assembly was monitored by measuring turbidity at 350 nm (A350) for 2 hrs using SpectraMax M6 Multi-Mode Microplate Reader recording spectrophotometer. Control experiments were done by monitoring A350 for buffer alone with 0.5 μM Abl2 and Abl2 fragments without tubulin. 4 μl reactions at 10 min were taken out and visualized using electron microscopy (EM) of negatively stained samples. A 400-mesh copper grid (Ted Pella, Redding, CA) overlaid with a very thin continuous carbon layer was gently glow discharged and 4 μl of the diluted protein was applied to the grid. After a 30s adsorption, the reactions were blotted away from the grid with filter paper (Whatman No.1) leaving a thin layer of solution on the grid. 4 μl of 2% uranyl-acetate solution were applied to the grid for 30s before blotting twice. After blotting, the grid was left to dry for 2 min. The negative stain sample of Abl2 was imaged using a Tecnai12 transmission electron microscope (TEM) and images recorded on a Gatan CCD camera at ~−2–3 μm defocus.
Construction and preparation of flow chambers for TIRF imaging
Glass microfluidic chambers were constructed as described previously in Johnson-Chavarria et al. (2011)84. Briefly, inlet and outlet ports were generated into polydimethylsiloxane (PDMS) molds using a blunt tip needle. Holes were drilled into a glass cover slide with a diamond tip bit. Plasma cleaning was used to bond PDMS molds onto the cover slides where PDMS ports and holes meet.
Coverslips were cleaned with the following solutions, all incubated in a sonicator for 15 min (unless otherwise stated) and washed with ddH2O in between steps: 2% Hellmanex, ddH2O, 90% ethanol, 1M HCl (1 hr to overnight). To extensively rinse off HCl, coverslips were washed with ddH2O 3X for 15 min per step. Coverslips were then cleaned with 0.22 μm filtered 1M KOH for 30 min, followed by ddH2O, and 100% ethanol for 60 min to overnight. Coverslips were dried in a 55°C incubator and silanized with 300 ml dichlorodimethyl silane in 300 ml hexane solution for 35–45 min. They were subsequently sonicated in hexanes for 15 min. Coverslips were air dried and stored in sterile 50 ml Falcon tubes at −20°C until use. Immediately before use, TIRF chambers were assembled using parafilm, with rectangular cutouts to allow for flow between inlet and outlet ports; and was sealed between clean coverslips and the PDMS chamber with heat (via pressing the chamber with coverslip-side down onto surface of a 100°C heat block for ~5–10 s).
Flow chambers were prepared by washing in BRB80 and incubating 1 mg*ml−1 biotin-BSA (0.22 μm filtered in BRB80) for 5–10 min. Chambers were blocked with 2% Pluronic F-127 and allowed to incubate at RT for 30–45 min, followed by washing with BRB80 and functionalizing with 50 mg*ml−1 neutravidin (0.22 μm filtered in BRB80). After 5 min incubation, flow chambers were washed with BRB80 and perfused with biotin GMPCPP-MT seeds. Density of seeds were checked under the scope. Chambers were washed with BRB80 to flow out unattached seeds.
Microtubule dynamics assay
Microtubule seeds were prepared by mixing 10% biotin, 1% rhodamine-labelled 10 μM tubulin in BRB80 buffer, supplemented with 1 mM GMPCPP. The seed mixture was incubated in a 37°C water bath for 30–45 min, followed by centrifugation in a TLA100 rotor at 80K rpm for 5 min at 37°C. Single-cycled seeds were resuspended in warm BRB80 buffer and ready for use. Imaging buffer consisted of 10.5 μM 10% rhodamine-labelled tubulin, 1 mM GTP, 0.02% methylcellulose, 1X anti-blink cocktail (1% β-ME, 40 mM glucose, 250 nM glucose oxidase, 64.5 nM catalase, 1 mM Trolox, BRB80), and BRB80. All proteins were centrifuged in TLA100 rotor for 5 min at 80K rpm, 4°C prior to use to remove aggregates. In conditions with Abl2-eGFP, Abl2Δ688-790, or Abl2-688-924-eGFP, salt concentration was adjusted such that the final [KCl] in the imaging buffer was 50 mM. Reaction mixture was pipetted up and down 3X prior to perfusing into flow chamber. Dynamic MTs were allowed to grow, equilibrating in the chamber for 5 min prior to image acquisition. The objective was maintained at 37°C with an objective heater. Images were acquired at 1 fps for 25min, 2×2 binning with 300 ms integration times per channel, using the Nikon Elements software; and a Nikon ECLIPSE Ti Series Inverted Microscope equipped with a 100X TIRF 1.49NA oil objective and Andor Zyla 4.2 sCMOS camera. Dynamic instability parameters were analyzed from kymographs generated from Multi Kymograph using ‘read velocities from tsp’ plug-in (tsp050706.txt macro written by J. Rietdorf and A. Seitz, EMBL). Events were scored as pausing if the pause duration lasted at least 10s. At least 3 technical replicates (at least one different TIRF chamber per independent experiment) were conducted for each condition. MT lifetime distributions were fitted to a gamma distribution.
Microtubule segmentation assay and analysis
10% biotinylated rhodamine GMPCPP-MT seeds were prepared as described above. Seeds were flowed into the chamber and allowed to incubate for a minute prior to BRB80 wash out. 11 μM 10% labelled Alexa647 tubulin in 1X anti-blink cocktail supplemented with 1 mM GTP and 0.02% methylcellulose was perfused into the flow chamber to generate GDP segments. Dynamic MTs were allowed to grow for 10 minutes at 37°C. To generate GMPCPP-caps without inducing catastrophe of existing MTs, 10% rhodamine-labelled 11 μM tubulin was perfused into the flow chamber in 1X anti-blink cocktail supplemented with 2 mM GMPCPP and 0.02% methylcellulose. GMPCPP-caps were allowed to grow for at least 5 minutes prior to 3X BRB80 washout to remove excess soluble tubulin in the flow chamber. 0.5 μM Abl2-eGFP, 0.5 μM 557-C-eGFP, 0.5 μM EB1-eGFP, or 25 nM tau-eGFP was mixed in 1X anti-blink cocktail supplemented with 0.02% methylcellulose and 50 mM KCl and perfused in flow chamber. Binding was allowed to equilibrate in the flow chamber for 5 minutes prior to 1X BRB80 washout. Images were acquired at 2×2 binning with 300 ms integration times per channel using the Nikon TIRF 1.49NA oil objective and Andor Zyla 4.2 sCMOS camera.
Nucleotide state preference (NSP) was quantified by first making 3 pixel-wide line scans across a single filament. Fluorescence intensity per unit of length was linearly normalized per channel. Occupancies of EB1, tau 2N4R, 557-C, and Abl2 were determined by taking the fraction of the fluorescence intensity of the 488 nm channel over that of the 561 nm (red, GMPCPP) or 647 nm (pseudo-colored blue, GDP). NSP of the MBP on either GMPCPP- or GDP-segments for that single filament was recorded as the mean ratio of GMPCPP occupancy divided by the mean ratio of GDP occupancy. NSP measurements are shown on log10 scale. Values above 0 indicate preference for GMPCPP; values below 0 indicate preference for GDP.
Preparation of damaged MTs and lattice repair assay
100 μl mixture of 2% biotinylated, 10% rhodamine-labeled 20 μM tubulin was thawed on ice and incubated with 1 mM GTP. The mixture was allowed to polymerize in 37°C water bath for 30–45 min. MTs were centrifuged in a TLA100 rotor for 5 min at 37°C and resuspended in warm BRB80 buffer supplemented with 10 μM paclitaxel (taxol). Taxol-stabilized MTs were separated into 2 mixtures: 1) one 50 μl stored at RT (23°C) overnight; 2) the remaining 50 μl stored in 37°C.
The following day, the MTs were centrifuged for 5 min at 80K rpm, 37°C; and resuspended in warm BRB80 supplemented with 10 μM taxol.The 2 mixtures were separated into 2 25 μl Eppendorf tubes. Alexa647-tubulin and Abl2-eGFP were clarified for 5 min at 80K rpm, 4°C. 2 μM Alexa647-tubulin and 10 mM GTP were added into each 25 μl mixture of MTs. 1 μM Abl2-eGFP was added into one of 23°C and 37°C-stored MT reactions. Reactions were left to incubate in a 37°C water bath for 3 hrs prior to imaging. For damaged boundary analysis assays where 0.5 μM of Abl2-eGFP was added in absence of Alexa647-tubulin, reactions were left to incubate in flow chambers for 10 min prior to imaging. To assess the extent of lattice damage visually, a parallel set of non-fluorescent taxol-stabilized MTs were stored overnight at 23°C and 37°C and supplemented with 10 μM taxol the following morning for negative stain EM. Reactions were diluted 3-fold in warm 1X BRB80 buffer. 4 μl per reaction were applied onto glow-discharged grids.
After 45s adsorption, the reactions were blotted away from the grid with filter paper (Whatman No.1) leaving a thin layer of solution on the grid. 4 μl of 2% uranyl-acetate solution were applied to the grid for 30 s before blotting twice. After blotting, the grid was left to dry. Negative stain samples of damaged MTs were imaged using a Tecnai12 TEM scope and images recorded on a Gatan CCD camera at ~−0.8–1.5 μm defocus. The same qualitative assessment using negativestain EM for recruitment of 1 μM non-fluorescent Abl2 to damaged lattice sites in the presence of 2 μM non-fluorescent tubulin dimers was conducted.
TIRF chambers were functionalized as described above. 25 μl of each reaction (control and +Abl2 conditions) containing MTs stored at either 23°C or 37°C were perfused into the chamber. Warm BRB80 buffer was flowed into the chamber to wash out unattached taxol-MTs. Still images for the green (Abl2), red (MT; pseudo-colored magenta), and far red (tubulin reporter; pseudo-colored cyan) channels were acquired using single-band emission filter sets for 488, 561, and 638 nm, with 2×2 binning 300 ms acquisition times on the Nikon ECLIPSE Ti Series Inverted microscope.
To score for damaged MTs at either temperature, 3-pixel wide line scans across the entire length of rhodamine-MTs and tubulin reporter were recorded. User-defined MT and tubulin reporter intensity thresholds were used as parameters in custom-written MATLAB script to determine borders of MT segments that are structurally damaged, and whether tubulin reporter was present or not. MT segments of mean fluorescence intensity lower than user-defined MT-intensity threshold were scored as ‘damaged’. If the mean intensity of tubulin reporter of the equivalent damaged MT length is at least the user-defined reporter intensity threshold, the reporter length, reporter fraction, and frequency of reporter incorporation for that one MT were recorded.
For Abl2 localization and border analysis, MT segments of mean fluorescence intensity at least the user-defined MT threshold value were determined as ‘healthy’. Along these ‘healthy’ segments, borders are defined as edges of healthy segments that border damaged segments (of mean intensity values less than user-defined threshold). Border length was quantified as being 1/5 of the total healthy MT segment away from either terminus, with the lattice length quantified as the middle 3/5 of the MT segment. Localization of 0.5 μM Abl2-eGFP was quantified by taking the mean fluorescence intensity of the green channel along sub-sections of the healthy MT segment that were either borders or lattices.
Microtubule severing assay
10% biotinylated, 10% rhodamine labelled GMPCPP-stabilized MTs were flowed into TIRF chambers, followed by flow-in and incubation of 1 μM Abl2-eGFP in imaging buffer (1X anti-blink cocktail, 0.02% MC, 50 mM KCl, and BRB80) for 5 min to allow for condensate formation. Flow chamber was washed with BRB80 buffer to remove non-bound Abl2. 90 nM spastin in imaging buffer supplemented with 2 mM ATP was introduced into the chamber and image acquisition at 0.34 fps for 15 min immediately followed. 3 pixel-wide line scans of MTs were recorded for MTs at the beginning of each image acquisition and fluorescence intensity decay traces were plotted and fit to a single exponential decay: (Equation 2).
Phase separation assay
Abl2-eGFP, 557-C-eGFP, and N-557-eGFP were thawed on ice and concentrated to final concentration of 10 μM or higher using 0.5 ml centrifugal filter units (EMD Millipore #UFC501096) equilibrated with Abl2 storage buffer (20 mM HEPES, 300 mM KCl, 5% glycerol, 1 mM DTT).
Concentrated proteins were centrifuged to remove aggregates in a TLA100 rotor at 80K rpm, 4°C for 5 min. Individual wells in Cellvis #1.5 glass-bottom 96-well plates were washed with BRB80 and blocked with 2% Pluronic F-127 for 30–45 min. 20 μl of imaging buffer (0.2% methylcellulose, 1X anti-blink cocktail, BRB80, 0.1–3 μM Abl2-eGFP, 557-C-eGFP or N-557-eGFP) was mixed on ice. 6.25 μl 16% Dextran-70 was added into the imaging buffer reaction to achieve final concentration of 5% dextran. The mixture was pipetted up and down 3X at RT and added into the well. Condensates were left to age for 10 min at RT prior to image acquisition. Epifluorescent imaging was performed using the Nikon ECLIPSE Ti Series Inverted microscope adapted to a mercury lamp source and a 50:50 beam splitter to toggle between TIRF and Epi modalities. Focus was set just above the coverslip at the bottom of the well (z-height ≈ 3795–3850mm) where condensates have sedimented and settled.
To calculate partition coefficients and score for phase-separated condensation, we thresholded still images of sedimented Abl2 coacervates by filtering out particles for sizes 1−∞ and those with circularity values of 0.1–100. We then analyzed particles using Otsu, black and white thresholding, followed by color inversion. Circular ROIs were generated to quantify background values. Mean intensities of detected particles and background were calculated for each condition. If mean(particles) ≥ mean(background), i.e. PC ≥ 4, the particle was scored to be phase-separated condensate under the particular Abl2 concentration and salt concentration condition. For Abl2-tubulin co-condensation analysis, all partitioning experiments were maintained at 50 mM KCl, 5% dextran. Tubulin PCs were fitted to a sigmoidal equation, (Equation 3).
Live-cell imaging of COS-7 cells transfected with mCherry-Cry2 constructs
Abl2 was cloned in frame upstream of mCherry-Cry2 in pHR-HNRNPA1C-mCh-Cry2WT and the entire Abl2-mCherry-Cry2 or mCherry-Cry2 were cloned into the pN1-eGFP vector, replacing the eGFP. COS-7 cells were plated at 0.5*106 cells in 6 cm dishes and transfected with 6 μg of pN1-mCherry-Cry2 or pN1-Abl2-mCherry-Cry2 using a calcium phosphate transfection, as per Pear et al85. 12 hrs after transfection, transfected COS-7 cells were plated on 60 mm #1.5 glass dishes with 30 mm bottom wells (Cellvis, Mountainview, CA) that had been acid washed at 55°C overnight and coated with 50 μg*ml−1 poly-d-lysine for 20 min at room temperature and 10 μg*ml−1 fibronectin in phosphate-buffered saline (PBS) for 1 hr at 37°C. Prior to imaging, medium was exchanged into HBSS supplemented with 2% FBS. All live-cell imaging was conducted in a heated stage maintained at 37°C with CO2. Pre blue light activation recording was done for 1 min.
Activation period recording was done for 5 min, with blue light being turned ‘on’ and ‘off’ every other frame interchanging with the 561 nm filter. Recording was continued for 10 min after blue light exposure. Videos for each period were all taken at 2 fps and acquired using a Nikon CSU-W1 SoRa spinning disk confocal, with 35% laser power (LP) for 561 (mCherry), and 50% LP for 488 (blue light activation) nm channels, at 200 ms exposure time each.
Estimation of intracellular levels of mCherry-Cry2 constructs
Recombinant 6XHis-mCherry was used to estimate intracellular concentrations of mCherry-Cry2 constructs expressed in transfected COS-7 cells. 35 mm #1.5 glass bottom dishes (MatTek, Ashland, MA) were coated with 35 μg*ml−1 poly-D-lysine in 10 mM HEPES, 1X PBS for 30 min at RT. Dishes were then washed twice with PBS and coated in 250 μL each of 0.01, 0.05, 0.1, 0.25, and 0.5 mg*ml−1 6XHis-mCherry diluted in PBS. Images were acquired using the same imaging conditions as done for live-cell imaging of COS-7 cells transected with mCherry-Cry2 constructs (35% LP for 561 nm, 200 ms exposure with PFS setting on). Average mean intensity per pixel (a.u.*pixel−1) values were plotted against increasing concentrations of 6XHis-mCherry to acquire a line of best fit. ROIs that outlined the cells were generated and mean intensity*pixel−1 values were collected to extrapolate the intracellular concentrations based on the standard curve, knowing that MW6XHis-mCherry ≈ 26.6 kDa.
Circular dichroism of Abl2 557-C
Purified His-tag free Abl2 557-C was purified into storage buffer containing 20 mM HEPES pH 7.25, 300 mM KCl, 5% glycerol, 1 mM DTT. CD spectra were collected at a constant temperature of 4°C using a Chirascan CD spectrometer (Applied Photophysics) at 0.5 nm wavelength step size and 0.5 s per wavelength point. CD spectra of the gel filtration buffer were collected to background subtract from the sample spectra. Averages of 2 spectra from 280 to 190 nm were calculated per replicate for duplicate technical replicates.
FRAP of Abl2 and tubulin in solution
Images were acquired using a Cellvis #1.5 glass-bottom 96-well plate. Condensates were prepared and aged as described above, maintained at 50 mM KCl. Prior to photobleaching, images were acquired every second for 30 seconds to obtain an average pre-bleach fluorescence intensity value. Photobleaching was carried out using 20% power of the 405 nm laser line, with 20 μs dwell time at a single focal plane near top of the condensate. An ROI was chosen as a circular region within the condensate. Intensity of each ROI was recorded every 10s (~0.14 fps) over a 10–15 min acquisition period. Recovery in photobleached ROIs was normalized to changes in background intensity to account for global bleaching. Images were acquired using Nikon CSU-W1 SoRa spinning disk confocal microscope equipped 100X oil objective. Triplicate technical replicates of Abl2 and duplicate replicates of tubulin FRAP curves were fitted to double exponential decay: (Equation 4).
FRAP of Abl2 on MTs
Preparation of biotinylated MT seeds and functionalization of TIRF chambers were described as above. 10% biotinylated 10% rhodamine-labelled GMPCPP-stabilized seeds were flowed into the chamber and unattached seeds were washed out with BRB80. Imaging buffer (1 μM Abl2-eGFP, 1X anti-blink cocktail, 50 mM KCl, 0.02% methylcellulose, and BRB80) was perfused into the flow chamber and left to incubate at RT for 5 min to allow condensates to form on MTs. Prior to photobleaching, images were acquired at 1 fps for 15 seconds for both 488 and 561 nm channels. Photobleaching was carried out using 20% laser power of the 405 nm laser line, with 20 μs dwell time. Intensity of each ROIs were recorded at ~0.37 fps over a 10 min acquisition period. Images were acquired using Nikon Elements software; and a Nikon Ti2-E inverted microscope equipped with a 100X 1.49NA TIRF objective, stage top Piezo for 405 nm FRAP laser, and a Photometrics Prime 95B CMOS camera. FRAP curves of MTs from 4 TIRF chambers were fitted to Eqn. 4.
Microtubule nucleation observation using confocal microscopy
8 μM 10% Alexa Fluor 647-labeled porcine brain tubulin in BRB80 was incubated with the MT polymerization buffer (2 mM of GTP, 1 mM DTT, 10% glycerol), 1 μM Abl2-eGFP, 3% dextran at 37°C. Reactions were added to a 384-well glass-bottom plate (Corning, Corning, NY), which was acid-washed and coated with 2% F-127 for 30 min. Tubulin nucleation was monitored using Nikon CSU-W1 SoRa spinning disk confocal 30 min and 1 hr after the start of the reactions.
Fixed-cell sample preparation
Cells were plated on 12 mm #1.5 coverslips (Warner Instruments, Hamden, CT) in 3 cm TC-treated dishes (Corning, Corning, NY) the night prior, such that they were 80–90% confluent the morning of the experiment. Coverslips were acid washed at 55°C overnight and coated with 50 μg*ml−1 poly-d-lysine for 20 min at room temperature and 10 μg*ml−1 fibronectin in PBS for 1 hr at 37°C. Cells were treated with 10 μM nocodazole for 60 min, and the coverslips were taken out at 5 min after washout with complete medium.
Cells were fixed with 4% paraformaldehyde in cytoskeleton buffer (10 mM MES pH 6.1, 138 mM KCl, 3 mM MgCl2, 2 mM EGTA, 0.32 M sucrose) for 20 min at room temperature and then permeabilized with permeabilization buffer (0.5% Triton X-100 in PBS) for 20 min. The fixed cells were blocked in blocking buffer (2% BSA, 0.1% Triton X-100 in PBS) for 1 hr at RT, followed by primary and antibody incubation. Primary α-tubulin (DM1a) and α-GFP antibodies were used at 1:500 dilution and cells were left to stain overnight at 4°C. Cells were washed 3x with 0.05% Tween-20 in PBS on a shaker at RT. DAPI was used at 1:1000 to label nuclei; Alexa488 goat anti-chicken secondary Ab (Invitrogen) was used at 1:500 to label GFP; and Alexa647 goat anti-mouse secondary Ab (Invitrogen) was used at 1:500 to label α-tubulin. All antibodies were diluted in blocking buffer, and cells were left to stain at RT for ~1 hr. Cells were washed 3x with 0.05% Tween-20 in PBS on a shaker at RT prior to mounting.
MT recovery analysis
Cell areas and MT recoveries were imaged using Nikon CSU-W1 SoRa spinning disk confocal, with 5% LP for 405 (nuclei), 488 (eGFP), and 647 (tubulin) nm channels, at 200 ms exposure time each. Samples were blinded prior to imaging. Images were Z-projected with “Max Intensity”. Only mononucleated cells were chosen for analysis. Cell boundaries were defined using polygonal selection to measure cell areas. A background region within the selected cells with no MTs was measured, with mean background intensity recorded as Ibg. Ibg *3 was used as the threshold to quantify for MTs recovered upon post-nocodazole treatment. Regions throughout the cell of fluorescence intensity above the threshold near the MTOC were measured to determine the amount of MTs recovered. At least 25 cells from 2–3 experimental replicates were quantified for each assay condition (n ≥ 25) by a reviewer blinded to sample identity.
SEC-MALS and Stokes radius analyses
To measure the particle size of purified Abl2 and 557-C in solution, we used an experimental setup including an upstream size exclusion chromatography (SEC, Superdex 200 increase 10/300 GL) coupled with a downstream multi-angle light scattering (MALS), which includes a multiple light scattering detector (DAWN HeleosII, Wyatt Technology Corporation) and a refractive index detector (Optilab T-rEX, Wyatt Technology). Each sample injection consisted of 0.5 to 2 mg of purified protein sample in buffer containing 20 mM HEPES pH 7.25, 5% glycerol, 100 mM KCl, 1 mM DTT. The flow rate was set at 0.4 ml*min−1. Data were recorded at 1 second intervals and processed using ASTRA software (Wyatt Technology). Stokes radius analyses were performed as described previously86.
Cryo-EM grid preparation and data collection
On-grid incubation method was used to prepare cryo-EM samples of MT decorated with 6XHis-tagged Abl2-557-1090. In brief, glow-discharged cryo-EM grids (Quantifoil Au 2/1, 300 mesh) were loaded into a Vitrobot Mark IV (ThermoFisher Scientific). The chamber was set at 15°C and 95% humidity. 2 μL 6 μM GMPCPP-stabilized MT was applied onto the grid and incubated for 30 seconds. After that, 2 μL 20 μM 6XHis-Abl2-557-1090 was added and incubated for 60 seconds. The grid was then blotted for 3–6 seconds and plunged into liquid ethane.
MTs with or without 557–1090 data were collected at the Yale Science Hill CryoEM facility using a Glacios microscope (Thermo Fisher Scientific) operated at 200 keV. The images were collected with a K2 direct electron detector (Gatan) operating in super-resolution mode, at a magnification of 36,000 corresponding to a pixel size of 1.143 Å. Data collection was automated by SerialEM software with a defocus range −1.5 μm to −2.7 μm. In total, 720 movies for MT only and 1780 movies for Abl2-MT were collected and each movie was dose-fractionated to 40 frames with a total dose of 40 e-/Å2 (Figure S4, Table S1).
Cryo-EM image processing
Cryo-EM data processing workflows are outlined in Figure S4. Recorded movies were pre-processed using cryoSPARC Live including patch motion correction and patch CTF estimation. CryoSPARC template picker was used to pick MT segments and rounds of 2D classification were used to remove junk particles. For MT 3D reconstruction, a published 14-protofilament MT map (EMD-7973) was used, and low-pass filtered to 6 Å for homogenous refinement. The reconstructed maps were used for the comparison of MT with (84, 826 particles) or without (24, 485 particles) Abl2 (Figures 4, S4).
To improve the resolution and map quality of Abl2-MT reconstruction, protofilament refinement protocol55 was applied to the dataset. After this, 1,187,564 protofilament particles were subjected to local 3D refinement and focused 3D classification (Figure S4). Map statistics can be found in Table S1. All cryo-EM maps were convoluted with a Gaussian function which has a standard deviation of 3 Å before comparison. ChimeraX was used for the Gaussian filtering and figure preparation.
Isoform abundance profiling for ABL2/Abl2 across adult human and mouse organs
We downloaded the SMART-seq2-based single cell transcriptomic data profiling 20 adult mouse organs from Tabula Muris87 and conducted RSEM analysis88 to measure the abundance of Abl2 isoforms (Figure S6, Table S2). Specifically, we first built the genome reference by running rsem-prepare-reference on the mouse GRCm39 genome assembly and the GENCODE vM32 gene annotation. The built reference was then piped into rsem-calculate-expression function together with the STAR algorithm for alignment and isoform abundance quantification. We extracted the TPM values from the RSEM outputs and log-transformed the values for visualization using R ggplot2 package.
In measuring the ABL2 isoform abundance in the GTEx human bulk-tissue RNA-seq data across 30 organs, we directly utilized the processed expression data from the GTEx Portal89 followed by visualization using R ggplot2 package (Figure S6, Table S3). The GTEx also applied RSEM in the isoform abundance quantification, which enabled direct comparisons of the transcript expression patterns between human and mouse.
Quantification and Statistical Analysis
Fiji was used for all image processing and analysis. MATLAB and GraphPad Prism 9 were used for quantification analysis. At least triplicate technical replicates were performed for each experiment unless otherwise stated. Details on the statistical test are presented in the figure legends. Graphs show mean ± SD, unless otherwise stated in legends. P-values < 0.05 are considered as statistically significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |||
|---|---|---|---|---|---|
| Antibodies | |||||
| Mouse monoclonal anti-Tubulin (clone DM1A) | Sigma-Aldrich | Cat#T9026; RRID: AB_477593 | |||
| Chicken polyclonal anti-GFP | Novus Biologicals | Cat# NB100-1614-0.02ml | |||
| Mouse monoclonal anti-Abl2 (Ar11) | Hybridoma from Dr. Peter Davies, Albert Einstein Medical College, Bronx, NY | N/A | |||
| Goat anti-Chicken IgY (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor™ Plus 488 | Invitrogen | Cat#A32931 | |||
| Goat anti-Mouse IgG (H+L), Superclonal™ Recombinant Secondary Antibody, Alexa Fluor™ 647 | Invitrogen | Cat#A28181 | |||
| Bacterial and virus strains | |||||
| One Shot TOP10 Chemically Competent E. coli | Invitrogen | Cat#C404010 | |||
| BL21 Competent Cells | Agilent | Cat#200133 | |||
| DH10Bac Competent Cells | Invitrogen | Cat#10361012 | |||
| 3rd generation lentivirus produced in 293T cells to express eGFP, Abl2-eGFP, or Abl2Δ688-790-eGFP in COS-7 cells | This paper | N/A | |||
| Chemicals, peptides, and recombinant proteins | |||||
| Paclitaxel | Sigma-Aldrich | Cat#T7191 | |||
| Guanosine-5’-[(α,β)-methyleno]triphosphate, Sodium (GMPCPP) | Jena Bioscience | Cat#NU-405L | |||
| Nocodazole | Sigma-Aldrich | Cat#M1404–2mg; CAS: 31430-18-9 | |||
| Dextran from Leuconostoc spp. Mr ~70,000 | Sigma-Aldrich | Cat#31390–25G | |||
| Zeocin Selection Reagent | Gibco | Cat#R25005 | |||
| Deposited data | |||||
| cryoEM-map of microtubule only reconstruction | This paper | EMDB: EMD-41167 | |||
| cryoEM-map of microtubule-Abl2 reconstruction | This paper | EMDB: EMD-41169 | |||
| cryoEM-map of protofilament reconstruction of Abl2+MT | This paper | EMDB: EMD-41170 | |||
| cryoEM-map of protofilament reconstruction of Abl2+MT class 1 | This paper | EMDB: EMD-41173 | |||
| cryoEM-map of protofilament reconstruction of Abl2+MT class 2 | This paper | EMDB: EMD-41176 | |||
| cryoEM-map of protofilament reconstruction of Abl2+MT class 3 | This paper | EMDB: EMD-41177 | |||
| Experimental models: Cell lines | |||||
| Sf9 insect cells | ATCC | CRL-1711 | |||
| Hi Five insect cells | Thermo Fisher | B85502 | |||
| 293T cells | ATCC | CRL-3216 | |||
| COS-7 cells | ATCC | CRL-1651 | |||
| Abl2 KO COS-7 cells | Hu et al.38 | N/A | |||
| Abl2 KO COS-7 cells separately stably expressing eGFP, Abl2-eGFP, and Abl2Δ688-790-eGFP | This paper | N/A | |||
| Recombinant DNA | |||||
| FUGW for lentivirus generation | Lois et al.90 | Addgene#14883; RRID: Addgene_14883 | |||
| FUGW derived vector to stably express eGFP, Abl2-eGFP, or Abl2Δ688-790-eGFP | This paper | N/A | |||
| Abl2, Abl2-eGFP, Abl2 fragments, or Abl2 fragments-eGFP cloned into pFastBac1 | This paper | N/A | |||
| pGEX-6p1-GST-eGFP | This paper | N/A | |||
| pRK793 TEV S219V | The laboratory of Dr. Titus Boggon | N/A | |||
| pGEX-PP-Spastin(87–616)DeltaExon4 | Han et al. | Addgene#128794; RRID: Addgene_128794 | |||
| EB1-eGFP | The laboratory of Dr. Jonathon Howard | N/A | |||
| FUW-tau 2N4R | The laboratory of Dr. Elizabeth Rhoades | N/A | |||
| pHR-HNRNPA1C-mCh-Cry2WT | Shin et al.48 | Addgene #101226; RRID: Addgene_101226 | |||
| Software and algorithms | |||||
| ImageJ/Fiji | Schneider et al.91 | https://imagej.nih.gov/ij/ | |||
| EMBOSS | Madeira et al.92 | https://www.bioinformatics.nl/cgi-bin/emboss/ | |||
| PONDR-FIT | Xue et al.93 | https://www.disprot.org/ | |||
| Diso-Pred3 | Jones and Cozzetto et al.94 | http://bioinf.cs.ucl.ac.uk/psipred/ | |||
| Circular Dichroism algorithms (CONTIN, VARSLC, CDSSTR) | Provencher and Glöckner95; Compton and Johnson96; Sreerama and Woody97 | http://dichroweb.cryst.bbk.ac.uk/html/links.shtml | |||
| AlphaFold2 | Jumper et al.98 | https://www.deepmind.com/ | |||
| ChimeraX v1.4 | Pettersen et al.99 | https://www.rbvi.ucsf.edu/chimerax/ | |||
| Coot v.0.9.5 | Brown et al.100 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ | |||
| cryoSPARC v3.3.2 | Punjani et al101 | https://cryosparc.com/ | |||
| Protofilament refinement package | Debs et al.55 | https://gitlab.com/gedebs371/protofilament-refinement/-/tree/smooth_stable | |||
| SerialEM v.3.8.5 | Schorb et al.102 | https://bio3d.colorado.edu/SerialEM/ | |||
| RSEM v1.3.3 | Li and Dewey88 | https://deweylab.github.io/RSEM/ | |||
| STAR 2.7.11a | Dobin et al.103 | https://github.com/alexdobin/STAR | |||
| Matlab | The Mathworks, Inc. | https://nl.mathworks.com/products/matlab.html | |||
| Nikon Elements Software | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements | |||
| Other | |||||
| Superdex 75 Increase 10/300 GL | Cytiva | Cat#29148721 | |||
| Superdex 200 Increase 10/300 GL | Cytiva | Cat#28990944 | |||
| Octet® NTA Biosensors | Sartorius | Cat#18–5101 | |||
| BLItz | ForteBio | https://intellicyt.com/products/fortebio/ | |||
| Carbon support Film 5–6nm on Square 400 mesh Cu grids | Electron Microscopy Sciences | Cat#CF400-Cu | |||
| R2/1, 300 mesh gold, QUANTIFOIL grids | Electron Microscopy Sciences | Cat#258376 | |||
| Nikon ECLIPSE Ti Series inverted microscope equipped with CFI Apo 100X TIRF 1.49 NA oil objective and Andor Zyla 4.2 sCMOS camera | Nikon, Andor Technology |
https://www.microscope.healthcare.nikon.com/products/inverted-microscopes/eclipse-ti-series; https://www.microscope.healthcare.nikon.com/products/optics/cfi-apochromat-tirf-series |
|||
| Nikon CSU-W1 SoRa spinning disk confocal microscope | Nikon | https://www.microscope.healthcare.nikon.com/products/confocal-microscopes/csu-series/csu-w1-sora | |||
Highlights:
Abl2 binds tubulin dimers and undergoes liquid-liquid phase separation (LLPS)
Co-condensation of Abl2 and tubulin promotes MT nucleation under LLPS conditions
Abl2 binds expanded MT lattices and bridges subunits along a protofilament
Abl2 promotes rescue and facilitates MT reassembly after nocodazole washout in cells
Acknowledgments
This work was supported by the National Institutes of Health grants R21 NS112121, R01 MH115939, R01 NS105640, and R56MH122449 (to A.J.K.), R35GM142959 (to K.Z.), U01 MH122678 and U01 MH124619 (to N.S.), S10OD023603 (to Fred Sigworth for purchase of the Glacios microscope); an American Heart Association Predoctoral Fellowship (to W.L.), and a National Science Foundation Graduate Research Fellowship Program (to D.D.). We would like to thank lab members Josie E. Bircher, Noële Certain, and Pauline Lining Pan for providing thoughtful feedback on the manuscript; Xianyun Ye for her assistance in purifying DNA; and Amanda T Jeng for designing CRISPR-Cas9 oligos for stably knocking out endogenous Abl2 in COS-7 cells. We thank Elizabeth Rhoades for the template FUW-tau 2N4R construct, and Joe Howard for pETM-11-EB1-eGFP. We also greatly appreciate the Titus Boggon Lab for providing access to the BLItz instrument. Finally, we would like to thank Jennifer Kelly of the Yale Flow Cytometry Core for assisting with fluorescence-activated cell sorting. We apologize to the many stellar colleagues whose work we could not cite due to space restrictions. The schematic for Figure 6 was generated using https://biorender.com.
Inclusion and Diversity
We support inclusive, diverse, and equitable conduct of research. One or more of the authors of this paper self-identifies as living with a disability. While citing references, scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References:
- 1.Kirschner M, and Mitchison T (1986). Beyond self-assembly: From microtubules to morphogenesis. Cell 45, 329–342. 10.1016/0092-8674(86)90318-1. [DOI] [PubMed] [Google Scholar]
- 2.Desai A, and Mitchison TJ (1997). Microtubule Polymerization Dynamics. Annual Review of Cell and Developmental Biology 13, 87–117. 10.1146/annurev.cellbio.13.1.83. [DOI] [PubMed] [Google Scholar]
- 3.Goodson HV, Valetti C, and Kreis TE (1997). Motors and membrane traffic. Curr Opin Cell Biol 9, 18–28. 10.1016/s0955-0674(97)80147-0. [DOI] [PubMed] [Google Scholar]
- 4.Mitchison TJ (1988). Microtubule dynamics and kinetochore function in mitosis. Annu Rev Cell Biol 4, 527–549. 10.1146/annurev.cb.04.110188.002523. [DOI] [PubMed] [Google Scholar]
- 5.Petry S (2016). Mechanisms of Mitotic Spindle Assembly. Annual Review of Biochemistry 85, 659–683. 10.1146/annurev-biochem-060815-014528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Teixido-Travesa N, Roig J, and Luders J (2012). The where, when and how of microtubule nucleation - one ring to rule them all. J Cell Sci 125, 4445–4456. 10.1242/jcs.106971. [DOI] [PubMed] [Google Scholar]
- 7.Mitchison T, and Kirschner M (1984). Dynamic instability of microtubule growth. Nature 312, 237–242. 10.1038/312237a0. [DOI] [PubMed] [Google Scholar]
- 8.Akhmanova A, and Steinmetz MO (2015). Control of microtubule organization and dynamics: two ends in the limelight. Nature Reviews Molecular Cell Biology 16, 711–726. 10.1038/nrm4084. [DOI] [PubMed] [Google Scholar]
- 9.Triclin S, Inoue D, Gaillard J, Htet ZM, DeSantis ME, Portran D, Derivery E, Aumeier C, Schaedel L, John K, et al. (2021). Self-repair protects microtubules from destruction by molecular motors. Nature Materials 20, 883–891. 10.1038/s41563-020-00905-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andreu-Carbó M, Fernandes S, Velluz M, Kruse K, and Aumeier C (2022). Motor usage imprints microtubule stability along the shaft. Developmental Cell 57, 5–18. 10.1016/j.devcel.2021.11.019. [DOI] [PubMed] [Google Scholar]
- 11.Schaedel L, John K, Gaillard J, Nachury MV, Blanchoin L, and Théry M (2015). Microtubules self-repair in response to mechanical stress. Nature Materials 14, 1156–1163. 10.1038/nmat4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reid TA, Coombes C, and Gardner MK (2017). Manipulation and quantification of microtubule lattice integrity. Biology Open 6, 1245–1256. 10.1242/bio.025320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schaedel L, Triclin S, Chrétien D, Abrieu A, Aumeier C, Gaillard J, Blanchoin L, Théry M, and John K (2019). Lattice defects induce microtubule self-renewal. Nature Physics 15, 830–838. 10.1038/s41567-019-0542-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Forges H, Pilon A, Cantaloube I, Pallandre A, Haghiri-Gosnet A, Perez F, and Poüs C (2016). Localized Mechanical Stress Promotes Microtubule Rescue. Current Biology 26, 3399–3406. 10.1016/j.cub.2016.10.048. [DOI] [PubMed] [Google Scholar]
- 15.Lawrence EJ, Arpag G, Arnaiz C, and Zanic M (2021). SSNA1 stabilizes dynamic microtubules and detects microtubule damage. eLife 10, e67282. 10.7554/eLife.67282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van den Berg CM, Volkov VA, Schnorrenberg S, Huang Z, Stecker KE, Grigoriev I, Gilani S, Frikstad KM, Patzke S, Zimmerman T, et al. (2023). CSPP1 stabilizes growing microtubule ends and damaged lattices from the luminal side. Journal of Cell Biology 222, e202208062. 10.1083/jcb.202208062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aher A, Rai D, Schaedel L, Gaillard J, John K, Liu Q, Altelaar M, Blanchoin L, Thery M, and Akhmanova A (2020). CLASP Mediates Microtubule Repair by Restricting Lattice Damage and Regulating Tubulin Incorporation. Current Biology 30. 10.1016/j.cub.2020.03.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Al-Bassam J, Kim H, Brouhard GJ, van Oijen A, Harrison SC, and Chang F (2010). CLASP Promotes Microtubule Rescue by Recruiting Tubulin Dimers to the Microtubule. Developmental Cell 19, 245–258. 10.1016/j.devcel.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.King MR, and Petry S (2020). Phase separation of TPX2 enhances and spatially coordinates microtubule nucleation. Nature Communications 11. 10.1038/s41467-019-14087-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rale MJ, Kadzik RS, and Petry S (2018). Phase Transitioning the Centrosome into a Microtubule Nucleator. Biochemistry 57, 30–37. 10.1021/acs.biochem.7b01064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aumeier C, Schaedel L, Gaillard J, John K, Blanchoin L, and Théry M (2016). Self-repair promotes microtubule rescue. Nature Cell Biology 18, 1054–1064. 10.1038/ncb3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maan R, Reese L, Volkov VA, King MR, van der Sluis EO, Andrea N, Evers WH, Jakobi AJ, and Dogterom M (2022). Multivalent interactions facilitate motor-dependent protein accumulation at growing microtubule plus-ends. Nature Cell Biology 25, 68–78. 10.1038/s41556-022-01035-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meier SM, Farcas A, Kumar A, Ijavi M, Bill RT, Stelling J, Dufresne ER, Steinmetz MO, and Barral Y (2022). Multivalency ensures persistence of a +TIP body at specialized microtubule ends. Nature Cell Biology 25, 56–67. 10.1038/s41556-022-01035-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song X, Yang F, yang T, Wang Y, Ding M, Li L, Xu P, Liu S, Dai M, Chi C, et al. (2022). Phase separation of EB1 guides microtubule plus-ends dynamics. Nature Cell Biology 25, 79–91. 10.1038/s41556-022-01033-4. [DOI] [PubMed] [Google Scholar]
- 25.Jijumon AS, Bodakuntla S, Genova M, Bangera M, Sackett V, Besse L, Maksut F, Henriot V, Magiera MM, Sirajuddin M, and Janke C (2022). Lysate-based pipeline to characterize microtubule-associated proteins uncovers unique microtubule behaviours. Nature Cell Biology 24, 253–267. 10.1038/s41556-021-00825-4. [DOI] [PubMed] [Google Scholar]
- 26.Khatri A, Wang J, and Pendergast AM (2016). Multifunctional Abl kinases in health and disease. Journal of Cell Science 129, 9–16. 10.1242/jcs.175521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rogers EM, Spracklen AJ, Bilancia CG, Sumigray KD, Allred SC, Nowotarski SH, Schaefer KN, Ritchie BJ, and Peifer M (2016). Abelson kinase acts as a robust, multifunctional scaffold in regulating embryonic morphogenesis. Molecular Biology of the Cell 27, 2613–2631. 10.1091/mbc.E16-05-0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bradley WD, and Koleske AJ (2009). Regulation of cell migration and morphogenesis by Abl-family kinases: emerging mechanisms and physiological contexts. Journal of Cell Science 122, 3441–3454. 10.1242/jcs.039859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peacock JC, Miller AL, Bradley WD, Rodriguez OC, Webb DJ, and Koleske AJ (2007). The Abl-related Gene Tyrosine Kinase Acts Through p190RhoGAP to Inhibit Actomyosin Contractility and Regulate Focal Adhesion Dynamics Upon Adhesion to Fibronectin. Molecular Biology of the Cell 18, 2860–2872. 10.1091/mbc.e07-01-0075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mader CC, Oser M, Magalhaes MAO, Bravo-Cordero JJ, Condeelis J, Koleske AJ, and Gil-Henn H (2011). An EGFR-Src-Arg-Cortactin Pathway Mediates Functional Maturation of Invadopodia and Breast Cancer Cell Invasion. Cancer Research 71, 1730–1741. 10.1158/0008-5472.CAN-10-1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boyle SN, Michaud GA, Schweitzer B, Predki PF, and Koleske AJ (2007). A Critical Role for Cortactin Phosphorylation by Abl-family Kinases in PDGF-induced Dorsal-Wave Formation. Current Biology 17, 445–451. 10.1016/j.cub.2007.01.057. [DOI] [PubMed] [Google Scholar]
- 32.Hu H, Bliss JM, Wang Y, and Colicelli J (2005). RIN1 Is an ABL Tyrosine Kinase Activator and a Regulator of Epithelial-Cell Adhesion and Migration. Current Biology 15, 815–823. 10.1016/j.cub.2005.03.049. [DOI] [PubMed] [Google Scholar]
- 33.Miller ALM, Wang Y, Mooseker MS, and Koleske AJ (2004). The Abl-related gene (Arg) requires its F-actin microtubule cross-linking activity to regulate lamellipodial dynamics during fibroblast adhesion. Journal of Cell Biology 165, 407–420. 10.1083/jcb.200308055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee H, Engel U, Rusch J, Scherrer S, Sheard K, and Vactor DV (2004). The Microtubule Plus End Tracking Protein Orbit/MAST/CLASP Acts Downstream of the Tyrosine Kinase Abl in Mediating Axon Guidance. Neuron 42, 913–926. 10.1016/j.neuron.2004.05.020. [DOI] [PubMed] [Google Scholar]
- 35.Lowery LA, Lee H, Lu C, Murphy R, Obar RA, Zhai B, Schedl M, Van Vactor D, and Zhan Y (2010). Parallel Genetic and Proteomic Screens Identify Msps as a CLASP–Abl Pathway Interactor in Drosophila. Genetics 185. 10.1534/genetics.110.115626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lawrence EJ, Chatterjee S, and Zanic M (2023). CLASPs stabilize the pre-catastrophe intermediate state between microtubule growth and shrinkage. Journal of Cell Biology 222, e202107027. 10.1083/jcb.202107027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo W, Demidov V, Shen Q, Girão H, Chakraborty M, Maiorov A, Ataullakhanov FI, Lin C, Maiato H, and Grishchuk EL (2023). CLASP2 recognizes tubulins exposed at the microtubule plus-end in a nucleotide state-sensitive manner. Science Advances 9, eabq5404. 10.1126/sciadv.abq5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu Y, Lyu W, Lowery LA, and Koleske AJ (2019). Regulation of MT dynamics via direct binding of an Abl family kinase. Journal of Cell Biology 218, 3986–3997. 10.1083/jcb.201812144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang G, Dong Q, Bai Y, Gu J, Tao Q, Yue J, Zhou R, Niu X, Zhu L, Song C, et al. (2022). c-Abl kinase-mediated phosphorylation of γ-tubulin promotes γ-tubulin ring complexes assembly and microtubule nucleation. Journal of Biological Chemistry 298, 101778. 10.1016/j.jbc.2022.101778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Akhmanova A, and Steinmetz MO (2008). Tracking the Ends: A Dynamic Protein Network Controls the Fate of Microtubule Tips. Nature Reviews Molecular Cell Biology 9, 309–322. 10.1038/nrm2369. [DOI] [PubMed] [Google Scholar]
- 41.Tan R, Lam AJ, Tan T, han J, Nowakowski DW, Vershinin M, Simó S, Ori-McKenney KM, and McKenney RJ (2019). Microtubules gate tau condensation to spatially regulate microtubule functions. Nature Cell Biology 21, 1078–1085. 10.1038/s41556-019-0375-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hernández-Vega A, Braun M, Scharrel L, Jahnel M, Wegmann S, Hyman BT, Alberti S, Diez S, and Hyman AA (2017). Local Nucleation of Microtubule Bundles through Tubulin Concentration into a Condensed Tau Phase. Cell Reports 20, 2304–2312. 10.1016/j.celrep.2017.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin Y, Currie SL, and Rosen MK (2017). Intrinsically disordered sequences enable modulation of protein phase separation through distributed tyrosine motifs. Journal of Biological Chemistry 292, 19110–19120. 10.1074/jbc.M117.800466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang J, Choi J, Holehouse AS, Lee HO, Zhang X, Jahnel M, Maharana S, Lemaitre R, Pozniakovsky A, Drechsel D, et al. (2018). A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell 174, 688–699. 10.1016/j.cell.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Speer SL, Stewart CJ, Sapir L, Harries D, and PIelak GJ (2022). Macromolecular Crowding Is More than Hard-Core Repulsions. Annual Review of Biophysics 51, 267–300. 10.1146/annurev-biophys-091321-071829. [DOI] [PubMed] [Google Scholar]
- 46.Alberti S, Gladfelter AS, and Mittag T (2019). Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 176, 419–434. 10.1016/j.cell.2018.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feng Z, Chen X, Wu X, and Zhang M (2019). Formation of biological condensates via phase separation: Characteristics, analytical methods, and physiological implications. Journal of Biological Chemistry 294, 14823–14835. 10.1074/jbc.REV119.007895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shin Y, Berry J, Pannucci N, Haataja MP, Toettcher JE, and Brangwynne CP (2017). Spatiotemporal Control of Intracellular Phase Transitions Using Light-Activated optoDroplets. Cell 168, 159–171. 10.1016/j.cell.2016.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Siahaan V, Krattenmacher J, Hyman AA, Diez S, Hernández-Vega A, Diez S, Hernández-Vega A, Lansky Z, and Braun M (2019). Kinetically distinct phases of tau on microtubules regulate kinesin motors and severing enzymes. Nature Cell Biology 21, 1086–1092. 10.1038/s41556-019-0374-6. [DOI] [PubMed] [Google Scholar]
- 50.Manka SW, and Moores CA (2018). The role of tubulin–tubulin lattice contacts in the mechanism of microtubule dynamic instability. Nature Structural & Molecular Biology 25, 607–615. 10.1038/s41594-018-0087-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Siahaan V, Tan R, Humhalova T, Libusova L, Lacey SE, Tan T, Dacy M, Ori-McKenney KM, McKenney RJ, Braun M, and Lansky Z (2022). Microtubule lattice spacing governs cohesive envelope formation of tau family proteins. Nature Chemical Biology 18, 1224–1235. 10.1038/s41589-022-01096-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Estévez-Gallego J, Josa-Prado F, Ku S, Buey RM, Balaguer FA, Prota AE, Lucena-Agell D, Kamma-Lorger C, Yagi T, Iwamoto H, et al. (2020). Structural model for differential cap maturation at growing microtubule ends. eLife 9, 1–26. 10.7554/eLife.50155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alushin GM, Lander GC, Kellogg EH, Zhang R, Baker D, and Nogales E (2014). High resolution microtubule structures reveal the structural transitions in αβ–tubulin upon GTP hydrolysis. Cell 157, 1117–1129. 10.1016/j.cell.2014.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zanic M, Stear JH, Hyman AA, and Howard J (2009). EB1 recognizes the nucleotide state of tubulin in the microtubule lattice. PLoS One 4, e7585. 10.1371/journal.pone.0007585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Debs GE, Cha M, Liu X, Huehn AR, and Sindelar CV (2020). Dynamic and asymmetric fluctuations in the microtubule wall captured by high-resolution cryoelectron microscopy. Proc Natl Acad Sci U S A 117, 16976–16984. 10.1073/pnas.2001546117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang R, Alushin GM, Brown A, and Nogales E (2015). Mechanistic Origin of Microtubule Dynamic Instability and Its Modulation by EB Proteins. Cell 162, 849–859. 10.1016/j.cell.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reid TA, Coombes C, Mukherjee S, Goldblum RR, White K, Parmar S, McClellan M, Zanic M, Courtemanche N, and Gardner MK (2019). Structural state recognition facilitates tip tracking of EB1 at growing microtubule ends. eLife 8, e48117. 10.7554/eLife.48117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tropini C, Roth EA, Zanic M, Gardner MK, and Howard J (2012). Islands Containing Slowly Hydrolyzable GTP Analogs Promote Microtubule Rescues. PLoS One 7, e30103. 10.1371/journal.pone.0030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dimitrov A, Quesnoit M, Moutel S, Cantaloube I, Poüs C, and Perez F (2008). Detection of GTP-Tubulin Conformation in Vivo Reveals a Role for GTP Remnants in Microtubule Rescues. Science 322, 1353–1356. doi: 10.1126/science.1165401. [DOI] [PubMed] [Google Scholar]
- 60.Gazzola M, Schaeffer A, Butler-Hallissey C, Friedl K, Vianay B, Gaillard J, Leterrier C, Blanchoin L, and Théry M (2023). Microtubules self-repair in living cells. Current Biology 33, 122–133. 10.1016/j.cub.2022.11.060. [DOI] [PubMed] [Google Scholar]
- 61.Tasic B, Yao Z, Graybuck LT, Smith KA, Nguyen TN, Bertagnolli D, Goldy J, Garren E, Economo MN, Viswanathan S, et al. (2018). Shared and distinct transcriptomic cell types across neocortical areas. Nature 563, 72–78. 10.1038/s41586-018-0654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rice LM, Moritz M, and Agard DA (2021). Microtubules form by progressively faster tubulin accretion, not by nucleation-elongation. Journal of Cell Biology 220, e202012079. 10.1083/jcb.202012079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brouhard GJ, and Rice LM (2018). Microtubule dynamics: an interplay of biochemistry and mechanics. Nature Reviews Molecular Cell Biology 19, 451–463. 10.1038/s41580-018-0009-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Höög JL, Huisman SM, Sebö-Lemke Z, Sandblad L, McIntosh JR, Antony C, and Brunner D (2011). Electron tomography reveals a flared morphology on growing microtubule ends. Journal of Cell Science 124, 693–698. 10.1242/jcs.072967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McIntosh JR, O’Toole E, Morgan G, Austin J, Ulyanov E, Ataullakhanov F, and Gudimchuk N (2018). Microtubules grow by the addition of bent guanosine triphosphate tubulin to the tips of curved protofilaments. Journal of Cell Biology 217, 2691–2708. 10.1083/jcb.201802138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kellogg EH, Hejab NMA, Poepsel S, Downing KH, DiMaio F, and Nogales E (2018). Near-atomic model of microtubule-tau interactions. Science 360, 1242–1246. 10.1126/science.aat1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ferro LS, Fang Q, Eshun-Wilson L, Fernandes J, Jack A, Farrell DP, Golcuk M, Huijben T, Costa K, Gur M, et al. (2022). Structural and functional insight into regulation of kinesin-1 by microtubule-associated protein MAP7. Science 375, 326–331. 10.1126/science.abf6154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bigman LS, and Levy Y (2020). Tubulin tails and their modifications regulate protein diffusion on microtubules. Proceedings of the National Academy of Sciences 117, 8876–8883. 10.1073/pnas.1914772117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bigman LS, and Levy Y (2020). Protein Diffusion on Charged Biopolymers: DNA versus Microtubule. Biophysical Journal 118, 3008–3018. 10.1016/j.bpj.2020.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hinrichs MH, Jalal A, Brenner B, Mandelkow E, Kumar S, and Scholz T (2012). Tau Protein Diffuses along the Microtubule Lattice. Journal of Biological Chemistry 287, 38559–38568. 10.1074/jbc.M112.369785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miesch J, Wimbish RT, Velluz M, and Aumeier C (2021). Phase separation of +TIP-networks regulates microtubule dynamics. bioRxiv. 10.1101/2021.09.13.459419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen D, and Huang S (2001). Nucleolar Components Involved in Ribosome Biogenesis Cycle between the Nucleolus and Nucleoplasm in Interphase Cells. The Journal of Cell Biology 153, 169–176. 10.1083/jcb.153.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J, Jülicher F, and Hyman AA (2009). Germline P Granules Are Liquid Droplets That Localize by Controlled Dissolution/Condensation. Science 324, 1729–1732. 10.1126/science.1172046. [DOI] [PubMed] [Google Scholar]
- 74.Kanaan NM, Hamel C, Grabinski T, and Combs B (2020). Liquid-liquid phase separation induces pathogenic tau conformations in vitro. Nature Communcations 11. 10.1038/s41467-020-16580-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Taylor NO, Wei M, Stone HA, and Brangwynne CP (2019). Quantifying Dynamics in Phase-Separated Condensates Using Fluorescence Recovery After Photobleaching. Biophysical Journal 117, 1285–1300. 10.1016/j.bpj.2019.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Boeynaems S, Alberti S, Fawzi NL, Mitag T, Polymenidou M, Rousseau F, Schymkowitz J, Shorter J, Wolozin B, Van Den Bosch L, et al. (2018). Protein Phase Separation: A New Phase in Cell Biology. Trends in Cell Biology 28, 420–435. 10.1016/j.tcb.2018.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Safari MS, King MR, Brangwynne CP, and Petry S (2021). Interaction of spindle assembly factor TPX2 with importins-α/β inhibits protein phase separation. Journal of Biological Chemistry 297, 100998. 10.1016/j.jbc.2021.100998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang K, Lyu W, Yu J, and Koleske AJ (2018). Abl2 is recruited to ventral actin waves through cytoskeletal interactions to promote lamellipodium extension. Molecular Biology of the Cell 29, 2863–2873. 10.1091/mbc.E18-01-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Castoldi M, and Popov AV (2003). Purification of brain tubulin through two cycles of polymerization-depolymerization in a high-molarity buffer. Protein Expression & Purification 32, 83–88. 10.1016/S1046-5928(03)00218-3. [DOI] [PubMed] [Google Scholar]
- 80.Peloquin J, Komarova Y, and Borisy G (2005). Conjugation of fluorophores to tubulin. Nature Methods 2, 299–303. 10.1038/nmeth0405-299. [DOI] [PubMed] [Google Scholar]
- 81.Campbell JN, and Slep KC (2011). αβ-Tubulin and Microtubule-Binding Assays. Methods in Molecular Biology 777, 87–97. 10.1007/978-1-61779-252-6_6. [DOI] [PubMed] [Google Scholar]
- 82.Spector JO, Vemu A, and Roll-Mecak A (2020). In Vitro Microtubule Dynamics Assays Using Dark-Field Microscopy 10.1007/978-1-0716-0219-5_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pollard TD (2010). A Guide to Simple and Informative Binding Assays. Molecular Biology of the Cell 21, 4061–4067. 10.1091/mbc.e10-08-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Johnson-Chavarria EM, Tanyeri M, and Schroeder CM (2011). A Microfluidic-based Hydrodynamic Trap for Single Particles. Journal of Visualized Experiments 21, 2517. 10.3791/2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pear WS, Nolan GP, Scott ML, and Baltimore D (1993). Production of high-titer helper-free retroviruses by transient transfection. Proceedings of the National Academy of Sciences 90, 8392–8396. 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.La Verde V, Dominici P, and Astegno A (2017). Determination of Hydrodynamic Radius of Proteins by Size Exclusion Chromatography. Bio-Protocol 7. 10.21769/BioProtoc.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schaum N, Karkanias J, Neff NF, May AP, Quake SR, Wyss-Coray T, Darmanis S, Batson J, Botvinnik O, Chen MB, et al. (2018). Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372. 10.1038/s41586-018-0590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12. 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lonsdale J, Thomas J, Salvatore M, and al., e. (2013). The Genotype-Tissue Expression (GTEx) project. Nature Genetics 45, 580–585. 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lois C, Hong EJ, Pease S, Brown EJ, and Baltimore D (2002). Germline Transmission and Tissue-Specific Expression of Transgenes Delivered by Lentiviral Vectors. Science 295, 868–872. 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- 91.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nature Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Madeira F, Pearce M, Tivey ARN, Basutkar P, Lee J, Edbali O, Madhusoodanan N, Kolesnikov A, and Lopez R (2022). Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Research 50, W276–W279. 10.1093/nar/gkac240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xue B, Dunbrack RL, Williams RW, Dunker AK, and Uversky VN (2010). PONDR-FIT: A Meta-Predictor of Intrinsically Disordered Amino Acids. Biochimica et Biophysica Acta 1804, 996–1010. 10.1016/j.bbapap.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jones DT, and Cozzetto D (2014). DISOPRED3: precise disordered region predictions with annotated protein-binding activity. Bioinformatics 31, 857–863. 10.1093/bioinformatics/btu744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Provencher SW, and Glöckner J (1981). Estimation of Globular Protein Secondary Structure from Circular Dichroism. Biochemistry 20, 33–37. 10.1021/bi00504a006. [DOI] [PubMed] [Google Scholar]
- 96.Compton LA, and Johnson WC Jr (1986). Analysis of protein circular dichroism spectra for secondary structure using a simple matrix multiplication. Analytical Biochemistry 155, 155–167. 10.1016/0003-2697(86)90241-1. [DOI] [PubMed] [Google Scholar]
- 97.Sreerama N, and Woody RW (2000). Estimation of Protein Secondary Structure from Circular Dichroism Spectra: Comparison of CONTIN, SELCON, and CDSSTR Methods with an Expanded Reference Set. Analytical Biochemistry 287, 252–260. 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 98.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, and Ferrin TE (2018). UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci 27, 14–25. 10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Brown A, Long F, Nicholls RA, Toots J, Emsley P, and Murshudov G (2015). Tools for macromolecular model building and refinement into electron cryo-microscopy reconstructions. Acta Crystallographica Section D Biological Crystallography 71, 136–153. 10.1107/S1399004714021683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296. 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- 102.Schorb M, Haberbosch I, Hagen WJH, Schwab Y, and Mastronarde DN (2019). Software tools for automated transmission electron microscopy. Nature Methods 16, 471–477. 10.1038/s41592-019-0396-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is available in the main text or supplemental information.
Custom-written MATLAB scripts for lattice damage analysis are available at https://github.com/koleskelab/lattice_damage.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.