

Abstract

Modern organic synthesis requires methodologies that bring together abundant feedstock chemicals in a mild and efficient manner. To aid in this effort, we have developed a multicomponent radical hydroxyarylation reaction that utilizes aryl halides, olefins, and O2 as the reaction components. Crucial to this advance was an oxidative, rather than a reductive, approach to aryl radical generation, which enables reaction tolerance to O2. This methodology displays a broad functional group tolerance with a variety of functionalized aryl halides and a broad array of olefins. Development of this methodology enables rapid access to biologically relevant hydroxyaryl products from simple, commercially available starting materials.
Keywords: photoredox, multicomponent coupling, catalysis, olefin functionalization, radical
Multicomponent coupling reactions, especially those that bring together simple and abundant feedstock chemicals under mild, functional group-tolerant conditions, hold exceptional potential in creating molecular complexity, advancing sustainable synthesis, and generating chemical libraries.1 Within this context, a catalytic photoredox-driven union of aryl halides, olefins, and O2 to deliver the olefin hydroxyarylation product represents an important and previously unrealized transformation. The development of such a reaction enables rapid access to hydroxyaryl motifs that can be found in natural products, agrochemicals, and pharmaceutical agents (Figure 1, top).2
Figure 1.

Rational for pursuing a multicomponent radical hydroxyarylation of olefins using aryl halides and O2.
The conceptual challenge associated with this transformation is that the generation of aryl radical intermediates from aryl halides is classically considered a reductive process, and the reaction conditions are often incompatible with O2 (Figure 1, middle).3 Indeed, numerous photoredox reactions that operate in a reductive manifold frequently necessitate air-free conditions to achieve optimal yields.4 However, silyl radical-mediated halogen atom transfer (XAT) is an attractive approach to aryl radical generation from aryl halides5 and has been shown to tolerate O2,6 though no instances of utilizing O2 as a reaction component for C–O bond formation have been reported to this point.
Based on this observation, we hypothesized that the oxidative activation of silanols would be highly compatible with aryl radical generation and olefin addition in the presence of O2.6a,6b,7 Furthermore, we recognized that nucleophilic radicals are known to react with triplet O2, and the differential nucleophilicity of an sp2-aryl radical and an sp3-alkyl radical could be utilized for sequencing bond formation in a multicomponent reaction.8 We expected the less nucleophilic sp2-radical to preferentially react with an olefin substrate, while the resulting more nucleophilic sp3-radical is polarity-matched for subsequent capture by O2, leading to clean and efficient multicomponent coupling.
Prior work in this area shows several examples of multicomponent olefin oxyarylation reactions, though these reactions require 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) or metal-assisted trapping of the sp3-radical intermediate.9 To date, only a few reports have described the aryl radical reactivity in the presence of O2. Such reported reactions require aryl hydrazines,10 aryl diazoniums,11 or aryl boronates9d,12 as aryl radical precursors and are often combined with stoichiometric metal reductants, limiting their synthetic utility. Prior to this study, no catalytic multicomponent hydroxyarylation of olefins with aryl halides and O2 has been reported.
Our reaction design is depicted in Figure 2A. Oxidation of tris(trimethylsilyl)silanol ((TMS)3SiOH) by a photocatalyst (PC) initiates a radical Brook rearrangement to silyl radical intermediate I. Polarity-matched halogen-atom transfer (XAT) between I and aryl halide II results in aryl radical III. Trapping of the aryl radical with an olefin forges a C–C bond and alkyl radical intermediate IV. This radical engages with triplet O2 to form the C–O bond and peroxyl radical species V, which is reduced by the photocatalyst radical anion to deliver peroxide VI. Conversion of VI to hydroxy product VIII requires an additional equivalent of reductant. We envision that silanol VII is formed from the hydrolysis of the halogenated silyl species resulting from XAT of I and II. We propose that silanol VII is oxidized by the excited-state photocatalyst, undergoing a second radical Brook rearrangement and generating another equivalent of photocatalyst radical anion to reduce peroxide VI to the alcohol product VIII.
Figure 2.
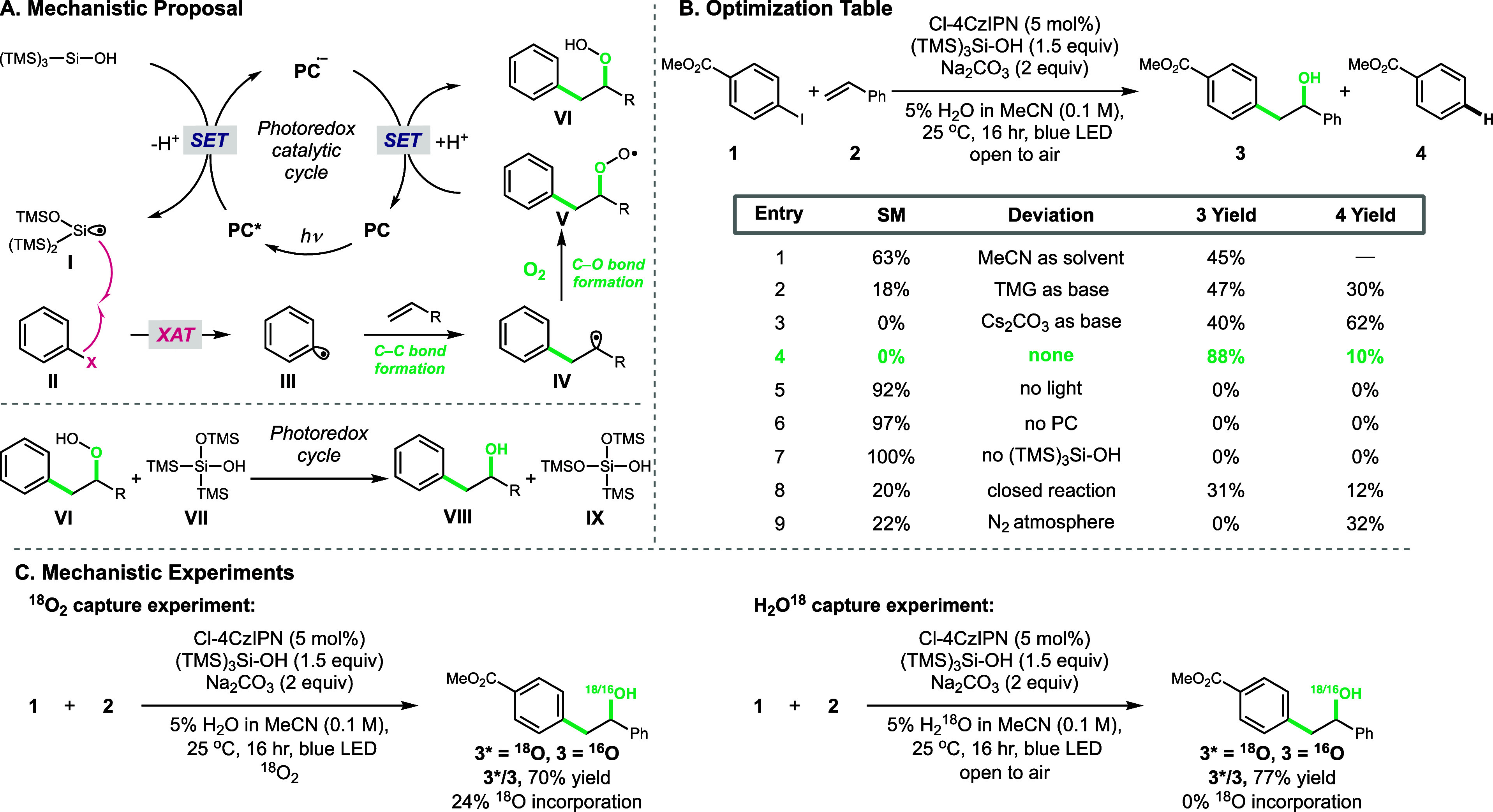
(A) Proposed mechanism for the formation of the observed hydroxyarylation products. (B) Optimization of the hydroxyarylation of olefins with aryl halides and O2. Conditions are as follows: 1 (0.1 mmol), 2 (0.3 mmol), Na2CO3 (0.2 mmol), (TMS)3SiOH (0.15 mmol), solvent (0.1 M), blue LEDs, and open to air at room temperature for 16 h. (C) Mechanistic experiments to support radical trapping of O2 rather than radical–polar crossover and capture of H218O.
With this mechanism in mind, we note that the activation of (TMS)3SiOH to form the halogen-abstracting silyl radical requires a highly oxidizing photocatalyst (Ep = +1.54 V vs SCE),7 so we chose a metal-free photocatalyst Cl-4CzIPN (E1/2red [*PC/PC•– ] = +1.71 vs SCE)7a to begin our studies. In our initial experiment, we investigated the reaction of 4-methyl iodobenzoate as the aryl radical precursor (1), styrene as the radical acceptor (2), and (TMS)3SiOH as the XAT reagent in the presence of the photocatalyst Cl-4CzIPN, and sodium carbonate. Subjecting these reagents to irradiation by blue LEDs in a MeCN solution open to air produced hydroxyarylated product 3 in 45% yield, with the starting material composing the rest of the reaction mass balance (Figure 2B, entry 1). In an effort to push the reaction to complete starting material consumption, we conducted experiments exploring the base solubility, as base is required to generate the silyl radical abstractor. Organic base tetramethylguanidine (TMG) increased the starting material consumption, although a large amount of hydrodehalogenation product 4 was observed (Figure 2B, entry 2). Similarly, cesium carbonate as base led to full consumption of the starting material, but hydrodehalogenation outcompeted hydroxyarylation (Figure 2B, entry 3). Adding water as a cosolvent to the original conditions aided sodium carbonate solubility, facilitating full starting material consumption and increasing the hydroxyarylation yield to 88% with minimal hydrodehalogenation (10%, Figure 2B, entry 4). Control experiments revealed that light, photocatalyst, and XAT reagent are all necessary components for product formation (Figure 2B, entries 5–7).
To probe our hypothesis that C–O bond formation arises from O2 capture, we conducted experiments investigating how the reaction atmosphere impacts product formation. Varying the optimized conditions so that the reaction is conducted in a closed reaction vessel rather than open to air reduced the yield of 3 to 31% (Figure 2B, entry 8). Additionally, under an inert N2 atmosphere, no 3 was observed. Conducting the experiment under an atmosphere of 18O2 resulted in a 70% yield of 3 and 3* with 24% 18O2 incorporation, giving strong evidence to support C–O bond formation resulting from O2 capture. To rule out a potential mechanism where the radical intermediate IV is oxidized to radical cation and C–O bond formation results in the nucleophilic addition of H2O, we replaced the H2O cosolvent with H218O. No 18O 3* was detected by mass spectroscopy, giving further evidence that C–O bond formation arises from radical capture by O2.
We next investigated the scope of the reaction, beginning with the scope of aryl iodides as aryl radical precursors. A variety of para-substituted electron-poor, -neutral, and -rich aromatics all reacted smoothly in good to excellent yields (Scheme 1, 3–19, 78–96%). Although nitro groups have been reported to be photoactive under blue light irradiation, we obtained a good yield of nitro-containing hydroxyaryl product 6 (62%).13 Notably, unprotected phenol 12 was compatible (51%), though increased yields could be obtained with phenol protection (13, 87%). Protected aniline 14 reacted smoothly in 77% yield. Given the selectivity for aryl iodides, we explored the reaction tolerance for aromatic substituents that could be utilized for orthogonal reactivity. Pinacol boronate (Bpin)-substituted arene produced a mixture of hydroxyarylated product 15 in 25% yield and hydroxyarylated phenol 12 in 40% yield where 12 likely arises from oxidation and hydrolysis of the aryl-Bpin under photoredox conditions.14 Switching from Bpin to the less redox-sensitive N-methyliminodiacetic acid (MIDA)-substituted borane increased the yield to 70% of 16. Electrophilic cross-coupling handles TfO-, Cl-, and Br-substituted arenes were tolerated, and products were obtained in good yields (17–19, 76–90%). Expanding beyond para-substituted arenes, meta-substitution was well tolerated with electron-rich, -poor, and reactive handles, all producing good yields (20–22, 65–95%). Additionally, ortho-substitution was well tolerated with both mono- and disubstituted arenes (23–28, 36–90%). Previous hydroxyarylations have limited utility in functionalizing heteroaryl species.9−12 Under our XAT conditions, pyridines could be functionalized at the 2-, 3-, and 4-positions in moderate yields (29–31, 42–76%). Additional heterocycles such as pyrazine 32, thiophene 33, and indole 34 all produced hydroxyarylated products in 51–62% yields.
Scheme 1. Scope of the Multicomponent Radical Hydroxyarylation of Olefins with Aryl Halides and O2.
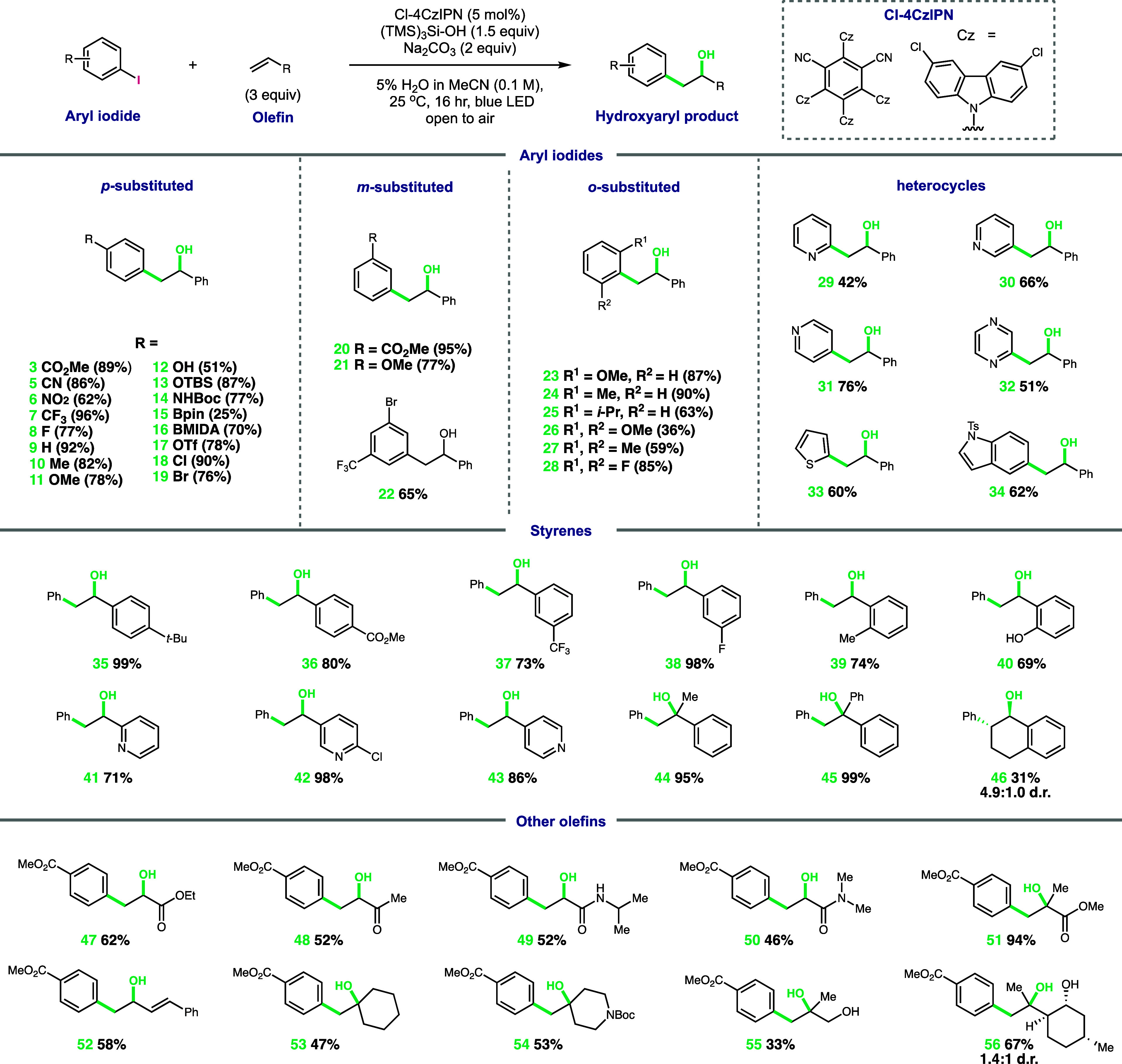
Conditions are as follows: aryl halide (1 equiv), olefin (3 equiv), (TMS)3SiOH (1.5 equiv), Na2CO3 (2 equiv), 5% H2O in MeCN (0.1 M), blue LEDs, and open to air at room temperature for 16 h.
With the aryl iodide scope established, we investigated the range of olefin radical acceptors that can be applied to our conditions. We began by exploring styrene substitution and its effect on the hydroxyarylation yield. Electron-poor, -neutral, and -rich styrenes substituted at the ortho-, meta-, and para-positions, all gave good to excellent yields, resulting in a variety of benzylic hydroxylated products (35–46, 74–99%). Again, phenol protection was not required (40, 69%), and heterocycles, such as, 2-, 3-, and 4-vinylpyridines, all reacted in good-to-excellent yields (41–43, 71–96%). Investigating the substitution of the styrene olefin revealed α-substituted styrenes gave excellent yields (44 and 45, 95 and 99%, respectively), likely associated with the increased stability of tertiary-substituted radical intermediates. Conversely, β-substitution reduced the hydroxyarylated product yield (46, 31%), likely due to additional steric demands at the site of aryl radical addition to the olefin. Expanding beyond styrenes, we investigated the reactivity of Michael acceptors and their ability to generate synthetically useful α-hydroxy carbonyl compounds. Acrylates, enones, and acrylamides were all competent with the α-substitution of the olefin, again increasing the yield (47–51, 46–94%). Aryl radical addition to a conjugated diene formed secondary allylic alcohol 52 in 58% yield. Additionally, unactivated 1,1-disubstitued olefins reacted to give a variety of tertiary alcohols. Cyclohexane 53, medicinally relevant piperidine 54, β-hydroxy alcohol 55, and functionalized chiral pool substrate 56 (from (−)-isopulegol) were all obtained in moderate yields (33–67%).
To further demonstrate the applicability of this methodology in complex molecule synthesis, we conducted experiments to perform this reaction on scale. Early on in our studies, we observed a decreased yield moving from the optimization scale (0.1 mmol) to the substrate scope scale (0.5 mmol, Table S2). This effect was largely associated with the stir rate, where we observed that smooth “vortexing” of the reaction mixture throughout the reaction time was crucial for optimal and reproducible yields. Scaling from 0.5 to 2 mmol also required slight tuning of the reaction conditions (Table S3). Though the organic photocatalyst Cl-4CzIPN provided good and reproducible yields on scales ≤0.5 mmol, its poor solubility in MeCN/H2O became an issue when scaling the reaction, presumably due to reduced light penetration. Alternatively, the photocatalyst [Ir(dFCF3ppy)2(5,5′-dCF3bpy)]PF6 has similar oxidizing properties to Cl-4CzIPN (E1/2red [*IrIII/IrII] = +1.68 V vs SCE)6b and an improved solubility profile in our reaction solvent. With this photocatalyst and a slightly longer reaction time, we scaled the hydroxyarylation reaction to 2 mmol without an appreciable decrease in yield (86%, Figure 3A). With the access to millimole quantities of hydroxyaryl product 3, we subjected 3 to enzyme-catalyzed kinetic resolution. Using commercially available Pseudomonas stutzeri lipase, enantioenriched benzylic alcohols could be accessed in just two steps from 1 and 2 (Figure 3A).15
Figure 3.

(A) Rapid access to enantioenriched 1,2-diarylalcohols on mmol scale. (B) Diversification of the aliphatic alcohol 54 hydroxyaryl product.
Additionally, we highlight the ability to diversify alcohol products into a range of medicinally relevant motifs. Using a deoxygenative alkylation protocol reported by MacMillan and co-workers, 54 could be transformed into quaternary carbon containing unnatural amino acid 59 (25% yield, Figure 3B).16 The free hydroxyl group of 54 could be utilized for a Yu C–H annulation to deliver spirocyclic benzofuran/piperidine heterocycle 60 in 33% yield.17 Finally, functionalization of the benzylic site through Du Bois C–H amination produced synthetically useful oxazolidinone 61 (Figure 3B).18 In all cases, the reported yields represent reactions run under the originally reported conditions. No attempts to optimize for this particular substrate were made, highlighting the ability to diversify without the need for significant extra experiments.
In summary, we present the first example of a hydroxyarylation of alkenes using aryl halides and O2. Our methodology has significant potential for industrial impact given it utilizes abundant feedstock chemicals, displays a diverse substrate scope, and is operationally simple to set up. In addition to our methodology, providing rapid access to biologically relevant hydroxyaryl scaffolds, these products can undergo a variety of transformations to build molecular complexity and introduce new motifs. Investigations to develop new reactions using our O2-tolerant oxidative aryl radical generation conditions are ongoing.
Acknowledgments
Financial support for this work was provided by the National Institutes of Health (GM129495), and NMR data were collected on instruments obtained with support from the National Science Foundation (CHE-1521620). We thank Jack C. Sharland and the Huw M. L. Davies Lab at Emory University for providing some styrene starting materials. We thank AiLing Yu and the Cora E. MacBeth lab at Emory University for assistance with the 18O2 experiment. We thank Meito Sangyo for providing PSL for the kinetic resolution experiment.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.3c05988.
Experimental procedures, characterization data, and spectra (PDF)
Author Contributions
The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- a Zarganes-Tzitzikas T.; Chandgude A. L.; Dömling A. Multicomponent reactions, Union of MCRs and beyond. Chem. Rec. 2015, 15, 981–996. 10.1002/tcr.201500201. [DOI] [PubMed] [Google Scholar]; b Coppola G. A.; Pillitteri S.; Van der Eycken E. V.; You S.-L.; Sharma U. K. Multicomponent reactions and photo/electrochemistry join forces: atom economy meets energy efficiency. Chem. Soc. Rev. 2022, 51, 2313–2382. 10.1039/D1CS00510C. [DOI] [PubMed] [Google Scholar]; c Quazi S.; Rashid M. T.; Malik J. A.; Gavas S. The discovery of novel antimicrobial agents through the application of isocyanide-based multicomponent reactions. Antibiotics 2023, 12, 849. 10.3390/antibiotics12050849. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]; d Mardjan M. I. D.; Mayooufi A.; Parrain J.-L.; Thibonnet J.; Commeiras L. Straightforward access to a great diversity of complex biorelevant γ-lactams thanks to a tunable cascade multicomponent process. Org. Process Res. Dev. 2020, 24, 606–614. 10.1021/acs.oprd.9b00438. [DOI] [Google Scholar]; e Reguera L.; Rivera D. G. Multicomponent reaction toolbox for peptide macrocyclization and stapling. Chem. Rev. 2019, 119, 9836–9860. 10.1021/acs.chemrev.8b00744. [DOI] [PubMed] [Google Scholar]
- a van der Zanden S. Y.; Qiao X.; Neefjes J. New insights into the activities and toxicities of the old anticancer drug doxorubicin. FEBS J. 2021, 288, 6095–6111. 10.1111/febs.15583. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Matteelli A.; Carvalho A. C.; Dooley K. E.; Kritski A. TMC207: the first compound of a new class of potent anti-tuberculosis drugs. Future Microbio. 2010, 5, 849–858. 10.2217/fmb.10.50. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Rogawski M. A.; Löscher W. The neurobiology of antiepileptic drugs. Nat. Rev. Neurosci. 2004, 5, 553–564. 10.1038/nrn1430. [DOI] [PubMed] [Google Scholar]; d Parker J. E.; Warrilow A. G. S.; Cools H. J.; Fraaije B. A.; Lucas J. A.; Rigdova K.; Griffiths W. J.; Kelly D. E.; Kelly S. L. Prothioconazole and prothioconazole-desthio activities against candida albicans sterol 14-a-demethylase. Appl. Environ. Microbiol. 2013, 79, 1639–1645. 10.1128/AEM.03246-12. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Deecher D. C.; Beyer C. E.; Johnston G.; Bray J.; Shah S.; Abou-Gharbia M.; Andree T. H. Desvenlafaxine succinate: a new serotonin and norepinephrine reuptake inhibitor. J. Pharmacol. Exp. Ther. 2006, 318, 657–665. 10.1124/jpet.106.103382. [DOI] [PubMed] [Google Scholar]
- a Nelleborg P.; Lund H.; Eriksen J. Photochemical vs. electrochemical electron-transfer reactions. one-electron reduction of aryl halides by photoexcited anion radicals. Tetrahedron Lett. 1985, 26, 1773–1776. 10.1016/S0040-4039(00)98335-7. [DOI] [Google Scholar]; b Cowper N. G. W.; Chernowsky C. P.; Williams O. P.; Wickens Z. K. Potent reductants via electron-primed photoredox catalysis: unlocking aryl chlorides for radical coupling. J. Am. Chem. Soc. 2020, 142, 2093–2099. 10.1021/jacs.9b12328. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hendy C. M.; Smith G. C.; Xu Z.; Lian T.; Jui N. T. Radical chain reduction via carbon dioxide radical anion (CO2•–). J. Am. Chem. Soc. 2021, 143, 8987–8992. 10.1021/jacs.1c04427. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Nguyen J. D.; D’Amato E. M.; Narayanam J. M. R.; Stephenson C. R. J. Engaging unactivated alkyl, alkenyl and aryl iodides in visible-light-mediated free radical reactions. Nat. Chem. 2012, 4, 854–859. 10.1038/nchem.1452. [DOI] [PubMed] [Google Scholar]; e Molander G. A., Reductions with samarium(II) iodide. In Organic Reactions; Wiley, 2004; Vol. 211; p 367. [Google Scholar]; f Meyer A. U.; Slanina T.; Yao C.-J.; König B. Metal-free perfluoroarylation by visible light photoredox catalysis. ACS Catal. 2016, 6, 369–375. 10.1021/acscatal.5b02410. [DOI] [Google Scholar]
- a Ghosh I.; Ghosh T.; Bardagi J. I.; König B. Reduction of aryl halides by consecutive visible-light induced electron transfer processes. Science 2014, 346, 725–728. 10.1126/science.1258232. [DOI] [PubMed] [Google Scholar]; b Tintori G.; Fall A.; Assani N.; Zhao Y.; Bergé-Lefranc D.; Redon S.; Vanelle P.; Broggi J. Generation of powerful electron donors by water-assisted decarboxylation of benzimidazolium carboxylates. Org. Chem. Front. 2021, 8, 1197–1205. 10.1039/D0QO01488E. [DOI] [Google Scholar]; c Yu D.; To W. P.; Tong G. S. M.; Wu L.-L.; Kaai-Tung C.; Du L.; Phillips D. L.; Liu Y.; Che C.-M. Luminescent tungsten(VI) complexes as photocatalysts for light-driven C-C and C-B bond formation reactions. Chem. Sci. 2020, 11, 6370–6382. 10.1039/d0sc01340d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliá F.; Constantin T.; Leonori D. Applications of halogen atom transfer (XAT) for the generation of carbon radicals in synthetic photochemistry and photocatalysis. Chem. Rev. 2022, 122, 2292–2352. 10.1021/acs.chemrev.1c00558. [DOI] [PubMed] [Google Scholar]
- a Dow N. W.; Cabré A.; MacMillan D. W. C. A general N-alkylation platform via copper metallaphotoredox and silyl radical activation of alkyl halides. Chem. 2021, 7, 1827–1842. 10.1016/j.chempr.2021.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lavagnino M. N.; Liang T.; MacMillan D. W. C. HARC as an open-shell strategy to bypass oxidative additions in Ullman-Goldberg couplings. Proc. Natl. Acad. Sci. U. S. A. 2020, 117 (35), 21058–21064. 10.1073/pnas.2011831117. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Devery J. J. III; Nguyen J. D.; Dai C.; Stephenson C. R. J. Light-mediated reductive debromination of unactivated alkyl and aryl bromides. ACS Catal. 2016, 6, 5962–5967. 10.1021/acscatal.6b01914. [DOI] [Google Scholar]
- a Wiles R. J.; Phelan J. P.; Molander G. A. Metal-free defluorinative arylation of trifluoromethyl alkenes via photoredox catalysis. Chem. Commun. 2019, 55, 7599–7602. 10.1039/C9CC04265B. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Le C.; Chen T. Q.; Liang T.; Zhang P.; MacMillan D. W. C. A radical approach to the copper oxidative addition problem: Trifluoromethylation of bromoarenes. Science 2018, 360, 1010–1014. 10.1126/science.aat4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tang C.; Qiu X.; Cheng Z.; Jiao N. Molecular oxygen-mediated oxygenation reactions involving radicals. Chem. Soc. Rev. 2021, 50, 8067–8101. 10.1039/D1CS00242B. [DOI] [PubMed] [Google Scholar]; b Parsaee F.; Senarathna M. C.; Kannangara P. B.; Alexander S. N.; Arche P. D. E.; Welin E. R. Radical philicity and its role in selective organic transformations. Nat. Rev. Chem. 2021, 5, 486–499. 10.1038/s41570-021-00284-3. [DOI] [PubMed] [Google Scholar]; c De Vleeschouwer F.; Van Speybroeck V.; Waroquier M.; Geerlings P.; De Proft F. Electrophilicity and nucleophilicity index for radicals. Org. Lett. 2007, 9, 2721–2724. 10.1021/ol071038k. [DOI] [PubMed] [Google Scholar]
- a Hartmann M.; Li Y.; Studer A. Transition-metal-free oxyarylation of alkenes with aryl diazonium salts and TEMPONa. J. Am. Chem. Soc. 2012, 134, 16516–16519. 10.1021/ja307638u. [DOI] [PubMed] [Google Scholar]; b Hartmann M.; Li Y.; Mück-Lichtenfeld C.; Studer A. Generation of aryl radicals through reduction of hypervalent iodine(III) compounds with TEMPONa: radical alkene oxyarylation. Chem.—Eur. J. 2016, 22, 3485–3490. 10.1002/chem.201504852. [DOI] [PubMed] [Google Scholar]; c Hopkinson M. N.; Sahoo B.; Glorius F. Dual photoredox and gold catalysis: intermolecular multicomponent oxyarylation of alkenes. Adv. Synth. Catal. 2014, 356, 2794–2800. 10.1002/adsc.201400580. [DOI] [Google Scholar]; d Wang Y.; Lin C.; Zhang Z.; Shen L.; Zou B. Directed nickel-catalyzed selective arylhydroxylation of unactivated alkenes under air. Org. Lett. 2023, 25, 2172–2177. 10.1021/acs.orglett.3c00085. [DOI] [PubMed] [Google Scholar]
- a Kindt S.; Jasch H.; Heinrich M. R. Manganese(IV)-mediated hydroperoxyarylation of alkenes with aryl hydrazines and dioxygen from air. Chem.—Eur. J. 2014, 20, 6251–6255. 10.1002/chem.201400064. [DOI] [PubMed] [Google Scholar]; b Taniguchi T.; Zaimoku H.; Ishibashi H. A mild oxidative aryl radical addition into alkenes by aerobic oxidation of arylhydrazines. Chem.—Eur. J. 2011, 17, 4307–4312. 10.1002/chem.201003060. [DOI] [PubMed] [Google Scholar]
- Kindt S.; Wicht K.; Heinrich M. R. Thermally induced carbohydroxylation of styrenes with aryldiazonium salts. Angew. Chemie. Int. Ed. 2016, 55, 8744–8747. 10.1002/anie.201601656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Dickschat A.; Studer A. Radical addition of arylboronic acids to various olefins under oxidative conditions. Org. Lett. 2010, 12, 3972–3974. 10.1021/ol101818k. [DOI] [PubMed] [Google Scholar]; b Wang D.-M.; She L.-Q.; Yuan H.; Wu Y.; Tang Y.; Wang P. Ligand-enabled Ni(II)-catalyzed hydroxylarylation of alkenes with molecular oxygen. Angew. Chem., Int. Ed. 2023, 62, e202304573 10.1002/anie.202304573. [DOI] [PubMed] [Google Scholar]
- Wise D. E.; Parasram M. Photoexcited nitroarenes as anaerobic oxygen atom transfer reagents. Synlett 2023, 34, 1655–1661. 10.1055/s-0042-1751443. [DOI] [Google Scholar]
- Zou Y.-Q.; Chen J.-R.; Liu X.-P.; Lu L.-Q.; Davis R. L.; Jo̷rgensen K. A.; Xiao W.-J. Highly efficient aerobic oxidative hydroxylation of arylboronic acids: photoredox catalysis using visible light. Angew. Chem., Int. Ed. 2012, 51, 784–788. 10.1002/anie.201107028. [DOI] [PubMed] [Google Scholar]
- Kim M.-J.; Choi K. Y.; Kim S.; Kim D.; Han K.; Ko S.-B.; Park J. Highly enantioselective dynamic kinetic resolution of 1,2-diarylethanols by a lipase-ruthenium couple. Org. Lett. 2008, 10, 1295–1298. 10.1021/ol800163z. [DOI] [PubMed] [Google Scholar]
- Wang J. Z.; Sakai H. A.; MacMillan D. W. C. Alcohols as alkylating agents: photoredox-catalzyed conjugate alkylation via in situ deoxygenation. Angew. Chem., Int. Ed. 2022, 61, e202207150 10.1002/anie.202207150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Dai H.-X.; Yu J.-Q. Pd(II)-catalyzed hydroxyl-directed C–H activation/C–O cyclization: expedient construction of dihydrobenzofurans. J. Am. Chem. Soc. 2010, 132, 12203–12205. 10.1021/ja105366u. [DOI] [PubMed] [Google Scholar]
- Espino C. G.; Du Bois J. A Rh-catalzyed C-H insertion reaction for the oxidative conversion of carbamates to oxazolidinones Angew. Chem. Int. Ed. 2001, 40, 598–600. . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.