

ABSTRACT
Natural killer (NK) cells emerged as a promising effector population that can be harnessed for anti-tumor therapy. In this work, we constructed NK cell engagers (NKCEs) based on NKp30-targeting single domain antibodies (sdAbs) that redirect the cytotoxic potential of NK cells toward epidermal growth factor receptor (EGFR)-expressing tumor cells. We investigated the impact of crucial parameters such as sdAb location, binding valencies, the targeted epitope on NKp30, and the overall antibody architecture on the redirection capacity. Our study exploited two NKp30-specific sdAbs, one of which binds a similar epitope on NKp30 as its natural ligand B7-H6, while the other sdAb addresses a non-competing epitope. For EGFR-positive tumor targeting, humanized antigen-binding domains of therapeutic antibody cetuximab were used. We demonstrate that NKCEs bivalently targeting EGFR and bivalently engaging NKp30 are superior to monovalent NKCEs in promoting NK cell-mediated tumor cell lysis and that the architecture of the NKCE can substantially influence killing capacities depending on the NKp30-targeting sdAb utilized. While having a pronounced impact on NK cell killing efficacy, the capabilities of triggering antibody-dependent cellular phagocytosis or complement-dependent cytotoxicity were not significantly affected comparing the bivalent IgG-like NKCEs with cetuximab. However, the fusion of sdAbs can have a slight impact on the NK cell release of immunomodulatory cytokines, as well as on the pharmacokinetic profile of the NKCE due to unfavorable spatial orientation within the molecule architecture. Ultimately, our findings reveal novel insights for the engineering of potent NKCEs triggering the NKp30 axis.
KEYWORDS: ADCC, antibody engineering, bispecific antibody, EGFR, NK cell engager, NKp30, paratope, protein engineering, single domain antibody, valency, VHH
Introduction
Natural killer (NK) cells represent an important part of the first line of defense of the innate immune system and can be recruited to the tumor site through pro-inflammatory chemokines produced by other immune cells.1 NK cells have the ability to detect and destroy malignant or infected cells by integrating positive and negative signals.2 Positive signals are usually provided by a set of germline-encoded activating receptors that typically sense stress-induced ligands on other cells.3–6 Negative signals are mediated by self-major histocompatibility complex (MHC) class I ligands with either killer cell immunoglobulin-like receptor (KIR) family members or NKG2A.7
In recent years, NK cell-mediated immunotherapy emerged as a promising approach for cancer therapy.1 Consequently, several different molecules are currently being tested with different modes of action in clinical trials.1,8 Related to this, the redirection of NK cells to the tumor site in conjunction with the conditional activation of the cytotoxic and immunomodulatory potential evolved as one very promising therapeutic approach. In this context, a strategy for the conditional activation of NK cells relies in the engagement of the low-affinity Fc γ receptor IIIa (FcγRIIIa/CD16a) with target cells opsonized by IgG antibodies via the Fc region. This mechanism, referred to as antibody-dependent cell-mediated cytotoxicity (ADCC), is considered to be an important mechanism of action of numerous therapeutic antibodies.9–13 Notwithstanding, several conditions impede the capacity of an antibody to elicit ADCC for instance, FcγRIIIa polymorphism, shedding or internalization of FcγRIIIa from the NK cell surface, and competition of the therapeutic antibody with serum IgG.14–17 Consequently, alternative approaches for NK cell redirection were developed, in which activating receptors on the NK cell are harnessed for conditional activation, such as NKp30, NKp46 or NKG2D.18–23
We have previously described the generation of a panel of bispecific NKp30 × EGFR NK cell engagers (NKCEs) harboring NKp30-specific single domain antibodies (sdAbs)/VHHs targeting different epitopes on the extracellular domain (ECD) of the NKp30 receptor.18 For tumor targeting, we exploited the Fab fragment of a humanized version of cetuximab. We were able to demonstrate that potencies of constructed bispecifics in triggering NK cell-mediated lysis of high EGFR-expressing tumor cells were strongly dependent on the targeted epitope on NKp30. VHHs targeting a similar epitope as the natural ligand of NKp30 displayed higher cytotoxic capacities when reformatted as NKCE than sdAb-derived paratopes addressing different epitopes on NKp30. Likewise, it was shown for T cell-engaging antibodies that the cytotoxic potential relies on the targeted epitope of the antigen on the tumor cell. Consequently, depending on the addressed epitope, different architectures are favorable in terms of T cell redirection.24–26 Besides the targeted epitope on both, the receptor on the effector cell and the tumor-associated antigen (TAA) as well as the spatial orientation of the individual paratopes within the molecule, it is well described that other attributes, such as inherent affinities and valencies, are also important factors that need to be fine-tuned for the generation of potent effector cell engagers displaying an adequate safety profile.27–29
In this study, we systematically investigated how antibody architecture impacts NK cell engagement of EGFR-targeting NKCEs based on two different NKp30-targeting sdAbs. To this end, we exploited a VHH (VHH1) sharing a similar epitope on NKp30 with B7-H6, the natural ligand of this activating receptor, and a second VHH (VHH2) addressing a non-competing epitope on NKp30.18 We demonstrate here that, depending on the incorporated VHH as well as on EGFR densities on tumor cells, bivalent triggering of NKp30 enhanced the killing capacities of the NKCEs. Additionally, bivalent targeting of EGFR was superior in mediating NK cell lysis of EGFR-expressing tumor cells compared with monovalent TAA binding. Consequently, bivalent triggering of both, EGFR and NKp30, enabled the most robust conditional activation of NK cells, which could be further enhanced by additional FcγRIIIa binding molecules. Intriguingly, depending on the targeted epitope on NKp30 and the spatial orientation of the incorporated VHHs within the molecules, huge differences were observed with regard to NK cell activation, tumor cell lysis and immunomodulatory cytokine release. Moreover, single dose pharmacokinetics (PK) studies in wildtype mice exhibited slightly reduced serum half-life and hence clearance of the symmetric NKCEs compared to cetuximab, although the behavior can still be considered as IgG-like. On the contrary, the efficacy of the cetuximab-like molecules in antibody-dependent cellular phagocytosis (ADCP) and complement-dependent cytotoxicity (CDC) was not affected by the VHH locations in the novel NKCEs.
Results
Biophysical and initial biochemical evaluation of bispecific NKp30 × EGFR engagers with different architectures and valencies resulted in multifaceted set of functional molecules
The molecule architecture and valency of antigen binding paratopes may significantly impact the cytotoxic capacity of antibody derivatives. Therefore, antibody design and valencies were varied for NKp30 and EGFR binding domains to identify favorable molecule parameters having a positive impact on killing efficacy of NKp30 × EGFR bispecific NKCEs. Moreover, to assess paratope specific attributes, two distinct NKp30 binding VHHs were used, of which VHH2 targets a different epitope than NKp30s natural ligand B7-H6, while VHH1 competes with B7-H6 for NKp30 binding.18 In order to generate asymmetric antibodies, we used the strand-exchanged engineered domain (SEED) heterodimerization technology, which facilitates heavy chain (HC) dimerization due to beta-strand exchanges of IgG and IgA isotypes in the respective HCs, referred to as AG and GA chain.30 Varying valencies of the scrutinized NKCEs included the comparison of monovalent (mv) and bivalent (bv) SEEDbodies as well as IgG-based molecules, either applied to the NKp30 binding domain (VHH), the EGFR binding domain (hu225 Fab) or both (Figure 1). Additionally, the impact of the NK cell-engaging VHH location was tested in the backbone of wildtype hu225_IgG (bv) with the VHH fused to the N- or C-terminus of the light chain (LC) or to the C-terminus of the HC, consequently resulting in two NKp30 binding domains (bv) (Figure 1).
Figure 1.
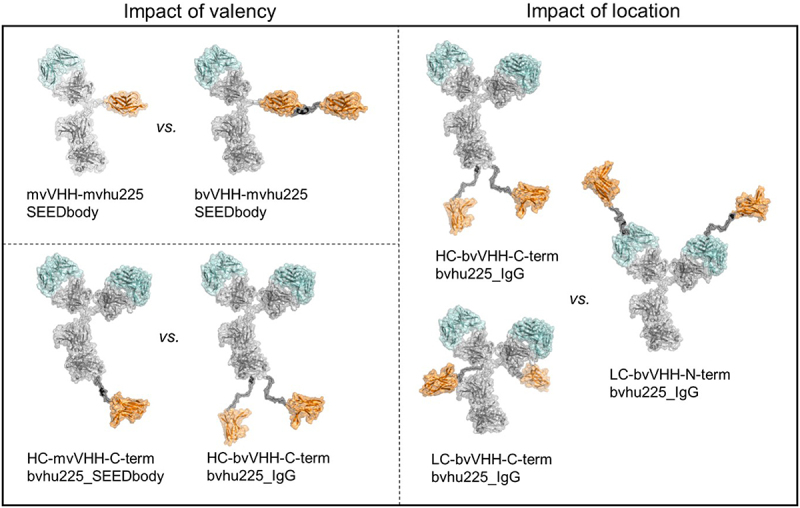
Antibody architecture of bispecific NKp30 × EGFR NKCEs. Structural models of the molecule formats tested in functional assays for their killing capacities of EGFR-expressing cell lines by engagement of human NK cells. NKp30 binding VHH sdAbs (orange) were either used to generate asymmetric bispecific SEEDbody or symmetric bispecific IgG-based NKCEs all harboring one or two humanized Fab entities of cetuximab (hu225, cyan). The impact of monovalent (mv) vs. bivalent (bv) engagement was investigated for NKp30 in combination with mv EGFR (upper left) as well as bv EGFR (lower left) targeting with the depicted VHH-mvhu225_SEEDbody and HC-VHH-C-term-bvhu225 molecules (left panel). To investigate the impact of VHH location in bivalent NKp30- and EGFR-engaging molecules, the IgG-based NKCEs in the right panel were compared. Molecule schemes were generated using PyMol software version 2.3.0.
All molecules possessing a modified IgG CH2 domain devoid of binding to FcγRs and complement protein C1q (referred to as eff-), as well as possessing a wildtype IgG CH2 region (referred to as eff+), were tested. Transient transfection of Expi293 cells resulted in a broad range of expression titers ranging from lower double (12.1 mg/L for bvVHH1-mvhu225_SEEDbody) to triple digit (296.8 mg/L for LC-bvVHH2-N-term-bvhu225_IgG) milligram-per-liter scale, identifying the N-terminal LC fusion of the VHHs as the most favorable format in regards of expression yields (Table 1). Additionally, the IgG CH2 domain, meaning either eff- or eff+ versions of otherwise identical molecules, did not impact the expressed protein amounts. Notably, however, the asymmetric NKCEs demonstrated overall reduced expression yields compared to the symmetric IgG-like molecules (Table 1). Furthermore, engrafting VHH1 also resulted in overall reduced production yields compared to VHH2, underlying the significance of attuning the utilized paratopes with the protein architecture to ensure sufficient producibility of NKCEs.
Table 1.
Biochemical and biophysical properties of bispecific NKCEs.
| Molecule | MW (kDa) | SEC monomeric fraction [%] | Yield [mg/L] | Target | KD [M] | Kon [1/Ms] | Koff [1/s] |
|---|---|---|---|---|---|---|---|
| mvVHH1-mvhu225_SEEDbody eff- | 112.2 | 91.2 | 106.9 | EGFR | 4.45E–09 | 4.20E + 05 | 1.87E–03 |
| NKp30 | 1.18E–10 | 1.07E + 06 | 1.27E–04 | ||||
| mvVHH1-mvhu225_SEEDbody eff+ | 112.5 | 92.3 | 75.9 | ||||
| bvVHH1-mvhu225_SEEDbody eff- | 126.7 | 88.6 | 32.2 | EGFR | 3.90E–09 | 4.66E + 05 | 1.82E–03 |
| NKp30 | 4.30E–10 | 5.34E + 05 | 2.29E–04 | ||||
| bvVHH1-mvhu225_SEEDbody eff+ | 126.9 | 75.5 | 12.1 | ||||
| HC-mvVHH1-C-term-bvhu225_SEEDbody eff- | 160.1 | 86.4 | 32.1 | EGFR | 3.88E–09 | 3.18E + 05 | 1.23E–03 |
| NKp30 | 3.23E–11 | 1.22E + 06 | 3.95E–05 | ||||
| HC-mvVHH1-C-term-bvhu225_SEEDbody eff+ | 160.4 | 85.5 | 33.5 | ||||
| HC-bvVHH1-C-term-bvhu225_IgG eff- | 173.9 | 93.4 | 19.0 | EGFR | 4.89E–09 | 3.20E + 05 | 1.57E–03 |
| NKp30 | 3.60E–10 | 5.08E + 05 | 1.83E–04 | ||||
| HC-bvVHH1-C-term-bvhu225_IgG eff+ | 174.1 | 92.5 | 21.0 | ||||
| LC-bvVHH1-C-term-bvhu225_IgG eff- | 173.9 | 89.2 | 73.5 | EGFR | 4.21E–09 | 3.05E + 05 | 1.28E–03 |
| NKp30 | 4.61E–10 | 5.20E + 05 | 2.40E–04 | ||||
| LC-bvVHH1-C-term-bvhu225_IgG eff+ | 174.1 | 88.6 | 70.6 | ||||
| LC-bvVHH1-N-term-bvhu225_IgG eff- | 173.9 | 94.0 | 205.0 | EGFR | 2.27E–09 | 2.68E + 05 | 6.09E–04 |
| NKp30 | 5.43E–10 | 5.57E + 05 | 3.03E–04 | ||||
| LC-bvVHH1-N-term-bvhu225_IgG eff+ | 174.1 | 92.2 | 205.8 | ||||
| mvVHH2-mvhu225_SEEDbody eff- | 112.7 | 91.7 | 94.8 | EGFR | 3.16E–09 | 4.90E + 05 | 1.55E–03 |
| NKp30 | 7.45E–10 | 2.63E + 05 | 1.96E–04 | ||||
| mvVHH2-mvhu225_SEEDbody eff+ | 112.5 | 86.3 | 92.8 | ||||
| bvVHH2-mvhu225_SEEDbody eff- | 127.2 | 88.8 | 82.6 | EGFR | 4.37E–09 | 4.88E + 05 | 2.13E–03 |
| NKp30 | 1.65E–09 | 2.13E + 05 | 3.50E–04 | ||||
| bvVHH2-mvhu225_SEEDbody eff+ | 127.4 | 82.4 | 108.4 | ||||
| HC-mvVHH2-C-term-bvhu225_SEEDbody eff- | 160.4 | 94.8 | 80.8 | EGFR | 4.08E–09 | 2.95E + 05 | 1.21E–03 |
| NKp30 | 2.42E–09 | 4.29E + 05 | 1.04E–03 | ||||
| HC-mvVHH2-C-term-bvhu225_SEEDbody eff+ | 160.6 | 95.1 | 87.1 | ||||
| HC-bvVHH2-C-term -bvhu225_IgG eff- | 174.4 | 95.5 | 92.6 | EGFR | 4.73E–09 | 3.37E + 05 | 1.60E–03 |
| NKp30 | 5.21E–10 | 2.54E + 05 | 1.32E–04 | ||||
| HC-bvVHH2-C-term -bvhu225_IgG eff+ | 174.7 | 95.9 | 75.2 | ||||
| LC-bvVHH2-C-term-bvhu225_IgG eff- | 174.4 | 92.2 | 80.4 | EGFR | 3.69E–09 | 3.49E + 05 | 1.29E–03 |
| NKp30 | 3.64E–09 | 1.28E + 05 | 4.65E–04 | ||||
| LC-bvVHH2-C-term-bvhu225_IgG eff+ | 174.7 | 91.9 | 76.1 | ||||
| LC-bvVHH2-N-term-bvhu225_IgG eff- | 174.4 | 98.2 | 281.7 | EGFR | 1.70E–09 | 3.00E + 05 | 5.10E–04 |
| NKp30 | 1.72E–09 | 2.95E + 05 | 5.07E–04 | ||||
| LC-bvVHH2-N-term-bvhu225_IgG eff+ | 174.7 | 98.7 | 296.8 | ||||
Analytical size exclusion chromatography (SEC) post protein A purification to determine aggregation propensities indicated favorable biophysical properties of most of the engineered NKCE molecules. In more detail, except for both bvVHH-mvhu225_SEEDbody eff+ molecules (i.e., bvVHH1-mvhu225_SEEDbody eff+ with 75.7% and bvVHH2-mvhu225_SEEDbody eff+ with 82.4%), SEC profiles for all proteins showed target monomer peaks above 85% (Table 1; exemplified SEC profiles shown in Supplementary Figure S2). The sample purities were also visualized by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) with subsequent Coomassie staining, which parallelly allowed for molecular mass confirmation of the intact proteins and the respective protein chains (Supplementary Figure S1).
For the initial biochemical characterization, simultaneous binding of the NKCEs to EGFR and NKp30 was analyzed using biolayer interferometry (BLI). To this end, the NKCEs were loaded on the sensor tips via their constant HC domains followed by successive association of recombinant human (rh) EGFR ECD and rh NKp30 ECD. This revealed simultaneous binding properties for all generated bispecific NKCEs on the protein level (Supplementary Figure S3). To scrutinize binding capabilities of the paratopes, the respective affinities were assessed using the eff- NKCEs, exhibiting no significant loss of binding capacity for any paratope in the tested NKCE architectures (Table 1, Supplementary Figure S4). Moreover, to assess the effect of bivalent receptor targeting, we performed inverted kinetics measurements on the BLI Octet Red System. While the strictly monovalent binding molecules retain their respective affinities (i.e., mvVHH-mvhu225_SEEDbodies eff- for EGFR and NKp30, bvVHH-mvhu225_SEEDbodies eff- for EGFR only and HC-mvVHH-C-term-bvhu225_SEEDbodies eff- for NKp30 only) or exhibit even slightly reduced affinities due to measurement artifacts, bivalent binding to recombinant protein demonstrates a significant increase in apparent affinities against both targets, EGFR and NKp30, indicating a favorable binding behavior for the generated bivalent molecules due to occurring avidity effects (Table 1, Supplementary Figure S5).
Tumor cell killing triggered by bivalent NKp30 engagement is superior to monovalent targeting
To assess the influence of NK activating receptor-engaging valencies, NK cell-mediated tumor cell killing induced by mv vs. bv NKp30 engagement in combination with mv EGFR targeting was compared for high EGFR-expressing A431 epidermoid tumor cells as well as on moderate EGFR-expressing A549 non-small cell lung cancer cells (Figure 2).31 All molecules, i.e., mv, bv as well as eff- and eff+ SEEDbodies, triggered significant killing of A431 and A549 tumor cells by mononuclear cell (MNC)-derived unstimulated NK cells compared to the one-armed (oa)-hu225_SEEDbody eff- control molecule binding to EGFR, but lacking an NK cell binding domain (Figure 2). While no significant impact on maximum killing levels was observed for the comparison of the two VHHs, both as mv and bv NKp30 binding NKCEs, at least a trend toward higher efficacies for VHH2 could be seen (Figure 2). Notably, the half-maximal killing obtained for the B7-H6 non-competing VHH2-harboring eff- SEEDbodies were generally not as potent as observed for the B7-H6 competing VHH1 molecules (3.5- to 39-fold lower EC50 values, Table 2). For both cell lines, the bvVHH1-mvhu225_SEEDbody eff- achieved almost 3-fold improved killing compared to the monovalent NKp30 binding molecule mvVHH1-mvhu225_SEEDbody eff- with EC50 values of 2.2 pM vs. 5.7 pM (A431) and 7.3 pM vs. 19.7 pM (A549) (Figure 2(a), Table 2). For the VHH2-based SEEDbodies, lysis of EGFR-expressing tumor cells was up to 4-fold enhanced by bivalent NKp30 engagement with the bvVHH2-mvhu225_SEEDbody eff- (A431, EC50 = 7.8 pM; A549, EC50 = 191.5 pM) compared to mvVHH2-mvhu225_SEEDbody eff- (A431, EC50 = 21.8 pM; A549, EC50 = 774.6 pM) (Figure 2(a) and Table 2). The data indicate that NKp30 activation by bivalent B7-H6-competing as well as non-competing VHH sdAbs incorporated in monovalent EGFR binding SEEDbodies had a positive impact on NK cell-mediated killing of EGFR-expressing tumor cells, although not on maximum lysis, which was more pronounced for the less potent NKp30-engaging VHH2.
Figure 2.
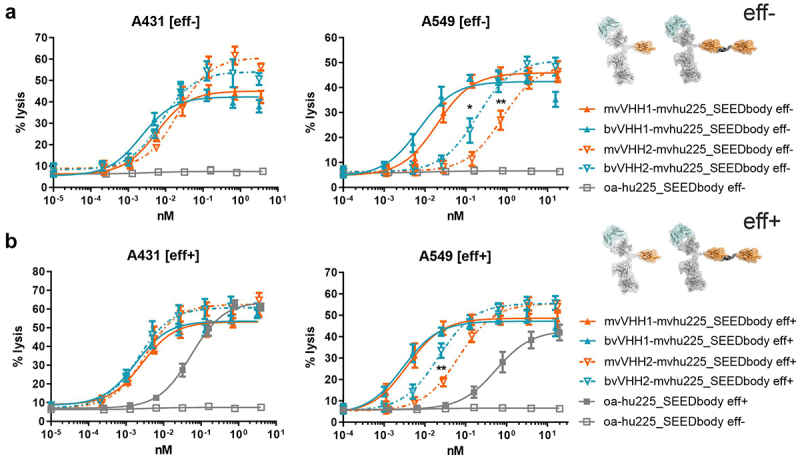
Tumor cell lysis of bivalent NKp30 addressing molecules is enhanced compared to monovalent NKp30 × EGFR binding NKCEs and augmented by concomitant FcγRIIIa activation. Standard 4 h chromium release assays were performed with A431 (left graphs) and A549 cells (right graphs) cells and NK cells from healthy donors as effector cells at an E:T ratio of 10:1 and increasing concentrations of (a) eff- or (b) eff+ VHH SEEDbodies. VHH1-containing molecules (B7-H6 competing NKp30 sdAb) are displayed as filled characters and with solid lines while VHH2-harboring (B7-H6 non-competing NKp30 binding sdAb) NKCEs are displayed as open characters and with dotted lines. Monovalent NKp30 engagers (mv, orange) were compared to bivalent NKp30 engagers (bv, blue). One-armed EGFR binding SEEDbodies lacking the NKp30 activating VHH sdAb (oa-hu225_SEEDbody eff-; oa-hu225_SEEDbody eff+) were used as controls and are shown in gray (eff- as open characters; eff+ as filled characters). Mean values ± SEM of 4 independent experiments with different donors are shown, *p < 0.05%, **p ≤ 0.01, ***p ≤ 0.001, mv vs. bv, two-way ANOVA with Šidák-test. EC50 values are summarized in table 2.
Table 2.
Killing potencies of eff- vs. eff+ as well as mv vs. bv NKp30-targeting SEEDbodies monovalently engaging EGFR.
| mvVHH1-mvhu225_ SEEDbody |
bvVHH1-mvhu225_ SEEDbody |
improvement (mv vs. bv NKp30 binding) | mvVHH2-mvhu225_SEEDbody | bvVHH2-mvhu225_ SEEDbody |
improvement (mv vs. bv NKp30 binding) | |
|---|---|---|---|---|---|---|
| A431 | ||||||
| EC50 eff- (pM) | 5.7 | 2.2 | 2.6-fold | 21.8 | 7.8 | 2.8-fold |
| EC50 eff+ (pM) | 2.7 | 1.8 | 1.5-fold | 3.2 | 2.1 | 1.5-fold |
| improvement (eff+ vs. eff-) |
2.1-fold | 1.2-fold | 6.8-fold | 3.7-fold | ||
| A549 | ||||||
| EC50 eff- (pM) | 19.7 | 7.3 | 2.8-fold | 774.6 | 191.5 | 4.0-fold |
| EC50 eff+ (pM) | 3.7 | 2.9 | 1.3-fold | 67.2 | 21.7 | 3.1-fold |
| improvement (eff+ vs. eff-) |
5.3-fold | 2.5-fold | 11.5-fold | 8.8-fold |
As we were able to show in previous experiments that concomitant engagement of NKp30 and FcγRIIIa on unstimulated NK cells resulted in significantly improved tumor cell killing,18 we additionally analyzed the impact of an effector-competent SEEDbody backbone (eff+) in our monovalent and bivalent NKp30-engaging SEEDbodies on NK cell-mediated target cell lysis. Of note, concomitant engagement of NKp30 and FcγRIIIa resulted in improved half-maximal killing of up to 11.5-fold (Figure 2, Table 2). The eff+ VHH1_SEEDbodies (mv and bv NKp30-engaging) enhanced lysis of EGFR-expressing tumor cells 2.1-fold (mvVHH1-mvhu225_SEEDbody eff+ EC50 = 2.7 pM vs. mvVHH1-mvhu225_SEEDbody eff- EC50 = 5.7 pM) and 1.2-fold (bvVHH1-mvhu225_SEEDbody eff+ EC50 = 1.8 pM vs. bvVHH1-mvhu225_SEEDbody eff- EC50 = 2.2 pM) for A341 cells, as well as 5.3-fold (mvVHH1-mvhu225_SEEDbody eff+ EC50 = 3.7 pM vs. mvVHH1-mvhu225_SEEDbody eff- EC50 = 19.7 pM) and 2.5-fold (bvVHH1-mvhu225_SEEDbody eff+ EC50 = 2.9 pM vs. bvVHH1-mvhu225_SEEDbody eff- EC50 = 7.3 pM) for A549 cells (Figure 2, Table 2). Importantly, in this experimental setting bivalent NKp30 binding did not markedly enhance tumor cell killing compared to monovalent NKp30 engagement (bv vs. mv; A431 cells: 1.5-fold and A549 cell: 1.3-fold). In contrast, for the less potent, B7-H6 non-competing VHH2 bivalent NKp30 binding resulted in a 3.1-fold increased potency (bvVHH2-mvhu225_SEEDbody eff+ EC50 = 21.7 pM vs. mvVHH2-mvhu225_SEEDbody eff+ EC50 = 67.2 pM) against lower EGFR-expressing A549 tumor cells, whereas against high EGFR-expressing A431 cells it was only slightly enhanced by 1.5-fold (Figure 2(b), Table 2).
Comparing the eff- and eff+ VHH2 SEEDbodies, killing capacities could be increased notably by concomitant engagement of NKp30 and FcγRIIIa up to 6.8-fold for the monovalent NKp30 binding molecules (mvVHH2-mvhu225_SEEDbody eff+ EC50 = 3.2 pM vs. mvVHH2-mvhu225_SEEDbody eff- EC50 = 21.8 pM) and 3.7-fold for the bivalent NKp30 binding molecules (bvVHH2-mvhu225_SEEDbody eff+ EC50 = 2.1 pM vs. bvVHH2-mvhu225_SEEDbody eff- EC50 = 7.8 pM) for A431 cells, and significantly for A549 cells with an 11.5-fold increase for the monovalent (mvVHH2-mvhu225_SEEDbody eff+ EC50 = 67.2 pM vs. mvVHH2-mvhu225_SEEDbody eff- EC50 = 774.6 pM) and an 8.8-fold increase for the bivalent (bvVHH2-mvhu225_SEEDbody eff+ EC50 = 21.7 pM vs. bvVHH2-mvhu225_SEEDbody eff- EC50 = 191.5 pM) molecules (Figure 2(b), Table 2). Consequently, these data suggest that bivalent engagement of NKp30 in combination with monovalent EGFR binding is superior to monovalent NKp30 engagement and that concomitant activation of the FcγRIIIa/CD16a can further enhance NK cell-mediated tumor cell killing, especially for less potent, B7-H6 non-competing NKp30 paratopes.
Bivalent NKp30 combined with bivalent EGFR engagement augments NK cell-mediated tumor cell killing
Besides the valency of binding to the activating NKp30 receptor on NK cells, the TAA valency could also significantly impact the cytolytic capacity of effector cell-engaging molecules. To get first insights into whether valency of EGFR binding may represent another parameter to improve the cytotoxic activity of our novel NKCEs and to eliminate the concern that the Fc SEEDbody composition of the molecules alters the capability to induce NK cell-mediated tumor cell killing, cetuximab, its humanized IgG version (hu225_IgG eff+), the SEEDbody version (hu225_SEEDbody eff+), and the monovalent EGFR binding SEEDbody (oa-hu225_SEEDbody eff+) were tested side-by-side in standard 4 h chromium release assays with A431 and A549 cells. All bivalent EGFR binding molecules triggered tumor cell lysis to a similar extent, while the monovalent EGFR binding molecule showed an up to 13-fold higher EC50 value (Supplementary Figure S6). Since bivalent EGFR binding antibodies activating NK cells solely via FcγRIIIa were in general more potent than monovalent molecules, we next investigated the impact of bivalent EGFR binding on NKp30 activation of NK cells. Therefore, either VHH1 or VHH2 were fused to one or both C-terminal ends of effector-silenced, bivalent EGFR binding molecules (SEEDbody format for mvVHH, IgG for bvVHH, Figures 1 and 3). All tested molecules were potent in killing the high EGFR-expressing tumor cell line A431, showing EC50 values between 0.9 pM (HC-bvVHH1-C-term-bvhu225_IgG) and 51 pM (HC-mvVHH2-C-term-bvhu225_SEEDbody) (Table 3). Although the maximum tumor cell lysis was slightly higher on A431 cells for the molecules harboring VHH2, the half-maximal killing was superior for the molecules carrying the VHH1 in the respective NKCE formats, i.e., HC-mvVHH1-C-term-bvhu225_SEEDbody eff- EC50 = 5.1 pM vs. HC-mvVHH2-C-term-bvhu225_SEEDbody eff- EC50 = 51 pM and HC-bvVHH1-C-term-bvhu225_IgG eff- EC50 = 0.9 pM vs. HC-bvVHH2-C-term-bvhu225_IgG eff- EC50 = 5.2 pM (Figure 3(a), Table 3). With moderate EGFR-expressing tumor cell line A549 the molecules employing VHH1 still potently lysed tumor cells (HC-mvVHH1-C-term-bvhu225_SEEDbody eff- EC50 = 11.2 pM and HC-bvVHH1-C-term-bvhu225_IgG eff- EC50 = 2.9 pM), while NKCEs with VHH2 showed drastically impaired tumor cell lysis, emphasizing the crucial role of the targeted NKp30 epitope for NKCE efficiency, especially with lower antigen expressing tumor cells. Bivalent NKp30 engagement was considerably superior to the monovalent NKp30 engagement provoking an up to 5.6-fold and 9.8-fold EC50 enhancement for VHH1 and VHH2, respectively. (Figure 3, Table 3).
Figure 3.
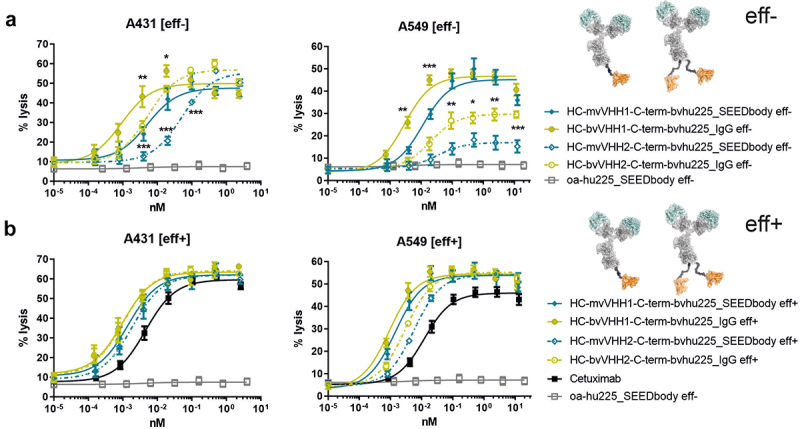
Bivalent NKp30 engagement increases killing potencies of bivalent EGFR binding NKCEs, outperforming cetuximab when concomitantly engaging FcγRIIIa. Cytotoxic capacities of the indicated NKCEs were measured in standard 4 h chromium release assays with A431 (left graph) and A549 (right graph) target cells and NK cells from healthy donors at an E:T ratio of 10:1. Increasing concentrations of the respective (a) eff- or (b) eff+ molecules fused with one (mv, blue) or two (bv, green) B7-H6 competing VHH1 (filled characters and solid lines) or non-competing VHH2 (open characters and dotted lines) sdAbs at the C-terminus of hu255_IgG or hu225_SEEDbody were used. One-armed eff- SEEDbody lacking the NKp30 activating VHH sdAb (oa-hu225_SEEDbody eff-, gray) and cetuximab (black) were used as controls. Mean values ± SEM of 4 independent experiments with different donors are shown, *p < 0.05%, **p ≤ 0.01, ***p ≤ 0.001, mv vs. bv, two-way ANOVA with Šidák-test. Killing potencies are summarized in table 3.
Table 3.
Killing potencies of eff- vs. eff+ as well as mv vs. bv NKp30 targeting NKCEs bivalently binding EGFR.
| cetuximab | HC-mvVHH1-C-term-bvhu225 _SEEDbody | HC-bvVHH1-C-term-bvhu225_IgG | improvement (mv vs. bv NKp30 binding) |
HC-mvVHH2-C-term-bvhu225 _SEEDbody | HC-bvVHH2-C-term-bvhu225_IgG | improvement (mv vs. bv NKp30 binding) |
|
|---|---|---|---|---|---|---|---|
| A431 | |||||||
| EC50 eff- (pM) | - | 5.1 | 0.9 | 5.6-fold | 51 | 5.2 | 9.8-fold |
| EC50 eff+ (pM) | 4.5 | 1.4 | 1.0 | 1.4-fold | 1.9 | 1.0 | 1.9-fold |
| improvement (eff+ vs. eff-) | 3.6-fold | - | 26.8-fold | 5.2-fold | |||
| A549 | |||||||
| EC50 eff- (pM) | - | 11.2 | 2.9 | 3.9-fold | 32.6 | 19.8 | 1.6-fold |
| EC50 eff+ (pM) | 12.1 | 1.4 | 1.0 | 1.4-fold | 5.6 | 3.0 | 1.9-fold |
| improvement (eff+ vs. eff-) |
8.0-fold | 2.9-fold | 5.8-fold | 6.6-fold |
Similar to the monovalent EGFR binding molecules (Figure 2), we tested the impact of NK cell activation by concomitant NKp30 and FcγRIIIa engagement on bivalent EGFR binding NKCEs (Figure 3(b)). Cetuximab, activating NK cells exclusively via FcγRIIIa, was used as control. Intriguingly, all NKp30 and FcγRIIIa activating molecules were more potent than cetuximab against both tumor cell lines, with the most pronounced EC50 improvement of 12.1-fold exhibited by HC-bvVHH1-C-term-bvhu225_IgG eff+ on A549 tumor cells (EC50 = 1.0 pM vs. cetuximab EC50 = 12.1 pM) (Figure 3(b), Table 3). Bivalent NKp30 engagement in combination with NK cell activation via FcγRIIIa slightly improved half-maximal killing of high and moderate EGFR-expressing A431 and A549 tumor cells by 1.4-fold and 1.9-fold, respectively, depending on the VHH used (Table 3). Compared to the eff- NKCEs, concomitant engagement of FcγRIIIa resulted in markedly increased A431 tumor cell killing, with 26.8-fold and 5.2-fold enhancement for monovalent and bivalent NKp30 binding HC-VHH2-C-term-bvhu225 eff+ molecules, respectively, as well as a 3.6-fold enhancement for HC-mvVHH1-C-term-bvhu225_SEEDbody eff+, while no further increase was obtained for the bivalent HC VHH1-based molecule (EC50 eff+ = 1.0 pM vs. EC50 eff- = 0.9 pM, Figure 3, Table 3). Interestingly, killing of the high EGFR-expressing A431 cells triggered by monovalent and bivalent VHH2-fused eff+ molecules was eventually as potent as observed for the VHH1 fusions, with EC50 values between 1.0 pM and 1.9 pM (Table 3). For A549 tumor cells expressing lower EGFR antigen levels, potency enhancement was observed for all eff+ molecules compared to the eff- versions, ranging from 2.9-fold for HC-bvVHH1-C-term-bvhu225_IgG to 8.0-fold for HC-mvVHH1-C-term-bvhu225_SEEDbody (Figure 3, Table 3). Importantly, the maximum lysis of A549 tumor cells could also be increased by simultaneously engaging NKp30 and FcγRIIIa compared to cetuximab irrespective of the VHH used and the presence of one or two NKp30 binding VHH, with the most prominent recovery of killing capacities for the C-terminal HC VHHs fusion molecules (Figure 3(b)). Taken together, the data indicate that bivalent fusion of NKp30-activating VHHs in combination with bivalent EGFR engagement is beneficial for killing of EGFR-expressing tumor cells, rendering this bivalent x bivalent format favorable for further investigations.
Location of NKp30-specific VHHs determines the tumor cell killing efficacy of hu225-IgG molecules
In addition to antibody valency, the overall antibody architecture is a critical parameter for the efficacy of an antibody because it, for example, determines the space between target and effector cell and potentially the formation of the immunological synapse. Therefore, we analyzed bivalently targeting EGFR and NKp30 IgG-based molecules side-by-side. To this end, the NKp30 binding VHH sdAbs were either engrafted N- or C-terminally of the LC or C-terminally of the HC (Figure 1).
While A431 cells were lysed similarly effectively by the three eff- molecules harboring the B7-H6-competing VHH1 domain, with LC-bvVHH1-C-term-bvhu225_IgG eff- being slightly more effective (EC50 = 0.3 pM vs. LC-bvVHH1-N-term-bvhu225_IgG eff- EC50 = 0.7 pM vs. HC-bvVHH1-C-term-bvhu225_IgG eff- EC50 = 1.1 pM), substantial differences were observed between the molecules employing B7-H6 non-competing VHH2 (Figure 4(a), Table 4). Killing efficacies and potencies against A431 tumor cells with both C-and N-terminally fused LC-bvVHH2-bvhu225_IgG eff- molecules (C-term, EC50 = 53.3 pM and N-term, EC50 = 35.9 pM) were substantially decreased compared to HC-bvVHH2-C-term-bvhu225_IgG eff- EC50 = 5.1 pM, Table 4).
Figure 4.
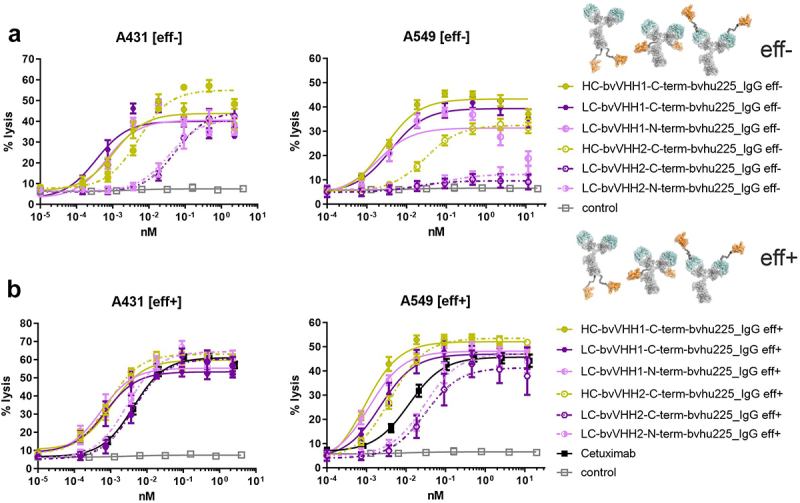
Impact of VHH location on killing capacity differs depending on NKp30 binding epitope of the incorporated VHH. Standard 4 h chromium release assays were performed with A431 (left graphs) and A549 (right graphs) target cells and NK cells from healthy donors as effector cell population at an E:T ratio of 10:1 and increasing concentrations of the respective (a) eff- or (b) eff+ hu225_IgG antibodies. NKp30-specific VHHs were fused to the N- or C-terminus of the light chains (LC; N-term, pink; C-term, purple) or the C-terminus of the heavy chains (HC; C-term, ocher) employing two NKp30 binding sdAbs either competing with the natural ligand B7-H6 binding site (VHH1, solid lines) or not (VHH2, dotted lines). One-armed eff- SEEDbody lacking the NKp30 activating VHH (gray) and cetuximab (black) were used as controls. Mean values ± SEM of at least 3 independent experiments are shown. EC50 values are summarized in Table 4.
Table 4.
Killing potencies of eff- vs. eff+ bivalent NKp30 × EGFR IgG-based NKCEs with differently located VHHs.
| cetuximab | HC-bvVHH1-C-term-bvhu225 IgG | LC-bvVHH1-N-term-bvhu225 IgG | LC-bvVHH1-C-term-bvhu225 IgG | HC-bvVHH2-C-term-bvhu225 IgG | LC-bvVHH2-N-term-bvhu225 IgG | LC-bvVHH2-C-term-bvhu225 IgG | |
|---|---|---|---|---|---|---|---|
| A431 | |||||||
| EC50 eff- (pM) | - | 1.1 | 0.7 | 0.3 | 5.1 | 35.9 | 53.3 |
| EC50 eff+ (pM) | 4.2 | 1.1 | 0.5 | 0.7 | 0.8 | 2.9 | 4.3 |
| improvement (eff+ vs. eff-) | - | 1.4-fold | 2.3-fold | 6.4-fold | 12.4-fold | 12.4-fold | |
| A549 | |||||||
| EC50 eff- (pM) | - | 2.8 | 1.8 | 3.9 | 28.9 | - | - |
| EC50 eff+ (pM) | 11.8 | 0.9 | 1.2 | 2.2 | 4.1 | 29.4 | 30.8 |
| improvement (eff+ vs. eff-) |
3.1-fold | 1.5-fold | 1.8-fold | 7-fold | - | - |
In terms of killing moderate EGFR-expressing cell line A549, the three eff- molecules employing VHH1 again induced comparable half-maximal killing with EC50 values between 1.8 pM and 3.9 pM (Figure 4(a), Table 4), but exhibited differences in maximum lysis, showing a pronounced reduction in the overall killing capacities when VHH1 was engrafted N-terminally onto the LC (Figure 4(a)). Interestingly, while the HC-bvVHH2-C-term-bvhu225_IgG eff- molecule showed some reduction in efficacy and potency (EC50 = 28.9 pM), both C- and N-terminally fused LC-bvVHH2-bvhu225_IgG eff- VHH2 molecules engaging NK cells solely via NKp30 were almost completely ineffective against A549 cells (Figure 4(a)). Although the maximum lysis could largely be recovered when used as eff+ NKCEs, these VHH2-based molecules did not achieve EC50 values of cetuximab (EC50 = 11.8 pM vs. N-term LC EC50 = 29.4 pM vs. C-term LC EC50 = 30.8 pM) (Figure 4(b)), indicating that LC fusions of VHH2 in combination with bivalent EGFR targeting in an effector-competent IgG backbone may even hamper activation of NK cells via FcγRIIIa (Figure 4(b)). In contrast, the bivalent HC-bvVHH2-C-term-bvhu225_IgG eff+ was 2.9-fold more potent compared to cetuximab of (EC50 = 4.1 pM vs. EC50 cetuximab = 11.8 pM). Similarly, the VHH1 engrafted eff+ molecules demonstrated enhanced EC50 values compared to cetuximab ranging between 0.9 pM (HC-bvVHH1-C-term-bvhu225_IgG eff+) and 2.2 pM (LC-bvVHH1-C-term-bvhu225_IgG eff+) in terms of A549 killing (Table 4). The most active VHH1-based molecule HC-bvVHH1-C-term-bvhu225_IgG eff+ thereby achieved a more than 13-fold effective half-maximal A549 tumor cell killing compared to cetuximab (EC50 = 0.9 pM vs. EC50 cetuximab = 11.8 pM, Figure 4(b), Table 4) and a 3.1-fold increased potency compared to its eff- counterpart (EC50 eff+ = 0.9 pM vs. EC50 eff- = 2.8 pM).
Comparable cytotoxic capacity against A431 cells was observed for these VHH1 engrafted eff+ molecules, all showing an improved EC50 compared to cetuximab with a maximum enhancement for LC-bvVHH1-N-term-bvhu225_IgG eff+ of 8.4-fold (EC50 = 0.5 pM vs. EC50 cetuximab = 4.2 pM, Table 4). The effect was similar for moderate EGFR-expressing A549 cells, although not as pronounced (Figure 4(b)). Regarding the VHH2 molecules, the C- and N-terminally fused LC-bvVHH2-bvhu225_IgG eff+ molecules showed significantly improved potencies of 12.4-fold each, resulting in EC50 values of 2.9 pM for the N-terminal and 4.3 pM for the C-terminal VHH orientation, although they were still slightly less effective than HC-bvVHH2-bvhu225_IgG eff+ with an EC50 of 0.8 pM (Figure 4(b), Table 4).
Realizing concomitant engagement of FcγRIIIa and NKp30 in a single molecule by using an appended IgG design may increase the risk of triggering fratricide by cross-linking NK cells, therefore we tested our NKCEs in standard chromium release assays using autologous NK cells of healthy donors as effector and target cells without addition of EGFR-positive tumor cells. Of note, all generated molecules for this study triggered only negligible fratricide (Supplementary Figure S7). Importantly, HC-bvVHH1-C-term-bvhu225_IgG eff+ and HC-bvVHH2-bvhu225_IgG eff+ being the most effective molecules in triggering NK cell-mediated killing of tumor cells showed no fratricide (Supplementary Figure S7).
Taken together, the most robust and favorable molecule architecture for the herein scrutinized bispecific NKp30 × EGFR NKCEs might be the symmetric bivalent C-terminal HC fusion (HC-bvVHH-C-term-bvhu225_IgG). However, the data also indicate that NK cell-mediated killing significantly differ between B7-H6 competing and non-competing sdAb paratopes and are strongly influenced by the chosen NKCE architecture.
NK cell release of immunomodulatory cytokines is influenced by the bispecific NKp30 × EGFR NKCE format
Besides focusing on NK cell-mediated tumor cell lysis, we analyzed the capacity to trigger immunomodulatory interferon-γ (IFN-γ) and tumor-necrosis factor (TNF) cytokine release, as this represents another important NK cell function mediated by the symmetric bivalent IgG-based NKCEs concomitant engaging NKp30 and FcγRIIIa on NK cells in comparison to reference molecule cetuximab (Figure 5). To this end, NK cells were incubated with high EGFR-expressing A431 target cells for 24 h prior to testing the supernatants for released IFN-γ and TNF. While all NKCE formats were able to induce NK cell-mediated cytokine release (mean IFN-γ release 67.33-107.1 pg/ml and TNF 122.4-176.6 pg/ml), differences were observed between the distinct formats. For the molecules harboring VHH1, the highest NK cell-mediated IFN-γ release was observed with LC-bvVHH1-C-term-bvhu225_IgG eff+ (103.3 pg/ml) and TNF release with LC-bvVHH1-N-term-bvhu225_IgG eff+ (146.6 pg/ml) (Figure 5(a)). Regarding VHH2-harboring NKCEs, the highest release of IFN-γ and TNF were measured for the HC VHH fusion, HC-bvVHH2-C-term-bvhu225_IgG eff+ (107.1 pg/ml and 176.6 pg/ml, respectively).
Figure 5.
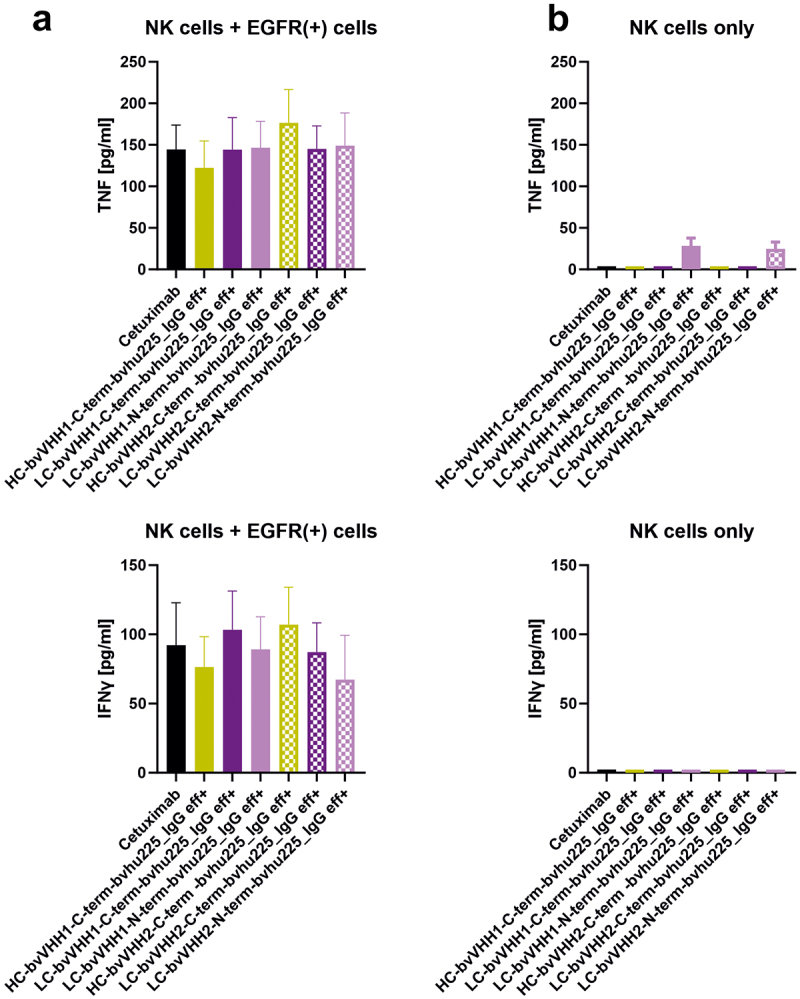
NKp30-specific eff+ NKCEs mediate release of immunomodulatory cytokines. Bivalent NKp30 × EGFR IgG-like eff+ NKCEs were compared with cetuximab (black) for their ability to induce NK cell-mediated TNF (upper graphs) and IFN-γ release (lower graphs). Differences in cytokine release between C-terminal HC (ocher), C-terminal LC (purple) as well as N-terminal LC (pink) fusions were determined for VHH1 (solid bars) and VHH2 (dotted bars) with (a) NK cells cocultured with EGFR-expressing A431 cells or (b) NK cells without any other cells for 24 h, respectively. Graphs show column bar plots ± SEM of 4 individual experiments.
Compared to cetuximab, similar levels of TNF release were measured for the LC VHH fusions (cetuximab = 144.5 pg/ml vs. LC C-term VHH1 = 144.3 pg/ml vs. LC N-term VHH1 = 146.6 pg/ml vs. LC C-term VHH2 = 145.1 pg/ml vs. LC N-term VHH2 = 149.1 pg/ml). While a reduced level of TNF release was measured for the HC-bvVHH1-C-term-bvhu225_IgG eff+ (cetuximab = 144.5 pg/ml vs. HC C-term VHH1 = 122.4 pg/ml), an increased level of TNF release could be seen for the HC-bvVHH2-C-term-bvhu225_IgG eff+ (cetuximab = 144.5 pg/ml vs. HC C-term VHH2 = 176.6 pg/ml). Regarding the IFN-γ secretion, similar levels of IFN-γ release compared to cetuximab could be observed for two LC VHH fusions (cetuximab = 92.22 pg/ml vs. LC N-term VHH1 = 89.13 pg/ml vs. LC C-term VHH2 = 87.18 pg/ml). A reduced level of IFN-γ release compared to cetuximab was measured for two of the eff+ bivalent IgG-based NKCEs (cetuximab = 92.22 pg/ml vs. HC C-term VHH1 = 76.40 pg/ml vs. LC N-term VHH2 = 67.33 pg/ml). Only the LC-bvVHH1-C-term-bvhu225_IgG eff+ with 103.3 pg/ml and the HC-bvVHH2-C-term-bvhu225_IgG eff+ with 107.1 pg/ml showed an increased level of IFN-γ release compared to cetuximab with 92.22 pg/ml (Figure 5(a)).
However, these minor differences in cytokine release were not statistically significant between the tested NKCEs. Moreover, to unravel if our novel molecules may induce non-directional cytokine release, the NKCEs were incubated solely with NK cells. Of note, only the NKCEs with LC VHH fusion induced minor amounts of TNF secretion while no IFN-γ release was observed (Figure 5(b)). This is in line with our finding that only minimal fratricide of NK cells in the absence of EGFR-positive tumor cells is induced by our novel IgG-like NKCEs (Supplementary Figure S7). Consequently, these data indicate that secretion of systemically relevant immunomodulatory cytokines, i.e., IFN-γ and TNF, by NK cells can only be modified to a minor extend by the overall antibody architecture of NKp30-engaging IgG-like NKCEs and that concomitant NKp30 and FcγRIIIa engagement results in comparable IFN-γ and TNF cytokine release as induced by FcγRIIIa engagement of cetuximab.
ADCP and CDC capacities of NKCEs are not affected by VHH location
To analyze whether fusion of VHH to hu225_IgG eff+ molecules have an impact on other known effector mechanisms of cetuximab, CDC and ADCP assays were conducted. To this end, tumor cell phagocytosis was tested using human macrophages derived from peripheral blood monocytes of healthy donors and ADCP established EGFR-expressing human breast carcinoma cell line MDA-MB-468.32 Tumor cells were stained with pHrodo inducing a red fluorescence signal when target cells are phagocytosed. Consequently, red object counts per image were measured constantly every 20 min for 8 h by live cell imaging analyses. The data revealed similar induction of phagocytosis for all tested bv x bv eff+ NKCEs irrespective of VHH location and characteristics of the VHH itself, i.e., B7-H6 competing or non-competing paratope, and comparable behavior to clinically applied antibody cetuximab. As expected, significantly lower ADCP were observed for the samples using an IgG isotype control and cells without antibodies (Figure 6(a)).
Figure 6.
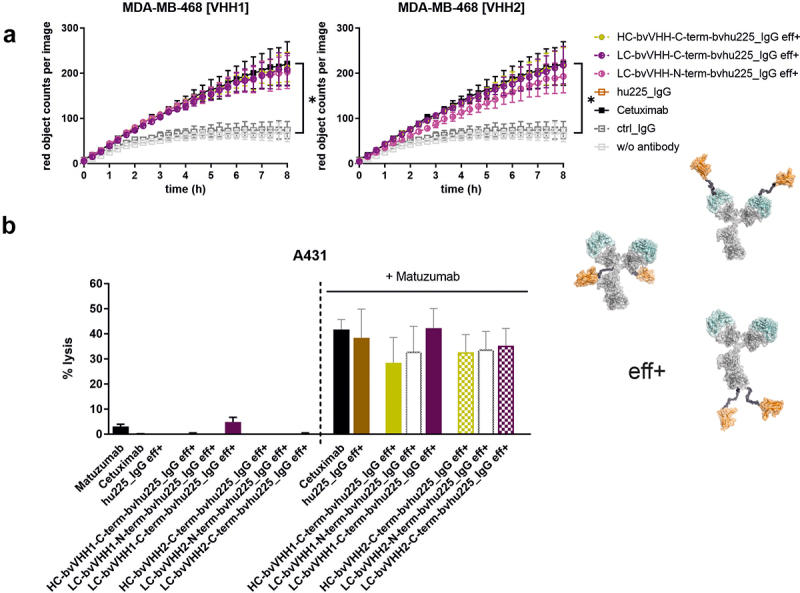
Bivalent NKp30 × EGFR targeting IgG-based NKCEs demonstrate significant ADCP and CDC activity. (a) Phagocytosis of EGFR-expressing MDA-MB-468 tumor cells was measured over 8 h by live cell imaging analyses using macrophages derived from healthy donors at an E:T ratio of 1:1 with 10 µg/ml eff+, bivalent EGFR-targeting hu225_IgG NKCEs carrying either VHH1 (left graph) or VHH2 (right graph) NKp30-engaging sdAbs. For both VHHs, the NKp30-directed paratopes were either fused C-terminally at the HC (ocher), C-terminally at the LC (purple) or N-terminally at the LC (pink). Reference proteins cetuximab and an IgG isotype control were included. Mean values of red object counts per image representing phagocytosed tumor cells ± SEM of 3 independent experiments are shown. (b) CDC of EGFR-expressing A431 tumor cells was measured in standard 3 h chromium release assays using human serum (25% v/v) from healthy donors in the presence of 10 µg/ml effector-functional (eff+) symmetric bivalent NKp30 × EGFR NKCEs employing either VHH1 (solid bars) or VHH2 (dotted bars) solely (left part of the graph) or in combination with 10 µg/ml matuzumab, a humanized IgG1 antibody targeting a non-cross-blocking epitope of EGFR (right part of the graph). Cetuximab in combination with matuzumab was included as positive control. Mean values ± SEM of 3 independent experiments are shown, *p < 0.05%, NKCE or cetuximab vs. w/o antibody, two-way ANOVA with Šidák-test.
Moreover, the impact of VHH location in the herein generated IgG-based molecules (eff+) on CDC capacity was analyzed in standard 3 h chromium release assays using human serum of healthy donors (25% v/v) and A431 tumor cells expressing high levels of EGFR. Since it was shown before that cetuximab is only capable of inducing CDC in combination with a second EGFR-targeting IgG antibody binding to a non-overlapping epitope on EGFR,33 combinations with matuzumab were included as positive controls. Cetuximab and all eff+ hu225_IgGs mediated potent and comparable CDC of A431 tumor cells in combination with matuzumab (Figure 6(b)). As expected, no considerable CDC was observed for all NKCEs without the addition of matuzumab, while lysis induced by the combination of matuzumab and cetuximab was comparable to the ones achieved for our novel eff+ IgG-based molecules (Figure 6(b)). This clearly demonstrates that LC or HC fusions of distinct NKp30-specific VHHs to an IgG backbone do not impact ADCP as well as CDC capacities of the novel bivalent EGFR x NKp30-engaging antibodies.
Bispecific NKp30 × EGFR NKCEs demonstrate IgG-like PK profilesinvivo
To investigate whether serum half-life and clearance of the eff+ NKCEs is altered by the antibody architecture, the PK of bivalent VHH1-carrying NKCEs were studied in three female C57BL/6N mice per group with single doses of 3 mg/kg. No rapid clearance was seen for the tested eff+ NKCEs in mice plasma, implying an intact neonatal Fc receptor (FcRn) recycling for all molecules in vivo (Figure 7). However, the IgG-based NKCEs, irrespective of the location of the fused sdAbs, exhibited faster clearance compared to cetuximab (Cl = 0.257ml/h/kg), i.e., HC-bvVHH1-C-term-bvhu225_IgG eff+ Cl = 0.375 ml/h/kg vs. LC-bvVHH1-C-term-bvhu225_IgG eff+ Cl = 0.413 ml/h/kg vs. LC-bvVHH1-N-term-bvhu225_IgG eff+ Cl = 0.470 ml/h/kg (Table 5). These observations are in line with previously published data describing IgG Fc-fusion molecules demonstrating a shorter half-life than monoclonal IgG molecules.29 Nevertheless, considering the clearance as well as terminal area under the curve (AUClast) values for the tested NKCEs (HC-bvVHH1-C-term-bvhu225_IgG eff+ AUClast = 6.5 × 106 h*ng/ml vs. LC-bvVHH1-C-term-bvhu225_IgG eff+ AUClast = 5.4 × 106 h*ng/ml vs. LC-bvVHH1-N-term-bvhu225_IgG eff+ AUClast = 5.2 × 106 h*ng/ml), these molecules still demonstrate IgG-like PK behavior.
Figure 7.
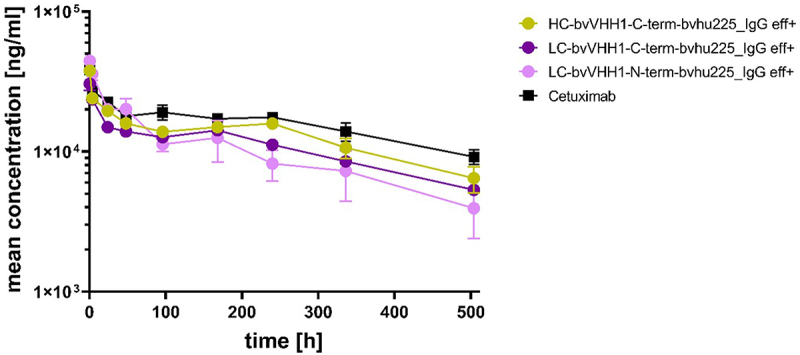
VHH location induce different pharmacokinetic profiles of bispecific EGFR x NKp30 NKCEs. Mouse plasma concentrations of bispecific eff+ NKCEs and cetuximab were measured over time after single intravenous administration of 3 mg/kg of the indicated molecules. Mean concentrations ± SEM of 3 specimen per sample are shown for cetuximab (black), HC-bvVHH1-bvhu225_IgG eff+ (ocher), LC-bvVHH1-N-term-bvhu225_IgG eff+ (pink) and LC-bvVHH1-C-term-bvhu225_IgG eff+ (purple) as determined by ELISA.
Table 5.
PK properties of eff+ bivalent NKp30 × EGFR IgG-based NKCEs with differently located VHH in C57BL/6N mice. The percentage of extrapolated area under the curve (AUCinf) was generally high for total antibody and therefore AUCinf as well as clearance (CL) and volume of distribution at steady state (Vss) calculated using AUCinf should be considered cautiously. Half-life (t1/2) was derived from the 3 terminal data points.
| t1/2 [h] | Cmax [ng/mL] | AUCinf [h*ng/mL] | CL [mL/h/kg] | Vss [mL/kg] | |
|---|---|---|---|---|---|
| HC-bv-VHH1-C-term-bvhu225_IgG eff+ | 206 | 37800 | 8390000 | 0.357 | 123 |
| LC-bv-VHH1-C-term-bvhu225_IgG eff+ | 239 | 30500 | 7260000 | 0.413 | 152 |
| LC-bv-VHH1-N-term-bvhu225_IgG eff+ | 214 | 44500 | 6390000 | 0.470 | 141 |
| cetuximab | 281 | 38400 | 11700000 | 0.257 | 112 |
Discussion
NK cell-engaging bi- or multi-specific antibodies represent promising agents in cancer immunotherapy. Although the concept appears straight-forward, the design of potent agents could be demanding. It is well appreciated that the antibody architecture has a direct impact on antibody effector functions.29 Consequently, various parameters of an antibody need to be fine-tuned in order to generate an optimal molecule for in vivo application.27 A large number of different antibody formats that could be used as backbones for antibody design are available, but general rules in designing molecules have not yet been formulated, making the design process a critical part in the generation of bsAbs.28,34–37 Here, we investigated the impact of antibody architecture on cytolytic capacity, cytokine release, and PK of novel NKCEs triggering NKp30 and targeting EGFR on tumor cells. Our data demonstrate that the specific design, valency and NKp30 epitope recognized by the antibody have a significant impact on NKCEs characteristics.
Our results clearly show that bivalent target antigen binding on tumor cells is beneficial in terms of triggering tumor cell killing by NK cells. This is in line with previous findings by other groups. For example, 20- to 30-fold enhanced NK cell-mediated tumor cell killing through bivalent target antigen binding was described by Kellner and coworkers for a bispecific antibody targeting CD19 and FcγRIIIa.38 In line with this, Reusch and colleagues were able to show that TandAbs bivalently binding target and effector cells showed 10- to 15-fold lower EC50 values for NK cell-mediated tumor cell lysis compared to diabodies with monovalent binding.39
To additionally assess paratope specific attributes, we have used two distinct NKp30 addressing VHHs, of which one targets an epitope not overlapping with NKp30’s natural ligand B7-H6, while the other VHH competes with B7-H6 for NKp30 binding.18 In a previous study as well as in the current study, we found that B7-H6-competing VHH1 molecules were in general more effective in triggering NK cell-mediated tumor cell lysis than NKCEs harboring the B7-H6 non-competing VHH2. The differences became even more prominent when the eff- IgG-like NKCEs were compared. Molecules carrying the VHH2 fused to the LC almost lost their ability to trigger NK cell-dependent tumor cell lysis of lower EGFR-expressing cell lines, while the IgG-like NKCEs carrying the VHH1 showed potent killing irrespective of the antibody architecture. These kinds of observations were described before for T cell-engaging antibodies, where the addressed epitope played a critical role in the design of the antibody architecture.24–26
Irrespective of the design and paratope, molecules bivalently binding NKp30 showed a higher capacity of triggering tumor cell lysis by NK cells than molecules monovalently binding NKp30. These data may indicate that strong cross-linking of NKp30 is beneficial for triggering a cytolytic NK cell response. Moreover, activating more than one pathway to redirect NK cells to eliminate tumor cells has been demonstrated previously.20,40,41 To achieve this, different approaches can be applied. In earlier studies, we were able to show that combinations of monoclonal antibodies and NK cell-engaging bifunctional fusion proteins showed synergistic effects in terms of tumor cell lysis.41 Especially triggering the FcγRIIIa by the antibodies’ Fc domain in parallel to activating NK cell receptors such as NKG2D or NKp30 by bifunctional fusion proteins showed strong synergistic effects.18,21 Here, we integrated all functionalities in one molecule by using an IgG/IgG-like backbone with an active Fc domain. We were able to demonstrate that our molecules triggering both FcγRIIIa and NKp30 mediated the strongest anti-tumor effects and were significantly more potent than molecules triggering only one of the two pathways. Gauthier and colleagues and in a more recent study also our group described a similar strategy in triggering FcγRIIIa and NKp46 on NK cells, leading to a more potent antitumor activity compared to clinically approved antibodies.19,42 Splitting the two functionalities on separate molecules has the advantage that different combinations could easily be tested and novel bifunctional molecules can be combined with already approved therapeutic antibodies.41 On the other hand, integrating all functionalities on one molecule as presented here has the advantage that, for in vivo application, only one NKCE has to be clinically developed, rendering this strategy the potentially preferred option.
Nevertheless, integration of different functionalities in terms of engaging different activating receptors such as NKp30 and FcγRIIIa by using an active Fc domain in combination with a VHH antibody activating NKp30 in one molecule is not trivial. Besides the desired effect of dual activation, inducing fratricide of NK cells could be an issue when different NK cells are cross-linked by the dual activating molecules. Interestingly, molecule designs with NKp30 VHHs fused to the LC showed minimal fratricide, while C-terminal Fc fusion did not show any signs of fratricide. A possible explanation may be that, due to the flexibility of the hinge region, binding of the two receptors is possible when VHHs are fused to the LC, while this is not the case when fused to the C-terminus of the HC. Whether engager-induced fratricide plays a critical role in an in vivo situation needs to be further addressed. Here, carefully designed in vivo models and/or three-dimensional ex vivo tumor models may provide additional critical information for selecting a molecule with the most favorable architecture. The design of such models is challenging since there is no NKp30 homologue in the mouse and NK cells do not have a sufficient half-life in mice.
Integration of an effector-competent Fc portion not only allows engagement of FcγRIIIa in parallel to NKp30 on NK cells, but also activation of other effector populations such as macrophages expressing activating FcγR.43 Our data clearly demonstrate that the IgG-like NKCEs were still capable of inducing phagocytosis of tumor cells by macrophages. Since there is growing evidence that myeloid cells play an important role as effector cells in antibody therapy and that there is crosstalk between NK cells and myeloid cells, this specific feature may be beneficial when applied in vivo.43,44 In addition, other Fc-mediated effector functions such as CDC are not compromised by the molecule design, offering opportunities for further engineering the IgG-like NKCEs.
Finally, bispecific antibodies harboring Fc domains usually exhibit a prolonged in vivo half-life compared to Fc-less molecule designs such as the tandem scFv format used for the generation of the clinically approved bispecific antibody blinatumomab. While blinatumomab is cleared within 1-2 h and has to be applied by continuous infusion over several weeks, more recently approved half-life extended bispecific antibodies such as teclistamab show a half-life of several days, comparable to non-engineered IgG antibodies.45–47 Our data demonstrate that the novel bispecific NKp30 × EGFR NKCEs described here show a shorter half-life compared to wildtype IgG1, but a significantly prolonged half-life compared to Fc-less formats. Interestingly, the bispecific antibodies with the NKp30-specific VHH fused to the LC showed a slightly reduced in vivo half-life compared to the molecule with the VHH antibody fused to the C-terminus of the HC. This is maybe unexpected since the FcRn binding site is located in the interface of the CH2-CH3 domain. It is so far not resolved why our VHH LC fusions compromise in vivo half-life.
Although designed to efficiently engage NK cells as effector cells as a main mode of action, the molecules presented here may also trigger so-called direct effector mechanisms. Cetuximab has been demonstrated to inhibit signaling, receptor down-regulation and tumor cell growth by binding to domain three of EGFR. We did not address the down-regulation of the EGFR after activation in this study, but would expect similar behavior for our constructs bivalently binding EGFR. In a previously published study, we evaluated the potential of different molecules to inhibit the EGFR downstream signaling.18 Cetuximab inhibited the AKT phosphorylation with an EC50 of approximately 0.5 nM. On the other hand, EGFR x NKp30 NKCEs monovalently binding EGFR displayed a significantly reduced inhibition capacity. This capacity was similar to the monovalent EGFR-targeting molecule not triggering NKp30 with an EC50 of 154.9 nM, indicating that monovalent formats show a reduced inhibition of the EGFR. Although not formally proven, we expect similar behavior of bivalent molecules described here.
The herein presented investigations support the notion that the targeted epitope of an activating immune cell receptor, valencies of receptor, and TAA targeting in combination with the spatial orientation of the individual paratopes within the molecular architecture are important factors impacting killing capacities, immunomodulatory cytokine release, and in vivo half-life that ultimately need to be considered when designing NKCEs. In conclusion, our work confirmed earlier findings that, for example, bivalency has a favorable impact on the cytolytic capacity of antibody derivatives. In this respect our findings may be applicable to other target antigens and suggest that a bi-/multivalent design may be a good starting point in designing bispecific effector cell engagers. Nevertheless, significant differences were observed between the two VHHs targeting different epitopes of the NKp30 receptor. Therefore, to a certain extent, the testing matrix we applied here may have to be applied for each new set of antibodies, especially if they bind to distinct fine epitopes.
Materials and methods
Protein expression and purification
Two NKp30-specific VHHs were genetically fused in different orientations either directly to the humanized version of cetuximab (hu225) or onto a separate SEED-modified antibody chain. In more detail, in order to generate asymmetric NKCEs the NKp30-specific VHHs (VHH1 and VHH2) were fused either as single VHH or in tandem arrangement N-terminally to the hinge region of the SEED AG chain or monovalently to the C-terminus of the SEED AG chain with concomitant hu225 Fab N-terminal fusion to the same antibody chain. Transfection together with the corresponding hu225 N-terminally fused to the SEED GA chain then resulted in the assessed SEED-based bsAbs. For the symmetric, IgG-like NKCE formats, the VHHs were fused monovalently either N- or C-terminally to the LC of hu225 or to the C-terminus of the HC of hu225. All bsAbs were produced with a modified IgG CH2 domain devoid of binding to FcγRs and complement protein C1q (eff-, LALA-PG mutations) as well as a wildtype IgG CH2 region (eff+). For the recombinant production of all NKCEs scrutinized in this study, pTT5-derived expression vectors were used, allowing for the transient expression in Expi293 cells according to the manufacturer’s instructions (Thermo Fisher Scientific). Antibody-containing supernatants were harvested by centrifugation five days post transfection prior to purification via MabSelect antibody purification chromatography resin (GE Healthcare). Afterwards, buffer exchange to phosphate-buffered saline (PBS) pH 6.8 was performed overnight using Pur-A-LyzerTM Maxi 3500 Dialysis Kit (Sigma-Aldrich). After sterile filtration with Ultrafree®-CL GV 0.22 µm centrifugal devices (Merck Millipore), the antibody concentrations were measured using Nanodrop ND-1000 (Peqlab). Analytical SEC analyses were conducted to determine sample purities (% target monomer peaks) using 7.5 µg protein per sample on a TSKgel UP-SW3000 column (2 µm, 4.6 × 300 mm, Tosoh Bioscience) in an Agilent HPLC 1260 Infinity system with a flow rate of 0.35 ml/min. Visualization of protein purities and verification of molecular masses were performed via SDS-PAGE (Thermo Fisher Scientific) under reducing and non-reducing conditions, loading 1 µg prepared protein sample per lane (4-12% Bis-Tris gel), respectively, and separating for 90 min at 200 V prior Coomassie staining. Endotoxin levels of the final protein preparations were determined according to standard procedures. All tested protein preparations showed levels < 0.001 EU/µg.
Biolayer interferometry
For simultaneous binding experiments as well as kinetic measurements, the Octet RED96 system (ForteBio, Pall Life Science) was employed using 25°C and 1000 rpm agitation settings. To assess the simultaneous binding properties of the NKCEs, the bsAbs were loaded at 10 μg/ml in PBS for 4 min to anti-human CH1 tips (FAB2G) followed by 1 min of sensor rinsing in kinetics buffer (KB; PBS + 0.1% Tween20 and 1% bovine serum albumin, BSA). Association of rh EGFR ECD (100 nM, produced in-house) was conducted for 100 s in KB, followed by an additional association step for another 100 s with rh NKp30 ECD (100 nM, Acro, NC3-H5228) in KB. For binding kinetic measurements, the NKCEs were loaded for 2 min on anti-human Fc (AHC) biosensors at 10 μg/ml in PBS followed by 1 min of sensor rinsing in KB. The association to human NKp30 ECD was measured as serial 1:2 dilution from 100 nM to 1.56 nM for 300 s followed by dissociation for 300 s in KB. Further binding kinetics involving possible avidity effects of the NKCEs were evaluated by immobilizing 5 µg/ml rh NKp30 ECD or rh EGFR ECD in PBS on anti-penta His (HIS1K) biosensors for 2 min, followed by 1 min of sensor rinsing in KB. Association of the respective bispecific NKCE in KB was then subsequently recorded for 300 s using a 1:2 serial dilution from 100 nM to 1.56 nM prior 300 s dissociation in KB. In each experiment, a negative control using an irrelevant antigen was included. Furthermore, one reference value was measured incubating the antibody in KB instead of the antigen.
Data was fitted and analyzed with ForteBio data analysis software 8.0 using a 1:1 binding model after Savitzky-Golay filtering.
Cell culture
EGFR-expressing tumor cell lines A549, A431 and MDA-MB-468 were obtained from DSMZ and cultured at 6% atmospheric CO2 and 37°C in either Dulbecco’s Modified Eagle’s Medium (DMEM, 41965–062) or RPMI 1640 Glutamax-I medium (11835–030) supplemented with 10% FCS (10270–106), 100 U/ml penicillin and 100 mg/ml streptomycin (15140–163) (D10+ and R10+; all components were purchased from Thermo Fisher Scientific). EGFR-negative ExpiCHO cells were cultivated in ExpiCHO expression medium (both Thermo Fisher Scientific) at 5% atmospheric CO2, 80% humidity, 36.5°C temperature and 80 rpm agitation with 25 mm shaking amplitude.
Tumor cell killing assays
Experiments were approved by the Ethics Committee of the Christian-Albrechts-University of Kiel (Kiel, Germany) and were in accordance with the Declaration of Helsinki. Isolation of mononuclear cells (MNCs) and serum from healthy donors was performed as described elsewhere.48,49 Isolated NK cells were maintained overnight at 6% atmospheric CO2 and 37°C at a density of 2 × 106 cells/ml in R10+ medium after the isolation by negative selection using NK cell isolation kit (Miltenyi Biotec, 130-092-657). Cytotoxicity was analyzed in standard 3 h (for CDC) and 4 h (ADCC) 51Cr release assays performed in 96-well microtiter plates in a total volume of 200 μl as previously described.50 Isolated NK cells were used as effector cells at effector-to-target cell (E:T) ratios of 10:1, while human serum (25% v/v) served as a source of complement. Bispecific NKp30 × EGFR NKCEs and cetuximab were compared at equivalent concentrations. Percent lysis was calculated from counts per minute (cpm) as follows: % lysis = (experimental cpm-basal cpm)/(maximal cpm-basal cpm)x100.
Live cell imaging analyses/phagocytosis assays
Human M0 macrophages expressing FcγRs were generated from MNCs of healthy donors by incubation with monocyte-attachment medium (PromoCell, C-28051) for 30 min at 37°C and subsequent cultivation in X-VIVO 15 medium (Lonza, BEBP02-061Q) supplemented with 50 ng/ml M-CSF (PeproTech, 300–25). ADCP was determined by seeding 4 × 104 macrophages/well in a 96-well flat-bottom plate, rested for 1 h at room temperature for cell adherence prior addition of 4 × 104 pH-sensitive, red fluorescently labeled (pHrodo dye, Thermo Fisher Scientific, P36600) target cells (E:T ratio of 1:1) and the indicated antibodies in saturating concentrations of 10 µg/ml. The IncuCyte high-throughput fluorescence microscopy system (Sartorius) was utilized to record images of the fluorescent cells every 20 min for 8 h in total while incubating the cells at 37°C. Phagocytosis was determined as the red object counts per image over time.
Cytokine release assay
MNC-derived NK cells from healthy donors were isolated with NK Cell Isolation Kit (human, Miltenyi Biotec) and incubated overnight in complete AIM V Medium supplemented with 100 U/ml rh IL-2 (R&D Systems). Next day, 2.5 × 103 A431 or CHO target cells were seeded in a 384-well microtiter plates (Greiner Bio-One) and incubated for 3 h prior addition of NK cells at an E:T ratio of 5:1. Afterwards, the indicated NKCEs were added to a final concentration of 50 nM and incubated for 24 h with the target cells or with NK cells only. Finally, the supernatants were analyzed using human IFN-γ or TNF-α HTRF kits (Cisbio) according to the manufacturer’s instructions. Plates were measured with a PHERAstar FSX device (BMG Labtech). Resulting data were analyzed using the MARS software (v.3.32; BMG Labtech) enabling a four-parameter logistic (4PL 1/y2) model fitting of the standard curve.
PK study
The PK of cetuximab and bispecific NKp30 × EGFR NKCEs as hu225_IgG eff+ fusion proteins were studied in three female C57BL/6N mice (9-11 weeks old), respectively. Single doses of 3 mg/kg were administered intravenously into the tail vein and 20 µl of blood was collected at 0.5, 4, 24, 48, 96, 168, 240, 336 and 504 h after dosing in heparin-coated tubes. Plasma concentrations of the molecules were measured via ELISA using a ligand-binding assay with a lower limit of quantification of 50 ng/mL as described previously.51 Biotin SP-conjugated AffiniPure goat anti-human IgG (Fcγ fragment specific, Jackson Immuno Research, 109-065-098) was used as capture reagent and SULFO-tag labeled AffiniPure goat anti-human IgG (Fcγ fragment specific, Jackson Immuno Research, 109-005-098) was used as a detection reagent.
Molecular modeling
To create structural models of the full-length IgGs and VHHs, the antibody modeler tool in the molecular modeling software package MOE (Molecular Operating Environment 2020.09: Chemical Computing Group Inc.; 2020) was utilized. Different architectures of the bispecific IgG-VHH constructs were built by adding linkers via MOE’s protein builder, followed by a conformational search of the linker via MOE’s linker modeler. Finally, an energy minimization was performed, treating the linker as flexible and the IgG and VHH domains as rigid bodies. Visualization of 3D structures was done with PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.).
Data processing and statistical analysis
If not otherwise indicated, graphical and statistical analyses were performed using GraphPad Prism 9 software. P-values were calculated applying two-way ANOVA with Šidák-test posttest as recommended, or the student’s t-test when appropriate. p ≤ 0.05 were regarded as statistically significant.
Supplementary Material
Acknowledgments
We gratefully acknowledge Britta von Below and Anja Muskulus for excellent technical assistance. We further thank Kerstin Hallstein, Laura Unmuth, Sigrid Auth, Vanessa Siegmund, and Dirk Mueller-Pompalla for experimental support. Furthermore, we would like to thank Mantas Malisauskas (DMPK-NBE), Luca Marchiando (DMPK-NBE), Elisa Bertotti (DMPK-NBE), Patrizia Tavano (DMPK-NBE), Andrea Paoletti (DMPK-NBE) and Klaus-Georg Gerth (DMPK-NCE) for the support regarding the PK study.
Funding Statement
The work was supported by research funds within the clinical research unit CATCH-ALL funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 444949889 to MP.
Disclosure statement
ASB, CLG, SK, MP and KK received research funding from Merck KGaA. LP, KK, BL, BR, AE, YX, SK, RZ, LT, SP and SZ are or were employees at either Merck Healthcare KGaA or EMD Serono. Besides, this work was conducted in the absence of any further commercial interest.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2024.2315640
References
- 1.Myers JA, Miller JS.. Exploring the NK cell platform for cancer immunotherapy. Nat Rev Clin Oncol. 2021;18(2):85–17. PMID: 32934330. doi: 10.1038/s41571-020-0426-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chiossone L, Dumas PY, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol. 2018;18(11):671–88. PMID: 30209347. doi: 10.1038/s41577-018-0061-z. [DOI] [PubMed] [Google Scholar]
- 3.Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, Yokoyama WM, Ugolini S. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331(6013):44–49. PMID: 21212348. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moretta L, Bottino C, Cantoni C, Mingari MC, Moretta A. Human natural killer cell function and receptors. Curr Opin Pharmacol. 2001;1(4):387–91. PMID: 11710737. doi: 10.1016/s1471-4892(01)00067-4. [DOI] [PubMed] [Google Scholar]
- 5.Quatrini L, Della Chiesa M, Sivori S, Mingari MC, Pende D, Moretta L. Human NK cells, their receptors and function. Eur J Immunol. 2021;51(7):1566–79. PMID: 33899224. doi: 10.1002/eji.202049028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barrow AD, Martin CJ, Colonna M. The natural cytotoxicity receptors in health and disease. Front Immunol. 2019;10:909. PMID: 31134055. doi: 10.3389/fimmu.2019.00909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9(5):503–10. PMID: 18425107. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- 8.Minetto P, Guolo F, Pesce S, Greppi M, Obino V, Ferretti E, Sivori S, Genova C, Lemoli RM, Marcenaro E. Harnessing NK cells for cancer treatment. Front Immunol. 2019;10:2836. PMID: 31867006. doi: 10.3389/fimmu.2019.02836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bibeau F, Lopez-Crapez E, Di Fiore F, Thezenas S, Ychou M, Blanchard F, Lamy A, Penault-Llorca F, Frebourg T, Michel P. et al. Impact of FcγRIIa-FcγRIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J Clin Oncol. 2009;27(7):1122–29. PMID: 19164213. doi: 10.1200/JCO.2008.18.0463 [DOI] [PubMed] [Google Scholar]
- 10.Seidel UJ, Schlegel P, Lang P. Natural killer cell mediated antibody-dependent cellular cytotoxicity in tumor immunotherapy with therapeutic antibodies. Front Immunol. 2013;4:76. PMID: 23543707. doi: 10.3389/fimmu.2013.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang DS, Wei XL, Wang ZQ, Lu YX, Shi SM, Wang N, Qiu MZ, Wang FH, Wang RJ, Li YH. et al. FcγRIIA and IIIA polymorphisms predict clinical outcome of trastuzumab-treated metastatic gastric cancer. Onco Targets Ther. 2017;10:5065–76. PMID: 29089776. doi: 10.2147/OTT.S142620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darwich A, Silvestri A, Benmebarek MR, Mouries J, Cadilha B, Melacarne A, Morelli L, Supino D, Taleb A, Obeck H. et al. Paralysis of the cytotoxic granule machinery is a new cancer immune evasion mechanism mediated by chitinase 3-like-1. J Immunother Cancer. 2021;9(11):e003224. PMID: 34824159. doi: 10.1136/jitc-2021-003224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beano A, Signorino E, Evangelista A, Brusa D, Mistrangelo M, Polimeni MA, Spadi R, Donadio M, Ciuffreda L, Matera L. Correlation between NK function and response to trastuzumab in metastatic breast cancer patients. J Transl Med. 2008;6(1):25. PMID: 18485193. doi: 10.1186/1479-5876-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Romee R, Foley B, Lenvik T, Wang Y, Zhang B, Ankarlo D, Luo X, Cooley S, Verneris M, Walcheck B. et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood. 2013;121(18):3599–608. doi: 10.1182/blood-2012-04-425397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coënon L, Villalba M. From CD16a biology to antibody-dependent cell-mediated cytotoxicity improvement. Front Immunol. 2022;13:13. doi: 10.3389/fimmu.2022.913215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cecchetti S, Spadaro F, Lugini L, Podo F, Ramoni C. Functional role of phosphatidylcholine-specific phospholipase C in regulating CD16 membrane expression in natural killer cells. Eur J Immunol. 2007;37(10):2912–22. PMID: 17899539. doi: 10.1002/eji.200737266. [DOI] [PubMed] [Google Scholar]
- 17.Preithner S, Elm S, Lippold S, Locher M, Wolf A, da Silva AJ, Baeuerle PA, Prang NS, Silva AJD. High concentrations of therapeutic IgG1 antibodies are needed to compensate for inhibition of antibody-dependent cellular cytotoxicity by excess endogenous immunoglobulin G. Mol Immunol. 2006;43(8):1183–93. PMID: 16102830. doi: 10.1016/j.molimm.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 18.Klausz K, Pekar L, Boje AS, Gehlert CL, Krohn S, Gupta T, Xiao Y, Krah S, Zaynagetdinov R, Lipinski B. et al. Multifunctional NK cell–engaging antibodies targeting EGFR and NKp30 elicit efficient tumor cell killing and proinflammatory cytokine release. J Immunol. 2022;209(9):1724–35. PMID: 36104113. doi: 10.4049/jimmunol.2100970 [DOI] [PubMed] [Google Scholar]
- 19.Gauthier L, Morel A, Anceriz N, Rossi B, Blanchard-Alvarez A, Grondin G, Trichard S, Cesari C, Sapet M, Bosco F. et al. Multifunctional natural killer cell engagers targeting NKp46 trigger protective tumor immunity. Cell. 2019;177(7):1701–13 e1716. PMID: 31155232. doi: 10.1016/j.cell.2019.04.041 [DOI] [PubMed] [Google Scholar]
- 20.Pekar L, Klausz K, Busch M, Valldorf B, Kolmar H, Wesch D, Oberg HH, Krohn S, Boje AS, Gehlert CL. et al. Affinity maturation of B7-H6 translates into enhanced NK cell–mediated tumor cell lysis and improved proinflammatory cytokine release of bispecific immunoligands via NKp30 engagement. J Immunol. 2021;206(1):225–36. PMID: 33268483. doi: 10.4049/jimmunol.2001004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raynaud A, Desrumeaux K, Vidard L, Termine E, Baty D, Chames P, Vigne E, Kerfelec B. Anti-NKG2D single domain-based antibodies for the modulation of anti-tumor immune response. Oncoimmunology. 2020;10(1):1854529. PMID: 33457075. doi: 10.1080/2162402X.2020.1854529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ellwanger K, Reusch U, Fucek I, Wingert S, Ross T, Muller T, Schniegler-Mattox U, Haneke T, Rajkovic E, Koch J. et al. Redirected optimized cell killing (ROCK®): a highly versatile multispecific fit-for-purpose antibody platform for engaging innate immunity. MAbs. 2019;11(5):899–918. PMID: 31172847. doi: 10.1080/19420862.2019.1616506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Demaria O, Gauthier L, Vetizou M, Blanchard Alvarez A, Vagne C, Habif G, Batista L, Baron W, Belaid N, Girard-Madoux M. et al. Antitumor immunity induced by antibody-based natural killer cell engager therapeutics armed with not-alpha IL-2 variant. Cell Rep Med. 2022;3(10):100783. PMID: 36260981. doi: 10.1016/j.xcrm.2022.100783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bluemel C, Hausmann S, Fluhr P, Sriskandarajah M, Stallcup WB, Baeuerle PA, Kufer P. Epitope distance to the target cell membrane and antigen size determine the potency of T cell-mediated lysis by BiTE antibodies specific for a large melanoma surface antigen. Cancer Immunol Immunother. 2010;59(8):1197–209. PMID: 20309546. doi: 10.1007/s00262-010-0844-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen W, Yang F, Wang C, Narula J, Pascua E, Ni I, Ding S, Deng X, Chu ML, Pham A. et al. One size does not fit all: navigating the multi-dimensional space to optimize T-cell engaging protein therapeutics. MAbs. 2021;13(1):1871171. PMID: 33557687. doi: 10.1080/19420862.2020.1871171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li J, Stagg NJ, Johnston J, Harris MJ, Menzies SA, DiCara D, Clark V, Hristopoulos M, Cook R, Slaga D. et al. Membrane-proximal epitope facilitates efficient T cell synapse formation by anti-FcRH5/CD3 and is a requirement for myeloma cell killing. Cancer Cell. 2017;31(3):383–95. PMID: 28262555. doi: 10.1016/j.ccell.2017.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boder ET, Jiang W. Engineering antibodies for cancer therapy. Annu Rev Chem Biomol Eng. 2011;2(1):53–75. PMID: 22432610. doi: 10.1146/annurev-chembioeng-061010-114142. [DOI] [PubMed] [Google Scholar]
- 28.Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs. 2017;9(2):182–212. PMID: 28071970. doi: 10.1080/19420862.2016.1268307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goulet DR, Atkins WM. Considerations for the design of antibody-based therapeutics. J Pharm Sci. 2020;109(1):74–103. PMID: 31173761. doi: 10.1016/j.xphs.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davis JH, Aperlo C, Li Y, Kurosawa E, Lan Y, Lo KM, Huston JS. Seedbodies: fusion proteins based on strand-exchange engineered domain (SEED) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies. Protein Eng Des Sel. 2010;23(4):195–202. PMID: 20299542. doi: 10.1093/protein/gzp094. [DOI] [PubMed] [Google Scholar]
- 31.Schlaeth M, Berger S, Derer S, Klausz K, Lohse S, Dechant M, Lazar GA, Schneider-Merck T, Peipp M, Valerius T. Fc-engineered EGF-R antibodies mediate improved antibody-dependent cellular cytotoxicity (ADCC) against KRAS-mutated tumor cells. Cancer Sci. 2010;101:1080–88. PMID: 20331636. doi: 10.1111/j.1349-7006.2010.01505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baumann N, Rosner T, Jansen JHM, Chan C, Marie Eichholz K, Klausz K, Winterberg D, Muller K, Humpe A, Burger R. et al. Enhancement of epidermal growth factor receptor antibody tumor immunotherapy by glutaminyl cyclase inhibition to interfere with CD47/signal regulatory protein alpha interactions. Cancer Sci. 2021;112:3029–40. PMID: 34058788. doi: 10.1111/cas.14999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dechant M, Weisner W, Berger S, Peipp M, Beyer T, Schneider-Merck T, Lammerts van Bueren JJ, Bleeker WK, Parren PW, van de Winkel JG. et al. Complement-dependent tumor cell lysis triggered by combinations of epidermal growth factor receptor antibodies. Cancer Research. 2008;68(13):4998–5003. PMID: 18593896. doi: 10.1158/0008-5472.CAN-07-6226 [DOI] [PubMed] [Google Scholar]
- 34.Tiller KE, Tessier PM. Advances in antibody design. Annu Rev Biomed Eng. 2015;17(1):191–216. PMID: 26274600. doi: 10.1146/annurev-bioeng-071114-040733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol. 2015;67(2):95–106. PMID: 25637431. doi: 10.1016/j.molimm.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 36.Ma J, Mo Y, Tang M, Shen J, Qi Y, Zhao W, Huang Y, Xu Y, Qian C. Bispecific antibodies: from research to clinical application. Front Immunol. 2021;12:626616. PMID: 34025638. doi: 10.3389/fimmu.2021.626616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elshiaty M, Schindler H, Christopoulos P. Principles and Current clinical landscape of multispecific antibodies against cancer. Int J Mol Sci. 2021;22(11):5632. PMID: 34073188. doi: 10.3390/ijms22115632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kellner C, Bruenke J, Stieglmaier J, Schwemmlein M, Schwenkert M, Singer H, Mentz K, Peipp M, Lang P, Oduncu F. et al. A novel CD19-directed recombinant bispecific antibody derivative with enhanced immune effector functions for human leukemic cells. J Immunother (1991). 2008;31:871–84. PMID: 18833000. doi: 10.1097/CJI.0b013e318186c8b4. [DOI] [PubMed] [Google Scholar]
- 39.Reusch U, Burkhardt C, Fucek I, Le Gall F, Le Gall M, Hoffmann K, Knackmuss SH, Kiprijanov S, Little M, Zhukovsky EA. A novel tetravalent bispecific TandAb (CD30/CD16A) efficiently recruits NK cells for the lysis of CD30+ tumor cells. MAbs. 2014;6(3):728–39. PMID: 24670809. doi: 10.4161/mabs.28591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Colomar-Carando N, Gauthier L, Merli P, Loiacono F, Canevali P, Falco M, Galaverna F, Rossi B, Bosco F, Caratini M. et al. Exploiting natural killer cell engagers to control pediatric B-cell precursor acute lymphoblastic leukemia. Cancer Immunol Res. 2022;10(3):291–302. PMID: 35078821. doi: 10.1158/2326-6066.CIR-21-0843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kellner C, Gunther A, Humpe A, Repp R, Klausz K, Derer S, Valerius T, Ritgen M, Bruggemann M, van de Winkel JG. et al. Enhancing natural killer cell-mediated lysis of lymphoma cells by combining therapeutic antibodies with CD20-specific immunoligands engaging NKG2D or NKp30. Oncoimmunology. 2016;5(1):e1058459. PMID: 26942070. doi: 10.1080/2162402X.2015.1058459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lipinski B, Arras P, Pekar L, Klewinghaus D, Boje AS, Krah S, Zimmermann J, Klausz K, Peipp M, Siegmund V. et al. NKp46-specific single domain antibodies enable facile engineering of various potent NK cell engager formats. Protein Sci. 2023;32:e4593. PMID: 36775946. doi: 10.1002/pro.4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weiskopf K, Weissman IL. Macrophages are critical effectors of antibody therapies for cancer. MAbs. 2015;7:303–10. PMID: 25667985. doi: 10.1080/19420862.2015.1011450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou J, Zhang S, Guo C. Crosstalk between macrophages and natural killer cells in the tumor microenvironment. Int Immunopharmacol. 2021;101:108374. PMID: 34824036. doi: 10.1016/j.intimp.2021.108374. [DOI] [PubMed] [Google Scholar]
- 45.Pillarisetti K, Powers G, Luistro L, Babich A, Baldwin E, Li Y, Zhang X, Mendonca M, Majewski N, Nanjunda R. et al. Teclistamab is an active T cell–redirecting bispecific antibody against B-cell maturation antigen for multiple myeloma. Blood Adv. 2020;4(18):4538–49. PMID: 32956453. doi: 10.1182/bloodadvances.2020002393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goldsmith SR, Streeter S, Covut F. Bispecific Antibodies for the Treatment of Multiple Myeloma. Curr Hematol Malig Rep. 2022;17(6):286–97. PMID: 36029366. doi: 10.1007/s11899-022-00675-3. [DOI] [PubMed] [Google Scholar]
- 47.Liu L. Pharmacokinetics of monoclonal antibodies and Fc-fusion proteins. Protein Cell. 2018;9(1):15–32. PMID: 28421387. doi: 10.1007/s13238-017-0408-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Repp R, Kellner C, Muskulus A, Staudinger M, Nodehi SM, Glorius P, Akramiene D, Dechant M, Fey GH, van Berkel PH. et al. Combined Fc-protein- and Fc-glyco-engineering of scFv-Fc fusion proteins synergistically enhances CD16a binding but does not further enhance NK-cell mediated ADCC. J Immunol Methods. 2011;373(1–2):67–78. PMID: 21855548. doi: 10.1016/j.jim.2011.08.003 [DOI] [PubMed] [Google Scholar]
- 49.Klausz K, Berger S, van Bueren JJ L, Derer S, Lohse S, Dechant M, van de Winkel JG, Peipp M, Parren PW, Valerius T. et al. Complement-mediated tumor-specific cell lysis by antibody combinations targeting epidermal growth factor receptor (EGFR) and its variant III (EGFRvIII). Cancer Sci. 2011;102(10):1761–68. PMID: 21718386. doi: 10.1111/j.1349-7006.2011.02019.x. [DOI] [PubMed] [Google Scholar]
- 50.Rosskopf S, Eichholz KM, Winterberg D, Diemer KJ, Lutz S, Munnich IA, Klausz K, Rosner T, Valerius T, Schewe DM. et al. Enhancing CDC and ADCC of CD19 antibodies by combining Fc protein-engineering with Fc glyco-engineering. Antibodies (Basel). 2020;9(4):63. PMID: 33212776. doi: 10.3390/antib9040063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaempffe A, Dickgiesser S, Rasche N, Paoletti A, Bertotti E, De Salve I, Sirtori FR, Kellner R, Konning D, Hecht S. et al. Effect of conjugation site and technique on the stability and pharmacokinetics of antibody-drug conjugates. J Pharm Sci. 2021;110:3776–85. PMID: 34363839. doi: 10.1016/j.xphs.2021.08.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.