

Abstract
Background/Aims.
In cholangiocarcinoma, early metastatic spread via lymphatic vessels often precludes curative therapies. Cholangiocarcinoma invasiveness is fostered by an extensive stromal reaction, enriched in cancer-associated fibroblasts (CAF) and lymphatic endothelial cells (LEC). Cholangiocarcinoma cells recruit and activate CAF by secreting PDGF-D. Here we investigated the role of PDGF-D and liver myofibroblasts in promoting lymphangiogenesis in cholangiocarcinoma.
Methods.
Human cholangiocarcinoma specimens were immunostained for podoplanin (LEC marker), α-SMA (CAF), VEGF-A, VEGF-C, and their cognate receptors (VEGFR2, VEGFR3). VEGF-A and VEGF-C secretion (ELISA) was evaluated in human fibroblasts obtained from primary sclerosing cholangitis explants stimulated by PDGF-D, with/without the PDGFRβ inhibitor imatinib. Using human LEC incubated with conditioned medium from PDGF-D-stimulated fibroblasts (with/without imatinib), we assessed migration (Boyden chambers), 3-D vascular assembly (AngioTool), trans-endothelial electric resistance and trans-endothelial migration of cholangiocarcinoma cells (EGI-1). In Fischer-344 rats transplanted with a syngeneic cholangiocarcinoma cell line, we studied effects of selective CAF depletion induced by the BH3 mimetic navitoclax on LEC density and lymphnode metastases.
Results.
In cholangiocarcinoma specimens, CAF and LEC were closely adjacent. CAF expressed VEGF-A and VEGF-C, while LEC expressed VEGFR2 and VEGFR3. Upon PDGF-D stimulation, fibroblasts secreted increased levels of VEGF-C and VEGF-A. Fibroblasts, stimulated by PDGF-D induced LEC recruitment and 3-D assembly, increased LEC monolayer permeability, and promoted trans-endothelial EGI-1 migration. These effects were all suppressed by imatinib. In the rat model of cholangiocarcinoma, navitoclax-induced CAF depletion markedly reduced lymphatic vascularization and lymphnode metastases.
Conclusion.
PDGF-D stimulates VEGF-C and VEGF-A production by fibroblasts, resulting in expansion of the lymphatic vasculature and tumor cell intravasation. This process critical for the early metastatization of cholangiocarcinoma, may be blocked by inducing CAF apoptosis or by inhibiting PDGF-D-induced axis.
Keywords: cholangiocytes, lymphatic endothelial cells, tumor reactive stroma, VEGF-C, VEGFR3
Graphical abstract
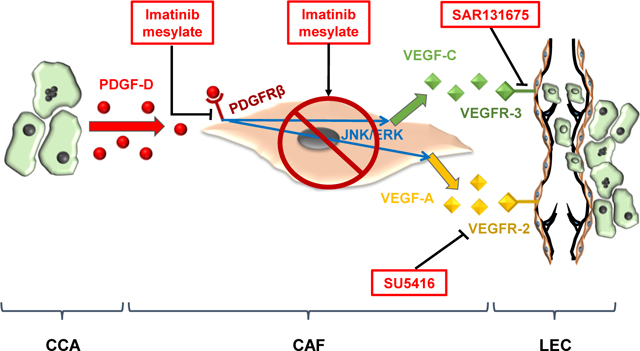
LAY SUMMARY
Cholangiocarcinoma (CCA) is a highly malignant cancer affecting the biliary tree. In CCA, a rich stromal reaction densely populated by cancer-associated fibroblasts promotes early metastatic spread. Here we show that CCA-derived PDGF-D recruiting fibroblasts, also stimulates them to secrete VEGF-A and VEGF-C. These vascular growth factors kindle both lymphatic vascularization and tumour cell vascular invasion. Thus, targeting fibroblasts or PDGF-D-induced signals may represent an effective tool to block tumor-associated lymphangiogenesis and reduce CCA invasiveness.
INTRODUCTION
Cholangiocarcinoma (CCA) originates from the intrahepatic or extrahepatic bile ducts and unfortunately, carries a very poor prognosis.1 Despite of its increasing incidence, effective treatment options for CCA are scarce and limited to surgical resection or liver transplantation in few highly selected patients.1 Less than one third of patients are eligible for curative surgery at the time of diagnosis due to a proclivity for early lymph node metastatization.1,2 Although mechanisms promoting CCA invasiveness are still unclear,3 the lymphatic vessels that develop within the tumor provide an important initial route of metastatic dissemination. Indeed, several lines of evidence indicate that the expansion of the lymphatic bed correlates with both increased metastatization and poor prognosis in CCA.4,5
Tumor-associated lymphangiogenesis is driven by a number of soluble mediators, including vascular endothelial growth factor (VEGF)-A, VEGF-C, VEGF-D, angiopoietin (Ang)-1 and Ang-2, together with their cognate receptors VEGFR2 (for VEGF-A, VEGF-C and VEGF-D), VEGFR3 (for VEGF-C and VEGF-D), and Tie2 (for angiopoietins).6,7 In CCA, the inflammatory cells and fibroblasts of the tumor microenvironment represent the main source of VEGF.8 In fact, as in other ductal carcinomas with pronounced invasiveness (e.g., breast and pancreatic cancer),9,10 growth of tumoral bile ducts occurs in close contiguity with a rich stromal reaction, termed tumor reactive stroma, mainly composed of cancer-associated fibroblasts (CAF), tumor-associated macrophages, and lymphatic endothelial cells (LEC).11,12
Within the tumor reactive stroma, a multitude of paracrine signals is exchanged between the cancer and its stromal compartment, aimed at fostering local invasiveness and metastatic spread of the epithelial counterpart.8,13 CAF are the most abundant cell type in the tumor stroma in CCA, and recently, we demonstrated that they are locally recruited by malignant cholangiocytes that secrete platelet-derived growth factor (PDGF)-D.14 PDGF-D is, in fact, specifically produced by CCA cells upon hypoxic stimulus, and binds its cognate receptor PDGFRβ expressed by CAF.14 Furthermore, the concept that CAF are essential drivers of CCA growth has been highlighted by the observation that in a syngeneic rat model of CCA, selective CAF depletion from the tumor microenvironment by the pro-apoptotic BH3 mimetic navitoclax, a specific inhibitor of the anti-apoptotic Bcl2 proteins, suppressed tumor growth and improved animal survival.15
In the present study, we hypothesized that in addition to promoting CAF accumulation within the tumor stroma, PDGF-D stimulated their pro-lymphangiogenic abilities, eventually inducing the chemotaxis of LEC and their assembly in a proper vascular system favoring CCA cell intravasation. Furthermore, we tested the hypothesis that depletion of CAF by navitoclax would reduce the lymphatic vascularization of the tumor mass and lymphatic dissemination in vivo.
MATERIALS AND METHODS
Cell lines
Human lymphatic endothelial cells (LEC, purchased from ScienCell™) and human male EGI-1 cells (PDGF-D expressing extrahepatic CCA cell line, purchased from Deutsche Sammlung von Mikroorganismen und Zellkulturen) were employed for in vitro experiments. Activated stromal fibroblasts were isolated from human liver explants of primary sclerosing cholangitis (n=3), as previously published.14 Freshly isolated liver myofibroblasts were characterized by checking morphology and by evaluating their immunophenotype, including alpha-smooth muscle actin (α-SMA), PDGFRβ, vimentin, podoplanin, VEGFR2 and VEGFR-3, and compared either with LEC or CAF, including a primary cell line obtained from a human sample of resected intrahepatic CCA, and a commercially available cell line derived from renal carcinoma (CellBiologics) (Supplemental Fig.2 and Supplemental Fig.3). EGI-1 cells were characterized by expression of cytokeratin (K) 7, K19, EpCAM (clone HEA125), E-cadherin and β-catenin. Following experiments were run in cultured cells with <10 passages. Mycoplasma contamination was excluded using a specific biochemical test (Lonza). See Supplemental Materials for details.
Human tissue samples and immunohistochemistry
Formalin-fixed, paraffin-embedded (FFPE) histological samples of surgically resected intrahepatic CCA (n=6, 3M/3F) and hepatocellular carcinoma (HCC) (n=6, 4M/2F) were obtained from archival tissues of Treviso Regional Hospital (Italy). Further details are given in Supplemental Methods and Supplemental Table 1 and 2.
Syngeneic rat model of CCA
To evaluate whether targeting CAF affects tumor lymphangiogenesis and lymphatic spread in vivo, we used the syngeneic rat model of CCA, generated by intrahepatic injection of neoplastic, highly malignant cholangiocytes (BDEneu rat cells) into adult Fischer-344 male rats (Harlan). Rats were housed in a barrier facility with 12 h light-dark cycle with free access to water and standard mouse chow, in environmentally enriched cages with 2 rats for cage at a temperature of 21°C. After allowing tumor growth for two weeks, animals were randomly assigned to 2 experimental groups, treated with A) navitoclax [5mg/kg] or B) vehicle by intraperitoneum injection once daily for 10 consecutive days to selectively deplete CAF from the tumor stroma and then sacrificed (Supplemental Fig.4A).15 Cryosections of these rat CCA tumors with (n=6) and without (n=6) navitoclax treatment were stained by immunohistochemistry and dual immunofluorescence for PDGF-D, α-SMA, Lyve-1 (LEC marker) and CD31 (blood vessel marker) (same protocol as above). Slides were evaluated with respect to PDGF-D expression by CCA cells (immunohistochemistry), α-SMA density and Lyve-1+ and CD31+ microvascular density following CAF depletion (dual immunofluorescence). After sacrificing the rats, the abdominal cavity was opened by star-shaped incision. High-resolution photographs of the peritoneal cavity and para-aortic region were taken to assess for lymph node metastasis. Metastatic lymph nodes in both regions were counted by two independent observers (JM, AM). These studies were performed in accordance with and approved by the Institutional Animal Care and Use Committee (JM, AM, CF).15
Xenograft mouse model of CCA
To evaluate whether targeting LEC affects tumor lymphangiogenesis in vivo, we used the xenograft model of CCA. This was generated by intraportal injection of EGI-1 cells (500.000 cells suspended in PBS 100 μl) in male CD17/lcr-Prkdc severe combined immunodeficient (SCID) mice (6–8 weeks old; Charles River Laboratories), after transduction with a lentiviral vector encoding the firefly luciferase gene to enable detection of tumor engraftment by in vivo bioluminescence imaging before starting treatment.14 SCID mice were housed in our Specific Pathogen Free (SPF) animal facility in Allentown IVC cages (floor area 542 cm2) with 6 mice per cage. All mice received water and food ad libitum and were kept under a 12 h light/dark cycle in a well-ventilated room at an approximate temperature of 22°C. Mice acclimatized for a minimum of 7 days and a maximum of 15 days prior to be randomly assigned to treatment or vehicle groups.
Once tumor engraftment in the liver of EGI-1 cells was confirmed, mice were randomly divided into 3 experimental groups: a) controls (vehicle only, n=6); b) SU5416 (VEGFR2 inhibitor), at the dosage of 12.5mg/kg/die, by i.p. injection using micro-osmotic pumps (Alzet 1004, Durec) (n=6); c) SAR131675 (VEFGFR3 inhibitor), (100mg/kg/die), by oral gavage (n=5). At the end of a 3-week treatment, mice were sacrificed and FFPE liver tissue sections were evaluated for LMVD by immunohistochemistry for Lyve-1. See Supplemental Methods and Supplemental Fig.4B for further details. Procedures involving animals and their care were conform to the institutional guidelines that comply with national and international laws and policies (EEC Council Directive 86/609, OJ L 358, December 12, 1987), and approved by the Ethical Committee of the University of Padua (CEASA).
Quantification of lymphangiogenic growth factors
ELISA was performed from liver myofibroblasts and CAF culture media following incubation with recombinant human (rh) PDGF-D (100ng/ml, 24h, R&D Systems) to quantify secretion of the lymphangiogenic growth factors VEGF-A, VEGF-C, VEGF-D, Ang-1 or Ang-2. See Supplemental Material for details.
Western blotting (WB) was performed to assess the immunophenotype of LEC, liver myofibroblasts and CAF as above reported, and activation of p44/42, JNK and PI3K/AKT/mTOR pathway in LEC exposed to fibroblast conditioned medium after PDGF-D stimulation. Details of WB experiments are reported in Supplemental Methods, and Supplemental Table 2.
In vitro assessment of lymphangiogenesis.
Effects of conditioned medium harvested from fibroblasts after PDGF-D stimulation and from EGI-1 cells on LEC transwell migration and 3-D tube formation assay were studied as reported in Supplemental Materials. Details of conditioned medium preparation are given in Supplemental Methods. These experiments were performed with/without antagonism of PDGFRβ (on fibroblasts), VEGFR2 and VEGFR3 (on LEC). VEGF-A and VEGF-C served as positive controls. To see if PDGF-D could exert some direct effects on LEC, LEC transwell migration was also evaluated following exposure to PDGF-D at the same conditions (100ng/ml, 24h).
In vitro assessment of lymphatic intravasation by CCA cells.
Effects of conditioned medium harvested from fibroblasts after PDGF-D stimulation were studied even on transendothelial electric resistance (TEER) and transendothelial migration (TrEM) of EGI-1 cells transduced with a lentiviral vector encoding EGFP reporter gene, as previously described.14 In these conditions, we also tested the effects of binding VEGF-A and VEGF-C to prevent ligand interactions with its cognate receptor, by bevacizumab (250μg/ml)16 and anti-VEGF-C antibody (10μg/ml)17. See Supplemental Methods.
MTS assay was performed to check toxicity on LEC of all inhibitors (SU5416, SAR131675, navitoclax, bevacizumab) and to see if conditioned medium of PDGF-D-stimulated fibroblasts, VEGF-A, and VEGF-C induced LEC proliferation. See Supplemental Methods.
Statistical Analysis
Results are shown as the mean ± standard deviation. Statistical comparisons were performed using Student’s t-test (Origin8 software, OriginLab). A p value <0.05 was considered significant.
Number of experiments are reported in Supplemental Table 2.
RESULTS
CCA specimens show a rich lymphatic vascularization.
To evaluate the extent of tumor-associated lymphangiogenesis and angiogenesis, we quantified microvascular density of podoplanin+ and Lyve-1+ lymphatic vessels (LMVD) and of CD34+ blood vessels (BMVD) in human CCA samples. We compared results obtained in CCA with those obtained in HCC, a primary liver malignancy typically characterized by a rich blood vascularization. Clinical data of both cohorts of patients are shown in Supplemental Table 3. As compared to HCC, where extensive and strong BMVD and negligible LMVD were observed, CCA showed a marked increase in LMVD, but a less remarkable BMVD (Fig.1A-C). These findings confirm that expansion of the lymphatic vasculature is a defining feature of the typically desmoplastic CCA, in contrast to the scarcity of lymphatic vessels observed in HCC. Importantly, since podoplanin was reported to be displayed by CAF, by dual immunofluorescence for podoplanin and Lyve-1 we showed high coincident expression of the two markers with scant extra-lymphatic expression of podoplanin in CCA, thus confirming that in CCA, podoplanin is highly specific to identify the lymphatic vasculature (Supplemental Fig.1).
Fig.1: In CCA, lymphatic microvascular density (LMVD) is much more preponderant than in HCC, at variance with blood microvascular density (BMVD).

(A-B) In human archival paraffin sections, LMVD was more extensively represented in CCA compared to HCC, as shown by IHC for podoplanin and Lyve-1 (lymphatic endothelial cell marker). (C) On the contrary, BMVD, evaluated as number of CD34+ (blood endothelial cell marker) cells, was increased in HCC samples. Right-side the plots, representative pictures of podoplanin+ (A), Lyve-1+ (B), and CD34+ (C) structures are shown for CCA and HCC; some faint expression of podoplanin is expressed also by CAF. n=6; *p<0.01, using two-tail t test. Original magnification: 200x.
In human CCA specimens, CAF and LEC are in close vicinity and reciprocally express VEGF ligands and receptors.
We then investigated the spatial relationships between the lymphatic vasculature and CAF in CCA. Double immunostaining for α-SMA (CAF marker) and podoplanin (LEC marker) revealed that CAF and LEC are in close vicinity within the tumor reactive stroma of human CCA (Fig.2A). By dual immunofluorescence, we further observed that CAF expressed VEGF-A and VEGF-C, whereas their cognate receptors VEGFR2 and VEGFR3 were displayed by LEC (Fig.2B-E and Supplemental Fig.5). Some immunostaining for VEGF-A and VEGF-C could also be observed in inflammatory cells populating the tumor stroma. Conversely, in tumoral bile ducts, VEGF-A expression was patchy, and much weaker as compared with CAF, whereas VEGF-C was constantly negative (Fig.2F,G and Supplemental Fig.5). Overall, these data are consistent with the hypothesis that paracrine signals between CAF and LEC are responsible for the generation of a rich lymphatic plexus within the tumor microenvironment.
Fig.2: In human CCA specimens, lymphatic endothelial cells (LEC) closely align with CAF.
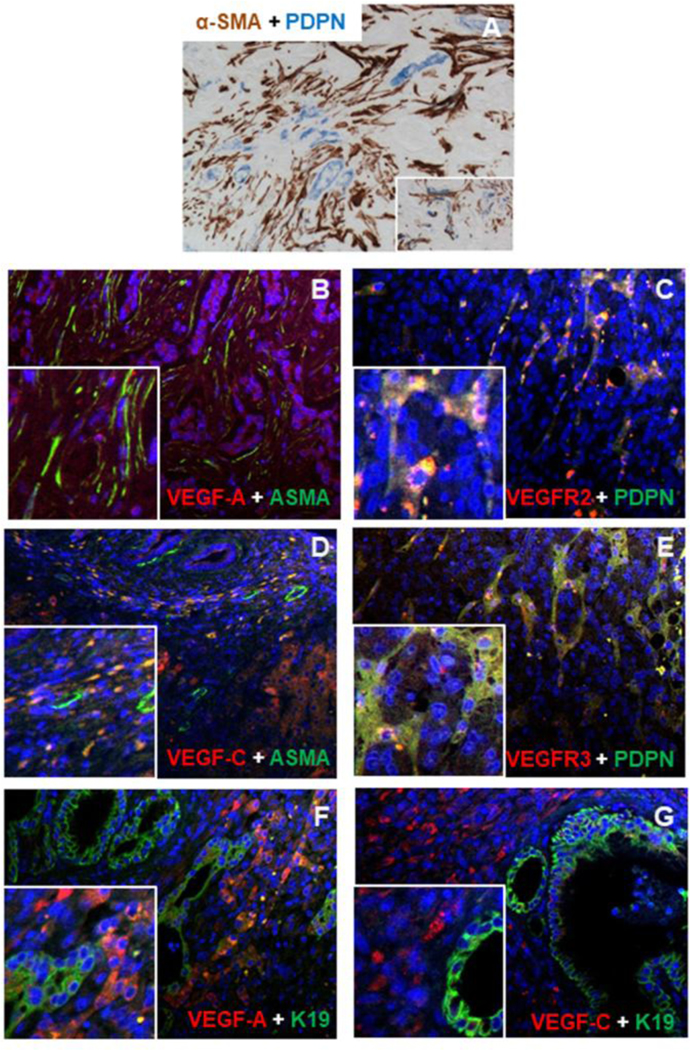
(A) Within the stroma of CCA, LEC were localized in close proximity to CAF, as shown by dual IHC for podoplanin (blue) and α-SMA (brown). (B, D) α-SMA+ CAF (green) expressed VEGF-A and VEGF-C (red), while (C, E) podoplanin+ LEC (green) reciprocally expressed VEGFR2 and VEGFR3 (red). (F) In malignant cholangiocytes (K19, green), expression of VEGF-A was weak and uneven (red), whilst (G) VEGF-C (red) was constantly not expressed. Original magnification: A-G, 200x; Insets: 400x.
Immunophenotype of liver fibroblasts, CAF and LEC.
By WB, we characterized distinctive profiles of liver fibroblasts, CAF and LEC, with expression of α-SMA, PDGFRβ, vimentin, and podoplanin by fibroblasts and CAF, and of podoplanin, vimentin, VEGFR2 and VEGFR-3 by LEC, though weak expression of podoplanin was present even on fibroblasts and CAF (Supplemental Fig.2 and Supplemental Fig.3A).
PDGF-D stimulates secretion of VEGF-A and VEGF-C, but not of VEGF-D, Ang-1 and Ang-2 by human fibroblasts.
To assess whether PDGF-D could provide fibroblasts with pro-lymphangiogenic functions, we stimulated primary human fibroblasts with PDGF-D, and evaluated the secretion of VEGF-A, VEGF-C, VEGF-D, Ang-1 and Ang-2, by ELISA. We found that PDGF-D-treated fibroblasts had a significant and markedly increased secretion of both VEGF-A and VEGF-C; on the other hand, Ang-1 was insensitive to PDGF-D stimulation and remained at much lower levels (Table 1). Of note, VEGF-D and Ang-2 secretion was never detectable. Notably, upon PDGF-D-stimulation, CAF from both CCA and renal carcinoma increased VEGF-A and VEGF-C secretion levels of an extent comparable to fibroblasts (Supplemental Fig.3B) that thus we used in the following experiments for their easy handling.
Table 1.
Assessment of lymphangiogenic growth factors secreted in the supernatant by cultured human fibroblasts exposed to PDGF-D.
| Ctrl | PDGF-D | |
|---|---|---|
| Ang-1 | 190.57±84.68 | 162.27±84.10 |
| Ang-2 | ND | ND |
| VEGF-A | 188.51±87.56 | 1152.48±297.11** |
| VEGF-C | 696.47±119.20 | 1715.30±579.83** |
| VEGF-D | ND | ND |
p<0.01 vs Ctrl; ND, not detectable
ERK and JNK signaling mediates VEGF-A and VEGF-C secretion by fibroblasts stimulated with PDGF-D.
We next examined the signaling pathways that mediate VEGF-A and VEGF-C secretion by fibroblasts, upon PDGFRβ activation by PDGF-D. VEGF-A and VEGF-C were measured in supernatants from fibroblasts challenged with PDGF-D, with or without inhibitors of PDGFRβ (imatinib mesylate), or of its downstream effectors ERK (U0126) and JNK (SP600125). PDGFRβ antagonism and ERK or JNK inhibition significantly counteracted the increase in VEGF-A and VEGF-C secretion induced by PDGF-D, with comparable efficacy (Fig.3A,B). Overall, these data indicate that two distinct pathways downstream of PDGFRβ, one dependent on ERK and another one dependent on JNK, are activated in fibroblasts by PDGF-D, and cooperate to modulate both VEGF-A and VEGF-C secretion.
Fig.3: PDGF-D-stimulated secretion of VEGF-A and VEGF-C by human fibroblasts is dependent on ERK and JNK activation.

(A, B) While evaluating the intracellular signaling mediating VEGF-A and VEGF-C secretion by fibroblasts, we found that ERK and JNK inhibition abrogated the stimulatory effects of PDGF-D on both VEGF-A (A) and VEGF-C (B) secretory levels, similarly to the PDGFRβ inhibitor, imatinib mesylate (IM). n=5–7 experiments in duplicate; **p<0.01 vs Ctrl; ^p<0.05 vs PDGF-D; ^^p<0.01 vs PDGF-D, using two-tail t test.
Conditioned medium from PDGF-D-activated fibroblasts is a strong stimulator of LEC migration.
LEC recruitment is a prerequisite for tumor lymphangiogenesis.7 To understand whether PDGF-D empowered fibroblasts with the ability to attract LEC, we studied LEC migration in Boyden chambers. We found that conditioned medium from fibroblasts treated with PDGF-D significantly increased the migration of LEC compared to controls, an effect completely abolished by treating fibroblasts with imatinib. The promigratory effect of the conditioned medium from PDGF-D-stimulated fibroblasts on LEC was comparable to that exerted by VEGF-A or VEGF-C, whilst no direct effect by PDGF-D on LEC migration was observed (Fig.4A,B). Of note, WB analysis confirmed that LEC express VEGFR2 and VEGFR3, but not PDGFRβ, consistent with their complete lack of response to PDGF-D (Supplemental Fig.3A). Moreover, LEC migratory effects were independent of proliferation, since conditioned medium from PDGF-D-stimulated fibroblasts, as well as VEGF-A and VEGF-C exerted only a slight proliferative effect on LEC (Supplemental Fig.6A-C). LEC exposed to fibroblast conditioned medium upon PDGF-D stimulation did not show any activation of the p44/42 MAPK, JNK, and PI3K/AKT/mTOR pathways (Supplemental Fig.7A-E).
Fig.4: Upon PDGF-D stimulation, cultured human fibroblasts promote LEC recruitment, in vitro.

(A) Conditioned medium (CM) from fibroblasts exposed to PDGF-D ((PDGF-D) CM), potently stimulated LEC migration, an effect prevented by PDGFRβ antagonism with imatinib mesylate (IM) ((PDGF-D+IM) CM). This effect was reproduced by VEGF-A and VEGF-C, but not by the PDGF-D itself. (B) Representative pictures of Boyden Chamber inserts (M = 200x). n=4–11 experiments; **p<0.01 vs Ctrl; ^^p<0.01 vs (PDGF-D) CM, using two-tail t test.
Conditioned medium from PDGF-D-activated fibroblasts but not from EGI-1 cells stimulates tube formation in three-dimensional cultures of LEC.
After showing the recruiting effect of conditioned medium on LEC, we sought to evaluate whether PDGF-D-stimulated fibroblasts could also drive the generation of tubular vascular structures. To this end, we generated 3-D cultures of LEC, and exposed them to condition medium from either PDGF-D-stimulated fibroblasts or EGI-1 cells. Then, we performed an AngioTool software-based tubulization assay, whereby we evaluated the length of vascular branches, the number of junctions between branches and the percentage of area covered by vessels.18 Compared to controls, PDGF-D-stimulated fibroblast conditioned medium significantly increased all three readouts of vasculogenesis, similarly to VEGF-A and VEGF-C, while EGI-1 conditioned medium was ineffective, except for a minimal increase in the vessel area. Importantly, effects of PDGF-D-stimulated fibroblast conditioned medium were abrogated by antagonizing either PDGFRβ on fibroblasts, or VEGFR2 (by SU5416) and VEGFR3 (by SAR131675) on LEC (Fig.5A-D). Of note, SU5416 and SAR131675 were not cytotoxic on LEC at the chosen experimental doses, as shown by a dose-response MTS assay (Supplemental Fig.8A,B). It is important to remember that VEGF-C can act by binding to either VEGFR2 or VEGFR3, whereas VEGF-A only binds to VEGFR2.19 Altogether, these data argue for the concept that following PDGF-D activation, fibroblasts potently stimulate lymphangiogenesis, which is not affected by EGI-1 cells.
Fig.5: Upon PDGF-D stimulation, liver fibroblasts conversely from EGI-1 cells exert multiple lymphangiogenetic functions, inducing lumen formation, tubular branching and tubular lengthening of cultured LEC, in vitro.

(A) Representative micrographs of LEC tubulization and branching in a fibronectin/matrigel sandwich for the main treatment conditions. (B-D) CM from PDGF-D-treated fibroblasts ((PDGF-D) CM) induced 3-D cultured LEC to increase significantly the vessel area (B), the vessel length (C) and the number of junctions (D), with respect to controls. These effects were significantly attenuated by PDGFRβ antagonism in PDGF-D-treated fibroblasts ((PDGF-D + IM) CM), as well as by pre-treatment of LECs with inhibitors of VEGFR2 ((PDGF-D) CM + αVEGFR2) or VEGFR3 ((PDGF-D) CM + αVEGFR3). Similar effects to CM from PDGF-D-treated fibroblasts were obtained with VEGF-A and VEGF-C, but not with CM from EGI-1 cells, which induced only a slight increase in the vessel area (B). n=6–14 experiments; *p<0.05 vs Ctrl; **p<0.01 vs Ctrl; ^p<0.05 vs (PDGF-D) CM; ^^p<0.01 vs (PDGF-D) CM, using two-tail t test. Original magnification: 200x.
Conditioned medium from PDGF-D-activated fibroblasts enhances the permeability of LEC monolayers.
The high permeability of the lymphatic vasculature caused by defective tight junctions is conducive to tumor cell invasion.20 Thus, to evaluate the effect of PDGF-D-stimulated fibroblasts on the permeability of lymphatic vessels, we allowed LEC to become confluent, and then measured TEER across the endothelial monolayer. We found that TEER was significantly impaired by conditioned medium from PDGF-D-stimulated fibroblasts (Fig.6A), and this effect could be blocked by treatment of fibroblasts with imatinib. Interestingly, VEGF-C also increased the permeability of LEC monolayer, whereas VEGF-A did not. Consistently, blocking of VEGFR3 but not VEGFR2 on LEC exposed to conditioned medium from PDGF-D-stimulated fibroblasts restored the TEER to the basal levels, similarly to the reversion induced by anti-VEGF-C and anti-VEGF-A antibodies (Fig.6A). Overall, these results suggest that fibroblasts activated by PDGF-D can perturb the integrity of the lymphatic endothelial barrier by secreting VEGF-C, which is indeed capable of triggering the formation of intercellular gaps between adjacent LEC.7
Fig.6: Upon PDGF-D stimulation, liver fibroblasts reduce trans-endothelial resistance of LEC monolayers (TEER) and stimulate trans-endothelial migration (TrEM) of CCA cells (EGI-1-EGFP).

(A) CM from PDGF-D-treated fibroblasts ((PDGF-D) CM) dramatically impaired the integrity of the lymphatic endothelial barrier, more effectively than VEGF-C and VEGF-A. This effect was only partially counteracted by the concomitant treatment with αVEGFR2 ((PDGF-D) CM + αVEGFR2), but completely abrogated by the supplementation with αVEGFR3 ((PDGF-D) CM + αVEGFR3) or with bevacizumab ((PDGF-D) CM + Bev) or anti-VEGF-C ((PDGF-D) CM + αVEGF-C), as well as by PDGFRβ antagonism in PDGF-D-treated fibroblasts ((PDGF-D + IM) CM). (B) In TrEM experiments, CM from PDGF-D-treated fibroblasts ((PDGF-D) CM) enabled the CCA cell line EGI-1-EGFP to cross the LEC monolayer (similar to VEGF-A and VEGF-C), an effect blunted by the treatment of PDGF-D-stimulated fibroblasts with PDGFRβ antagonist ((PDGF-D + IM) CM), or with bevacizumab ((PDGF-D) CM + Bev) or anti-VEGF-C ((PDGF-D) CM + αVEGF-C), or by the treatment of LEC with αVEGFR3 ((PDGF-D) CM + αVEGFR3), but not with αVEGFR2 ((PDGF-D) CM + αVEGFR2). For TEER, n=4–15 experiments in duplicate. For TrEM, n=3–4; *p<0.05 vs Ctrl, **p<0.01 vs Ctrl, ^p<0.05 vs (PDGF-D) CM, ^^p<0.01 vs (PDGF-D) CM, using two-tail t test.
Conditioned medium from PDGF-D-activated fibroblasts induces trans-endothelial migration of CCA cells across LEC monolayer.
After showing that the permeability of LEC monolayers increased upon exposure to conditioned medium from PDGF-D-treated fibroblasts, we sought to determine whether the migration of CCA cells through the endothelial barrier was promoted as well. We seeded EGFP-expressing EGI-1 cells on top of a LEC monolayer and added CM from PDGF-D-activated fibroblasts. The number of trans-LEC migrated CCA cells was considerably increased in the CM-treated group, compared with controls. This effect was almost completely inhibited by antagonizing either PDGFRβ on fibroblasts, or VEGFR3 (but not VEGFR2) on LEC, and by neutralizing VEGF-A and VEGF-C (Fig.6B, and Supplemental Fig.9). Toxicity of the different compounds on LEC was excluded by MTS (Supplemental Fig.8A-C)
Depletion of CAF by navitoclax markedly decreases the lymphatic vascularization and reduces lymph node metastases in a syngeneic rat model of CCA.
To confirm the pivotal role played by CAF in directing tumor lymphangiogenesis in vivo, we used a well-established syngeneic rat model of CCA.15,21,22 This model shows an intense expression of PDGF-D by CCA cells. Selective depletion of CAF through apoptosis induced by navitoclax was accompanied by a significant decrease in the LMVD but not in the BMVD compared to control rats (Fig.7A), without affecting the expression of PDGF-D on CCA cells (Supplemental Fig.10). This effect was accompanied by a decrease in lymph node metastases that was significant at the peritoneal region, and with a tendency towards significance at the para-aortic region (Fig.7B). Noteworthy, possible toxicity-related effects on hypovascularization by navitoclax were clearly ruled out by MTS assay (Supplemental Fig.8D). These data support the concept that CAF may represent a valuable target to inhibit lymphangiogenesis and lymphatic dissemination in CCA.
Fig.7: In a syngeneic rat model of CCA, targeting CAF by navitoclax associates to a decreased lymphatic vascularization and lymph node metastasisation.

(A) In Fischer 344 male rats transplanted with BDE-neu rat CCA cells, selective depletion of CAF by navitoclax was accompanied by a significant decrease in Lyve-1+ LEC without affecting CD31+ blood endothelial cells compared to untreated rats. Up-sided, representative images of CCA sections, with dual immunofluorescence for CD31 (red) and Lyve-1 (green), show the stark differences in lymphatic and blood vessels between navitoclax and vehicle groups. (B) Concomitantly, navitoclax led to a reduction in the number of lymph node metastases that was significant at the peritoneal region (p<0.05), and close to significance at the paraortic region (p=0.068). (n=6 for each group). Original magnification: 100x. **p<0.01 vs Vehicle, using two-tail t test.
VEGFR2 and VEGFR3 antagonism markedly decreases the lymphatic vascularization in a xenograft mouse model of CCA.
To see if interference with the PDGF-D-driven sequence involving CAF and LEC was translationally significant to hamper tumor-associated lymphangiogenesis in vivo, we used a xenograft model of CCA in SCID mice generated by intraportal injection of human EGI-1 cells secreting PDGF-D.15,21,22 Antagonism of both VEGFR2 (SU5416) and VEGFR3 (SAR131675) led to a significant decrease in the LMVD compared to controls (Supplemental Fig.11). Again, a possible toxicity of both compounds on LEC was excluded by MTS (Supplemental Fig.8A-B). These data provide in vivo evidence that acting on the cross-talk mechanisms governed by PDGF-D represents a useful strategy to halt CCA-associated lymphangiogenesis.
CONCLUSIONS
The generation of an extensive lymphatic vascularization within the tumor reactive stroma is a key event in CCA progression. Indeed, early metastatic dissemination through the lymphatic vessels occurs in 60–70% of CCA patients, often impairing efficacy of curative treatments.23,24 However, the molecular mechanisms modulating lymphangiogenesis in CCA are still poorly understood, and more generally, tumor-associated lymphangiogenesis itself is still a major knowledge gap in cancer research.
Epithelial-mesenchymal interactions are potent enhancers of CCA aggressiveness. PDGF-D is an important candidate in this cross-talk, due to its key role in the activation of stromal cells and generation of the tumor reactive stroma, as well as its potential druggability.11,14 PDGF-D plays a repertoire of functions related to tumor-promotion, by fostering cancer cell proliferation and invasiveness, fibroblast recruitment and activation, and aberrant extracellular matrix deposition.15,25–27 As we have previously shown, PDGF-D expression is up-regulated in malignant cholangiocytes under hypoxic conditions.14 In this study, we provide evidence that in CCA, cancer cell-derived PDGF-D triggers a multi-step paracrine sequence centered around CAF. This paracrine signaling leads to recruitment of LEC into the tumor microenvironment, as well as to their assembly into highly branched and leaky tubular structures. These newly formed lymphatic vessels can be easily invaded by cancer cells resulting in early metastatic dissemination. However, it must be underlined that malignant cholangiocytes can secrete also PDGF-B, that like PDGF-D binds PDGFRβ28, possibly cooperating to the pro-lymphangiogenic activities of PDGF-D.
First, we observed that CCA samples displayed a striking expansion of the lymphatic vascular bed, as compared to HCC. These findings support the notion that the lymphatic vasculature represents a preferential route for CCA cells to escape from the primary site of growth. Moreover, we found that the lymphatic structures within the CCA stroma were closely surrounded by CAF. This physical proximity is most likely related to the reciprocal expression of VEGF-A and VEGF-C (which were negligibly expressed in malignant cholangiocytes) by CAF and of their cognate receptors VEGFR2 and VEGFR3 by LEC, suggesting an intimate cross talk between the two cell types. It is interesting to note that a similar pattern of expression was found in other desmoplastic epithelial cancers, such as colorectal29 and ovarian30 carcinomas. However, other studies performed in the South-Eastern Asia reported VEGF-C expression by malignant cholangiocytes in the intrahepatic variant of CCA31, although the unique environmental risk factors of CCA in this geographic area might account for the discrepancy with our findings.
In an effort to reproduce the biological interactions occurring within the tumor microenvironment, we next stimulated human fibroblasts with recombinant PDGF-D. The pro-secretory effects of PDGF family members (especially, PDGF-B) on VEGF ligands have been documented in various cell types, including hepatic stellate cells32 and pulmonary fibroblasts.33 However, no study has focused on the pro-lymphangiogenic properties of PDGF-D, the PDGF isoform specifically expressed by CCA cells. In this study, we demonstrated that PDGF-D potently elicits the secretion of VEGF-C and VEGF-A by myofibroblasts, without affecting the release of other lymphangiogenic growth factors, such as VEGF-D, Ang-1, and Ang-2. The PDGF-D-dependent expression of both VEGF isoforms by fibroblasts was mediated by two pathways, namely ERK and JNK. Thus, these data add another piece to the puzzle of pleiotropic functions played by CAF in response to PDGF-D: CAF are not only activated, but can also induce tumor lymphangiogenesis.
To address this concept further, we assessed the influence of conditioned medium from PDGF-D-stimulated fibroblasts on LEC motility, vascular assembly and permeability. In order to rule out confounding effects possibly related to the presence of residual exogenous PDGF-D, we preliminarily evaluated the expression of PDGFRβ on LEC. We found that LEC did not express PDGFRβ, while strongly expressing both VEGFR2 and VEGFR3. PDGFRβ expression on LEC of murine and rat origin and only at the mRNA level was reported by Cao.34 Other studies in mice reported PDGFRβ expression on LEC from large lymphatic vessels, whilst it was negative on LEC lining the small vessels.35
To evaluate the lymphangiogenic properties of CM from PDGF-D-treated fibroblasts, we employed the classic Boyden chamber system and the AngioTool software, which allows a reproducible quantification of different morphological and spatial parameters of newly formed vessels, including vessel length, the percentage of area covered by vessels, and the number of branch points.36 Cultured LEC exposed to CM from PDGF-D-stimulated fibroblasts were recruited to assemble branched vascular networks, with a negligible effect on proliferation. The larger surface area covered by lymphatic structures increases the likelihood of contact with tumor cells, thereby favoring their access to the lymphatic system, an important factor for lymph node metastatisation.7,37 Notably, conversely from liver myofibroblasts, conditioned medium from EGI-1 cells did not play significant pro-lymphangiogenic functions.
In our model, enhanced lymphangiogenesis was dependent on the sequential activation of PDGFRβ on fibroblasts and of VEGFR2 or VEGFR3 on LEC. In this regard, it is worth noting that the blockade of PDGFRβ resulted in a stronger inhibition of vasculogenesis compared to the specific antagonism of VEGFR2 or VEGFR3. These observations hinted at the possibility that additional soluble factors released from PDGF-D-stimulated fibroblasts might act in concert with VEGF-A and VEGF-C in order to promote tumor lymphangiogenesis.
Although the correlation between the expression of lymphangiogenic growth factors and the propensity of the tumor to develop lymph node metastasis has been described in different cancer types,38–40 the molecular mechanisms driving the invasion of tumor cells into lymphatic vessels remain elusive. Thus, to evaluate whether the pro-lymphangiogenic functions of CAF could also promote the entry of invasive cancer cells into the lymphatic system, we assessed the trans-endothelial electric resistance across LEC monolayers, and migration of CCA cells across the LEC monolayers. Indeed, LEC monolayers challenged with conditioned medium from PDGF-D-stimulated fibroblasts showed an increased permeability compared to controls consistent with the recent observation that in colorectal cancer, VEGF-C makes the lymphatic endothelial barrier looser by weakening tight and adherent junctions.41 Furthermore, CCA cells were able to cross LEC monolayers more easily upon endothelial cell exposure to conditioned medium from PDGF-D-stimulated fibroblasts. Noteworthy, both effects were reverted by blocking either PDGFRβ on fibroblasts or VEGFR3 on LEC, or by neutralizing VEGF-A and VEGF-C, but not blocking VEGFR2. This suggests that albeit VEGF-C and VEGF-A possess partially overlapping functions in the initial establishment of the lymphatic vascular network, the increase in endothelial permeability is primarily an effect of VEGF-C. Taken together, these data demonstrate that PDGF-D is indeed a key factor secreted by CCA cells to recruit myofibroblasts and then turn them into potent pro-lymphangiogenic players, essential for permitting the lymphatic invasion and dissemination of tumor cells.
To confirm that CAF are central actors in tumor lymphangiogenesis in CCA, we used a syngeneic rat model of CCA.21,22 This animal model is characterized by a highly desmoplastic tumor mass expressing PDGF-D and an early metastatization. Selective depletion of CAF was achieved by treating rats with the anti-apoptotic protein inhibitor navitoclax.15 Ability of navitoclax to selectively target CAF is dependent on their distinctive expression profile of Bcl-2 family members (increased expression of Bax proteins and absence of Mcl-1 expression) compared with quiescent fibroblasts and CCA cells.15 Importantly, CAF depletion was paralleled by a stark decrease in the lymphatic vascular system embedded in the tumor mass, coupled with a significant reduction in lymph node metastases at the peritoneal region, without affecting the blood vascularization. This suggests that lymphatics strictly depend on cues originating from CAF, whereas tumoral neoangiogenesis can be sustained by signals derived from other cell types. Turning to a xenograft model of CCA induced by intraportal injection of EGI-1 cells, we found that therapeutic inhibition of the PDGF-D-driven multistep mechanism at the level of VEGFR2 (SU5416) and VEGFR3 (SAR131675) expressed by LEC significantly dampened the tumor-associated lymphangiogenesis.
In conclusion, our results unveil the presence of a paracrine loop within the tumor reactive stroma of CCA, unleashed by PDGF-D, involving CCA cells, CAF and LEC, and able to orchestrate tumor-associated lymphangiogenesis and tumor cell intravasation (see graphical abstract). This process is a pre-requisite for the dissemination of cancer cells to the regional lymph nodes, which is an early, frequent and feared event in CCA, precluding potential curative therapies. This working model provides a series of putative molecular targets for therapeutic interventions aimed at halting the metastatic dissemination of CCA. Several molecules are available to interfere with the different arms of the described paracrine mechanism, encompassing VEGFR inhibitors, PDGFR inhibitors, and inhibitors of CAF-specific antiapoptotic proteins.
Supplementary Material
HIGHLIGHTS.
CCA are rich in stroma containing CAF and lymphatic vessels.
PDGF-D released by tumoral ducts attracts and activates liver fibroblasts to secrete VEGF-C/VEGF-A.
Lymphangiogenesis and lymphatic invasion are driven by VEGF-A/-C released by liver myofibroblasts.
Targeting liver myofibroblasts in vivo inhibits tumor-associated lymphangiogenesis and lymph node metastases.
These studies identify new possible molecular targets for the treatment of CCA.
ACKNOWLEDGEMENTS:
We thank Dr. Elisabetta Zulato for her valuable support in performing experiments with SCID mice.
Financial support statement:
LF was supported by the University of Padua, Progetti di Ricerca di Dipartimento (PRID) 2017; MC, and MS were supported by FAQC grant #2016-ATESP-0569, University of Milan-Bicocca; CS, RF, and MS were supported by Liver Center Cellular and Molecular Physiology and Morphology Core Facilities P30, DK034989-Silvio O. Conte Digestive Diseases Research Core Center, and RO1 DK079005 grants.
Footnotes
Conflict of interest statement: authors have nothing to disclose
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Banales JM, Cardinale V, Carpino V, Marzioni M, Andersen JB, Invernizzi P, et al. Expert consensus document: Cholangiocarcinoma: current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat Rev Gastroenterol Hepatol 2016;13:261–280. [DOI] [PubMed] [Google Scholar]
- 2.DeOliveira ML, Cunningham SC, Cameron JL, Kamangar F, Winter JM, Lillemoe KD, et al. Cholangiocarcinoma: thirty-one-year experience with 564 patients at a single institution. Ann Surg 2007;245:755–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brivio S, Cadamuro M, Fabris L, Strazzabosco M. Molecular mechanisms driving cholangiocarcinoma invasiveness: an overview. Gene Expr 2017; Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thelen A, Scholz A, Weichert W, Wiedenmann B, Neuhaus P, Gessner R, et al. Tumor-associated angiogenesis and lymphangiogenesis correlate with progression of intrahepatic cholangiocarcinoma. Am J Gastroenterol 2010;105:1123–1132. [DOI] [PubMed] [Google Scholar]
- 5.Luo X, Yuan L, Wang Y, Ge R, Sun Y, Wei G. Survival Outcomes and Prognostic Factors of surgical therapy for all potentially resectable intrahepatic cholangiocarcinoma: a large single-center cohort study. J Gastrointest Surg 2014;18:562–572. [DOI] [PubMed] [Google Scholar]
- 6.Cao Y. Opinion: emerging mechanisms of tumor lymphangiogenesis and lymphatic metastasis. Nat Rev Cancer 2005;5:735–743. [DOI] [PubMed] [Google Scholar]
- 7.Stacker SA, Williams SP, Karnezis T, Shayan R, Fox SB, Achen MG. Lymphangiogenesis and lymphatic vessel remodelling in cancer. Nat Rev Cancer 2014;14:159–172. [DOI] [PubMed] [Google Scholar]
- 8.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer 2006;6:392–401. [DOI] [PubMed] [Google Scholar]
- 9.Qiao A, Gu F, Guo X, Zhang X, Fu L. Breast cancer-associated fibroblasts: their roles in tumor initiation, progression and clinical applications. Front Med 2016;10:33–40. [DOI] [PubMed] [Google Scholar]
- 10.Pan B, Liao Q, Niu Z, Zhou L, Zhao Y. Cancer-associated fibroblasts in pancreatic adenocarcinoma. Future Oncol 2015;11:2603–2610. [DOI] [PubMed] [Google Scholar]
- 11.Cadamuro M, Stecca T, Brivio S, Mariotti V, Fiorotto R, Spirli C, et al. The deleterious interplay between tumor epithelia and stroma in cholangiocarcinoma. Biochim Biophys Acta. 2018;1864:1435–1443. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mertens JC, Rizvi S, Gores GJ. Targeting cholangiocarcinoma. Biochim Biophys Acta. 2018;1864:1454–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vaquero J, Guedj N, Clapéron A, Nguyen Ho-Bouldoires TH, Paradis V, Fouassier L. Epithelial-mesenchymal transition in cholangiocarcinoma: From clinical evidence to regulatory networks. J Hepatol. 2017;66:424–441. [DOI] [PubMed] [Google Scholar]
- 14.Cadamuro M, Nardo G, Indraccolo S, Dall’olmo L, Sambado L, Moserle L, et al. Platelet-Derived Growth Factor and Rho GTPases regulate recruitment of Cancer-Associated Fibroblasts in Cholangiocarcinoma. Hepatology 2013;58:1042–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mertens JC, Fingas CD, Christensen JD, Smoot RL, Bronk SF, Werneburg NW, et al. Therapeutic effects of deleting cancer-associated fibroblasts in cholangiocarcinoma. Cancer Res 2013;73:897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carneiro A, Falcão M, Azevedo I, Falcão Reis F, Soares R. Multiple effects of bevacizumab in angiogenesis: implications for its use in age-related macular degeneration. Acta Ophthalmol. 2009;87:517–523. [DOI] [PubMed] [Google Scholar]
- 17.Timoshenko AV, Rastogi S, Lala PK. Migration-promoting role of VEGF-C and VEGF-C binding receptors in human breast cancer cells. Br J Cancer. 2007;97:1090–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kazenwadel J, Secker GA, Betterman KL, Harvey NL. In Vitro Assays Using Primary Embryonic Mouse Lymphatic Endothelial Cells Uncover Key Roles for FGFR1 Signalling in Lymphangiogenesis. PLoS One 2012;7:e40497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer 2008;8:579–591. [DOI] [PubMed] [Google Scholar]
- 20.Duong T, Koopman P, Francois M. Tumor lymphangiogenesis as a potential therapeutic target. J Oncol 2012;2012:204946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lai GH, Zhang Z, Shen XN, Ward DJ, Dewitt JL, Holt SE, et al. erbB-2/neu transformed rat cholangiocytes recapitulate key cellular and molecular features of human bile duct cancer. Gastroenterology 2005;129:2047–2057. [DOI] [PubMed] [Google Scholar]
- 22.Sirica AE, Zhang Z, Lai GH, Asano T, Shen XN, Ward DJ, et al. A novel “patient-like” model of cholangiocarcinoma progression based on bile duct inoculation of tumorigenic rat cholangiocyte cell lines. Hepatology 2008;47:1178–1190. [DOI] [PubMed] [Google Scholar]
- 23.Yamaguchi K, Chijiiwa K, Saiki S, Shimizu S, Takashima M, Tanaka M. Carcinoma of the extrahepatic bile duct: mode of spread and its prognostic implications. Hepatogastroenterology 1997;44:1256–1261. [PubMed] [Google Scholar]
- 24.Uenishi T, Hirohashi K, Kubo S, Yamamoto T, Yamazaki O, Kinoshita H. Clinicopathological factors predicting outcomes after resection of mass-forming intrahepatic cholangiocarcinoma. Br J Surg 2001;88:969–974. [DOI] [PubMed] [Google Scholar]
- 25.Ustach CV, Taube ME, Hurst NJ Jr, Bhagat S, Bonfil RD, Cher ML, et al. A potential oncogenic activity of platelet-derived growth factor d in prostate cancer progression. Cancer Res 2004;64:1722–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu L, Tong R, Cochran DM, Jain RK. Blocking platelet-derived growth factor-D/platelet-derived growth factor receptor beta signaling inhibits human renal cell carcinoma progression in an orthotopic mouse model. Cancer Res 2005;65:5711–5719. [DOI] [PubMed] [Google Scholar]
- 27.Borkham-Kamphorst E, van Roeyen CR, Ostendorf T, Floege J, Gressner AM, Weiskirchen R. Pro-fibrogenic potential of PDGF-D in liver fibrosis. J Hep 2007;46:1064–1074. [DOI] [PubMed] [Google Scholar]
- 28.Fingas CD, Bronk SF, Werneburg NW, Mott JL, Guicciardi ME, Cazanave SC, et al. Myofibroblast-derived PDGF-BB promotes Hedgehog survival signaling in cholangiocarcinoma cells. Hepatology. 2011;54:2076–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang P, Hong JW, Ubukata H, Liu G, Katano M, Motohashi G, et al. Myofibroblasts Correlate with Lymphatic Microvessel Density and Lymph Node Metastasis in Early-stage Invasive Colorectal Carcinoma. Anticancer Res 2005;25:2705–2712. [PubMed] [Google Scholar]
- 30.Zhang Y, Tang H, Cai J, Zhang T, Guo J, Feng D, et al. Ovarian cancer-associated fibroblasts contribute to epithelial ovarian carcinoma metastasis by promoting angiogenesis, lymphangiogenesis and tumor cell invasion. Cancer Lett 2011;303:47–55. [DOI] [PubMed] [Google Scholar]
- 31.Aishima S, Nishihara Y, Iguchi T, Taguchi K, Taketomi A, Maehara Y, et al. Lymphatic spread is related to VEGF-C expression and D2–40-positive myofibroblasts in intrahepatic cholangiocarcinoma. Mod Pathol. 2008;21:256–264. [DOI] [PubMed] [Google Scholar]
- 32.Lu Y, Lin N, Chen Z, Xu R. Hypoxia-induced secretion of platelet-derived growth factor-BB by hepatocellular carcinoma cells increases activated hepatic stellate cell proliferation, migration and expression of vascular endothelial growth factor-A. Mol Med Rep 2015;11:691–697. [DOI] [PubMed] [Google Scholar]
- 33.Nauck M, Roth M, Tamm M, Eickelberg O, Wieland H, Stulz P, et al. Induction of vascular endothelial growth factor by platelet-activating factor and platelet-derived growth factor is downregulated by corticosteroids. Am J Respir Cell Mol Biol 1997;16:398–406. [DOI] [PubMed] [Google Scholar]
- 34.Cao R, Björndahl MA, Religa P, Clasper S, Garvin S, Galter D, et al. PDGF-BB induces intratumoral lymphangiogenesis and promotes lymphatic metastasis. Cancer Cell 2004;6:333–345. [DOI] [PubMed] [Google Scholar]
- 35.Kodama M, Kitadai Y, Sumida T, Ohnishi M, Ohara E, Tanaka M, et al. Expression of platelet-derived growth factor (PDGF)-B and PDGF-receptor β is associated with lymphatic metastasis in human gastric carcinoma. Cancer Sci 2010;101:1984–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zudaire E, Gambardella L, Kurcz C, Vermeren S. A computational tool for quantitative analysis of vascular networks. PLoS One 2011;6:e27385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leu AJ, Berk DA, Lymboussaki A, Alitalo K, Jain RK. Absence of functional lymphatics within a murine sarcoma: a molecular and functional evaluation. Cancer Res 2000;60:4324–4327. [PubMed] [Google Scholar]
- 38.Mandriota SJ, Jussila L, Jeltsch M, Compagni A, Baetens D, Prevo R, et al. Vascular endothelial growth factor-C-mediated lymphangiogenesis promotes tumour metastasis. EMBO J 2001;20:672–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Skobe M, Hawighorst T, Jackson DG, Prevo R, Janes L, Velasco P, et al. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med 2001;7:192–198. [DOI] [PubMed] [Google Scholar]
- 40.Karnezis T, Shayan R, Caesar C, Roufail S, Harris NC, Ardipradja K, et al. VEGF-D promotes tumor metastasis by regulating prostaglandins produced by the collecting lymphatic endothelium. Cancer Cell 2012;21:181–195. [DOI] [PubMed] [Google Scholar]
- 41.Tacconi C, Correale C, Gandelli A, Spinelli A, Dejana E, D’Alessio S, et al. Vascular endothelial growth factor C disrupts the endothelial lymphatic barrier to promote colorectal cancer invasion. Gastroenterology 2015;148:1438–1451.e8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.