

ABSTRACT
Liver-specific ten-eleven translocation (Tet) methylcytosine dioxygenases 2 and 3 (Tet2 plus Tet3)-deficient hepatitis B virus (HBV) transgenic mice fail to support viral biosynthesis. The levels of viral transcription and replication intermediates are dramatically reduced. Hepatitis B core antigen is only observed in a very limited number of pericentral hepatocytes in a pattern that is similar to glutamate-ammonia ligase (Glul), a β-catenin target gene. HBV transcript abundance in adult Tet-deficient mice resembles that observed in wild-type neonatal mice. Furthermore, the RNA levels of several β-catenin target genes including Glul, Lhpp, Notun, Oat, Slc1a2, and Tbx3 in Tet-deficient mice were also similar to that observed in wild-type neonatal mice. As HBV transcription is regulated by β-catenin, these findings support the suggestion that neonatal Tet deficiency might limit β-catenin target gene expression, limiting viral biosynthesis. Additionally, HBV transgene DNA displays increased 5-methylcytosine (5mC) frequency at CpG sequences consistent with neonatal Tet deficiency being responsible for decreased developmental viral DNA demethylation mediated by 5mC oxidation to 5-hydroxymethylcytosine, a process that might be responsible for the reduction in cellular β-catenin target gene expression and viral transcription and replication.
IMPORTANCE
Chronic hepatitis B virus (HBV) infection causes significant worldwide morbidity and mortality. There are no curative therapies available to resolve chronic HBV infections, and the small viral genome limits molecular targets for drug development. An alternative approach to drug development is to target cellular genes essential for HBV biosynthesis. In the liver, ten-eleven translocation (Tet) genes encode cellular enzymes that are not essential for postnatal mouse development but represent essential activities for viral DNA demethylation and transcription. Consequently, Tet inhibitors may potentially be developed into therapeutic agents capable of inducing and/or maintaining HBV covalently closed circular DNA methylation, resulting in transcriptional silencing and the resolution of chronic viral infection.
KEYWORDS: hepatitis B virus (HBV), ten-eleven translocation (Tet) methylcytosine dioxygenases, DNA methyltransferase (Dnmt), viral biosynthesis
INTRODUCTION
Chronic hepatitis B virus (HBV) infection is a major clinical problem that lacks a curative therapy (1). Current treatments include interferon and nucleos(t)ide analog therapies that inhibit viral reverse transcriptase but fail to clear nuclear HBV covalently closed circular DNA (cccDNA), the transcriptional template for the synthesis of the viral RNAs (2–4). The HBV 3.5-kb pregenomic RNA is the template for reverse transcription that results in the synthesis of HBV relaxed circular (rc) DNA within cytoplasmic viral capsids (5–9). These mature capsids can cycle back to the nucleus to maintain the pool of HBV cccDNA or become enveloped by the hepatitis B surface antigen and bud into the lumen of the endoplasmic reticulum (5–9). Subsequently, virions are transported through the Golgi apparatus and secreted into the circulation where they can infect additional hepatocytes (5–9). Reverse transcriptase inhibitors limit cytoplasmic HBV rcDNA synthesis but fail to eliminate nuclear HBV cccDNA, which appears to be a highly stable viral replication intermediate (3, 4). Therefore, therapies designed to cure chronic HBV infections must eliminate or inactivate HBV cccDNA as a transcriptional template. Methylation of HBV cccDNA represents a mechanism that might be considered as an approach toward silencing the transcriptional potential of this nuclear viral replication intermediate that could lead to the resolution of chronic HBV infections.
HBV transgenic mice do not support viral transcription and replication at birth, but postnatal viral biosynthesis is initiated within the first day of life, reaching maximal levels by about 4 weeks of age (10, 11). The HBV transgene DNA is fully methylated at birth and is progressively demethylated through liver maturation (10). The level of viral biosynthesis is inversely correlated with the degree of HBV DNA methylation, suggesting that viral transcription is silenced when viral cytosine residues present in CpG dinucleotide sequences are modified from C to 5-methylcytosine (5mC) (10, 12). FoxA-deficient HBV transgenic mice, lacking wild-type levels of this pioneer transcription factor, fail to support viral biosynthesis presumably because the hepatic HBV transgene DNA is essentially fully methylated outside of the CpG island region spanning approximately nucleotide coordinates 1,000–2,000 (12). This observation demonstrated that FoxA is required for the postnatal demethylation of HBV transgene DNA, which permits the initiation of viral transcription and subsequent HBV replication. Demethylation of the HBV transgene DNA throughout postnatal liver development is associated with the appearance of 5-hydroxymethylcytosine (5hmC) that persists in adult mice (10). The conversion of 5mC to 5hmC indicates that demethylation of the HBV transgene DNA requires the oxidation of 5mC by ten-eleven translocation (Tet) methylcytosine dioxygenases, primarily by Tet2 and Tet3 in the liver (10, 13, 14). These observations suggested that the developmental recruitment of Tet enzymes to the HBV transgene DNA by FoxA results in the demethylation and subsequent transcription of the viral genome, leading to the initiation and maintenance of hepatic HBV biosynthesis in this small animal model of chronic viral infection (10, 12).
In the current study, the suggestion that FoxA directly or indirectly recruits Tet enzymes to the HBV transgene DNA to mediate demethylation and subsequent transcriptional activation was evaluated. Liver-specific Tet-deficient HBV transgenic mice were produced, and viral RNA, protein synthesis, and replication were evaluated. Tet2-null Tet3-null hepatocytes failed to support all aspects of HBV biosynthesis in a manner that was very similar to that observed in FoxA-deficient HBV transgenic mice (12). The downregulation of several β-catenin target gene transcripts resembled that of the HBV transcripts, suggesting that the expression of these genes might be controlled in a similar manner. Direct evaluation of the DNA methylation status of the HBV transgene DNA indicated that the loss of Tet2 plus Tet3 resulted in a major increase in viral DNA methylation with the associated loss of HBV biosynthesis. These observations are consistent with findings observed in FoxA-deficient mice (12). Together, these results indicate that liver-specific FoxA and Tet deficiencies display very similar phenotypes and support the suggestion that FoxA recruits Tet enzymes, resulting in demethylation of the HBV transgene DNA. This permits the postnatal expression of viral RNAs and subsequent HBV biosynthesis (10, 12). Liver-specific Tet-deficient mice develop normally and display only limited physiological alterations (15) while failing to support HBV biosynthesis. This suggests that specific Tet inhibitors might represent a new class of antiviral therapeutics. Furthermore, Tet inhibitors could potentially lead to HBV cccDNA methylation with subsequent transcriptional silencing, which may resolve chronic HBV infections.
RESULTS
Characterization of Tet gene expression in HBVTet2(fl/fl)AlbCre, HBVTet3(fl/fl)AlbCre, and HBVTet2(fl/fl)Tet3(fl/fl)AlbCre HBV transgenic mice
Tet-deficient HBV transgenic mice [HBVTet2(fl/fl)AlbCre(+), HBVTet3(fl/fl)AlbCre(+), and HBVTet2(fl/fl)Tet3(fl/fl)AlbCre(+)] were viable and displayed no overt phenotype (15). The levels of the Tet RNAs in the livers of the wild-type HBV transgenic mice [HBVTet2(fl/fl)AlbCre(−), HBVTet3(fl/fl)AlbCre(−), and HBVTet2(fl/fl)Tet3(fl/fl)AlbCre(−)] were compared with the levels of the Tet RNAs in the Tet-deficient HBV transgenic mice [HBVTet2(fl/fl)AlbCre(+), HBVTet3(fl/fl)AlbCre(+), and HBVTet2(fl/fl)Tet3(fl/fl)AlbCre(+)], respectively (Fig. 1A through D). Tet1 RNA is essentially absent from adult liver tissue (15). Comparisons of Tet2 and Tet3 RNA levels detected using probes targeting regions outside of the deleted exons indicate that the liver-specific deletion of functional regions of either or both Tet genes does not affect the total level of these transcripts observed in liver tissue (Fig. 1A and C). Using probes within the deleted exons, Tet2 RNA was reduced by twofold to threefold in the HBV transgenic mice with any Tet2(fl/fl)AlbCre(+) genotype (Fig. 1B). Tet3 RNA was reduced by fivefold to eightfold in the HBV transgenic mice with any Tet3(fl/fl)AlbCre(+) genotype (Fig. 1D). The modest reduction in the Tet RNAs in these mice likely reflects gene deletion within hepatocytes, but not within nonparenchymal cells, including biliary epithelial, endothelial, Kupffer, and stellate cells, plus the intrinsically low level of adult liver Tet transcripts (15). In contrast, Ctnnb1 (β-catenin) and glyceraldehyde 3-phosphate dehydrogenase (Gapdh) RNA levels appear to be largely insensitive to changes in Tet protein abundance (Fig. 1E and F).
Fig 1.

Effects of Tet deletion on liver Tet2, Tet3, Ctnnb1, and Gapdh transcript levels in HBV transgenic mice. Quantitative analysis of (A) Tet2 (with probe outside deleted exons 8–10), (B) Tet2 (with probe within deleted exons 8–10), (C) Tet3 (with probe outside deleted exon 2), (D) Tet2 (with probe within deleted exon 2), (E) Ctnnb1, and (F) Gapdh transcripts by NanoString gene expression analysis in HBV transgenic mice. Individual (circles) and mean (line) transcript levels are indicated for male (M), female (F), homozygous Tet2(fl/fl) alleles (Tet2), homozygous Tet3(fl/fl) alleles (Tet3), homozygous Tet2(fl/fl)Tet3(fl/fl) alleles (Tet2 and Tet3), Cre-negative allele [Cre(−)], and Cre-positive allele [Cre(+)] HBV transgenic mice. Statistical differences (adjusted P values; Tukey’s multiple comparison test) in the levels of the transcripts between Cre(−) and Cre(+) HBV transgenic mice are indicated (GraphPad Prism 9.4.1).
Effect of Tet gene deletion on liver size and intracellular HBcAg in HBV transgenic mice
In contrast to the Tet2(fl/fl)AlbCre(+) and Tet3(fl/fl)AlbCre(+) mice, the HBVTet2(fl/fl)Tet3(fl/fl)AlbCre(+) mice displayed mild hepatomegaly (Fig. 2A) as reported previously (15). The livers of these mice were 20%−40% larger than their AlbCre-negative wild-type controls. Immunohistochemical analysis of the livers of HBV transgenic mice lacking Tet expression in hepatocytes, HBVTet2(fl/fl)Tet3(fl/fl)AlbCre(+), shows greatly reduced central vein hepatitis B core antigen (HBcAg) staining compared to all control and single Tet-null mice, Tet2(fl/fl)Cre(−), Tet3(fl/fl)Cre(−), Tet2(fl/fl)Tet3(fl/fl)AlbCre(−), Tet2(fl/fl)AlbCre(+), and Tet3(fl/fl)AlbCre(+) (Fig. 2B). These observations indicate that Tet2 and Tet3 are largely functionally redundant with regard to HBV biosynthesis in hepatocytes. Consequently, all subsequent analyses were performed using the liver-specific Tet2-null plus Tet3-null HBV transgenic mice. Interestingly, the staining of glutamate-ammonia ligase (Glul), which is limited to central vein proximal hepatocytes, was also largely absent in these mice. HBV and Glul transcription are both regulated by β-catenin, suggesting that Tet deficiency might be affecting β-catenin signaling despite the absence of any effects on Cnntb1 RNA levels in these mice (Fig. 1E) (16–19). Furthermore, the HBcAg and Glul staining observed in HBVTet2(fl/fl)Tet3(fl/fl)AlbCre(+) mice could also reflect incomplete Tet gene deletion in a limited number of hepatocytes located close to the central vein (Fig. 2C).
Fig 2.

Effects of Tet deletion on liver size, serum HBeAg, and intracellular HBcAg in HBV transgenic mice. (A) Quantitative analysis of liver size in HBV transgenic mice. Individual (circles) and mean (line) liver sizes are indicated for male (M), female (F), homozygous Tet2(fl/fl) alleles (Tet2), homozygous Tet3(fl/fl) alleles (Tet3), homozygous Tet2(fl/fl)Tet3(fl/fl) alleles (Tet2 and Tet3), Cre-negative allele [Cre(−)], and Cre-positive allele [Cre(+)] HBV transgenic mice. Statistical differences (adjusted P values; Tukey’s multiple comparison test) in liver size between Cre(−) and Cre(+) HBV transgenic mice are indicated (GraphPad Prism 10.0.3). (B) Immunohistochemical staining of liver HBcAg and Glul located abundantly in the centrolobular hepatocytes except in Tet-deficient HBV transgenic mice, HBVTet2(fl/fl)Tet3(fl/fl)Cre(+).
Effect of Tet gene deletion on viral transcription and replication in HBV transgenic mice
HBV transgenic mice that lacked liver-specific expression of Tet2 plus Tet3 [HBVTet2(fl/fl)Tet3(fl/fl)AlbCre(+)] were examined for their steady state levels of HBV transcripts by analysis of total liver RNA (Fig. 3A through C). The steady-state levels of the HBV 3.5- and 2.1-kb transcripts in the livers of the HBV transgenic mice lacking Tet expression, HBVTet2(fl/fl)Tet3(fl/fl)AlbCre(+), were reduced fourfold to sevenfold relative to their littermate controls, HBVTet2(fl/fl)Tet3(fl/fl)AlbCre(−) (Fig. 3A through C). Serum hepatitis B e antigen (HBeAg), encoded by the HBV 3.5-kb precore RNA, was reduced to a similar extent (Fig. 3C). Furthermore, the level of expression of the HBV RNAs observed in the adult HBVTet2(fl/fl)Tet3(fl/fl)Cre(+) mice is similar to that observed in neonatal wild-type mice (Fig. 3A and B). This observation is consistent with the suggestion that viral transcription failed to increase normally during postnatal development due to the neonatal deletion of the Tet genes and the subsequent absence of Tet activity during this stage of liver maturation. Given the role of Tet2 and Tet3 enzyme activity in DNA demethylation in the liver (15), it appears probable that postnatal HBV DNA demethylation ceased shortly after birth as a result of the absence of Tet function. The resulting loss of viral transcription resulted in reduced HBV RNA and a dramatic loss of HBV replication intermediates, which were reduced 10- to 20-fold in HBVTet2(fl/fl)Tet3(fl/fl)Cre(+) mice compared to their littermate controls, HBVTet2(fl/fl)Tet3(fl/fl)Cre(−) (Fig. 3D).
Fig 3.

Effects of Tet deletion on liver viral transcript and replication intermediate levels in HBV transgenic mice. Quantitative analysis of (A) HBV 3.5-kb RNA and (B) total HBV RNA (3.5-kb plus 2.1-kb RNA) by NanoString gene expression analysis in HBV transgenic mice. Individual (circles) and mean (line) transcript levels are indicated for male (M), female (F), homozygous Tet2(fl/fl)Tet3(fl/fl) alleles (Tet2 and Tet3), Cre-negative allele [Cre(−)], and Cre-positive allele [Cre(+)] HBV transgenic mice. Statistical differences (adjusted P values; Tukey’s multiple comparison test) in the levels of the transcripts between Cre(−) and Cre(+) HBV transgenic mice are indicated (GraphPad Prism 10.0.3). Transcript levels from adult Tet-deficient HBV transgenic mice (left panel) are compared to the changes in gene expression observed throughout postnatal liver development in wild-type HBV transgenic mice (right panel). Arrows indicate the average levels of transcript observed in HBVTet2(fl/fl)Tet3(fl/fl)Cre(−) (top arrow) and HBVTet2(fl/fl)Tet3(fl/fl)Cre(+) (bottom arrow) mice. (C) RNA (Northern) filter hybridization analyses of liver transcripts isolated from HBV transgenic mice are shown. Noncontiguous lanes from a single analysis are presented. The probes used were HBVayw genomic DNA plus Gapdh cDNA. The Gapdh transcript was used as an internal control for the quantitation of the HBV 3.5-kb RNA. Serum HBeAg levels encoded by the HBV 3.5-kb precore RNA are also indicated. (D) DNA (Southern) filter hybridization analyses of liver replication intermediates isolated from HBV transgenic mice are shown. Noncontiguous lanes from a single analysis are presented. The probe used was HBVayw genomic DNA. The HBV transgene (Tg) was used as an internal control for the quantitation of the HBV replication intermediates. RC, HBV relaxed circular replication intermediates; SS, HBV single-stranded replication intermediates.
Effect of Tet gene deletion on β-catenin target gene expression in HBV transgenic mice
The observation that Glul protein expression was downregulated in HBVTet2(fl/fl)Tet3(fl/fl)Cre(+) mice as a result of the loss of Tet expression (Fig. 2C) made it of interest to determine if the transcription of additional β-catenin target genes was altered in liver-specific Tet-deficient transgenic mice (Fig. 4). Initially, Glul RNA levels in these mice were evaluated (Fig. 4A). As observed with HBV transcript levels, the level of expression of the Glul RNA in the adult HBVTet2(fl/fl)Tet3(fl/fl)Cre(+) mice was similar to that observed in wild-type neonatal mice (Fig. 4A). Examination of the expression of five additional β-catenin target genes revealed a similar pattern of transcript levels to that observed with HBV and Glul RNAs (Fig. 4B through F). These findings indicate that the levels of multiple β-catenin target gene transcripts fail to increase normally throughout postnatal liver development in the absence of Tet enzyme activity, despite expressing normal amounts of β-catenin RNA (Fig. 1E). The mechanism controlling this developmentally stalled transcriptional program is unclear but may involve DNA methylation, which was previously shown to govern HBV transcription throughout liver development (10, 12).
Fig 4.
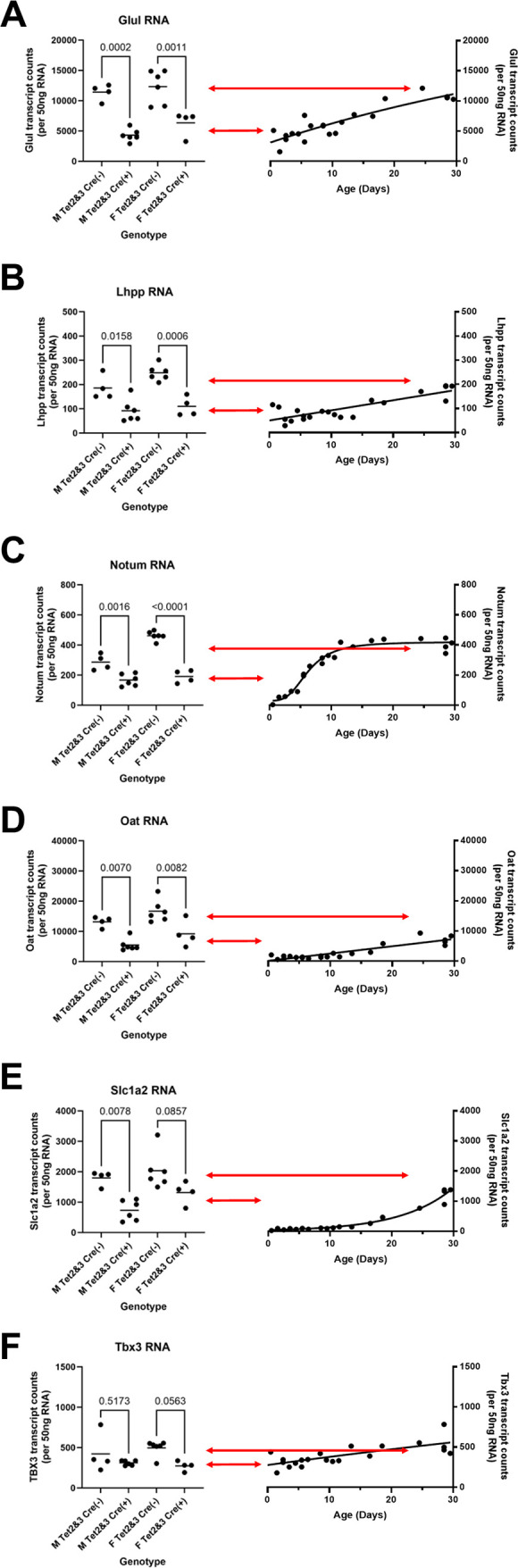
Effects of Tet deletion on liver β-catenin target gene transcript levels in HBV transgenic mice. Quantitative analysis of (A) Glul, (B) Lhpp, (C) Notum, (D) Oat, (E) Slc1a2, and (F) Tbx3 transcripts by NanoString gene expression analysis in HBV transgenic mice. Individual (circles) and mean (line) transcript levels are indicated for male (M), female (F), homozygous Tet2(fl/fl)Tet3(fl/fl) alleles (Tet2 and Tet3), Cre-negative allele [Cre(−)], and Cre-positive allele [Cre(+)] HBV transgenic mice. Statistical differences (adjusted P values; Tukey’s multiple comparison test) in the levels of the transcripts between Cre(−) and Cre(+) HBV transgenic mice are indicated (GraphPad Prism 10.0.3). Transcript levels from adult Tet-deficient HBV transgenic mice (left panel) are compared to the changes in gene expression observed throughout postnatal liver development in wild-type HBV transgenic mice (right panel). Arrows indicate the average levels of transcript observed in HBVTet2(fl/fl)Tet3(fl/fl)Cre(−) (top arrow) and HBVTet2(fl/fl)Tet3(fl/fl)Cre(+) (bottom arrow) mice.
Effect of liver-specific et gene deletion on HBV DNA methylation in the livers of HBV transgenic mice
Liver-specific deletion of Tet genes results in the loss of HBV biosynthesis (Fig. 3). Given the known role of Tet enzymes in DNA demethylation, the effect of neonatal Tet gene deletion on HBV transgene DNA methylation levels was evaluated (Fig. 5). Bisulfite genomic sequencing of the HBV transgene DNA from wild-type and liver-specific Tet-deficient HBV transgenic mice was performed and demonstrated that the loss of Tet resulted in reduced HBV DNA demethylation (Fig. 5). The HBV DNA amplicon spanning nucleotide coordinates 1215–1629 contains 38 CpG sites (Fig. 5) located within the CpG island and, as such, is hypomethylated (12, 20). In this region, the loss of Tet expression was estimated to modestly increase HBV DNA methylation by about 7%–11% depending upon the sex of the mice (Fig. 5).
Fig 5.

Effects of Tet deletion on liver viral DNA methylation in HBV transgenic mice. Percentages of CpG DNA methylation at each of the 11, 38, and 13 sites located within HBV nucleotide coordinates 341–711, 1215–1629, and 2264–2474, respectively, for the viral genome from the livers of HBV transgenic mice are shown. (A) Male and (B) female HBVTet2(fl/fl)Tet3(fl/fl)Cre(−) and HBVTet2(fl/fl)Tet3(fl/fl)Cre(+) mice. The viral transcription initiation sites for the X gene, the precore/pregenomic, the large surface antigen, and the middle/major surface antigen transcripts plus the CpG island are located at nucleotide coordinates 1310, 1785/1821, 2809, and 3159/3178 plus 1000–2000, respectively.
The HBV DNA amplicons spanning nucleotide coordinates 341–711 and 2164–2474 contain 11 and 13 CpG sites, respectively (Fig. 5). The deletion of both Tet2 and Tet3 genes increased HBV transgene DNA methylation by 28%–40%, indicating that these regions of the viral genome were particularly sensitive to Tet activity (Fig. 5). Therefore, it seems likely that the higher levels of DNA methylation mediated by the loss of both Tet2 and Tet3 throughout postnatal liver development were responsible for the observed loss of HBV biosynthesis (Fig. 3). Additionally, the level and pattern of methylation observed in the adult Tet-deficient HBV transgenic mice (Fig. 5) are similar to that observed in wild-type neonatal HBV transgenic mice (10), suggesting that the postnatal loss of Tet enzymes prevents further viral DNA demethylation.
DISCUSSION
The value of the HBV transgenic mouse model of chronic infection is the ability to identify host genes that modulate viral biosynthesis in the liver. Relevant host transcription factors include Hnf1α that limits HBV cccDNA production (3, 21), Hnf4α that is essential for HBV RNA production (11), and FoxA that limits epigenetic transcriptional silencing by developmental viral genomic DNA demethylation (10, 12). Furthermore, this model system permits the critical role of hepatocyte maturation on HBV biosynthesis to be examined throughout postnatal liver developmental (10). However, this model does not permit viral infection that is restricted to hepatoma cell lines expressing the HBV receptor, sodium taurocholate cotransporting polypeptide, and primary human hepatocytes (PHHs) (22). Reconstitution of mouse liver with PHHs does permit HBV infection with limited viral biosynthesis (23, 24). However, the absence of reliable approaches for genetic manipulation of PHH currently limits analysis of host genes regulating viral biosynthesis using these model systems. The absence of significant levels of viral cccDNA in the wild-type HBV transgenic mouse model of chronic infection (25) also limits its utility to study the regulation of cccDNA biosynthesis. In contrast, the role of host genes in HBV cccDNA synthesis may be evaluated in the context of the HNF1α-null HBV transgenic mouse where viral cccDNA is readily detectable (3, 21).
In the HBV transgenic mouse model of chronic infection, viral biosynthesis is initiated neonatally and increases throughout postnatal liver development (10, 11). During postnatal hepatocyte maturation, HBV transgene DNA is progressively demethylated with viral transcription and replication negatively correlating with the level of viral DNA methylation (10). Additionally, liver-specific FoxA-deficient HBV transgenic mice that display only limited FoxA3 expression fail to demethylate HBV DNA and do not support HBV biosynthesis (12). A detailed examination of HBV DNA demethylation throughout liver development in wild-type HBV transgenic mice demonstrated that 5mC was actively modified to 5hmC from birth and also continued in adult HBV transgenic mice to maintain viral biosynthesis (10). Conversion of 5mC to 5hmC is mediated by Tet enzymes and represents a mechanism of active DNA demethylation that has been implicated in the postnatal expression of a subset of liver-specific genes, a hepatocyte maturation process that is not essential for mouse viability. Indeed, the liver-specific loss of postnatal Tet expression results in only a mild phenotype (15). In combination, these observations led to a basic understanding of the mechanism of induction of postnatal liver HBV biosynthesis in this model of chronic viral infection (10, 12). Specifically, late embryonic or early neonatal binding of FoxA to their binding sites in the viral genome leads to the recruitment of Tet enzymes that convert 5mC to 5hmC (10, 12). Tet enzymes may further oxidize 5hmC to 5-formylcytosine and 5-carboxylcytosine leading to their active elimination and replacement through the base-excision repair pathway (13, 14). These modifications are replication-independent and may represent a strategy for the maintenance of viral biosynthesis in the adult liver, given the observed presence of 5hmC in adult HBV transgene DNA. Concurrently, 5hmC may limit subsequent DNA methylation of hemi-methylated DNA by DNA methyltransferase 1 (D1), leading to passive DNA demethylation as a result of subsequent replication cycles (26, 27). Regardless of the precise mechanism of FoxA-mediated demethylation, the presence of 5hmC residues in HBV DNA indicates that Tet enzymes play a role in this process (10).
To determine the role of Tet enzymes in the developmental demethylation of HBV DNA, liver-specific Tet-deficient HBV transgenic mice were generated and characterized (Fig. 1). Tet2 RNA and Tet3 RNA were reduced twofold to threefold and fivefold to eightfold, respectively, in the livers of these mice. Notably, intracellular HBcAg was significantly reduced in Tet-deficient HBV transgenic mice, HBV(+/−)Tet2(fl/fl)Tet3(fl/fl)AlbCre(+), lacking expression of both Tet2 and Tet3 enzymes in hepatocytes (Fig. 2), suggesting that these two enzymes have redundant function within the liver with regard to viral protein synthesis. Similarly, these Tet-deficient HBV transgenic mice display a fourfold to sevenfold and 10- to 20-fold reduction in viral transcripts and replication intermediates, respectively (Fig. 3). Interestingly, the level of viral transcripts detected in these mice is similar to that observed in wild-type neonatal HBV transgenic mice, consistent with the suggestion that the early developmental loss of Tet activity prevents the normal postnatal Tet-dependent induction of viral RNA synthesis associated with normal liver development (Fig. 3). Furthermore, several cellular β-catenin target genes displayed a similar developmental reduction in RNA synthesis (Fig. 4). Given that HBV transcription is also regulated by β-catenin (16, 17), it suggests there might be a Tet-dependent mechanism responsible for the coordinate postnatal regulation of a group of β-catenin target genes. However, this mode of gene regulation is independent of β-catenin transcript abundance as this RNA is not modulated by Tet activity throughout liver maturation (Fig. 1E).
The observation that Tet deficiency results in the loss of HBV RNA and DNA synthesis was consistent with the prior suggestion that liver-specific postnatal Tet-dependent demethylation of the HBV transgene DNA was responsible for HBV biosynthesis in this model of chronic infection (10, 12). This developmental mechanism of HBV biosynthesis induction was verified by directly examining HBV DNA methylation levels in hepatocyte-specific Tet-deficient HBV transgenic mice (Fig. 5). This analysis indicated that both Tet2 and Tet3 contributed to HBV DNA demethylation that is required for viral transcription and subsequent HBV replication. These observations support the model where FoxA recruits Tet enzymes, directly or indirectly, to HBV genomic DNA resulting in demethylation, RNA synthesis, and viral replication (10, 12). As viral methylation and demethylation appear to persist throughout liver maturation (10), the relative levels of D and Tet activities determine the steady-state level of HBV DNA methylation. Indeed, studies examining the methylation of HBV cccDNA during natural chronic infection in man have demonstrated that viral DNA displays various levels of 5mC modification, showing that it is a DNMT substrate in chronic carriers (28–30). Additionally, it strongly supports the contention that de novo methylation of nuclear HBV cccDNA derived from unmethylated cytoplasmic rcDNA must be continually occurring during chronic viral infection. Consequently, small molecular weight compounds capable of activating DNMT or inhibiting T enzyme activities could influence total viral DNA methylation and, hence, HBV biosynthesis. Specifically, during natural infection, T inhibitors might limit HBV cccDNA demethylation while permitting DNMT to progressively increase the level of viral DNA methylation and, hence, limit or silence viral transcription with only limiting effect on nonessential cellular gene expression (Fig. 4) (15), which would potentially be reversible upon cessation of therapy. Silencing nuclear HBV cccDNA transcription should, over time, lead to the elimination of viral biosynthesis and resolution of chronic infection, a major worldwide health concern (31, 32).
MATERIALS AND METHODS
Transgenic mice
The production and characterization of the HBV transgenic mouse lineage 1.3.32 have been described (25). These HBV transgenic mice contain a single copy of the terminally redundant, 1.3-genome length copy of the HBVayw genome integrated into the mouse chromosomal DNA. High levels of HBV replication occur in the livers of adult mice. The mice used were heterozygous for the HBV transgene and were maintained on the SV129 genetic background (33).
The production and characterization of the floxed Tet2, Tet2(fl/fl), floxed Tet3, Tet3(fl/fl), and albumin Cre [lineage B6.Cg-Tg(Alb-cre)21Mgn/J, Jackson Laboratory] transgene (AlbCre) mice have been described (15, 34–36). The HBVTet2(fl/fl)AlbCre(+), HBVTet3(fl/fl)AlbCre(+), and HBVTet2(fl/fl)Tet3(fl/fl)AlbCre(+) mice do not express Tet2 and Tet3 in the liver after Cre-mediated excision of Tet2 exons 8–10 and Tet3 exon 2, respectively, located between the loxP sites. The mice used in the breeding experiments were homozygous for their respective alleles and maintained on either the C57BL6 [Tet2(fl/fl) and Tet3(fl/fl)] or SV129 (AlbCre) genetic backgrounds.
HBV transgenic mice (lineage 1.3.32) were bred with mice carrying the floxed Tet2 and floxed Tet3 alleles and the AlbCre transgene to generate HBVTet2(fl/fl)AlbCre(+), HBVTet3(fl/fl)AlbCre(+), and HBVTet2(fl/fl)Tet3(fl/fl)AlbCre(+) transgenic mice. Littermate HBVTet2(fl/fl)AlbCre(−), HBVTet3(fl/fl)AlbCre(−), and HBVTet2(fl/fl)Tet3(fl/fl)AlbCre(−) transgenic mice without the AlbCre transgene were used as controls.
HBV transgenic mice were fed with normal rodent chow, and water was available ad libitum. Mice were sacrificed at 8–12 weeks, and liver tissue was frozen in liquid nitrogen and stored at –70°C prior to DNA and RNA extraction. All animal experiments were Institutional Animal Care and Use Committee approved and performed according to institutional guidelines with the University of Illinois at Chicago Institutional Biosafety Committee (number: 22-056) and Animal Care Committee approval (number: 22-143). All animal procedures were performed in the College of Medicine Research Building at the University of Illinois at Chicago and adhere to the policies of the National Institutes of Health Office of Laboratory Animal Welfare, the standards of the Animal Welfare Act, the Public Health Service Policy, and the Guide for the Care and Use of Laboratory Animals.
HBV DNA and RNA analysis
Total DNA and RNA were isolated from the liver of HBV transgenic mice as described previously (37, 38). Protein-free DNA was isolated in an identical manner to the total DNA except that the proteinase K digestion was omitted (21) . DNA (Southern) filter hybridization analyses were performed using 10 µg of HindIII digested DNA (38). Filters were probed with 32P-labeled HBVayw genomic DNA (39) to detect HBV sequences. RNA (Northern) filter hybridization analyses were performed using 10 µg of total cellular RNA as described previously (38). Filters were probed with 32P-labeled HBVayw genomic DNA to detect HBV sequences and mouse Gapdh cDNA to detect the Gapdh transcript used as an internal control (40). Filter hybridization analyses were quantified by phosphorimaging using a Molecular Dynamics Typhoon 8600 Phosphor Imager system.
NanoString nCounter gene expression transcript counting was used to quantify the levels of Ctnnb1, Gapdh, Glul, Lhpp, Notum, Oat, Slc1a2, Tbx3, Tet2, Tet3, and HBV 3.5-kb and total HBV 3.5- plus 2.1 kb transcript levels in 50-ng mouse liver RNA using a specifically designed code set. Data were quality controlled and normalized using the nSolver Analysis Software 4.0 (NanoString Technologies). RNA expression levels were normalized to mouse Cnot1, Rps29, Slc9a8, Mrps5, and Scarb1 RNA controls.
Serum HBeAg and immunohistochemistry of liver HBcAg
HBeAg analysis was performed using 2 µL of mouse serum and the HBe enzyme-linked immunosorbent assay as described by the manufacturer (Epitope Diagnostics). The level of antigen was determined in the linear range of the assay. Liver tissue samples were fixed in sodium phosphate-buffered formalin (Fisher), embedded in paraffin, and sectioned (5 µm). Immunohistochemical detection of HBcAg in paraffin-embedded mouse liver sections was performed using a polyclonal rabbit anti-HBcAg primary antiserum (Dako, B0586). Glul was detected with a monoclonal mouse anti-Glul primary antibody (BD Biosciences, 610517). Immunohistochemical slides were developed using the BOND Polymer Refine Detection System (Leica Biosystems).
DNA methylation analysis
Bisulfite treatment of protein-free genomic DNA for methylation analysis was performed using the EZ DNA Methylation-Lightning Kit (D5030; Zymo Research, Inc., Irvine, CA, USA) according to the manufacturer’s instructions. Three major viral amplicons covering 62 of the 99 CpG sequences in the HBV DNA genome were targeted using PCR amplification of the bisulfite-treated DNA, followed by sequencing of the amplicons on an Illumina MiSeq instrument. Preparation of DNA for high-throughput amplicon sequencing was performed in two PCR stages in a protocol termed targeted amplicon sequencing as described previously (41, 42). The primer pairs targeting the bisulfite-converted HBV DNA were (i) 5′-ACACTGACGACATGGTTCTACACCCCCACTAACTAAAAC-3′ (oligo CS1FP2; HBV nucleotide coordinates 1198–1214) and 5′-TACGGTAGCAGAGACTTGGTCTGGGTAATATTTGGTGG-3′ (oligo CS2RP2; HBV nucleotide coordinates 1645–1630), (ii) 5′-ACACTGACGACATGGTTCTACAAGTTATAGAGTATTTGGTGT-3′ (oligo CS1FP5a; HBV nucleotide coordinates 2244–2263) and 5′-TACGGTAGCAGAGACTTGGTCTCCCAATAAAATTCCCCA-3′ (oligo CS2RP5a; HBV nucleotide coordinates 2491–2475), and (iii) 5′-ACACTGACGACATGGTTCTACAACCTCCAATCACTCAC-3′ (oligo CS1FP9; HBV nucleotide coordinates 325–340) and 5′-TACGGTAGCAGAGACTTGGTCTGTTAAATAGTGGGGGAAAG-3′ (oligo CS2RP9; HBV nucleotide coordinates 730–712) (12). Bisulfite conversion, library preparation, and sequencing were performed at the Genomics and Microbiome Core Facility at Rush University. Data analysis was performed as described previously (12), and demultiplexed raw FASTQ files are accessible on the Gene Expression Omnibus (GEO) under GEO ID GSE242448.
ACKNOWLEDGMENTS
We thank Luca G. Guidotti and Francis V. Chisari (the Scripps Research Institute, La Jolla, CA, USA) for providing the HBV transgenic mice and Dr. Anjana Rao (La Jolla Institute for Allergy and Immunology, La Jolla, CA, USA) for providing the mice with floxed Tet2 and Tet3 alleles.
This work was supported by the National Institutes of Health grants R01 AI170785 and R01 CA238328. M.M.-C. was supported in part by NCATS through grant UL1TR002003.
Contributor Information
Alan McLachlan, Email: mclach@uic.edu.
J.-H. James Ou, University of Southern California, Los Angeles, California, USA.
REFERENCES
- 1. Dienstag JL. 2008. Hepatitis B virus infection. N Engl J Med 359:1486–1500. doi: 10.1056/NEJMra0801644 [DOI] [PubMed] [Google Scholar]
- 2. Lucifora J, Protzer U. 2016. Attacking hepatitis B virus cccDNA - the holy grail to hepatitis B cure. J Hepatol 64:S41–S48. doi: 10.1016/j.jhep.2016.02.009 [DOI] [PubMed] [Google Scholar]
- 3. Anderson AL, Banks KE, Pontoglio M, Yaniv M, McLachlan A. 2005. Alpha/beta interferon differentially modulates the clearance of cytoplasmic encapsidated replication intermediates and nuclear covalently closed circular hepatitis B virus (HBV) DNA from the livers of hepatocyte nuclear factor 1α-null HBV transgenic mice. J Virol 79:11045–11052. doi: 10.1128/JVI.79.17.11045-11052.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moraleda G, Saputelli J, Aldrich CE, Averett D, Condreay L, Mason WS. 1997. Lack of effect of antiviral therapy in nondividing hepatocyte cultures on the closed circular DNA of woodchuck hepatitis virus. J Virol 71:9392–9399. doi: 10.1128/JVI.71.12.9392-9399.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Seeger C, Mason WS. 2015. Molecular biology of hepatitis B virus infection. Virology 479–480:672–686. doi: 10.1016/j.virol.2015.02.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tuttleman JS, Pourcel C, Summers J. 1986. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell 47:451–460. doi: 10.1016/0092-8674(86)90602-1 [DOI] [PubMed] [Google Scholar]
- 7. Summers J, Mason WS. 1982. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 29:403–415. doi: 10.1016/0092-8674(82)90157-x [DOI] [PubMed] [Google Scholar]
- 8. Oropeza CE, Tarnow G, Sridhar A, Taha TY, Shalaby RE, McLachlan A. 2020. The regulation of HBV transcription and replication. Adv Exp Med Biol 1179:39–69. doi: 10.1007/978-981-13-9151-4_3 [DOI] [PubMed] [Google Scholar]
- 9. Will H, Reiser W, Weimer T, Pfaff E, Büscher M, Sprengel R, Cattaneo R, Schaller H. 1987. Replication strategy of human hepatitis B virus. J Virol 61:904–911. doi: 10.1128/jvi.61.3.904-911.1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Oropeza CE, Tarnow G, Taha TY, Shalaby RE, Hyde MV, Maienschein-Cline M, Green SJ, McLachlan A. 2021. Relative DNA methylation and demethylation efficiencies during postnatal liver development regulate hepatitis B virus biosynthesis. J Virol 95:e02148-20. doi: 10.1128/JVI.02148-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li L, Oropeza CE, Sainz B, Uprichard SL, Gonzalez FJ, McLachlan A. 2009. Developmental regulation of hepatitis B virus biosynthesis by hepatocyte nuclear factor 4α. PLoS ONE 4:e5489. doi: 10.1371/journal.pone.0005489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McFadden VC, Shalaby RE, Iram S, Oropeza CE, Landolfi JA, Lyubimov AV, Maienschein-Cline M, Green SJ, Kaestner KH, McLachlan A. 2017. Hepatic deficiency of the pioneer transcription factor FoxA restricts hepatitis B virus biosynthesis by the developmental regulation of viral DNA methylation. PLoS Pathog 13:e1006239. doi: 10.1371/journal.ppat.1006239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shen L, Wu H, Diep D, Yamaguchi S, D’Alessio AC, Fung H-L, Zhang K, Zhang Y. 2013. Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell 153:692–706. doi: 10.1016/j.cell.2013.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang Y, Zhang D, Li Q, Liang J, Sun L, Yi X, Chen Z, Yan R, Xie G, Li W, Liu S, Xu B, Li L, Yang J, He L, Shang Y. 2016. Nucleation of DNA repair factors by FoxA1 links DNA demethylation to transcriptional pioneering. Nat Genet 48:1003–1013. doi: 10.1038/ng.3635 [DOI] [PubMed] [Google Scholar]
- 15. Reizel Y, Sabag O, Skversky Y, Spiro A, Steinberg B, Bernstein D, Wang A, Kieckhaefer J, Li C, Pikarsky E, Levin-Klein R, Goren A, Rajewsky K, Kaestner KH, Cedar H. 2018. Postnatal DNA demethylation and its role in tissue maturation. Nat Commun 9:2040. doi: 10.1038/s41467-018-04456-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tarnow G, McLachlan A. 2021. β-catenin signaling regulates the in vivo distribution of hepatitis B virus biosynthesis across the liver lobule. J Virol 95:e0078021. doi: 10.1128/JVI.00780-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tarnow G, McLachlan A. 2022. Selective effect of β-catenin on nuclear receptor-dependent hepatitis B virus transcription and replication. Virology 571:52–58. doi: 10.1016/j.virol.2022.04.006 [DOI] [PubMed] [Google Scholar]
- 18. Sekine S, Lan B-A, Bedolli M, Feng S, Hebrok M. 2006. Liver‐specific loss of β‐catenin blocks glutamine synthesis pathway activity and cytochrome p450 expression in mice. Hepatology 43:817–825. doi: 10.1002/hep.21131 [DOI] [PubMed] [Google Scholar]
- 19. Cadoret A, Ovejero C, Terris B, Souil E, Lévy L, Lamers WH, Kitajewski J, Kahn A, Perret C. 2002. New targets of β-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene 21:8293–8301. doi: 10.1038/sj.onc.1206118 [DOI] [PubMed] [Google Scholar]
- 20. Straussman R, Nejman D, Roberts D, Steinfeld I, Blum B, Benvenisty N, Simon I, Yakhini Z, Cedar H. 2009. Developmental programming of CpG island methylation profiles in the human genome. Nat Struct Mol Biol 16:564–571. doi: 10.1038/nsmb.1594 [DOI] [PubMed] [Google Scholar]
- 21. Raney AK, Eggers CM, Kline EF, Guidotti LG, Pontoglio M, Yaniv M, McLachlan A. 2001. Nuclear covalently closed circular viral genomic DNA in the liver of hepatocyte nuclear factor 1α-null hepatitis B virus transgenic mice. J Virol 75:2900–2911. doi: 10.1128/JVI.75.6.2900-2911.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, Huang Y, Qi Y, Peng B, Wang H, Fu L, Song M, Chen P, Gao W, Ren B, Sun Y, Cai T, Feng X, Sui J, Li W. 2012. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 1:e00049. doi: 10.7554/eLife.00049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bissig K-D, Wieland SF, Tran P, Isogawa M, Le TT, Chisari FV, Verma IM. 2010. Human liver chimeric mice provide a model for hepatitis B and C virus infection and treatment. J Clin Invest 120:924–930. doi: 10.1172/JCI40094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Winer BY, Huang T, Low BE, Avery C, Pais M-A, Hrebikova G, Siu E, Chiriboga L, Wiles MV, Ploss A. 2017. Recapitulation of treatment response patterns in a novel humanized mouse model for chronic hepatitis B virus infection. Virology 502:63–72. doi: 10.1016/j.virol.2016.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guidotti LG, Matzke B, Schaller H, Chisari FV. 1995. High-level hepatitis B virus replication in transgenic mice. J Virol 69:6158–6169. doi: 10.1128/jvi.69.10.6158-6169.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de Mendoza A, Lister R, Bogdanovic O. 2020. Evolution of DNA methylome diversity in eukaryotes. J Mol Biol 432:1687–1705. doi: 10.1016/j.jmb.2019.11.003 [DOI] [PubMed] [Google Scholar]
- 27. Wu X, Zhang Y. 2017. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet 18:517–534. doi: 10.1038/nrg.2017.33 [DOI] [PubMed] [Google Scholar]
- 28. Kim J-W, Lee SH, Park YS, Hwang J-H, Jeong S-H, Kim N, Lee DH. 2011. Replicative activity of hepatitis B virus is negatively associated with methylation of covalently closed circular DNA in advanced hepatitis B virus infection. Intervirology 54:316–325. doi: 10.1159/000321450 [DOI] [PubMed] [Google Scholar]
- 29. Jain S, Chang T-T, Chen S, Boldbaatar B, Clemens A, Lin SY, Yan R, Hu C-T, Guo H, Block TM, Song W, Su Y-H. 2015. Comprehensive DNA methylation analysis of hepatitis B virus genome in infected liver tissues. Sci Rep 5:10478. doi: 10.1038/srep10478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vivekanandan P, Thomas D, Torbenson M. 2009. Methylation regulates hepatitis B viral protein expression. J Infect Dis 199:1286–1291. doi: 10.1086/597614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schweitzer A, Horn J, Mikolajczyk RT, Krause G, Ott JJ. 2015. Estimations of worldwide prevalence of chronic hepatitis B virus infection: a systematic review of data published between 1965 and 2013. The Lancet 386:1546–1555. doi: 10.1016/S0140-6736(15)61412-X [DOI] [PubMed] [Google Scholar]
- 32. Ott JJ, Stevens GA, Groeger J, Wiersma ST. 2012. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine 30:2212–2219. doi: 10.1016/j.vaccine.2011.12.116 [DOI] [PubMed] [Google Scholar]
- 33. Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, Gonzalez FJ. 1995. Targeted disruption of the α isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol 15:3012–3022. doi: 10.1128/MCB.15.6.3012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, Magnuson MA. 1999. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using cre recombinase. J Biol Chem 274:305–315. doi: 10.1074/jbc.274.1.305 [DOI] [PubMed] [Google Scholar]
- 35. Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, Tsangaratou A, Rajewsky K, Koralov SB, Rao A. 2011. Ten-eleven-translocation 2 (Tet2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci USA 108:14566–14571. doi: 10.1073/pnas.1112317108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ko M, An J, Pastor WA, Koralov SB, Rajewsky K, Rao A. 2015. TET proteins and 5-methylcytosine oxidation in hematological cancers. Immunol Rev 263:6–21. doi: 10.1111/imr.12239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chomczynski P, Sacchi N. 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162:156–159. doi: 10.1006/abio.1987.9999 [DOI] [PubMed] [Google Scholar]
- 38. Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual. 2 ed. Cold Spring Harbor Laboratory Press, New York. [Google Scholar]
- 39. Galibert F, Mandart E, Fitoussi F, Tiollais P, Charnay P. 1979. Nucleotide sequence of the hepatitis B virus genome (subtype ayw) cloned in E. coli. Nature 281:646–650. doi: 10.1038/281646a0 [DOI] [PubMed] [Google Scholar]
- 40. Sabath DE, Broome HE, Prystowsky MB. 1990. Glyceraldehyde-3-phosphate dehydrogenase mRNA is a major interleukin 2-induced transcript in a cloned T-helper lymphocyte. Gene 91:185–191. doi: 10.1016/0378-1119(90)90087-8 [DOI] [PubMed] [Google Scholar]
- 41. Bybee SM, Bracken-Grissom H, Haynes BD, Hermansen RA, Byers RL, Clement MJ, Udall JA, Wilcox ER, Crandall KA. 2011. Targeted amplicon sequencing (TAS): a scalable next-gen approach to multilocus, multitaxa phylogenetics. Genome Biol Evol 3:1312–1323. doi: 10.1093/gbe/evr106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Green SJ, Venkatramanan R, Naqib A. 2015. Deconstructing the polymerase chain reaction: understanding and correcting bias associated with primer degeneracies and primer-template mismatches. PLoS One 10:e0128122. doi: 10.1371/journal.pone.0128122 [DOI] [PMC free article] [PubMed] [Google Scholar]