

Abstract
Single-cell sequencing-based methods for profiling gene transcript levels have revealed substantial heterogeneity in expression levels among morphologically indistinguishable cells. This variability has important functional implications for tissue biology and disease states such as cancer. Mapping of epigenomic information such as chromatin accessibility, nucleosome positioning, histone tail modifications and enhancer–promoter interactions in both bulk-cell and single-cell samples has shown that these characteristics of chromatin state contribute to expression or repression of associated genes. Advances in single-cell epigenomic profiling methods are enabling high-resolution mapping of chromatin states in individual cells. Recent studies using these techniques provide evidence that variations in different aspects of chromatin organization collectively define gene expression heterogeneity among otherwise highly similar cells.
Multicellular organisms comprise specialized tissues that perform distinct physiological functions. Although they possess identical or near-identical genomic DNA sequences, these tissues are differentiated in function by maintaining distinct gene expression profiles. However, even morphologically homogeneous and clonally derived cell populations have been found to exhibit cell-to-cell variations in gene expression and stimulus responses1–4. This cellular heterogeneity has been detected in many organisms and developmental contexts5–9 and has important roles in the biology of tissues and disease states such as cancer7,10–12. It is also often linked to expression heterogeneity of phenotypically relevant genes. For example, expression heterogeneity among embryonic stem cells can alter differentiation characteristics13. Furthermore, expression heterogeneity among pathogen cells or cancer cells can be relevant to human disease. Transcriptional variation within a population of the unicellular malaria pathogen Plasmodium falciparum is potentially important for adaptive responses to the host organism14. Cancer cells sampled from the same tumour can exhibit substantial heterogeneity in morphology and gene expression, and these differences are relevant to treatment and progression of the disease15–17. In particular, cancer stem cells exhibit specific gene expression, differentiation and proliferation characteristics that contribute to tumorigenesis and therapeutic resistance18–20. These examples highlight how profiling cell-to-cell variations in gene expression and linking these differences to specific cellular properties promises to enhance our understanding of development and disease.
The basis of cell-to-cell heterogeneity in gene expression is a field of active investigation, and multiple mechanisms are thought to contribute to this phenomenon. Gene expression is a multifaceted process that begins with transcription of part of the genomic DNA template into messenger RNA. Transcription occurs in ‘bursts’, which are short transcriptionally active states punctuated by longer transcriptionally silent states21–23. Stochastic transcriptional bursting may contribute to variations in expression levels among similar cells23,24. In addition, expression heterogeneity could arise from cell-to-cell differences in gene activation and repression. In most cells, only a subset of genes are transcriptionally active at any given time. These include ‘housekeeping’ genes that are constitutively expressed as well as genes that are relevant to the cell’s current environment and developmental state. Whether or not a gene is transcribed is determined by the binding of proteins such as transcription factors and initiators to genomic regulatory elements such as promoters and enhancers25–27. In eukaryotes, access to these regulatory elements is controlled, in part, by the surrounding chromatin context, including nucleosome positioning and composition, histone tail modifications and 3D structural interactions28–30. These aspects of chromatin state are often referred to as epigenetic marks due to their persistent nature and the powerful effects they exert on transcription of associated genes (FIG. 1). Gene expression heterogeneity among individual cells likely arises from a combination of these phenomena as well as other uncharacterized mechanisms.
Fig. 1 |. Epigenetic landscapes of transcribed and silent genes in eukaryotes.

Diagram depicting the epigenetic landscapes present at transcriptionally repressed and active chromatin. a | Transcriptionally repressed, enzymatically inaccessible cis-regulatory elements are characterized by regularly spaced nucleosome arrays enriched for DNA methylation and specific histone modifications, such as trimethylation of lysine 27 on histone 3 (H3K27me3). b | Transcriptionally active genes exhibit enzymatically accessible cis-regulatory elements with positioned, flanking nucleosomes enriched for specific histone modifications such as acetylation. CCCTC-binding factor (CTCF) and cohesin are depicted facilitating a chromatin contact between an active enhancer and an active promoter. 5mC, 5-methylcytosine; Pol II, Polymerase II; TSS, transcription start site.
Experimental methods that operate on samples comprising thousands or millions of cells (‘bulk-cell’ methods) provide a single aggregate or average signal for the cell population. Thus, they are not suitable for resolving cell-to-cell differences in gene expression and epigenetic marks. Emerging techniques are capable of single-cell profiling of epigenetic marks such as chromatin accessibility, nucleosome positioning, DNA methylation, histone post-translational modifications and enhancer–promoter interactions either singly (TABLE 1) or in combination (TABLE 2). In this Review, we summarize recent studies that use single-cell methods to shed light on the nature of gene expression heterogeneity and whether phenotypically homogeneous populations of cells exhibit variations in chromatin states31–34.
Table 1 |.
Data types generated by selected single-omics methods
| Data type | Bulk-cell methods | Single-cell methods |
|---|---|---|
| Gene expression | RNA-seq41,166,167 DNA microarrays37–39 |
scRNA-seq168 |
| DNA accessibility | DNase-seq52 ATAC-seq54 |
scDNase-seq57 scATAC-seq55,56 |
| Nucleosome positioning | MNase-seq77 | scMNase-seq78 |
| Histone modifications | ChIP–seq88,89 ACT-seq103 CUT&RUN169 CUT&TAG104 ChIL-seq102 |
scChIP–seq99 scChIC–seq100 iACT-seq103 scCUT&RUN101 CUT&TAG104 CoBATCH105 scChIL-seq102 |
| DNA methylation | BS-seq170 RRBS171 MeDIP-seq172 MethylCap-seq173 |
scBS-seq119 scRRBS174 |
| Chromatin contacts | Hi-C136 Hi-ChIP175 ChIA-PET176 |
scHi-C121,125,143–146 |
ACT-seq, antibody-guided chromatin tagmentation sequencing; ATAC-seq, assay for transposase-accessible chromatin sequencing; BS-seq, bisulfite sequencing; ChIA-PET, chromatin interaction analysis by paired-end tag sequencing; ChIC–seq, chromatin immunocleavage followed by sequencing; ChIL-seq, chromatin integration labelling sequencing; ChIP–seq, chromatin immunoprecipitation followed by sequencing; CoBATCH, combinatorial barcoding and targeted chromatin release; CUT&RUN, cleavage under targets and release using nuclease; CUT&TAG, cleavage under targets and tagmentation; DNase-seq, DNase I hypersensitive site sequencing; Hi-C, high-throughput chromosome conformation capture; iACT-seq, indexed antibody-guided chromatin tagmentation sequencing; MeDIP-seq, methylated DNA immunoprecipitation sequencing; MethylCap-seq, methyl-CpG binding domain-based capture and sequencing; MNase-seq, micrococcal nuclease digestion deep sequencing; RNA-seq, RNA sequencing; RRBS, reduced representation bisulfite; sc, single-cell.
Table 2 |.
Selected single-cell multi-omics methods and the types of epigenomic data they provide
| Method | Data set 1 | Data set 2 | Data set 3 |
|---|---|---|---|
| Paired-seq60 | Gene expression | DNA accessibility and nucleosome position | – |
| sci-CAR63 | Gene expression | DNA accessibility and nucleosome position | – |
| SNARE-seq64 | Gene expression | DNA accessibility and nucleosome position | – |
| scM&T123 | Gene expression | DNA methylation | – |
| scNMT-seq84 | Gene expression | DNA accessibility and nucleosome position | DNA methylation |
| scTrio-seq122 | Gene expression | DNA methylation | Copy number variation |
| scNOMe-seq83 | DNA accessibility and nucleosome position | DNA methylation | – |
| scCOOL-seq85 | DNA accessibility and nucleosome position | DNA methylation | Copy number variation |
| Methyl-HiC125 | DNA methylation | Chromatin contacts | – |
| sc-m3c-seq121 | DNA methylation | Chromatin contacts | – |
paired-seq, parallel analysis of individual cells for RNA expression and DNA accessibility by sequencing; scCOOL-seq, single-cell chromatin overall omic-scale landscape sequencing; sci-CAR, single-cell combinatorial indexing of chromatin accessibility and mRNA; sc-m3c-seq, single-nucleus methyl-3C sequencing; scM&T, single-cell methylome and transcriptome sequencing; scNMT-seq, single-cell nucleosome, methylation and transcription sequencing; scNOMe-seq, single-cell nucleosome occupancy and methylome sequencing; scTrio-seq, single-cell triple omics sequencing; SNARE-seq, single-nucleus chromatin accessibility and mRNA expression sequencing.
Measuring cellular heterogeneity
Bulk-cell methods measure differences between populations.
Cell populations from different tissues of the same organism exhibit phenotypic heterogeneity due to both genetic and epigenetic factors. Genetic differences among human cells include variations in ploidy number, such as haploid gametes and polyploid muscle cells. Epigenetic differences, which refer to mechanisms that operate above the level of the genome sequence, help to produce specialized cell types with distinct functions among cells containing identical genetic information. For example, differences in covalent modification of histone tails help to functionally distinguish T helper cell variants from naive CD4 T cells in mice35. Such specialization of cell and tissue types is broadly necessary for the functioning of complex multicellular life.
Epigenetic specialization of tissues affects cellular characteristics by influencing the expression levels and/or potential of genes associated with those traits. Understanding variations in gene expression among tissues and cell types as well as the functional differences they confer has become a prominent theme in modern molecular biology. Conventional genome-wide techniques for measuring gene expression, such as DNA microarrays and next-generation RNA sequencing (RNA-seq), are performed on bulk-cell samples comprising thousands or millions of cells and provide average profiles of gene expression. Similarly, bulk-cell epigenomic methods such as chromatin immunoprecipitation followed by sequencing (ChIP–seq) provide average profiles of histone modifications of many cells. Although these bulk-cell methods have provided valuable information on how gene expression and enrichment of epigenetic marks differ between different populations of cells, such methods lack the resolution required to resolve cell-to-cell heterogeneity within a population (FIG. 2).
Fig. 2 |. Single-cell techniques provide greater resolution than bulk-cell methods.
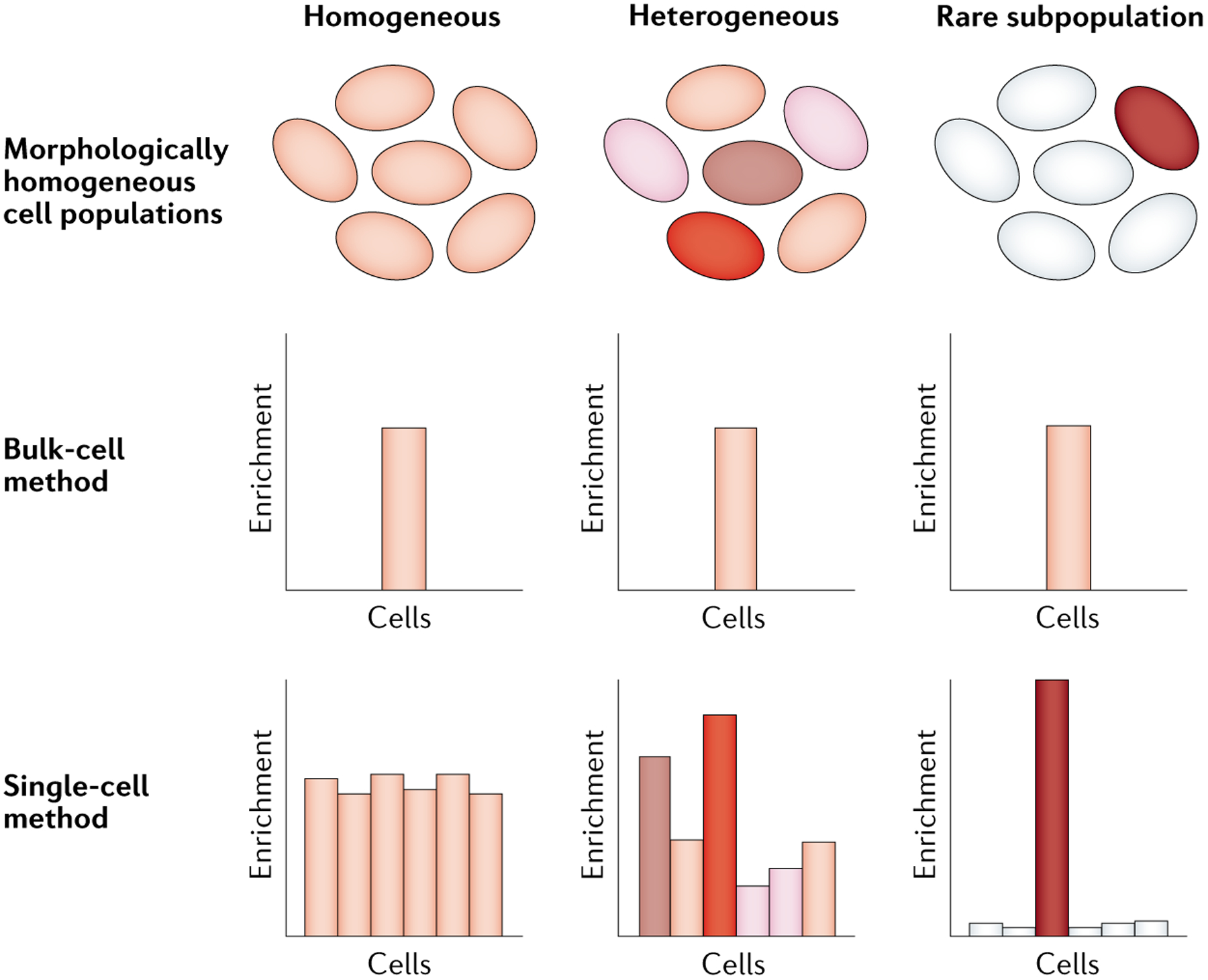
Three morphologically homogeneous cell populations exhibiting varying patterns of enrichment for a particular epigenetic characteristic, with darker shading indicating greater enrichment. Bulk-cell analysis generates a population-average value and fails to differentiate between the different scenarios. By contrast, single-cell approaches can readily distinguish variable levels of enrichment (middle) from rare subpopulations (right) and homogeneous enrichment (left).
Single-cell methods measure differences within populations.
Cell-to-cell heterogeneity in gene expression often correlates with functional differences in cellular developmental potential or disease characteristics7,10–12,15–20. For example, human induced pluripotent stem cells contain subpopulations of cells that exhibit differential expression of genes relating to pluripotency and cell fate decisions7. Similarly, expression heterogeneity of the important pluripotency genes Rex1 and Oct4 revealed subpopulations of mouse embryonic stem cells that were primed for differentiation into distinct cell lineages1. Because expression heterogeneity is relevant to cellular differentiation, development and disease, it is important to understand cell-to-cell variation of expression within a population and the mechanisms by which this variability arises. Although gene expression was successfully measured in individual cells in the 1990s36, these studies often relied on laborious methods such as microinjection of primers and enzymes into manually dissociated cells. Over time, the throughput and resolution of single-cell techniques have been dramatically enhanced through improvements in microarray technologies37–39 and the advent of RNA-seq40,41. In particular, sequencing-based methods are capable of providing whole-transcriptome profiles of gene transcription in thousands of individual cells simultaneously. Indeed, the widespread application of single-cell RNA-seq (scRNA-seq) has revealed that transcriptional heterogeneity among highly similar cells seems to be a common phenomenon among eukaryotes, bacteria and clonally derived cell populations1–4,8,42.
Epigenetic mechanisms, such as variations in chromatin states, present attractive candidates for contributors to cellular gene expression heterogeneity. Over the past decade, sequencing-based methods have been developed that can generate genome-wide profiles of epigenetic marks in single cells (TABLE 1). These methods measure the epigenomic differences among individual cells within a population and enable multiple applications that would be difficult or unfeasible using bulk-cell approaches. For example, epigenomic changes that occur across the cell cycle can be directly analysed in passaging cultures without the need for chemical perturbations. Additionally, subtle plasticity of epigenomic states within a cell population, such as among individual stem cells that exhibit variable priming for differentiation, necessitate the use of single-cell techniques. Finally, rare cellular subtypes within a larger tissue or culture can be identified in an unbiased manner using single-cell approaches without the need for antibody or fluorescence cell labelling. These advantages also make single-cell epigenomic methods ideal for investigating the mechanisms that underlie gene expression variability among cells.
Although single-cell methods can reveal cell-to-cell heterogeneity for individual epigenetic marks, these methods are unable to simultaneously profile multiple marks alongside transcript levels in the same single cells. Thus, these techniques can only suggest the presence of correlations between epigenetic phenomena and transcript levels without interrogating those relationships directly. To address this limitation, investigators are pioneering ‘multi-omics’ methods that are capable of co-profiling an epigenetic mark alongside gene expression and/or additional aspects of chromatin state (TABLE 2).
Limitations and auxiliary methods.
Sequencing-based single-cell techniques for RNA expression and epigenomic modifications are dependent on amplification of target sequences. Thus, they can be subject to experimental noise arising from amplification bias, differences in library sizes, missing data or low RNA capture rates43,44. Although multiple computational techniques have been developed that are capable of mitigating these sources of noise43–45, a current limitation of scRNA-seq is that quantification of transcripts is often limited to those genes that are most highly expressed, with accurate quantification occurring for generally only 5,000–9,000 of the most highly expressed transcripts per cell46. Fluorescence-based, non-sequencing methods such as fluorescence in situ hybridization can serve as parallel methods for validation of scRNA-seq findings1,3,14. In particular, single-molecule RNA fluorescence in situ hybridization provides a complementary approach for single-cell transcript analysis that can accurately quantify lowly expressed genes due to its sensitivity47. Unlike scRNA-seq data, there are no practical imaging-based techniques to validate findings from single-cell chromatin accessibility, DNA methylation and histone modification experiments. Most of these data sets are benchmarked by comparison with the corresponding bulk-cell data with regard to signal enrichment at enhancers or promoters (see the following sections for details). Notably, however, imaging-based analysis of 3D structure may now independently validate some single-cell 3D chromatin interactions identified using high-throughput chromosome conformation capture (Hi-C)48–50.
Chromatin accessibility
Chromatin accessibility varies from cell to cell.
Chromatin comprises regions that are highly condensed and inaccessible as well as regions that are more loosely packaged. The accessibility of DNA within chromatin, particularly at cis-regulatory regions such as enhancers and promoters, impacts how amenable a given gene is to transcription, and thus plays an important part in promoting or repressing gene expression51. Chromatin accessibility can be measured genome-wide using DNase I hypersensitive site sequencing (DNase-seq), which identifies accessible DNA based on susceptibility to digestion by the enzyme DNase I52. DNase I hypersensitive sites (DHSs) often represent nucleosome-depleted cis-regulatory elements that are permissive for the binding of transcription factors and other regulatory proteins53. Chromatin accessibility is also measured using the assay for transposase-accessible chromatin sequencing (ATAC-seq), a method analogous to DNase-seq that measures accessibility based on susceptibility to Tn5 transposase54. Both DNase-seq and ATAC-seq have been adapted for analysis of single cells and are abbreviated as scDNase-seq and scATAC-seq when used for this purpose55–57 (TABLE 1). Early scATAC-seq methods represented a trade-off between cell throughput and read density per cell55,56. However, a recently published scATAC-seq method is capable of profiling tens of thousands of single cells at an improved read density of ~100,000 reads per cell58. Compared with previous scATAC-seq methods, scDNase-seq offers much higher read coverage (~350,000 reads per cell) but in only dozens of cells57. As most peaks detected by DNase-seq and ATAC-seq overlap, we will use the term ‘accessible chromatin regions’ to refer to both DHSs detected by DNase-seq and accessible chromatin sites detected by ATAC-seq in the following discussions.
Substantial heterogeneity in chromatin accessibility has been observed among different cells. A quantitative comparison revealed that about 25% of accessible chromatin regions are distinct between two single cells57. Accessible chromatin regions that contained low read densities in bulk-cell data were found to be accessible at low and variable frequencies in single cells, whereas strongly accessible chromatin regions that were associated with multiple active histone marks in bulk cells exhibited more robust accessibility in single cells57, suggesting that histone modifications may contribute to the robust accessibility of active transcriptional regulatory elements. Not surprisingly, gene promoters that exhibited low cell-to-cell variation in accessibility were significantly enriched in constitutive housekeeping gene functions such as transcription and RNA processing.
Variability in chromatin accessibility is likely to be functional.
It could be hypothesized that cell-to-cell heterogeneity in chromatin accessibility arises from technical variation in experiments and is therefore an artefact. This possibility can be investigated using correlations between heterogeneity in gene expression and in chromatin accessibility. For example, accessibility variations arising from technical noise would not be predicted to correlate with gene expression heterogeneity. However, strong positive correlations have been observed between cell-to-cell variations in chromatin accessibility at gene regulatory elements and variations in expression of the associated genes56,57, which support a functional role for heterogeneity of chromatin accessibility. Moreover, studies that examine chromatin accessibility and gene expression in tandem often reveal correlations that would not be uncovered otherwise. For example, the read density of accessible chromatin regions in bulk-cell data was found to correlate with mRNA levels up to a low threshold, beyond which expression was insensitive to further density increases57. This observation indicates that a minimum level of accessibility is sufficient to facilitate binding of transcriptional machinery. In single cells, promoters and enhancers of highly transcribed genes almost uniformly overlapped with accessible chromatin regions57, demonstrating that the association between transcript levels and chromatin accessibility is broadly applicable to individual cells. Furthermore, single-cell chromatin accessibility data have been successfully used to separate human white blood cells into major clusters corresponding to the known B cell, T cell and monocyte cell types59. These results not only indicate that the variation in chromatin accessibility in these single-cell data contains important biological information, which is not attainable by bulk-cell methods, but also demonstrate the unique features and applications of single-cell methods.
Separate analyses of gene expression and chromatin accessibility at single-cell resolution have revealed a positive correlation between their variations56,57, and co-profiling of chromatin accessibility and mRNA in the same cell using multi-omics methods (TABLE 2) provides direct evidence that these variations are related60–64. For example, a recently developed method applies scATAC-seq to the genomic DNA and scRNA-seq to the mRNA from the same cell61. When used on human primary immune cells, this revealed a direct relationship between mRNA expression and chromatin accessibility in each individual cell without the need to draw correlations between unrelated data sets61. Co-profiling of gene expression and chromatin accessibility also facilitates direct correlations and computational testing of the relationships between cis-regulatory elements and the genes they control60,62. For example, one study identified over 30,000 new regulatory relationships from single-cell gene expression and chromatin accessibility data obtained from adult mouse cerebral cortex60. Distal regulatory relationships are more easily identified using both data types, and the combined use of distal and proximal regulatory elements more robustly predicted gene expression compared with the use of proximal elements alone63. The ability to identify and describe these functional relationships underscores the value of single-cell multi-omics profiling.
Unlike scRNA-seq data, which can be directly validated by methods such as RNA fluorescence in situ hybridization1,3,14,47, there are no parallel methods to validate the cell-to-cell variation in accessibility of a specific chromatin region found from scDNase-seq or scATAC-seq data sets. Even so, benchmarking of scDNase-seq or scATAC-seq data can be accomplished by comparing the pooled single-cell data with bulk-cell ‘gold standard’ data from sources such as the Encyclopedia of DNA Elements (ENCODE)65. Furthermore, scATAC-seq and scDNase-seq data quality can be independently evaluated by performing computational clustering analysis of mixed cell populations using the single-cell data. For example, as mentioned above, human white blood cells generated clusters corresponding to B cell, T cell and monocyte cell types, demonstrating that scATAC-seq provided meaningful biological information59.
Cell cycle and transcription factors underlie variable chromatin accessibility.
Recent studies suggest that cellular variations in chromatin accessibility likely arise from a combination of asynchronicity in the cell cycle stage and differences in transcription factor expression and/or binding. For example, scATAC-seq performed on passaging K562 human leukaemic cells revealed heterogeneity in the ATAC-seq signal within genomic regions that differ in replication timing during the cell cyle56. These observations suggest that changes in the DNA content during replication contribute to variable ATAC-seq signals in passaging cell cultures. Other studies have shown that variability in the expression and/or binding of specific transcription factors is highly correlated with heterogeneity of chromatin accessibility at associated binding sites, and this relationship is independent of cell cycle effects. For example, scATAC-seq performed on leukaemic K562 cells revealed heterogeneous expression of the sequence-specific transcription factors GATA1 and GATA2 (REF.56), which are important for development and self-renewal of many types of blood cells in vertebrates66,67. Binding motifs for these factors were statistically associated with heterogeneity in chromatin accessibility independent of cell cycle effects. Another scATAC-seq study performed in mouse cardiac progenitor cells observed that chromatin accessibility was associated with binding of the transcription factors ISL1 and NKX2–5 (REF.68). Furthermore, human immune cells exhibited significant heterogeneity in accessibility at the binding sites of developmentally relevant transcription factors including AP-1, FOS and JUN in memory T cells as well as CEBP and PU.1 in monocytes69. Based on the extent of heterogeneity in chromatin accessibility observed in these immune cells, it is likely that these populations exist in a ‘phenotypic continuum’ rather than in a set of distinct chromatin states69.
Although most sequence-specific transcription factors are able to recognize their associated DNA motifs only at accessible chromatin sites, some GATA-family transcription factors and PU.1 are members of a category of transcription factors known as ‘pioneer factors’ that are able to bind to their sequence motifs in closed regions as well70–72. Binding of pioneer factors to their target motifs in closed and heterochromatic regions can result in the formation of accessible chromatin sites73,74. Thus, the heterogeneity in chromatin accessibility among immune cells detailed above is likely to arise, in part, from the observed expression heterogeneity of pioneer factors such as GATA1, GATA2 and PU.1. By contrast, the majority of transcription factors that do not possess pioneering activity are likely to be dependent on changes in chromatin accessibility rather than vice versa. Thus, the studies described above are consistent with a model in which heterogeneity in the binding and/or expression of pioneering transcription factors contributes to heterogeneity in chromatin accessibility and in the binding of other sequence-specific transcription factors (FIG. 3).
Fig. 3 |. scATAC-seq and scDNase-seq measure the dynamics of accessible chromatin sites.

a | Assay for transposase-accessible chromatin sequencing (ATAC-seq) and DNase I hypersensitive site sequencing (DNase-seq) use enzymes (Tn5 transposase and DNase I, respectively) that cut DNA but can only do so at accessible chromatin sites. The activity of these enzymes produces fragments for sequencing that correspond to open chromatin. b | Model of interactions between transcription factors and accessible chromatin. Closed chromatin sites can be bound by sequence-specific pioneer transcription factors. Pioneer transcription factor binding facilitates the opening of chromatin and the creation of a new accessible site. Chromatin-modifying enzymes and other transcription factors that may not possess pioneering activity are able to bind to the accessible site. scATAC-seq, single-cell ATAC-seq; scDNase-seq, single-cell DNase-seq.
Implications of variable chromatin accessibility for development and disease.
Studies of heterogeneity in chromatin accessibility are generating important insights into disease and development. For example, many cancers exhibit high levels of cell-to-cell epigenetic heterogeneity, which may drive the evolution of the cancer cells and/or progression of the disease. In the human leukaemic K562 cell line, it was observed using scRNA-seq that the cell surface marker gene CD24 exhibited heterogeneity in expression levels, and that high CD24 expression correlated with high expression of GATA2 (REF.75). Cells exhibiting high levels of CD24 expression were isolated using fluorescence-activated cell sorting, and subsequent scATAC-seq analysis revealed increased chromatin accessibility at GATA2 binding motifs and at genes associated with maintenance of a haematopoietic progenitor state in these cells relative to cells expressing lower levels of CD24 (REF.75). These results suggest that this subset of K562 cells were more ‘stem-like’ than the rest of the population. The presence of a less-differentiated subpopulation is relevant for studies of how cancer stem cells contribute to treatment resistance and disease recurrence in patients15. In another study, scATAC-seq data revealed considerable heterogeneity in chromatin accessibility within populations of mouse excitatory neurons and kidney tubule cells, and this variance correlated with the location of a cell within its parent tissue76. This observation suggests that epigenetic heterogeneity within solid tissues arises in part from responses to the cells’ tissue microenvironment. These examples highlight the potential of methods for investigating chromatin accessibility heterogeneity to provide future biological insights.
Nucleosome positioning
Heterogeneity in nucleosome positioning depends on genomic context.
The positioning of nucleosomes relative to the DNA sequence has a central role in the organization of gene regulatory features. Nucleosome organization can be probed on a genome-wide scale using micrococcal nuclease digestion deep sequencing (MNase-seq), which uses micrococcal nuclease (MNase) to digest accessible DNA and sequence the remaining protein-bound DNA fragments77. This method directly sequences nucleosome-bound regions, in contrast to the nucleosome-free DHSs that are sequenced by DNase-seq and ATAC-seq (FIG. 4a). MNase-seq has recently been adapted for single-cell analysis (scMNase-seq)78 (TABLE 1), and single-cell profiles of nucleosome organization are enhancing our understanding of the relationships between nucleosome positioning, chromatin accessibility and gene expression.
Fig. 4 |. scMNase-seq reveals nucleosome positioning dynamics.
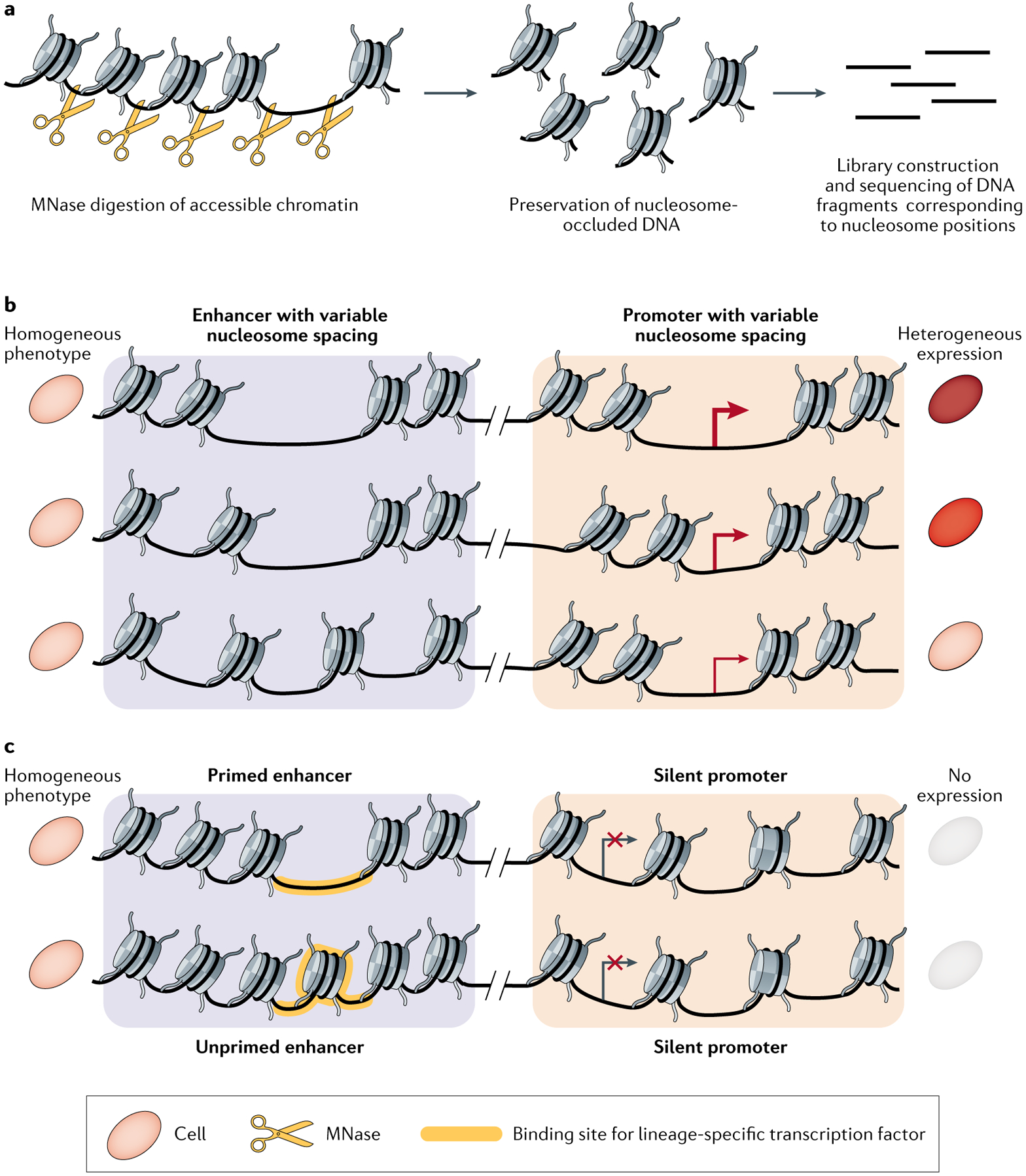
a | Diagram depicting how nucleosome-bound DNA sequences are isolated for sequencing using micrococcal nuclease digestion deep sequencing (MNase-seq). Open chromatin and linker DNA is digested using micrococcal nuclease (MNase), and the resulting nucleosome-occluded DNA fragments are isolated and sequenced. b | Illustration of variable nucleosome positioning around accessible chromatin sites among a morphologically homogeneous cell population. The spacing mode of nucleosomes flanking the enhancer informs expression levels of the associated gene. Arrow thickness represents elevated levels of transcription. c | Illustration of enhancer priming in a morphologically homogeneous population. The yellow-shaded region represents nucleosome depletion at a lineage-specific transcription factor binding motif. Such epigenetic priming, which can produce cells with different developmental potentials, is not detectable using single-cell RNA sequencing because the gene is not expressed in either cell. scMNase-seq, single-cell MNase-seq.
Early studies using microarray-based DNA foot-printing revealed that highly transcribed genes typically contain a nucleosome-free region upstream of the transcription start site and a regularly positioned ‘+1 nucleosome’ downstream of it79,80. Using MNase-seq on human T cells, it was found that binding of RNA Polymerase II (Pol II) to promoters results in nucleosome positioning at sites surrounding the transcription start sites of active genes77, and nucleosome reorganization at promoters and enhancers is associated with gene activation77 (FIG. 1). This phenomenon has been confirmed in multiple tissue types and organisms77,81,82 and was found to be highly consistent between individual cells using scMNase-seq78. Furthermore, scMNase-seq has provided insights into patterns of nucleosome positioning at transcriptionally silent genomic regions78. Whereas bulk-cell MNase-seq failed to resolve the nucleosome organization pattern of heterochromatin and promoters of silent genes, scMNase-seq revealed that these regions bear arrays of nucleosomes that are regularly spaced but randomly positioned relative to the underlying DNA sequence78 (FIG. 1). These regular arrays of nucleosomes potentially arise from chromatin remodelling and assembly factors that propagate a repressive chromatin architecture irrespective of the underlying genomic sequence. Notably, this random positioning of arrays is a source of heterogeneity between cells and stands in marked contrast to the promoters and enhancers of active genes that bear nucleosomes that are positioned relative to the underlying DNA sequence. scMNase-seq has also provided information about nucleosome positioning around DHSs. Two distinct positioning patterns were observed: one with an average distance of ~190 bp between the flanking nucleosomes and another with an average distance of 300 bp78. Within a cell population, more than 80% of DHSs exhibited both spacing types, revealing considerable heterogeneity (FIG. 4b). Furthermore, this cell-to-cell variation in nucleosome positioning is positively correlated with variations in DHSs and target gene expression. Thus, DHSs can be characterized both by the extent to which they are accessible and by their mode of nucleosome spacing, both of which contribute to cellular heterogeneity78.
Heterogeneous nucleosome positioning reveals lineage priming.
Cell populations can be computationally clustered based on single-cell data sets such as those from scMNase-seq. Interestingly, clustering analyses based on nucleosome positioning have uncovered examples of cellular subpopulations that exhibit distinct epigenetic patterns in the absence of corresponding gene expression differences. For example, mouse naive CD4 T cells, which were purified based on cell surface marker expression patterns, exhibited distinct cell clusters that showed nucleosome depletion patterns similar to either T helper 1 (TH1) or TH2 cells at their cell type-specific enhancers, but neither TH1-specific nor TH2-specific genes were expressed in these cells78. Motif analysis revealed that nucleosome losses at TH1 and TH2 enhancers are specifically associated with motifs for the cell type-specific transcription factors RELA and GATA3, respectively. These results suggest that over 40% of naive CD4 T cells are epigenetically primed for differentiation to either TH1 or TH2 cells (FIG. 4c). These subpopulations would not be detectable using clustering analysis of scRNA-seq data because the TH1 or TH2-specific genes are only primed but not yet transcribed. Furthermore, they would not be revealed by bulk-cell epigenetic analysis because bulk-cell assays provide only average profiles for the entire population of cells. Consistent with the possible epigenetic priming observed in mouse naive CD4 T cells, 40% of cultured mouse embryonic stem cells exhibited nucleosome losses at embryoid body-specific enhancers78. These cells also exhibited heterogeneity of nucleosome positioning that corresponded to genes linked to endoderm or mesoderm markers, suggesting that embryonic stem cells are differentially primed for various lineages such as myeloid and neural tube fates. This differential priming and heterogeneity in nucleosome positioning may result in part from cell-to-cell variability in expression and binding of lineage-specific transcription factors (FIG. 2). However, it is challenging to resolve whether differential expression or binding of a given transcription factor is a cause or an indirect effect of differential lineage priming among individual cells.
Whereas scMNase-seq simultaneously measures nucleosome position and chromatin accessibility, several other techniques can additionally measure DNA methylation and/or RNA levels in the same cell83–85 (TABLE 2) and reveal new insights into chromatin dynamics at a single-cell level. For example, a study that used multi-omics profiling of DNA methylation and chromatin accessibility revealed that inhibition of Pol II resulted in attenuation of gene-proximal nucleosome-free regions in pre-implantation mouse embryos85. These regions were enriched for binding motifs associated with essential transcription factors such as SP1 and E2F4, supporting a causative role for transcription in generating and/or maintaining these proximal nucleosome-free regions. Furthermore, tandem profiling of nucleosome positioning and DNA methylation in individual human K562 cells and GM12878 lymphoblastoid cells revealed that nucleosome-free regions were depleted for DNA methylation83. This inverse correlation was found to trend increasingly negative during mouse stem cell differentiation84, suggesting a potential relationship between chromatin accessibility, loss of methylation and lineage priming.
Collectively, the studies discussed in this section support a model in which cell-to-cell variation in nucleosome positioning at active promoters and enhancers links transcriptional heterogeneity to that of chromatin states and informs cell fate decisions through lineage priming.
Histone modifications
Different histone modifications are associated with different chromatin states.
Chromatin states are often described using the presence or absence of histone tail modifications. These post-translational covalent modifications are deposited by dedicated epigenetic machinery and exert dramatic effects on chromatin structure and gene expression29,30,86,87. Chromatin regions enriched in these epigenetic marks exhibit functional characteristics that are not revealed by mapping nucleosome organization alone. Histone modifications have predominantly been profiled using ChIP–seq88,89, which has revealed distinct patterns in which different histone modifications are enriched at transcriptionally active versus transcriptionally repressed chromatin90,91 (FIG. 1). For example, methylation of lysine 4 on histone 3 (H3K4me) is enriched at active genes, whereas H3K27me is enriched at silent genes92. Genes associated with both H3K4me and H3K27me, termed ‘bivalent modifications’, may be primed for future activation or repression depending on cell surface signalling events93,94. Histone tails can also be acetylated to facilitate gene activation, and deacetylation is conversely associated with gene silencing95–97. However, an unbiased genome-wide survey of acetylation profiles revealed that both histone acetyltransferases and histone deacetylases are modestly enriched at many silent gene promoters98. Further investigation suggested a model in which H3K4 is methylated to prime silent promoters, followed by a dynamic cycle of acetylation and deacetylation by transient binding of acetylation machinery. This concerted action of histone methylation, acetylation and deacetylation prevents Pol II from binding to these genes but poises them for future activation98.
Single-cell analyses of histone modifications can identify cell subpopulations.
Following the first report on profiling histone modification at single-cell resolution using single-cell ChIP–seq99, several more recent techniques have been developed including single-cell chromatin immunocleavage followed by sequencing (scChIC–seq)100, single-cell cleavage under targets and release using nuclease (scCUT&RUN)101 and single-cell chromatin integration labelling sequencing (ChIL-seq)102. Methods using Tn5 transposase-mediated tagmentation have also been developed, such as indexed antibody-guided chromatin tagmentation sequencing (iACT-seq)103, cleavage under targets and tagmentation (CUT&TAG)104 and combinatorial barcoding and targeted chromatin release (CoBATCH)105 (TABLE 1). When combined with split-pool barcoding, these methods can dramatically increase the throughput of mapping histone marks in single cells from dozens or hundreds to tens of thousands103.
There are no established parallel methods to validate the variation in histone modifications at a specific genomic locus detected by the above single-cell techniques. These data sets are typically validated by comparing the pooled single-cell data with the gold standard bulk-cell ChIP–seq data and whether they are suitable for computational clustering of cell populations into distinct cell types. If known subpopulations of cells exist within the sample population, such clustering analyses can serve as independent verification that the method is providing useful biological information. For example, clustering analysis performed on profiles of trimethylation of lysine 4 on H3 (H3K4me3) obtained using scChIC–seq in human white blood cells robustly produced clusters corresponding to characterized cell types including B cells, T cells, natural killer cells and monocytes100. Single-cell profiling of acetylation of lysine 27 on H3 (H3K27ac) in mouse endothelial cells by CoBATCH was similarly able to correctly cluster the cells based on their tissue of origin105. These studies reveal that single-cell mapping of histone modifications can identify cellular subpopulations with a similar level of power as the well-established method of clustering based on scRNA-seq data. Furthermore, they support the robustness of biological insights gained using single-cell histone modification mapping.
Single-cell analyses of histone modifications reveal cellular heterogeneity.
The important roles of histone modifications in setting up the chromatin states critical for transcription suggest that cell-to-cell variations in histone modifications contribute to gene expression heterogeneity. Indeed, multiple single-cell studies have identified cellular heterogeneity in histone marks and have established correlations between this heterogeneity and gene expression levels. For example, H3K27ac exhibited variable enrichment in mouse endothelial cells that corresponded to expression of sequence-specific transcription factors linked to specific cell lineages105. Measurements of the histone modification dimethylation of lysine 4 on H3 (H3K4me2), which is associated with gene promoters and enhancers, exhibited considerable variation within a population of mouse embryonic stem cells99. This variability was observed both at pluripotency-related gene enhancers and at transcriptionally repressed genes, and these data sets were capable of resolving three distinct subpopulations of embryonic stem cells99. In addition, single-cell H3K4me3 profiles from various human and mouse cell types exhibited remarkable cellular heterogeneity within each cell type, and this heterogeneity was found to significantly correlate with cellular heterogeneity of gene expression100. In the T cell subset, this heterogeneity seems to be related to differential expression of the transcription factors BCL11B, which is specific to naive T cells, and PRDM1, which is specific to TH1 cells100. Taken together, these studies suggest that heterogeneity in histone modifications may often be correlated with varying degrees of affinity for specific lineage fates among cell populations. Interestingly, histone modifications in immune cells were found to exhibit increasing heterogeneity with age, suggesting a possible change or deterioration in the mechanisms that govern epigenetic heterogeneity and/or immune cell differentiation in the elderly106.
Histone modifications have also been found to be associated with heterogeneity in nucleosome organization. For example, cross-referencing of scMNase-seq data with bulk-cell ChIP–seq data in mouse embryonic stem cells revealed that genomic regions enriched for histone marks associated with active chromatin, such as H3K4me1, H3K4me3, H3K27ac, H3K9ac and H2AZ, exhibited a high degree of positional uniformity compared with regions enriched for the heterochromatic histone modification H3K27me3 (REF.78). Promoters exhibiting bivalent enrichment of active and repressive histone modifications were found to be associated with heterogeneous chromatin accessibility84. These correlations suggest that histone modifications inform nucleosome organization or, alternatively, that both processes are jointly influenced by the same fundamental underlying mechanisms, which ultimately affects the observed cellular heterogeneity in gene expression.
Ideally, the correlations described above between histone tail modifications and other types of epigenomic data sets would be directly tested using multi-omics methods. However, multi-omics approaches that are capable of mapping histone modifications alongside gene expression, nucleosome positioning or chromatin accessibility are not yet available. Such methods would enable, among other applications, direct testing of gene expression from chromatin landscapes enriched for specific histone modifications in the same single cell. Thus, these types of multi-omics methods are needed for elucidating the mechanisms that regulate the cell-to-cell variations in histone modifications and their functional implications.
DNA methylation
DNA methylation dynamics and heterogeneity revealed by bulk-cell methods.
The term DNA methylation usually refers to the modified nucleotide 5-methylcytosine (5mC), which was the first epigenetic factor to be identified and whose discovery predated our understanding of DNA as the material of heredity107,108. Bulk-cell methods for genome-wide mapping of 5mC have provided extensive insights into the distribution and dynamics of DNA methylation. Although 5mC is commonly found within all sequence contexts in flowering plants, mammalian DNA is primarily enriched for 5mC at sequences in which a cytosine is immediately followed by a guanine in the 5′ to 3′ direction109. Genomic regions that exhibit unusually elevated frequencies of these CpG sites are known as ‘CpG islands’, which are present at over two-thirds of gene promoters and can serve as epigenetic regulatory switches that restrict gene expression when methylated110. Gene silencing mediated by methylation of promoter CpG islands can occur both during normal development and during tumorigenesis111–113. Compared with the relatively plastic transcriptional repression conferred by histone modifications, gene silencing via promoter DNA methylation is more durable and persistent114. Perhaps as a consequence of this stability, this is the primary epigenetic silencing mechanism used for repression of endogenous transposons, imprinted genes and pluripotency-related genes in somatic cells114.
Although most DNA methylation is relatively stable, whole-genome bisulfite sequencing (BS-seq) of bulk cells has revealed that many enhancers and transcription factor binding sites exhibit dynamic methylation states that vary in enrichment among different human cell and tissue types115. The binding of DNA-associated proteins at these sites is thought to compete with DNA methyl-transferase activity to generate variable methylation patterns115,116.
Single-cell and multi-omics approaches reveal DNA methylation heterogeneity.
The advent of single-cell methods for genome-wide 5mC profiling (TABLE 1), including single-cell BS-seq, has revealed substantial DNA methylation heterogeneity among both mouse and human cells117–119. For example, the use of allele-specific reporters revealed that DNA methylation variability at regulatory elements directly affects transcription of associated genes and contributes to gene expression heterogeneity among cells120. Relative to other epigenomic data types, there are many multi-omics techniques suitable for investigating functional interactions between DNA methylation heterogeneity, gene expression and, occasionally, other epigenetic marks in single cells83–85,121–125 (TABLE 2). As a consequence, many such functional interactions have been detailed. For example, promoter methylation was found to correlate with transcriptional silencing, but methylation at distal regulatory elements exhibited a balance of positive and negative correlations with expression of associated genes123. These observations reveal that DNA methylation likely plays differing roles at gene promoters and enhancers. Interestingly, heterogeneous methylation of distal regulatory elements was linked to heterogeneous gene expression using single-cell methylome and transcriptome sequencing (scM&T), revealing a functional link between DNA methylation and gene expression variability123. Although computational clustering of the scM&T data identified substantial correlations between methylation and gene expression, clustering patterns unique to either DNA methylation or expression data were also present123. This observation reveals that DNA methylation and gene expression are complementary but distinct phenomena. Another study also used scM&T to co-profile DNA methylation and gene expression in mouse muscle stem cells124. This study found that DNA methylation heterogeneity at gene promoters was linked to greater levels of expression heterogeneity in the associated genes. The concurring observations made in these studies strongly support a functional interaction between gene expression heterogeneity and DNA methylation heterogeneity in multiple cell types.
Several methods are available for co-profiling DNA methylation and other epigenetic factors, such as nucleosome positioning and chromatin contacts, in the same single cells (TABLE 2). Data obtained using these methods have provided fascinating information about the roles of DNA methylation in chromatin biology. For example, single-nucleus methyl-chromatin conformation capture sequencing (sn-m3C-seq) was used to co-profile 5mC and chromatin conformation in individual human brain prefrontal cortex cells121. This method leverages 5mC profiles to accurately cluster cells based on cell type, and subsequently enables identification of cell type-specific features of chromatin organization. Using this approach, cell type-specific chromatin loops were identified as well as correlations between cell type-specific contacts and DNA methylation enrichment121. A similar study used methyl-HiC to co-profile 5mC and chromatin contacts in individual cells125, revealing coordination of DNA methylation states among distal genomic regions that were in close spatial proximity in nuclei of mouse embryonic stem cells. DNA methylation has also been examined alongside nucleosome positioning: single-cell chromatin overall omic-scale landscape sequencing (scCOOL-seq) performed on cells from multiple mouse pre-implantation embryos revealed strong epigenetic distinctions between individuals85. For example, heterogeneity in both 5mC distributions and nucleosome positioning was greater between embryos than among cells from the same embryo, suggesting that care must be taken when interpreting studies in which single-cell epigenomic data sets are collected from multiple animals or human patients. In summary, these multi-omics approaches that profile DNA methylation alongside gene expression and other epigenetic features provide unique insights into chromatin biology and functional relationships involving DNA methylation heterogeneity. Furthermore, they highlight the benefits that will be obtained from future extension of multi-omics methods to other epigenomic data types.
Enhancer–promoter interactions
3C-based methods map chromatin contacts and enhancer–promoter interactions.
In mammalian genomes, cis-regulatory elements direct transcription of genes and are often located many kilobases away from the genes they regulate. Among these regulatory elements are enhancers, which can promote expression of target genes across long genomic distances containing multiple unassociated genes126. Mammalian genomes contain many times more enhancers than genes127, illustrating the important role that these regulatory systems play in gene expression28. In particular, precise spatial and temporal gene expression during development is often achieved using complex networks of enhancers128. Enhancers are able to regulate transcription by forming physical interactions with gene promoters via chromatin looping and folding129,130. Such enhancer–promoter interactions are examples of long-range contacts that occur as a result of the spatial arrangement of chromatin. Individual pairwise contacts can be quantified in bulk-cell samples using chromatin conformation capture (3C)131. Derivative methods have expanded the number of loci that can be simultaneously examined132–135 — culminating with Hi-C136, a sequencing-based method capable of genome-wide quantification of long-range chromatin interactions. Computational analysis of bulk-cell Hi-C data has facilitated mapping of enhancer–promoter interactions and is thus helping to clarify the regulatory relationships that govern gene expression137–140. For example, many enhancer–promoter interactions have been found to occur concomitantly with gene expression and are abolished when genes are repressed141. The functional nature of these contacts is supported by strong correlations with enrichment of active histone modifications and transcription factor binding142.
Bulk-cell approaches for mapping chromatin contacts provide sample-average signals and cannot resolve cell-to-cell differences or heterogeneity. To investigate the spatial arrangement of chromatin on a single-cell level, various methods based on Hi-C have been introduced121,125,143–146 (TABLE 1). Using these techniques, substantial heterogeneity in chromatin contacts has been observed in various cell types, which correlated with distinct developmental states. For example, a study using single-cell Hi-C observed that genome organization varied dramatically among individual mouse embryonic stem cells based on the cell cycle stage143, revealing a layer of complexity to genome organization that it would not be possible to observe in a passaging culture using bulk-cell samples. Single-cell techniques have additionally enabled researchers to dissect the contributions of chromatin contacts to developmental events in stem cells and during fertilization of oocytes. For example, computational clustering of multi-omics Hi-C and DNA methylation data from mouse embryonic stem cells revealed the presence of an embryonic stem cell subset that exhibited a methylome pattern that was associated with embryonic limb development125, raising the possibility that these cells are epigenetically primed for specific differentiation programmes. Furthermore, single-nucleus interactome mapping revealed a spatial reorganization of chromatin that occurs during the oocyte-to-zygote transition in mice and contrasted these organizational states with those of somatic cells144. The sensitivity provided by single-cell techniques was necessary to characterize this developmental transition as it occurs at the single-cell stage. The results of these studies highlight the biological insights that can be gained using the sensitivity and resolution provided by single-cell mapping of chromatin contacts. Importantly, imaging-based parallel techniques have been developed that are capable of validating the chromatin interactions identified using sequencing-based methods. For example, Hi-M, a high-throughput, high-resolution, high-coverage, microscopy-based technique, can simultaneously visualize transcriptional activity and chromosome organization in individual cells of intact Drosophila embryos49. Similarly, multiple super high-resolution microscopy studies are providing information about chromatin folding and interactions on the single-locus, kilobase scale147–149. Although they are relatively new, these highly sensitive imaging-based approaches represent an exciting frontier in the mapping of chromatin contacts and may facilitate co-visualization of multiple types of epigenomic data in the future.
CTCF facilitates enhancer–promoter interactions and limits expression heterogeneity.
Enhancer–promoter contacts are regulated in cis by DNA regulatory elements and in trans by chromatin-binding factors. Among the most studied trans factors are the cohesin complex and CTCF150–152. CTCF is well-established as a key regulator in the formation of long-range chromatin contacts and the structure of larger topologically associating domains, and is enriched at the boundaries between topologically associating domains or between condensed and open chromatin regions79,153–155. Deletion of CTCF-binding sites can disrupt insulation between these domains155,156, and a single-cell Hi-C study revealed that CTCF/cohesin-mediated chromatin loops are heterogeneous among individual cells146. In addition to a role in the structural organization of chromatin domains, recent data suggest that CTCF also contributes to the more dynamic enhancer–promoter interactions142. An unbiased analysis of chromatin interactions using the three-enzyme Hi-C (3eHi-C) assay in the mouse EL4 cell line revealed positive correlations between CTCF binding, interactivity of regulatory regions and enhancer activity142. Furthermore, CTCF-binding sites were found to be interspersed with enhancer elements, and active gene promoters exhibited greater degrees of interaction with CTCF-binding sites than did inactive promoters142. Interestingly, although there were only very modest decreases in gene expression levels upon CTCF knockdown, the depletion of CTCF resulted in significantly increased heterogeneity in expression of the T cell-specific genes Gata3, Thy1, Cd28 and Cd5. In addition, CriSPr–Cas9-mediated deletion of specific CTCF-binding sites at the Th1, Cd5 and Runx3 loci compromised the respective enhancer–promoter interaction and resulted in increased cell-to-cell variation of their expression142. Taken together, these observations support a role for enhancer–promoter contacts in restricting gene expression heterogeneity of developmental genes among cell populations157.
How might CTCF contribute to enhancer–promoter interactions and control of gene expression variation? Considering that CTCF and cohesin interact physically and functionally158–160, and that CTCF-binding sites and enhancers are interspersed with each other in the genome142, one plausible mechanism is that CTCF binds to regions near enhancers and promoters and brings these elements into proximity via interactions with cohesin. As a consequence, this would increase the local ‘concentration’ of enhancer and promoter elements, which would favour enhancer–promoter interactions and induce expression of target genes (FIG. 5). In this model, decreases in CTCF binding and enhancer–promoter interactions would decrease the effectiveness and consistency of transcriptional activation and increase the variation of target gene expression.
Fig. 5 |. Single-cell Hi-C-based techniques measure the variability of enhancer–promoter contacts.
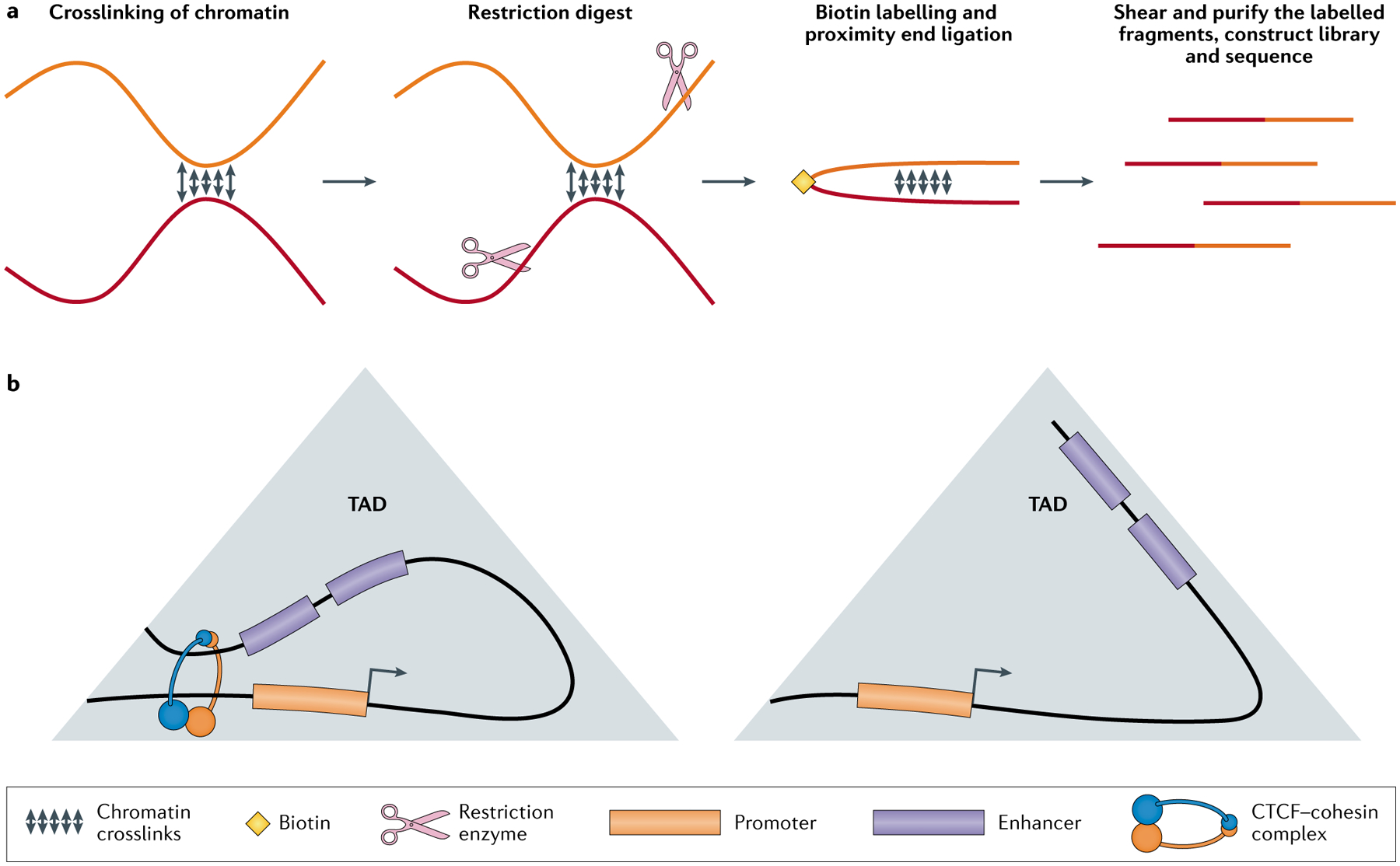
a | Simple depiction of how a chromatin contact is captured by Hi-C. Distal genomic regions in close spatial proximity are indicated using different colours. The chromatin is crosslinked to preserve the interactions, followed by a sequence-specific restriction digest to cut the chromatin into fragments. The cut ends are then ligated together and labelled with biotin. Finally, the crosslinking is reversed and the DNA fragments are purified using the biotin label and sequenced. b | Depiction of topologically associating domains (TADs), which are chromatin regions comprising many intra-regional spatial interactions. TAD exhibiting enhancer–promoter contacts facilitated by CCCTC-binding factor (CTCF) and cohesin. CTCF and cohesin bring distal genomic elements together into domains containing elevated local concentrations of enhancer (orange) and promoter (blue) elements (left). Absence of CTCF-mediated and cohesin-mediated interactions affects transcriptional heterogeneity without disrupting overall TAD structure (right). Hi-C, high-throughput chromosome conformation capture.
Expression of developmental genes occurs in an intermittent pulsing pattern that transitions between active and inactive transcriptional states at irregular intervals161,162. This sporadic activation was found to contribute to gene expression heterogeneity between cells162. Furthermore, the kinetics of these transcriptional bursts are highly gene-specific and controlled by non-random factors such as nearby cis-regulatory elements and the characteristics of the surrounding chromatin environment23,163,164. In a recent single-molecule imaging study in live breast cancer cells, transcription of the oestrogen-dependent gene TFF1 exhibited remarkably heterogeneous expression levels across different cells upon treatment with oestradiol, even in the presence of saturating amounts of the hormone23. Interestingly, this heterogeneity was found to correspond with a pronounced variability in the duration of the transcriptionally inactive state. Deletion of the proximal enhancer upstream of TFF1 resulted in reduced expression of the gene owing to twofold fewer transcription bursts compared with the parental cell line. This result suggests that enhancer–promoter contacts are integral for controlling the frequency with which a transcriptionally active state occurs in developmentally regulated genes, and are thus important for limiting gene expression heterogeneity among cell populations23. However, it is not clear how the transcriptionally inactive state of TFF1 remains unresponsive to oestradiol stimulation whereas the active state can respond with remarkably increased frequency of transcription bursts. One possibility is that the response may require a particular histone modification pattern. This idea is supported by the observation that chemical inhibition of the oestrogen-specific chromatin binding protein TRIM24 (REF.165), which blocks bromodomain binding to acetylated histones, caused a threefold reduction of TFF1 induction23. Together, these studies support the importance of enhancer–promoter interactions in controlling gene expression heterogeneity among cells. Furthermore, they illustrate the power of parallel non-sequencing approaches to test hypotheses generated by genome-wide, sequencing-based methods.
Conclusion and future perspectives
An increasing body of data indicate that cellular heterogeneity in gene expression does not simply reflect transient noise in transcription, but instead has a basis in epigenetic states and has been implicated in various biological processes23,56,57,100,120,123,124,142. In particular, cell-to-cell variation in chromatin accessibility, nucleosome positioning, histone modifications and DNA methylation at enhancers and promoters is positively correlated with gene expression variation in established cell lines as well as primary tissues. Additionally, long-distance chromatin interactions exhibit substantial variations across different single cells125,143. Among these interactions are CTCF-dependent enhancer–promoter contacts, which may play an important role in restricting gene expression heterogeneity23,146. Although the mechanisms by which epigenetic variations are regulated remain to be determined in many cases, it is clear that DNA-binding transcription factors are important contributors. For example, pioneer transcription factors such as GATA2 may bind to nucleosome-occupied DNA, open up chromatin and facilitate transcriptional activation73,74. Thus, cellular heterogeneity in expression and/or binding of these factors likely results in heterogeneity of chromatin accessibility, nucleosome positioning and histone modifications, and may be responsible for epigenetic variations associated with lineage priming and cell fate determination. By contrast, cell surface signalling-activated factors such as AP-1 may simply recognize the variably accessible chromatin regions and be responsible for differential response to signalling in terminally differentiated cells. Taken together, the interplay of various transcription factors and different aspects of chromatin organization in each cell may collectively define transcriptional outputs of individual genes, and hence the observed cellular heterogeneity in gene expression.
Current single-cell epigenomic techniques suffer from data loss. As a consequence, even though single-cell epigenomic data sets are powerful resources for clustering analyses and for revealing cellular heterogeneity based on the collection of a large number of target sites, they have only very limited power in providing information on individual target sites. Thus, future studies will need to improve the recovery of chromatin target sites in various single-cell epigenomic assays, which will help to reduce technical variations between different single cells and facilitate understanding of cellular heterogeneity not only at a whole cell level but also at individual sites. The ability to assay epigenetic heterogeneity at specific genomic sites will enhance the utility of single-cell methods in understanding processes such as differential priming for cell fate decisions among populations of stem cells, human immune response against pathogens and drug responses during disease treatment.
Multi-omics techniques provide information on two or more features of gene expression and/or chromatin states in the same cell, which could facilitate correlation analysis of the features under investigation. New multi-omics techniques such as those that measure both RNA and histone modifications or chromatin-binding proteins in the same cell will be needed to investigate how cis and trans factors control cellular epigenomic heterogeneity and how they contribute to transcriptional regulation. In combination with genome editing tools such as CRISPR–Cas9 to control various cis elements and trans factors, single-cell epigenomics assays could be applied to test the causality of chromatin modifications and cellular heterogeneity in various systems.
Rapid advances are being made in the throughput and resolution of experimental techniques to map chromatin landscapes in single cells. Moving forward, these techniques promise to provide detailed profiles of cellular states and will facilitate a much deeper understanding of the underlying mechanisms that control gene expression heterogeneity.
Acknowledgements
The research in the authors’ laboratory is supported by the Division of Intramural Research, National Heart, Lung, and Blood Institute, National Institutes of Health.
Glossary
- Chromatin
genomic material found in the cell nucleus comprising DNA in complex with structural proteins such as histones.
- Epigenetic marks
Changes in chromatin including covalent modifications of DNA and histones that are persistent and often associated with changes in gene expression.
- Chromatin immunoprecipitation followed by sequencing
(ChiP–seq). A commonly used method for profiling epigenetic marks or chromatin binding proteins. The method is based on the affinity of an antibody for its target antigen(s).
- Heterochromatic
Pertaining to heterochromatin, which is tightly packaged chromatin that is associated with transcriptional repression and enriched for characteristic epigenetic marks such as trimethylation of lysine 9 at histone 3 (H3K9me3).
- Fluorescence-activated cell sorting
A method used to separate and distribute a cellular subpopulation from a larger sample based on fluorescence, typically accomplished via expression of a fluorophore transgene or via labelling with a fluorescent antibody.
- Bivalent modifications
genomic regions exhibiting co-enrichment of epigenetic marks considered to be transcriptionally activating and transcriptionally repressive. in particular, they often refer to dual enrichment of the histone modifications trimethylation of lysine 4 at histone 3 (H3K4me3) and H3K27me3.
- Tagmentation
The use of a transposase to fragment DNA and append sequence tags to the resulting fragments in a single experimental step.
- Split-pool barcoding
A procedure that uses libraries of sequence tags to obtain information at a single-cell level without the need for sorting of individual cells. This is accomplished by splitting samples into small aliquots, adding a barcode and pooling them. This process is repeated two or three times to ensure that each cell is highly likely to receive a unique combination of barcodes.
- Imprinted genes
genes that exhibit allele-specific or allele-biased expression depending on the parent of origin.
- CRISPR–Cas9
A method for genome engineering that is derived from the CriSPr system of antiviral defence in bacteria.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Toyooka Y, Shimosato D, Murakami K, Takahashi K & Niwa H Identification and characterization of subpopulations in undifferentiated ES cell culture. Development 135, 909–918 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Chang HH, Hemberg M, Barahona M, Ingber DE & Huang S Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature 453, 544–547 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bumgarner SL et al. Single-cell analysis reveals that noncoding RNAs contribute to clonal heterogeneity by modulating transcription factor recruitment. Mol. Cell 45, 470–482 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shalek AK et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 510, 363–369 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rutledge EA, Benazet JD & McMahon AP Cellular heterogeneity in the ureteric progenitor niche and distinct profiles of branching morphogenesis in organ development. Development 144, 3177–3188 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morales M & Margolis EB Ventral tegmental area: cellular heterogeneity, connectivity and behaviour. Nat. Rev. Neurosci 18, 73–85 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Nguyen QH et al. Single-cell RNA-seq of human induced pluripotent stem cells reveals cellular heterogeneity and cell state transitions between subpopulations. Genome Res. 28, 1053–1066 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y et al. Bacterial single cell whole transcriptome amplification in microfluidic platform shows putative gene expression heterogeneity. Anal. Chem 91, 8036–8044 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang TQ, Xu ZG, Shang GD & Wang JW A single-cell RNA sequencing profiles the developmental landscape of arabidopsis root. Mol. Plant 12, 648–660 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Calbo J et al. A functional role for tumor cell heterogeneity in a mouse model of small cell lung cancer. Cancer Cell 19, 244–256 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Tellez-Gabriel M, Ory B, Lamoureux F, Heymann MF & Heymann D Tumour heterogeneity: the key advantages of single-cell analysis. Int. J. Mol. Sci 17, 2142 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao Q et al. Single-cell transcriptome analyses reveal endothelial cell heterogeneity in tumors and changes following antiangiogenic treatment. Cancer Res. 78, 2370–2382 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Wen L & Tang F Single-cell sequencing in stem cell biology. Genome Biol. 17, 71 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walzer KA, Fradin H, Emerson LY, Corcoran DL & Chi JT Latent transcriptional variations of individual Plasmodium falciparum uncovered by single-cell RNA-seq and fluorescence imaging. PLoS Genet. 15, e1008506 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes an interesting example of how gene expression heterogeneity is relevant for human infectious diseases.
- 15.Dagogo-Jack I & Shaw AT Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol 15, 81–94 (2018). [DOI] [PubMed] [Google Scholar]; This article reviews tumour cell heterogeneity and its implications for cancer treatments.
- 16.Heppner GH Tumor heterogeneity. Cancer Res. 44, 2259–2265 (1984). [PubMed] [Google Scholar]
- 17.Meacham CE & Morrison SJ Tumour heterogeneity and cancer cell plasticity. Nature 501, 328–337 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eun K, Ham SW & Kim H Cancer stem cell heterogeneity: origin and new perspectives on CSC targeting. BMB Rep. 50, 117–125 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng H et al. Single-cell analysis reveals cancer stem cell heterogeneity in hepatocellular carcinoma. Hepatology 68, 127–140 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prasetyanti PR & Medema JP Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 16, 41 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tunnacliffe E & Chubb JR What is a transcriptional burst? Trends Genet. 36, 288–297 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Brouwer I & Lenstra TL Visualizing transcription: key to understanding gene expression dynamics. Curr. Opin. Chem. Biol 51, 122–129 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Rodriguez J et al. Intrinsic dynamics of a human gene reveal the basis of expression heterogeneity. Cell 176, 213–226.e18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes a fascinating use of live-cell imaging to dissect transcription dynamics.
- 24.Harper CV et al. Dynamic analysis of stochastic transcription cycles. PLoS Biol. 9, e1000607 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lambert SA et al. The human transcription factors. Cell 175, 598–599 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Haberle V & Stark A Eukaryotic core promoters and the functional basis of transcription initiation. Nat. Rev. Mol. Cell Biol 19, 621–637 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cramer P Organization and regulation of gene transcription. Nature 573, 45–54 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Schoenfelder S & Fraser P Long-range enhancer–promoter contacts in gene expression control. Nat. Rev. Genet 20, 437–455 (2019). [DOI] [PubMed] [Google Scholar]; This review article illustrates the concepts of chromatin contacts and how they facilitate control of gene expression.
- 29.Schuettengruber B, Bourbon HM, Di Croce L & Cavalli G Genome regulation by polycomb and trithorax: 70 years and counting. Cell 171, 34–57 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Venkatesh S & Workman JL Histone exchange, chromatin structure and the regulation of transcription. Nat. Rev. Mol. Cell Biol 16, 178–189 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Cheow LF et al. Single-cell multimodal profiling reveals cellular epigenetic heterogeneity. Nat. Methods 13, 833–836 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Gay L, Baker AM & Graham TA Tumour cell heterogeneity. F1000Research 5, 238 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grosselin K et al. High-throughput single-cell ChIP–seq identifies heterogeneity of chromatin states in breast cancer. Nat. Genet 51, 1060–1066 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Linker SM et al. Combined single-cell profiling of expression and DNA methylation reveals splicing regulation and heterogeneity. Genome Biol. 20, 30 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei G et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 30, 155–167 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eberwine J et al. Analysis of gene expression in single live neurons. Proc. Natl Acad. Sci. USA 89, 3010–3014 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article presents an interesting look at the origins of single-cell gene expression measurements.
- 37.Klein CA et al. Combined transcriptome and genome analysis of single micrometastatic cells. Nat. Biotechnol 20, 387–392 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Lennon GG & Lehrach H Hybridization analyses of arrayed cDNA libraries. Trends Genet. 7, 314–317 (1991). [DOI] [PubMed] [Google Scholar]
- 39.Kurimoto K et al. An improved single-cell cDNA amplification method for efficient high-density oligonucleotide microarray analysis. Nucleic Acids Res. 34, e42 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Emrich SJ, Barbazuk WB, Li L & Schnable PS Gene discovery and annotation using LCM-454 transcriptome sequencing. Genome Res. 17, 69–73 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lister R et al. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 133, 523–536 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Erickson KE, Otoupal PB & Chatterjee A Gene expression variability underlies adaptive resistance in phenotypically heterogeneous bacterial populations. ACS Infect. Dis 1, 555–567 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Eraslan G, Simon LM, Mircea M, Mueller NS & Theis FJ Single-cell RNA-seq denoising using a deep count autoencoder. Nat. Commun 10, 390 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bai YL, Baddoo M, Flemington EK, Nakhoul HN & Liu YZ Screen technical noise in single cell RNA sequencing data. Genomics 112, 346–355 (2020). [DOI] [PubMed] [Google Scholar]
- 45.Li R & Quon G scBFA: modeling detection patterns to mitigate technical noise in large-scale single-cell genomics data. Genome Biol. 20, 193 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ziegenhain C et al. Comparative analysis of single-cell RNA sequencing methods. Mol. Cell 65, 631–643.e4 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A & Tyagi S Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 5, 877–879 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bintu B et al. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 362, eaau1783 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cardozo Gizzi AM et al. Microscopy-based chromosome conformation capture enables simultaneous visualization of genome organization and transcription in intact organisms. Mol. Cell 74, 212–222.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Liu M et al. Multiplexed imaging of nucleome architectures in single cells of mammalian tissue. Nat. Commun 11, 2907 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klemm SL, Shipony Z & Greenleaf WJ Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet 20, 207–220 (2019). [DOI] [PubMed] [Google Scholar]
- 52.Song L & Crawford GE DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb. Protoc 2010, pdb.prot5384 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu C, Bingham PM, Livak KJ, Holmgren R & Elgin SC The chromatin structure of specific genes: I. Evidence for higher order domains of defined DNA sequence. Cell 16, 797–806 (1979). [DOI] [PubMed] [Google Scholar]
- 54.Buenrostro JD, Giresi PG, Zaba LC, Chang HY & Greenleaf WJ Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cusanovich DA et al. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes a pioneering example of how split-pool barcoding can be used to profile epigenetic marks in single cells.
- 56.Buenrostro JD et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jin W et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature 528, 142–146 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lareau CA et al. Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nat. Biotechnol 37, 916–924 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mezger A et al. High-throughput chromatin accessibility profiling at single-cell resolution. Nat. Commun 9, 3647 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhu C et al. An ultra high-throughput method for single-cell joint analysis of open chromatin and transcriptome. Nat. Struct. Mol. Biol 26, 1063–1070 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reyes M, Billman K, Hacohen N & Blainey PC Simultaneous profiling of gene expression and chromatin accessibility in single cells. Adv. Biosyst 3, 1900065 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu L et al. Deconvolution of single-cell multi-omics layers reveals regulatory heterogeneity. Nat. Commun 10, 470 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cao J et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380–1385 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen S, Lake BB & Zhang K High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat. Biotechnol 37, 1452–1457 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Davis CA et al. The Encyclopedia of DNA Elements (ENCODE): data portal update. Nucleic Acids Res. 46, D794–D801 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Crispino JD & Horwitz MS GATA factor mutations in hematologic disease. Blood 129, 2103–2110 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lentjes MHFM et al. The emerging role of GATA transcription factors in development and disease. Expert. Rev. Mol. Med 18, e3 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jia G et al. Single cell RNA-seq and ATAC-seq analysis of cardiac progenitor cell transition states and lineage settlement. Nat. Commun 9, 4877 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Satpathy AT et al. Transcript-indexed ATAC-seq for precision immune profiling. Nat. Med 24, 580–590 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bossard P & Zaret KS GATA transcription factors as potentiators of gut endoderm differentiation. Development 125, 4909 (1998). [DOI] [PubMed] [Google Scholar]
- 71.Barozzi I et al. Coregulation of transcription factor binding and nucleosome occupancy through DNA features of mammalian enhancers. Mol. Cell 54, 844–857 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tremblay M, Sanchez-Ferras O & Bouchard M GATA transcription factors in development and disease. Development 145, dev164384 (2018). [DOI] [PubMed] [Google Scholar]
- 73.Cirillo LA et al. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell 9, 279–289 (2002). [DOI] [PubMed] [Google Scholar]
- 74.Iwafuchi-Doi M & Zaret KS Pioneer transcription factors in cell reprogramming. Genes Dev. 28, 2679–2692 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Litzenburger UM et al. Single-cell epigenomic variability reveals functional cancer heterogeneity. Genome Biol. 18, 15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cusanovich DA et al. A single-cell atlas of in vivo mammalian chromatin accessibility. Cell 174, 1309–1324.e18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schones DE et al. Dynamic regulation of nucleosome positioning in the human genome. Cell 132, 887–898 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lai B et al. Principles of nucleosome organization revealed by single-cell micrococcal nuclease sequencing. Nature 562, 281–285 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yuan GC et al. Genome-scale identification of nucleosome positions in S. cerevisiae. Science 309, 626–630 (2005). [DOI] [PubMed] [Google Scholar]
- 80.Ozsolak F, Song JS, Liu XS & Fisher DE High-throughput mapping of the chromatin structure of human promoters. Nat. Biotechnol 25, 244–248 (2007). [DOI] [PubMed] [Google Scholar]
- 81.Lai WKM & Pugh BF Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nat. Rev. Mol. Cell Biol 18, 548–562 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hughes AL & Rando OJ Mechanisms underlying nucleosome positioning in vivo. Annu. Rev. Biophys 43, 41–63 (2014). [DOI] [PubMed] [Google Scholar]
- 83.Pott S Simultaneous measurement of chromatin accessibility, DNA methylation, and nucleosome phasing in single cells. eLife 6, e23203 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Clark SJ et al. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat. Commun 9, 781 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Guo F et al. Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells. Cell Res. 27, 967–988 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boland MJ, Nazor KL & Loring JF Epigenetic regulation of pluripotency and differentiation. Circ. Res 115, 311–324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Atlasi Y & Stunnenberg HG The interplay of epigenetic marks during stem cell differentiation and development. Nat. Rev. Genet 18, 643–658 (2017). [DOI] [PubMed] [Google Scholar]
- 88.Barski A et al. High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 (2007). [DOI] [PubMed] [Google Scholar]
- 89.Mikkelsen TS et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang Z, Schones DE & Zhao K Characterization of human epigenomes. Curr. Opin. Genet. Dev 19, 127–134 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bannister AJ & Kouzarides T Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hyun K, Jeon J, Park K & Kim J Writing, erasing and reading histone lysine methylations. Exp. Mol. Med 49, e324 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bernstein BE et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–326 (2006). [DOI] [PubMed] [Google Scholar]
- 94.Roh TY, Cuddapah S, Cui K & Zhao K The genomic landscape of histone modifications in human T cells. Proc. Natl Acad. Sci. USA 103, 15782–15787 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang Z et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet 40, 897–903 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Graff J & Tsai LH Histone acetylation: molecular mnemonics on the chromatin. Nat. Rev. Neurosci 14, 97–111 (2013). [DOI] [PubMed] [Google Scholar]
- 97.Marmorstein R & Zhou MM Writers and readers of histone acetylation: structure, mechanism, and inhibition. Cold Spring Harb. Perspect. Biol 6, a018762 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang Z et al. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 138, 1019–1031 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rotem A et al. Single-cell ChIP–seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol 33, 1165–1172 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes the early use of drop fluidics to profile histone modifications in single cells, which has formed the basis of multiple commercially available technologies.
- 100.Ku WL et al. Single-cell chromatin immunocleavage sequencing (scChIC-seq) to profile histone modification. Nat. Methods 16, 323–325 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hainer SJ, Boskovic A, McCannell KN, Rando OJ & Fazzio TG Profiling of pluripotency factors in single cells and early embryos. Cell 177, 1319–1329.e11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Harada A et al. A chromatin integration labelling method enables epigenomic profiling with lower input. Nat. Cell Biol 21, 287–296 (2019). [DOI] [PubMed] [Google Scholar]
- 103.Carter B et al. Mapping histone modifications in low cell number and single cells using antibody-guided chromatin tagmentation (ACT-seq). Nat. Commun 10, 3747–3747 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article outlines the application of split-pool barcoding to profiling histone modifications.
- 104.Kaya-Okur HS et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun 10, 1930 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang Q et al. CoBATCH for high-throughput single-cell epigenomic profiling. Mol. Cell 76, 206–216.e7 (2019). [DOI] [PubMed] [Google Scholar]
- 106.Cheung P et al. Single-cell chromatin modification profiling reveals increased epigenetic variations with aging. Cell 173, 1385–1397.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Johnson TB & Coghill RD Researches on pyrimidines. C111. The discovery of 5-methyl-cytosine in tuberculinic acid, the nucleic acid of the tubercle bacillus. J. Am. Chem. Soc 47, 2838–2844 (1925). [Google Scholar]; This article provides an interesting historical look at the discovery of 5mC prior to our understanding of DNA’s role in genetics.
- 108.Hotchkiss RD The quantitative separation of purines, pyrimidines, and nucleosides by paper chromatography. J. Biol. Chem 175, 315–332 (1948). [PubMed] [Google Scholar]
- 109.Feng S et al. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl Acad. Sci. USA 107, 8689–8694 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Deaton AM & Bird A CpG islands and the regulation of transcription. Genes Dev. 25, 1010–1022 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ferreira HJ & Esteller M CpG islands in cancer: heads, tails, and sides. Methods Mol. Biol 1766, 49–80 (2018). [DOI] [PubMed] [Google Scholar]
- 112.Ando M et al. Chromatin dysregulation and DNA methylation at transcription start sites associated with transcriptional repression in cancers. Nat. Commun 10, 2188 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Greenberg MVC & Bourc’his D The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol 20, 590–607 (2019). [DOI] [PubMed] [Google Scholar]
- 114.Reik W Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 447, 425–432 (2007). [DOI] [PubMed] [Google Scholar]
- 115.Ziller MJ et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 500, 477–481 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Stadler MB et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 480, 490–495 (2011). [DOI] [PubMed] [Google Scholar]
- 117.Farlik M et al. Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics. Cell Rep. 10, 1386–1397 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hui T et al. High-resolution single-cell DNA methylation measurements reveal epigenetically distinct hematopoietic stem cell subpopulations. Stem Cell Rep. 11, 578–592 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Smallwood SA et al. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods 11, 817–820 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Song Y et al. Dynamic enhancer DNA methylation as basis for transcriptional and cellular heterogeneity of ESCs. Mol. Cell 75, 905–920.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lee DS et al. Simultaneous profiling of 3D genome structure and DNA methylation in single human cells. Nat. Methods 16, 999–1006 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hou Y et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 26, 304–319 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Angermueller C et al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat. Methods 13, 229–232 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hernando-Herraez I et al. Ageing affects DNA methylation drift and transcriptional cell-to-cell variability in mouse muscle stem cells. Nat. Commun 10, 4361 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Li G et al. Joint profiling of DNA methylation and chromatin architecture in single cells. Nat. Methods 16, 991–993 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kvon EZ et al. Genome-scale functional characterization of Drosophila developmental enhancers in vivo. Nature 512, 91–95 (2014). [DOI] [PubMed] [Google Scholar]
- 127.Dunham I et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Furlong EEM & Levine M Developmental enhancers and chromosome topology. Science 361, 1341–1345 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Robson MI, Ringel AR & Mundlos S Regulatory landscaping: how enhancer–promoter communication is sculpted in 3D. Mol. Cell 74, 1110–1122 (2019). [DOI] [PubMed] [Google Scholar]
- 130.Deng W et al. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell 149, 1233–1244 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dekker J, Rippe K, Dekker M & Kleckner N Capturing chromosome conformation. Science 295, 1306–1311 (2002). [DOI] [PubMed] [Google Scholar]
- 132.Dostie J et al. Chromosome conformation capture carbon copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 16, 1299–1309 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Simonis M et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat. Genet 38, 1348–1354 (2006). [DOI] [PubMed] [Google Scholar]
- 134.Würtele H & Chartrand P Genome-wide scanning of HoxB1-associated loci in mouse ES cells using an open-ended chromosome conformation capture methodology. Chromosome Res. 14, 477–495 (2006). [DOI] [PubMed] [Google Scholar]
- 135.Zhao Z et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra-and interchromosomal interactions. Nat. Genet 38, 1341–1347 (2006). [DOI] [PubMed] [Google Scholar]
- 136.Lieberman-Aiden E et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ron G, Globerson Y, Moran D & Kaplan T Promoter–enhancer interactions identified from Hi-C data using probabilistic models and hierarchical topological domains. Nat. Commun 8, 2237–2237 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Xu H, Zhang S, Yi X, Plewczynski D & Li MJ Exploring 3D chromatin contacts in gene regulation: the evolution of approaches for the identification of functional enhancer–promoter interaction. Comput. Struct. Biotechnol. J 18, 558–570 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lu L et al. Robust Hi-C maps of enhancer–promoter interactions reveal the function of non-coding genome in neural development and diseases. Mol. Cell 79, 521–534.e15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Schoenfelder S et al. Divergent wiring of repressive and active chromatin interactions between mouse embryonic and trophoblast lineages. Nat. Commun 9, 4189 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bonev B et al. Multiscale 3D genome rewiring during mouse neural development. Cell 171, 557–572.e24 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ren G et al. CTCF-mediated enhancer–promoter interaction is a critical regulator of cell-to-cell variation of gene expression. Mol. Cell 67, 1049–1058.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study informs much of our discussion of how CTCF-mediated chromatin contacts affect gene expression heterogeneity.
- 143.Nagano T et al. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature 547, 61–67 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Flyamer IM et al. Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature 544, 110–114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ramani V et al. Massively multiplex single-cell Hi-C. Nat. Methods 14, 263–266 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Stevens TJ et al. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 544, 59–64 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Fabre PJ et al. Nanoscale spatial organization of the HoxD gene cluster in distinct transcriptional states. Proc. Natl Acad. Sci. USA 112, 13964 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Boettiger AN et al. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 529, 418–422 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cook GW et al. Structural variation and its potential impact on genome instability: novel discoveries in the EGFR landscape by long-read sequencing. PLoS ONE 15, e0226340 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Rao SS et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gosalia N, Neems D, Kerschner JL, Kosak ST & Harris A Architectural proteins CTCF and cohesin have distinct roles in modulating the higher order structure and expression of the CFTR locus. Nucleic Acids Res. 42, 9612–9622 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Jia Z et al. Tandem CTCF sites function as insulators to balance spatial chromatin contacts and topological enhancer–promoter selection. Genome Biol. 21, 75 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tang Z et al. CTCF-mediated human 3D genome architecture reveals chromatin topology for transcription. Cell 163, 1611–1627 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Cuddapah S et al. Global analysis of the insulator binding protein CTCF in chromatin barrier regions reveals demarcation of active and repressive domains. Genome Res. 19, 24–32 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Nora EP et al. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 169, 930–944.e22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Narendra V et al. CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science 347, 1017–1021 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ren G & Zhao K CTCF and cellular heterogeneity. Cell Biosci. 9, 83 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Busslinger GA et al. Cohesin is positioned in mammalian genomes by transcription, CTCF and Wapl. Nature 544, 503–507 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Li Y et al. The structural basis for cohesin–CTCF-anchored loops. Nature 578, 472–476 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hansen AS, Pustova I, Cattoglio C, Tjian R & Darzacq X CTCF and cohesin regulate chromatin loop stability with distinct dynamics. eLife 6, e25776 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Chubb JR, Trcek T, Shenoy SM & Singer RH Transcriptional pulsing of a developmental gene. Curr. Biol 16, 1018–1025 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Raj A, Peskin CS, Tranchina D, Vargas DY & Tyagi S Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 4, e309 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Suter DM et al. Mammalian genes are transcribed with widely different bursting kinetics. Science 332, 472–474 (2011). [DOI] [PubMed] [Google Scholar]
- 164.Nicolas D, Phillips NE & Naef F What shapes eukaryotic transcriptional bursting? Mol. Biosyst 13, 1280–1290 (2017). [DOI] [PubMed] [Google Scholar]
- 165.Tsai WW et al. TRIM24 links a non-canonical histone signature to breast cancer. Nature 468, 927–932 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Bainbridge MN et al. Analysis of the prostate cancer cell line LNCaP transcriptome using a sequencing-by-synthesis approach. BMC Genomics 7, 246 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Nagalakshmi U et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320, 1344–1349 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Tang F et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382 (2009). [DOI] [PubMed] [Google Scholar]
- 169.Skene PJ & Henikoff S An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife 6, e21856 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Cokus SJ et al. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452, 215–219 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Meissner A et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 33, 5868–5877 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Weber M et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat. Genet 37, 853–862 (2005). [DOI] [PubMed] [Google Scholar]
- 173.Brinkman AB et al. Whole-genome DNA methylation profiling using MethylCap-seq. Methods 52, 232–236 (2010). [DOI] [PubMed] [Google Scholar]
- 174.Guo H et al. Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome Res. 23, 2126–2135 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Mumbach MR et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 13, 919–922 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wei CL et al. A global map of p53 transcription-factor binding sites in the human genome. Cell 124, 207–219 (2006). [DOI] [PubMed] [Google Scholar]