

Abstract
Throughout the body, T cells monitor MHC-bound ligands expressed on the surface of essentially all cell types. MHC ligands that trigger a T cell immune response are referred to as T cell epitopes. Identifying such epitopes enables tracking, phenotyping, and stimulating T cells involved in immune responses in infectious disease, allergy, autoimmunity, transplantation, and cancer. The specific T cell epitopes recognized in an individual are determined by genetic factors such as the MHC molecules the individual expresses, in parallel to the individual’s environmental exposure history. The complexity and importance of T cell epitope mapping have motivated the development of computational approaches that predict what T cell epitopes are likely to be recognized in a given individual or in a broader population. Such predictions guide experimental epitope mapping studies and enable computational analysis of the immunogenic potential of a given protein sequence region.
Keywords: T cells, immune epitopes, machine learning, databases, benchmarking
INTRODUCTION
T cells scan MHC ligands presented to them on the surface of nucleated cells (expressing MHC class I molecules) and on professional antigen-presenting cells and other cells of the lymphoid lineage (expressing both MHC class I and II molecules). This allows T cells to detect antigens derived from pathogens as well as the presence of abnormal self-antigens expressed by cancer cells (Figure 1a). Complexes of MHC molecules and their ligands are generated by antigen-processing and -presentation pathways consisting of a series of enzymatic events involving specialized organelles and processes, which are distinct for MHC class I and MHC class II. As a first approximation, class I molecules sample the interior of each cell, while class II molecules provide a window to what proteins and peptides are present in the extracellular environment. MHC ligands that trigger a T cell immune response are referred to as epitopes. T cells recognizing an epitope can exert direct effector functions such as the production of inflammatory or regulatory cytokines, cytotoxicity, and providing help to B cells regulating the development and maturation of antibody responses. Upon recognition of an epitope, T cells proliferate to form an effector population that can detect the same epitope on other cells and can form long-lived memory populations that enable the host to rapidly respond to subsequent encounters of the same epitope (1). Thus, T cell epitope recognition is a critical step in the formation and recall of adaptive immune responses.
Figure 1.
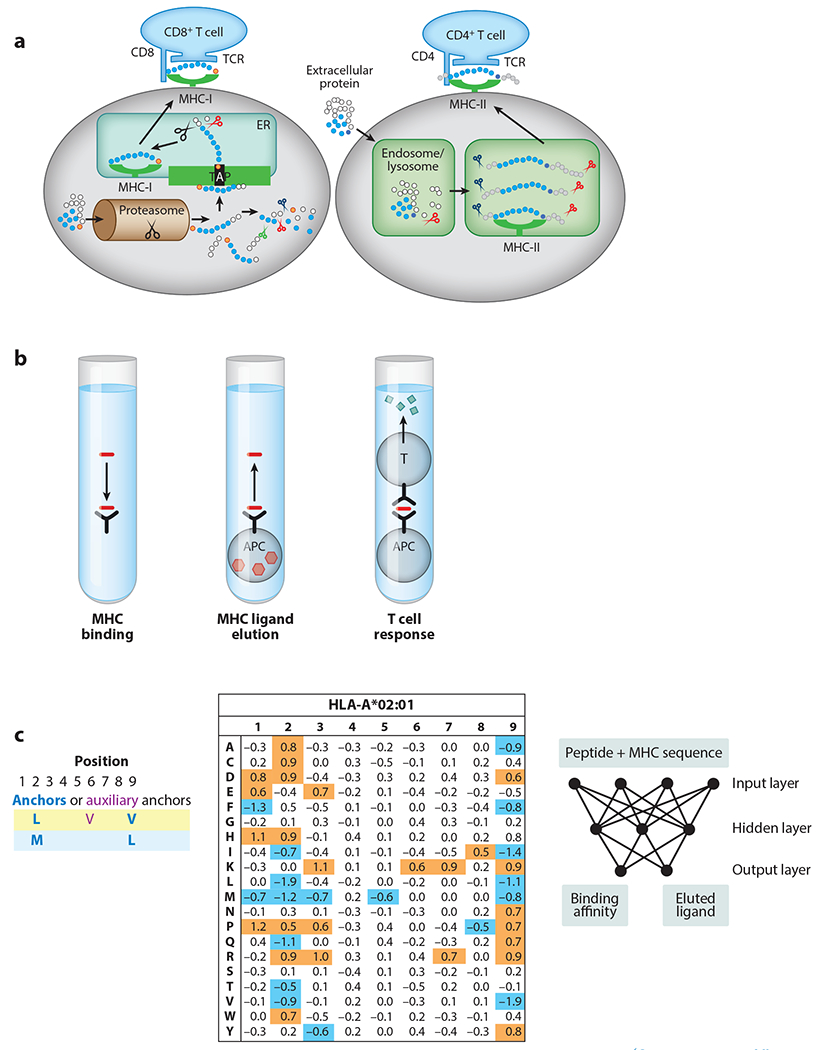
Overview of the biological process, experimental assessment, and computational prediction of T cell epitope recognition. (a) Overview of the main cellular mechanisms involved in antigen processing, presentation, and recognition of T cell epitopes that have been included in computational predictions. (Left) MHC class I–restricted T cell epitopes primarily arise from intracellular antigens that are cleaved by the proteasome and transported into the ER by TAP, where they can bind to MHC class I molecules that get transported to the cell surface, where they can be recognized by CD8+ T cells. Proteins and peptides are depicted as beads-on-a-string, with red circles indicating amino acids that are C-terminal residues of peptides presented by MHC molecules. In contrast, MHC class II–restricted T cell epitopes (right) are primarily derived from extracellular proteins taken up by professional APCs that are cleaved in lysosomal vesicles, where they can bind to MHC class II molecules, be transported to the cell surface, and be recognized by CD4+ T cells. Dark purple circles indicate amino acids at the C-terminal end of the core binding to MHC-II. (b) Three main categories of experimental assays have been utilized to characterize the steps involved in antigen processing and recognition of T cell epitopes. (Left) MHC binding assays that determine the affinity of a synthetic peptide to a specific MHC molecule. (Middle) MHC ligand elution assays that isolate and identify peptides bound to MHC molecules on the cell’s surface as a result of natural antigen processing and presentation. (Right) T cell epitope recognition assays, in which the ability of T cells to interact with and/or respond to a candidate epitope is determined. (c) Approaches to the computational prediction of T cell epitopes, starting with pioneering use of MHC motifs such as SYFPEITHI (left) (47), in which allowed amino acids at anchor positions (blue bolded) and at auxiliary anchor positions (purple) were identified based on a heuristic analysis. This was followed by machine learning approaches that were explicitly trained on quantitative data such as BIMAS (middle) (50), where numeric values would be assigned for each of the 20 conventional amino acids (rows) at each position in a 9-residue peptide (columns), so that they best reproduce measured binding affinities for a set of peptides that were previously tested (the training data). Finally, current neural networks approaches have custom architectures that allow training on combined data from multiple MHC alleles and from both MHC binding and elution data, such as the recent NetMHCpan version 4.0 (right) (127). Abbreviations: APC, antigen-presenting cell; ER, endoplasmic reticulum; TAP, transporter associated with antigen processing; TCR, T cell receptor.
The identification of epitopes enables tracking, phenotyping, and stimulating T cells. Epitopes can be used to detect the magnitude and cytokine polarization of epitope-specific T cell responses in an input sample based on cytokine secretion assays such as ELISPOTs or ELISAs. They can be used in flow cytometry and mass cytometry assays to detect and phenotype epitope-specific T cells based on intracellular cytokine-staining assays, or to isolate and characterize them in single-cell RNA-seq assays and emerging technologies such as CITE-seq. These experimental techniques have provided an improved mechanistic understanding of T cells involved in different disease contexts. Furthermore, detection of epitope-specific T cells has been used in diagnostic applications (2) and to deimmunize proteins used as biological drugs (3–5). Additional interest in T cell epitopes has arisen in the context of cancer immunotherapy, where the number of potential T cell epitopes in a tumor has been proposed as a marker of success for checkpoint blockade treatments, and where tumor-specific epitopes are being used to induce tumor-specific T cell responses (6). These practical applications of T cell epitopes have continued to drive efforts to improve methods to identify them.
There are three main categories of assays that have been used to dissect the mechanistic steps involved in T cell epitope formation and recognition (Figure 1b). The first is assays measuring MHC binding in vitro. This directly determines which peptides have the potential to be presented to T cells and can provide quantitative affinity data (7–9). The second is assays detecting MHC ligands presented on cells by elution of such ligands and their detection by mass spectrometry. This allows us to factor in the influence of antigen processing in the generation of the ligand before and after MHC binding (10–14). Third is assays measuring T cell epitope recognition of an epitope. This directly reads out the type and magnitude of T cell responses to epitopes in a specific individual (15–19).
A key challenge in identifying T cell epitopes is that their recognition varies substantially between individuals. One factor driving this variability is that the genes encoding for MHC molecules (called HLA in humans) are the most polymorphic in the human genome. Different MHC molecules have distinct binding specificities, which results in them presenting different MHC ligands to T cells. As a result, different individuals in the human population will present different epitopes, and pathogens will find it difficult to evade recognition completely. In addition to MHC polymorphism, what T cell epitopes are recognized is also shaped by the exposure history of an individual. Reencounter of an epitope will favor activation of memory cell responses, rather than induce de novo responses. Overall, this means that what specific T cell epitopes are recognized in a given individual will be impacted by both genetic and environmental factors.
Given the importance and complexity of T cell epitope mapping, there has been a continued interest to develop computational prediction methods that aid in the identification of T cell epitopes. These prediction methods have evolved from the identification of amino acid motifs in peptides that correspond to their MHC binding residues, to the advent of quantitative MHC binding affinity predictions using machine learning approaches, to the current state-of-the-art predictions that utilize custom neural network architectures that are capable of integrating information from MHC binding and MHC ligand elution data across multiple MHC molecules (Figure 1c).
This review focuses on the development of computational methods for T cell epitope prediction, how these methods have been shaped by the experimental data available, the best practices in practical applications, and remaining challenges to the field. We restrict our scope to T cells recognizing peptide epitopes presented by classical MHC class I and class II molecules. This is not to diminish the importance of nonpeptidic or posttranslationally modified epitopes, but it reflects that essentially all current T cell epitope predictions target conventional peptide epitopes.
A BRIEF HISTORY OF THE DISCOVERY OF T CELL EPITOPES AND THEIR MHC RESTRICTION
Several Annual Review of Immunology articles have covered the topics of antigen processing and presentation, their relationship to MHC molecules and epitope generation, and epitope recognition by the T cell receptor (TCR) repertoire (7, 8, 10–21). Here, the history of how the mechanisms of T cell epitope recognition were discovered is briefly recapped to introduce the vocabulary still in use today and the different experimental methods that form the basis of T cell epitope discovery. Two different Nobel prizes were awarded to recognize the seminal observation that immune responsiveness is regulated by genes encoded in the MHC locus, which are associated with high allelic polymorphism: one to Snell, Daussett, and Benacerraf in 1980 “for their discoveries concerning genetically determined structures on the cell surface that regulate immunological reactions,” and one to Doherty and Zinkernagel in 1996 “for their discoveries concerning the specificity of the cell mediated immune defense” (22, 23). In this context, it was stated that T cell recognition is MHC restricted, as conventional T cells recognize a particular combination of a given antigen and a specific MHC. The molecular basis for this MHC restriction was much debated. Some investigators thought that this must reflect the fact that T cells carried two receptors, one for MHC and one for antigen; others argued that it was most likely that a single receptor recognized the combination of both (24). In the 1970s, parallel investigations by Gell, Benaceraff, and Ishizaka compared the capacity of B cells and T cells to distinguish between native and denatured forms of the same antigen (25). They found that antibodies derived from animals immunized with native antigen reacted strongly against native antigen but failed to react with denatured antigen. In contrast, T cells broadly cross-reacted with both forms, suggesting that while antibodies are very dependent on the 3D structure of antigen for recognition, T cell cross-reactivity is dictated solely by the primary amino acid sequence of a protein. The point was proven by observations from Grey, Kappler, and Marrack for CD4 (26, 27) and expanded by the Townsend group for CD8 T cells (14, 28), showing that T cells recognize a peptide fragment derived from their antigen of specificity, the epitope. Soon after, Unanue’s and Grey’s groups demonstrated specific binding of epitopes to purified MHC in vitro, and showed that the binding pattern to different alleles matched their known MHC restriction (29, 30).
THE CONCEPT OF MHC MOTIFS AND THE EXPERIMENTAL METHODS TO DETERMINE THEM
In the late 1980s several groups developed approaches to predict which peptides might be epitopes. DeLisi & Berzofsky (31) proposed that T cell epitopes might be predicted on the propensity to form amphipathic α helices, and Rothbard and colleagues proposed a short 4- to 5-residue hydrophobic stretch as a predictor (32). While neither of these approaches held up well with larger data sets, they opened the field for further development of new prediction approaches. In retrospect, the missing insight was that separate predictors for different class I and class II allelic variants are necessary, as each MHC allele is associated with a different binding specificity. This became increasingly clear through studies showing that MHC variants have different epitope binding capacity, which predicted T cell responsiveness (33), and that peptide binding specificity is determined by the presence of specific amino acid patterns (34). Systematic studies revealed that while certain positions in the peptide could be substituted with almost any amino acid, other positions would only tolerate limited substitutions with closely related amino acids in terms of side chain properties. These positions were termed main anchor residues of the epitopes. It was further shown that these anchor residues were found with similar spacing in different epitopes restricted by the same MHC. Therefore, these residues were termed anchor positions, and the sum of the anchor positions spacing and specificity was called the MHC ligand motif.
The physical basis of MHC ligand motifs was first hinted at by earlier data from McDevitt and coworkers (35), which had shown that the MHC residues polymorphic in different allelic variants clustered in hypervariable regions reminiscent of what was previously shown for antibody molecules. It was hypothesized that these hypervariable regions formed epitope-binding sites in the MHC molecule and that the anchor positions within MHC peptide ligands were bound to this site. This was shown to be exactly the case when Wiley and associates solved the crystal structures of HLA A2 first (36) and HLA DR1 shortly after (37). The MHC molecules in these structures were found to have characteristic pockets that explained the spacing and residue specificity of the MHC ligand motifs.
The definition and refinement of MHC motifs received a significant boost through the advent of experimental techniques to isolate naturally processed and presented MHC ligands. Studies by Nathenson, Rammensee, and Bevan had shown that the exact natural ligand recognized by T cells could be recovered from purified MHC (38–41). Rammensee’s group took this observation one step further, by sequencing by Edman degradation pooled class I ligands, and showing that they were remarkably homogeneous in size and that certain main anchor positions were conserved and associated with limited chemical diversity (42). Elution of natural class I ligands provided a powerful method to define class I MHC ligands motifs that was simple, conceptually elegant, and technically powerful, resulting in definition in a brief time of tens of motifs for different allelic variants. This methodology proved effective for class I MHC, but less so for class II molecules, which have a peptide-binding groove that is open at both sides. As a result, the anchor positions of MHC class II–bound peptides are not in frame, rendering sequencing of pooled ligands more difficult to interpret. To overcome the limitations of pooled ligand sequencing, eluted peptides were separated by chromatography and individual ligands identified by mass spectrometry approaches (43, 44), resulting in the direct identification of peptide ligands presented on cells—a technique that has continued to be improved in throughput and sensitivity to this day, providing a true wealth of information and insight into the natural ligands of MHC molecules.
MHC BINDING PREDICTIONS BASED ON MOTIFS AND OTHER HEURISTIC APPROACHES
The era of computational T cell epitope predictions was initiated in 1989 by Sette and colleagues (45), who described a computer program that used MHC allele–specific motifs to identify potential ligands in a protein sequence. By the mid-1990s, the motifs associated with many class I and class II MHCs were defined at a variable level of resolution. The simplest canonical motifs were based on the determination of the main anchor residues and their relative spacing. It became apparent, however, that such motifs were an oversimplification, with only about a third of peptides containing the canonical motif being able to bind MHC, and many ligands binding MHC not containing the exact motif. This was reconciled by taking into account additional auxiliary (or secondary) anchor positions that could influence binding, albeit in a less pronounced fashion than the primary anchor positions (46). Several approaches were developed that aimed at producing a quantitative score, related to the predicted binding affinity or to the probability of binding. Essentially these methods were based on a matrix that for each position assigned a heuristic numerical value corresponding to the expected impact of the peptide carrying that specific amino acid. The various values for each position were then combined to derive a final score for a given peptide/MHC combination. Popular scores were the SYFPEITHI score, which was based on the analysis of ligand elution data (47), and the average relative binding (ARB) matrices (48), which were based on measured binding data from single substitution analogs of known ligands.
THE ADVENT OF MACHINE LEARNING TO PREDICT MHC BINDING
Driven by the success of the heuristic predictions, more advanced supervised machine learning approaches were soon proposed. Such approaches consist of training an algorithm based on labeled input data, such as sets of peptides with measured binding affinities, to generate a function that approximately reproduces the input data by learning patterns that are not defined a priori and that are capable of predicting how new data should be labeled (49). The first such method applied to MHC binding predictions was the BIMAS (50) model proposed by Parker et al. (50), in which coefficients of a matrix defining the contribution of different residues in a peptide to binding to HLA-A*02:01 were fitted by linear regression to experimentally measured half-lives (Figure 1c). This matrix model implicitly assumes independent contributions of each residue in a peptide to the overall binding affinity, and it provided robust predictions of the affinity of previously untested peptides. A website hosted at the US National Institutes of Health (NIH) (unfortunately retired in 2019) made predictions for HLA-A*02:01 and several other alleles publicly available, along with the underlying prediction matrices, which set an important positive precedent for making computational predictions accessible and reproducible for the community at large.
More complex prediction models for MHC class I peptide binding that allowed for nonlinear interactions were also proposed, including artificial neural networks (ANN) (51–54), hidden Markov models (HMM) (55, 56), and QSAR (quantitative structure-affinity relationship)-based regression models (57). While these early models demonstrated reasonable success in reproducing the data used for their development, their usefulness for epitope discovery was often limited due to their low allelic coverage (most methods were trained and evaluated on data covering one or two MHC molecules) and the low number of data points available for model construction. Specifically, the low number of data points was a critical problem for complex prediction methods that require determining many parameters, which makes them prone to overfitting and overestimation of model performance on a small data set. In contrast, the performance of the simple model underlying the BIMAS predictions held up remarkably well given the limited input data available used to generate them. The originally unexpected finding that simple linear methods outperformed more complex nonlinear predictions was further dissected in the development of the stabilized matrix method (SMM) (58), which explicitly separated linear contributions of each residue in a peptide to binding, and nonlinear pair-interaction terms quantifying the impact of two specific residues at different positions, and which used regularization to avoid overfitting. This approach showed that some pair-interactions are reproducibly found in data sets that are large enough, but that their strength is at least an order of magnitude lower than the direct contributions to binding of individual peptide residues.
There is a physical explanation for why simple linear models of peptide–MHC interactions can provide accurate predictions of measured binding affinities: Peptides conventionally bind to MHC molecules in an extended conformation, where every residue in an MHC-bound peptide has a defined position in the MHC binding groove. Thus, as a first approximation, each amino acid in a peptide contributes independently to the overall peptide binding affinity. This largely fixed structural configuration of peptide binding also explains why computational approaches that explicitly model the 3D structure of MHC–ligand complexes and their physicochemical interactions have not provided prediction performances superior to those of sequence-based machine learning approaches for MHC binding (59), in contrast to the success (or even requirement) of 3D modeling for other ligand interactions, such as those inducing conformational changes in ligand-binding proteins (60).
EPITOPE DATABASES AS A SOURCE OF DATA TO TRAIN MACHINE LEARNING ALGORITHMS
The performance of machine learning algorithms increases with the amount of data available to train them, which makes data set assembly an essential step in tool development. Epitope databases that compile records from publications and other data sources in a consistent format make this task much easier. Several pioneering databases were initiated starting in the 1990s. Among those still available today are the HIV molecular immunology database, led by the Korber lab, which catalogs T cell and B cell epitopes in HIV viruses (61); and the SYFPEITHI database, led by the Rammensee lab, which catalogs eluted MHC ligands, pool sequencing motifs, and T cell epitopes from any source (47); and the MHCBN database, led by the Raghava lab, which in addition to MHC binding and T cell epitope data also contains transporter associated with antigen processing (TAP) binding data (62). In 2003, the Immune Epitope Database (IEDB) (63) was initiated as a repository for epitope-related data curated from the literature as well as for data generated by large-scale T cell and B cell epitope discovery contracts funded by the NIH. As of today, the IEDB is led by the Sette and Peters labs, contains over 2,000,000 experiments curated from over 20,000 references (64), and is accompanied by a companion site providing access to epitope prediction and analysis tools (65), many of which were developed in the Nielsen lab. The most recent major addition to epitope-related databases is SysteMHC (66), which captures MHC ligand elution data identified by mass spectrometry and provides access to both raw data from multiple labs and (re)analyzed data run through a consistent pipeline (67). All of these database efforts compile data from different sources in a consistent format, which enables the training and evaluation of machine learning predictions.
THE VALUE OF BENCHMARKING TO UNRAVEL DIFFERENCES IN PREDICTION METHOD PERFORMANCES FOR MHC BINDING TO GUIDE TOOL DEVELOPERS AND USERS
With a proliferation of different prediction methods, the field was challenged by a lack of objective metrics allowing comparisons of their performance. A systematic attempt to address this issue was a benchmark of publicly available prediction methods conducted by us using data assembled in the course of the initial construction of the IEDB. This benchmark utilized a data set of quantitative peptide binding to a panel of mouse, human, macaque, and chimpanzee MHC molecules, most of which were previously unpublished (68). For three prediction methods, the underlying algorithms were directly available to us, so cross validated performances of the algorithms could be obtained. These algorithms were ARB, SMM, and NetMHC, the latter being referred to as ANN (artificial neural network) in the paper, as it was a neural network designed based on the NetMHC algorithm (69) but retrained on benchmark data. Two main conclusions were drawn from this study: First, all three methods when retrained on the large benchmark data set outperformed the earlier published web servers. This demonstrated that the size of the data set used for training plays a critical role in determining the predictive power of a given prediction method, suggesting that not only machine learning algorithmic advances but also persistent retraining on newly available data sets is required for tools to have optimal predictive power. Second, the NetMHC-based method performed best overall, with highest performance in 30 out of 46 data sets that could be compared between all three methods, while the SMM approach had the highest performance on 16 data sets, and the heuristic ARB method did not score highest on any data set.
Interestingly, NetMHC showed the most dominant outperformance on smaller data sets. This conflicts with the assumption that its ability to predict nonlinear interactions is the basis of its superior performance, as predicting nonlinear interactions should depend highly on having large training data sets available. In fact, no method has been able to identify quantitative nonlinear contributions to MHC binding beyond what has been published for SMM. Closer examination of how NetMHC performed well in predicting binding when trained on small data sets revealed that a key advantage was the way it presented peptides to the neural network. The naive approach to encode a peptide of length N is to generate a binary vector with N × 20 entries corresponding to the N positions in the peptide and the 20 canonical amino acids. NetMHC does not use binary vectors but rather encodes peptides using BLOSUM matrices that implicitly provide information on amino acid similarity to the network. This allowed the NetMHC algorithm to extrapolate patterns of binding to residue types not found in the training set. Motivated by this, a Bayesian Prior was added to the SMM method that essentially teaches it how similar different amino acids are, which significantly improved the ability of the algorithm to predict MHC binding on small data sets [SMM-PMBEC (70)]. Overall, these observations indicate that there is not a simple dichotomy between linear versus nonlinear predictions that explains differences in prediction performance for MHC class I binding predictions, but that additional factors, such as the encoding of biological knowledge in the peptide presentation, are key to generating high-performance predictions.
For MHC class II, similar efforts were dedicated to the development of peptide-binding prediction methods, but the challenge was substantially greater due to the open binding groove of MHC class II molecules. This allows peptides to protrude outside of the binding groove and makes alignment of binding peptides essential to identifying the common binding core. The list of machine learning frameworks proposed to resolve this challenge is long and includes HMM (71), SVM (72, 73), Gibbs sampling (74), and ANN (75, 76) among others. A decade ago, benchmarking studies showed that machine learning–based models achieved the highest predictive power (77, 78) and that overall prediction performance was lower than for MHC class I, but that it could be improved by making consensus predictions, similar to what was done for MHC class I before (79). Over the years, the ANN-based framework NNAlign (76) used to develop the NetMHCII (and NetMHCIIpan; see below) methods (80–82) has been continuously refined (83–88). And MHC class II binding predictions have broadly caught up to where MHC class I binding predictions were a decade ago They achieve area-under-the-curve (AUC) values of 0.87 (80), while for MHC class I AUC can be as high as >0.98 when identifying binders from peptide sets that were not preselected, but such data sets are increasingly rare (89), as most peptides tested experimentally for binding today are preselected based on predicted binding to avoid obvious nonbinders.
DEVELOPMENT OF PAN-MHC BINDING PREDICTION METHODS
The experimental data available for different MHC molecules are highly uneven, with some alleles being very well studied, such as HLA-A*02:01, while many other alleles have never been studied in binding assays at all. Given that the frequency of HLA alleles can vary substantially between different ethnicities, and given that there is an interest in rarely expressed alleles that are associated with specific diseases, it is desirable to generate accurate predictions for all HLA alleles. However, with over 10,000 allelic variants of HLA molecules described in the IMGT-HLA database (90), the conventional approach of generating large data sets for each allele and training allele-specific prediction algorithms on each data set is not feasible. To resolve this, pan-specific prediction methods were proposed that can predict binding for MHC molecules not characterized experimentally. The first method to successfully do this was TEPITOPE (91), which could predict binding to 51 prevalent HLA-DR alleles. TEPITOPE is based on the construction of virtual matrices that characterize the binding profile of a given HLA-DR molecule by comparing residues in its sequence that are forming a binding pocket to pockets from other MHC molecules where the binding specificity has been defined. This approach achieved solid prediction performances and demonstrated for the first time that the specificity of an MHC molecule that has never been experimentally characterized can be computationally predicted.
The first computational method to implement pan-specific predictions for MHC class I molecules was NetMHCpan (92). This method was inspired by the work of Brusic and coworkers (93), who complemented the peptide binding information used to train a prediction model with information about the amino acids defining the MHC binding groove, which allowed utilizing binding data generated from different MHC molecules to train a single neural network. NetMHCpan expanded this to make predictions for MHC molecules that had never been tested and demonstrated that this could greatly improve the ability to make accurate predictions for alleles characterized with limited or even no binding data. Later, other pan-specific approaches for MHC class I such as ADT (94), KISS (95), and PickPocket (96) were proposed, each implementing different representations of the MHC binding environment to allow for the development of pan-specific prediction models. Independent benchmarking subsequently demonstrated the superior performance of NetMHCpan for prediction of peptide binding, MHC ligands, and CD8 epitopes (97). For MHC class II, later approaches similar to that of NetMHCpan described above were proposed, including MultiRTA (98), MHCIIMulti (99), and NetMHCIIpan (100), each of which represented the MHC binding environment along with the peptide in a machine learning approach to enable true pan-specificity covering all class II proteins of known sequence.
PREDICTING NATURALLY PROCESSED AND PRESENTED MHC LIGANDS
For a peptide epitope to be recognized by T cells, it has to bind to an MHC molecule. Prior to that, it has to be generated by the antigen-processing and -presentation pathway. Several studies have been performed to predict steps in the MHC class I antigen-processing pathway, including proteasomal cleavage (101, 102) and TAP transport efficiency (103, 104). These studies showed that the steps involved in antigen processing have specificities that can be learned from experimental data and applied to identify MHC ligands. However, the specificity of proteasome cleavage and TAP transport is much less selective than MHC binding. Several methods have integrated the prediction of different steps in antigen processing and presentation to allow for improved prediction of MHC class I ligands and T cell epitopes (105–108). These approaches were able to achieve consistent but minor improvements in predictive power for epitope identification compared to state-of-the-art methods for MHC binding alone (109). Comparing the specificity of the proteasome, TAP and MHC suggested a simple explanation for this: MHC molecules appear to have (co)evolved to be able to bind peptides that are efficiently generated by the proteasome and transported by TAP. This means that incorporating the specificity of these antigen-processing steps into a prediction algorithm does not significantly improve the specificity of the results (102).
While the impact of the antigen-processing machinery on the sequence composition of MHC class I ligands is masked by the overlapping MHC binding specificity, in contrast, it does have an apparent impact on the peptide length distribution of presented MHC ligands. While binding assays reveal that different HLA class I molecules favor distinct peptide lengths, ligand elution profiles show a much narrower distribution of peptide lengths, strongly favoring peptides of length 9 (110, 111). This narrower distribution can be explained by the peptide length preference resulting from antigen-processing steps, which limits the ligands available for binding to MHC (111), which has been previously postulated (112–114).
For MHC class II, a different set of cellular and biochemical processes are operational in determining how the antigen-processing machinery shapes the ligand repertoire. These processes have also been studied and characterized in detail (10, 18, 115, 116), but until recently, they had not been incorporated into computational prediction methods. Large-scale data sets of MHC class II–restricted eluted ligands made it apparent that there is indeed a sequence motif characteristic of N-terminal and C-terminal residues of processed ligands, consistent with the termini being generated through proteolytic cleavage with specific motifs (117, 118), and that incorporation of these cleavage signals benefits the prediction of MHC class II ligands. However, T cell epitopes are typically discovered by testing synthetic peptides, and for class II–restricted peptides, their ends can be normally extended or trimmed without impacting T cell recognition. This is because the epitope core residues directly interacting with the MHC molecule are also neighboring or close to the residues that contact the TCR (although examples of TCR interaction with the residues flanking the binding core have been described, such as in Reference 119). Thus, the termini of MHC class II T cell epitopes are not well defined, which is also apparent from MHC class II ligand elution data, which often results in ladders of peptides (44) that share a common core of typically nine residues binding the MHC molecule. This multitude of possible peptide ligands that the antigen-processing machinery can generate for any given binding core might explain why it has not been feasible to use the antigen-processing motifs to significantly improve T cell epitope prediction. Overall, there is symmetry in that both MHC class I and class II antigen processing follow deterministic steps that can be successfully predicted in isolation, but incorporating these steps into T cell epitope predictions as a separate selective step does not result in notable performance gains beyond MHC binding.
COMBINING MHC BINDING AND ELUTION DATA TO IMPROVE PREDICTION PERFORMANCE
The elution of MHC ligands naturally processed and presented by antigen-presenting cells and their identification by mass spectrometry have rapidly advanced in recent years, and it is now possible to routinely generate data sets with thousands of MHC ligands. As described above, efforts to learn motifs unrelated to MHC binding that enable identification of naturally processed and presented ligands and can be transferred to T cell epitope predictions have been disappointing. However, large-scale ligand elution data are still highly useful to improve the prediction of MHC binding overall. The analysis and interpretation of MHC eluted ligand data to improve MHC ligand prediction can be challenging if the ligands are eluted from cells expressing multiple MHC molecules and thus do not have well-defined MHC restriction. Experimental approaches to address this include the use of mono-allelic cell lines, such as in Abelin et al. (120), or the use of cell lines expressing a secreted form of specific MHC molecules, which was pioneered by the Hildebrand group (121). However, the use of such cell lines is not always possible, and computational approaches have been proposed to deconvolute data gathered in the context of multiple MHC molecules. Bassani-Sternberg et al. (122) demonstrated how the unsupervised Gibbs clustering approach developed by Andreatta et al. (123) could be elegantly used to deconvolute MHC class I ligand data. Later, Gfeller and colleagues extended this approach and suggested a framework for deciphering and annotating HLA-I motifs based on co-occurrence of alleles across large MHC ligand data sets (124). Other studies applied binding prediction methods to infer the MHC restriction of each ligand (125). Independent of the approach utilized, the analyses result in long lists of MHC ligands and their putative MHC restriction.
The availability of large MHC ligand data sets allowed training machine learning algorithms that demonstrated high performance in particular for the prediction of other MHC eluted ligands but also to a lesser extent for T cell epitopes (120, 126, 127). While this shows that MHC ligand data are a rich source of information, there are downsides in that the numbers of alleles covered are still comparably low (although this is rapidly changing), and more importantly, MHC ligand elution data are not quantitative in contrast to MHC binding data. Given that these two types of data measure overlapping characteristics, it is desirable to develop prediction algorithms that can benefit from both MHC binding and MHC ligand elution data. This was implemented in NetMHCpan version 4.0 by Jurtz et al. (127), which took MHC binding data covering 130 MHC class I alleles and MHC ligand elution data covering 55 alleles and combined these to train a single neural network with a novel architecture that outputs both predicted binding affinity and likelihood of being an eluted ligand for a given peptide, which enables the combined training. This approach had better performance than models trained on each data set separately for both for class I and class II (117, 127). An alternative approach for integrating MHC binding and MHC ligand data was implemented in the MHCFlurry tool (128), where the discordance between the qualitative eluted ligand and quantitative binding affinity data was handled using measurement inequalities in the machine learning cost function. This approach also demonstrated improved performance in particular for prediction of ligand elution data, further supporting that combining MHC binding and elution data generates superior prediction models.
A recently proposed novel approach to utilize MHC ligand elution data from cell lines expressing multiple MHC molecules is to make the assignment of MHC restriction to individual peptides a concurrent step in the training of a pan-allele ligand predictor, which was implemented as an extension of the NNAlign framework described above. This extension is capable of taking a mixed training set composed of single-allele (peptides assigned to single MHCs) and multiple-allele data (peptides with multiple options for MHC assignments) as inputs and fully deconvoluting the individual MHC restriction of all sequences while simultaneously training a pan-specific MHC binding predictor covering the binding specificities of all the MHCs present in the training set (129). This promises to be the next conceptual advance for prediction of both MHC class I and class II, as it allows compiling even larger combined data sets from both MHC binding and MHC ligand elution experiments. It has to be stressed again that integrating MHC ligand elution data sets does not seem to provide insights into fundamentally distinct properties of ligands in contrast to binding, but that the main advantage is simply the increase in the amount of data that can be used for training, which provides for a more refined understanding of the MHC binding motif.
IDENTIFYING T CELL EPITOPES USING MHC BINDING PREDICTIONS
The ultimate goal of most MHC binding and MHC ligand processing predictions is the identification of T cell epitopes. These applications require translating how differences in predicted MHC binding affinity or in the probabilities of being an MHC ligand relate to T cell recognition. The first systematic assessment that compared MHC class I binding affinity to T cell epitope recognition revealed that an affinity measurement of IC50 < 500 nM is a useful threshold to identify ~90% of class I restricted T cell epitopes (130). While this first assessment was largely based on data for HLA-A*02:01, a much larger data set of T cell epitopes covering diverse MHC alleles has become available from the Immune Epitope Database (IEDB) (64). Analysis of the IEDB data set confirmed the usefulness of 500 nM as a general threshold that captured about 85% of all epitopes when epitopes from all alleles were considered together (131). However, it also revealed significant variability of this threshold’s performance when epitopes restricted by individual HLA alleles are considered separately. The frequency of peptides that are predicted to bind at <500 nM varies substantially between MHC alleles, reflecting the difference in permissiveness of their binding motifs. Alleles that have a high frequency of binding peptides showed clustering of T cell epitopes at the higher end of the binding range, while alleles that had few predicted binders showed more T cell epitopes at lower affinity ranges. Incorporating these findings into epitope candidate selection can be achieved by using HLA allele–specific binding affinity cutoffs. However, concerns about study bias and the desire to have epitope candidates for different HLA alleles equally represented support a different approach of using a percentile ranking system. Such percentile ranks are established by predicting IC50 values for peptides from a large set of protein sequences for each MHC allele of interest, and establishing buckets that identify the top 0.1 percentile of IC50 values, the 0.1–0.2 percentile, and so on. Any predicted IC50 value can then be transformed into percentile values using these buckets. Based on the analysis of MHC class I–restricted epitope data, we would consider 2% to be a minimum predicted binding affinity (covering >95% of epitopes), 1% covering >80% percent of all epitopes, and 0.5% a threshold for high-affinity binders that are enriched for T cell epitopes. These thresholds were confirmed to be applicable for the identification of MHC class I–restricted neoepitopes in cancer cells (132, 133), and in a large-scale comparison of different prediction methods to identify epitopes derived from vaccinia virus (134).
For MHC class II molecules, an IC50 < 1,000 nM threshold was established using the same methodology used to establish the 500 nM threshold for class I (135). A thorough evaluation of MHC class II allele–specific thresholds using percentile cutoffs or IC50 values remains to be performed.
THE IMPACT OF MHC/HLA POLYMORPHISM: WHICH ALLELES TO CONSIDER
As different MHC alleles can have very different binding specificities, it is necessary to define which alleles are considered when making T cell epitope predictions. Importantly, the answer to this question will strongly depend on the application. If a study is testing candidate epitopes that are intended to cover a broader human population, it is necessary to cover a sufficient number of alleles expressed by most individuals in that population. This can be achieved by covering representative alleles of different supertypes of HLA molecules. Such supertypes of HLA molecules have been defined based on grouping together MHC alleles that share similar binding specificity, and they include ten major MHC class I (136) and ten MHC class II (137) supertypes. Alternatively, peptides can be assessed for their ability to cover a panel of alleles that represent all MHC molecules expressed in a significant frequency worldwide. While the supertype concept is useful to explain broad MHC binding patterns, we prefer to pick peptides predicted to bind to specific MHC alleles. We have found that approximately 25–30 HLA alleles for both class I (138) and class II (139) provide coverage for the most common allelic variants expressed in most well-studied ethnicities.
For MHC class II, we found that promiscuous peptides, defined as those capable of binding multiple common HLAs, are often dominant and account for approximately 50% of the total response (140). We further found that due to the high cross-reactivity between alleles, predicting peptides on the basis of the median MHC binding for a limited set of HLA alleles representative of main binding patterns was most effective in predicting responses of patient populations exposed to various pathogens or allergens (141). In the case of HLA class I, development of a similar single predictor has not yet been achieved, perhaps because of the more limited cross-reactivity across the main class I supertypes.
In contrast, if the goal of a study is to define epitopes for a specific human individual, the MHC alleles expressed by that host should be the focus. This is where the value of pan-allelic prediction approaches that are able to make predictions for all MHC alleles (including understudied ones) has greatly improved the ability to perform such personalized predictions. This is of particular importance in cancer for the discovery and evaluation of neoepitopes, which are inherently personal to a specific host (142, 143).
PREDICTIONS OF T CELL IMMUNOGENICITY
Prediction of which peptides are not just MHC binders or eluted ligands, but are immunogenic, meaning they trigger a T cell response, is highly desirable but also highly challenging. T cell receptors are generated in stochastic processes, and substantial differences in TCR repertoires exist between individuals. Despite the stochasticity of the TCR repertoire, it is possible (and likely) that at least on average, some residues or residue combinations in an MHC ligand that face the TCR are more likely to induce a response than others. For MHC class I, it was indeed possible to derive a score based on amino acid composition that separates MHC-binding peptides of similar affinity into immunogenic and nonimmunogenic peptides (144). However, while this separation was statistically significant, it was far from perfect. Similar results were obtained for MHC class II (145).
OUTLIERS ARE REAL. AND THEY ARE OUTLIERS
The advent of high-throughput MHC ligand identification by mass spectrometry has not only improved the ability to predict such ligands, but it has also led to the discovery of a number of highly unusual peptide ligands that would not have previously been expected to be presented but that have been reproducibly identified by different groups. This includes peptides that are not simple cleavage products of protein sequences but appear to have been spliced together after protein expression (146, 147). Another unexpected finding has been the identification of peptides binding to MHC class I molecules that extend past the expected termini, several of which have been confirmed by X-ray crystallography (148–152). For C-terminally extended peptides, which appear to be more common, it was shown that certain amino acids following the C-terminal anchor residues in a peptide are capable of inducing structural changes in MHC molecules that open up the C-terminal pocket and allow for extension of the peptide out of the pocket (150, 152, 153). Comprehensive profiling of HLA class I alleles in Reference 153 revealed that the ability to bind such C-terminal extended ligands is shared by at least 8 of 54 studied alleles. Traditional MHC binding prediction approaches will likely miss unconventional peptide ligands such as these, and this has to be taken into consideration when applying them for epitope discovery. At the same time, it is important to not throw out the baby with the bathwater: The majority of T cell epitopes discovered so far do not require peptide splicing or changes in the structural conformation of MHC molecules. When algorithms are used to down select which peptides to test for T cell recognition of the most likely targets, it is appropriate to prioritize conventional candidates, while keeping in mind that such candidates do not represent the totality of possible recognized targets.
THE IMPACT OF EXPOSURE HISTORY, SEQUENCE CONSERVATION, AND CROSS-REACTIVITY ON T CELL EPITOPE RECOGNITION IN HUMANS
Humans are continuously exposed to foreign antigens, resulting in the generation of a pool of memory T cells whose epitope specificity was shaped by prior exposures. These memory T cells can rapidly re-expand when they encounter the epitope again. Importantly, an epitope may be contained in a different antigen than the original one, and it can still be recognized even if not 100% conserved. For example, preexisting T cell immunity to the pandemic 2009 influenza strain was found in blood samples from individuals gathered years prior, which confirmed that T cells could recognize epitopes in the more conserved proteins of the pandemic strain (154). Such cross-reactive responses were shown to be predictive of protection from symptomatic disease (155). Similarly, in the case of dengue virus, individuals that were infected by viruses with two different serotypes show a skewing toward recognition of epitopes that are conserved, and therefore cross-reactive between the two strains, compared to individuals that have been infected only once (139). In the case of pollen allergens, where the exposure history of individuals cannot be readily ascertained, epitopes conserved across different pollen allergens have a higher likelihood of being recognized (156), suggesting again that repeated exposures to the same epitopes drives the dominant T cell specificity.
Conservation of epitopes can also dampen their recognition by T cells. It is expected that epitopes that are found conserved in proteins from the host will not be recognized by T cells, as such self-reactive T cells should have been negatively selected during maturation. For humans, such reduced recognition of self-peptides could indeed be confirmed, but to a much lesser degree than expected (157), confirming that negative selection is not a straightforward yes/no process (158). In addition to tolerance of self-proteins due to negative selection, epitopes highly conserved across bacterial species, including those making up the human microbiome, could also be less recognized to avoid chronic inflammatory processes. Indeed, there is evidence for increased tolerance of epitopes from Mycobacterium tuberculosis that were conserved across the microbiome to be less frequently recognized (157), although this finding could not be universally confirmed in other systems (159). Importantly, T cell epitope recognition is heavily shaped by the antigens in which an epitope is found. This can be incorporated into T cell epitope prediction schemes (160): If the goal is to identify epitopes recognized in a viral species, peptides contained in only one isolate need to be avoided. If the goal is to identify epitopes that could be used as diagnostics for specific infections, epitopes conserved in other antigens need to be avoided. And so on. Several tools to assess the conservation and sequence overlap of epitopes exist to facilitate such study designs in the IEDB (161, 162).
CURRENT CHALLENGES FOR THE FIELD
While a lot of progress has been made in the development of T cell epitope predictions, a number of challenges remain. Some of these are incremental, but nevertheless important: The utility of HLA allele–specific thresholds needs to be further explored when applied to the de novo prediction of epitopes. More generally, for MHC class II–restricted epitopes some groups have reported poor results of epitope predictions (163), which are at odds with our experience and need to be more thoroughly investigated. Broadly speaking, the performance of all algorithms needs to be (re)assessed for the ability to identify T cell epitopes in data sets that are large-scale, cover multiple alleles, and were generated in a consistent fashion. This will enable clear recommendations for what methods and thresholds to use for predictions in practice.
In addition to the need for incremental changes, several new challenges have emerged that could significantly shape the T cell epitope prediction field in the future. Three of these that we consider particularly important are the following. (a) First is integration of RNA expression data into epitope predictions. It is obvious that a peptide that is not expressed cannot be recognized. But the relevant thresholds and kinetics of expression that impact which antigens are most visible to the immune system remain to be determined. (b) Second are TCR-specific epitope predictions. New technologies have enabled routine sequencing of epitope-specific TCRs, and such data are now becoming available in the IEDB and other databases (164). Several pioneering methods have established that it is possible in principle to determine what epitope is recognized by a given TCR in a controlled setting (165,166). The ultimate goal of such methods is the de novo identification of an epitope given a TCR sequence from a T cell of unknown specificity. With enough data available, it should be possible to achieve this. (c) Third is prediction of neoepitopes that arise from somatic mutations in cancer cells as targets of T cell responses. In this context, factors not previously considered for traditional epitope predictions become relevant, such as clonality and expression level of the mutation. Common to all of these challenges is the need to provide community-accepted data sets and metrics that allow comparison of different prediction approaches in an unbiased fashion. If one thing is for certain, it is that this challenge will remain.
ACKNOWLEDGMENTS
This work was supported through funding from NIH contract 75N93019C00001 for the Immune Epitope Database.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Paul WE. 2008. Fundamental Immunology. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins [Google Scholar]
- 2.Baumgartner JD, O’Brien TX, Kirkland TN, Glauser MP, Ziegler EJ. 1987. Demonstration of cross-reactive antibodies to smooth gram-negative bacteria in antiserum to Escherichia coli J5. J. Infect. Dis 156:136–43 [DOI] [PubMed] [Google Scholar]
- 3.De Groot AS, Scott DW. 2007. Immunogenicity of protein therapeutics. Trends Immunol. 28:482–90 [DOI] [PubMed] [Google Scholar]
- 4.Mazor R, King EM, Pastan I. 2018. Strategies to reduce the immunogenicity of recombinant immunotoxins. Am. J. Pathol 188:1736–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sauna ZE, Lagasse D, Pedras-Vasconcelos J, Golding B, Rosenberg AS. 2018. Evaluating and mitigating the immunogenicity of therapeutic proteins. Trends Biotechnol. 36:1068–84 [DOI] [PubMed] [Google Scholar]
- 6.Schumacher TN, Schreiber RD. 2015. Neoantigens in cancer immunotherapy. Science 348:69–74 [DOI] [PubMed] [Google Scholar]
- 7.Madden DR. 1995. The three-dimensional structure of peptide-MHC complexes. Annu. Rev. Immunol 13:587–622 [DOI] [PubMed] [Google Scholar]
- 8.McDevitt HO. 2000. Discovering the role of the major histocompatibility complex in the immune response. Annu. Rev. Immunol 18:1–17 [DOI] [PubMed] [Google Scholar]
- 9.Sidney J, Southwood S, Moore C, Oseroff C, Pinilla C, et al. 2013. Measurement of MHC/peptide interactions by gel filtration or monoclonal antibody capture. Curr. Protoc. Immunol 100:18.3.1–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blum JS, Wearsch PA, Cresswell P. 2013. Pathways of antigen processing. Annu. Rev. Immunol 31:443–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pamer E, Cresswell P. 1998. Mechanisms of MHC class I–restricted antigen processing. Annu. Rev. Immunol 16:323–58 [DOI] [PubMed] [Google Scholar]
- 12.Rammensee HG, Falk K, Rotzschke O. 1993. Peptides naturally presented by MHC class I molecules. Annu. Rev. Immunol 11:213–44 [DOI] [PubMed] [Google Scholar]
- 13.Rock KL, Goldberg AL. 1999. Degradation of cell proteins and the generation of MHC class I-presented peptides. Annu. Rev. Immunol 17:739–79 [DOI] [PubMed] [Google Scholar]
- 14.Townsend A, Bodmer H. 1989. Antigen recognition by class I-restricted T lymphocytes. Annu. Rev. Immunol 7:601–24 [DOI] [PubMed] [Google Scholar]
- 15.Garcia KC, Teyton L, Wilson IA. 1999. Structural basis of T cell recognition. Annu. Rev. Immunol 17:369–97 [DOI] [PubMed] [Google Scholar]
- 16.Rossjohn J, Gras S, Miles JJ, Turner SJ, Godfrey DI, McCluskey J. 2015. T cell antigen receptor recognition of antigen-presenting molecules. Annu. Rev. Immunol 33:169–200 [DOI] [PubMed] [Google Scholar]
- 17.Rudolph MG, Stanfield RL, Wilson IA. 2006. How TCRs bind MHCs, peptides, and coreceptors. Annu. Rev. Immunol 24:419–66 [DOI] [PubMed] [Google Scholar]
- 18.Unanue ER, Turk V, Neefjes J. 2016. Variations in MHC class II antigen processing and presentation in health and disease. Annu. Rev. Immunol 34:265–97 [DOI] [PubMed] [Google Scholar]
- 19.Yewdell JW, Bennink JR. 1999. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu. Rev. Immunol 17:51–88 [DOI] [PubMed] [Google Scholar]
- 20.Marrack P, Scott-Browne JP, Dai S, Gapin L, Kappler JW. 2008. Evolutionarily conserved amino acids that control TCR-MHC interaction. Annu. Rev. Immunol 26:171–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shastri N, Schwab S, Serwold T. 2002. Producing nature’s gene-chips: the generation of peptides for display by MHC class I molecules. Annu. Rev. Immunol 20:463–93 [DOI] [PubMed] [Google Scholar]
- 22.Nobel Found. 2019. The Nobel Prize in Physiology or Medicine 1980. The Nobel Prize. https://www.nobelprize.org/prizes/medicine/1980/summary/
- 23.Nobel Found. 2019. The Nobel Prize in Physiology or Medicine 1996. The Nobel Prize. https://www.nobelprize.org/prizes/medicine/1996/summary/
- 24.Matzinger P. 1981. A one-receptor view of T-cell behaviour. Nature 292:497–501 [DOI] [PubMed] [Google Scholar]
- 25.Berzofsky JA. 1980. Immune response genes in the regulation of mammalian immunity. In Biological Regulation and Development, Vol 2, ed. Goldberger RF, pp. 467–94. Boston, MA: Springer [Google Scholar]
- 26.Shimonkevitz R, Colon S, Kappler JW, Marrack P, Grey HM. 1984. Antigen recognition by H-2-restricted T cells. II. A tryptic ovalbumin peptide that substitutes for processed antigen. J. Immunol 133:2067–74 [PubMed] [Google Scholar]
- 27.Shimonkevitz R, Kappler J, Marrack P, Grey H. 1983. Antigen recognition by H-2-restricted T cells. I. Cell-free antigen processing. J. Exp. Med 158:303–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Townsend AR, Gotch FM, Davey J. 1985. Cytotoxic T cells recognize fragments of the influenza nucleoprotein. Cell 42:457–67 [DOI] [PubMed] [Google Scholar]
- 29.Babbitt BP, Allen PM, Matsueda G, Haber E, Unanue ER. 1985. Binding of immunogenic peptides to Ia histocompatibility molecules. Nature 317:359–61 [DOI] [PubMed] [Google Scholar]
- 30.Buus S, Colon S, Smith C, Freed JH, Miles C, Grey HM. 1986. Interaction between a “processed” ovalbumin peptide and Ia molecules. PNAS 83:3968–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeLisi C, Berzofsky JA. 1985. T-cell antigenic sites tend to be amphipathic structures. PNAS 82:7048–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rothbard JB, Townsend A, Edwards M, Taylor W. 1987. Pattern recognition among T-cell epitopes. Haematol. Blood Transfus 31:324–31 [DOI] [PubMed] [Google Scholar]
- 33.Buus S, Sette A, Colon SM, Miles C, Grey HM. 1987. The relation between major histocompatibility complex (MHC) restriction and the capacity of Ia to bind immunogenic peptides. Science 235:1353–58 [DOI] [PubMed] [Google Scholar]
- 34.Sette A, Buus S, Colon S, Smith JA, Miles C, Grey HM. 1987. Structural characteristics of an antigen required for its interaction with Ia and recognition by T cells. Nature 328:395–99 [DOI] [PubMed] [Google Scholar]
- 35.Benoist CO, Mathis DJ, Kanter MR, Williams VE 2nd, McDevitt HO. 1983. Regions of allelic hypervariability in the murine A alpha immune response gene. Cell 34:169–77 [DOI] [PubMed] [Google Scholar]
- 36.Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC. 1987. Structure of the human class I histocompatibility antigen, HLA-A2. Nature 329:506–12 [DOI] [PubMed] [Google Scholar]
- 37.Brown JH, Jardetzky TS, Gorga JC, Stern LJ, Urban RG, et al. 1993. Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 364:33–39 [DOI] [PubMed] [Google Scholar]
- 38.Pamer EG, Harty JT, Bevan MJ. 1991. Precise prediction of a dominant class I MHC-restricted epitope of Listeria monocytogenes. Nature 353:852–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rotzschke O, Falk K, Deres K, Schild H, Norda M, et al. 1990. Isolation and analysis of naturally processed viral peptides as recognized by cytotoxic T cells. Nature 348:252–54 [DOI] [PubMed] [Google Scholar]
- 40.Rotzschke O, Falk K, Wallny HJ, Faath S, Rammensee HG. 1990. Characterization of naturally occurring minor histocompatibility peptides including H-4 and H-Y. Science 249:283–87 [DOI] [PubMed] [Google Scholar]
- 41.Van Bleek GM, Nathenson SG. 1990. Isolation of an endogenously processed immunodominant viral peptide from the class I H-2Kb molecule. Nature 348:213–16 [DOI] [PubMed] [Google Scholar]
- 42.Falk K, Rotzschke O, Stevanovic S, Jung G, Rammensee HG. 1991. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature 351:290–96 [DOI] [PubMed] [Google Scholar]
- 43.Henderson RA, Cox AL, Sakaguchi K, Appella E, Shabanowitz J, et al. 1993. Direct identification of an endogenous peptide recognized by multiple HLA-A2.1-specific cytotoxic T cells. PNAS 90:10275–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hunt DF, Michel H, Dickinson TA, Shabanowitz J, Cox AL, et al. 1992. Peptides presented to the immune system by the murine class II major histocompatibility complex molecule I-Ad. Science 256:1817–20 [DOI] [PubMed] [Google Scholar]
- 45.Sette A, Buus S, Appella E, Smith JA, Chesnut R, et al. 1989. Prediction of major histocompatibility complex binding regions of protein antigens by sequence pattern analysis. PNAS 86:3296–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruppert J, Sidney J, Celis E, Kubo RT, Grey HM, Sette A. 1993. Prominent role of secondary anchor residues in peptide binding to HLA-A2.1 molecules. Cell 74:929–37 [DOI] [PubMed] [Google Scholar]
- 47.Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. 1999. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 50:213–19 [DOI] [PubMed] [Google Scholar]
- 48.Bui HH, Sidney J, Peters B, Sathiamurthy M, Sinichi A, et al. 2005. Automated generation and evaluation of specific MHC binding predictive tools: ARB matrix applications. Immunogenetics 57:304–14 [DOI] [PubMed] [Google Scholar]
- 49.Kotsiantis SB. 2007. Supervised machine learning: a review of classification techniques. In Proceedings of the 2007 conference on Emerging Artificial Intelligence Applications in Computer Engineering: Real Word AI Systems with Applications in eHealth, HCI, Information Retrieval and Pervasive Technologies, ed. Maglogiannis I, Karpouzis K, Wallace M, Soldatos J, pp. 3–24. Amsterdam: IOS [Google Scholar]
- 50.Parker KC, Bednarek MA, Coligan JE. 1994. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J. Immunol 152:163–75 [PubMed] [Google Scholar]
- 51.Adams HP, Koziol JA. 1995. Prediction of binding to MHC class I molecules. J. Immunol. Methods 185:181–90 [DOI] [PubMed] [Google Scholar]
- 52.Brusic V, Rudy G, Harrison LC. 1994. Prediction of MHC binding peptides using artificial neural networks. In Complex Systems: Mechanism of Adaptation, ed. Stonier RJ, Yu XH, pp. 253–60. Amsterdam: IOS [Google Scholar]
- 53.Milik M, Sauer D, Brunmark AP, Yuan L, Vitiello A, et al. 1998. Application of an artificial neural network to predict specific class I MHC binding peptide sequences. Nat. Biotechnol 16:753–56 [DOI] [PubMed] [Google Scholar]
- 54.Nielsen M, Lundegaard C, Worning P, Lauemoller SL, Lamberth K, et al. 2003. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 12:1007–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mamitsuka H. 1998. Predicting peptides that bind to MHC molecules using supervised learning of hidden Markov models. Proteins 33:460–74 [DOI] [PubMed] [Google Scholar]
- 56.Zhang C, Bickis MG, Wu FX, Kusalik AJ. 2006. Optimally-connected hidden Markov models for predicting MHC-binding peptides. J. Bioinform. Comput. Biol 4:959–80 [DOI] [PubMed] [Google Scholar]
- 57.Doytchinova IA, Flower DR. 2001. Toward the quantitative prediction of T-cell epitopes: coMFA and coMSIA studies of peptides with affinity for the class I MHC molecule HLA-A*0201. J. Med. Chem 44:3572–81 [DOI] [PubMed] [Google Scholar]
- 58.Peters B, Tong W, Sidney J, Sette A, Weng Z. 2003. Examining the independent binding assumption for binding of peptide epitopes to MHC-I molecules. Bioinformatics 19:1765–72 [DOI] [PubMed] [Google Scholar]
- 59.Zhang H, Wang P, Papangelopoulos N, Xu Y, Sette A, et al. 2010. Limitations of Ab initio predictions of peptide binding to MHC class II molecules. PLOS ONE 5:e9272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grant BJ, Gorfe AA, McCammon JA. 2010. Large conformational changes in proteins: signaling and other functions. Curr. Opin. Struct. Biol 20:142–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Korber BTM, Moore JP, Brander C, Walker BD, Haynes BF, Koup R. 1998. HIV Molecular Immunology Compendium. Los Alamos, NM: Los Alamos Natl. Lab. Theor. Biol. Biophys. [Google Scholar]
- 62.Bhasin M, Singh H, Raghava GP. 2003. MHCBN: a comprehensive database of MHC binding and non-binding peptides. Bioinformatics 19:665–66 [DOI] [PubMed] [Google Scholar]
- 63.Peters B, Sidney J, Bourne P, Bui HH, Buus S, et al. 2005. The immune epitope database and analysis resource: from vision to blueprint. PLOS Biol. 3:e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, et al. 2019. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 47:D339–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dhanda SK, Mahajan S, Paul S, Yan Z, Kim H, et al. 2019. IEDB-AR: immune epitope database—analysis resource in 2019. Nucleic Acids Res. 47:W502–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shao W, Pedrioli PGA, Wolski W, Scurtescu C, Schmid E, et al. 2018. The SysteMHC Atlas project. Nucleic Acids Res. 46:D1237–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lill JR, van Veelen PA, Tenzer S, Admon A, Caron E, et al. 2018. Minimal Information About an Immuno-Peptidomics Experiment (MIAIPE). Proteomics 18:e1800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Peters B, Bui HH, Frankild S, Nielson M, Lundegaard C, et al. 2006. A community resource benchmarking predictions of peptide binding to MHC-I molecules. PLOS Comput. Biol 2:e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M. 2008. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res. 36:W509–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim Y, Sidney J, Pinilla C, Sette A, Peters B. 2009. Derivation of an amino acid similarity matrix for peptide: MHC binding and its application as a Bayesian prior. BMC Bioinform. 10:394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Noguchi H, Kato R, Hanai T, Matsubara Y, Honda H, et al. 2002. Hidden Markov model-based prediction of antigenic peptides that interact with MHC class II molecules. J. Biosci. Bioeng 94:264–70 [DOI] [PubMed] [Google Scholar]
- 72.Cui J, Han LY, Lin HH, Zhang HL, Tang ZQ, et al. 2007. Prediction of MHC-binding peptides of flexible lengths from sequence-derived structural and physicochemical properties. Mol. Immunol 44:866–77 [DOI] [PubMed] [Google Scholar]
- 73.Salomon J, Flower DR. 2006. Predicting class II MHC-peptide binding: a kernel based approach using similarity scores. BMC Bioinform. 7:501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nielsen M, Lundegaard C, Worning P, Hvid CS, Lamberth K, et al. 2004. Improved prediction of MHC class I and class II epitopes using a novel Gibbs sampling approach. Bioinformatics 20:1388–97 [DOI] [PubMed] [Google Scholar]
- 75.Brusic V, Rudy G, Honeyman G, Hammer J, Harrison L. 1998. Prediction of MHC class II-binding peptides using an evolutionary algorithm and artificial neural network. Bioinformatics 14:121–30 [DOI] [PubMed] [Google Scholar]
- 76.Nielsen M, Lund O. 2009. NN-align: An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinform. 10:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lin HH, Zhang GL, Tongchusak S, Reinherz EL, Brusic V. 2008. Evaluation of MHC-II peptide binding prediction servers: applications for vaccine research. BMC Bioinform. 9(Suppl. 12):S22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang P, Sidney J, Dow C, Mothe B, Sette A, Peters B. 2008. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLOS Comput. Biol 4:e1000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Moutaftsi M, Peters B, Pasquetto V, Tscharke DC, Sidney J, et al. 2006. A consensus epitope prediction approach identifies the breadth of murine TCD8+–cell responses to vaccinia virus. Nat. Biotechnol 24:817–19 [DOI] [PubMed] [Google Scholar]
- 80.Jensen KK, Andreatta M, Marcatili P, Buus S, Greenbaum JA, et al. 2018. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 154:394–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Karosiene E, Rasmussen M, Blicher T, Lund O, Buus S, Nielsen M. 2013. NetMHCIIpan-3.0, a common pan-specific MHC class II prediction method including all three human MHC class II isotypes, HLA-DR, HLA-DP and HLA-DQ. Immunogenetics 65:711–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nielsen M, Justesen S, Lund O, Lundegaard C, Buus S. 2010. NetMHCIIpan-2.0—Improved pan-specific HLA-DR predictions using a novel concurrent alignment and weight optimization training procedure. Immunome Res. 6:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Andreatta M, Jurtz VI, Kaever T, Sette A, Peters B, Nielsen M. 2017. Machine learning reveals a non-canonical mode of peptide binding to MHC class II molecules. Immunology 152:255–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Andreatta M, Karosiene E, Rasmussen M, Stryhn A, Buus S, Nielsen M. 2015. Accurate pan-specific prediction of peptide-MHC class II binding affinity with improved binding core identification. Immunogenetics 67:641–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Andreatta M, Nielsen M. 2016. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics 32:511–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Andreatta M, Schafer-Nielsen C, Lund O, Buus S, Nielsen M. 2011. NNAlign: a web-based prediction method allowing non-expert end-user discovery of sequence motifs in quantitative peptide data. PLOS ONE 6:e26781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nielsen M, Andreatta M. 2016. NetMHCpan-3.0: improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 8:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nielsen M, Andreatta M. 2017. NNAlign: a platform to construct and evaluate artificial neural network models of receptor-ligand interactions. Nucleic Acids Res. 45:W344–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kim Y, Sidney J, Buus S, Sette A, Nielsen M, Peters B. 2014. Dataset size and composition impact the reliability of performance benchmarks for peptide-MHC binding predictions. BMC Bioinform. 15:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Robinson J, Halliwell JA, Hayhurst JD, Flicek P, Parham P, Marsh SG. 2015. The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res. 43:D423–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sturniolo T, Bono E, Ding J, Raddrizzani L, Tuereci O, et al. 1999. Generation of tissue-specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA class II matrices. Nat. Biotechnol 17:555–61 [DOI] [PubMed] [Google Scholar]
- 92.Nielsen M, Lundegaard C, Blicher T, Lamberth K, Harndahl M, et al. 2007. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLOS ONE 2:e796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang GL, Khan AM, Srinivasan KN, August JT, Brusic V. 2005. MULTIPRED: a computational system for prediction of promiscuous HLA binding peptides. Nucleic Acids Res. 33:W172–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jojic N, Reyes-Gomez M, Heckerman D, Kadie C, Schueler-Furman O. 2006. Learning MHC I–peptide binding. Bioinformatics 22:e227–35 [DOI] [PubMed] [Google Scholar]
- 95.Jacob L, Vert JP. 2008. Efficient peptide-MHC-I binding prediction for alleles with few known binders. Bioinformatics 24:358–66 [DOI] [PubMed] [Google Scholar]
- 96.Zhang H, Lund O, Nielsen M. 2009. The PickPocket method for predicting binding specificities for receptors based on receptor pocket similarities: application to MHC-peptide binding. Bioinformatics 25:1293–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang L, Udaka K, Mamitsuka H, Zhu S. 2012. Toward more accurate pan-specific MHC-peptide binding prediction: a review of current methods and tools. Brief Bioinform. 13:350–64 [DOI] [PubMed] [Google Scholar]
- 98.Bordner AJ, Mittelmann HD. 2010. MultiRTA: a simple yet reliable method for predicting peptide binding affinities for multiple class II MHC allotypes. BMC Bioinform. 11:482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pfeifer N, Kohlbacher O. 2008. Multiple instance learning allows MHC class II epitope predictions across alleles. In Algorithms in Bioinformatics, ed. Crandall KA, Lagergren J, pp. 210–21. Berlin, Heidelberg: Springer [Google Scholar]
- 100.Nielsen M, Lundegaard C, Blicher T, Peters B, Sette A, et al. 2008. Quantitative predictions of peptide binding to any HLA-DR molecule of known sequence: NetMHCIIpan. PLOS Comput. Biol 4:e1000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Eggers M, Boes-Fabian B, Ruppert T, Kloetzel PM, Koszinowski UH. 1995. The cleavage preference of the proteasome governs the yield of antigenic peptides. J. Exp. Med 182:1865–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nielsen M, Lundegaard C, Lund O, Kesmir C. 2005. The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics 57:33–41 [DOI] [PubMed] [Google Scholar]
- 103.Bhasin M, Raghava GP 2004. Analysis and prediction of affinity of TAP binding peptides using cascade SVM. Protein Sci. 13:596–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Peters B, Bulik S, Tampe R, Van Endert PM, Holzhutter HG. 2003. Identifying MHC class I epitopes by predicting the TAP transport efficiency of epitope precursors. J. Immunol 171:1741–49 [DOI] [PubMed] [Google Scholar]
- 105.Doytchinova IA, Guan P, Flower DR. 2006. EpiJen: a server for multistep T cell epitope prediction. BMC Bioinform. 7:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hakenberg J, Nussbaum AK, Schild H, Rammensee HG, Kuttler C, et al. 2003. MAPPP: MHC class I antigenic peptide processing prediction. Appl. Bioinform 2:155–58 [PubMed] [Google Scholar]
- 107.Larsen MV, Lundegaard C, Lamberth K, Buus S, Brunak S, et al. 2005. An integrative approach to CTL epitope prediction: a combined algorithm integrating MHC class I binding, TAP transport efficiency, and proteasomal cleavage predictions. Eur. J. Immunol 35:2295–303 [DOI] [PubMed] [Google Scholar]
- 108.Tenzer S, Peters B, Bulik S, Schoor O, Lemmel C, et al. 2005. Modeling the MHC class I pathway by combining predictions of proteasomal cleavage, TAP transport and MHC class I binding. Cell Mol. Life Sci 62:1025–37 [DOI] [PubMed] [Google Scholar]
- 109.Stranzl T, Larsen MV, Lundegaard C, Nielsen M. 2010. NetCTLpan: pan-specific MHC class I pathway epitope predictions. Immunogenetics 62:357–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gfeller D, Guillaume P, Michaux J, Pak HS, Daniel RT, et al. 2018. The length distribution and multiple specificity of naturally presented HLA-I ligands. J. Immunol 201:3705–16 [DOI] [PubMed] [Google Scholar]
- 111.Trolle T, McMurtrey CP, Sidney J, Bardet W, Osborn SC, et al. 2016. The length distribution of class I-restricted T cell epitopes is determined by both peptide supply and MHC allele-specific binding preference. J. Immunol 196:1480–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chang SC, Momburg F, Bhutani N, Goldberg AL. 2005. The ER aminopeptidase, ERAP1, trims precursors to lengths of MHC class I peptides by a “molecular ruler” mechanism. PNAS 102:17107–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Saveanu L, Carroll O, Lindo V, Del Val M, Lopez D, et al. 2005. Concerted peptide trimming by human ERAP1 and ERAP2 aminopeptidase complexes in the endoplasmic reticulum. Nat. Immunol 6:689–97 [DOI] [PubMed] [Google Scholar]
- 114.Wenzel T, Eckerskorn C, Lottspeich F, Baumeister W. 1994. Existence of a molecular ruler in proteasomes suggested by analysis of degradation products. FEBS Lett. 349:205–9 [DOI] [PubMed] [Google Scholar]
- 115.Neefjes J, Jongsma ML, Paul P, Bakke O. 2011. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol 11:823–36 [DOI] [PubMed] [Google Scholar]
- 116.Sant AJ, Chaves FA, Leddon SA, Tung J. 2013. The control of the specificity of CD4 T cell responses: thresholds, breakpoints, and ceilings. Front. Immunol 4:340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Barra C, Alvarez B, Paul S, Sette A, Peters B, et al. 2018. Footprints of antigen processing boost MHC class II natural ligand predictions. Genome Med. 10:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Paul S, Karosiene E, Dhanda SK, Jurtz V, Edwards L, et al. 2018. Determination of a predictive cleavage motif for eluted major histocompatibility complex class II ligands. Front. Immunol 9:1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Carson RT, Vignali KM, Woodland DL, Vignali DA. 1997. T cell receptor recognition of MHC class II-bound peptide flanking residues enhances immunogenicity and results in altered TCR V region usage. Immunity 7:387–99 [DOI] [PubMed] [Google Scholar]
- 120.Abelin JG, Keskin DB, Sarkizova S, Hartigan CR, Zhang W, et al. 2017. Mass spectrometry profiling of HLA-associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity 46:315–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Prilliman K, Lindsey M, Zuo Y, Jackson KW, Zhang Y, Hildebrand W. 1997. Large-scale production of class I bound peptides: assigning a signature to HLA-B* 1501. Immunogenetics 45:379–85 [DOI] [PubMed] [Google Scholar]
- 122.Bassani-Sternberg M, Pletscher-Frankild S, Jensen LJ, Mann M. 2015. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol. Cell Proteom 14:658–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Andreatta M, Lund O, Nielsen M. 2013. Simultaneous alignment and clustering of peptide data using a Gibbs sampling approach. Bioinformatics 29:8–14 [DOI] [PubMed] [Google Scholar]
- 124.Bassani-Sternberg M, Chong C, Guillaume P, Solleder M, Pak H, et al. 2017. Deciphering HLA-I motifs across HLA peptidomes improves neo-antigen predictions and identifies allostery regulating HLA specificity. PLOS Comput. Biol 13:e1005725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Murphy JP, Konda P, Kowalewski DJ, Schuster H, Clements D, et al. 2017. MHC-I ligand discovery using targeted database searches of mass spectrometry data: implications for T-cell immunotherapies. J. Proteome Res 16:1806–16 [DOI] [PubMed] [Google Scholar]
- 126.Bassani-Sternberg M, Gfeller D. 2016. Unsupervised HLA peptidome deconvolution improves ligand prediction accuracy and predicts cooperative effects in peptide-HLA interactions. J. Immunol 197:2492–99 [DOI] [PubMed] [Google Scholar]
- 127.Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. 2017. NetMHCpan-4.0: improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol 199:3360–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.O’Donnell TJ, Rubinsteyn A, Bonsack M, Riemer AB, Laserson U, Hammerbacher J. 2018. MHCflurry: Open-source class I MHC binding affinity prediction. Cell Syst. 7:129–32.e4 [DOI] [PubMed] [Google Scholar]
- 129.Alvarez B, Reynisson B, Barra C, Buus S, Ternette N, et al. 2019. NNAlign_MA: MHC peptidome deconvolution for accurate MHC binding motif characterization and improved T cell epitope predictions. bioRxiv 550673. 10.1101/550673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sette A, Vitiello A, Reherman B, Fowler P, Nayersina R, et al. 1994. The relationship between class I binding affinity and immunogenicity of potential cytotoxic T cell epitopes. J. Immunol 153:5586–92 [PubMed] [Google Scholar]
- 131.Paul S, Weiskopf D, Angelo MA, Sidney J, Peters B, Sette A. 2013. HLA class I alleles are associated with peptide-binding repertoires of different size, affinity, and immunogenicity. J. Immunol 191:5831–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bjerregaard AM, Nielsen M, Jurtz V, Barra CM, Hadrup SR, et al. 2017. An analysis of natural T cell responses to predicted tumor neoepitopes. Front. Immunol 8:1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kosaloglu-Yalcin Z, Lanka M, Frentzen A, Logandha Ramamoorthy Premlal A, Sidney J, et al. 2018. Predicting T cell recognition of MHC class I restricted neoepitopes. Oncoimmunology 7:e1492508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Paul S, Croft NP, Purcell AW, Tscharke DC, Sette A, et al. 2019. Benchmarking predictions of MHC class I restricted T cell epitopes. bioRxiv 694539. 10.1101/694539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Southwood S, Sidney J, Kondo A, del Guercio MF, Appella E, et al. 1998. Several common HLA-DR types share largely overlapping peptide binding repertoires. J. Immunol 160:3363–73 [PubMed] [Google Scholar]
- 136.Sidney J, Peters B, Frahm N, Brander C, Sette A. 2008. HLA class I supertypes: a revised and updated classification. BMC Immunol. 9:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Greenbaum J, Sidney J, Chung J, Brander C, Peters B, Sette A. 2011. Functional classification of class II human leukocyte antigen (HLA) molecules reveals seven different supertypes and a surprising degree of repertoire sharing across supertypes. Immunogenetics 63:325–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.McKinney DM, Southwood S, Hinz D, Oseroff C, Arlehamn CS, et al. 2013. A strategy to determine HLA class II restriction broadly covering the DR, DP, and DQ allelic variants most commonly expressed in the general population. Immunogenetics 65:357–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Weiskopf D, Angelo MA, de Azeredo EL, Sidney J, Greenbaum JA, et al. 2013. Comprehensive analysis of dengue virus-specific responses supports an HLA-linked protective role for CD8+ T cells. PNAS 110:E2046–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Oseroff C, Sidney J, Kotturi MF, Kolla R, Alam R, et al. 2010. Molecular determinants of T cell epitope recognition to the common Timothy grass allergen. J. Immunol 185:943–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Paul S, Lindestam Arlehamn CS, Scriba TJ, Dillon MB, Oseroff C, et al. 2015. Development and validation of a broad scheme for prediction of HLA class II restricted T cell epitopes. J. Immunol. Methods 422:28–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Marty Pyke R, Thompson WK, Salem RM, Font-Burgada J, Zanetti M, Carter H. 2018. Evolutionary pressure against MHC class II binding cancer mutations. Cell 175:416–28.e13. Erratum. 2018. Cell 175(7):1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Marty R, Kaabinejadian S, Rossell D, Slifker MJ, van de Haar J, et al. 2017. MHC-I genotype restricts the oncogenic mutational landscape. Cell 171:1272–83.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Calis JJ, Maybeno M, Greenbaum JA, Weiskopf D, De Silva AD, et al. 2013. Properties of MHC class I presented peptides that enhance immunogenicity. PLOS Comput. Biol 9:e1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Dhanda SK, Karosiene E, Edwards L, Grifoni A, Paul S, et al. 2018. Predicting HLA CD4 immunogenicity in human populations. Front. Immunol 9:1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Faridi P, Li C, Ramarathinam SH, Vivian JP, Illing PT, et al. 2018. A subset of HLA-I peptides are not genomically templated: evidence for cis- and trans-spliced peptide ligands. Sci. Immunol 3:eaar3947. [DOI] [PubMed] [Google Scholar]
- 147.Liepe J, Marino F, Sidney J, Jeko A, Bunting DE, et al. 2016. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science 354:354–58 [DOI] [PubMed] [Google Scholar]
- 148.Collins EJ, Garboczi DN, Wiley DC. 1994. Three-dimensional structure of a peptide extending from one end of a class I MHC binding site. Nature 371:626–29 [DOI] [PubMed] [Google Scholar]
- 149.Li X, Lamothe PA, Walker BD, Wang JH. 2017. Crystal structure of HLA-B*5801 with a TW10 HIV Gag epitope reveals a novel mode of peptide presentation. Cell Mol. Immunol 14:631–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.McMurtrey C, Trolle T, Sansom T, Remesh SG, Kaever T, et al. 2016. Toxoplasma gondii peptide ligands open the gate of the HLA class I binding groove. eLife 5:e12556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pymm P, Illing PT, Ramarathinam SH, O’Connor GM, Hughes VA, et al. 2017. MHC-I peptides get out of the groove and enable a novel mechanism of HIV-1 escape. Nat. Struct. Mol. Biol 24:387–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Remesh SG, Andreatta M, Ying G, Kaever T, Nielsen M, et al. 2017. Unconventional peptide presentation by major histocompatibility complex (MHC) class I allele HLA-A*02:01: BREAKING CONFINEMENT. J. Biol. Chem 292:5262–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Guillaume P, Picaud S, Baumgaertner P, Montandon N, Schmidt J, et al. 2018. The C-terminal extension landscape of naturally presented HLA-I ligands. PNAS 115:5083–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Greenbaum JA, Kotturi MF, Kim Y, Oseroff C, Vaughan K, et al. 2009. Pre-existing immunity against swine-origin H1N1 influenza viruses in the general human population. PNAS 106:20365–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sridhar S, Begom S, Bermingham A, Hoschler K, Adamson W, et al. 2013. Cellular immune correlates of protection against symptomatic pandemic influenza. Nat. Med 19:1305–12 [DOI] [PubMed] [Google Scholar]
- 156.Westernberg L, Schulten V, Greenbaum JA, Natali S, Tripple V, et al. 2016. T-cell epitope conservation across allergen species is a major determinant of immunogenicity. J. Allergy Clin. Immunol 138:571–78.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bresciani A, Paul S, Schommer N, Dillon MB, Bancroft T, et al. 2016. T-cell recognition is shaped by epitope sequence conservation in the host proteome and microbiome. Immunology 148:34–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Klein L, Kyewski B, Allen PM, Hogquist KA. 2014. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat. Rev. Immunol 14:377–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Carrasco Pro S, Lindestam Arlehamn CS, Dhanda SK, Carpenter C, Lindvall M, et al. 2018. Microbiota epitope similarity either dampens or enhances the immunogenicity of disease-associated antigenic epitopes. PLOS ONE 13:e0196551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Moise L, Beseme S, Tassone R, Liu R, Kibria F, et al. 2016. T cell epitope redundancy: cross-conservation of the TCR face between pathogens and self and its implications for vaccines and autoimmunity. Expert Rev. Vaccines 15:607–17 [DOI] [PubMed] [Google Scholar]
- 161.Bui HH, Sidney J, Li W, Fusseder N, Sette A. 2007. Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinform. 8:361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Dhanda SK, Vaughan K, Schulten V, Grifoni A, Weiskopf D, et al. 2018. Development of a novel clustering tool for linear peptide sequences. Immunology 155:331–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chaves FA, Lee AH, Nayak JL, Richards KA, Sant AJ. 2012. The utility and limitations of current Web-available algorithms to predict peptides recognized by CD4 T cells in response to pathogen infection. J. Immunol 188:4235–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Mahajan S, Vita R, Shackelford D, Lane J, Schulten V, et al. 2018. Epitope specific antibodies and T cell receptors in the immune epitope database. Front. Immunol 9:2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Glanville J, Huang H, Nau A, Hatton O, Wagar LE, et al. 2017. Identifying specificity groups in the T cell receptor repertoire. Nature 547:94–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Jurtz VI, Jessen LE, Bentzen AK, Jespersen MC, Mahajan S, et al. 2018. NetTCR: sequence-based prediction of TCR binding to peptide-MHC complexes using convolutional neural networks. bioRxiv 433706. 10.1101/433706 [DOI] [Google Scholar]