

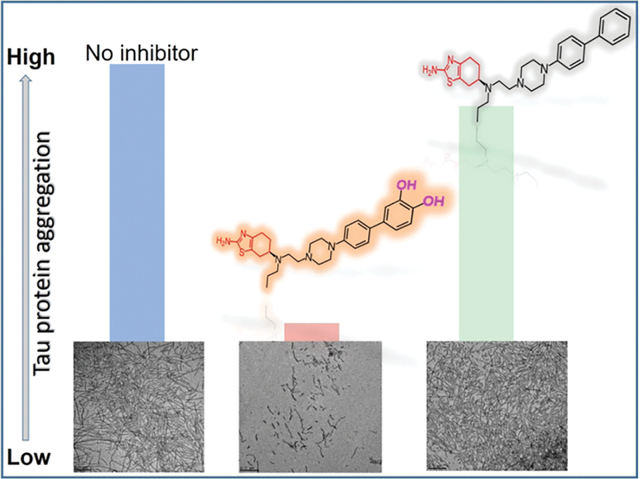
Abstract
Neurodegeneration leads to variety of diseases which are linked to aberrant protein or peptide aggregation, as a one possible mechanism. Hence, small drug molecules targeting aggregation are of interest. Tau protein aggregation is one of the biomarkers of neurodegenerative diseases and is a viable drug target. Toward multifunctional inhibitors, we aim to incorporate structural elements in a potential drug in order to preserve dopamine agonist activity, which elevates disease symptoms associated with motor skills, and promote inhibitory activity against aggregation of the full-length tau (2N4R, tau441) protein. In our design, we introduced various moieties (catechol, non-catechol, biphenyl, piperazine, and thiazole) to determine which functional group leads to the greatest aggregation inhibition of tau. In vitro, tau aggregation was induced by heparin and monitored by using fluorescence aggregation assay, transmission electron microscopy and 4,4′-Dianilino-1,1′-binaphthyl-5,5′-disulfonic acid dipotassium salt (Bis-ANS) fluorescence spectroscopy. The catechol containing compounds, D-519 and D-520, prevented aggregation of tau. By contrast, non-catechol and thiazole containing compounds (D-264 and D-636) were poor inhibitors. The Bis-ANS studies revealed that the potent inhibitors bound solvent-exposed hydrophobic sites. Based on the density functional theory calculations on inhibitors tested, the compounds characterized with the high polarity and polarizability were more effective aggregation inhibitors. These findings could lead to the development of small multifunctional drug inhibitors for the treatment of tau-associated neurodegeneration.
Keywords: Tau protein, aggregation, neurodegeneration, Alzheimer’s disease, Dopamine agonist
Graphical Abstract
1. Introduction
Tau is a structural neuronal protein which stabilizes microtubules (MT) and maintains cell integrity and viability. Due to post-translational modifications of tau, tau detaches from MT, and MT subsequently disaggregate, leading to neuronal cell death.1 In turn, modified tau is prone to aggregation into a variety of structures, including oligomers, fibrils, paired-helical filaments and neurofibrillary tangles. In addition, cytotoxic tau oligomers are involved in cell-to-cell spreading of tau resulting in aggregation of endogenous tau and toxicity. Hence, tau protein aggregation is a feature of the diseased state in Alzheimer’s Disease (AD) and other tauopathies, rather than normal state, and is a viable drug target.
The nonpeptidic small molecule tau aggregation inhibitors (TAGIs) are promising drugs against AD and tauopathies. Several classes of small-molecules have been designed and tested as inhibitors of tau aggregation, and include benzothiazole, hydroquinoline, aminothienopyridazine, N-phenylamine, phenylthiazolylhydrazide, rhodanines, polyphenols, and phenothiazines.2–20 The common feature of these inhibitors is the presence of aromatic and hydrophobic groups which may bind hydrophobic region of tau protein and prevent its aggregation. The hydrophobic aromatic rings or heteroaromatic rings may bind protein, prevent conformational change from random coil to β-sheet structure, and promote off-pathway aggregation. The molecular interactions between the hydroxyl groups in polyphenols and hydrophobic regions of tau may also prevent fibril formation. In addition to non-covalent interactions between tau and inhibitors, the irreversible inhibitor binding, in some instances, suggests covalent bonding between tau protein and the inhibitor. Hence, both non-covalent and covalent interactions between tau and inhibitors are likely. In addition to molecular interactions with tau protein, the inhibitors (such as polyphenols) may also exhibit antioxidant and radical quenching properties. Hence, a number of aromatic hydroxyl or reactive groups in inhibitors, protein binding affinity and binding site, and inhibitor antioxidant activity may contribute to the effectiveness of the inhibitor on tau aggregation. We have shown that D2/D3 receptor agonists acted as aggregation inhibitors of α-synuclein, which is associated with Parkinson’s disease.21 In addition, therapeutic agents for the symptomatic treatment of Parkinson’s disease (levodopa or dopamine agonists) are used to improve motor symptoms in tau-related diseases, termed tauopathies.22 For example, compounds such as D2/D3 dopamine receptor agonists used to target Parkinson’s disease, have been prescribed to patients with tauopathies. The lower levels of dopamine, dopamine 1 receptor and dopamine 2 receptors were observed in patients with AD compared to control.23 Other studies have suggested that targeting dopaminergic activity is a viable target in pharmacological development targeting AD.24,25 The DA-D2 agonist rotigotine, a dopaminergic drug, showed beneficial effects on cognitive domains in AD patients.26 While the role of dopamine receptor agonists on motor symptoms in tauopathies has been identified, little is known about the role of such compounds on tau aggregation.
Herein, we describe the use of unique class of multivalent inhibitors which contain an aromatic core, thiazole moiety, and catechol group toward inhibition of heparin-induced aggregation of the longest isoform of tau protein (tau441, 2N4R). The efficiency of the inhibitors was compared to methylene blue as well to control for compounds which lacked the catechol or thiazole moieties. The fluorescence aggregation assay was used to screen inhibitory activity of all compounds and IC50 values were determined. The hydrophobicity of tau aggregates was evaluated using the Bis-ANS fluorescence probe. The aggregate morphologies were characterized using transmission electron microscopy. Density functional calculations were used to determine polarity, polarizability and electrostatic potentials of all compounds in order to shed light on the mechanisms of inhibition.
2. Results and Discussion
2.1. Design strategy
The in vitro aggregation of tau441 protein was induced by the anionic inducer heparin (72 h, 37 °C).27,28 The aggregate formation was detected by using fluorescence spectroscopy, including fluorescence aggregation assay and Bis-ANS assay, and transmission electron microscopy (TEM). The following multifunctional D2/D3 dopamine agonists were tested as potential aggregation inhibitors: (S)-4’-(4-(2-((2-amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6yl) (propyl)amino)ethyl)piperazin-1yl)biphenyl-3,4-diol (D-519), (S)-4’-(4-(2-((5-hydroxy-1,2,3,4-tetrahydronaphtalen-2-yl) (propyl)amino)ethyl)piperazin-1-yl)biphenyl-3,4-diol (D-520), (S)-N6-(2-(4-(biphenyl-4-yl) piperazin-1-yl)ethyl)-N6-propyl-4,5,6,7-tetrahydrobenzo[d]thiazole-2,6-diamine (D-264), and (S)-N6-(2-(4-(9H-carbazol-3-yl)piperazin-1-yl)ethyl)-N6-propyl-4,5,6,7-tetrahydrobenzo [d] thiazole-2,6-diamine (D-636) (Fig. 1). The structural variation of compounds allows rank ordering inhibitors based on their activity, and identification of key functional groups for inhibition. All compounds tested contain the central fragment, piperazine motif. Both compounds, D-519 and D-520, contain the biphenyl linker with catechol terminal group. However, D-519 contains the thiazole group which was replaced by a phenol moiety in D-520. Compound D-264 is a D-519 analogue which lacks the catechol moiety. Similarly, compound D-636 is an analogue of D-519 but contains carbazole group instead of biphenyl linker and catechol terminal group. We have previously reported on the synthesis and inhibitory activity of D-519 and D-520 as effective aggregation inhibitors of α-synuclein in vitro and in vivo Drosophila model.21 In addition, the compound D-264 was found to strongly activate GIRK channels and MAPK coupled to D2 and D3 dopamine receptors in AtT-20 cells. It is also a potent inducer of cell proliferation.29 In animal models of Parkinson’s Disease, D-264 strongly prevented neurodegeneration caused by the selective neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and the ubiquitin-proteasome system (UPS) inhibitor lactasyn.30 The 3,7-bis(dimethylamino)phenothiazine-5-ium chloride (Methylene blue, MB) is a known inhibitor of tau aggregation and was used as a control for comparison with D2/D3 agonists.19,31–33
Fig. 1.
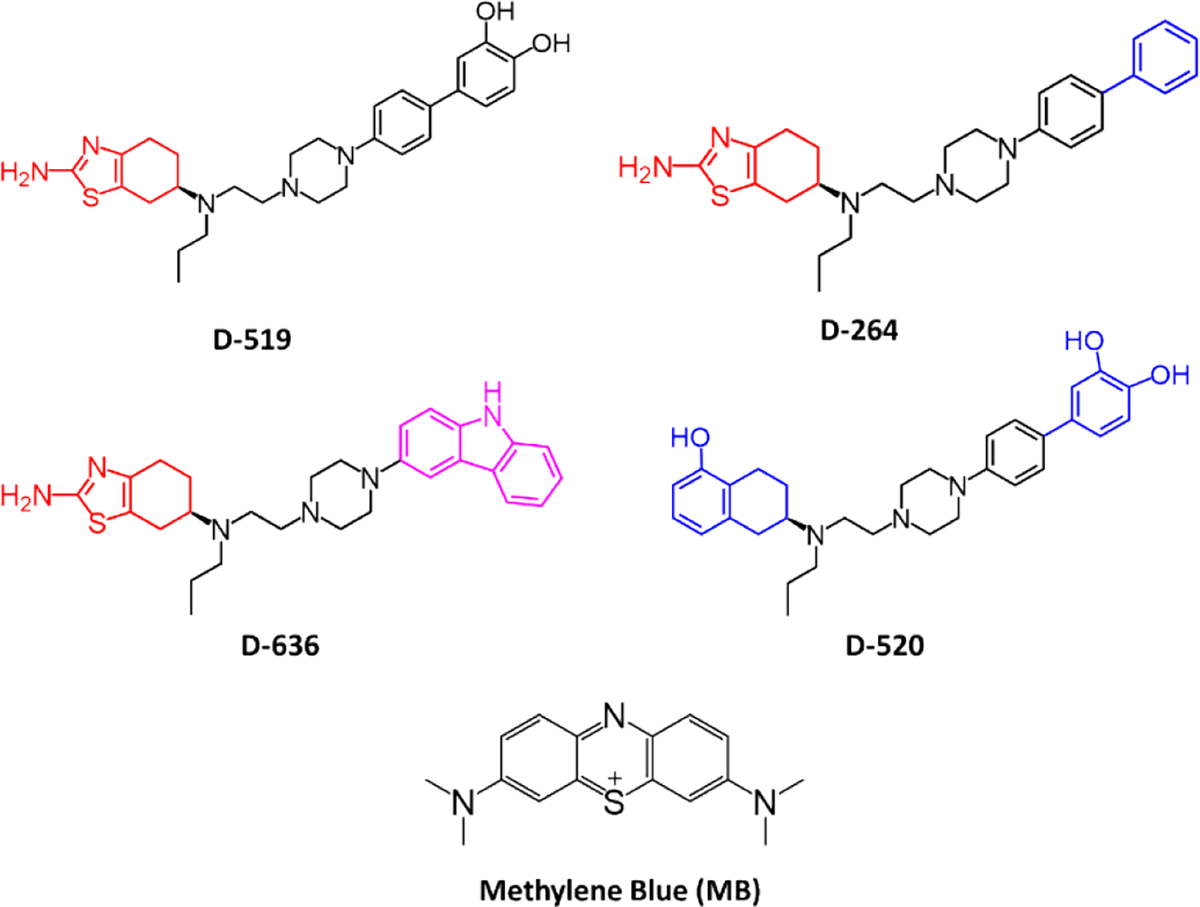
Chemical structures of aggregation inhibitors tested.
2.2. Evaluation of inhibitor activity on β-sheet formation during tau aggregation
The Amyloidogenic aggregation of peptides or proteins, including tau protein, is characterized by the β-sheet formation which can be detected using fluorescence aggregation probes.34,35 In order to monitor the β-sheet formation in heparin-induced aggregation of tau441, the Proteostat fluorescence aggregation assay was employed.36 The non-aggregated tau441 protein exhibited low fluorescence intensity due to the lack of β-sheet structure in the highly disordered protein (~1000 a.u.) even after 72 h of incubation at 37 °C (Figure S1). The increase in the fluorescence intensity (~8000 a.u.) was indicative of heparin-induced tau441 aggregation via β-sheet after 72 h of ageing. Next, the heparin-induced tau441 aggregation (for 72 h at 37 °C) was carried out with the specific inhibitor compound at various concentrations (Fig. 2A). The decrease in fluorescence intensity was observed with increasing MB concentrations as seen in Figure 2A, indicating that MB is an effective tau aggregation inhibitor. Previous studies with tau have shown similar effects with MB in vitro.37 Compounds D-519 and D-520 reduced fluorescence intensity to a similar extent as MB (Fig. 2A). By contrast, the compounds lacking catechol moiety, D-264 and D-636, were ineffective in inhibiting tau aggregation at the concentrations tested.
Fig. 2.
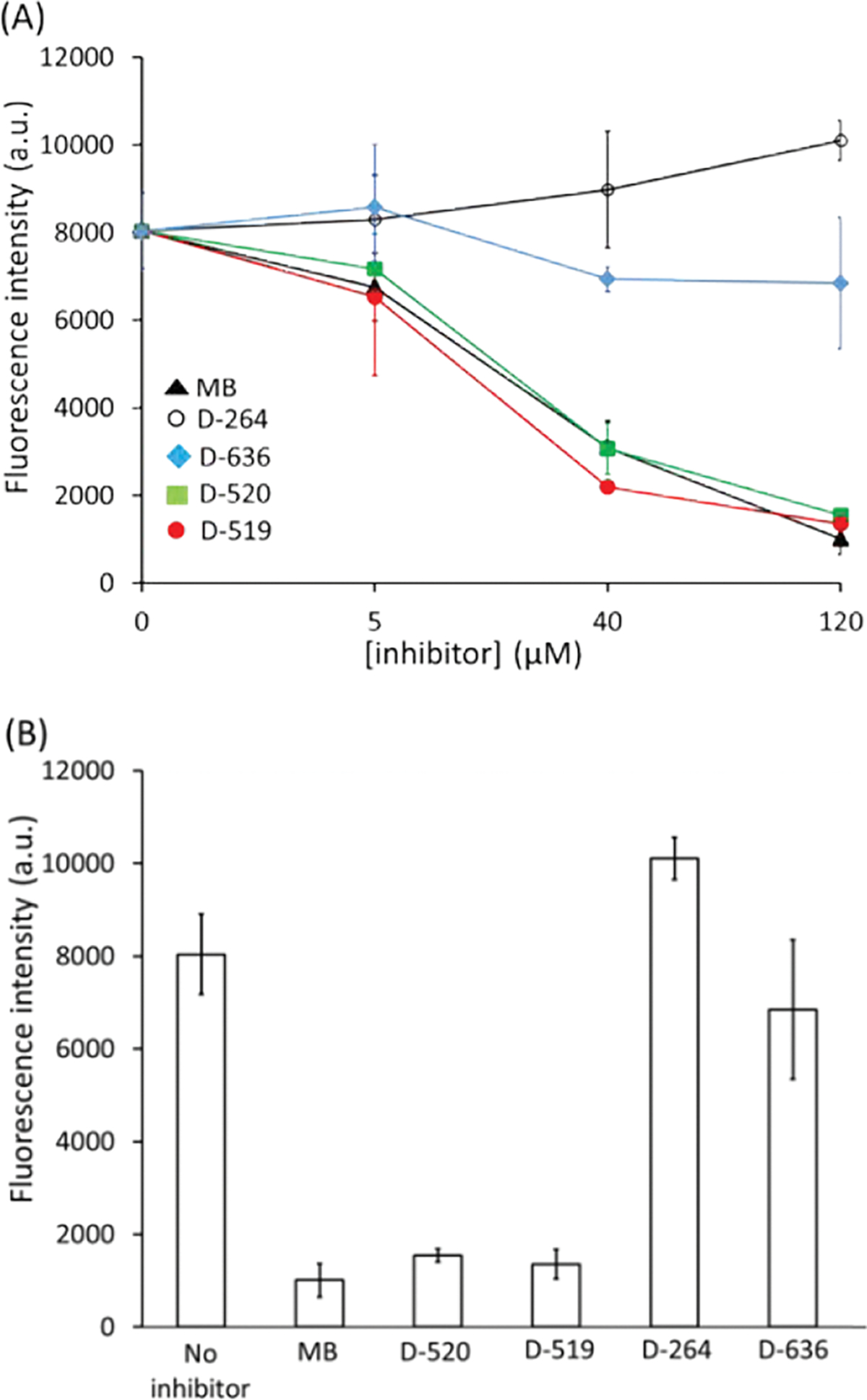
Plot of fluorescence aggregation intensity (A) as a function of inhibitor concentrations ([inhibitor] = 0 – 120 μM), (B) as function of inhibitor type ([tau441] = 21 μM; [inhibitor] = 120 μM; ageing time = 72 h; ageing temperature = 37 °C; error bars represent standard deviation associated with the triplicate measurements).
At the highest inhibitor concentration (120 μM), the fluorescence intensities for all samples were compared. Fig. 2B shows that MB, D-520 and D-519 produced low fluorescence intensities which was similar to that of non-aggregated tau (~1000 a.u.) indicating almost complete reduction in β-sheet formation and aggregation. Compound D-264 produced high fluorescence (~9000 a.u.) indicating high content of β-sheet due to tau aggregation. Similarly, D-636 induced high fluorescence intensity (~7000 a.u.) which was similar to that of aggregated tau in the absence of inhibitor.
The fluorescence aggregation assay indicated inhibition of β-sheet formation during heparin-induced tau aggregation in the presence of catechol-containing inhibitors, D-519 and D520, while the inhibitors which lacked this critical functional group were ineffective in inhibiting aggregation.
From the fluorescence aggregation data, the IC50 values were determined for all compounds: MB (31 μM), D-519 (21 μM), and D-520 (25 μM). The D-264 and D-636 compounds were non-inhibitors. The IC50 values were largely equimolar to tau protein (21 μM) used in the assay. Other inhibitors, such as aminothienopyridazine, were also reported to be in the range of stoichiometric equivalence with tau.5 At 100 μM, D-520 inhibited aggregation of α-synuclein, which was similar to the efficacy to reference drug rifampicin.21 The D-520 also inhibited cytotoxicity of aggregated of α-synuclein, but D-520 was not cytotoxic at 20 μM after longer cell incubation.21
2.3. Bis-ANS hydrophobicity assay
Bis-ANS probe is used to determine hydrophobicity content of a protein in solution. The fluorescence intensity at λem = 510 nm for Bis-ANS in the absence of protein is low. However, in the presence of a protein which contains solvent exposed hydrophobic site, the Bis-ANS-associated fluorescence increases. ANS-based fluorescence probes have been used for evaluation of tau protein aggregation, and in increase in fluorescence was observed for heparin-induced aggregation of tau compared to the non-aggregated tau which exhibited low fluorescence intensity.38
The Bis-ANS fluorescence intensity was measured in the 400–600 nm range. The Bis-ANS spectra collected in the presence of non-aggregated tau and aggregated tau in the presence of inhibitors are depicted in Fig. 3A. The fluorescence intensity associated with the Bis-ANS and non-aggregated tau was much greater (~2200 a.u.) (Fig. 3A) than the tau-free Bis-ANS solution which exhibited low fluorescence (~80 a.u.). The high fluorescence due to the non-aggregated tau indicates the presence of solvent-exposed hydrophobic pockets. The aggregated tau exhibited a slight increase in Bis-ANS fluorescence (~2800 a.u.) (Fig. 3A). Notably, the addition of MB, D-519, and D-520 compounds during heparin-induced tau aggregation resulted in a reduction of Bis-ANS signal intensity which was lower than that of non-aggregated tau. By contrast, compounds D-264 and D-636 resulted in intensities similar to that of non-aggregated tau. The intensity of Bis-ANS fluorescence band at λmax = 510 nm was also plotted as a function of inhibitor type at the highest inhibitor concentration (120 μM) (Fig. 3B). The highest fluorescence was observed for heparin induced tau aggregation (~2800 a.u.). In the presence of MB, the fluorescence dramatically decreased (~400 a.u.). Similar reduction in the fluorescence was seen with D-519 (~1000 a.u.) and D-520 (~1500 a.u.). Addition of other compounds (D-264 and D-636) had little effect on Bis-ANS fluorescence intensity. In the absence of Bis-ANS, all compounds tested did not exhibit fluorescence at 510 nm and were unlikely to contribute to fluorescence observed in the Bis-ANS assay.
Fig. 3.
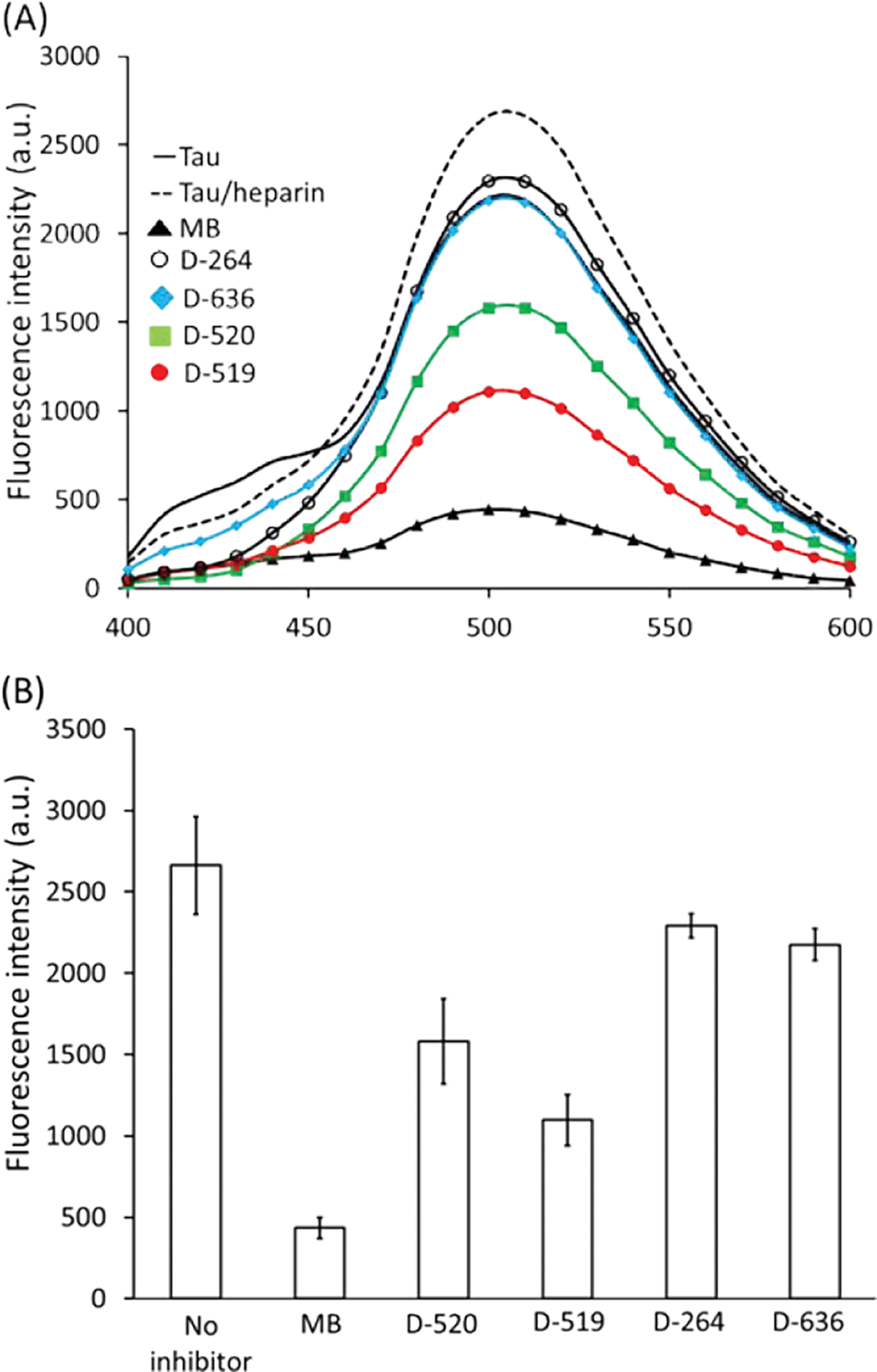
Bis-ANS (A) fluorescence (average curves) and (B) fluorescence intensity at 510 nm of heparin-induced aggregated tau441 in the absence or presence of inhibitors ([tau441] = 21 μM; [inhibitor] = 120 μM; ageing time = 72 h; ageing temperature = 37°C; [Bis-ANS] = 100μM; error bars represent standard deviation associated with the triplicate measurements).
The lowering of fluorescence with certain compounds, such as MB, D-519 and D-520, may be due to inhibition of tau aggregation as well as due to competitive displacement of Bis-ANS from hydrophobic sites in non-aggregated tau. In order to probe if compounds act as Bis-ANS competitors for binding to tau hydrophobic sites, the control studies were carried out with non-aggregated tau and the specific inhibitors in the absence of heparin. The Bis-ANS fluorescence associated with non-aggregated tau was much greater than that of non-aggregated tau with MB. The drop in fluorescence (95%) with MB, indicates that MB interacts and binds tau protein at similar site as Bis-ANS. Similarly, D-519 and D-520 when co-present with non-aggregated tau induced a significant decrease in fluorescence compared to the non-aggregated tau. The lowering of Bis-ANS fluorescence in the presence of these inhibitors may indicate that they bind to sites on tau protein and compete with Bis-ANS. In this context, Bis-ANS may serve as a probe of tau interactions with other ligands, including potential inhibitors. The use of ANS probe for competition studies with other protein and ligands has been previously reported.39 Hence, Bis-ANS probe has allowed for determination of the inhibitor binding sites on tau by MB, as well as catechol containing inhibitors, D-519 and D-520. The inhibitor binding mode via hydrophobic solvent exposed sites on tau may contribute to the anti-aggregation properties of such compounds. Hence, the inhibitor binding to hydrophobic sites of tau monomer may prevent further tau fibrillization. This is further supported by non-inhibitory compounds, such as D-264 and D-636, which did not displace hydrophobic probe as evidenced by the high fluorescence when mixed with non-aggregated tau. The Bis-ANS data show that monomeric and aggregated tau exhibited similar hydrophobicity. The Bis-ANS probe is known to bind monomeric proteins, rather than aggregates, due to steric hindrance.40 However, if the binding sites are similar between monomeric and aggregated protein then Bis-ANS will bind to both and produce similar fluorescence. The potential binding site of MB and catechol inhibitors was determined to be similar to that of Bis-ANS probe, predominantly, solvent exposed hydrophobic sites. Unlike Bis-ANS which binds surface (solvent exposed) hydrophobic sites on protein including β-sheet if accessible, the proteostat fluorescence dye binds β-sheet which make up the fibrils. Hence, an increase in proteostat fluorescence dye, which is analogues to rotational Thioflavin T dye, was observed upon β-sheet formation and protein aggregation. Bis-ANS does not bind solely to β-sheet of fibrils and hence is much less sensitive to aggregation.
2.4. TEM imaging of aggregates
The heparin-induced tau aggregation produces the short fibrils and network of filaments which can be imaged by TEM.41,42 TEM was used to monitor inhibitor effectiveness and characterize aggregate morphologies as a function of inhibitor concentrations. In the absence of compounds, the heparin-induced aggregated tau was characterized by short and long fibrils and fibrillar networks (100–1000 nm length, and 25–35 nm diameter) (Fig. 4). Notably, no aggregates were observed for the non-aggregated tau as expected for the highly soluble and disordered protein. After heparin-induced tau aggregation in the presence of inhibitors, the aggregate morphologies were also characterized by TEM. The short and long fibrils and fibrillar networks were observed in the presence of D-264 and D-636 at the highest inhibitor concentration (120 μM) (Fig. 4). However, the addition of catechol-containing compounds D-519 and D-520 induced a significant reduction in fibril formation, indicating their ability as aggregation inhibitors. Notably, some residual shorter filaments were still observed even at highest inhibitor concentration. Similarly, MB inhibitor reduced fibrilization of tau. TEM analysis clearly shows reduction in fibril formation by tau in the presence of potent inhibitors such as MB, D-519 and D-520. The reduction in fibril formation observed by TEM is in agreement with the fluorescence aggregation data.
Fig. 4.
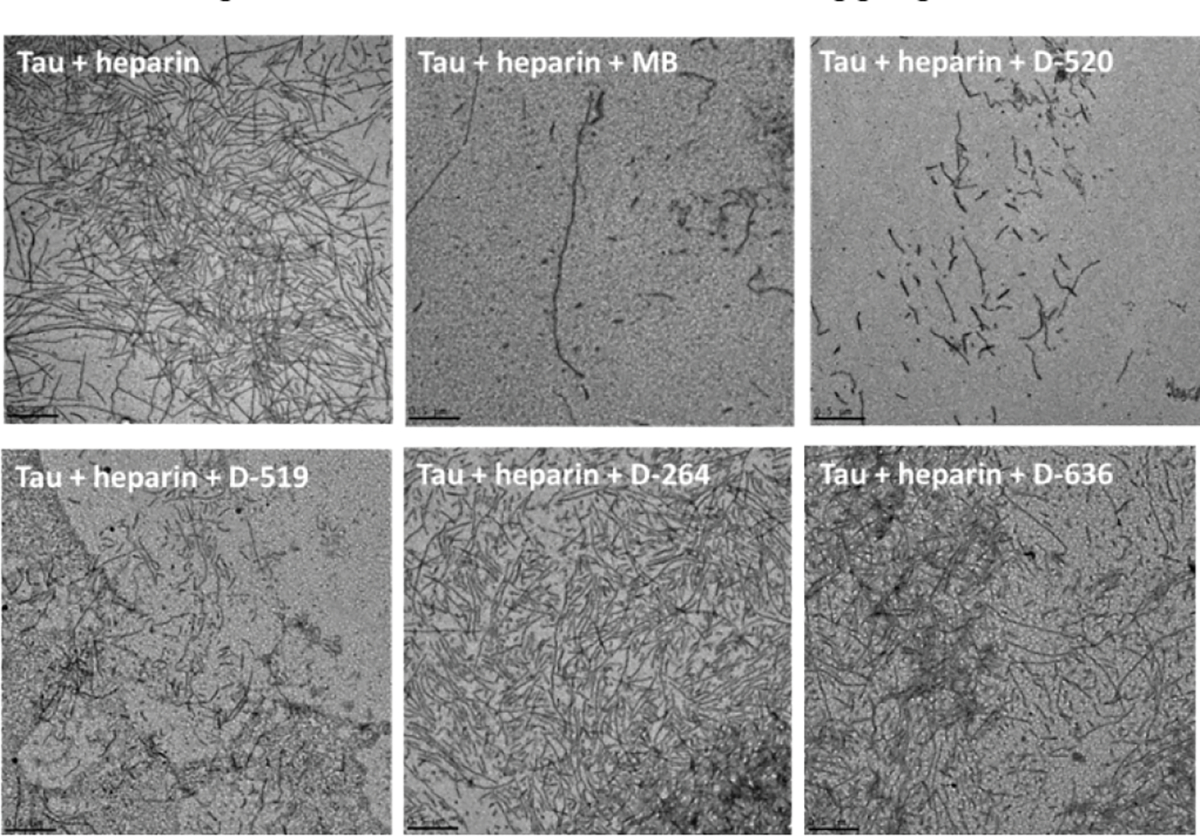
TEM images of heparin-induced tau441 aggregates in the absence or presence of MB, D-520, D-519, D-264, and D-636 ([compound] = 120 μM; ageing time = 72 h; ageing temperature = 37 C°).
2.5. Theoretical calculations of inhibitors
Density-functional theory (DFT) has been successfully applied in drug design for modeling interactions between drug and receptor, to determine stability of drug/receptor complex, to elucidate drug action mechanisms, among other applications. Herein, DFT was used to calculate electronic properties of inhibitors tested. HOMO and LUMO energy levels, electrostatic surface potentials, and dipole moments were determined from optimized structures. The B3LYP with a 6–311++G(d,p) basis set was employed for DFT calculations. In MB, both HOMO and LUMO levels are delocalized over the entire MB molecule. D-519, D-520 and D-264 all have the HOMOs and LUMOs localized on the aromatic biphenyl portion of the molecule. In addition, the catechol moieties of D-519 and D-520 also contribute to the HOMOs and LUMOs. In D-636, the HOMO and LUMO are localized on the aromatic thiazine moiety.
Electrostatic potential maps were generated by mapping the potential energy and density surfaces of each structure (Fig. 5). Compounds D-264 and D-636 exhibited electron depletion (electropositive: blue) at the exocyclic amine group only, while the electropositive regions in D-519 and D-520 included the exocyclic amine as well as the catechol. Both compounds also possess an electron accepting hydroxyl groups (electronegative: red) at the catechol moiety. By contrast, MB was largely electropositive across its structure. Hence, electrostatic potentials may forecast the reactive sites for electrophilic and nucleophilic attack. Red color area is indicative of favorable site for electrophilic attach, and blue color represents the favorable sites for nucleophilic attack.
Fig. 5.

Semi-transparent electrostatic potential maps for all inhibitors tested (color-coded red to blue).
The chemical hardness, softness, and potential values depend on the energy gap of HOMO-LUMO.43 The difference between the orbital energies of HOMO and LUMO is an important parameter that can determine the reactivity or stability of molecules.43 The HOMO-LUMO gap values for all inhibitors were calculated to be ~ 4.5 eV, with the exception of MB (~ 2.5 eV). The lowest gap for MB may indicate highest softness and higher chemical reactivity. The dipole moments were rank ordered from the highest to the lowest: D-519, D-636, D-520, D-264, and MB. The most effective inhibitors, D-519 and D-520, were characterized by larger dipole moments compared to non-inhibitors (non-catechol compounds). The size of dipole moment may influence the solubility of the compound in polar environments, such that compounds with greater dipole moment will exhibit greater interactions with polar groups (such as those in tau protein).44 Polarizability is the ability of a molecule to form an instantaneous dipoles in the presence of an external fields. The polarizability is an ideal factor for promoting van der Waals interactions with aromatic side chains of proteins.45 Other non-ionic compounds may also exhibit high polarizability due to the highly conjugated network of π electrons. The catechol inhibitors were characterized by greater polarizability compared to non-catechol analogues (Fig. 5). Recent DFT on tau-amyloid sequence VQIVYK revealed that potent inhibitors of tau aggregation participate in H-bonding with Q amino acid side chain within this sequence.46 Hence, inhibitors with H-bonding ability, such as catechol compounds, may interfere with intermolecular interactions within amyloidogenic structure and lead to disaggregation. The topological polar surface area (TPSA) and partition coefficient (log P) are useful parameters for prediction of drug transport properties and molecular hydrophobicity, respectively. Hydrophobicity of a compounds affects drug absorption, bioavailability, drug-receptor interactions, metabolism, and toxicity. These parameters are used in Structure Activity Relationships studies and rotational drug design. The interactive molecular calculator was used to predict TPSA and log P values of all compounds in this study. The reference compound used was aspirin for which TPSA and log P were predicted to be 63 and 1.4, respectively, which matched well with the Pubchem reported values. The log P values for D-519, D-520, D-264, D-636, and MB were calculated to be 4.8, 5.9, 5.8, 5.5, and 3.3, respectively. The positive value of log P indicates a higher compound concentration in the lipid phase due to its greater lipophilicity. The TPSA parameters for D-519, D-520, D-264, D-636, and MB were calculated to be 90, 70, 48, 65, and 19, respectively. For the molecule to penetrate the blood-brain barrier, a TPSA should be less than 90. To improve log P and TPSA values of this class of molecules further chemical modifications are required.
3. Conclusions
Catechol-based compounds were effective in inhibiting in vitro heparin-induced aggregation of full-length tau441 protein. The inhibitory activity with such molecules was similar to that of a well-known aggregation inhibitor MB. The greater electron polarizability, dipole moment and hydrogen bonding may contribute to the efficacy of catechol-based compounds.
Inhibition mechanisms based on non-covalent electrostatic interactions have been demonstrated for other phenols, such as tolcapone, ventacapone, and nitrocatechol, which inhibited tau-derived hexapeptide 306VQIVYK311 aggregation.47 The tolcapone and entacapone participated in the electrostatic interactions with Lys (K) residues of tau, and prevented β-sheet formation.47 The catechol-containing compound, such as 3,4-dihydroxy-5-nitrophenol, formed polar contacts with Lys residues of tau hexapeptide β-sheet, and subsequently inhibiting aggregation.48 Hydrogen bonding interactions between procyanidins family of compounds and Thr residues of tau peptide also contributed to inhibition.48
The disulfide crosslinking was proposed to be a necessary step in tau oligomerization and ultimate aggregation. Tau441 contains Cys291 and Cys322 residues which can form intermolecular disulfide bonds with neighboring tau molecules leading to aggregation.49 The mechanism of tau aggregation inhibition by MB was ascribed to the chemical modification of tau, specifically to an oxidation of native Cys residues.9,50–51 Other aminothienopyridazine molecules produced oxidation of Cys residues of tau which may have contributed to their inhibitory efficacies.9,52 Inhibitors which contain the catechol motif may bind to and cap Cys residues of tau and prevent its aggregation by hindering interactions between tau molecules.53
The catechol/quinone containing compounds may covalently bind to proteins and inhibit fibrillization.48,53–56 For example, quinone analogs formed adducts with α-Synuclein peptide, thereby inhibiting fibrillization. Some quinones may form covalent bonds with nucleophilic residues, including Cys, Lys or N-terminal NH2 group of the protein. The (−)-epigallocatechin gallate (EGCG), a green tea polyphenol, covalently bound to tau peptide K18ΔK280 and prevented its aggregation.48 However, EGCG was a poor inhibitor of tau protein aggregation (IC50>200 μM), despite the presence of multiple phenolic moieties. The reduced EGCG inhibitory activity was attributed to its greater binding affinity to regions of tau outside the R repeat domain which do not participate in aggregation.
Similar mechanisms mentioned above may be proposed for D-519 and D-520 with tau protein presented in this work. The non-inhibitory compounds, such as D-264 and D-636, which lack hydroxyl groups, were ineffective as tau aggregation inhibitors. In addition, the compound (N6-propyl-4,5,6,7-tetrahydro-benzothiazole-2,6-diamine (S-pramipexole), which contains only thiazolyl unit without biphenyl or carbazoyl group, was a non-inhibitor as well. By contrast, the compound 6-propylamino-5,6,7,8-tetrahydro-naphthalen-1-ol (D-686), which lacks biphenyl or catechol moieties, was a promoter of tau aggregation. The compound D-519 also contained a thiazine moiety, in addition to the catechol group, and was an effective tau aggregation inhibitor. However, the compounds with a thiazine group (D-264 and D-636) were non-inhibitors due to the lack of catechol functionality. Hence, data indicate that catechol containing compounds are promising inhibitors of in vitro aggregation of tau protein. Given their dopamine activity and aggregation inhibition properties D2/D3 molecules are viable drug candidates and need to be explored further. Additional studies are required to evaluate activity of such compounds in preventing cytotoxicity of tau oligomers and aggregates. For these promising compounds to become therapeutics, further chemical functionalization and optimization are required to improve their bioavailability and ultimately their ability to cross the blood-brain barrier and target tauopathies.
4. Experimental section
4.1. Chemicals
Tau441 protein (recombinant tau441, 2N4R) was purchased as a lyophilized powder from rPeptide (GA, USA). It was hydrated with deionized (DI) water to a concentration of 1 mg/mL prior to use. A stock solution of tau buffer, pH 6.8, was prepared using 0.5 mM ethylene glycol-O, O’- bis (2-aminoethyl)-N, N, N’, N’-tetraacetic acid (EGTA), obtained from Alfa Aesar (Heysham, England), 50 mM 2-(N-5-morpholino) ethanesulfonic acid (MES) and 100 mM sodium chloride (NaCl), purchased from Fisher Scientific (NY, USA). The pH was adjusted using sodium hydroxide (NaOH) obtained from Fisher Scientific (NY, USA). Dimethyl sulfoxide (DMSO) was purchased from J. T. Baker Chemical Co. (Phillipsburg, NJ, USA). Heparin sodium salt from porcine intestinal mucosa was purchased from Sigma-Aldrich (St. Luis, MO, USA). A stock solution of Tris buffer, pH 7.4, was prepared using Tris ultrapure from Amresco (Solon, OH, USA) in DI water. 4,4´-dianilino-1,1´-binaphtyl-5,5´-disulfonic acid dipotassium salt (Bis-ANS) and MB were purchased from Sigma Aldrich (St. Louis, MO, USA). Proteostat Protein Aggregation Assay kit was purchased from Enzo Life Sciences, Inc (Farmingdale, New York, USA). Uranyl acetate (1 %), glutaraldehyde (2 %), and Formvar carbon film on 200 mesh nickel grids were purchased from Electron Microscopy Science (Hatfield, PA, USA). Compounds tested were dissolved in DMSO, then diluted to various concentrations in tau buffer, pH 6.8.
4.2. Aggregation of tau with heparin
Tau protein aggregation was induced with heparin, in the absence of potential inhibitors. 30 μL of tau (1 mg/mL = 21.79 μM) were mixed with 1.5 μL heparin (108.9 μM) and DMSO (instead of potential inhibitor) (% of DMSO solution depended on potential inhibitor concentration and ranged from 0.11– 5.5 % initial DMSO solution in tau buffer). Tau final tau concentration was 909 μg/mL (19.8 μM) and heparin final concentration was 4.95 μM). Samples were incubated for 72 h at 37 °C. After 72 h, 10 μL aliquots were taken for Proteostat aggregation assay, Bis-ANS assay and TEM.
4.3. Aggregation inhibition studies
Tau protein aggregation was induced with heparin in the presence of potential inhibitors. 30 μL of tau (1 mg/mL = 21.79 μM) were mixed with 1.5 μL heparin (108.9 μM) and 1.5 μL potential inhibitor (110 μM, 880 μM, 2.64 mM, 5.5 mM). Tau final tau concentration was 909 μg/mL (19.8 μM), heparin final concentration was 4.95 μM and potential inhibitor final concentration was 5 μM, 40 μM, 120 μM or 250 μM. Samples were incubated for 72 h at 37 °C. After 72 h, 10 μL aliquots were taken for Proteostat aggregation assay, Bis-ANS assay and Transmission Electron Microscopy imaging.
4.4. Proteostat fluorescence aggregation assay
The fluorescence aggregation assay was carried out using the Proteostat aggregation assay kit. The sample-to-detection reagent ratio was 10:1 % v/v. The positive and negative controls (aggregated and native lysozyme, respectively) were used as per the Proteostat aggregation protocol. The sample solutions were loaded (3 μL onto each well) onto a Take3 micro-volume plate. Fluorescence was acquired using the H1 synergy microplate reader from Biotek at an excitation of 550 nm and emission of 600 nm. Each condition was repeated in triplicate. The IC50 values were determined by fitting the experimental data to a Non-linear curve fit (growth/sigmoidal) and dose response curve by using OriginPro 8 software.
4.5. Bis-ANS fluorescence assay
The following control samples were prepared for fluorescence analysis: a) 10 μL tau protein at a final concentration of 909 μg/mL with 0.5 μL tau buffer and 0.5 μL DMSO (at various %, as described above), b) 10.5 μL tau buffer with 0.5 μL DMSO, c) 10 μL tau buffer with 0.5 μL heparin and 0.5 μL DMSO, d) Bis-ANS 100 μM, e) 10 μL tau buffer with 0.5 μL heparin and 0.5 μL potential inhibitor in DMSO. A stock solution of 200 μM Bis-ANS was prepared in 20 mM Tris buffer (pH 7.4). The fluorescence associated with Bis-ANS was measured by H1 synergy Biotek microplate reader and Take3 micro-volume plate. Samples were incubated for 72 hours at 37 °C. To 10 μL of each sample were added 10 μL of Bis-ANS, producing a final concentration of Bis-ANS at 100 μM. The mixture was mixed and incubated at room temperature for 60 minutes. Measurements were carried out at 25 °C using excitation wavelength at 350 nm and emission spectra were recorded between 400 and 600 nm.
4.6. Transmission electron microscopy
Transmission electron microscopy (TEM) was carried out on the FEI Morgagni M268D transmission electron microscope (Eye Research Institute, Oakland University) or JEOL 2200FS (Japan Electron Optics Laboratories) (Michigan State University). 10 μL of each sample were deposited on a Formvar-carbon coated 200 mesh nickel grids and allowed to absorb for 2 hours in ambient light. The grids were washed with DI water and blotted dry, then loaded with 2 % glutaraldehyde solution for 5 minutes. The grids were washed again with DI water, blotted dry and loaded with 1 % Uranyl Acetate solution for 5 minutes. Finally, they were washed with DI water and examined at 22000x, 56000x, and 140000x magnification to obtain an overall evaluation of the samples. Duplicate grids were made for each sample.
4.7. Theoretical calculations
Calculations were performed using Density Functional Theory (DFT) with the B3LYP functional and 6–311++G(d,p)basis set using the Gaussian 09 suite of programs. The highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO), and electrostatic potential maps were generated for all compounds using the cubegen utility. The prediction of log P and TPSA was carried out by using Molinspiration software available freely online (https://www.molinspiration.com/). For 50.5% of molecules logP is predicted with error < 0.25, for 80.2% with error < 0.5 and for 96.5% with error < 1.0. Only for 3.5% of structures logP is predicted with error > 1.0.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health/National Institutes of General Medical Sciences (R15 GM11905301, S. M.; C. W.) and by National Institute of Neurological Disorders and Stroke/National Institute of Health (NS047198, AKD). We are grateful to Dr. Victoria Kimler from Eye Research Institute (Oakland University) and Dr. Alicia Withrow from Michigan State University for TEM imaging. The authors also recognize the Oakland University for the 2017 Provost Graduate Students Research award to I.Z. Calculations were done T.M and I.R. at the Portland Institute for Computational Science which is funded by the NSF Grant # DMS-1624776 and the ARO under US Army Federal Grant # W911NF-15–0590.
Footnotes
Supplementary Material
Supplementary material includes fluorescence spectroscopy, TEM data, DFT calculations.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Ann. Rev. Neurosci. 2001; 24:1121–1159. [DOI] [PubMed] [Google Scholar]
- 2.Keri RS, Patil MR, Patil SA, Budagumpi S. A Comprehensive review in current developments of benzothiazole-based molecules in medicinal chemistry. ChemInform, 2015; 89: 207–51. [DOI] [PubMed] [Google Scholar]
- 3.Honson NS, Jensen JR, Abraha A, Hall GF, Kuret J. Small-molecule mediated neuroprotection in an in situ model of tauopathy. Neurotox. Res. 2009; 15: 274–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shi CJ, Peng W, Zhao JH, Yang HL, Qu LL, Wang C, Kong LY, Wang XB. Usnic acid derivatives as tau-aggregation and neuroinflammation inhibitors. Eur. J. Med. Chem. 2020; 187: 111961. [DOI] [PubMed] [Google Scholar]
- 5.Ballatore C, Brunden KR, Piscitelli F, James MJ, Crowe A, Yao Y, Hyde E, Trojanowski JQ, Lee VM, Smith AB. Discovery of brain-penetrant, orally bioavailable aminothienopyridazine inhibitors of tau aggregation. J. Med. Chem. 2010; 53: 3739–3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ballatore C, Crowe A, Piscitelli F, James M, Lou K, Rossidivito G, Yao Y, Trojanowski JQ, Lee VM, Brunden KR, Smith AB 3rd. Aminothienopyridazine inhibitors of tau aggregation: Evaluation of structure–activity relationship leads to selection of candidates with desirable in vivo properties. Bioorg. Med. Chem. 2012; 20: 4451–4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okuda M, Hijikuro I, Fujita Y, Teruya T, Kawakami H, Takahashi T, Sugimoto H. Design and synthesis of curcumin derivatives as tau and amyloid beta dual aggregation inhibitors. Bioorg. Med. Chem. 2016; 26: 5024–5028. [DOI] [PubMed] [Google Scholar]
- 8.Moir M, Chua SW, Reekie T, Martin AD, Ittner A, Ittner LM, Kassiou M. Ring-opened aminothienopyridazines as novel tau aggregation inhibitors. Med. Chem. Comm. 2017; 8: 1275–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crowe A, James MJ, Lee VM, Smith AB, Trojanowski JQ, Ballatore C, Brunden KR. Aminothienopyridazines and methylene blue affect tau fibrillization via cysteine oxidation. J. Biol. Chem. 2013; 288: 11024–11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pickhardt M, Neumann T, Schwizer D, Callaway K, Vendruscolo M, Schenk D, George-Hyslop P, Mandelkow EV, Dobson CM, McConlogue L, Mandelkow E, Toth G. Identification of small molecule inhibitors of tau aggregation by targeting monomeric tau as a potential therapeutic approach for tauopathies. Curr. Med. Chem. 2015; 9: 814–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baggett DW, Nath A. The rational discovery of a tau aggregation inhibitor. Biochemistry. 2018; 57: 6099–6107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bulic B, Pickhardt M, Mandelkow E, Mandelkow E. Tau protein and tau aggregation inhibitors. Neuropharmacology, 2010; 59: 276–289. [DOI] [PubMed] [Google Scholar]
- 13.Pickhardt M, Larbig G, Khlistunova I, Coksezen A, Meyer B, Mandelkow EM, Schmidt B, Mandelkow E. Phenylthiazolylhydrazide and its derivatives are potent inhibitors of tau aggregation and toxicity in vitro and in cells. Biochemistry, 2007; 46: 10016–10023. [DOI] [PubMed] [Google Scholar]
- 14.Bulic B, Pickhardt M, Khlistunova I, Biernat J, Mandelkow E, Mandelkow E, Waldmann H. Rhodanine-based tau aggregation inhibitors in cell models of tauopathy. Angew. Chem. Int. Ed. 2007; 119: 9375–9379. [DOI] [PubMed] [Google Scholar]
- 15.Cai N, Chen J, Bi D, Gu L, Yao L, Li X, Li H, Xu H, Hu Z, Liu Q, Xu X. Specific degradation of endogenous tau protein and inhibition of tau fibrillation by Tanshinone IIA through the ubiquitin-proteasome pathway. J. Agric. Food Chem. 2020; 68: 2054–2062. [DOI] [PubMed] [Google Scholar]
- 16.Congdon EE, Sigurdsson EM. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018; 14: 399–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ksiezak-Reding H, Ho L, Santa-Maria I, Diaz-Ruiz C, Wang J, Pasinetti GM. Ultrastructural alterations of Alzheimer’s disease paired helical filaments by grape seed-derived polyphenols. Neurobiol. Aging, 2012; 33: 1427–1439. [DOI] [PubMed] [Google Scholar]
- 18.Paranjape SR, Riley AP, Somoza AD, Oakley CE, Wang CCC, Prisinzano TE, Oakley BR, Gamblin TC. ACS Chem. Neurosci. 2015; 6: 751–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Seidler PM, Boyer DR, Rodriguez MR, Sawaya MR, Cascio D, Murray K, Gonne T, Eisenberg DS. Structure-based inhibitors of tau aggregation. Nat. Chem. 2018; 10: 170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mohamed T, Hoang T, Jelokhani-Niaraki M, Rao PPN. Tau-derived-hexapeptide 306VQIVYK311 aggregation inhibitors: nitrocatechol moiety as a pharmacophore in drug design. ACS Chem. Neurosci. 2013; 4: 1559–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Modi G, Voshavar C, Gogoi S, Shah M, Antonio T, Reith ME, Dutta AK. Multifunctional D2/D3 agonist D-520 with high in vivo efficacy: modulator of toxicity of alpha-synuclein aggregate. ACS Chem. Neurosci. 2014; 8: 700–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karakaya T, Fuber F, Prvulovic D, Hampel H. Pharmacological treatment of mild cognitive impairment as a prodromal syndrome of Alzheimeŕs disease Curr. Treatments Opt. Neurol. 2012; 14: 126–136. [Google Scholar]
- 23.Pan X, Kaminga AC, Wen SW, Wu X, Acheampong K, Liu A. Dopamine and dopamine receptors in Alzheimer’s disease: A systematic review and network meta-analysis. Frontier: Aging Neurosci. 2019; 11:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mitchell RA, Herrmann N, Lanctôt KL. The role of dopamine in symptoms and treatment of apathy in Alzheimer’s disease. CNS Neurosci. Ther. 2011; 17: 411–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martorana A, DiLorenzo F, Esposito Z, Lo GT, Bernardi G. Dopamine D₂-agonist rotigotine effects on cortical excitability and central cholinergic transmission in Alzheimer’s disease patients. Neuropharmacology, 2013; 64: 108–113. [DOI] [PubMed] [Google Scholar]
- 26.Koch G, Di Lorenzo F, Bonnì S, Giacobbe V, Bozzali M, Caltagirone C. Dopaminergic modulation of cortical plasticity in Alzheimer’s disease patients. Neuropsychopharm. 2014; 39: 2654–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeganathan S, Bergen MV, Mandelkow E, Mandelkow E. The Natively unfolded character of tau and its aggregation to Alzheimer-like paired helical filaments. Biochemistry, 2008; 47: 10526–10539. [DOI] [PubMed] [Google Scholar]
- 28.Barghorn S, Zheng-Fischhöfer Q, Ackmann M, Biernat J, Bergen MV, Mandelkow E, Mandelkow E. Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry, 2000; 39: 11714–11721. [DOI] [PubMed] [Google Scholar]
- 29.Kuzhikandathil EV, Cote S, Santra S, Dutta AK. Interaction of D3 preferring agonist (−)-N 6-(2-(4-(biphenyl-4-yl)piperazin-1-yl)ethyl)-N 6-propyl-4,5,6,7-tetrahydrobenzo[d]thiazole-2,6-diamine (D-264) with cloned human D2L, D2S, and D3 receptors: Potent stimulation of mitogen-activated protein kinases and G protein-coupled inward rectifier potassium channels. Naunyn-Schmiedebergs Arch. Pharmacol. 2012; 386: 97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li C, Biswas S, Li X, Dutta AK, Le W. Novel D3 dopamine receptor-preferring agonist D-264: Evidence of neuroprotective property in Parkinsons disease animal models induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and lactacystin. J. Neurosci. Res. 2010; 88: 2513–2523. [DOI] [PubMed] [Google Scholar]
- 31.Wischik CM, Harrington C, Storey J. TauRx global phase 3 trial in Alzheimer’s disease with tau aggregation inhibitor LMTX. Neurobio. Aging, 2014; 35: S26–S26. [Google Scholar]
- 32.Wischik CM, Staff RT, Wischik DJ, Bentham P, Murray AD, Storey JM, Kook KA, Harrington CR. Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate Alzheimer’s disease. J. Alz. Dis. 2015; 44: 705–720. [DOI] [PubMed] [Google Scholar]
- 33.Wischik CM, Bentham P, Wischik DJ, Seng KM. O3–04-07: Tau aggregation inhibitor (TAI) therapy with rember™ arrests disease progression in mild and moderate Alzheimer’s disease over 50 weeks. Alz. Dementia, 2008; 4. [Google Scholar]
- 34.Barghorn S, Zheng-Fischhöfer Q, Ackmann M, Biernat J, Bergen MV, Mandelkow E, Mandelkow E. Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry, 2000; 39: 11714–11721. [DOI] [PubMed] [Google Scholar]
- 35.Barghorn S, Mandelkow E. Toward a unified scheme for the aggregation of tau into Alzheimer paired helical filaments. Biochemistry, 2002; 41: 14885–14896. [DOI] [PubMed] [Google Scholar]
- 36.Esteves-Villanueva JO, Trzeciakiewicz H, Loeffler DA, Martić S. Effects of tau domain-specific antibodies and intravenous immunoglobulin on tau aggregation and aggregate degradation. Biochemistry, 2015; 54: 293–302. [DOI] [PubMed] [Google Scholar]
- 37.Hattori M, Sugino E, Minoura K, In Y, Sumida M, Taniguchi T, Tomoo K, Ishida T. Different inhibitory response of cyanidin and methylene blue for filament formation of tau microtubule-binding domain. Biochem. Biophys. Res. Commun. 2008; 374: 158–163. [DOI] [PubMed] [Google Scholar]
- 38.Jangholi A, Ashrafi-Kooshk MR, Arab SS, Riazi G, Mokhtari F, Poorebrahim M, Mahdiuni H, Kurgano BI, Moosavi-Mohavedi AA, Khodarahmi R. Appraisal of role of the polyanionic inducer length on amyloid formation by 412-residue 1N4R Tau protein: A comparative study. Arch. Biochem. Biophys. 2016; 609: 1–19. [DOI] [PubMed] [Google Scholar]
- 39.De S, Girigoswami A, Das S. Fluorescence probing of albumin–surfactant interaction. J. Coll. Inter. Sci. 2005; 285: 562–573. [DOI] [PubMed] [Google Scholar]
- 40.Lindgren M, Sörgjerd K, Hammarström P. Detection and characterization of aggregates, prefibrillar amyloidogenic oligomers, and protofibrils using fluorescence spectroscopy. Biophys. J. 2005; 88: 4200–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu S, Brunden KR, Trojanowski JQ, Lee VM. Characterization of tau fibrillization in vitro. Alz. Dementia, 2010; 6: 110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Al-Hilaly YK, Pollack SJ, Vadukul DM, Citossi F, Rickard JE, Simpson M, Storey JMD, Harrington CR, Wischik CMR, Serpell LC. Alzheimer’s disease-like paired helical filament assembly from truncated tau protein is independent of disulfide crosslinking. J. Mol. Biol. 2017; 429: 3650–3665. [DOI] [PubMed] [Google Scholar]
- 43.Buyukuslu H, Akdogan M, Yildirim G, Parlak C. Ab initio Hartree-Fock and density functional theory study on characterization of 3-(5-methylthiazol-2-yldiazenyl)-2-phenyl-1H-indole. Spectrochim Acta A. 2010; 75: 1362–9 [DOI] [PubMed] [Google Scholar]
- 44.Plumley JA, Dannenberg JJ. The importance of hydrogen bonding between glutamine side chains to the formation of amyloid VQIVYK parallel beta-sheets: an ONIOM DFT/AM1 study. J. Am. Chem. Soc. 2010; 132: 1758–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nayeem A, Krystek S Jr, Stouch T. An assessment of protein-ligand binding site polarizability. Biopolymers. 2003; 70: 201–211. [DOI] [PubMed] [Google Scholar]
- 46.Lindgren M, Sörgjerd K, Hammarström P. Detection and characterization of aggregates, prefibrillar amyloidogenic oligomers, and protofibrils using fluorescence spectroscopy. Biophys. J. 2005; 88: 4200–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mohamed T, Hoang T, Jelokhani-Niaraki M, Rao PPN. Tau-derived-hexapeptide 306VQIVYK311 aggregation inhibitors: nitrocatechol moiety as a pharmacophore in drug design. ACS Chem. Neurosci. 2013; 4: 1559–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wobst HJ, Sharma A, Diamond MI, Wanker EE, Bieschke J. The green tea polyphenol (−)-epigallocatechin gallate prevents the aggregation of tau protein into toxic oligomers at substoichiometric ratios. FEBS Lett. 2014; 589: 77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schweers O, Mandelkow EM, Biernat J, Mandelkow E. Oxidation of cysteine-322 in the repeat domain of microtubule-associated protein tau controls the in vitro assembly of paired helical filaments. Proc. Nat. Acad. Sci. USA. 1995; 92: 8463–8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Noto LD, Deture MA, Purich DL. Disulfide-cross-linked tau and MAP2 homodimers readily promote microtubule assembly. Mol. Cell Biol. Res. Commun. 1999; 2: 71–76. [DOI] [PubMed] [Google Scholar]
- 51.Necula M, Breydo L, Milton S, Kayed R, Veer WE, Tone P, Glabe CG. Methylene blue inhibits Amyloid Aβ oligomerization by promoting fibrillization. Biochemistry, 2007; 46: 8850–8860. [DOI] [PubMed] [Google Scholar]
- 52.Akoury E, Pickhardt M, Gajda M, Biernat J, Mandelkow E, Zweckstetter M. Mechanistic basis of phenothiazine-driven inhibition of tau aggregation. Angew. Chem. Int. Ed. 2013; 52: 3511–3515. [DOI] [PubMed] [Google Scholar]
- 53.Soeda Y, Yoshikawa M, Almeida OF, Sumioka A, Maeda S, Osada H, Kondoh Y, Saito A, Miyasaka T, Kimura T, Suzuki M, Koyama H, Yoshiike Y, Sugimoto H, Ihara Y, Takashima A. Toxic tau oligomer formation blocked by capping of cysteine residues with 1,2-dihydroxybenzene groups. Nat. Commun. 2015; 6: 10216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang J, Ho L, Zhao W, Ono K, Rosensweig C, Chen L, Humala N, Teplow DB, Pasinetti GM. Grape-derived polyphenolics prevent Abeta oligomerization and attenuate cognitive deterioration in a mouse model of Alzheimer’s disease. J. Neurosci. 2008; 28: 6388–6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ono K, Condron MM, Ho L, Wang J, Zhao W, Pasinetti GM, Teplow DB. Effects of grape seed-derived polyphenols on Amyloid β-protein self-assembly and cytotoxicity. J. Biol. Chem. 2008; 283: 32176–32187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gueroux M, Laguerre M, Szlosek-Pinaud M, Fouquet E, Pianeta I. Polyphenols and Alzheimer’s disease: Tau/polyphenol interactions investigated by NMR and molecular modelling. Nutrit. Aging, 2012; 1: 201–206. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.