

ABSTRACT
The identification of mechanisms capable of modifying genetic information by the addition of covalent RNA modifications distinguishes a level of complexity in gene expression which challenges key long-standing concepts of RNA biology. One of the current challenges of molecular biology is to properly understand the molecular functions of these RNA modifications, with more than 170 different ones having been identified so far. However, it has not been possible to map specific RNA modifications at a single-cell resolution until very recently. This review will highlight the technological advances in single-cell methodologies aimed at assessing and testing the biological function of certain RNA modifications, focusing on m6A. These advances have allowed for the development of novel strategies that enable the study of the ‘epitranscriptome’. Nevertheless, despite all these improvements, many challenges and difficulties still need fixing for these techniques to work efficiently.
KEYWORDS: Single-cell, epitranscriptome, RNA modifications, m6A, Inosine
The complex process of gene expression begins with regulating the access to specific DNA sequences. This genetic information is then copied into RNA molecules, which can be classified into two extensive groups: coding RNAs (cRNAs, also known as messenger RNAs, mRNAs), information of which is translated into proteins, and non-coding RNAs (ncRNAs). Additionally, the alternative splicing of an RNA molecule expands its intrinsic information in multiple isoforms, together with the fact that each RNA nucleoside can be chemically modified. Even though the roles of most RNA modifications remain unknown, the ubiquity of these in many species points to them as being evolutionary conserved molecular toolboxes that may allow for a rapid response to environmental challenges and to control the flow of genetic information. These RNA modifications comprise what is now known as the ‘epitranscriptome’. So far, more than 170 RNA modifications have been described [1]. Abundant RNAs such as transfer RNA (tRNA) and ribosomal RNA (rRNA) are mapped to most of the known epitranscriptomic modifications. Consequently, our knowledge about the molecular function of RNA modifications has been mostly moulded by research carried out on rRNAs and tRNAs. However, owing to the emergence of innovative next-generation sequencing (NGS) technologies, the field of epitranscriptomics has experienced significant growth in the past decade, particularly in studying RNA modifications present in mRNA molecules.
The identification of cell-type-specific and intricate patterns of RNA splicing and the large presence of RNA editing events demonstrated that RNA molecules need to be furtherly modified in order for them to exert their correct functions [2,3]. Internal modifications of mRNA like N6-methyladenosine (m6A), ribose-methylation (2’-O-Me), pseudouridine () and methyl-5-cytosine (m5C), and inosine (I), although having been uncovered for more than 50 years [4–8], were not accessible to molecular research until the emergence of the aforementioned sequencing technologies (Figure 1).
Figure 1.
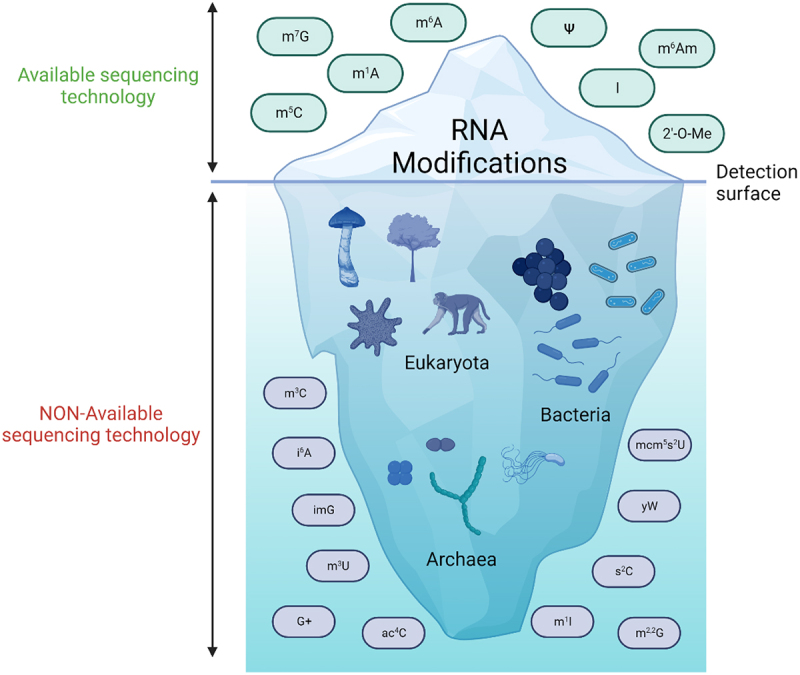
RNA modifications detected and those yet to be found. Better sequencing technologies have allowed the identification of millions of new modification sites in all types of RNAs. Considering that > 170 different types of modified ribonucleotides have been found until now [9], it is to be expected that these novel technologies will result in a big increase in identifiable RNA modifications which may be now hidden below the detection threshold. Created with BioRender.com [10].
Decades ago, epitranscriptomic modifications were only quantified in terms of abundancy. Liquid chromatography coupled with mass spectrometry (LC-MS/MS) allowed for the discovery and quantification of many epitranscriptomic marks [11,12]. Unfortunately, knowing only the abundance of these RNA modifications did not allow for a proper determination of their biological function. In order to address the role of epitranscriptomic modifications, it’s crucial to understand not just the relative abundance of a modification, but also its specific location within the various RNA species. The position of these modifications has only been tackled in the last decade, thanks to the emergence of NGS technologies.
Adapted NGS technologies have demonstrated that nearly every RNA species (including low-expressed RNAs) are accompanied by certain RNA modifications, while physicochemical detection methods (LC-MS/MS, nuclear magnetic resonance, colorimetric assays, etc.) are only capable of reporting the presence or lack of a modified nucleoside [13]. The accessibility of these sequencing technologies has also transformed our capacity to obtain transcriptome-wide outlines on RNA modifications. These specialized protocols have already been developed for pseudouridine (Pseudo-seq [14,15], 2′-O-methylations (RiboMeth-seq [16,17] m5C (Bisulfite-converted RNA-seq [18,19], inosine [RNA-seq with A-to-G variant calling [20], m1A (m1A-seq/m1A-ID-seq [21,22]] and m6A (m6A-seq/MeRIP-Seq [23,24], among others (Figure 1).
Further analyses of NGS data suggested an enrichment of specific RNA modifications at functional sequence features, particularly in mRNAs like untranslated regions (UTRs), retained introns, exons and transcriptional start sites [13]. For example, m6A is mapped mostly within the last exon in nearly all mRNAs [25]. In contrast, m1A was highly present in 5’ UTR and in the vicinity of start codons of mouse and human mRNAs [21,22]. On the other hand, m5C mapped at 3’ and 5’ UTR in mRNAs of an extremely unstable cancer cell line [18], at sites of translation start in mouse embryonic stem cells (mESCs) and whole brain tissues [26], in coding sequences from various mouse tissues [27,28] and in Arabidopsis [29]. Other NGS data also unmasked how most of the A-to-I RNA editing events happen in mobile element-derived sequences [30], and also in mRNA, controlling transcript stability and localization [31].
The addition of m6A in mRNA happens co-transcriptionally in the nucleus, within nuclear speckles specifically, where active mRNA splicing and transcription take place [32,33]. Inside the nucleus, the WMM complex (the WTAP-METTL3-METTL14 m6A methyltransferase complex) modifies different regions of nascent mRNAs. Around 70% of the m6A in mRNA is situated in the 3’UTR region and near the STOP codon [23,24]. m6A can also be located in introns, exons and, less frequently, in 5’UTR. Depending on where m6A is localized in an mRNA molecule and its interactions with various m6A readers, this modification can exert different functions, many of them undefined (Figure 2). Current antibody-based sequencing methods to detect m6A, like MeRIP-Seq, do not allow to pinpoint this modification at single-nucleotide resolution, but new antibody-free technologies, like the ‘glyoxal and nitrite-mediated deamination of unmethylated adenosines sequencing’ technique (GLORI-seq), are achieving single-nucleotide m6A resolution levels [34].
Figure 2.
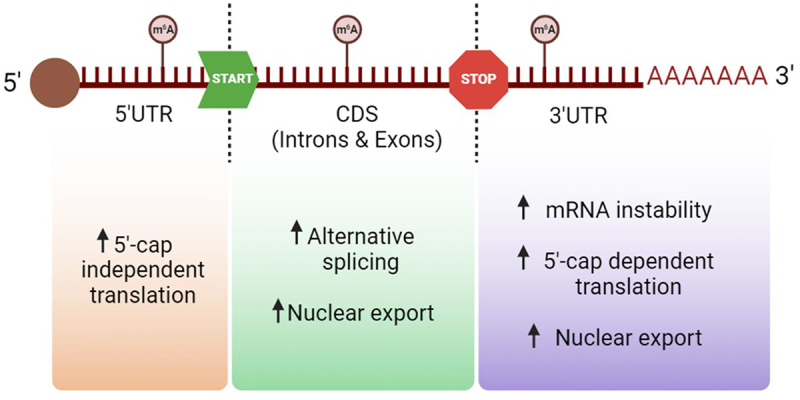
Biological effects of m6A modification in human mRNA depending on its position. Depending on where the modification m6A is added onto the mRNA molecule it may enhance different consequences, such as translation, splicing, its exportation or even an increase of the molecule’s instability. Created with BioRender.com [10].
Thanks to these new technologies, the field of epitranscriptomics has advanced enormously in the last decade, disclosing not only the different biological functions of many types of RNA modifications, but their role in different human diseases, such as obesity, neurological disorders, and cancer [35]. Unfortunately, there is a crucial aspect these NGS technologies cannot tackle: the great dynamism and heterogeneity of RNA modifications in a specific microenvironment. For all these reasons, we need single-cell technologies.
Single-cell RNA sequencing has uncovered the level of heterogeneity that the transcriptome of single cells can possess in an otherwise homogeneous cell group or tissue. This contributed to our knowledge about fate, cell identity, and function in the context of pathology and normal biology [36,37]. The advancement of single-cell RNA sequencing was continued by single-cell genome sequencing, which has generated new understandings about the genomic stability and variations that take place in physiology and disease- like in reproductive medicine, cancer, or microbial genetics [38]. Different scRNA-seq technologies are already available, such as 10X [39], SMART-seq [40], SORT-seq [41], C1-CAGE [42], etc. These methodologies usually differ between reverse transcription, amplification, and cDNA synthesis strategies, and whether they accommodate sequence-specific barcodes (UMIs) or the possibility of processing pooled samples [43]. In 2017, two new approaches were established to measure simultaneously single cells mRNA and expression of protein, known as REAP-seq [44], and CITE-seq [45]. Additionally, the Tapestry Platform is the only technology capable of providing both phenotype and genotype data from the same single cell [46]. In addition, the recently developed spatial transcriptomics can employ unique positional barcodes to identify and visualize the distribution of RNA in RNA sequencing of tissue sections [47].
On the other hand, methods for analysing the single-cell epigenome can distinguish between open and closed chromatin and determine the position of nucleosomes across the genome [48–54]. In addition, new single-cell chromatin immunoprecipitation sequencing (scChIP-seq) technologies allow us to study how different transcription factors bind to the genome. Lastly, some DNA modifications, like methylation (5mC), hydroxymethylation (5hmC), and formylation (5fC), can be identified at a single-cell level by sequencing in most parts of the genome, along with single-nucleotide resolution [55–59]. These modifications are linked, for instance, with transcriptional repression (5mC) or activation (5hmC and 5fC) of gene promoters and enhancers. Thus, currently, we can research most of the epigenetic dimensions with single-cell resolution. However, within the realm of epitranscriptomics, single-cell technologies are still in their infancy. In fact, at present there only exist single-cell technologies designed for analysing two type RNA modifications: m6A and inosine.
Single-cell technologies for mapping m6A
Epitranscriptomics is an essential component of gene expression, and the methylation of adenosine at the N6 position (m6A) is the most abundantly found in mRNA [60]. However, the present strategies used for m6A detection need high levels of input RNA and, consequently, every transcriptome-wide m6A profiling study until now has mapped m6A in bulk populations that consist of thousands or millions of cells.
Following the first studies of m6A mapping methods from 2012 (m6A RNA immunoprecipitation and sequencing (MeRIP – seq [24]), various bulk methodologies have been designed: antibody-based PA-m6A-seq [61], miCLIP [62], m6A-CLIP [25] and m6A-LAIC-seq [63] and antibody-free DART-seq [64], MAZTER-seq [65], m6A-REF-seq [66] and m6A-SEAL [67]. Methods based on immunoprecipitation (IP) do not supply m6A stoichiometry nor single-nucleotide resolution. However, they can estimate the position based on the RRACH motif and can be employed for differential enrichment analysis with tools like DESeq2. The mapping of m6A usually demands big amounts of input material. The minimum amount of starting material reported to date is 10 ng of total RNA when using the DART-seq technique [64]. Despite these progressions, there is an ample need for strongly sensitive and single cell m6A mapping methods that can be used in primary cell types. Up until now, only five technologies have been developed for this purpose: m6AISH-PLA, Epitranscriptome profiling, scDART-seq, scm6A-seq and picoMeRIP – seq, (Table 1 and Figure 3).
Table 1.
List of single-cell technologies being used to study m6A RNA modification, with the correspondent cell types it was used on and a small explanation of the various methodologies.
| Single-cell methods used to study RNA modification: m6A | |||
|---|---|---|---|
| Method name | Single cells used on | Method | Ref. |
| m6AISH-PLA | Hepa 1–6 Cell Line | m6A-specific in situ hybridization mediated proximity ligation assay for cellular imaging of m6A RNA. It utilizes two proximity probes to target m6A-specific RNA and m6A methylation, followed by ligation and in situ rolling circle amplification (RCA) | [68] |
| Epitranscriptome profiling of microscopy-based, low-input samples and individual cells | MUTZ3 leukaemia cells | mRNAs from cell lysates on oligo-dT-coated coverslips are captured, individual m6A-immunobloated transcripts are visually detected and sequenced without being amplified. Finally, a nanoscale machine allows the isolation of individual cells, and thus, relate cell surface markers and gene expression signatures to single-cell m6A modification states | [69] |
| scDART-seq | HEK293T cell line | Deamination adjacent to RNA modification targets, cells expressing inducible APOBEC1-YTH | [70] |
| scm6A-seq | In single oocytes/blastomeres of cleavage-stage embryos | RNA fragmentation and ligation, labelling each twice and parallelized single-cell seq | [71] |
| picoMeRIP-seq | In mouse embryonic stem (mES) cells and in single zebrafish zygotes, single mouse oocytes and preimplantation embryos | RNA fragmentation, sonication, and RNA immunoprecipitation with validated anti-m6A | [72] |
Figure 3.
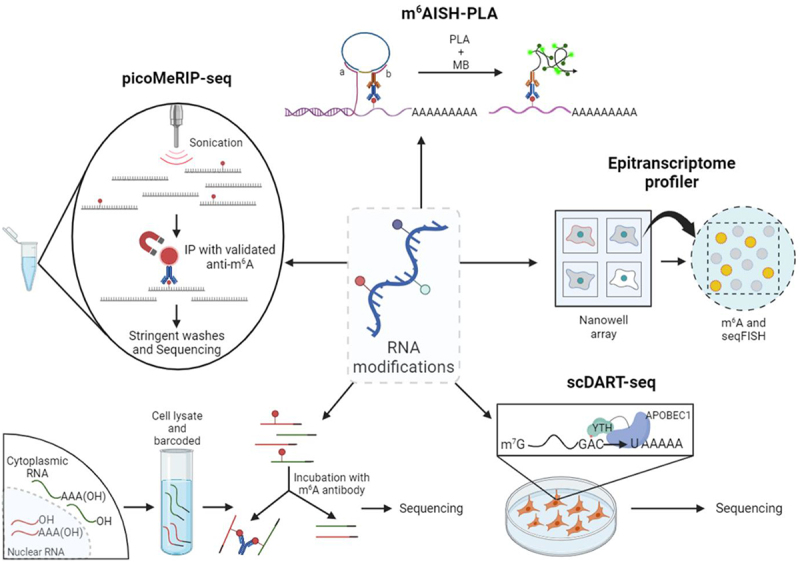
Single-cell m6A sequencing technologies. From the top in a clockwise manner, small explanation of all existing techniques: m6AISH-PLA, epitranscriptome profiler, scDART-seq, scm6A-seq and picoMeRIP-seq. Created with BioRender.com [10].
The first single-cell epitranscriptomic mapping method to ever be used was m6A-specific in situ hybridization mediated proximity ligation assay (m6AISH-PLA) [68], which allows the identification of m6A modification at certain locations in RNA and to image m6A RNA at single-molecule resolution. This methodology presented some advantages from pre-existing techniques: it can locate m6A at specific places in mRNA by two recognition events of m6A modification and RNA sequence, and its isothermal and mild conditions are useful to maintain cellular morphology and structure, helping to analyse m6A modified RNAs in the tissues, and their one-target-one-amplicon amplification allows the quantification of m6A RNA with single-molecule resolution, providing rigorous quantitative and spatial measurements. However, m6AISH-PLA has low throughput (cell number ≥ 100) and it’s not capable of distinguishing m6A sites located within a short sequence range because of the limitation of PLA in physical distance.
In addition, a microscopy-based technology to measure gene expression, cell surface markers, and m6A modification in single cells and at single-molecule resolution was also developed [69]. Various innovations in nanowell technology were combined, together with low-quality digital gene expression (LQ-DGE), image registration, and sequential fluorescence in situ hybridization (seqFISH) to produce data containing many parameters from single cells. Essentially, an open platform was validated for multi-modal single-cell experiments. At a single-cell level, the use of nanowells has enabled to analyse the total polyA+ RNA content, and to quantify cell surface markers, absolute numbers of individual transcripts, and RNA modifications, all at the same time and from the same individual cells. Cellular barcodes are not necessary for this technique because of the steps of image registration and direct imaging between molecular imaging and cellular phenotyping, which prevents having to use difficult library preparation steps. It is possible that in future versions the nanowell technology will allow RNA density optimization and stimulate the integration with single-molecule sequencing methodologies, like LQ-DGE, which would expand the transcriptome-wide throughput and make possible for the measurements of single isoforms and allelic expression.
In general, this would seem like a flexible and effective method to measure the epigenetic m6A modification of mRNAs, their transcript quantity and surface proteins at a single-molecule and single-cell level. However, their platform to detect RNA modifications in individual molecules leans towards a binary interpretation, so it can detect its absence or presence over stoichiometry of the modification’s locations in a transcript. Also, in a transcript, despite the number of m6A-modified sites, the detection of antibody fluorescent signal is transformed into m6A-positive transcripts while the single-molecule image is being processed. This way, the assay differentiates the transcripts as those containing one or more modifications or those that are unmodified. Additionally, cross-reactivity of the m6A antibody and the cap-specific m6Am modification exist, and it could confuse the interpretation of the data from other systems. Thus, for this method, it is recommended to supplement the analysis of single-cell data with other assays like DART sequencing, m6Am-exo sequencing and/or LC-MS analysis, which means this technology, by itself, is not yet capable of differentiating m6A modification in single-cells appropriately.
Afterwards, single-cell DART-seq (scDART-seq) [70] was developed. In this technique, they induced APOBEC1-YTH (cytidine deaminase APOBEC1 fused to YT521-B homology domain) expression in HEK293T cells and performed droplet-based scRNA-seq and SMART-seq2 technology. scDART-seq was established as a robust m6A detection method in single cells and it can identify infrequently methylated mRNAs which could otherwise dodge detection by standard bulk m6A mapping approaches. This methodology is very promising for studying m6A modifications in mRNA from individual cells since it managed to discover that many m6A sites which only show modest stoichiometry estimates at the population level are quite abundant in subpopulations of individual cells. Additionally, it is also possible to cluster groups of cells based on their m6A abundance patterns which reveal m6A as a potential driving force for co-regulation of subpopulations of cells. Nevertheless, scDART-seq also has some important limitations such as the fact that it needs the expression of APOBEC1-YTH in cells or tissues of interest, and that, as with many m6A mapping approaches, it is possible for it to detect false-positive m6A sites. On the other hand, some sites can also be missed because of inaccessibility or low abundance of the APOBEC1-YTH protein.
Single-cell m6A sequencing (scm6A-seq) [71] technology can simultaneously profile the m6A methylome and transcriptome in single oocytes/blastomeres of cleavage-stage embryos. In this single-cell method, the RNA molecules from each cell must first be fragmented, and then be labelled with two rounds of barcoded DNA adapters for multiplexed single-cell sequencing. To manage that, the barcoded RNA molecules from different cells are pooled together and subjected to RNA-seq and m6A immunoprecipitation (m6A-IP). Thus, this methodology enables in-depth research of m6A functions and characteristics, and the results have provided valuable single-cell resolution resources to define the mechanism involved in gametogenesis and early embryonic development. In addition, scm6A-seq is more sensitive than other strategies, especially for low-abundant modified RNAs, since it minimizes the variance and batch effect (non-biological inter-sample variance) of enrichment efficiency among individual cells. By barcoding each single cell separately and then pooling together the different cells from different samples, the inter-sample variance due to batch effect is avoided, making it possible to compare the relative m6A level directly among individual cells. It can also detect m6A at single-cell resolution under natural conditions, without the need for exogenously engineered YTH-APOBEC1 gene expression. However, until now, this technique has only been employed on single oocytes/blastomeres of cleavage-stage embryos but not on single cells from other tissues.
Finally, picogram-scale m6A RNA immunoprecipitation and sequencing (picoMeRIP – seq) [72] has been the latest developed and benchmarked method for small-scale and single-cell m6A mapping, which does not require the use of specialized equipment, making it easier to be adopted by many laboratories. It is expected that this technique will allow the profiling of m6A in some limited cell types from various in vivo sources, like in biopsies from healthy and diseased tissues. Additionally, its sensitivity allows for single-oocyte and single-embryo studies, therefore, this method could reveal the m6A landscape of preimplantation embryos and human oocytes in connection to developmental defects and fertility. Still, some improvements are needed on picoMeRIP – seq since they also tried to apply it to single mES cells but could not acquire enough libraries for sequencing. Possibly in the future, higher-throughput analysis will be capable of further evaluating the heterogeneity between single cells.
Single-cell technologies for mapping inosine
On the other hand, inosine is a crucial RNA modification, resulting from the A-to-I RNA editing process, catalysed by the family of deaminases ADAR [73]. Inosine is usually deposited at the anticodon wobble position of many tRNA molecules, modulating the protein sequence, and in different positions of a wide range of mRNA molecules, modulating their localization, splicing and stability. Dysregulation in A-to-I editing has been seen to be related with several diseases, such as autoimmune diseases or even cancer progression [74].
Since inosine naturally pairs with a cytosine after RNA retrotranscription to cDNA, inosine is substituted as guanine after PCR. Thus, to detect inosine, a classic RNA-seq is usually performed, followed with a A-to-G variant calling. In a single-cell scenario, and similarly to the bulk counterpart, inosine detection can be performed with any scRNA-seq technology, such as SMART-seq, or any 3’-oriented sequencing protocols (CEL-seq, DROP-seq, In-DROP, or 10× Chromium). A-to-G variant calling is usually performed with recoding editing index (REI) algorithms, such as QEdit [75].
Future perspectives
The technological development of single-cell epitranscriptomic analysis is still in its early stages. At present, out of the over 170 RNA modifications identified to date, only two of them (m6A and inosine) can be analysed at the single-cell level. New single-cell epitranscriptomic technologies will soon emerge, enhancing our understanding of the diverse RNA modifications within the cellular heterogeneity of healthy and dysfunctional tissues.
Until now, all transcriptome-wide m6A profiling studies had examined this modification with bulk RNA-sequencing, a technology that captures only an ‘average’ of the expression profiles from thousands up to millions of cells. However, with this review, it was possible to highlight that there are essential characteristics of m6A and inosine biology which can only be studied properly via single-cell RNA-sequencing.
The field of single-cell epitranscriptomic sequencing is growing to be capable of investigating a broadening range of modifications, not only for m6A or inosine in cultured or early embryonic development cells but also for different types of RNA modifications and in various types of cells and samples. To do so, it would be imperative for these hypothetical future technologies to diminish the number of steps necessary for the sample preparation before sequencing, standardizing all the procedures (by establishing gold standards) and creating an automated protocol for single-cell epitranscriptomic sequencing, allowing to lower their cost and to be broadly used. Additionally, in the years to come, it would be also necessary for these techniques to allow for a higher-throughput analysis (improving the number of cells able to be isolated and processed), provide better coverage of the numbers of expressed genes (which is usually lower in single-cell technologies when compared with population-level ensemble measurements, bulk RNA-seq) and to solve the ‘drop-out’ problem, where weakly (and even moderately) expressed genes can be missed during sequencing, mostly caused because of the low resolution of these current techniques.
Funding Statement
We thank CERCA Programme/Generalitat de Catalunya for institutional support. The research leading to these results has received funding from MCIN/AEI/10.13039/501100011033/and the European Development Regional Fund, ‘A way to make Europe’ ERDF (project PID2021-125282OB-I00); Departament de Recerca i Universitats/Generalitat de Catalunya (2021 SGR 01494) and the Cellex Foundation.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- [1].Cappannini A, Ray A, Purta E, et al. Modomics: a database of RNA modifications and related information. 2023 Nucleic Acids Res. 2023;52(D1):D239–D244. doi: 10.1093/nar/gkad1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Fersht AR. Review lecture enzymic editing mechanisms and the genetic code. Proc R Soc Lond B Biol Sci. 1981;212(1189):351–379. [DOI] [PubMed] [Google Scholar]
- [3].Benne R, Van Den Burg J, Brakenhoff JPJ, et al. Major transcript of the frameshifted coxll gene from trypanosome mitochondria contains four nucleotides that are not encoded in the DNA. Cell. 1986;46(6):819–26. doi: 10.1016/0092-8674(86)90063-2 [DOI] [PubMed] [Google Scholar]
- [4].Desrosiers R, Friderici K, Rottman F.. Identification of methylated nucleosides in messenger RNA from novikoff hepatoma cells. Proc Nat Acad Sci. 1974;71(10):3971–3975. doi: 10.1073/pnas.71.10.3971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Perry RP, Kelley DE, LaTorre J. Synthesis and turnover of nuclear and cytoplasmic polyadenylic acid in mouse L cells. J Mol Biol. 1974;82(3):315–31. doi: 10.1016/0022-2836(74)90593-2 [DOI] [PubMed] [Google Scholar]
- [6].Wei CM, Gershowitz A, Moss B. Methylated nucleotides block 5’ terminus of HeLa cell messenger RNA. Cell. 1975;4(4):379–386. doi: 10.1016/0092-8674(75)90158-0 [DOI] [PubMed] [Google Scholar]
- [7].Cohn WE. Pseudouridine, a Carbon-Carbon Linked Ribonucleoside in Ribonucleic Acids: Isolation, Structure, and Chemical Characteristics. J Biol Chem. 1960;235(5):1488–98. doi: 10.1016/S0021-9258(18)69432-3 [DOI] [PubMed] [Google Scholar]
- [8].Amos H, Korn M. 5-methyl cytosine in the RNA of Escherichia coli. Biochim Biophys Acta. 1958;29(2):444–5. doi: 10.1016/0006-3002(58)90214-2 [DOI] [PubMed] [Google Scholar]
- [9].Esteve-Puig R, Bueno-Costa A, Esteller M. Writers, readers and erasers of RNA modifications in cancer. Cancer Lett. 2020;474:127–37. doi: 10.1016/j.canlet.2020.01.021 [DOI] [PubMed] [Google Scholar]
- [10].BioRender : 2023. BioRender; [cited 2023. Available from: https://www.biorender.com/.
- [11].Limbach PA, Crain PF, McCloskey JA. Summary: the modified nucleosides of RNA. Nucleic Acids Res. 1994;22(12):2183–96. doi: 10.1093/nar/22.12.2183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jora M, Lobue PA, Ross RL, et al. Detection of ribonucleoside modifications by liquid chromatography coupled with mass spectrometry. Biochim Biophys Acta, Gene Regul Mech. 2019;1862(3):280–90. doi: 10.1016/j.bbagrm.2018.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Schaefer M, Kapoor U, Jantsch MF. Understanding RNA modifications: the promises and technological bottlenecks of the ‘epitranscriptome’. Open Biol. 2017;7(5):170077. doi: 10.1098/rsob.170077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Schwartz S, Bernstein Douglas A, Mumbach Maxwell R, et al. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell. 2014;159(1):148–162. doi: 10.1016/j.cell.2014.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Carlile TM, Rojas-Duran MF, Zinshteyn B, et al. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature. 2014;515(7525):143–6. doi: 10.1038/nature13802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Birkedal U, Christensen-Dalsgaard M, Krogh N, et al. Profiling of ribose methylations in RNA by high-throughput sequencing. Angewandte Chemie. 2015;54(2):451–5. doi: 10.1002/anie.201408362 [DOI] [PubMed] [Google Scholar]
- [17].Marchand V, Blanloeil-Oillo F, Helm M, et al. Illumina-based RiboMethSeq approach for mapping of 2’-O-Me residues in RNA. Nucleic Acids Res. 2016;44(16):e135. doi: 10.1093/nar/gkw547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Squires JE, Patel HR, Nousch M, et al. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012;40(11):5023–33. doi: 10.1093/nar/gks144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Edelheit S, Schwartz S, Mumbach MR, et al. Transcriptome-wide mapping of 5-methylcytidine RNA modifications in bacteria, archaea, and yeast reveals m5C within archaeal mRnas. PLoS Genet. 2013;9(6):e1003602. doi: 10.1371/journal.pgen.1003602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Oakes E, Anderson A, Cohen-Gadol A, et al. Adenosine Deaminase That Acts on RNA 3 (ADAR3) Binding to Glutamate Receptor Subunit B Pre-mRNA Inhibits RNA Editing in Glioblastoma. J Biol Chem. 2017;292(10):4326–35. doi: 10.1074/jbc.M117.779868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dominissini D, Nachtergaele S, Moshitch-Moshkovitz S, et al. The dynamic N1-methyladenosine methylome in eukaryotic messenger RNA. Nature. 2016;530(7591):441–6. doi: 10.1038/nature16998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Li X, Xiong X, Wang K, et al. Transcriptome-wide mapping reveals reversible and dynamic N1-methyladenosine methylome. Nat Chem Biol. 2016;12(5):311–316. doi: 10.1038/nchembio.2040 [DOI] [PubMed] [Google Scholar]
- [23].Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–6. doi: 10.1038/nature11112 [DOI] [PubMed] [Google Scholar]
- [24].Meyer Kate D, Saletore Y, Zumbo P, et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3‘UTRs and near stop codons. Cell. 2012;149(7):1635–1646. doi: 10.1016/j.cell.2012.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ke S, Alemu EA, Mertens C, et al. A majority of m6A residues are in the last exons, allowing the potential for 3’ UTR regulation. Genes Dev. 2015;29(19):2037–2053. doi: 10.1101/gad.269415.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Amort T, Rieder D, Wille A, et al. Distinct 5-methylcytosine profiles in poly(A) RNA from mouse embryonic stem cells and brain. Genome Bio. 2017;18(1):1. doi: 10.1186/s13059-016-1139-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ortiz-Barahona V, Soler M, Davalos V, et al. Epigenetic inactivation of the 5-methylcytosine RNA methyltransferase NSUN7 is associated with clinical outcome and therapeutic vulnerability in liver cancer. Mol Cancer. 2023;22(1):83. doi: 10.1186/s12943-023-01785-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Janin M, Ortiz-Barahona V, de Moura MC, et al. Epigenetic loss of RNA-methyltransferase NSUN5 in glioma targets ribosomes to drive a stress adaptive translational program. Acta Neuropathol. 2019;138(6):1053–74. doi: 10.1007/s00401-019-02062-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].David R, Burgess A, Parker B, et al. Transcriptome-wide mapping of RNA 5-methylcytosine in Arabidopsis mRnas and noncoding RNAs. Plant Cell. 2017;29(3):445–60. doi: 10.1105/tpc.16.00751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bazak L, Haviv A, Barak M, et al. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014;24(3):365–76. doi: 10.1101/gr.164749.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chen J-W, Shrestha L, Green G, et al. The hitchhikers’ guide to RNA sequencing and functional analysis. Brief Bioinform. 2023;24(1). doi: 10.1093/bib/bbac529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ke S, Pandya-Jones A, Saito Y, et al. m(6)A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 2017;31(10):990–1006. doi: 10.1101/gad.301036.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhou KI, Shi H, Lyu R, et al. Regulation of Co-transcriptional pre-mRNA splicing by m(6)A through the low-complexity protein hnRNPG. Mol Cell. 2019;76(1):70–81.e9. doi: 10.1016/j.molcel.2019.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Liu C, Sun H, Yi Y, et al. Absolute quantification of single-base m6A methylation in the mammalian transcriptome using GLORI. Nature Biotechnol. 2023;41(3):355–66. doi: 10.1038/s41587-022-01487-9 [DOI] [PubMed] [Google Scholar]
- [35].Jonkhout N, Tran J, Smith MA, et al. The RNA modification landscape in human disease. RNA. 2017;23(12):1754–69. doi: 10.1261/rna.063503.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Stubbington MJT, Rozenblatt-Rosen O, Regev A, et al. Single-cell transcriptomics to explore the immune system in health and disease. Science. 2017;358(6359):58–63. doi: 10.1126/science.aan6828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lein E, Borm LE, Linnarsson S. The promise of spatial transcriptomics for neuroscience in the era of molecular cell typing. Science. 2017;358(6359):64–9. doi: 10.1126/science.aan6827 [DOI] [PubMed] [Google Scholar]
- [38].Gawad C, Koh W, Quake SR. Single-cell genome sequencing: current state of the science. Nat Rev Genet. 2016;17(3):175–88. doi: 10.1038/nrg.2015.16 [DOI] [PubMed] [Google Scholar]
- [39].Zheng GXY, Terry JM, Belgrader P, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8(1):14049. doi: 10.1038/ncomms14049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ramsköld D, Luo S, Wang YC, et al. Full-length mRNA-seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol. 2012;30(8):777–82. doi: 10.1038/nbt.2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Muraro MJ, Dharmadhikari G, Grün D, et al. A single-cell transcriptome atlas of the human pancreas. Cell Syst. 2016;3(4):385–94.e3. doi: 10.1016/j.cels.2016.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kouno T, Moody J, Kwon AT, et al. C1 CAGE detects transcription start sites and enhancer activity at single-cell resolution. Nat Commun. 2019;10(1):360. doi: 10.1038/s41467-018-08126-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Dal Molin A, Di Camillo B. How to design a single-cell RNA-sequencing experiment: pitfalls, challenges and perspectives. Brief Bioinform. 2018;20(4):1384–1394. doi: 10.1093/bib/bby007 [DOI] [PubMed] [Google Scholar]
- [44].Peterson VM, Zhang KX, Kumar N, et al. Multiplexed quantification of proteins and transcripts in single cells. Nature Biotechnol. 2017;35(10):936–9. doi: 10.1038/nbt.3973 [DOI] [PubMed] [Google Scholar]
- [45].Stoeckius M, Hafemeister C, Stephenson W, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. 2017;14(9):865–868. doi: 10.1038/nmeth.4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ruff DW, Dhingra DM, Thompson K, et al. High-throughput multimodal single-cell targeted DNA and surface protein analysis using the mission bio tapestri platform. In: Ooi A, editor. Single-cell protein analysis: methods and protocols. New York, NY: Springer US; 2022. pp. 171–188. [DOI] [PubMed] [Google Scholar]
- [47].Ståhl PL, Salmén F, Vickovic S, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016;353(6294):78–82. doi: 10.1126/science.aaf2403 [DOI] [PubMed] [Google Scholar]
- [48].Jin W, Tang Q, Wan M, et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature. 2015;528(7580):142–6. doi: 10.1038/nature15740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Buenrostro JD, Wu B, Litzenburger UM, et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523(7561):486–90. doi: 10.1038/nature14590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Cusanovich DA, Daza R, Adey A, et al. Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science. 2015;348(6237):910–4. doi: 10.1126/science.aab1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Guo F, Li L, Li J, et al. Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells. Cell Res. 2017;27(8):967–88. doi: 10.1038/cr.2017.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Pott S. Simultaneous measurement of chromatin accessibility, DNA methylation, and nucleosome phasing in single cells. Elife. 2017;6:e23203. doi: 10.7554/eLife.23203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Clark SJ, Argelaguet R, Kapourani C-A, et al. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat Commun. 2018;9(1):781. doi: 10.1038/s41467-018-03149-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Casado-Pelaez M, Bueno-Costa A, Esteller M. Single cell cancer epigenetics. Trends Cancer. 2022;8(10):820–38. doi: 10.1016/j.trecan.2022.06.005 [DOI] [PubMed] [Google Scholar]
- [55].Guo H, Zhu P, Wu X, et al. Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome Res. 2013;23(12):2126–35. doi: 10.1101/gr.161679.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Smallwood SA, Lee HJ, Angermueller C, et al. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat Methods. 2014;11(8):817–820. doi: 10.1038/nmeth.3035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Farlik M, Sheffield Nathan C, Nuzzo A, et al. Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics. Cell Rep. 2015;10(8):1386–1397. doi: 10.1016/j.celrep.2015.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Mooijman D, Dey SS, Boisset J-C, et al. Single-cell 5hmC sequencing reveals chromosome-wide cell-to-cell variability and enables lineage reconstruction. Nature Biotechnol. 2016;34(8):852–6. doi: 10.1038/nbt.3598 [DOI] [PubMed] [Google Scholar]
- [59].Zhu C, Gao Y, Guo H, et al. Single-cell 5-formylcytosine landscapes of mammalian early embryos and ESCs at single-base resolution. Cell Stem Cell. 2017;20(5):720–31.e5. doi: 10.1016/j.stem.2017.02.013 [DOI] [PubMed] [Google Scholar]
- [60].Zeng C, Huang W, Li Y, et al. Roles of METTL3 in cancer: mechanisms and therapeutic targeting. J Hematol Oncol. 2020;13(1):117. doi: 10.1186/s13045-020-00951-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Chen K, Lu Z, Wang X, et al. High-resolution N6-methyladenosine (m6A) map using photo-crosslinking-assisted m6A sequencing. Angewandte Chemie. 2015;54(5):1587–90. doi: 10.1002/anie.201410647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Linder B, Grozhik AV, Olarerin-George AO, et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12(8):767–772. doi: 10.1038/nmeth.3453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Molinie B, Wang J, Lim KS, et al. m6A-LAIC-seq reveals the census and complexity of the m6A epitranscriptome. Nat Methods. 2016;13(8):692–698. doi: 10.1038/nmeth.3898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Meyer KD. DART-seq: an antibody-free method for global m6A detection. Nat Methods. 2019;16(12):1275–1280. doi: 10.1038/s41592-019-0570-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Garcia-Campos MA, Edelheit S, Toth U, et al. Deciphering the m6A code via antibody-independent quantitative profiling. Cell. 2019;178(3):731–47.e16. doi: 10.1016/j.cell.2019.06.013 [DOI] [PubMed] [Google Scholar]
- [66].Zhang Z, Chen L-Q, Zhao Y-L, et al. Single-base mapping of m6A by an antibody-independent method. Sci Adv. 2019;5(7):eaax0250. doi: 10.1126/sciadv.aax0250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Wang Y, Xiao Y, Dong S, et al. Antibody-free enzyme-assisted chemical approach for detection of N6-methyladenosine. Nat Chem Biol. 2020;16(8):896–903. doi: 10.1038/s41589-020-0525-x [DOI] [PubMed] [Google Scholar]
- [68].Ren X, Deng R, Zhang K, et al. Single-cell imaging of m6A modified RNA using m6A-Specific in situ hybridization mediated proximity ligation assay (m6AISH-PLA). Angewandte Chemie. 2021;60(42):22646–51. doi: 10.1002/anie.202109118 [DOI] [PubMed] [Google Scholar]
- [69].Kim KL, van Galen P, Hovestadt V, et al. Systematic detection of m(6)A-modified transcripts at single-molecule and single-cell resolution. Cell Rep Methods. 2021;1(5):100061. doi: 10.1016/j.crmeth.2021.100061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Tegowski M, Flamand MN, Meyer KD. scDART-seq reveals distinct m6A signatures and mRNA methylation heterogeneity in single cells. Molecular Cell. 2022;82(4):868–78.e10. doi: 10.1016/j.molcel.2021.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Yao H, Gao C-C, Zhang D, et al. scm6A-seq reveals single-cell landscapes of the dynamic m6A during oocyte maturation and early embryonic development. Nat Commun. 2023;14(1):315. doi: 10.1038/s41467-023-35958-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Li Y, Wang Y, Vera-Rodriguez M, et al. Single-cell m6A mapping in vivo using picoMeRIP–seq. Nature Biotechnol. 2023. doi: 10.1038/s41587-023-01831-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Nishikura K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol. 2016;17(2):83–96. doi: 10.1038/nrm.2015.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Jain M, Jantsch MF, Licht K. The Editor’s I on disease development. Trends Genet. 2019;35(12):903–913. doi: 10.1016/j.tig.2019.09.004 [DOI] [PubMed] [Google Scholar]
- [75].Fonzino A, Pesole G, Picardi E. Profiling RNA editing in single cells. Methods Mol Biol. 2023;2584:347–370. [DOI] [PubMed] [Google Scholar]