


Abstract
Translation of an mRNA is generally divided into three stages: initiation, elongation and termination. The relative rates of these steps determine both the number and position of ribosomes along the mRNA, but traditional velocity sedimentation assays for the translational status of mRNA determine only the number of bound ribosomes. We developed a procedure, termed Ribosome Density Mapping (RDM), that uses site-specific cleavage of polysomal mRNA followed by separation on a sucrose gradient and northern analysis, to determine the number of ribosomes associated with specified portions of a particular mRNA. This procedure allows us to test models for translation and its control, and to examine properties of individual steps of translation in vivo. We tested specific predictions from the current model for translational control of GCN4 expression in yeast and found that ribosomes were differentially associated with the uORFs elements and coding region under different growth conditions, consistent with this model. We also mapped ribosome density along the ORF of several mRNAs, to probe basic kinetic properties of translational steps in yeast. We found no detectable decline in ribosome density between the 5′ and 3′ ends of the ORFs, suggesting that the average processivity of elongation is very high. Conversely, there was no queue of ribosomes at the termination site, suggesting that termination is not very slow relative to elongation and initiation. Finally, the RDM results suggest that less frequent initiation of translation on mRNAs with longer ORFs is responsible for the inverse correlation between ORF length and ribosomal density that we observed in a global analysis of translation. These results provide new insights into eukaryotic translation in vivo.
INTRODUCTION
The eukaryotic gene expression program is composed of many steps, from pre-mRNA transcription, processing and export out of the nucleus, to subcellular localization, translation and mRNA decay. Dissection of this program into each of its component steps and analysis of each of these steps is essential for complete understanding.
For translation, the most obvious and tractable parameter to measure is the number of ribosomes associated with the mRNA. The traditional method to determine the overall number of ribosomes on an mRNA involves separation of large complexes by velocity sedimentation through a sucrose gradient. The sedimentation position of an mRNA within the gradient is directly related to the number of ribosomes bound to it, as the mass of the ribosome dominates the sedimentation behavior of the complex [see (1) for technical details]. By this method, the existence of polysomes was identified (2) and changes in ribosomal association of mRNAs upon changes in conditions or in the genetic background have routinely been tested (3–6). Most recently, we and others have used polysome profiling in combination with DNA microarrays to obtain a global view of the translational status of mRNAs (7–14).
The translation of mRNA into protein is mechanistically divided into three successive stages: initiation, elongation and termination (Figure 1), with each of these stages further made up of multiple steps that use distinct sets of factors (15,16). In eukaryotes, initiation encompasses assembly of the 43S complex onto the mRNA, scanning toward and recognition of the start codon, and assembly of the active ribosome at this site; elongation includes codon by codon translocation of the ribosome along the mRNA in conjunction with amino-acyl tRNA selection and peptide bond formation; and termination involves the release of the ribosomal subunits and the mature protein from the mRNA. The relationship among the rates of these steps determines the rate of protein synthesis and the number and distribution of ribosomes on an mRNA. For example, slow initiation relative to elongation and termination will lead to a low density of ribosomes on the mRNA (Figure 1A); faster initiation relative to the scenario in Figure 1A will lead to an increase in the number of ribosomes on the mRNA (Figure 1B). Slower elongation relative to the scenario in Figure 1A will also lead to an increase in the number of ribosomes (Figure 1C).
Figure 1.

The number of ribosomes bound to an mRNA reflects the interplay between initiation, elongation and termination. The arrow thickness represents the relative rate constants for initiation (concave arrowheads), elongation (closed arrowheads) and termination (open arrowheads). Note the different number of bound ribosomes in (A–C).
Although some changes in rates of initiation, elongation and termination lead to changes in the number of associated ribosomes, as in Figure 1, there are many scenarios in which different underlying rates of initiation, elongation and termination give the same number of bound ribosomes. For example, Figure 1B and C both have higher number of bound ribosomes relative to Figure 1A, yet Figure 1B represents the outcome of increased initiation, and Figure 1C represents the outcome of slower elongation. Figure 2 provides additional examples. An mRNA with a slow elongation rate and one with fast elongation but with a strong pause site can have the same overall number of ribosomes (Figure 2A and B, respectively). Similarly, faster elongation than in Figure 2A but slower termination (Figure 2C) can lead to the same overall number of ribosomes as in Figure 2A. Finally, an mRNA with slow initiation could have the same overall number of ribosomes as an mRNA with fast initiation but with limited processivity [i.e. frequent dissociation of ribosomes during elongation (Figure 2D)].
Figure 2.

The position of ribosomes along an mRNA provides additional information about initiation, elongation and termination. Models for mRNAs with similar number of ribosomes but different distributions of ribosomes along the mRNA are presented. As in Figure 1, the arrow thickness represents the relative rate constant for steps. In (D), the downward arrows represent ribosomes dissociation due to limited processivity.
Although the same number of ribosomes is bound to the mRNA in each of the scenarios in Figure 2, the distribution of ribosomes along the mRNAs is different: a strong pause site would result in increased density of ribosomes immediately 5′ to the pause site (Figure 2B), slow termination would result in an increase in ribosomal density upstream of the termination site (Figure 2C), and ribosomal dissociation during elongation would decrease the ribosomal density towards the 3′ end of the mRNA (Figure 2D). Thus, an ability to assess the distribution of ribosomes along an mRNA would provide incisive information about the individual stages of translation and their variation among different mRNA and under different conditions.
In vitro, ribosomes accumulating at the initiation or termination sites can be detected by a ‘toeprinting’ procedure in which a radiolabeled primer is used to synthesize cDNA until the bound ribosome blocks the extension process (17,18). Pause sites along the mRNA can be detected by micrococcal digestion of mRNA associated with ribosomes followed by annealing to cDNA and primer extension reaction that will terminate at the position of the pause site (19); this assay relies on accumulation of ribosomes at a specific position on the mRNA and was used to identify pause sites in vitro on preprolactin mRNA induced by the signal recognition particle (20). Finally, ribosomal position can sometimes be inferred indirectly from the results of mutagenesis experiments (21–25).
We present here a general and direct method to identify the ribosomes in different regions along mRNAs isolated from translating cells. We have applied this method to several yeast mRNAs to test the highly developed model for translation control of GCN4 via uORFs and to provide insight into individual steps in translation.
MATERIALS AND METHODS
Yeast strains and growth conditions
To assay GCN4 ribosomal association under non-starvation conditions, S288c cells were grown to OD600 0.6–0.9 at 30°C in YPD medium (1% yeast extract/2% peptone/2% dextrose). Starvation conditions were imposed by growing cells in minimal medium (0.2% yeast nitrogen base, 0.5% ammonium sulfate and 2% dextrose) and adding the histidine analog 3-aminotriazole to a final concentration of 40 mM for 30 min. For analyses of all other mRNAs, BY4741 cells were grown to OD600 0.6–0.9 at 30°C in YPD.
RNase H treatment
mRNA associated with ribosomes was isolated from 100 ml of cells. Cells were treated with cycloheximide, immediately cooled and lysed, and resolved on a sucrose gradient as described previously (7), except that heparin was excluded from the sucrose gradient. Gradient fractions were collected into tubes containing DTT and RNasin (Promega) (final concentration: 15 mM and 500 U/ml, respectively) and selected fractions were immediately subjected to reaction with RNase H. To accomplish this, an aliquot of the polysomal fraction (400 μl) was mixed with 15 μl of 5 μM antisense oligodeoxynucleotide (ODN); annealing was performed at 37°C with slow cooling to room temperature over 20 min. RNase H (GibcoBRL) (5 U) and 100 μl 5× RNase H buffer (5× buffer is 0.1 M Tris–HCl, pH 7.5, 0.5 M KCl, 0.1 M MgCl2, 0.5 mM DTT and 2.5 mg/ml cycloheximide) were added and the mixture was incubated at 37°C. After 20 min, the volumes were increased to 1 ml with ice-cold LMD buffer (initial LMD buffer concentration: 20 mM Tris–HCl, pH 7.4, 140 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT, 0.1 mg/ml cycloheximide and 1 mg/ml heparin) and loaded on a sucrose gradient. Gradients and sedimentation were as described previously, including the presence of heparin (7). Eighteen fractions were collected into tubes containing 1 ml of 8 M guanidinium chloride, and RNA was precipitated by adding 2 ml ethanol and incubating overnight at −20°C. The precipitate was spun down and washed with 1 ml of 80% ethanol. Pellets were resuspended in 400 μl TE (10 mM Tris–HCl, pH 7.4, 1 mM Na·EDTA), and precipitated again by adding 0.1 vol of 3 M sodium acetate, pH 5.3, and 2.5 vol of ethanol. The final pellets were resuspended in 4.5 μl TE, pH 7.4, and the entire sample was analyzed by northern blot (11). Radioactive probes were prepared by random incorporation of 32P-labeled nucleotides into a PCR fragment homologous to the tested mRNA. Differences in the radioactive signals between the two cleavage products (Figure 4) are probably due to differences in their length, sequence or structure that lead to different hybridization efficiency.
Figure 4.

Testing the scanning and reinitiation model for GCN4 translation control by RDM. (A) Schematic structure of GCN4 mRNA. The four uORFs located on the 5′-leader are depicted as open boxes, and the GCN4 coding ORF is depicted as a hatched box. Numbers indicate distances (in nt) from the AUG and the arrow points to the cleavage position. (B and C) Polysomal RNA was isolated from cells grown in rich medium (B) or in minimal medium supplemented with 3-aminotriazole for 30 min (C). The GCN4 mRNA was cleaved with an antisense ODN complementary to sequence upstream of the AUG codon (the ODN is expected to cut at position −36). Cleavage reactions were separated on a sucrose gradient into 18 fractions, and the indicated fractions were analyzed by northern analysis. Gel migration distance of size markers is shown to the left and sedimentation positions of the 40S, 60S and ribosome–mRNA complexes are indicated at the bottom of each panel. Migration position of the cleaved fragments is shown to the right of each panel.
Sensitivity of RDM
Two factors determine the sensitivity of the procedure. One relates to the level of detection of a transcript, and the other to the precision with which we could determine the number of ribosomes bound to an RNA fragment. The northern protocol we used in this work (11) can successfully detect transcripts present at five or more copies per cell (26). Less abundant transcripts could potentially be detected via RT–PCR, but this assay was not used in the current experiments.
The precision with which we could determine the number of ribosomes bound to an mRNA fragment was determined by the resolution of the sucrose gradients (Figure 3B, upper panel). We could unambiguously determine if mRNA fragments contained either zero or one bound ribosome (GCN4 experiments, Figure 3B). The resolution decreases with increasing the numbers of bound ribosomes. For example, for mRNAs with ∼7 ribosomes bound, we estimate that the uncertainty is approximately ±1 ribosome. This estimate is based on the difference between the sum of the estimated number of ribosomes on the cleaved fragments and the estimated number of ribosomes on their corresponding full-length mRNA.
Figure 3.

Controls for RDM. (A) Specificity of the RNase H reactions. Antisense ODNs complementary to various positions of ADH1 or YEF3 were annealed to polysomal mRNA. The expected lengths (in nt) of the cleavage products are shown schematically at the top of each panel. Following annealing, RNase H was added and reactions proceeded for 20 min. Samples were then subjected to northern analysis using radiolabeled full-length DNA probe. Size markers are shown at the left and white asterisks indicate cleavage products. (−) No oligo added and the letters correspond to the schematic above. (B) No detectable ribosome dissociation during processing steps. The upper panel (first gradient) is an OD254 trace of a sucrose gradient from which two different polysomal fractions were collected. One fraction (mRNAs associated with 3–5 ribosomes) was resolved on a second gradient following incubation for 1 h at 37°C (second gradient, −RNase H panel) and the other fraction (mRNAs associated with 5–10 ribosomes) was annealed with an antisense ODN complementary to ADH1 around position 462, subjected to RNase H reaction, and then separated on a sucrose gradient (second gradient, +RNase H). The sedimentation positions of the 40S, 60S, 80S and polysomes are indicated.
In general, maximal sensitivity of RDM is achieved when abundant transcripts are analyzed and cleaved into fragments with three or fewer bound ribosomes.
Antisense oligodeoxynucleotides (ODNs)
The following antisense ODN were used in the RNase H cleavage reactions. The gene name and distance (in nt) from the initiation AUG to the center of the ODN:mRNA duplex is listed; negative numbers refer to positions 5′ of this AUG and positive numbers to position 3′ of it:
GCN4-(-)36, CTTGAGCAGACAAATTGGTAAACA;
PDC1-681, TCAGCCTTGACGTCGTGTCT;
PDC1-1109, TGGTTCCACATCCATTCTTGC;
YEF3-197, TTAGCAGCGGTCTTCTTGTCCT;
YEF3-1167, TTCGTGCAAGAAGATAGTCATGTATG;
YEF3-1510, CGGAAGTGTCAGAGTGAGTACCATC;
YEF3-2032, CAGTGATTTGTGGCTTAGAGGTACC;
YEF3-2757, TCAAGTGAGGTCTTTGCCATGT;
ADH1-312, CGTTACCCAATTCACAGTATTCACA;
ADH1-462, CGGTGATACCAGCACACAAGATGGG;
ADH1-612, CTTCCTTACCTTCACCACCGTCAAT;
ADH1-712, GTGAGCACCACCGTCAGTGGCCT;
HSP82-824, CTAGTCCACAAAGGCTTAGTCTTG;
HSP82-971, GGTGCTCTCTTTGGAATGAATAAG;
HSP82-1362, GTAACAACTTAGCCAAAGCAGCCC;
PDA1-648, CCTAAAGGAACCTGGGCACCCACGA; and
EF-2-1232, TGGACTTACCATGATCGACGTGA.
RESULTS AND DISCUSSION
The number of ribosomes on a particular mRNA is traditionally determined by separation of polysomal complexes on a sucrose gradient, followed by northern blot analysis of fractions through the gradient. As elaborated in the Introduction, incisive information about the individual steps of translation can be obtained from ribosome positions along an mRNA's open reading frame (ORF) (Figure 2). We therefore designed a procedure to determine the ribosome density of different regions of a particular mRNA. This procedure, termed Ribosome Density Mapping (RDM), is described in the first section below. We then apply RDM in subsequent sections to assess translational control of GCN4 expression and the rates and properties of translational steps.
Experimental procedure for RDM
The overall protocol for RDM is outlined in Scheme 1. We first use the standard technique of velocity sedimentation in a sucrose gradient to separate mRNAs based on the number of bound ribosomes (Scheme 1, step i). Cycloheximide addition and lysis on ice directly prior to sedimentation has been utilized in numerous investigations of in vivo translational status as this treatment is generally assumed to freeze translation and fix in place translating ribosomes. There is considerable circumstantial and correlative evidence that this protocol effectively halts translation and fixes translating ribosomes, thereby providing a snapshot of translation in vivo. Nevertheless, this evidence does not prove that the approach provides a direct readout of in vivo translational status. It particular, the effects of cycloheximide on the scanning 40S subunits or on the formation of initiating 80S complex are not clear. Techniques such as RDM that offer more highly refined and quantitative information in conjunction with mutagenesis and analysis of effects in vivo will provide new opportunities to test this basic assumption.
Scheme 1.

Outline of Ribosome Density Mapping Protocol.
Following translation arrest, fractions corresponding to a particular number of bound ribosomes, typically the most abundant fractions for the mRNA to be analyzed, are pooled (step ii). An antisense ODN complementary to a particular position within the target mRNA is then added together with RNase H to give specific cleavage of the target mRNA (step iii). The sample, which includes the cleaved mRNA along with all other mRNAs present in the pooled fraction, is then loaded onto a second sucrose gradient to separate the mRNA cleavage products based on the number of ribosomes bound to each (step iv). The expectation is that all mRNAs will sediment as they did in the first gradient with the exception of the single mRNA that was cleaved; the 5′- and 3′-fragments of this mRNA will migrate in the gradient at positions reflecting the number of ribosomes bound to each fragment. The position of the mRNA fragments in the gradient can then be determined by northern blot analysis (step v), and the number of bound ribosomes can be assigned with reference to control gradients in which the polysome peaks can be counted. Information about the ribosome distribution can also be obtained by direct comparison of the migration of the 5′- and 3′-fragments, avoiding any potential error due to uncertainty in determination of the number of ribosomes on each fragment. For example, cleavage in the middle of an mRNA will lead to three possible outcomes (Scheme 1, step v) depending on the ribosome density on each half.
Several controls were required to establish this procedure: (i) RNase H cleavage must be specific to the mRNA and the hybridization site specified by the antisense ODN; (ii) the cleaved fragments, which lack either the 3′-polyA tail or the 5′-cap structure, must be stable through the subsequent analysis; and (iii) ribosomes must remain bound to the cleaved fragments throughout the procedure. These controls, described below (Figure 3), and subsequent data confirm the reliability of the RDM procedure.
To establish the specificity of the RNase H cleavage, mRNA associated with 5–10 ribosomes was isolated following sucrose gradient sedimentation, and antisense ODNs complementary to different positions along various mRNAs were added together with RNase H (corresponding to Scheme 1, step iii). Reactions were subjected to northern blot analysis to detect the cleavage products. No cleavage products were observed in the absence of antisense ODNs for either YEF3 or ADH1 [Figure 3A, (−) lane in each panel]. In contrast, addition of ODNs complementary to YEF3 positions 197, 1180, 2045 or 2752 nt from the start codon (lanes a, b, c and d, respectively) or to ADH1 positions 312, 462 or 712 nt from the start codon (lanes a, b and d, respectively) yielded two prominent cleavage products of the expected sizes in each case. We tested 28 different ODNs for 9 different mRNAs, and all except one yielded accurate cleavage (Figures 4–6, and data not shown). Only the antisense ODN to ADH1 at position 612 gave additional bands of unexpected size (Figure 3A; ADH1, lane c) and therefore was not used further.
Figure 6.

Comparison of the ribosomal density at the ends of individual mRNAs. Polysomal mRNA was isolated from yeast cells and RNase H-cleaved in the presence of antisense ODNs complementary to positions at the 3′ end of the first third [(i) in each panel] and the second third [(ii) in each panel] of the coding region. Schematic representations of the coding region and the expected sizes of the cleavage products are shown for each mRNA. Samples were separated on sucrose gradients and, in all cases, 18 fractions were collected. The fractions indicated in each panel were subjected to northern analysis and hybridized with radiolabeled probes recognizing ADH1 (A), HSP82 (B), YEF3 (C) and PDC1 (D). Labeling is as in Figure 4. The graphs in (i) and (ii) of each panel show the phosphorimager quantification results of the 5′-cleavage product (blue) and the 3′-cleavage product (red), expressed as percent of total signal in the probed fractions. The graph in (iii) presents the signals of the short fragments from (i) (blue) and (ii) (red) with the overlap shown in purple.
To inhibit possible degradation of the cleavage products, an RNase inhibitor was added immediately upon collection of the fractions (Scheme 1, step ii). In no case was there a significant loss in hybridization signal (relative to uncut controls), or were multiple shorter fragments or substantial smears observed (Figures 3–6, and data not shown), as would be expected if the RNAs were subject to non-specific degradation. Thus, site-specific cleavage occurred with minimal non-specific degradation.
We next tested whether ribosomes dissociate from the mRNA during the procedure. The absorbance profiles (OD254) of sedimentation gradients after the RNase H reaction were compared with the corresponding profiles before the cleavage reaction (Figure 3B). mRNAs associated with 3–5 ribosomes were collected from a sucrose gradient, incubated for 1 h at 37°C without addition of antisense ODN, RNase H and LMD buffer, and resolved on a second sucrose gradient. The absorbance profile following this treatment (Figure 3B, middle panel) includes a single peak at the same position as in the first gradient. (Single ribosome resolution is not observed in the absorbance profile following the mock reaction. This may be due to the relatively high amount of sucrose in the sample leading to mixing during loading of the sample onto the gradient.) No absorbance peaks were observed at the positions corresponding to the size of ribosomal subunits or single ribosomes, as would be expected if ribosomes dissociated from the mRNA during the extended incubation at high temperature. We also collected mRNAs associated with ribosomes and subjected these samples to RNase H reaction using specific antisense ODNs. The lower panel of Figure 3B shows an example in which mRNA associated with more ribosomes (5–10 ribosomes) was collected and treated with RNase H and an antisense ODN complementary to ADH1 at position 462. The reaction products were then separated on a second sucrose gradient, and 18 fractions were collected across the gradient. The OD254 trace of the RNase H-treated sample showed one prominent peak at the expected position and no peaks at the sedimentation positions of 40S, 60S or 80S complexes. The ribosomes associated with the cleaved ADH1 fragments comprise a negligible fraction of the total polysomal pool so that no additional peak is detected by absorbance at 254 nm at their sedimentation position. The peak in the slowest migrating region, which was not observed in mock reactions (Figure 3B, middle panel), presumably arises from the added heparin, ODNs and enzyme. The absence of 40S, 60S or monosome complexes and the prominent polysomal peak at the same position as before the RNase H reaction demonstrate that there was no extensive ribosomal dissociation or association during the enzymatic reaction and subsequent separation.
The data also exclude the more drastic model in which the ribosomes dissociate during ODN annealing and RNase H treatment and then rebind randomly to the mRNA. This would lead to a substantial spreading of the OD254 in the subsequent sucrose gradient, which was not observed. Further, the ability to see asymmetric and highly specific ribosome positioning with GCN4 argues strongly against rearrangement during processing (following section and Figure 4).
Translational control of GCN4 expression via uORFs
Relative to the wealth of information available about regulation at the level of transcription, few examples of translational control have been identified, and much less is known about the underlying mechanisms. Perhaps the best-studied model for translation regulation in eukaryotes is GCN4, which has been the target of extensive genetic and biochemical studies by Hinnebusch and others (27–30). This work has led to a detailed model for translational control of GCN4 expression, involving upstream open reading frames (uORFs). Below, we briefly describe this model and tests, via RDM, of specific predictions arising from it.
GCN4 mRNA contains four short uORFs in its 5′-leader and a coding region of 281 amino acids (Figure 4A). According to the model for translational regulation of GCN4, when cells are growing in rich media small ribosomal subunits bind to the 5′ end of the mRNA, scan the 5′-leader and initiate translation at the first encountered ORF, uORF1 (Figure 4A). Following translation of uORF1, about half of the ribosomes dissociate while the other half remain bound and resume scanning, reinitiating at one of the next uORFs. These ribosomes have a high probability of dissociating after translating these uORFs, so that a negligible fraction of ribosomes reach the coding region and there is virtually no synthesis of GCN4 protein. When cells experience starvation conditions, some of the ribosomes that resume scanning after translating uORF1 skip the subsequent uORFs and reinitiate at the coding region. Reinitiation at the coding ORF is thought to be enhanced due to decrease in reinitiation and subsequent dissociation at uORFs 2, 3 and 4, with the decreased reinitiation at these uORFs resulting ultimately from phosphorylation of translation factor eIF2α (29–31). It is therefore predicted that ribosomes will be associated only with the 5′-leader when translation is repressed, and with the 5′-leader and the GCN4 coding ORF when translation is induced. As described below, we tested these and other predictions of this model.
When yeast cells are growing in rich media, the majority of polysomal GCN4 mRNA is associated with a single ribosome (∼60%) and there are almost no mRNAs with three or more ribosomes (6,7). We therefore first determined the position of the single ribosome that is associated with GCN4 mRNA under growth in rich media; the scanning model predicts a near-exclusive association with the uORFs. We isolated the fraction of mRNAs associated with a single ribosome by velocity sedimentation in a sucrose gradient, which, as expected, which, as expected, contained the majority of GCN4 mRNA (data not shown). The GCN4 mRNA was cleaved immediately upstream of the GCN4 ORF using an antisense ODN (ODN, −36) (Figure 4A). Cleavage products were then separated on a second sucrose gradient, 18 fractions were collected across the gradient, and the indicated fractions were analyzed by northern analysis (Figure 4B). Two bands are observed, corresponding in length to the two cleavage products. The cleavage product that corresponds to the GCN4 ORF sediments mainly free of assembled ribosomes (fractions 1–6), whereas the signal for 5′-leader fragment is almost exclusively ribosome-associated (fractions 7–9): <10% of GCN4 ORF fragments sediment with a ribosome, while >90% of the 5′-leader fragments are ribosome-associated. This result was confirmed using a different antisense ODN complementary to a nearby sequence (ODN, −6, data not shown); thus, the ribosome associated with GCN4 mRNA under growth in rich media is bound to the 5′-leader and not to the coding region of the mRNA on nearly all GCN4 mRNAs.
We next tested the ribosomal position under translationally derepressed conditions. Under these growth conditions, there is a sharp increase in the number of ribosomes associated with GCN4 mRNA; >70% of GCN4 mRNAs are associated with 3–7 ribosomes, and GCN4 mRNA with a single ribosome were barely detectable (data not shown).The scanning and reinitiation model predict that there should be ribosome occupancy of both the GCN4 ORF and the region containing the uORFs. The fractions of the sucrose gradient corresponding to 3–7 ribosomes were pooled, GCN4 mRNA was cleaved between the 5′-leader and the GCN4 ORF with the −36 antisense ODN, and the ribosomal association of the cleavage products was determined as before (Figure 4C). RDM analysis reveals that under starvation conditions, >80% of the coding fragments and >90% of the 5′-leader fragments are associated with ribosomes (fraction 7 and higher), and, moreover, that individual mRNA molecules have ribosomes associated with both the 5′-leader and the coding region.
The scanning and reinitiation model predicts ribosome association with uORF1 and also with uORFs 3 and 4 (22,32). Thus, ribosomes are expected on multiple uORFs along the GCN4 mRNA under repressed and derepressed growth conditions. To test this prediction, we isolated polysomes from cells grown under derepressed conditions and cleaved the GCN4 mRNA at two sites in the 5′-leader simultaneously (positions −214 and −36). Derepressed conditions were used because under these conditions, there are higher levels of GCN4 mRNA, thereby allowing the detection of the multiple fragments obtained from the double cleavage. This multiple cleavage scheme yields multiple fragments from the same mRNA, each sedimenting according to its ribosomal association, and thus allows finer mapping of the ribosomes on the 5′-leader. Almost all fragments that include uORFs 1 and 2, and a substantial fraction of the fragments that include uORFs 3 and 4 sediment as associated with ribosomes (data not shown). These data indicate that ribosomes are associated with uORFs 1 or 2 and uORFs 3 or 4 for a substantial fraction of GCN4 mRNAs. The association with multiple uORFs on the same mRNA is consistent with the scanning and reinitiation model. We attempted to test the prediction that the association of ribosomes with uORFs 3 and 4 is higher in cells grown under repressed conditions. Although we were able to determine that multiple ribosomes are associated with the uORFs region, however, we could not unequivocally determine whether the association at uORFs 3 and 4 is increased relative to derepressed conditions due to lower GCN4 mRNA levels under repressed growth conditions (data not shown).
In summary, our results demonstrate that RDM can efficiently detect changes in ribosomal association along a particular mRNA, and they confirm and extend several specific predictions from the scanning and reinitiation model developed from genetic studies and supported by prior in vitro translational toe-printing assays (27).
Exploring the stages of translation of yeast mRNAs by RDM
We used RDM to probe aspects of translation initiation, elongation and termination for yeast mRNAs with varying ORF lengths: ADH1 (1047 nt), PDA1 (1332 nt), PDC1 (1692 nt), HSP82 (2130 nt), EF-2 (2529 nt) and YEF3 (3135 nt). As described above (Scheme 1), polysomes corresponding to the predominant species for each mRNA were isolated from yeast cells through a sucrose gradient, and each target mRNA was cleaved independently at a single position within its coding region. Cleavage products were then separated on a second sucrose gradient, 18 fractions were collected across the gradient, and the indicated fractions were analyzed by northern blot (Figures 5 and 6).
Figure 5.

Comparison of the ribosomal density on two halves of individual mRNAs. Polysomal mRNA was isolated from yeast cells and cleaved with ODNs complementary to the positions near the center of each ORF (see text). Schematic representation of the coding region and the expected sizes of the cleavage products are shown for each mRNA (assuming cleavage in the center of the ODN:mRNA duplex). Samples were separated on sucrose gradients and, in all cases, 18 fractions were collected. The fractions indicated in each panel were subjected to northern analysis and hybridized with radiolabeled probes recognizing ADH1 (A), HSP82 (B), YEF3 (C), PDA1 (D) and EF-2 (E). On each northern blot, the open arrowheads indicate the full-length mRNA and the closed arrowheads indicate the two cleavage products. The assignment of the cleavage products as a 5′- or a 3′-fragment was based on the expected migration distance for an mRNA species corresponding to the length of the coding region and the untranslated regions (40). Labeling of the northern blots is as in Figure 4. The number of ribosomes associated with the full-length mRNA corresponds to the number of ribosomes in the fraction pooled from the first sucrose gradient for RNase H treatment. The graph in each panel shows the Phosphorimager quantifications of the 5′-cleavage product (blue) and the 3′-cleavage product (red), expressed as percent of total signal in the probed fractions. The overlap in the signal is shown in purple.
For each mRNA probed, three bands were observed in the northern blots, corresponding to the two cleavage products and a small amount of uncut mRNA. The full-length mRNA sedimented faster in the sucrose gradient than the cleavage products in all cases, as is expected for a species with more bound ribosomes, and this species serves as a useful control and marker. In all cases, the uncut mRNA migrated at the position expected based on determination of the number of bound ribosomes from northern and microarray analysis of the initial gradient [(7), and data not shown] and the number of ribosomes bound to the cleaved fragments summed, within error, to that for the uncut fragment.
Initially, the cleavage position was chosen close to the center of the ORF for each mRNA to provide the most sensitive readout for any 5′ or 3′ bias in ribosome density, i.e. a uniform density would give equivalent sedimentation of the 5′- and 3′-fragments, whereas a density difference would give different migration, as depicted in part v of Scheme 1. For each mRNA probed, cleavage near the center gave two products with sedimentation profiles that were identical within the limit of the resolution of the procedure (Figure 5). This can be qualitatively seen directly from the similar northern blot patterns and quantitatively from the near-perfect overlap of the graphical profiles of the band intensities. (The overlap is shown in purple in the panels above the northern blots.)
To further probe the distribution of ribosomes along the mRNA and to provide more stringent tests of models such as those in Figure 2, we performed mapping studies in which ADH1, PDC1, HSP82 and YEF3 were each cleaved at positions approximately one-third or two-thirds of the way from the 5′ end of the ORF (Figure 6). Three bands were again observed in each northern blot, corresponding to the two cleavage products and the uncut mRNA. As before, the full-length mRNA sedimented faster than the cleavage products in all cases. In this case, however, the two cleavage products, which differ substantially in their sizes, sedimented differently: in each case, the long cleavage product sedimented faster than the short fragment. This is most easily seen by comparing the quantitated profile for each fragment (red and blue areas in the graphs above each northern blot).
The pairs of cleavages for each mRNA yield products of similar lengths, but corresponding to opposite ends of the mRNA. They therefore allow accurate comparison of the ribosome density closer to the ends of ORFs. Comparison of the sedimentation for the two short fragments from the ends of the ORF (Figure 6, section iii in each panel) revealed that ribosome densities on the 5′- and 3′-thirds of each mRNAs were the same, within error. The implications of these observations for processivity and termination are discussed in the following sections.
Elongation processivity
It has been suggested, based on analyses in bacteria, that processivity of translation elongation is incomplete, with many ribosomes that initiate translation not completing translation of the entire ORF (33,34). Analogous premature dissociations in the mammalian cells have been suggested to produce aberrant proteins that serve as autoimmune antigens (35). Ribosome dissociation could occur at particular positions along the mRNA, due to a structural element or sequence motif. For example, certain sequences are known to induce ribosomal frameshifting with remarkable efficiency (36), and frameshifting followed by recognition of stop codon in the new coding frame would presumably result in ribosome dissociation. Ribosome dissociation could also occur uniformly along the mRNA, due to an inherent incomplete processivity of elongation. Either case would lead to lower density of ribosomes at the 3′ end of the mRNA compared to the 5′ end. For a given level of processivity, the ratio of the density of ribosomes at the 3′ and 5′ ends of an mRNA is expected to be an exponential function of the ORF length; longer ORFs are expected to have more dissociation events and therefore larger differences in the ribosome density at the 3′ and 5′ ends. Hence, comparing the number of ribosomes on both ends of long mRNAs provides the most sensitive probe for ribosomal dissociation.
Figure 6C (iii) presents the sedimentation profiles of fragments from both ends of YEF3, one of the longest yeast ORFs (3135 nt, longer than 92% of yeast ORFs). The sedimentation profiles of the fragments from both ends of the ORF are the same within error, indicating that a similar number of ribosomes are associated with the 5′ and 3′ ends of YEF3. Applying a model with a constant probability of dissociation for each elongation step, we can identify processivity levels that are consistent and inconsistent with these sedimentation profiles (see Methods Supplement and Supplemental Figure 1). The two fragments are expected to have similar sedimentation profiles only if processivity levels exceed 99.99% for YEF3 (Supplemental Figure 1A). Analogous analyses for the other yeast mRNAs also gave no indication of significant premature termination, although the lower limits were lower than that for YEF3 due to the shorter ORF length (lower limits of 99.8–99.99%, Supplemental Figure 1). These values suggest that translation elongation in yeast is highly processive for many or most yeast mRNAs. Nevertheless, more complex models, such as coincidentally slower elongation downstream of a structural element that promotes ribosome dissociation or incomplete processivity along with slow termination, remain possible. It will be of interest to further test this conclusion and to determine whether there are mutations or conditions associated with significantly lower processivity.
Termination rates
Translation termination, unlike elongation, requires release of the polypeptide chain and distinct set of trans-acting factors (37). Thus, the rate of termination could differ from that for elongation. If termination were slower than the average rate for elongation steps, the ribosome density at the 3′ end of an ORF would be greater than that for the rest of the ORF; the slower the termination step, the larger the expected increase in ribosome density at the 3′ end.
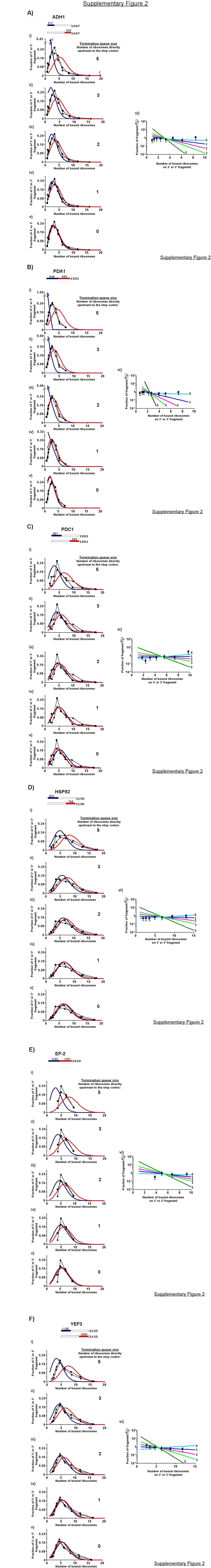
The most sensitive probe for an increase in ribosomal density upstream of the stop codon is a relatively short fragment from the 3′ end of the ORF, as its sedimentation will be affected the most by any queuing of ribosomes upstream to the stop codon. We therefore first examined the sedimentation profiles of the shortest fragment analyzed, the 335 nt fragment derived from cleavage of ADH1 at position 712 (Figure 6A). Comparing its sedimentation profile with the profile of a fragment of similar length (312 nt) from the 5′ end of the mRNA [Figure 6A (iii)] revealed similar sedimentation profiles, suggesting that termination is not sufficiently slow to give a build-up of ribosomes at the 3′-terminus. We simulated the sedimentation profiles of these fragments for situations with slow termination leading to queuing of one, two or more ribosomes upstream to the stop codon, and compared the simulated profiles to the experimental profiles (Supplemental Figure 2A). The experimental profile best fits a queue of less than one ribosome upstream to the stop codon [Supplemental Figure 2A (vi)]. Similar simulations for the rest of the mRNAs (Supplemental Figure 2B–F) reveal that the experimental profiles in all cases are consistent with an absence of a termination queue, although in certain instances a queue of one ribosome cannot be excluded (e.g., Supplemental Figure 2F). Thus, there is no indication of slower termination, and if ribosomes do pause at the termination site, they rarely fail to dissociate before the next ribosome reaches this site.
Assessing the basis for the inverse correlation between mRNA length and ribosomal density
The RDM experiments and results described above allow us to distinguish between models we previously proposed to account for unexpected results from a global analysis of translation (7). We observed that longer ORFs have lower ribosome density and proposed three mechanistic models to explain this inverse correlation: (i) Processivity is incomplete, because longer ORFs have more elongation steps and thus more opportunities for ribosome dissociation, their overall ribosome density would be lower. (ii) Slow termination rates relative to elongation leading to accumulation of ribosomes at the termination site. The additional ribosomes present at the 3′ end of mRNA of all lengths would increase the overall ribosome density of mRNAs with short ORF relative to these with long ORFs. (iii) Initiation is slower for longer mRNAs whereas elongation and termination rates are not correlated with length. This would lead to a general trend of larger spacing between ribosomes on longer mRNAs.
A different distribution of ribosomes along an mRNA is predicted for each of the above models. Models 1 and 2 both predict uneven distributions: a higher density of ribosomes at the 5′ end for model 1 (incomplete processivity) or a higher density at the 3′ end for model 2 (slow termination). Model 3 predicts an even distribution of ribosomes (see Supplemental Methods).
We fit each of these models to the observed inverse correlation between ribosome density and coding sequence length to provide quantitative constraints that could then be compared with the results from RDM (Figure 7). Model 1 gives a best fit to the correlation with a processivity of 99.3% per elongation step (Figure 7A). This is below the lower limit of 99.8% obtained for any mRNA from RDM described above and far below the limit of 99.99% for YEF3 (Supplemental Figure 1). Model 2 is also eliminated by the RDM data. Although accumulation of three ribosomes could account for the observed correlation in the slow termination model (Figure 7B, solid line), the RDM data suggests instead an accumulation of at most one ribosome, as described above (Supplemental Figure 2). For model 3, no specific mathematical form is necessitated; Figure 7C shows a simple exponential decrease in initiation rate with ORF length to demonstrate the magnitude of the trend required to account for the observed effect: the best fit corresponds to 2-fold decrease in initiation rate with each 1200 nt increase in ORF length (Figure 7C). The observed uniform distribution of ribosomes across the mRNA is predicted by this model, although the data do not provide direct information about variation in initiation rates with length. The origin of the apparently slower initiation of translation for longer mRNAs is not clear. It could arise as a result of the more distal polyA tail from the cap in longer mRNAs, as the polyA tail enhances translation initiation through interaction with initiation complex (38). Alternatively, or in addition, stable structures can inhibit translation initiation (39), and longer mRNAs might be more prone to self-structure than short mRNAs.
Figure 7.

Comparison of experimentally measured ribosomal densities to predictions from incomplete processivity, slow termination and slow initiation models. Ribosomal densities measured for 739 mRNAs by microarray analysis of polysomal fractions (7) are plotted versus ORF length (black circles). Each circle represents the average density for ORF lengths binned in 100 nt intervals (up to 4400 nt). Lines represent predicted density values by each model (the details of density calculations for each model are described in the Methods Supplement). (A) Incomplete processivity (model 1). Solid line represents the best fit to the experimentally measured densities with a processivity level of 99.30% per elongation step. Predicted values for higher processivity levels, corresponding to limits obtained from RDM of 99.8% processivity (short-dashes) and 99.99% processivity (long-dashes) are shown for comparison. (B) Slow termination (model 2). The solid line represents the best fit for the experimentally measured densities, which corresponding to a queue of three ribosomes at the 3′ end of the ORF. An example of the predicted density with queue of one ribosome is also shown (long dashes line). (C) Slow initiation (model 3). The line represents the best fit to the experimentally measured densities for a model in which initiation rates decrease exponentially with increase in mRNA length.
PERSPECTIVE
We have developed an efficient and general method to map ribosomal densities along individual mRNAs. Mapping the distribution of ribosomes along several yeast mRNAs has revealed properties of basic steps of translation in vivo and allowed tests of the mechanisms of GCN4 translational control. RDM will allow investigation of the features, factors and conditions that affect ribosome binding, dissociation and movement along the mRNA. In addition to general properties, the behavior of specific mRNAs that undergo translational regulation via upstream ORFs, frameshifting and other means can be probed. It should now be possible to investigate whether the relative rates of the three stages of translation are different under alternative growth conditions and in various mutant strains. These additional data should facilitate development and evaluation of quantitative models for translation and its control.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Supplementary Material
Acknowledgments
We thank A. Hinnebusch, J. Lorsch, P. Sarnow and members of the Herschlag laboratory for comments on the manuscript. This work was supported by grants from the American Heart Association (to D.H.) and the Howard Hughes Medical Institute (HHMI). P.O.B. is an HHMI investigator and Y.A. was an HHMI research associate. Funding to pay the Open Access publication charges for this article was provided by HHMI.
Conflict of interest statement. None declared.
REFERENCES
- 1.Rickwood D. Preparative Centrifugation. A Practical Approach, 2nd edn. Oxford, New York: 1992. [Google Scholar]
- 2.Warner J.R., Knopf P.M., Rich A. A multiple ribosomal structure in protein synthesis. Proc. Natl Acad. Sci. USA. 1963;49:122–129. doi: 10.1073/pnas.49.1.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ashe M.P., De Long S.K., Sachs A.B. Glucose depletion rapidly inhibits translation initiation in yeast. Mol. Biol. Cell. 2000;11:833–848. doi: 10.1091/mbc.11.3.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dickson L.M., Brown A.J. mRNA translation in yeast during entry into stationary phase. Mol. Gen. Genet. 1998;259:282–293. doi: 10.1007/s004380050814. [DOI] [PubMed] [Google Scholar]
- 5.Proweller A., Butler J.S. Ribosomal association of poly(A)-binding protein in poly(A)-deficient Saccharomyces cerevisiae. J. Biol. Chem. 1996;271:10859–10865. doi: 10.1074/jbc.271.18.10859. [DOI] [PubMed] [Google Scholar]
- 6.Tzamarias D., Roussou I., Thireos G. Coupling of GCN4 mRNA translational activation with decreased rates of polypeptide chain initiation. Cell. 1989;57:947–954. doi: 10.1016/0092-8674(89)90333-4. [DOI] [PubMed] [Google Scholar]
- 7.Arava Y., Wang Y., Storey J.D., Liu C.L., Brown P.O., Herschlag D. Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA. 2003;100:3889–3894. doi: 10.1073/pnas.0635171100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zong Q., Schummer M., Hood L., Morris D.R. Messenger RNA translation state: the second dimension of high-throughput expression screening. Proc. Natl Acad. Sci. USA. 1999;96:10632–10636. doi: 10.1073/pnas.96.19.10632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mikulits W., Pradet-Balade B., Habermann B., Beug H., Garcia-Sanz J.A., Mullner E.W. Isolation of translationally controlled mRNAs by differential screening. FASEB J. 2000;14:1641–1652. doi: 10.1096/fj.14.11.1641. [DOI] [PubMed] [Google Scholar]
- 10.Brown V., Jin P., Ceman S., Darnell J.C., O'Donnell W.T., Tenenbaum S.A., Jin X., Feng Y., Wilkinson K.D., Keene J.D., et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107:477–487. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- 11.Kuhn K.M., DeRisi J.L., Brown P.O., Sarnow P. Global and specific translational regulation in the genomic response of Saccharomyces cerevisiae to a rapid transfer from a fermentable to a nonfermentable carbon source. Mol. Cell. Biol. 2001;21:916–927. doi: 10.1128/MCB.21.3.916-927.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johannes G., Carter M.S., Eisen M.B., Brown P.O., Sarnow P. Identification of eukaryotic mRNAs that are translated at reduced cap binding complex eIF4F concentrations using a cDNA microarray. Proc. Natl Acad. Sci. USA. 1999;96:13118–13123. doi: 10.1073/pnas.96.23.13118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rajasekhar V.K., Viale A., Socci N.D., Wiedmann M., Hu X., Holland E.C. Oncogenic Ras and Akt signaling contribute to glioblastoma formation by differential recruitment of existing mRNAs to polysomes. Mol. Cell. 2003;12:889–901. doi: 10.1016/s1097-2765(03)00395-2. [DOI] [PubMed] [Google Scholar]
- 14.MacKay V.L., Li X., Flory M.R., Turcott E., Law G.L., Serikawa K.A., Xu X.L., Lee H., Goodlett D.R., Aebersold R., et al. Gene expression analyzed by high-resolution state array analysis and quantitative proteomics: response of yeast to mating pheromone. Mol. Cell Proteomics. 2004;3:478–489. doi: 10.1074/mcp.M300129-MCP200. [DOI] [PubMed] [Google Scholar]
- 15.Ramakrishnan V. Ribosome structure and the mechanism of translation. Cell. 2002;108:557–572. doi: 10.1016/s0092-8674(02)00619-0. [DOI] [PubMed] [Google Scholar]
- 16.Kapp L.D., Lorsch J.R. The molecular mechanics of eukaryotic translation. Annu. Rev. Biochem. 2004;73:657–704. doi: 10.1146/annurev.biochem.73.030403.080419. [DOI] [PubMed] [Google Scholar]
- 17.Hartz D., McPheeters D.S., Traut R., Gold L. Extension inhibition analysis of translation initiation complexes. Methods Enzymol. 1988;164:419–425. doi: 10.1016/s0076-6879(88)64058-4. [DOI] [PubMed] [Google Scholar]
- 18.Sachs M.S., Wang Z., Gaba A., Fang P., Belk J., Ganesan R., Amrani N., Jacobson A. Toeprint analysis of the positioning of translation apparatus components at initiation and termination codons of fungal mRNAs. Methods. 2002;26:105–114. doi: 10.1016/S1046-2023(02)00013-0. [DOI] [PubMed] [Google Scholar]
- 19.Wolin S.L., Walter P. Ribosome pausing and stacking during translation of a eukaryotic mRNA. EMBO J. 1988;7:3559–3569. doi: 10.1002/j.1460-2075.1988.tb03233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolin S.L., Walter P. Signal recognition particle mediates a transient elongation arrest of preprolactin in reticulocyte lysate. J. Cell Biol. 1989;109:2617–2622. doi: 10.1083/jcb.109.6.2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mueller P.P., Hinnebusch A.G. Multiple upstream AUG codons mediate translational control of GCN4. Cell. 1986;45:201–207. doi: 10.1016/0092-8674(86)90384-3. [DOI] [PubMed] [Google Scholar]
- 22.Abastado J.P., Miller P.F., Jackson B.M., Hinnebusch A.G. Suppression of ribosomal reinitiation at upstream open reading frames in amino acid-starved cells forms the basis for GCN4 translational control. Mol. Cell. Biol. 1991;11:486–496. doi: 10.1128/mcb.11.1.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Werner M., Feller A., Messenguy F., Pierard A. The leader peptide of yeast gene CPA1 is essential for the translational repression of its expression. Cell. 1987;49:805–813. doi: 10.1016/0092-8674(87)90618-0. [DOI] [PubMed] [Google Scholar]
- 24.Sagliocco F.A., Vega Laso M.R., Zhu D., Tuite M.F., McCarthy J.E., Brown A.J. The influence of 5′-secondary structures upon ribosome binding to mRNA during translation in yeast. J. Biol. Chem. 1993;268:26522–26530. [PubMed] [Google Scholar]
- 25.Yaman I., Fernandez J., Liu H., Caprara M., Komar A.A., Koromilas A.E., Zhou L., Snider M.D., Scheuner D., Kaufman R.J., et al. The zipper model of translational control: a small upstream ORF is the switch that controls structural remodeling of an mRNA leader. Cell. 2003;113:519–531. doi: 10.1016/s0092-8674(03)00345-3. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y., Liu C.L., Storey J.D., Tibshirani R.J., Herschlag D., Brown P.O. Precision and functional specificity in mRNA decay. Proc. Natl Acad. Sci. USA. 2002;99:5860–5865. doi: 10.1073/pnas.092538799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gaba A., Wang Z., Krishnamoorthy T., Hinnebusch A.G., Sachs M.S. Physical evidence for distinct mechanisms of translational control by upstream open reading frames. EMBO J. 2001;20:6453–6463. doi: 10.1093/emboj/20.22.6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Albrecht G., Mosch H.U., Hoffmann B., Reusser U., Braus G.H. Monitoring the Gcn4 protein-mediated response in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 1998;273:12696–12702. doi: 10.1074/jbc.273.21.12696. [DOI] [PubMed] [Google Scholar]
- 29.Hinnebusch A.G. Translational regulation of yeast GCN4. A window on factors that control initiator-tRNA binding to the ribosome. J. Biol. Chem. 1997;272:21661–21664. doi: 10.1074/jbc.272.35.21661. [DOI] [PubMed] [Google Scholar]
- 30.Dever T.E. Gene-specific regulation by general translation factors. Cell. 2002;108:545–556. doi: 10.1016/s0092-8674(02)00642-6. [DOI] [PubMed] [Google Scholar]
- 31.Geballe A.P., Sachs M.S. Translational control by upstream open reading frame. In: Sonenberg N., Hershey J.W.B., Mathews M.B., editors. Translational Control of Gene Expression. Cold Spring Harbor: CSHL Press; 2000. [Google Scholar]
- 32.Abastado J.P., Miller P.F., Hinnebusch A.G. A quantitative model for translational control of the GCN4 gene of Saccharomyces cerevisiae. New Biol. 1991;3:511–524. [PubMed] [Google Scholar]
- 33.Jorgensen F., Kurland C.G. Processivity errors of gene expression in Escherichia coli. J. Mol. Biol. 1990;215:511–521. doi: 10.1016/S0022-2836(05)80164-0. [DOI] [PubMed] [Google Scholar]
- 34.Tsung K., Inouye S., Inouye M. Factors affecting the efficiency of protein synthesis in Escherichia coli. Production of a polypeptide of more than 6000 amino acid residues. J. Biol. Chem. 1989;264:4428–4433. [PubMed] [Google Scholar]
- 35.Yewdell J.W., Anton L.C., Bennink J.R. Defective ribosomal products (DRiPs): a major source of antigenic peptides for MHC class I molecules? J. Immunol. 1996;157:1823–1826. [PubMed] [Google Scholar]
- 36.Farabaugh P.J., Qian Q., Stahl G. Programmed translational frameshifting, hopping and readthrough of termination codons. In: Sonenberg N., Hershey J.W.B., Mathews M.B., editors. Translational Control of Gene Expression. Cold Spring Harbor: CSHL Press; 2000. [Google Scholar]
- 37.Kisselev L., Ehrenberg M., Frolova L. Termination of translation: interplay of mRNA, rRNAs and release factors? EMBO J. 2003;22:175–182. doi: 10.1093/emboj/cdg017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sachs A.B. Physical and functional interactions between the mRNA cap structure and the polyA tail. In: Sonenberg N., Hershey J.W.B., Mathews M.B., editors. Translational Control of Gene Expression. Cold Spring Harbor: CSHL Press; 2000. [Google Scholar]
- 39.McCarthy J.E.G. Posttranscriptional control of gene expression in yeast. Microbiol. Mol. Biol. Rev. 1998;62:1492–1553. doi: 10.1128/mmbr.62.4.1492-1553.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hurowitz E.H., Brown P.O. Genome-wide analysis of mRNA lengths in Saccharomyces cerevisiae. Genome Biol. 2003;5:R2. doi: 10.1186/gb-2003-5-1-r2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}