

Summary
SARS-CoV-2 infection is silent or benign in most infected individuals, but causes hypoxemic COVID-19 pneumonia in about 10% of cases. We review here studies of the human genetics of life-threatening COVID-19 pneumonia, focusing on both rare and common variants. Large-scale genome-wide association studies have identified more than 20 common loci robustly associated with COVID-19 pneumonia with modest effect sizes, some implicating genes expressed in the lungs or leukocytes. The most robust association, on chromosome 3, concerns a haplotype inherited from Neanderthals. Sequencing studies focusing on rare variants with a strong effect have been particularly successful, identifying inborn errors of type I IFN immunity in 1–5% of unvaccinated patients with critical pneumonia, and their autoimmune phenocopy, autoantibodies against type I IFN, in another 15–20% of cases. Our growing understanding of the impact of human genetic variation on immunity to SARS-CoV-2 is enabling health systems to improve protection for individuals and populations.
Keywords: SARS-CoV-2, COVID-19 pneumonia, GWAS, Inborn errors of immunity, Type I interferons
Introduction
More than 600 million cases of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection and at least 6.5 million deaths from COVID-19 have already been recorded worldwide (1). The clinical manifestations of COVID-19 are highly variable, ranging from silent infection to life-threatening disease, typically beginning with pneumonia. Before effective anti-SARS-CoV-2 vaccines became available, ~ 3% of infected individuals developed critical COVID-19 pneumonia requiring supplementary high-flow oxygen (O2> 6 L/min), mechanical ventilation (non-invasive or by intubation), or extracorporeal membrane oxygenation (ECMO) (2), with an estimated infection fatality rate of about 0.5–1% (3, 4). Advanced age was, by far, the strongest predictor of COVID-19 severity at the time, with the risk of death doubling with every five years of age from childhood onward (3, 4). Unvaccinated men also have a 1.5 times greater risk of death than women (3, 5, 6). Ancestry, social status, and several comorbid conditions have been associated with higher disease severity and death rates, but with modest odds ratios (OR, typically <1.5, rarely >2) (5, 7–9). In the early days of the pandemic, it was already obvious that demographic and clinical factors did not entirely account for the marked inter-individual variability of COVID-19 clinical manifestations. The hypothesis of human genetic predisposition was most strongly supported by the rare cases of previously healthy young individuals being admitted to intensive care for respiratory failure. Other clinical presentations have emerged during the pandemic, including multisystem inflammatory syndrome (MIS) in children (MIS-C) (10) and adults (MIS-A) (11), “COVID-toes” (pernio) (12), and long-term neurocognitive, pulmonary, and musculoskeletal sequelae collectively referred to as “long COVID” or “post-acute COVID-19 syndrome” (13). Here again, clinicians were puzzled by the remarkable differences in clinical manifestations observed at population level and suggestive of a causal or modulating role for human genetic variation (2).
The development of anti-SARS-CoV-2 vaccines rapidly became a global health priority. The massive deployment of several effective vaccines, developed in less than a year, has indubitably altered the course of the pandemic, largely decreasing the risks of severe disease, hospitalization, and death in regions of high vaccine coverage (14). However, the success of vaccination has been jeopardized by the limited access to vaccination in lower-income countries, vaccine hesitancy, and the emergence of multiple variants of concern, such as Alpha (B1.1.7), Beta (B.1.351), Delta (B.1.617.2) and Omicron (BA.1, BA.2, BA.4, BA.5 and BQ.1), all of which are more transmissible than the original strain, and some of which increase the risk of severe disease (15–19) and/or immune escape (20). Moreover, even the most effective RNA vaccines do not prevent infection per se, sometimes resulting in “breakthrough” pneumonia in vaccinated individuals (21, 22). Hence, the fight against this disease continues, and humanity will benefit greatly from improvements in our understanding of the mechanisms of host defense and immune protection against SARS-CoV-2. Human genetic studies of infectious diseases can point to the primary cause of disease and provide invaluable mechanistic insights (23, 24). In this review, we will briefly introduce the field of human genetics of infectious diseases, and will describe the main genetic findings relating to susceptibility to COVID-19 pneumonia and its severity, with their downstream implications.
1. Human genetics of infectious diseases
For almost all infectious agents, the clinical manifestations of infection are highly variable, ranging from silent infection to lethal disease (25). The field of human genetics of infectious diseases aims to characterize the genetic variants accounting for this considerable interindividual variability. It was long held as a dominant paradigm that rare infections with weakly virulent microbes and/or multiple, recurrent infections in a single patient result from rare monogenic inborn errors of immunity (IEIs, also called primary immunodeficiencies [PIDs]), whereas common infections with more virulent pathogens are more influenced by the polygenic inheritance of common alleles (26). IEIs were discovered through individual-based studies focusing on a small number of patients, sometimes even a single patient, initially with sporadic infections. IEIs may be individually rare, but, collectively, they are more common than initially thought and their study can unravel general mechanisms of diseases that can be triggered by other causes (23, 24).
At population level, attempts to understand how human genetic variation modulates the individual response to more common pathogens began seven decades ago. One of the first major discoveries was the identification of multiple red blood cell abnormalities associated with a lower risk of severe malaria and subject to strong positive selection in populations living in regions in which malaria was endemic (27). However, host genetic studies with the objective of discovering variants were long hampered by technological limitations: the low throughput of DNA analysis methods at the time obliged researchers to use candidate gene approaches, unless they had access to family samples, which allowed more solid linkage studies. Most candidate gene studies had relatively small sample sizes and failed to account for population stratification or multiple testing, generating a flurry of false-positive results. Recent progress in large-scale genotyping and sequencing technology, coupled with dramatic improvements in bioinformatics and data science, have finally made it feasible to mine the full human genome for infectious disease-altering variants. Genome-wide association studies (GWAS) and deep sequencing analyses of individuals with unusually severe clinical presentations have both been used successfully, and in a highly complementary fashion, to explore the genetic architecture of human susceptibility to infections. GWAS, mostly based on genome-wide genotyping arrays, have identified many associations between common human genetic variants — generally defined as variants with a minor allele frequency of at least 1% — and complex traits or diseases. They have made it possible to identify chromosomal loci associated with the natural course of disease and responses to treatment in populations infected with HIV-1, hepatitis B and C viruses, Plasmodium falciparum, Mycobacterium tuberculosis and M. leprae, for example (25). However, the risk factors identified were no more than modest at individual level.
The recent development of next-generation sequencing technologies, making it possible to identify rare coding variants rapidly at genome-wide scale through whole-exome (WES) or whole-genome sequencing (WGS), has revolutionized the field of human genetics. It has accelerated the discovery of new disease-causing genes and provided molecular insight into the etiology of many IEIs (28, 29). Over the last two decades, we and others have demonstrated that rare monogenic IEIs underlie a growing number of life-threatening viral, bacterial, fungal, and parasitic infections in otherwise healthy individuals with normal resistance to other infectious agents (23, 24, 30, 31). Most of these IEIs are not Mendelian, as they frequently display incomplete penetrance. In addition, they are genetically heterogeneous, with both locus and allelic heterogeneity, with different mutations of several genes underlying the same infectious phenotype, but often united by the same signaling pathway (i.e. physiological homogeneity) (30). For example, Mendelian susceptibility to mycobacterial disease (MSMD) is caused by inborn errors of IFN-γ immunity (32–34), with mutations of 15 genes and 30 allelic forms already reported. Likewise, forebrain herpes simplex encephalitis (HSE) can be caused by IEI of TLR3-dependent type I IFN immunity resulting from mutations of eight genes (35, 36). Since 2001, rare monogenic defects have also been shown to underlie some common infectious diseases in rare patients, as exemplified by the identification of several patients with tuberculosis but no familial history of clinical MSMD carrying causal mutations of MSMD genes (37). These rare IEIs led to the recent discovery of a more common IEI, homozygosity for the P1104A TYK2 allele, underlying about 1% of cases of tuberculosis in populations of European descent (38–40).
Building on these achievements and long-standing international collaborations in human genomics dating back to the Human Genome Project, the HapMap project and the 1000 Genomes Project, the host genetic research community quickly came together in the early weeks of the COVID-19 pandemic, to search the human genome for potential clues relating to viral pathogenesis and host immune responses. Several consortia were created, including the COVID-19 Host Genetic Initiative (HGI, https://www.covid19hg.org) (41) and the Covid Human Genetic Effort (Covid-HGE, https://www.covidhge.com) (42). Existing resources were enhanced and redirected to facilitate the rapid analysis of large numbers of patients and controls. A prime example of this is provided by the GenOMICC study (Genetics Of Mortality In Critical Care, https://genomicc.org), which had been recruiting patients in the United Kingdom since 2015 for the study of emerging infections, sepsis and other forms of life-threatening illness. This study collected clinical data samples from thousands of critically ill COVID-19 patients and generated full genome sequence data in record time. The global effort was not restricted to the academic sector either: several direct-to-consumer companies used their large reservoir of clients (who agreed to be recontacted for research purposes) to perform genome-wide analyses of susceptibility to infection and symptom severity.
2. Human genetics of COVID-19 pneumonia
Numerous human genetic studies have investigated the genetic determinants of COVID-19 pneumonia. As discussed in the corresponding sections, large-scale GWAS approaches, mostly based on genome-wide SNP arrays and single-variant statistics assuming an additive genetic model, led to the identification of multiple common genetic variants associated with a modest increase in the risk of COVID-19 pneumonia. High-throughput DNA sequencing approaches, such as WES or WGS for the identification of rare genetic variants, have also been highly successful, resulting in the discovery of genes and pathways crucial for the control of SARS-CoV-2 infection through rare-variant aggregation tests followed by functional validation.
a. Common variants
Multiple large-scale GWAS have investigated the genetic factors associated with disease severity by comparing COVID-19 pneumonia patients stratified into various subgroups (hospitalized, requiring ventilation, admitted to the ICU, or deceased) with SARS-CoV-2 infected individuals with asymptomatic or mild clinical presentations, or untested individuals from the general population. More than 15 genomic regions harboring common variants (Figure 1) have already been robustly associated with COVID-19 pneumonia (43, 44). The strongest signal — and the very first to be reported, in the spring of 2020 — is that for the 3p21.31 locus, where a Neanderthal haplotype was found to be associated with an OR of 1.8 for severe infection (43, 45–48). The association with this locus has been replicated in several independent cohorts. The frequency of the lead SNP ranges from 1% in East Asians to 9% in Europeans and 23% in South Asians, but there is no evidence of between-origin heterogeneity (43). It has proved challenging to identify the causal genes and variants underlying this association, due to long-range linkage disequilibrium in the region and a high local density of genes with a known or putative role in immunity. In silico functional analyses identified LZTFL1 as the most probable candidate gene (49), but variants of the chemokine receptor genes CCR9, CXCR6 and XCR1 may also contribute to the association signal. Some of the additional loci implicated point to a role for known immune-related genes or genes known to be involved in lung function, as detailed below. Surprisingly, given the very high level of polymorphism of both class I and class II HLA genes and their demonstrated role in modulating multiple infectious diseases, the few associations identified in the HLA region have proved to be weak. It was not until very large meta-analyses were performed relatively late in the course of the pandemic that genome-wide significant association signals were confirmed for polymorphisms of both class I and class II HLA genes (43, 47, 48).
Figure 1. Genetic and immunological determinants of COVID-19 pneumonia.
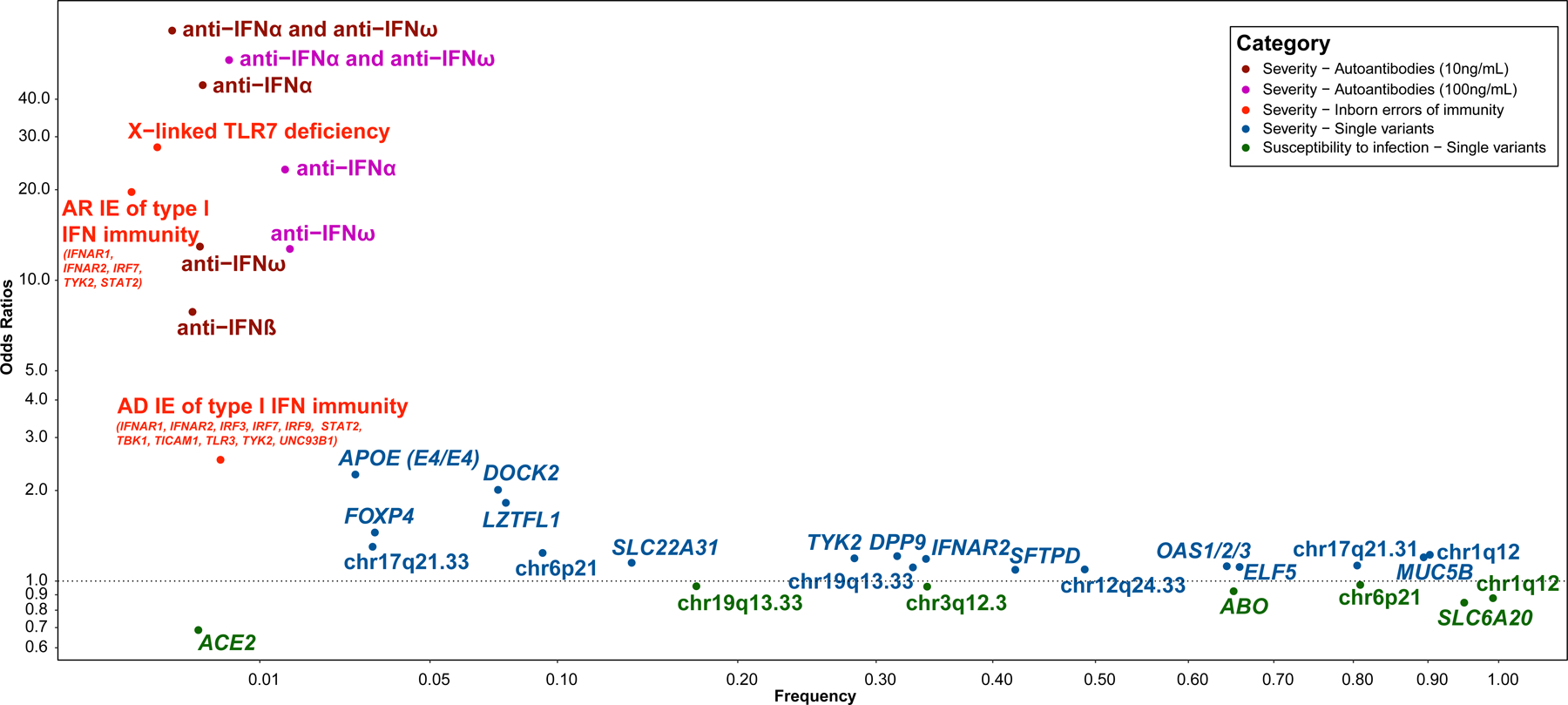
Odds ratios (OR) for the associations of auto-Abs against type I IFN, inborn errors of immunity (IEIs) and single genetic variants with the severity of COVID-19 pneumonia and resistance to SARS-CoV-2 infection are plotted according to risk factor frequency. For auto-Abs against type I IFN, the ORs and frequency were taken from (114). For X-linked TLR7 deficiency, the OR for the aggregated effect of biochemically proven loss of function (LOF) was taken from (74) and the cumulative frequency of biochemically proven LOF was taken from (76). For other IEIs, the OR for the aggregated effect of homozygous (autosomal recessive, AR) or heterozygous (autosomal dominant, AD) predicted LOF was taken from (74) and the corresponding cumulative frequencies were estimated from Gnomad v2.1.1. For single variants, the OR, assuming an additive model, and deleterious allele frequencies were taken from the most recent update of the COVID-19 Host Genetics Initiative GWAS study (43), except for the DOCK2 and APOE loci. The chromosomal region or closest gene is indicated. For the DOCK2 locus, the OR for the effect of the rs60200309-A variant on the severity of COVID-19 pneumonia under an additive model and allele frequency for rs60200309-A were taken from (156). For the APOE locus, the hazard ratio for the effect of APOE4 homozygosity as opposed to APOE3 homozygosity for COVID-19 mortality and the frequency of APOE4 homozygosity were taken from (157).
The loci pointing to a role for known immune-related genes include the IFNAR2 gene, encoding the interferon (IFN) alpha/beta receptor 2, for which associations with variants have been replicated in several studies (47, 50). Type I IFNs play a central role in the innate immune response to SARS-CoV-2, as demonstrated by the marked increase in the risk of life-threatening disease associated with rare loss-of-function variants of interferon-related genes and anti-IFN autoantibodies, as discussed below. It is, therefore, unsurprising that common variants of a subunit of the type I IFN receptor modulate the severity of infection, particularly as the top-ranking associated variants are expression quantitative trait loci (eQTL) for IFNRA2. An association with a TYK2 variant has been identified, which is particularly interesting, given the key role of TYK2 in infection and immunity. The common rs34536443 (p.Pro1104Ala) variant, which is protective against some autoimmune diseases (51) but increases the risk of tuberculosis in homozygous individuals (40), has been shown to be associated with a modest increase in the risk of severe COVID-19 (48). Rare loss-of-function TYK2 variants are discussed below. GWAS have also identified a common haplotype of Neanderthal origin encompassing the OAS1/2/3 genes on chromosome 12 (12q24.13) that is associated with the risk of hospitalization for COVID-19 (47, 48, 52, 53). The OAS proteins are cytosolic type I IFN-inducible antiviral proteins (54, 55). One of the candidate causal variants in this region is the OAS1 splice variant rs10774671. The minor and reference G allele at rs10774671, which provides weak protection against severe forms of COVID-19, encodes a longer and more active form of OAS1 (56).
In their most recent meta-analysis including more than 150,000 cases (43), the COVID-19 HGI reported multiple associations between disease severity and genes involved in normal lung function. First, a regulatory variant (rs35705950:G>T) mapping to the promoter region of MUC5B was found to be associated with a lower risk of hospitalization. The minor T allele is associated with higher levels of MUC5B mRNA and a higher risk of idiopathic pulmonary fibrosis (57). Second, a missense variant of SFTPD (rs721917:A>G, p.Met31Thr) was found to be associated with more severe respiratory symptoms. SFTPD encodes the surfactant protein D, and the same alternative allele was previously shown to have a negative impact on lung function (58) and to increase the risk of chronic obstructive pulmonary disease (59). Third, a missense variant of SLC22A31 (rs117169628:G>A, p.Pro256Leu) was also found to be associated with a higher risk of hospitalization. SLC22A31 encodes a solute carrier protein that is highly expressed in the lung. Finally, a strong association with severe disease was observed with an intronic variant of DPP9 (rs2109069:G>A) previously reported to increase the risk of idiopathic pulmonary fibrosis (60). These variants have very small effect sizes (with odds ratios between 0.8 and 1.2), but their study may improve our understanding of SARS-CoV-2 pathogenicity by shedding light on the underlying molecular mechanisms.
b. Rare variants
Type I IFN influenza susceptibility loci
Based on our discoveries over the last 20 years, we launched the Covid-HGE, an international consortium, with the aim of deciphering the human genetic and immunological basis of the various clinical manifestations of SARS-CoV-2 infection. The first breakthrough emerged from a study testing the hypothesis that candidate inborn errors of TLR3-, IRF7-, and IRF9-dependent type I IFN immunity previously shown to underlie life-threatening influenza pneumonia (2, 23, 24, 30, 31, 61–64) might also underlie critical COVID-19. We considered the three loci (IRF7, IRF9 and TLR3) for which germline mutations are causal for influenza pneumonia (62–64) and 10 other genes (IFNAR1, IFNAR2, IRF3, IKBKG, STAT1, STAT2, TBK1, TICAM1, TRAF3 and UNC93B1) encoding products biochemically and immunologically connected to the three core genes, for which deleterious genotypes have been shown to underlie other severe viral diseases (61). We screened a cohort of 659 patients with critical COVID-19 for rare variants predicted to be loss-of-function (pLOF) at these 13 type I IFN influenza susceptibility loci. We found a significant enrichment in these variants in patients with critical COVID-19 relative to 534 SARS-CoV-2-infected controls who remained asymptomatic or paucisymptomatic, with mild, self-healing, ambulatory disease (P = 0.01) (61). We also found that 23 (3.5%) of the patients with critical COVID-19 carried biochemically deleterious germline mutations of eight of the 13 genes. These patients included four unrelated previously healthy adults aged 25–50 years with autosomal recessive (AR) complete IRF7 or IFNAR1 deficiency. AR IFNAR1, IFNAR2, TBK1, and STAT2 deficiencies were subsequently reported in children with critical COVID-19, and AR TYK2 deficiency was identified in children with COVID-19 pneumonia (65–69).
Several other groups were unable to replicate our findings (44, 70–72). There are several possible reasons for this (73), two of which are particularly important. First, the key epidemiological factor driving COVID-19 severity was ignored: our international cohort was much younger than the other cohorts (mean age of 52 vs. 66 years). As discussed below, these inborn errors are much more frequent in patients under the age of 60 years (Figure 2). Second, the other groups did not test for auto-Abs against type I IFN, the most common determinant of critical COVID-19, especially in patients over 60 years old (discussed in a specific section below). More recently, we confirmed an enrichment in rare pLOF variants at the 13 type I IFN-related influenza susceptibility loci in an extended sample of 3269 patients with critical COVID-19 relative to 1373 controls with asymptomatic or mild infection (P = 2.1×10−4) (74). The addition of TYK2 strengthened the association signal, particularly if a recessive model was assumed. We also found that homozygous carriers of rare pLOF variants had a higher risk of life-threatening COVID-19 than heterozygotes, consistent with the expected higher penetrance of recessive IEIs than of dominant IEIs. In analyses restricted to rare in-frame nonsynonymous variants, we detected no significant enrichment in patients relative to controls. This result was not surprising, as we showed in a previous study (61) that less than 15% of the rare in-frame nonsynonymous variants at the 13 loci were biochemically proven LOF variants (bLOF), whereas all the pLOF variants were found to be bLOF variants. A similar trend was observed for other immune system genes, even when advanced in silico scoring systems, such as CADD score, were used to stratify the variants (39, 75–77). The study of in-frame nonsynonymous variants will therefore require the experimental characterization of all these variants.
Figure 2. Proportion of critical COVID-19 patients with IEIs and auto-Abs against type I IFN as a function of age.

Age-specific proportions of dominant and recessive IEIs among cases of critical COVID-19 pneumonia were estimated with Bayes theorem as a function of the probability of critical COVID-19 pneumonia for IEI carriers infected with SARS-CoV-2 (i.e. the penetrance). The age-specific frequency of IEIs in the general population and the age-specific critical infection rate were taken from (158). The age-specific frequency of IEIs in the general population was estimated from the frequency of IEIs at birth, assuming a non-specific mortality rate (i.e. not attributable to COVID-19) of 0 (panel A) or 1% (panel B) per year. Based on our previous findings (67, 159, 160), the frequency of IEIs at birth was set at 10−3 for dominant and 5 × 10−4 for recessive IEIs. We assumed a penetrance of 0.2 for dominant IEIs and 0.8 for recessive IEIs, consistent with the larger effect size estimated for recessive than for dominant IEIs. The age-specific proportions of patients with critical COVID-19 producing auto-Abs neutralizing low doses (100 pg/mL; gray line) or high doses (10 ng/mL; black line) of IFN-α and/or IFN-ω were taken from (114).
Unbiased screening for chromosome- and genome-wide rare variant burden
We hypothesized that the higher risk of critical COVID-19 in men than in women might be explained by X-linked disorders. We conducted an unbiased X chromosome-wide gene burden test on a cohort of 1,202 unrelated, autoantibody-negative, male patients with critical COVID-19 pneumonia and 331 men with asymptomatic or mild COVID-19. The most strongly associated gene, and the only gene remaining significant after accounting for the number of genes tested, was TLR7, with 21 unrelated patients carrying a very rare hemizygous nonsynonymous variant that was completely absent from controls (P = 3.5 × 10−5) (76). Sixteen of these 21 patients carried a TLR7 allele that was loss-of-function or hypomorphic. Biochemically proven TLR7 deficiency was identified in four additional male patients with critical COVID-19 (74), and four male patients with severe COVID-19 (67, 76) from the Covid-HGE cohort. Moreover, we confirmed the proposed diagnosis of TLR7 deficiency in nine of other 16 reported male patients (65, 78–82), with our biochemical assay (76). An enrichment in rare nonsynonymous TLR7 variants in patients with critical COVID-19 was reported in another two studies, but the variants were not disclosed and the diagnosis of TLR7 deficiency remains to be confirmed, especially in the women (70, 71). Overall, TLR7 deficiency was found to account for about 1% of cases of critical COVID-19 in men (74, 76). The penetrance of TLR7 deficiency for severe or critical COVID-19 among relatives of index cases was high, but incomplete, especially in children. Human TLR7 is an endosomal receptor of ribonucleic acids expressed by B cells and myeloid subsets. Its stimulation in plasmacytoid dendritic cells (pDCs) results in the production of large amounts of type I IFN (76). We showed that blood B-cell lines and myeloid cell subsets from patients with TLR7 deficiency did not respond to TLR7 stimulation and that the patients’ pDCs produced low levels of type I IFNs in response to SARS-CoV-2, further highlighting the essential role of type I IFN for protection against SARS-CoV-2 (Figure 3) (76).
Figure 3. Impaired type I IFN immunity underlies life-threatening COVID-19.
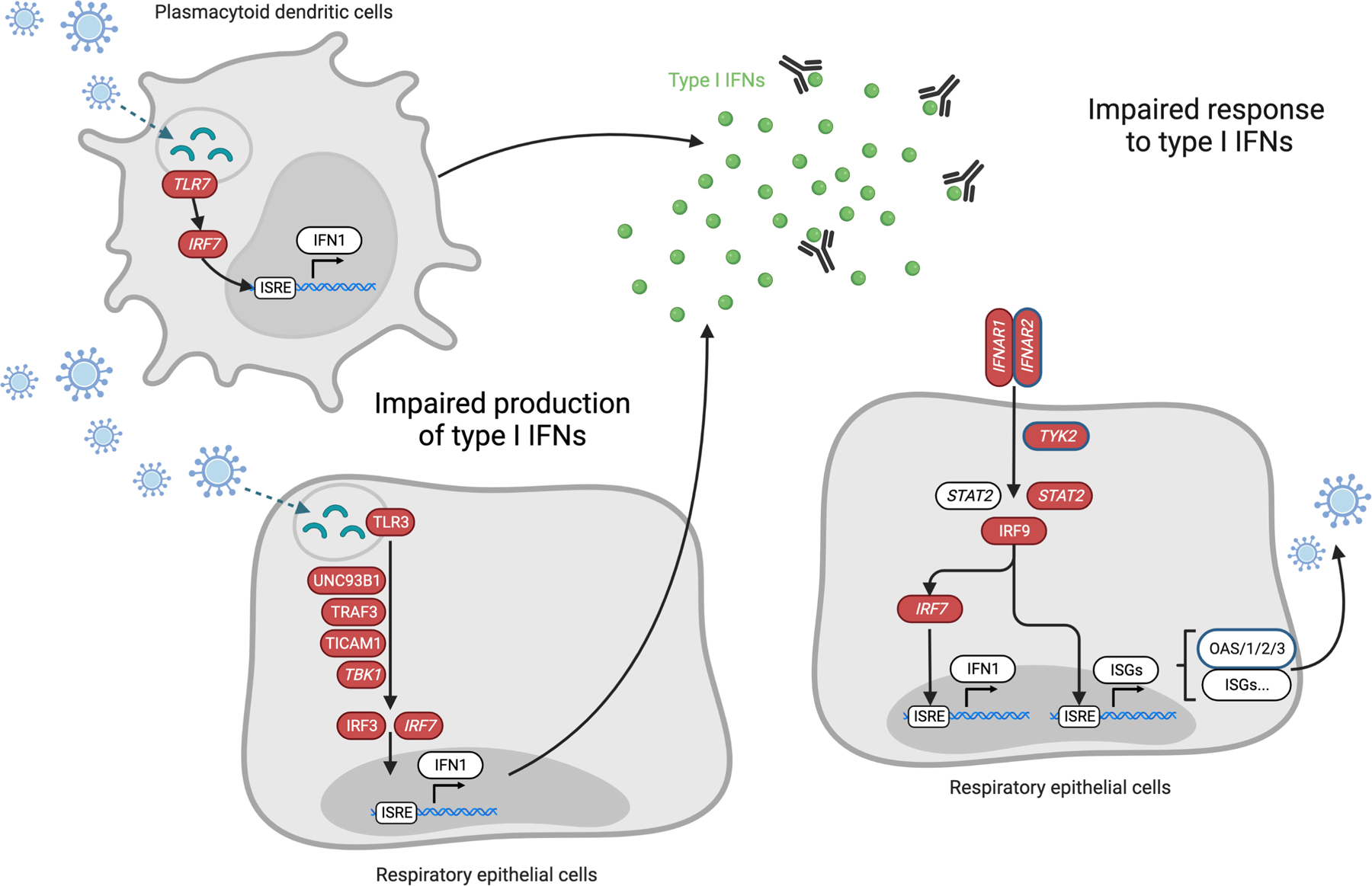
Nearly 20% of patients with life-threatening COVID-19 pneumonia have impaired production of or response to type I interferons (IFNs), due to inborn errors of immunity (IEIs, 1 to 5%) or to blockade of type I IFN activity by pre-existing neutralizing autoantibodies (~15%). IEI of type I IFN immunity (genes shown in red) have been identified and lead to: (1) an impaired production of type I IFNs (left) in respiratory epithelial cells (RECs) and/or in blood plasmacytoid dendritic cells (pDCs), or (2) an impaired response to type I IFNs (right) in RECs, in response to SARS-CoV-2 infection. Genes for which X-linked or autosomal recessive defects have been identified are in italic. Genes for which common variants have been associated by GWAS with severe COVID-19 are circled in blue.
Several large-scale sequencing studies have attempted to identify new genetic causes of severe COVID-19 pneumonia through unbiased genome-wide gene burden analyses (44, 70, 71, 74). None of the genes considered remained statistically significant after stringent correction for the number of genes and phenotypes tested. In addition to TLR7, for which association has consistently been reported across studies (70, 71, 74, 76, 78), two genes reached the less conservative exome-wide significance threshold of 2.5×10−6 in one study focusing on 5,085 individuals with critical COVID-19 and 571,737 controls, mostly uninfected, from the general population (70): MARK1 (P = 1.9×10−6) and RILPL1 (P = 2.4×10−6), with cases displaying an enrichment in pLOF variants. Nevertheless, these results require further investigation. It should be stressed that stringent correction for multiple testing, while necessary to avoid false positives, is a conservative strategy, and that a lack of formal statistical significance at genome-wide level does not exclude biological causality and medical significance. The burden of proof can be provided experimentally via biochemical, virological, and immunological experiments, as we previously did for TLR7 (76). Additional genes may be found by restricting the association analysis to variants proved experimentally to be deleterious.
c. Age-dependent genetic architecture
Inborn errors of type I IFN immunity are more frequent in younger patients (those under the age of 60 years) (61, 74, 76), an observation consistent with IEIs being generally more common in children (26, 83). Eighteen of the 23 patients (78%) first reported to carry biochemically deleterious germline mutations at eight type I IFN-related influenza susceptibility loci were under 60 years old (61). TLR7 deficiency was found to account for 1% of cases of critical COVID-19 in men and about 1.8% of cases of critical COVID-19 in male patients below the age of 60 years (76). In an extended sample of 3269 patients with critical COVID-19, we identified 57 patients carrying a rare predicted LOF variant at 14 type I IFN-related influenza susceptibility loci (including TYK2) or with biochemically proven TLR7 deficiency (74). These patients were significantly younger than the rest of the cohort of patients with critical COVID-19 (43.3 vs. 56 years old, p = 1.7×10−5) (74). Consistent with these results, we recently reported 12 children (~10%) from an international cohort of 112 pediatric patients hospitalized for COVID-19 pneumonia with biochemically complete recessive inborn errors of type I IFN immunity, including seven children with X-linked recessive TLR7 deficiency and five children with autosomal recessive IFNAR1, STAT2, or TYK2 deficiency (67). Interestingly, the effect of the major common genetic risk factor for severe COVID-19 pneumonia on chromosome 3 was also found to be more pronounced in individuals under the age of 60 years than in those over 60 years of age (odds of death or severe respiratory failure of 2.7 vs. 1.5; P interaction = 0.038) (84). A greater heritability of common SNPs has also been reported in patients under the age of 60 years than in those over 60 years of age (85).
Stronger genetic effects in young patients may partly reflect the greater contribution of other risk factors in the elderly, such as comorbid conditions and auto-antibodies against type I IFNs (which account for ~15% of critical cases in elderly patients, discussed in a specific section below), which become more frequent with increasing age. At the cellular level, aging is also associated with immunosenescence, which may contribute to a defective innate and adaptive response to SARS-CoV-2 infection, thereby conferring a non-specific predisposition to severe COVID-19 (86). At the molecular level, global type I IFN immunity in the blood (plasmacytoid dendritic cells) and respiratory tract (respiratory epithelial cells) has been shown to decline with age (87–90). The frequency of IEIs may also decline with age in the general population, because IEIs can underlie fatal illness due to influenza or other viruses, resulting in the premature death of affected individuals (2). Cohorts consisting mostly of patients are over the age of 60 years would, therefore, have a very low power to identify rare inborn errors, as illustrated in Figure 2, which shows the proportion of critical COVID-19 cases expected to be due to IEI as a function of age. Assuming a frequency of 10−3 for dominant IEIs and 5×10−4 for recessive IEIs and a penetrance for critical COVID-19 pneumonia of 0.2 and 0.8, respectively, the expected proportion of critical COVID-19 cases due to IEIs would be expected to decrease strongly with age, from more than 15% below the age of 30 years of age to less than 1% after the age of 60 years.
3. Human genetics of susceptibility to SARS-CoV-2 infection
It has been suggested that a fraction of the human population may possess intrinsic resistance to SARS-CoV-2 infection, but this remains unproven (91). Epidemiological observations suggest that some exposed individuals may indeed be resistant; in particular, there have been reports of highly exposed individuals remaining uninfected in the healthcare setting (92), and reports of households in which everyone except one of the spouses became infected (93). Genetic resistance to infection with specific pathogens is rare. The only validated examples in humans are autosomal recessive resistance to: [A] Plasmodium vivax, linked to a regulatory variant that modifies the GATA-1 binding site in the DARC promoter, thereby selectively preventing the expression of the Duffy antigen on red blood cells (94); [B] HIV-1, conferred by a 32 bp deletion in CCR5, the gene encoding the main HIV-1 coreceptor on CD4+ T cells (95–97); and [C] norovirus, due to FUT2 deficiency (non-secretor phenotype), which prevents the binding of the norovirus VPg capsid to FUT2 (98).
No highly penetrant protective variant against SARS-CoV-2 infection has yet been reported. However, GWAS has identified a few genetic factors as associated with a lower likelihood of infection at population level (Figure 1). One of the first genomic regions to be identified in COVID-19 host genetic studies was the ABO locus on chromosome 9 (45). A highly significant association with susceptibility to infection was later observed at the same locus for the SNP rs912805253 (OR ~ 0.9), both in the initial COVID-19 HGI meta-analysis (48) and in a large study by the direct-to-consumer genetics company 23andMe (99). Interestingly, a systematic review confirmed that the ABO association is mostly restricted to susceptibility to infection, with blood group O associated with a significantly lower susceptibility to infection than non-O blood groups (OR = 0.9), whereas most of the other reported genetic regions are associated with disease severity (100). The precise mechanism by which ABO blood status influences SARS-CoV-2 susceptibility remains unclear, but blood antigens are known to alter individual susceptibility to multiple pathogens (101), including other coronaviruses (102).
The angiotensin-converting enzyme 2 (ACE2) protein acts as a functional receptor for the spike glycoprotein of SARS-CoV-2 and other coronaviruses. ACE2 variants were thus suspected to play a role in modulating infectiousness. However, this gene is under strong negative selection. Putative functional variants are, therefore, rare and a very large number of study participants (>50,000 COVID-19 cases and >700,000 controls) were required to identify a relatively rare variant associated with protection against SARS-CoV-2 infection (rs190509934:T>C, frequency of the minor C allele: 0.3%, OR for the additive effect of each copy of the minor C allele = 0.69) (50). This variant, which is located 69 bp upstream from ACE2, is an eQTL for the gene: the C allele is associated with lower levels of mRNA, probably accounting for the lower level of susceptibility to infection. This association was replicated in the most recent HGI meta-analysis (43). Five additional loci were identified by GWAS as being more likely to be associated with susceptibility to SARS-CoV-2 infection than with disease severity (43, 103), but the effect sizes were, again, very modest (Figure 1) and the mechanisms involved remained mostly undefined. No inborn variant conferring strong resistance to SARS-CoV-2 infection has yet been identified. Human genetic studies of resistance to infection may benefit from large-scale host-viral interactome studies (61) and genome-wide CRISPR knockout (104–112) and activation (104, 112) screens, which can identify candidate genes influencing the viral life cycle. Such studies will require a specific strategy, particularly for the reliable identification of highly exposed subjects potentially resistant to infection (113).
4. Downstream implications of the genetic findings
Biological insight from genetic discoveries: auto-Abs neutralizing type I IFNs
The identification of type I IFN-related IEIs led to the almost simultaneous major discovery that pre-existing auto-Abs neutralizing type I IFNs account for about 15% of critical COVID-19 cases (114, 115). While searching for type I IFN-related IEIs in patients with critical COVID-19 pneumonia, we also hypothesized that autoimmune phenocopies of these IEIs might underlie critical COVID-19. Autoimmune phenocopies of IEIs of cytokines have already been described, in which patients with the same, or a similar infectious phenotype produce auto-Abs neutralizing the corresponding cytokines. Auto-Abs against cytokines have already been shown to underlie mycobacterial disease (type II IFN), mucocutaneous candidiasis (IL-17A/F), nocardiosis (GM-CSF), and staphylococcal disease (IL6) (116, 117). Auto-Abs neutralizing type I IFNs were known to occur in some patients receiving IFN therapy, and in patients with systemic lupus erythematosus, myasthenia gravis, thymoma, or autoimmune polyendocrine syndrome (APS-1) caused by germline mutations of AIRE, but they were not thought to confer a predisposition to viral diseases (2, 118). We first reported the presence of auto-Abs neutralizing high, supraphysiological concentrations (10 ng/mL, with plasma diluted 1/10) of IFN α2 and/or IFN ω in about 10% of 987 patients with critical COVID-19 pneumonia, but not in 663 individuals with asymptomatic or mild infection (115). This finding has been largely replicated worldwide in many studies (76, 119–134). We later detected auto-Abs neutralizing lower, more physiological concentrations (100 pg/mL, with plasma diluted 1/10) of IFN α2 and/or IFN ω in 13.6% of 3595 patients with life-threatening COVID 19 (114). This proportion increased in patients older than 65 years, reaching more than 20% in patients over 80 years old, and was greater in men than in women. Another 1% of patients with critical COVID-19 had auto-Abs neutralizing high concentrations of IFN-β.
Several lines of evidence strongly suggest that autoimmunity to type I IFN plays a causal role in life-threatening COVID-19 (135). For patients for whom plasma sampled before the pandemic was available, the auto-Abs were found to be present before SARS-CoV-2 infection (115). Patients with autoimmune polyendocrine syndrome type-1 (APS-1), who produce such auto-Abs from early childhood, were shown to be at very high risk of developing severe or critical COVID-19 pneumonia, especially after the age of 20 years (136). These auto-Abs neutralize the antiviral activity of type I IFNs against SARS-CoV-2 in vitro (115) and are found in vivo in the blood and in the respiratory tract of patients (123, 137, 138). Remarkably, these auto-Abs were also found in samples from a fraction of the general population before the pandemic. Their prevalence in the general population remains fairly stable until the age of 70 years (at ~1% for auto-Abs neutralizing low doses of IFN α2 and/or IFN ω), but sharply increases thereafter (reaching up to 6.3% after the age of 80 years) (114). Auto-Abs against type I IFNs strongly increase COVID-19 infection fatality rates (IFRs) in unvaccinated populations, especially those neutralizing both IFN-α2 and –ω (135). Screening for auto-Abs against type I IFN in patients infected with SARS-CoV-2, and even in uninfected individuals, is feasible and may be warranted. Individuals carrying such antibodies should be given high priority for vaccination against COVID-19. They may also benefit from specific care, such as the administration of monoclonal antibodies neutralizing the virus or early recombinant IFN-β therapy (139). The exact level of protection against severe COVID-19 pneumonia provided by COVID-19 vaccines in carriers of auto-Abs remains unclear. However, we recently detected auto-Abs against type I IFNs in 24% (10 of 48 tested) of fully vaccinated patients with normal antibody responses who developed critical breakthrough COVID-19 (140), suggesting that at least some of the carriers of these auto-Abs may not be fully protected by the vaccine. The same auto-Abs were subsequently shown to underlie other severe infectious manifestations, such as severe adverse reactions to yellow fever live attenuated viral vaccine (33) and critical influenza pneumonia (141). In critically ill COVID-19 patients, auto-Abs were also shown to increase the risk of herpesvirus reactivation, which has been associated with a poorer clinical outcome (142, 143).
Auto-Abs against type I IFNs can also be genetically driven and few IEIs are already known to underlie their production. The most striking example is the production of these auto-Abs from early childhood in nearly all patients with APS-1 due to germline deleterious variants of AIRE (4). They have also been reported in patients with immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX) due to deleterious variants of FOXP3, and combined immunodeficiency due to biallelic hypomorphic RAG1 or RAG2 variants (37–42). A feature common to these IEIs is that they affect T-cell tolerance. Interestingly, among the patients with auto-Abs against type I IFN and life-threatening COVID-19, we identified a woman with X-linked incontinentia pigmenti (IP), in which cells activate the same single X chromosome (cells having activated the X chromosome bearing the null mutation of NEMO dying during development) (27). A further study of 32 women with IP showed that 25% carried auto-Abs against type I IFNs, suggesting an X-linked germline genetic etiology for type I IFN auto-Abs (115). New IEIs underlying the production of auto-Abs against type I IFN are likely to be discovered in the future, particularly in young patients. It is also tempting to speculate that somatic mutations, which accumulate with aging in normal human tissues (144), may be partly responsible for the sharp increase in the prevalence of auto-Abs against type I IFNs after the age of 70 years.
Clinical implications: towards personalized medicine in infectious diseases
Deciphering the genetic architecture of susceptibility to SARS-CoV-2 infection and severe COVID-19 pneumonia is only the first step toward clinical implementation and improved healthcare. How can we translate the knowledge gained in the genomic screens described above into medically useful strategies? The first and most obvious answer to this question is through the identification of individuals at high risk. The rare deleterious variants of TLR7 and TLR3-dependent type I IFN immunity genes identified confer a massive increase in the risk of severe disease and account for a significant proportion of cases (e.g. 1% of men with critical disease carry a deleterious TLR7 variant). One could imagine screening programs, at population level, primary care centers or emergency rooms (at the time of early COVID-19 diagnosis), providing point-of-care testing for such variants (145). Carriers would then benefit from specific preventive and therapeutic measures. In particular, type I IFN might be useful in patients prone to severe COVID-19 and infected with the virus, provided it is administered at an early stage of infection. Along the same lines, the discovery that auto-antibodies against type I IFN account for 20% of COVID-19 deaths have clinical implications. The detection of such auto-Abs before or during early stages of infection is straightforward and warranted. Carriers of these auto-Abs should be vaccinated and given priority for booster injections and may benefit from specific treatments, such as IFN-β, mAbs neutralizing SARS-CoV-2, or plasma exchange (146, 147).
A second way forward in the clinical translation of these findings would be the development of polygenic risk scores (PRS), constructed by summing the effects of all genetic variants confirmed to be associated with a phenotype of interest. In recent years, PRS have been shown to have a high predictive value for a range of complex diseases, including cardiovascular, metabolic, and tumoral disorders (148). A few studies have attempted to build PRS for COVID-19, but with little success. Indeed, one of the main factors determining the predictive ability of PRS is the fraction of the phenotypic variance explained by the combination of selected variants, which remains low in COVID-19 host genetic studies of common variants (50, 84, 149). Nevertheless, some private companies already offer polygenic risk prediction for severe COVID-19. However, current tests have low discriminatory power at the individual level and variable accuracy depending on ancestry, making their clinical use questionable (150). Nevertheless, the assessment of patient risk based on a combination of demographic, clinical and genetic data has the potential to deliver more precise information that could prove useful for personalized health management.
5. Conclusion
The SARS-CoV-2 pandemic has highlighted the vast potential of human genomics research when it is performed at a large scale, in real time, and in a highly collaborative manner. One of its greatest successes has been the identification of a molecular explanation for about 20% of cases of critical COVID-19 pneumonia: inborn errors of type I IFN immunity in 1–5% of cases and auto-Abs against type I IFNs in 15–20% of cases. Other IEIs, related or unrelated to type I IFN, may also be involved. Future studies should build on the observations of the impact of SARS-CoV2 infection in individuals with previously known IEIs (151). Remarkably, common genetic variants with modest effect sizes were also identified in regions encompassing genes involved in type I IFN immunity. It is tempting to speculate that they may act as modifiers of the clinical expression of IEIs, which display high but incomplete penetrance. Most human genomics research studies have focused on COVID-19 pneumonia, with fewer considering resistance to SARS-CoV-2 infection. Interesting results are starting to emerge for MIS-C and already suggest a pathogenesis different from that of COVID-19 pneumonia (152–154). Future research studies should also encompass other COVID-19-related clinical manifestations, such as long-COVID (13) and COVID-toes (12), as well as severe adverse effects of vaccination (155) and breakthrough infections (140).
The lessons learned should be used to improve our collective capacity to confront other infectious threats. In particular, COVID-19 has illustrated the central importance of large-scale research infrastructures embedded in healthcare systems, facilitating the rapid collection and analysis of samples in times of crisis. It has also become clear that it is crucial to define disease outcomes clearly and to gather as much clinical data as possible to minimize patient misclassification, thereby maximizing statistical power to detect true genetic signals. Ultimately, a better understanding of the impact of human genetic variation on pathogen response will enable health systems to provide appropriate care to protect individuals and populations more efficiently against future infectious threats.
Acknowledgments
The Laboratory of Human Genetics of Infectious Diseases is supported by the Howard Hughes Medical Institute, The Rockefeller University, the St. Giles Foundation, the National Institutes of Health (NIH) (P01AI061093, R01AI088364, R01AI095983, R01AI127564, R01AI143810, R01AI163029, R01NS072381, R21AI137371, and U19AI162568), the National Center for Advancing Translational Sciences (NCATS), NIH Clinical and Translational Science Award (CTSA) program (UL1TR001866), the Fisher Center for Alzheimer’s Research Foundation, the Meyer Foundation, the JPB Foundation, the Robertson Therapeutic Development Fund, the Tri-Institutional Stem Cell Initiative fund (Tri-SCI), the French National Research Agency (ANR) under the “Investments for the Future” program (ANR-10-IAHU-01), the Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID), the French Foundation for Medical Research (FRM) (EQU201903007798), the French Ministry of Higher Education, Research, and Innovation (MESRI-COVID-19), the FRM and ANR GENCOVID project (ANR-20-COVI-0003), ANRS Nord-Sud (ANRS-COV05), ANR grants SEAeHostFactors (ANR-18-CE15-0020-02), LTh-MSMD-CMCD (ANR-18-CE93-0008), CNSVIRGEN (ANR-19-CE15-0009-01), GENVIR (ANR-20-CE93-003), GENMSMD (ANR-16-C17-005), AABIFNCOV (ANR-20-CO11-0001), and GenMIS-C (ANR-21-COVR-0039), the ANR-RHU program (ANR-21-RHUS-08), the European Union’s Horizon 2020 research and innovation program under grant agreement no. 824110 (EASI-genomics), the HORIZON-HLTH-2021-DISEASE-04 program under grant agreement 01057100 (UNDINE), the Square Foundation, Grandir - Fonds de solidarité pour l’enfance, the SCOR Corporate Foundation for Science, Fondation du Souffle, Institut National de la Santé et de la Recherche Médicale (INSERM), REACTing-INSERM, and the University Paris Cité. JF’s work on COVID is supported by the Ecole Polytechnique Fédérale de Lausanne (EPFL), the CHUV Foundation, and the Swiss National Science Foundation (grant #310030L_197721).
References
- 1.Dong E, Du H, Gardner L. 2020. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis 20: 533–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Q, Bastard P, Effort CHG, Cobat A, Casanova JL. 2022. Human genetic and immunological determinants of critical COVID-19 pneumonia. Nature 603: 587–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Driscoll M, Ribeiro Dos Santos G, Wang L, Cummings DAT, Azman AS, et al. 2021. Age-specific mortality and immunity patterns of SARS-CoV-2. Nature 590: 140–45 [DOI] [PubMed] [Google Scholar]
- 4.Team C-F. 2022. Variation in the COVID-19 infection-fatality ratio by age, time, and geography during the pre-vaccine era: a systematic analysis. Lancet 399: 1469–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bennett TD, Moffitt RA, Hajagos JG, Amor B, Anand A, et al. 2021. Clinical Characterization and Prediction of Clinical Severity of SARS-CoV-2 Infection Among US Adults Using Data From the US National COVID Cohort Collaborative. JAMA Netw Open 4: e2116901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takahashi T, Ellingson MK, Wong P, Israelow B, Lucas C, et al. 2020. Sex differences in immune responses that underlie COVID-19 disease outcomes. Nature 588: 315–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Navaratnam AV, Gray WK, Day J, Wendon J, Briggs TWR. 2021. Patient factors and temporal trends associated with COVID-19 in-hospital mortality in England: an observational study using administrative data. The Lancet Respiratory Medicine 9: 397–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ricoca Peixoto V, Vieira A, Aguiar P, Sousa P, Carvalho C, et al. 2021. Determinants for hospitalisations, intensive care unit admission and death among 20,293 reported COVID-19 cases in Portugal, March to April 2020. Euro Surveill 26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Williamson EJ, Walker AJ, Bhaskaran K, Bacon S, Bates C, et al. 2020. Factors associated with COVID-19-related death using OpenSAFELY. Nature 584: 430–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sancho-Shimizu V, Brodin P, Cobat A, Biggs CM, Toubiana J, et al. 2021. SARS-CoV-2-related MIS-C: A key to the viral and genetic causes of Kawasaki disease? J Exp Med 218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahmad F, Ahmed A, Rajendraprasad SS, Loranger A, Gupta S, et al. 2021. Multisystem inflammatory syndrome in adults: A rare sequela of SARS-CoV-2 infection. Int J Infect Dis 108: 209–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arkin LM, Moon JJ, Tran JM, Asgari S, O’Farrelly C, et al. 2021. From Your Nose to Your Toes: A Review of Severe Acute Respiratory Syndrome Coronavirus 2 PandemicAssociated Pernio. J Invest Dermatol 141: 2791–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brodin P, Casari G, Townsend L, O’Farrelly C, Tancevski I, et al. 2022. Studying severe long COVID to understand post-infectious disorders beyond COVID-19. Nat Med 28: 879–82 [DOI] [PubMed] [Google Scholar]
- 14.Moghadas SM, Vilches TN, Zhang K, Wells CR, Shoukat A, et al. 2021. The Impact of Vaccination on Coronavirus Disease 2019 (COVID-19) Outbreaks in the United States. Clin Infect Dis 73: 2257–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davies NG, Jarvis CI, Group CC-W, Edmunds WJ, Jewell NP, et al. 2021. Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7. Nature 593: 270–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nyberg T, Twohig KA, Harris RJ, Seaman SR, Flannagan J, et al. 2021. Risk of hospital admission for patients with SARS-CoV-2 variant B.1.1.7: cohort analysis. BMJ 373: n1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patone M, Thomas K, Hatch R, Tan PS, Coupland C, et al. 2021. Mortality and critical care unit admission associated with the SARS-CoV-2 lineage B.1.1.7 in England: an observational cohort study. Lancet Infect Dis 21: 1518–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Twohig KA, Nyberg T, Zaidi A, Thelwall S, Sinnathamby MA, et al. 2022. Hospital admission and emergency care attendance risk for SARS-CoV-2 delta (B.1.617.2) compared with alpha (B.1.1.7) variants of concern: a cohort study. Lancet Infect Dis 22: 35–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolter N, Jassat W, Walaza S, Welch R, Moultrie H, et al. 2022. Early assessment of the clinical severity of the SARS-CoV-2 omicron variant in South Africa: a data linkage study. Lancet 399: 437–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harvey WT, Carabelli AM, Jackson B, Gupta RK, Thomson EC, et al. 2021. SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol 19: 409–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuhlmann C, Mayer CK, Claassen M, Maponga T, Burgers WA, et al. 2022. Breakthrough infections with SARS-CoV-2 omicron despite mRNA vaccine booster dose. Lancet 399: 625–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bergwerk M, Gonen T, Lustig Y, Amit S, Lipsitch M, et al. 2021. Covid-19 Breakthrough Infections in Vaccinated Health Care Workers. N Engl J Med 385: 1474–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Casanova JL, Abel L. 2022. From rare disorders of immunity to common determinants of infection: Following the mechanistic thread. Cell 185: 3086–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Casanova JL, Abel L. 2021. Mechanisms of viral inflammation and disease in humans. Science 374: 1080–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gibbs KD, Schott BH, Ko DC. 2022. The Awesome Power of Human Genetics of Infectious Disease. Annu Rev Genet [DOI] [PubMed] [Google Scholar]
- 26.Alcais A, Quintana-Murci L, Thaler DS, Schurr E, Abel L, Casanova JL. 2010. Life-threatening infectious diseases of childhood: single-gene inborn errors of immunity? Ann N Y Acad Sci 1214: 18–33 [DOI] [PubMed] [Google Scholar]
- 27.Kwiatkowski DP. 2005. How malaria has affected the human genome and what human genetics can teach us about malaria. Am J Hum Genet 77: 171–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meyts I, Bosch B, Bolze A, Boisson B, Itan Y, et al. 2016. Exome and genome sequencing for inborn errors of immunity. J Allergy Clin Immunol 138: 957–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, et al. 2022. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Casanova JL, Abel L. 2020. The human genetic determinism of life-threatening infectious diseases: genetic heterogeneity and physiological homogeneity? Hum Genet 139: 681–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Casanova JL, Abel L. 2021. Lethal Infectious Diseases as Inborn Errors of Immunity: Toward a Synthesis of the Germ and Genetic Theories. Annu Rev Pathol 16: 23–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinez-Barricarte R, Markle JG, Ma CS, Deenick EK, Ramirez-Alejo N, et al. 2018. Human IFN-gamma immunity to mycobacteria is governed by both IL-12 and IL-23. Sci Immunol 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bustamante J 2020. Mendelian susceptibility to mycobacterial disease: recent discoveries. Hum Genet 139: 993–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosain J, Kong XF, Martinez-Barricarte R, Oleaga-Quintas C, Ramirez-Alejo N, et al. 2019. Mendelian susceptibility to mycobacterial disease: 2014–2018 update. Immunol Cell Biol 97: 360–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang SY. 2020. Herpes simplex virus encephalitis of childhood: inborn errors of central nervous system cell-intrinsic immunity. Hum Genet 139: 911–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bastard P, Manry J, Chen J, Rosain J, Seeleuthner Y, et al. 2021. Herpes simplex encephalitis in a patient with a distinctive form of inherited IFNAR1 deficiency. J Clin Invest 131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boisson-Dupuis S 2020. The monogenic basis of human tuberculosis. Hum Genet 139: 1001–09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boisson-Dupuis S, Ramirez-Alejo N, Li Z, Patin E, Rao G, et al. 2018. Tuberculosis and impaired IL-23-dependent IFN-gamma immunity in humans homozygous for a common TYK2 missense variant. Sci Immunol 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kerner G, Laval G, Patin E, Boisson-Dupuis S, Abel L, et al. 2021. Human ancient DNA analyses reveal the high burden of tuberculosis in Europeans over the last 2,000 years. Am J Hum Genet 108: 517–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kerner G, Ramirez-Alejo N, Seeleuthner Y, Yang R, Ogishi M, et al. 2019. Homozygosity for TYK2 P1104A underlies tuberculosis in about 1% of patients in a cohort of European ancestry. Proc Natl Acad Sci U S A 116: 10430–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Initiative C-HG. 2020. The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur J Hum Genet 28: 715–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Casanova JL, Su HC, Effort CHG. 2020. A Global Effort to Define the Human Genetics of Protective Immunity to SARS-CoV-2 Infection. Cell 181: 1194–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Initiative C-HG. 2022. A first update on mapping the human genetic architecture of COVID-19. Nature 608: E1–E10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kousathanas A, Pairo-Castineira E, Rawlik K, Stuckey A, Odhams CA, et al. 2022. Whole-genome sequencing reveals host factors underlying critical COVID-19. Nature 607: 97–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Severe Covid GG, Ellinghaus D, Degenhardt F, Bujanda L, Buti M, et al. 2020. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N Engl J Med 383: 1522–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zeberg H, Paabo S. 2020. The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature 587: 610–12 [DOI] [PubMed] [Google Scholar]
- 47.Pairo-Castineira E, Clohisey S, Klaric L, Bretherick AD, Rawlik K, et al. 2021. Genetic mechanisms of critical illness in COVID-19. Nature 591: 92–98 [DOI] [PubMed] [Google Scholar]
- 48.Initiative C-HG. 2021. Mapping the human genetic architecture of COVID-19. Nature 600: 472–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Downes DJ, Cross AR, Hua P, Roberts N, Schwessinger R, et al. 2021. Identification of LZTFL1 as a candidate effector gene at a COVID-19 risk locus. Nat Genet 53: 1606–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Horowitz JE, Kosmicki JA, Damask A, Sharma D, Roberts GHL, et al. 2022. Genome-wide analysis provides genetic evidence that ACE2 influences COVID-19 risk and yields risk scores associated with severe disease. Nat Genet 54: 382–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dendrou CA, Cortes A, Shipman L, Evans HG, Attfield KE, et al. 2016. Resolving TYK2 locus genotype-to-phenotype differences in autoimmunity. Sci Transl Med 8: 363ra149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huffman JE, Butler-Laporte G, Khan A, Pairo-Castineira E, Drivas TG, et al. 2022. Multi-ancestry fine mapping implicates OAS1 splicing in risk of severe COVID-19. Nat Genet 54: 125–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zeberg H, Paabo S. 2021. A genomic region associated with protection against severe COVID-19 is inherited from Neandertals. Proc Natl Acad Sci U S A 118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dong B, Xu L, Zhou A, Hassel BA, Lee X, et al. 1994. Intrinsic molecular activities of the interferon-induced 2–5A-dependent RNase. J Biol Chem 269: 14153–8 [PubMed] [Google Scholar]
- 55.Schwartz SL, Conn GL. 2019. RNA regulation of the antiviral protein 2’−5’-oligoadenylate synthetase. Wiley Interdiscip Rev RNA 10: e1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bonnevie-Nielsen V, Field LL, Lu S, Zheng DJ, Li M, et al. 2005. Variation in antiviral 2’,5’-oligoadenylate synthetase (2’5’AS) enzyme activity is controlled by a single-nucleotide polymorphism at a splice-acceptor site in the OAS1 gene. Am J Hum Genet 76: 623–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fadista J, Kraven LM, Karjalainen J, Andrews SJ, Geller F, et al. 2021. Shared genetic etiology between idiopathic pulmonary fibrosis and COVID-19 severity. EBioMedicine 65: 103277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shrine N, Guyatt AL, Erzurumluoglu AM, Jackson VE, Hobbs BD, et al. 2019. New genetic signals for lung function highlight pathways and chronic obstructive pulmonary disease associations across multiple ancestries. Nat Genet 51: 481–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hobbs BD, de Jong K, Lamontagne M, Bosse Y, Shrine N, et al. 2017. Genetic loci associated with chronic obstructive pulmonary disease overlap with loci for lung function and pulmonary fibrosis. Nat Genet 49: 426–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fingerlin TE, Murphy E, Zhang W, Peljto AL, Brown KK, et al. 2013. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet 45: 613–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gordon DE, Hiatt J, Bouhaddou M, Rezelj VV, Ulferts S, et al. 2020. Comparative host-coronavirus protein interaction networks reveal pan-viral disease mechanisms. Science 370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ciancanelli MJ, Huang SX, Luthra P, Garner H, Itan Y, et al. 2015. Infectious disease. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science 348: 448–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hernandez N, Melki I, Jing H, Habib T, Huang SSY, et al. 2018. Life-threatening influenza pneumonitis in a child with inherited IRF9 deficiency. J Exp Med 215: 2567–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lim HK, Huang SXL, Chen J, Kerner G, Gilliaux O, et al. 2019. Severe influenza pneumonitis in children with inherited TLR3 deficiency. J Exp Med 216: 2038–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Abolhassani H, Vosughimotlagh A, Asano T, Landegren N, Boisson B, et al. 2022. X-Linked TLR7 Deficiency Underlies Critical COVID-19 Pneumonia in a Male Patient with Ataxia-Telangiectasia. J Clin Immunol 42: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schmidt A, Peters S, Knaus A, Sabir H, Hamsen F, et al. 2021. TBK1 and TNFRSF13B mutations and an autoinflammatory disease in a child with lethal COVID-19. NPJ Genom Med 6: 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Q, Matuozzo D, Le Pen J, Lee D, Moens L, et al. 2022. Recessive inborn errors of type I IFN immunity in children with COVID-19 pneumonia. J Exp Med 219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Khanmohammadi S, Rezaei N, Khazaei M, Shirkani A. 2022. A Case of Autosomal Recessive Interferon Alpha/Beta Receptor Alpha Chain (IFNAR1) Deficiency with Severe COVID-19. J Clin Immunol 42: 19–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Abolhassani H, Landegren N, Bastard P, Materna M, Modaresi M, et al. 2022. Inherited IFNAR1 Deficiency in a Child with Both Critical COVID-19 Pneumonia and Multisystem Inflammatory Syndrome. J Clin Immunol 42: 471–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Butler-Laporte G, Povysil G, Kosmicki JA, Cirulli ET, Drivas T, et al. 2022. Exome-wide association study to identify rare variants influencing COVID-19 outcomes: Results from the Host Genetics Initiative. PLoS Genet 18: e1010367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kosmicki JA, Horowitz JE, Banerjee N, Lanche R, Marcketta A, et al. 2021. Pan-ancestry exome-wide association analyses of COVID-19 outcomes in 586,157 individuals. Am J Hum Genet 108: 1350–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Povysil G, Butler-Laporte G, Shang N, Wang C, Khan A, et al. 2021. Rare loss-of-function variants in type I IFN immunity genes are not associated with severe COVID-19. J Clin Invest 131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang Q, Cobat A, Bastard P, Notarangelo LD, Su HC, et al. 2021. Association of rare predicted loss-of-function variants of influenza-related type I IFN genes with critical COVID-19 pneumonia. J Clin Invest 131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Matuozzo D, Talouarn E, Marchal A, Manry J, Seeleuthner Y, et al. 2022. Rare predicted loss-of-function variants of type I IFN immunity genes are associated with life-threatening COVID-19. medRxiv: 2022.10.22.22281221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li J, Lei WT, Zhang P, Rapaport F, Seeleuthner Y, et al. 2021. Biochemically deleterious human NFKB1 variants underlie an autosomal dominant form of common variable immunodeficiency. J Exp Med 218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Koning R, Bastard P, Casanova JL, Brouwer MC, van de Beek D, with the Amsterdam UMCC-BI. 2021. Autoantibodies against type I interferons are associated with multi-organ failure in COVID-19 patients. Intensive Care Med 47: 704–06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lamborn IT, Jing H, Zhang Y, Drutman SB, Abbott JK, et al. 2017. Recurrent rhinovirus infections in a child with inherited MDA5 deficiency. J Exp Med 214: 1949–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fallerini C, Daga S, Mantovani S, Benetti E, Picchiotti N, et al. 2021. Association of Toll-like receptor 7 variants with life-threatening COVID-19 disease in males: findings from a nested case-control study. Elife 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mantovani S, Daga S, Fallerini C, Baldassarri M, Benetti E, et al. 2022. Rare variants in Toll-like receptor 7 results in functional impairment and downregulation of cytokine-mediated signaling in COVID-19 patients. Genes Immun 23: 51–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pessoa NL, Bentes AA, de Carvalho AL, de Souza Silva TB, Alves PA, et al. 2021. Case report: hepatitis in a child infected with SARS-CoV-2 presenting toll-like receptor 7 Gln11Leu single nucleotide polymorphism. Virol J 18: 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Solanich X, Vargas-Parra G, van der Made CI, Simons A, Schuurs-Hoeijmakers J, et al. 2021. Genetic Screening for TLR7 Variants in Young and Previously Healthy Men With Severe COVID-19. Front Immunol 12: 719115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.van der Made CI, Simons A, Schuurs-Hoeijmakers J, van den Heuvel G, Mantere T, et al. 2020. Presence of Genetic Variants Among Young Men With Severe COVID-19. JAMA 324: 663–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Casanova JL, Abel L. 2015. Disentangling inborn and acquired immunity in human twins. Cell 160: 13–5 [DOI] [PubMed] [Google Scholar]
- 84.Nakanishi T, Pigazzini S, Degenhardt F, Cordioli M, Butler-Laporte G, et al. 2021. Age-dependent impact of the major common genetic risk factor for COVID-19 on severity and mortality. J Clin Invest 131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cruz R, Almeida SD, Heredia ML, Quintela I, Ceballos FC, et al. 2022. Novel genes and sex differences in COVID-19 severity. Hum Mol Genet [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bartleson JM, Radenkovic D, Covarrubias AJ, Furman D, Winer DA, Verdin E. 2021. SARS-CoV-2, COVID-19 and the Ageing Immune System. Nat Aging 1: 769–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Splunter MV, Perdijk O, Fick-Brinkhof H, Floris-Vollenbroek EG, Meijer B, et al. 2019. Plasmacytoid dendritic cell and myeloid dendritic cell function in ageing: A comparison between elderly and young adult women. PLoS One 14: e0225825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schultze JL, Aschenbrenner AC. 2021. COVID-19 and the human innate immune system. Cell 184: 1671–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stark GR, Darnell JE Jr. 2012. The JAK-STAT pathway at twenty. Immunity 36: 503–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pierce CA, Sy S, Galen B, Goldstein DY, Orner E, et al. 2021. Natural mucosal barriers and COVID-19 in children. JCI Insight 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Andreakos E, Abel L, Vinh DC, Kaja E, Drolet BA, et al. 2022. A global effort to dissect the human genetic basis of resistance to SARS-CoV-2 infection. Nat Immunol 23: 159–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Martinez-Sanz J, Jimenez D, Martinez-Campelo L, Cruz R, Vizcarra P, et al. 2021. Role of ACE2 genetic polymorphisms in susceptibility to SARS-CoV-2 among highly exposed but non infected healthcare workers. Emerg Microbes Infect 10: 493–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Reukers DFM, van Boven M, Meijer A, Rots N, Reusken C, et al. 2022. High Infection Secondary Attack Rates of Severe Acute Respiratory Syndrome Coronavirus 2 in Dutch Households Revealed by Dense Sampling. Clin Infect Dis 74: 52–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tournamille C, Colin Y, Cartron JP, Le Van Kim C. 1995. Disruption of a GATA motif in the Duffy gene promoter abolishes erythroid gene expression in Duffy-negative individuals. Nat Genet 10: 224–8 [DOI] [PubMed] [Google Scholar]
- 95.Dean M, Carrington M, Winkler C, Huttley GA, Smith MW, et al. 1996. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science 273: 1856–62 [DOI] [PubMed] [Google Scholar]
- 96.Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, et al. 1996. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 86: 367–77 [DOI] [PubMed] [Google Scholar]
- 97.Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, et al. 1996. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 382: 722–5 [DOI] [PubMed] [Google Scholar]
- 98.Lindesmith L, Moe C, Marionneau S, Ruvoen N, Jiang X, et al. 2003. Human susceptibility and resistance to Norwalk virus infection. Nat Med 9: 548–53 [DOI] [PubMed] [Google Scholar]
- 99.Shelton JF, Shastri AJ, Ye C, Weldon CH, Filshtein-Sonmez T, et al. 2021. Trans-ancestry analysis reveals genetic and nongenetic associations with COVID-19 susceptibility and severity. Nat Genet 53: 801–08 [DOI] [PubMed] [Google Scholar]
- 100.Gutierrez-Valencia M, Leache L, Librero J, Jerico C, Enguita German M, Garcia-Erce JA. 2022. ABO blood group and risk of COVID-19 infection and complications: A systematic review and meta-analysis. Transfusion 62: 493–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cooling L 2015. Blood Groups in Infection and Host Susceptibility. Clin Microbiol Rev 28: 801–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cheng Y, Cheng G, Chui CH, Lau FY, Chan PK, et al. 2005. ABO blood group and susceptibility to severe acute respiratory syndrome. JAMA 293: 1450–1 [DOI] [PubMed] [Google Scholar]
- 103.Roberts GHL, Partha R, Rhead B, Knight SC, Park DS, et al. 2022. Expanded COVID-19 phenotype definitions reveal distinct patterns of genetic association and protective effects. Nat Genet 54: 374–81 [DOI] [PubMed] [Google Scholar]
- 104.Biering SB, Sarnik SA, Wang E, Zengel JR, Leist SR, et al. 2022. Genome-wide bidirectional CRISPR screens identify mucins as host factors modulating SARS-CoV-2 infection. Nat Genet 54: 1078–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Grodzki M, Bluhm AP, Schaefer M, Tagmount A, Russo M, et al. 2022. Genome-scale CRISPR screens identify host factors that promote human coronavirus infection. Genome Med 14: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhu Y, Feng F, Hu G, Wang Y, Yu Y, et al. 2021. A genome-wide CRISPR screen identifies host factors that regulate SARS-CoV-2 entry. Nat Commun 12: 961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang R, Simoneau CR, Kulsuptrakul J, Bouhaddou M, Travisano KA, et al. 2021. Genetic Screens Identify Host Factors for SARS-CoV-2 and Common Cold Coronaviruses. Cell 184: 106–19 e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Schneider WM, Luna JM, Hoffmann HH, Sanchez-Rivera FJ, Leal AA, et al. 2021. Genome-Scale Identification of SARS-CoV-2 and Pan-coronavirus Host Factor Networks. Cell 184: 120–32 e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wei J, Alfajaro MM, DeWeirdt PC, Hanna RE, Lu-Culligan WJ, et al. 2021. Genome-wide CRISPR Screens Reveal Host Factors Critical for SARS-CoV-2 Infection. Cell 184: 76–91 e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Daniloski Z, Jordan TX, Wessels HH, Hoagland DA, Kasela S, et al. 2021. Identification of Required Host Factors for SARS-CoV-2 Infection in Human Cells. Cell 184: 92–105 e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Baggen J, Persoons L, Vanstreels E, Jansen S, Van Looveren D, et al. 2021. Genome-wide CRISPR screening identifies TMEM106B as a proviral host factor for SARS-CoV-2. Nat Genet 53: 435–44 [DOI] [PubMed] [Google Scholar]
- 112.Rebendenne A, Roy P, Bonaventure B, Chaves Valadao AL, Desmarets L, et al. 2022. Bidirectional genome-wide CRISPR screens reveal host factors regulating SARS-CoV-2, MERS-CoV and seasonal HCoVs. Nat Genet 54: 1090–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Andreakos E, Abel L, Vinh DC, Kaja E, Drolet BA, et al. 2021. A global effort to dissect the human genetic basis of resistance to SARS-CoV-2 infection. Nat Immunol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bastard P, Gervais A, Le Voyer T, Rosain J, Philippot Q, et al. 2021. Autoantibodies neutralizing type I IFNs are present in ~4% of uninfected individuals over 70 years old and account for ~20% of COVID-19 deaths. Sci Immunol 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, et al. 2020. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Puel A, Bastard P, Bustamante J, Casanova JL. 2022. Human autoantibodies underlying infectious diseases. J Exp Med 219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ku CL, Chi CY, von Bernuth H, Doffinger R. 2020. Autoantibodies against cytokines: phenocopies of primary immunodeficiencies? Hum Genet 139: 783–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pozzetto B, Mogensen KE, Tovey MG, Gresser I. 1984. Characteristics of autoantibodies to human interferon in a patient with varicella-zoster disease. J Infect Dis 150: 707–13 [DOI] [PubMed] [Google Scholar]
- 119.Goncalves D, Mezidi M, Bastard P, Perret M, Saker K, et al. 2021. Antibodies against type I interferon: detection and association with severe clinical outcome in COVID-19 patients. Clin Transl Immunology 10: e1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shaw ER, Rosen LB, Cheng A, Dobbs K, Delmonte OM, et al. 2021. Temporal Dynamics of Anti-Type 1 Interferon Autoantibodies in COVID-19 Patients. Clin Infect Dis [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Savvateeva E, Filippova M, Valuev-Elliston V, Nuralieva N, Yukina M, et al. 2021. Microarray-Based Detection of Antibodies against SARS-CoV-2 Proteins, Common Respiratory Viruses and Type I Interferons. Viruses 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Troya J, Bastard P, Planas-Serra L, Ryan P, Ruiz M, et al. 2021. Neutralizing Autoantibodies to Type I IFNs in >10% of Patients with Severe COVID-19 Pneumonia Hospitalized in Madrid, Spain. J Clin Immunol 41: 914–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.van der Wijst MGP, Vazquez SE, Hartoularos GC, Bastard P, Grant T, et al. 2021. Type I interferon autoantibodies are associated with systemic immune alterations in patients with COVID-19. Sci Transl Med 13: eabh2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Vazquez SE, Bastard P, Kelly K, Gervais A, Norris PJ, et al. 2021. Neutralizing Autoantibodies to Type I Interferons in COVID-19 Convalescent Donor Plasma. J Clin Immunol 41: 1169–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wang EY, Mao T, Klein J, Dai Y, Huck JD, et al. 2021. Diverse functional autoantibodies in patients with COVID-19. Nature 595: 283–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Abers MS, Rosen LB, Delmonte OM, Shaw E, Bastard P, et al. 2021. Neutralizing type-I interferon autoantibodies are associated with delayed viral clearance and intensive care unit admission in patients with COVID-19. Immunol Cell Biol 99: 917–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chauvineau-Grenier A, Bastard P, Servajean A, Gervais A, Rosain J, et al. 2021. Autoantibodies neutralizing type I interferons in 20% of COVID-19 deaths in a French hospital. Res Sq [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Solanich X, Rigo-Bonnin R, Gumucio VD, Bastard P, Rosain J, et al. 2021. Pre-existing Autoantibodies Neutralizing High Concentrations of Type I Interferons in Almost 10% of COVID-19 Patients Admitted to Intensive Care in Barcelona. J Clin Immunol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Raadsen MP, Gharbharan A, Jordans CCE, Mykytyn AZ, Lamers MM, et al. 2022. Interferon-alpha2 Auto-antibodies in Convalescent Plasma Therapy for COVID-19. J Clin Immunol 42: 232–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chang SE, Feng A, Meng W, Apostolidis SA, Mack E, et al. 2021. New-onset IgG autoantibodies in hospitalized patients with COVID-19. Nat Commun 12: 5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ziegler CGK, Miao VN, Owings AH, Navia AW, Tang Y, et al. 2021. Impaired local intrinsic immunity to SARS-CoV-2 infection in severe COVID-19. Cell 184: 4713–33 e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Acosta-Ampudia Y, Monsalve DM, Rojas M, Rodriguez Y, Gallo JE, et al. 2021. COVID-19 convalescent plasma composition and immunological effects in severe patients. J Autoimmun 118: 102598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Carapito R, Li R, Helms J, Carapito C, Gujja S, et al. 2022. Identification of driver genes for critical forms of COVID-19 in a deeply phenotyped young patient cohort. Sci Transl Med 14: eabj7521. [DOI] [PubMed] [Google Scholar]
- 134.Eto S, Nukui Y, Tsumura M, Nakagama Y, Kashimada K, et al. 2022. Neutralizing Type I Interferon Autoantibodies in Japanese Patients with Severe COVID-19. J Clin Immunol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Manry J, Bastard P, Gervais A, Le Voyer T, Rosain J, et al. 2022. The risk of COVID-19 death is much greater and age dependent with type I IFN autoantibodies. Proc Natl Acad Sci U S A 119: e2200413119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bastard P, Orlova E, Sozaeva L, Levy R, James A, et al. 2021. Preexisting autoantibodies to type I IFNs underlie critical COVID-19 pneumonia in patients with APS-1. J Exp Med 218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lopez J, Mommert M, Mouton W, Pizzorno A, Brengel-Pesce K, et al. 2021. Early nasal type I IFN immunity against SARS-CoV-2 is compromised in patients with autoantibodies against type I IFNs. J Exp Med 218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sposito B, Broggi A, Pandolfi L, Crotta S, Clementi N, et al. 2021. The interferon landscape along the respiratory tract impacts the severity of COVID-19. Cell 184: 4953–68 e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bastard P, Zhang Q, Zhang SY, Jouanguy E, Casanova JL. 2022. Type I interferons and SARS-CoV-2: from cells to organisms. Curr Opin Immunol 74: 172–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bastard P, Vazquez S, Liu J, Laurie MT, Wang CY, et al. 2022. Vaccine breakthrough hypoxemic COVID-19 pneumonia in patients with auto-Abs neutralizing type I IFNs. Sci Immunol: eabp8966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhang Q, Pizzorno A, Miorin L, Bastard P, Gervais A, et al. 2022. Autoantibodies against type I IFNs in patients with critical influenza pneumonia. J Exp Med 219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Busnadiego I, Abela IA, Frey PM, Hofmaenner DA, Scheier TC, et al. 2022. Critically ill COVID-19 patients with neutralizing autoantibodies against type I interferons have increased risk of herpesvirus disease. PLoS Biol 20: e3001709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mathian A, Breillat P, Dorgham K, Bastard P, Charre C, et al. 2022. Lower disease activity but higher risk of severe COVID-19 and herpes zoster in patients with systemic lupus erythematosus with pre-existing autoantibodies neutralising IFN-alpha. Ann Rheum Dis [DOI] [PubMed] [Google Scholar]
- 144.Sun S, Wang Y, Maslov AY, Dong X, Vijg J. 2022. SomaMutDB: a database of somatic mutations in normal human tissues. Nucleic Acids Res 50: D1100–D08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Levy R, Zhang P, Bastard P, Dorgham K, Melki I, et al. 2021. Monoclonal antibody-mediated neutralization of SARS-CoV-2 in an IRF9-deficient child. Proc Natl Acad Sci U S A 118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.de Prost N, Bastard P, Arrestier R, Fourati S, Mahevas M, et al. 2021. Plasma Exchange to Rescue Patients with Autoantibodies Against Type I Interferons and Life-Threatening COVID-19 Pneumonia. J Clin Immunol 41: 536–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Vinh DC, Abel L, Bastard P, Cheng MP, Condino-Neto A, et al. 2021. Harnessing Type I IFN Immunity Against SARS-CoV-2 with Early Administration of IFN-beta. J Clin Immunol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, et al. 2018. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 50: 1219–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Huang QM, Zhang PD, Li ZH, Zhou JM, Liu D, et al. 2022. Genetic Risk and Chronic Obstructive Pulmonary Disease Independently Predict the Risk of Incident Severe COVID-19. Ann Am Thorac Soc 19: 58–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kaiser J 2021. DNA test to predict odds of severe COVID-19 draws scrutiny. Science 372: 1139. [DOI] [PubMed] [Google Scholar]
- 151.Tangye SG, consortium CHGE. 2022. Impact of SARS-CoV-2 infection and COVID-19 on patients with inborn errors of immunity. J Allergy Clin Immunol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lee D, Le Pen J, Dong B, Aquino Y, Ogishi M, et al. 2022. Inherited deficiency of the OAS-RNase L pathway in children with MIS-C. Submitted [Google Scholar]
- 153.Chou J, Platt CD, Habiballah S, Nguyen AA, Elkins M, et al. 2021. Mechanisms underlying genetic susceptibility to multisystem inflammatory syndrome in children (MIS-C). J Allergy Clin Immunol 148: 732–38 e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lee PY, Platt CD, Weeks S, Grace RF, Maher G, et al. 2020. Immune dysregulation and multisystem inflammatory syndrome in children (MIS-C) in individuals with haploinsufficiency of SOCS1. J Allergy Clin Immunol 146: 1194–200 e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bolze A, Mogensen TH, Zhang SY, Abel L, Andreakos E, et al. 2022. Decoding the Human Genetic and Immunological Basis of COVID-19 mRNA Vaccine-Induced Myocarditis. J Clin Immunol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Namkoong H, Edahiro R, Takano T, Nishihara H, Shirai Y, et al. 2022. DOCK2 is involved in the host genetics and biology of severe COVID-19. Nature 609: 754–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ostendorf BN, Patel MA, Bilanovic J, Hoffmann HH, Carrasco SE, et al. 2022. Common human genetic variants of APOE impact murine COVID-19 mortality. Nature [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Herrera-Esposito D, de Los Campos G. 2022. Age-specific rate of severe and critical SARS-CoV-2 infections estimated with multi-country seroprevalence studies. BMC Infect Dis 22: 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Asano T, Boisson B, Onodi F, Matuozzo D, Moncada-Velez M, et al. 2021. X-linked recessive TLR7 deficiency in ~1% of men under 60 years old with life-threatening COVID-19. Sci Immunol 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, et al. 2020. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 370 [DOI] [PMC free article] [PubMed] [Google Scholar]