

Abstract
Alzheimer’s disease (AD) is a neurodegenerative disease with heterogenous pathophysiological changes that develop years before the onset of clinical symptoms. These preclinical changes have generated considerable interest in identifying markers for the pathophysiological mechanisms linked to AD and AD related disorders (ADRD). Based on our prior work integrating cerebrospinal fluid (CSF) and brain proteome networks, we developed a reliable and high throughput mass spectrometry selected reaction monitoring (SRM) assay that targets 48 key proteins altered in CSF. To test the diagnostic utility of these proteins and compare them to existing AD biomarkers, CSF collected at baseline visits were assayed from 706 participants recruited from the Alzheimer’s Disease Neuroimaging Initiative (ADNI). We found the targeted CSF panel of 48 proteins (CSF 48 panel) performed at least as well as existing AD CSF biomarkers (Aβ42, tTau, and pTau181) for predicting clinical diagnosis, FDG PET, hippocampal volume, and measures of cognitive and dementia severity. Additionally, for each of those outcomes the CSF 48 panel plus the existing AD CSF biomarkers significantly improved diagnostic performance. Furthermore, the CSF 48 panel plus existing AD CSF biomarkers significantly improved predictions for changes in FDG PET, hippocampal volume, and measures of cognitive decline and dementia severity compared to either measure alone. A potential reason for these improvements is that the proteins in the CSF 48 panel reflect a range of altered biology previously observed in AD/ADRD. In conclusion, we show that the CSF 48 panel complements existing AD CSF biomarkers to improve diagnosis and predict future cognitive decline and dementia severity.
One Sentence Summary:
Cerebrospinal fluid proteins that reflect the diverse pathobiology of AD improve AD diagnosis and predictions of future decline.
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by cognitive decline and dementia in the presence of amyloid-β (Aβ) plaques and neurofibrillary tangles (NFTs) in the brain (1, 2). The presence of hallmark AD pathologies years before the onset of clinical symptoms has generated interest in markers to identify individuals at risk of progressive neurodegeneration (2–4). Imaging and cerebrospinal fluid (CSF) biomarkers that measure these hallmark AD pathologies include selective PET ligands that quantify and localize both amyloid plaques and tau pathologies in the brain and biochemical assays that measure CSF Aβ42, total Tau (tTau), and phospho-Tau181 (pTau181) abundance. Moreover, FDG PET and structural MRI are used as surrogate measures for neurodegeneration or synaptic loss in AD (5). Based on these findings, the AT(N) research framework has been proposed to classify AD based on the presence of amyloid plaques (A), neurofibrillary tangles (T), and neurodegeneration (N) (1).
While current biomarkers have provided critical advances, expansion of biomarkers for AD and AD-related disorders (ADRD) is important for several reasons. First, pathological complexity and heterogeneity is the rule, rather than the exception, with most AD dementia syndromes attributable to varying combinations of age-related pathologies, and amyloid plaque and neurofibrillary pathology responsible for dementia in only ~40% of cases (6–8). The ATN framework also anticipates future development of additional biomarkers, ATX(N), with X representing other pathophysiological mechanisms beyond amyloid and tau (1, 5). Second, biomarkers of amyloid and tau do not accurately predict the extent of cognitive impairment, with cognitively impaired individuals often showing normal amyloid PET and CSF amyloid, and 30–40% of cognitively unimpaired elderly individuals showing AD pathology (6–8). Third, biomarkers of the hallmark pathologies have limited prognostic ability for predicting disease progression. For these reasons, additional markers are needed for accurate tracking of a broader spectrum of pathophysiological mechanisms linked to AD.
Recent advances in large-scale molecular profiling technology have identified genetic, transcriptomic, and proteomic alterations in AD and led to the development of data-driven models of AD pathophysiology in postmortem human brain. In our prior work, we performed mass spectrometry (MS) based proteomics of ~2,000 brain tissues from the Accelerating Medicine Partnership for AD (AMP-AD) (9–15). To find suitable candidates for AD/ADRD markers, we recently used deep discovery-based proteomics on CSF samples to identify proteins alterations in CSF that reflect brain AD/ADRD pathophysiologies (16). Based on the CSF findings, we developed an accurate and reliable targeted MS assay using selected reaction monitoring (SRM) that measures a panel of 48 proteins with isotopically labeled peptide standards (17).
In this work, we tested the diagnostic and predictive utility of the CSF 48 panel in 706 individuals from the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Overall, the CSF 48 panel improved upon the ability of existing ATN biomarkers to monitor pathophysiological mechanisms strongly linked to AD and ADRD and improved prediction of disease progression, future cognitive decline and hippocampal atrophy compared to existing ATN measures.
RESULTS
All participants were recruited by ADNI study sites. Inclusion criteria in the current study were enrollment in either ADNI-2 or ADNI-GO and availability of baseline CSF. The dataset consisted of 706 eligible participants with an average age of 72.2 ± 7.3 years-old and 48% female (Table 1). The baseline diagnoses in ADNI were made by the investigators based on clinician judgment as described in Study Design and blinded to biomarker status, with participants assigned as cognitively normal (31%), mild cognitive impairment (MCI, 53%), and AD (16%). Using previously established CSF thresholds for CSF Aβ42 of less than 980 pg/ml and pTau181 greater than 21.8 pg/ml, samples were categorized into four groups: 36% A+T+, 18% A+T-, 16% A-T+, and 30% A-T- (Table 1).
Table 1.
Demographic and Clinical Characteristics of Study Participants.
| Control | MCI | Dementia | Overall | |
|---|---|---|---|---|
| No. Participants | 220 | 376 | 110 | 706 |
| Age at Enrollment | ||||
| Mean (SD) | 73.1 (6.04) | 71.1 (7.57) | 74.0 (8.31) | 72.2 (7.34) |
| Median [Min, Max] | 72.7 [56.2, 85.9] | 71.1 [55.0, 91.4] | 74.8 [55.9, 90.3] | 72.3 [55.0, 91.4] |
| Sex | ||||
| Female | 123 (55.9%) | 172 (45.7%) | 45 (40.9%) | 340 (48.2%) |
| Male | 97 (44.1%) | 204 (54.3%) | 65 (59.1%) | 366 (51.8%) |
| Clinical Diagnosis | ||||
| Control | 220 (100%) | 0 (0%) | 0 (0%) | 220 (31.2%) |
| MCI | 0 (0%) | 376 (100%) | 0 (0%) | 376 (53.3%) |
| Dementia | 0 (0%) | 0 (0%) | 110 (100%) | 110 (15.6%) |
| ATN Category | ||||
| A-T- | 91 (41.4%) | 116 (30.9%) | 3 (2.7%) | 210 (29.7%) |
| A-T+ | 53 (24.1%) | 57 (15.2%) | 6 (5.5%) | 116 (16.4%) |
| A+T- | 45 (20.5%) | 69 (18.4%) | 10 (9.1%) | 124 (17.6%) |
| A+T+ | 31 (14.1%) | 134 (35.6%) | 91 (82.7%) | 256 (36.3%) |
| APOE genotype | ||||
| e2-e2 | 0 (0%) | 1 (0.3%) | 0 (0%) | 1 (0.1%) |
| e2-e3 | 26 (11.8%) | 26 (6.9%) | 2 (1.8%) | 54 (7.6%) |
| e2-e4 | 3 (1.4%) | 5 (1.3%) | 1 (0.9%) | 9 (1.3%) |
| e3-e3 | 131 (59.5%) | 165 (43.9%) | 34 (30.9%) | 330 (46.7%) |
| e3-e4 | 53 (24.1%) | 139 (37.0%) | 48 (43.6%) | 240 (34.0%) |
| e4-e4 | 7 (3.2%) | 40 (10.6%) | 25 (22.7%) | 72 (10.2%) |
| MoCA | ||||
| Mean (SD) | 25.9 (2.46) | 23.4 (3.14) | 17.3 (4.76) | 23.2 (4.25) |
| Median [Min, Max] | 26.0 [19.0, 30.0] | 23.0 [14.0, 30.0] | 18.5 [4.00, 25.0] | 24.0 [4.00, 30.0] |
| Missing | 4 (1.8%) | 1 (0.3%) | 4 (3.6%) | 9 (1.3%) |
| CDR-SB | ||||
| Mean (SD) | 0.0500 (0.158) | 1.43 (0.853) | 4.60 (1.73) | 1.49 (1.74) |
| Median [Min, Max] | 0 [0, 1.00] | 1.25 [0.500, 4.50] | 4.50 [1.00, 10.0] | 1.00 [0, 10.0] |
The CSF 48 panel estimates baseline clinical diagnosis
The CSF 48 panel targets 62 peptides to measure 48 proteins (Fig. 1A, Table S1). Among the 48 proteins, 18 proteins were significantly increased in AD while 4 were significantly decreased in AD (p < 0.05, t-test, FDR corrected). To understand the diagnostic utility of the CSF 48 panel compared to canonical AD CSF biomarkers Aβ42, pTau181, and tTau, we used logistic regression to estimate differences between control and AD participants for each CSF analyte (Fig. 1A, Table S1). As expected, Aβ42, pTau181, and tTau were the top single estimators of AD diagnosis, with AUCs of 0.84, 95% CI [0.79, 0.8], 0.82, 95% CI [0.77, 0.87], and 0.80, 95% CI [0.75, 0.85], respectively. We found the most abundant proteins and the strongest individual classifiers of AD in the CSF 48 panel were tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein zeta (YWHAZ) and beta (YWHAB), 14-3-3 proteins, with AUCs of 0.78. These proteins have been previously linked to AD and Creutzfeldt-Jakob Disease (CJD) in CSF (16, 18, 19) and are associated with many aspects of brain function including neural signaling, neuronal synaptogenesis, and neurodifferentiation (20). We also found an increased abundance and AUC of 0.70 for SPARC related modular calcium binding 1 (SMOC1), a protein previously identified as a hub protein for the matrisomal/extracellular matrix (ECM)-associated co-expression module, with the strongest associations to AD global pathology in post-mortem brain (9, 10, 16). We also found modest increases in pyruvate kinase M1/2 (PKM1/2), malate dehydrogenase 1 (MDH1), enolase 1 (ENO1), and aldolase, fructose-bisphosphate A (ALDOA), proteins central to glycolysis, gluconeogenesis, and the citric acid cycle (Table S1). The proteins most decreased in abundance in AD CSF were VGF nerve growth factor inducible (VGF), and secretogranin II (SCG2), neurosecretory granins involved in axonal or synaptic vesicle transport, and NPTXR and NPTX2, proteins involved in glutamatergic synaptic transmission and implicated in synaptic plasticity and memory (Fig. 1A, Table S1). These data suggest that several proteins measured by the targeted approach could differentiate control and AD participants nearly as well as the gold standard CSF biomarkers.
Fig. 1. The CSF 48 panel estimates baseline cognitive FDG PET, hippocampal volume, cognitive status, and dementia severity in ADNI.
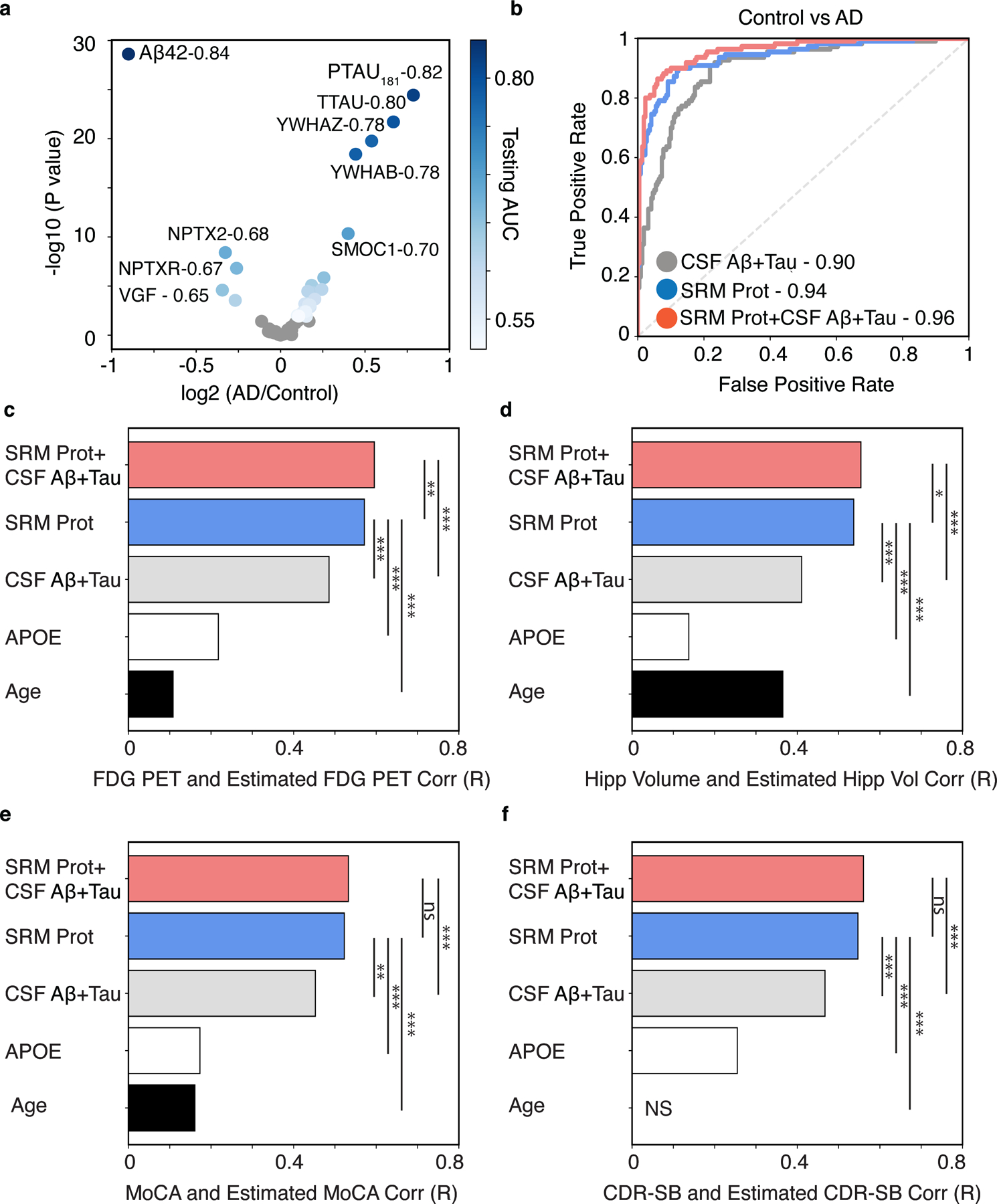
(A) Differential association analysis of all CSF analytes for clinical diagnosis of AD versus cognitively normal control. Analytes with FDR-adjusted significant association are shown in shades of blue that reflects their AUC comparing controls to AD dementia. Non-significant proteins are shown in grey (p > 0.05, FDR corrected). B) The cumulative performance of canonical AD CSF biomarkers (“CSF Aβ42+Tau”), the CSF 48 panel (“CSF 48”), and the existing AD CSF biomarkers plus the CSF protein panel (“CSF 48 + CSF Aβ42 + Tau”), estimated as the area under the curve (AUC) for clinical diagnosis of AD versus cognitively normal control. Bar plots show the Pearson correlation coefficients between observed and predicted values of (C) FDG PET, (D) Hippocampal Volume (“Hipp Volume”), (E) Montreal Cognitive Assessment (MoCA), (F) Clinical Dementia Rating scale Sum of Boxes (CDR-SB) for models using the following predictors: 1) the CSF protein panel plus existing AD CSF biomarkers plus (“CSF 48 + CSF Aβ42 + Tau”), 2) the CSF protein panel alone (“CSF 48”), 3) canonical AD CSF biomarkers alone (“CSF Aβ42+Tau”), 4) APOE E4 dose alone, or 5) Age alone. (*p < 0.05, **p < 0.01, ***p < 0.001. FDG-PET n = 703, Hippocampal Volume n = 640, MoCA n = 694, CDR-SB n = 704). NS (non-significant).
To understand the collective performance of the CSF 48 panel for predicting clinical AD dementia, we used penalized logistic regression to model the relationship between all proteins in the CSF 48 panel and AD clinical dementia. For comparison, the same model was fitted using the CSF 48 panel plus the canonical CSF biomarkers. For the CSF 48 panel, we estimated an AUC of 0.94, 95% CI [0.91, 0.97], while for canonical CSF biomarkers (Aβ42, pTau181, and tTau) we estimated an AUC of 0.90, 95% CI [0.86, 0.94] (Fig. 1B). The AUC for the CSF 48 panel was significantly higher than that for the canonical CSF biomarkers alone (p < 0.01, permutation procedure; Fig. 1B). Notably, the combination of the CSF 48 panel and canonical CSF biomarkers had the highest AUC of 0.96, 95% CI [0.94, 0.99] (Fig. 1B) and significantly improved the AUC compared to existing CSF biomarkers (p < 0.001, permutation procedure; Fig. 1B). These results demonstrate the cumulative ability of the CSF 48 panel to accurately differentiate clinical AD dementia as well or better than the gold standard markers of AD CSF biomarkers.
The CSF 48 panel accurately estimates baseline FDG PET and hippocampal volume
Synaptic dysfunction and neuronal loss occur many years before overt clinical AD dementia and strongly correlate with dementia severity, motivating the development of biomarkers for assessing synaptic function in AD. FDG-PET, a measure of the cerebral metabolic rate of glucose, and volumetric MRI, including hippocampal volume, have been used to reflect neurodegeneration; the “N” indicated in the ATN framework. To understand whether the proteins in the CSF 48 panel were associated with these changes in AD pathogenesis, we performed an association analysis which included synaptic proteins dysregulated in AD brain, FDG PET and MRI derived hippocampal volume, separately. We found that 20 of the proteins in the CSF 48 panel associated with FDG PET and 15 proteins associated with both FDG PET and hippocampal volume (p < 0.01, t-test, FDR corrected; Fig. S1). The individual CSF analyte most strongly associated with FDG PET and hippocampal volume was CSF Aβ42, with positive correlation coefficients of 0.43 and 0.35, respectively (Fig. S1). We found both FDG PET and hippocampal volume were positively associated with synaptic proteins decreased in AD, neuronal pentraxin 2 (NPTX2; FDG R=0.32; HV R=0.28), neuronal pentraxin receptor (NPTXR; FDG R=0.26; HV R=0.22), VGF (FDG R=0.24; HV R=0.13), and SCG2 (FDG R=0.2; HV R=0.14). We also found FDG PET and hippocampal volume were negatively associated with CSF pTau181 (FDG R=−0.36; HV R=−0.30), CSF tTau (FDG R=−0.34; HV R=−0.31), YWHAZ (FDG R=−0.34; HV R=−0.28), YWHAB (FDG R=−0.30; HV R=−0.26), and to a lesser extent SMOC1 (FDG R=−0.23; HV R=−0.14; Fig. S1). Notably, FDG PET and hippocampal volume showed even lower associations or were not associated with metabolic proteins PKM, ALDOA, calmodulin 2 (CALM2), and malate dehydrogenase 1 (MDH1; R < 0.15, Fig. S1). Hemoglobin A/B (HBB, HBA) and albumin (ALB), blood-based proteins serving as negative internal controls in this study, did not vary with FDG PET and hippocampal volume (Fig. S1). Together these data suggest that a reduction in FDG PET and hippocampal volume were linked to similar sets of proteins in CSF. The altered proteins indicate shared pathophysiologic changes, including reduced abundance of synaptic proteins and CSF Aβ42, increased abundance of the CSF tTau, pTau181, and two 14-3-3 proteins. In contrast, there were comparatively weak associations with metabolic proteins.
To assess the cumulative performance of the CSF 48 panel for estimating both FDG PET and hippocampal volume, we trained a penalized regression model for each of the following: the CSF 48 panel with and without canonical CSF biomarkers, CSF biomarkers alone, APOE genotype, and age. Canonical CSF biomarkers alone were able to predict FDG PET and hippocampal volume with an R of 0.49 and 0.41, respectively (FDG PET, p=6.4 × 10−43, Fig. 1C; hippocampal volume, p=2.1 × 10−27, Fig. 1D). However, we found the highest correlation between the combination of the CSF 48 panel and canonical biomarkers could estimate FDG PET and hippocampal volume with an R of 0.60 and 0.55 (FDG PET, p=1.2 × 10−68, Fig. 1D; hippocampal volume, p=9.1 × 10−53, Fig. 1D) that significantly outperformed canonical CSF biomarkers alone (p < 0.001, permutation procedure). The CSF 48 panel could predict FDG PET and hippocampal volume with an R of 0.57 and R of 0.54 (FDG PET, p=4.1 × 10−62, Fig. 1C; hippocampal volume, p=5.1 × 10−49, Fig. 1D), and outperformed age, APOE genotype, and canonical AD biomarkers (p < 0.001, permutation procedure). Since age was a significant estimator of hippocampal volume, we estimated hippocampal volume using age, CSF protein panel and canonical biomarkers (R of 0.58, p=1.0 × 10−58) and found significant improvement in performance compared to the CSF protein panel and canonical biomarkers alone (p < 0.001, permutation procedure).
The CSF 48 panel estimates baseline cognitive function and clinical measures of AD dementia severity
Next we examined how proteins in the CSF 48 panel were associated with cognitive measures and dementia symptom severity, which are generally not captured by canonical CSF biomarkers. We first performed an association analysis of proteins in the CSF 48 panel for the Montreal Cognitive Assessment (MoCA) and Clinical Dementia Rating scale Sum of Boxes (CDR-SB), separately. We found 24 proteins associated with the MoCA score and 19 proteins associated with CDR-SB (p < 0.01, t-test, FDR corrected; Fig. S1). As expected, the plasma proteins hemoglobin A/B (HBB, HBA) and albumin (ALB) did not vary with MoCA and CDR-SB (Fig. S1). To understand the collective performance of the CSF 48 panel for predicting either baseline MoCA and CDR-SB, we trained a penalized regression model as described above for FDG PET and hippocampal volume. The canonical CSF biomarkers estimated MoCA and CDR-SB with an R of 0.45 and 0.47, respectively (MoCA, p=2.4 × 10−36, Fig. 1E; CDR-SB, p=1.9 × 10−39, Fig. 1F), highlighting the limitations of amyloid and tau biomarkers in predicting cognitive status and dementia severity. The CSF 48 panel estimated MoCA and CDR-SB with an R of 0.52 and 0.55 for MoCA and CDR-SB, respectively (MoCA, p=6.6 × 10−50, Fig. 1E; CDR-SB, p=3.8 × 10−56, Fig. 1F), which was an improvement from canonical CSF biomarkers alone (p < 0.01, permutation procedure), APOE genotype (p <0.001, permutation procedure), and age (p <0.001, permutation procedure). The combination of the CSF 48 panel and canonical CSF biomarkers estimated MoCA and CDR-SB with R of 0.53 and 0.56 (MoCA, p=4.1 × 10−52, Fig. 1E; CDR-SB, p=2.2 × 10−59, Fig. 1F), outperforming the canonical CSF biomarkers alone (p <0.001, permutation procedure). Notably, the performance of the CSF 48 panel for predicting either the MoCA or CDR-SB was the same as the performance of the CSF 48 panel plus canonical AD biomarkers (p > 0.05, permutation procedure; Fig. 1E,F).
The CSF 48 panel predicts changes in cognition, dementia severity, and hippocampal volume
Longitudinal studies like ADNI provide a powerful resource to study AD trajectories and develop prognostic biomarkers to predict rates of progression. Canonical biomarkers of amyloid and tau have limited prognostic ability, likely because additional molecular mechanisms contribute to the vulnerability and resilience of individuals that underlies variability in disease progression. We examined whether the proteins in the CSF 48 panel and canonical CSF biomarkers could predict trajectories of cognition (MoCA), dementia severity (CDR-SB) and hippocampal volume. A minimum of at least three visits over a minimum of three years were required to estimate the trajectories for each participant (Table 2; Fig. S2). On average, controls showed a slower rate of cognitive decline compared to participants with MCI (MoCA, p=8.51× 10−3, t-test; Fig. S2) or AD (MoCA, p=1.4 × 10−25, t-test; Fig. S2). Similarly, rates of decline on CDR-SB scores were significantly lower in controls than participants with MCI (CDR-SB, p=5.36 × 10−6, t-test) and AD (CDR-SB, p=2.2 × 10−44, t-test). Testing for association between estimated trajectories and the individual proteins in the CSF 48 panel showed 24, 20, and 3 associated proteins with cognitive, dementia severity, and hippocampal changes, respectively (p < 0.01, t-test, FDR-corrected, Fig. 2A). Among the individual CSF analytes, CSF pTau181 was most strongly correlated with annual MoCA and CDR-SB change, R of −0.49 and 0.42, followed by CSF tTau, YWHAZ, YWHAB, and CSF Aβ42 (Fig. S1). CSF Aβ42 was most strongly correlated with annual hippocampal volume change with an R of 0.36, followed by CSF pTau181 (R=−0.33), YWHAZ (R=−0.31), CSF tTau (R=−0.30), YWHAB (R=−0.27), and SMOC1 (R=−0.22) (Fig. 2A, Fig. S1).
Table 2.
Trajectories of cognition, dementia severity, and hippocampal volume in ADNI participants.
| MoCA | CDR-SB | Hippocampal Volume (mm3/yr) | |
|---|---|---|---|
| No. Participants | 412 | 429 | 227 |
| Age at Enrollment | 71.4 ± 7.0 | 71.6 ± 7.0 | 70.5 ± 7.2 |
| Follow-Up Duration | 5.79 ± 1.8 | 5.85 ± 1.8 | 4.08 ± 0.55 |
| Number of Visits | 6.51 ± 1.5 | 6.58 ± 1.6 | 5.74 ± 0.95 |
| Annual Trajectory | −0.27 ± 0.90 | 0.30 ± 0.72 | −127 ± 113.2 |
Fig. 2. The CSF 48 panel predicts future change in cognition, dementia severity, and hippocampal volume.
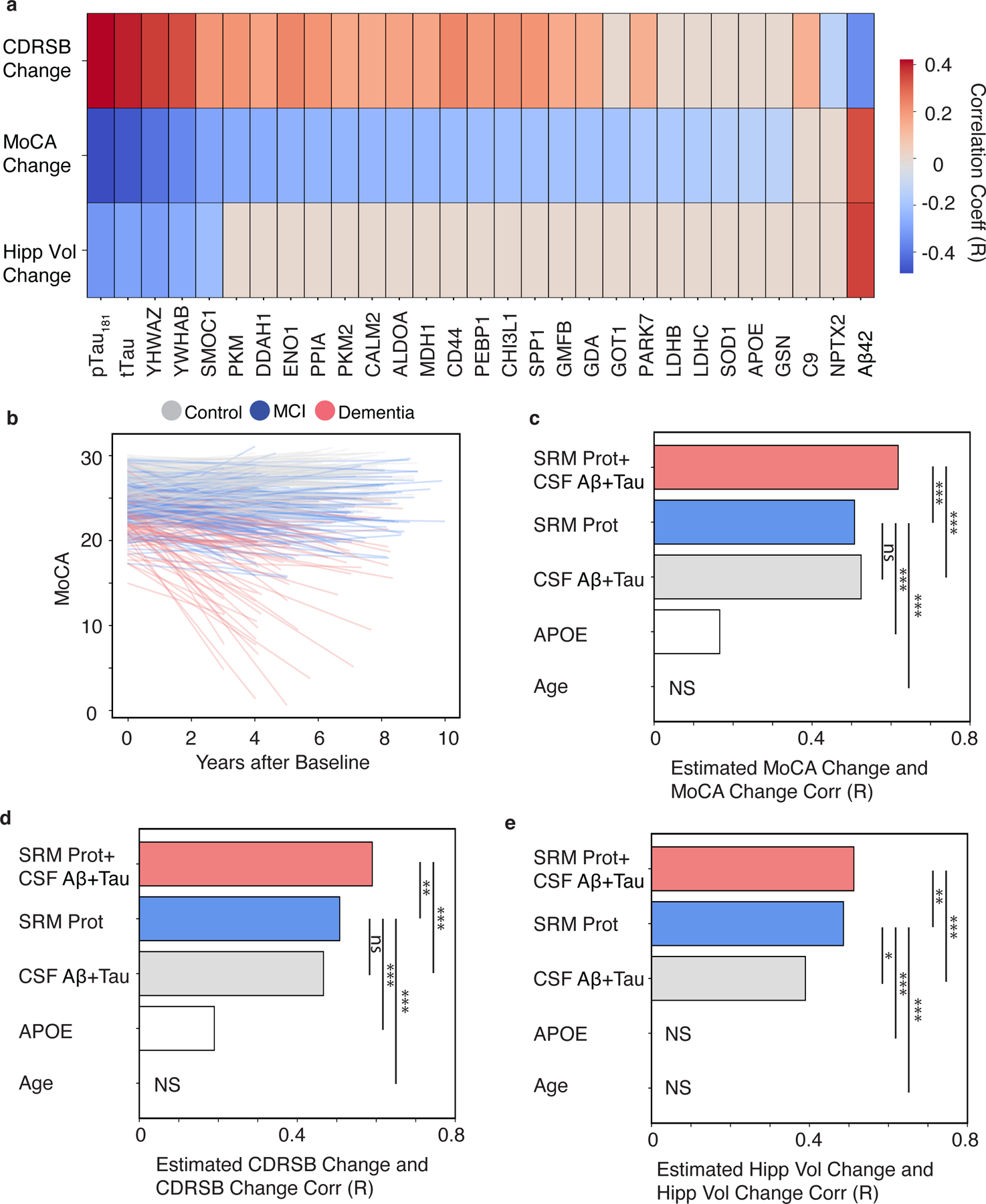
(A) A heatmap of FDR-adjusted Pearson correlations are shown for CSF analytes and change in CDR-SB, MoCA, or hippocampal volume. The CSF peptides are labeled as their respective gene symbols, and the strength and direction of correlation is shown by the red to blue scale and non-significant correlations are shown as grey. (B) Line plot of individual estimates of MoCA decline over time. The color of each line reflects the baseline clinical diagnosis. Bar plots show the Pearson correlation coefficients of between observed and predicted values. (C) MoCA, (D) CDR-SB, or (E) Hippocampal volume for models using the following predictors: 1) the CSF protein panel plus existing AD CSF biomarkers plus (“CSF 48+CSF Aβ42+Tau”), 2) the CSF protein panel alone (“CSF 48”), 3) canonical AD CSF biomarkers alone (“CSF Aβ42+Tau”), 4) APOE E4 dose alone, or 5) Age alone. (*p < 0.05, **p < 0.01, ***p < 0.001. MoCA N=412, CDR-SB n =429, Hippocampal Volume n = 227). NS (non-significant).
We next evaluated the collective prognostic potential of the CSF 48 panel compared to the canonical CSF biomarkers to predict each of the trajectories using a penalized multivariate linear regression model. First, annual MoCA change was estimated using both canonical CSF biomarkers (r=0.52, p =1. 98 × 10−30, Fig. 2B,C) and the CSF 48 panel (r=0.51, p =2.42 × 10−28, Fig. 2C), with no differences between the two measures (p > 0.05, permutation procedure). However, combining the CSF targeted peptides with canonical CSF biomarkers significantly improved cognitive trajectory prediction (r=0.62, p =9.12 × 10−45, Fig. 2C) compared to APOE genotype (p < 0.001, permutation procedure), canonical CSF biomarkers (p < 0.001, permutation procedure), or the CSF 48 panel alone (p < 0.001, permutation procedure). To predict rate of disease progression, annual CDR-SB change was also estimated using the canonical CSF biomarkers (r=0.47, p =1.51 × 10−24, Fig. 2D) and the CSF 48 panel (r=0.51, p =1.83 × 10−29, Fig. 2D). Combining the canonical biomarkers and the targeted panel improved the prediction of CDR-SB (r =0.59, p =1.44 × 10−41, Fig. 2D) compared to the panel (p < 0.01, permutation procedure) and existing biomarkers alone (p < 0.001, permutation procedure). For hippocampal volume trajectories, the model that combined the CSF 48 panel and canonical AD biomarkers resulted in the highest predicted correlation with observed change (r=0.51, p =1.4 × 10−16, Fig. 2E) and was a significant improvement compared to CSF protein panel alone (r=0.49, p=7.4 × 10−15) or canonical CSF biomarkers (r=0.39, p=1.2 × 10−9) alone using permutation (p < 0.001). Collectively, these results suggest the CSF 48 panel, reflecting additional pathophysiologies beyond amyloid and tau, provides substantial value when combined with the canonical CSF biomarkers for predictions of cognition, dementia severity, and hippocampal changes compared to existing CSF biomarkers alone.
The CSF 48 panel reveals distinct associations between existing CSF and PET biomarkers of AD
In vivo measurements of fibrillary amyloid in the brain using florbetapir (AV45) and other PET radioligands have emerged as important surrogate endpoints of AD pathophysiology (21) and are thought to reflect similar disease measures as CSF amyloid biomarkers due to their high concordance (1, 22, 23). Results of association testing between the CSF 48 panel and AV45 binding and the canonical CSF biomarkers Aβ42, p-tau, and t-tau are shown in Fig. 3A. AV45 binding was significantly associated with 25 CSF proteins (FDR p < 0.01, t-test), while CSF Aβ42 was associated with 21 proteins, with only 11 proteins showing an association with both CSF Aβ42 and AV45 (Fig. 3A, Fig. S1). CSF Aβ42 was positively associated with VGF and SCG2, neurosecretory granins involved in synaptic vesicle transport, and NPTXR and NPTX2, pentraxin associated proteins involved in glutamatergic synaptic transmission, indicating that low CSF Aβ42 was associated with decreased abundance of synaptic proteins (Fig. 3A). Notably, many of these synaptic proteins were not significantly associated with AV45 binding potential (p > 0.01, FDR corrected, t-test, Fig. 3A, Fig. S1). Rather, AV45 binding was most strongly associated with SMOC1, a matrisomal protein that strongly correlated with amyloid plaques and global pathology in AD brain (9, 10), and YWHAZ and YWHAB (Fig. 3A). In contrast to CSF Aβ42, AV45 binding was also positively associated with a host of proteins associated with glucose metabolism, including PKM, PKM2, CALM2, ALDOA, MDH1, and lactate dehydrogenase B (LDHB; Fig. 3A, Fig. S1). These results show a discordance between the two amyloid biomarkers within CSF peptide panel, with low CSF Aβ42 most strongly linked to synaptic proteins decreased in AD, and amyloid PET binding most strongly linked to matrisomal, 14-3-3 signaling, and metabolic proteins increased in AD.
Fig. 3. The CSF 48 panel estimates amyloid and tau biomarkers.
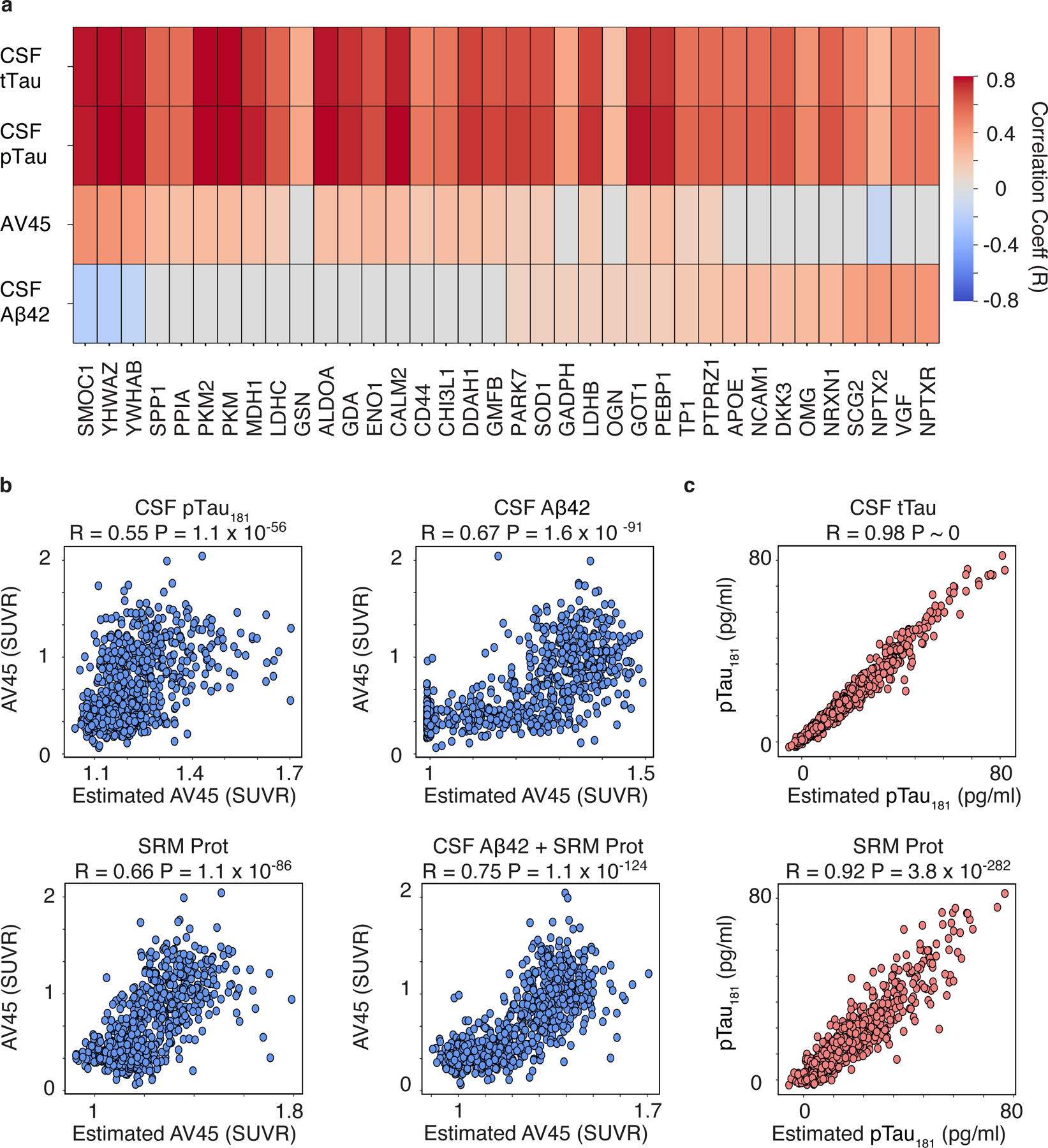
(A) Heatmaps of Pearson correlations are shown for CSF peptides that were significantly associated with one or more of the following outcomes after FDR-adjustment: AV45 SUVR, CSF Aβ42, CSF pTau181, and CSF tTau,. The significant CSF peptides are labeled as their respective gene symbols, and the strength and direction of the correlations are shown by the red to blue scale and non-significant correlations are shown as grey (FDR p < 0.01, t-test ) (B) Scatter plot showing Pearson correlations between the observed and predicated estimate of AV45 SUVR using CSF pTau181 (top left), CSF Aβ42 (top right), the CSF protein panel (“CSF 48”, bottom left), or the CSF protein panel plus CSF Aβ42 (“CSF Aβ42 + CSF 48”, bottom right). (C) Scatter plot showing Pearson correlations between the observed and predicated estimate of CSF pTau181 using either CSF tTau (top) or the CSF protein panel (“CSF 48”, bottom) as predictors.
In contrast to CSF Aβ42 and PET amyloid biomarkers, CSF tTau and pTau181 are thought to reflect related but distinct AD pathophysiology with CSF tTau reflecting the intensity of synaptic loss and neurodegeneration and pTau181 reflecting an AD-specific pathological state associated with paired helical filament tau formation (24). Association testing with the CSF 48 panel revealed significant positive associations with CSF tTau and CSF pTau181 with the same 36 proteins (p < 0.01, FDR corrected, t-test, Fig. 3A). CSF tTau and pTau181 were strongly associated with many proteins within our panel, reaching correlations of 0.70–0.80 (Fig. 3A). SMOC1, YWHAZ and YWHAB proteins all showed strong, positive associations with CSF tTau and pTau181. Both tau markers also showed strong, positive associations with neuronal proteins ontologically linked to cellular energy storage and metabolism (Fig. 3A) (16). PKM1/2, CALM2, ALDOA, and MDH1 are all proteins central to glycolysis, gluconeogenesis and the citric acid cycle, indicating the presence of tau pathology is tightly linked to altered glucose and energy metabolism in AD. CSF tau measures were also associated with aspartate aminotransferase GOT1, an important enzyme in amino acid metabolism, and guanine deaminase (GDA), an enzyme associated with purine metabolism and microtubule polymerization. These data suggest elevated CSF tTau and pTau181 may not only be linked to matrisomal dysfunction and impaired 14-3-3 signaling but also reflect widespread dysregulation across cellular energy and metabolism pathways.
The CSF 48 panel accurately estimates existing CSF and PET biomarkers of AD
Following the approach for other studied outcomes, regularized linear regression was used to model the relationships between AV45 standard uptake value ratio (SUVR) with canonical CSF biomarkers, the CSF 48 panel, and a combination of canonical CSF biomarkers and the CSF 48 panel, separately. The performance of the models was assessed by correlating the actual and estimated values for each predicted outcome. CSF pTau181 and CSF Aβ42 estimated AV45 SUVR with an R of 0.55 (p=1.1 × 10−56, Fig. 3B) and 0.67 (p=1.6 × 10−91, Fig. 3B), respectively. The CSF 48 panel collectively estimated AV45 SUVR with an R of 0.66 (p=1.1 × 10−86, Fig. 3B) and the CSF 48 panel combined with CSF Aβ42 improved the prediction to an R of 0.75 (p=1.1 × 10−124, Fig. 3B), which reflected a significant improvement over the targeted CSF protein panel alone, CSF pTau181 or CSF Aβ42 (p < 0.001, permutation procedure). Unsurprisingly, CSF tTau estimated CSF pTau181 well at R of 0.98 (p ~ 0, Figure 3C), and the CSF 48 panel estimated CSF pTau181 with an R of 0.92 (p=3.78 × 10−282, Fig. 3C). These results show the CSF 48 panel can accurately estimate AV45 binding, CSF Aβ42, and CSF pTau181. Interestingly, some of the discordance between the two amyloid biomarkers AV45 SUVR and CSF Aβ42, may be explained by different synaptic, matrisomal, and metabolic pathophysiology reflected by the targeted CSF peptides.
The CSF 48 panel accurately estimates baseline ATN status and improves estimation of cognitive decline
We next determined which proteins in the CSF 48 panel best classified ATN biomarker status and predicted changes in cognitive function or dementia. We stratified participants into those with and without evidence for both A and T pathologies (A+T+ vs. A-T-) based on the canonical AD CSF biomarkers. An association analysis of proteins in the CSF 48 panel in A+T+ versus A-T- individuals revealed 34 proteins were significantly increased in A+T+ compared to A-T- participants (p < 0.05, t-test, FDR-corrected, Fig. 4A), but no proteins were significantly decreased. SMOC1, YWHAZ, and YWHAB were the strongest differentiators of A+T+ and A-T- individuals, with AUCs ranging from 0.90–0.91 (Fig. 4A). The metabolic proteins PKM1/2, ALDOA and CALM2 could also differentiate A+T+ and A-T- individuals with high accuracy with AUCs of 0.82–0.85 (Fig. 4A, Table S2). Synaptic proteins VGF, NPTXR, and SCG2 were much weaker differentiators of A+T+ and A-T- individuals with AUCs ranging from 0.50–0.55 (Fig. 4A, Table S2). The combination of the proteins in the CSF 48 panel could estimate the differences between A+T+ and A-T- individuals with an AUC of 0.97, 95% CI [0.95, 0.98]. Because of the distinct proteomic profiles associating with CSF vs PET measures of amyloid shown earlier (Fig. 3), we also performed classification of amyloid PET positive (AV45+) and negative individuals (AV45-) using an SUVR of 1.1. The CSF 48 panel l could differentiate between AV45+ and AV45- individuals with an AUC of 0.89, 95% CI [0.86, 0.92]. For comparison, we also classified individuals who were CSF Aβ42+ (A+) and CSF Aβ42- (A-). The SRM proteome could separate these two populations with an AUC of 0.88, 95% CI [0.86, 0.91].
Fig. 4. The CSF 48 panel predicts future change in cognition and dementia severity among in patients with A+T+ biomarker status.
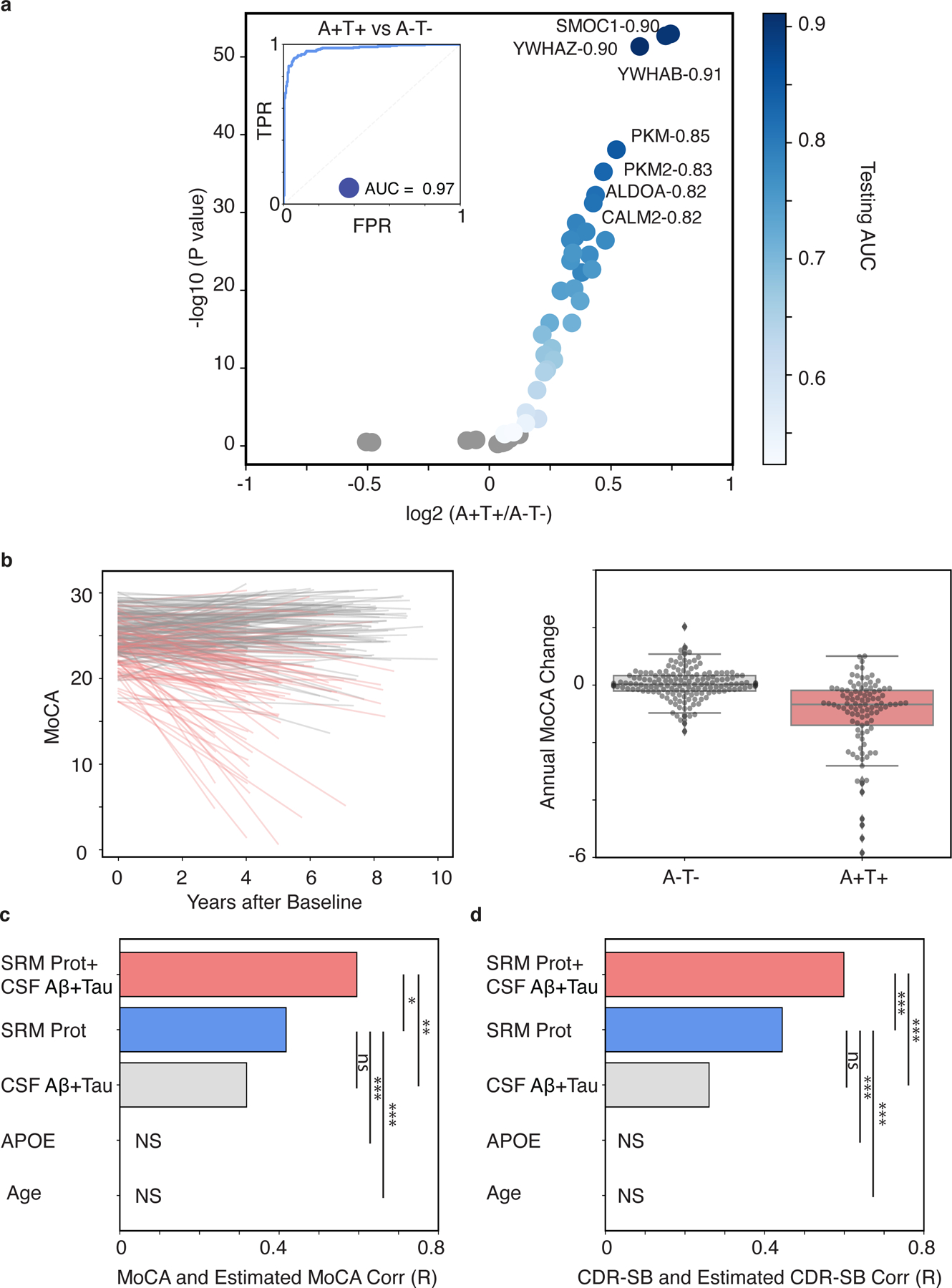
(A) Analytes with FDR-adjusted significant association are shown in shades of blue that reflect their AUC comparing A+T+ versus A-T- status. Non-significant proteins are shown in grey (p > 0.05, FDR corrected). (B) The left panel is a line plot of individual estimates of MoCA decline over time. The color of each line reflects the baseline A/T status. The right panel is a box plot of the annual MoCA change for individuals with baseline A-T- and A+T+. Bar plots show the Pearson correlation coefficients between observed and predicted values of (C) MoCA or (D) CDR-SB for models using the following predictors: 1) the CSF protein panel plus existing AD CSF biomarkers plus (“CSF-48+CSF Aβ42+Tau”), 2) the CSF protein panel alone (“CSF-48”), 3) canonical AD CSF biomarkers alone (“CSF Aβ42+Tau”), 4) APOE E4 dose alone, or 5) Age alone. (*p < 0.05, **p < 0.01, ***p < 0.001. MoCA, n = 101; CDR-SB, n = 113)
While the ATN framework has been useful in identifying individuals at risk of cognitive decline, AD dementia syndromes are attributable to varying combinations of pathologies and pathophysiological processes and therefore substantial heterogeneity may exist even among A+T+ individuals. Trajectories for cognitive decline and dementia severity were compared by baseline AT status. A-T- individuals showed a slower rate of cognitive decline compared to A+T+ individuals (MoCA, p=3.58 × 10−17, t-test; CDR-SB, p=5.32 × 10−16, t-test; Fig. 4B–C), which agrees with a prior study (25). For A+T+ individuals, we trained a multivariate linear regression model to predict trajectories of cognition and dementia severity using the CSF 48 panel, the canonical CSF biomarkers, and the combination. The canonical CSF biomarkers alone modestly predicted disease trajectory (MoCA r =0.32, p=1.18 × 10−3, CDR-SB r =0.26, p=5.33 × 10−3, Fig. 4C–D). The CSF 48 panel somewhat better predicted these trajectories (MoCA r =0.42, p=1.40 × 10−5, CDR-SB r =0.44, p=8.30 × 10−7, Fig. 4C–D), but without a significant difference (p > 0.05, permutation procedure). However, the combination of the CSF 48 panel plus the canonical CSF biomarkers was significant in predicting both MoCA (r =0.60, p=5.17 × 10−11) and CDR-SB trajectory (r =0.60, p=2.4 × 10−12, Fig. 4C–D). The CSF 48 panel plus canonical CSF biomarkers showed a significant improvement in prediction over the canonical biomarkers (p < 0.001, permutation procedure). Overall, the CSF 48 proteome improves prediction of cognitive trajectory and dementia severity decline in at risk individuals based on CSF amyloid and tau status.
DISCUSSION
Here, we tested the diagnostic and prognostic characteristics of a targeted CSF protein panel measured on baseline CSF from 706 ADNI participants. This work builds on prior studies of postmortem brain proteomics that identified brain protein networks consistently altered in AD brain and proteins alterations in CSF that reflect brain AD/ADRD pathophysiology (16). Here, we extended these findings using our recently developed SRM-MS assay with isotopically labeled peptide standards (17) to target and quantify 48 key proteins in baseline CSF samples from the ADNI study. The CSF 48 panel accurately predicted AD pathophysiology and disease as well as or better than the canonical CSF AD biomarkers, Aβ42, tTau, and Tau181, with many individual proteins and the panel providing additional diagnostic and prognostic utility. Interestingly, the CSF 48 panel generally showed improved performance for predicting baseline AD imaging biomarkers (FDG PET, hippocampal volume, and AV45 SUVR) and measures of cognition and dementia severity (MoCA and CDR-SB). Moreover, when the CSF 48 panel was combined with the canonical AD CSF biomarkers, we observed improved predictive capabilities. Notably, the CSF 48 panel showed ability to predict annual change in cognition (MoCA), dementia severity (CDR-SB), and neurodegeneration (hippocampal volume, FDG-PET) as well or better than traditional AD CSF biomarkers and an additive improvement over existing AD CSF biomarkers alone. Unsurprisingly, the ability of the CSF 48 panel to predict progression was more pronounced among individuals whose CSF was consistent with a higher risk for having underlying AD pathophysiology. Together, the combined diagnostic and prognostic information of the CSF 48 panel, which measures additional pathophysiological processes beyond amyloid and tau, may improve identification of those at risk for AD and future decline.
The CSF 48 panel incorporated proteins across a range of dementia-related biological pathways and therefore was able to identify heterogeneity across the various AD markers. Due to their high concordance across individuals, CSF Aβ42 and amyloid PET have been generally thought to reflect the same underlying pathological state and often used interchangeably as amyloid biomarkers (1, 22, 23). However, our findings revealed distinct proteomic signatures for CSF Aβ42 and amyloid PET. Low CSF Aβ42 most strongly linked to synaptic proteins decreased in AD while amyloid PET binding was most strongly linked to increased abundance of matrisomal, 14-3-3 signaling, and metabolic proteins in AD. Thus, we advocate against using these biomarkers interchangeably.
A theme that emerged from this study was the strong, positive association between CSF tTau and pTau181 with neuronal proteins ontologically linked to cellular energy storage and metabolism (16), with many metabolic proteins exhibiting correlation coefficients greater than 0.8. The correlation between CSF Tau and glycolytic proteins has been observed in other studies (26, 27). Metabolic proteins could differentiate the presence of increased CSF tTau and pTau181 with AUCs greater than 0.9. Notably, these metabolic proteins were not as strongly associated with CSF Aβ42, AV45, hippocampal atrophy, and surprisingly, FDG PET. Unlike SMOC1, YWHAZ, and YWHAB, which were also strongly associated with CSF tTau and pTau181, the metabolic proteins were poor estimators of clinical diagnosis. Together, our data suggested that CSF tTau and pTau181 are more tightly linked to cellular energy and metabolism compared to the other markers of AD. These findings also have potential biological implications that warrant future studies.
The CSF 48 panel was selected based on integration of large-scale brain and CSF protein networks, providing an unbiased approach that also sheds insights into the potential mechanisms underlying their roles in disease biology (10, 16). The divergence in CSF protein profiles associated CSF Aβ42 and amyloid PET, respectively, points towards distinct pathophysiologies linked to these two amyloid biomarkers. Unlike PET measures of fibrillar amyloid deposits in brain, low concentrations of CSF Aβ42 in AD were strongly associated with synaptic proteins that increased in CSF and decreased in brain (16). These changes begin early in the asymptomatic phases of disease (10, 16, 17) and together with the metabolic changes noted above, may reflect synaptic plasticity, microglial pruning and extrusion of synaptic material as we and others have discussed previously (16, 28). We speculate that reductions in CSF Aβ42 thus reflect changes in synaptic biology rather than deposition into plaques in brain. In contrast, amyloid PET was strongly associated with increases in proteins linked to the matrisome and 14-3-3 signaling, including SMOC1, YWHAZ, and YWHAB. SMOC1 is one of the most differentially expressed proteins in AD brain, and the hub protein in the matrisome module M42, which is highly correlated with AD neuropathology (r=0.75) and also contains Aβ42 and Apoe among its 32 protein members (9). Many of the proteins in this module bind heparin and are histologically associated with Aβ plaques (9, 29), potentially facilitating protein aggregation. As members of a synaptic module in brain (16), we speculate that the increased CSF abundance of 14-3-3 proteins reflect a neuritic response to Aβ42 deposition. Thus, we suggest that these biomarkers are good biofluid proxies for Aβ42 plaques in brain.
The CSF 48 panel also demonstrated specific proteins associated with FDG PET and hippocampal atrophy, currently considered biomarkers of neurodegeneration. Our results suggest that reductions in NPTX2 and NPTXR are associated with reduced FDG uptake and hippocampal volume loss. These results align with studies showing NPTX2 and NPTXR down-regulation prevents homeostatic scaling of excitatory synapses eventually leading to volume loss and cognitive dysfunction in AD 33–36. Surprisingly, FDG PET was more associated with decreased abundance of synaptic proteins rather than metabolic proteins, indicating that global measure of FDG uptake may reflect synaptic loss (or reduced synaptic activity) rather than brain glucose metabolism.
An advantage of this study was a large, well-characterized dataset consisting of a wide spectrum of individuals from ages 55–90 and across the US, that were not pre-selected based on discrete clinical categories or on the presence of amyloid and tau pathology. By using the well-characterized ADNI longitudinal dataset, several striking biological insights were identified. The CSF 48 panel significantly improved predictions for future declines in cognition, dementia severity, and hippocampal atrophy compared to the canonical AD CSF biomarkers and provided additional value when combined with existing CSF biomarkers for predicting these outcomes. Even among A+T+ individuals, we found the CSF 48 panel could nearly double the estimation of future cognitive decline and dementia risk. Future approaches to assess cognitive decline and dementia risk may therefore benefit from the incorporation of peptides such as those in our SRM panel representing multiple biological pathways.
Our study has limitations. The proteins in the CSF 48 panel were selected based on differences in abundance between control and AD cases, with these populations defined using Aβ42, tTau, and pTau181 thresholds (16). Thus, the selected proteins may be limited in their ability to find proteins relevant to clinical endpoints that are independent of amyloid and tau. In future studies, we plan to expand measured proteins that relate to clinical endpoints independent of ATN. Another limitation is that the ADNI cohort is not representative of the diversity of the population, and the canonical AD biomarkers show important racial differences that pose challenges to clinical translation in real world practice. Further investigation is needed in populations with greater disease-heterogeneity and racial/ethnic diversity to understand the generalizability of these findings. Despite these limitations, the CSF 48 panel improves upon existing ATN biomarkers to predict many pathophysiological mechanisms linked to AD and ADRD brain, distinguish pathophysiological mechanisms based on their proteomic signature, and improve the prediction of disease progression, future changes in cognition, dementia severity, and hippocampal volume.
MATERIALS AND METHODS
Study Design
The study was designed to identify whether a targeted CSF protein panel predicts future cognitive decline or dementia severity. To test the diagnostic utility of these proteins and compare them to existing AD biomarkers, CSF collected at baseline visits were assayed from 706 participants recruited from the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Samples were randomized and blinded for mass spectrometry analyses.
ADNI is a longitudinal, observational study with participant ages ranging from 55 to 90 designed to collect and validate biomarkers to predict progression to AD. ADNI was launched in 2003 as a public-private partnership with a primary goal to test whether serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and AD. Participant recruitment for ADNI is approved by the Institutional Review Board of each participating site. All ADNI participants undergo standardized diagnostic assessment that renders a clinical diagnosis of either control, MCI, or AD using standard research criteria (30). Control participants had no subjective memory complaints, tested normally on Logical Memory II of Weschler Memory Scale, had an MMSE between 24–30, and a CDR of 0 with memory box score of 0. MCI participants reported subjective memory concerns and exhibited abnormal memory function on Logical Memory II of Weschler Memory Scale, an MMSE between 24–30, and CDR of 0.5 with memory box score of at least 0.5. AD participants also exhibited subjective memory concerns but also met NINCDS/ARDA criteria for probable AD. AD participants also showed abnormal memory function on Logical Memory II subscale from the Weschler Memory Scale, an MMSE of 20–26, and CDR of 0.5 or 0.1. Inclusion criteria for the current study were enrollment in ADNI2 or ADNI GO and an available baseline CSF sample. Notably, there was no overlap between the 706 ADNI participants in this study and the Emory ADRC cohort that was used to develop the targeted SRM assay (17). Individuals in this study had CSF assessments for Aβ42, tTau, and pTau181 using the Elecsys immunoassay detection platform (Roche Diagnostics Corporation, Indianapolis, IN USA) by ADNI investigators (31). We used ADNI-established thresholds of CSF Aβ42 less than 980 pg/ml and pTau181 greater than 21.8 pg/ml to categorize individuals as either positive or negative for the respective measure (A+T+, A-T+, A+T-, and A-T-) (31). We also separated individuals into AV45+ and AV45- individuals using an SUVR of 1.1. To assess clinical outcomes, we used the Montreal Cognitive Assessment (MoCA) scores and CDR sum of boxes (CDR-SB).
Proteomic peptide measurement in CSF
The CSF protein panel measures 48 key proteins that were selected after evaluation of over 200 tryptic peptides considered from integration of the brain and CSF proteome network analysis (16, 17). Technical details of the discovery and validation of the selected peptides are described in Watson et al., 2022 (17), The participants in Watson et al., 2022 were distinct from the current study. The CSF protein panel targets 62 peptides to measure the 48 proteins. To select the most informative peptides for the 48 proteins a differential analysis of the baseline diagnosis of AD versus normal cognition was performed. For proteins with more than one peptide measured, we selected the peptide that most strongly associated with AD diagnosis for all subsequent analyses (Fig. 1A, Table S1). In brief, ADNI CSF aliquots were thawed and further aliquoted onto 9 shallow-well plates. On each plate, two pooled references were included that mimic AD-like (A+T+) and control-like (A-T-) CSF for quality control. In parallel, 50 µL each sample CSF and quality control aliquot were reduced, alkylated, and denatured with tris-2(-carboxyethyl)-phosphine (5 mM), chloroacetamide (40 mM), and sodium deoxycholate (1%) in triethylammonium bicarbonate buffer (100 mM) at 95°C for 10 min, followed by a 10-min cool down at room temperature. CSF proteins were digested with Lys-C (Wako; 0.5 µg; 1:100 enzyme to protein ratio) and trypsin (Promega; 5 µg; 1:10 enzyme to protein ratio) overnight at 37°C. After digestion, heavy labeled standards (15 µL per 50 µL CSF) were added to the peptide solutions followed by acidification with a 1% trifluoroacetic acid (TFA) and 10% formic acid (FA) solution to a final concentration of 0.1% TFA and 1% FA (pH ≤ 2). Sample plates were placed on an orbital shaker at 300 rpm for at least 10 minutes to ensure proper mixing. Plates were centrifuged (4680 rpm) for 30 minutes to pellet the precipitated surfactant. Peptides were desalted with Oasis PRiME HLB 96-well, 30mg sorbent per well, solid phase extraction (SPE) cleanup plates from Waters Corporation (Milford, MA) using a positive pressure system. Each SPE well was conditioned (500 µL methanol) and equilibrated twice (500 µL 0.1% TFA) before 500 µL 0.1% TFA and supernatant were added. Each well was washed twice (500 µL 0.1% TFA) and eluted twice (100 µL 50% acetonitrile/0.1% formic acid). All eluates were dried under centrifugal vacuum.
Each aliquot was reconstituted in 50 µL mobile phase A (0.1% FA). Resuspended peptides (20µL) were separated on an AdvanceBio Peptide Map Guard column (2.1×5mm, 2.7 μm, Agilent) connected to AdvanceBio Peptide analytical column (2.1×150mm, 2.7 μm, Agilent) by a 1290 Infinity II system (Agilent) and monitored on an TSQ Altis Triple Quadrupole mass spectrometer (Thermo Fisher Scientific). The sample was developed over a 14-min gradient using mobile phase A (MPA; 0.1% FA in water) and mobile phase B (B; 0.1% FA in acetonitrile) with flow rate at 0.4 mL/min. The gradient was from 2% to 24% B over 12.1 minutes, then from 24% to 80% over 0.2 min and held at 80% B for 0.7 min. The mass spectrometer was set to acquire data in positive-ion mode using single reaction monitoring acquisition. Three transitions were acquired for each target analyte cycle time set to 0.8 sec, Q1 resolution 0.7 FWHM, Q2 resolution 1.2 FWHM, and CID gas 1.5 mTorr. Total area ratios for each peptide were calculated by summing the area for each light (3) and heavy (3) transition and dividing the light total area by the heavy total area using Skyline. There were 9 total sample plates. Each plate was run independently with two quality control aliquots at the beginning, end, and after every 20 samples per plate.
Statistical analysis
Cognitive and Volumetric Trajectories
Cognitive trajectories were estimated by calculating the slope of the MoCA change from baseline for each participant with a minimum of three visits and a minimum follow-up three years. Cognitive trajectories 4SD above or below the mean were removed given the inherent variability associated with cognitive trajectories calculated from small number of visits. Trajectories for CDR-SB and hippocampal volume were calculated using the same approach for cognitive trajectory. The differences in cognitive, CDR-SB, and hippocampal volume trajectories by last cognitive diagnosis were compared using an unpaired, two-tailed, t-test.
Differential Expression and Correlational Analysis
All differential expression analysis was performed using an unpaired, two-tailed t-test for each outcome. Outcomes included AD clinical status and pairwise comparisons of individuals for published CSF Aβ42 and tTau threshold. Multiple hypothesis testing was accounted for using FDR adjusted p-value by the Benjamini-Hochberg method. We also used Pearson correlation to compare outcomes with individual peptide abundance. Hemoglobin A (HBA), B (HBB) and albumin (ALU) were not expected to vary with AD pathophysiology and, therefore, used as negative internal controls for differential expression and correlational analysis. The outcome variables of interest were CSF Aβ42, CSF tTau, CSF pTau181, AV45, FDG PET, hippocampal volume, MoCA, CDR-SB, annual MoCA change, annual CDR-SB change, and hippocampal volume change. Similar to the analysis of trajectories, outcomes outside 4 standard deviations from the mean were removed.
Classification and Regression Analysis
To test the predictive performance of each putative CSF protein for estimating clinical diagnosis, a logistic regression classifier (sklearn 0.24.2) was trained using a five-fold cross validation to classify individuals as cognitively normal or AD. Performance was assessed using the area under the true positive and false positive rate for the receiver operator curve. To determine the performance of demographic data, previously measured biomarkers, or the SRM CSF proteins generated by this study for estimating clinical diagnosis or dementia-related outcomes, we used multivariate logistic regression classifiers with elastic net regularization for dichotomous outcomes and multivariate linear regression with elastic net regularization for continuous outcomes. A five-fold cross validation to select the best L1-ratio for regularization was implemented to generate classification or regression estimates for all participants. Performance was assessed using a single area under the ROC curve for classification models and correlating the true and estimated outcomes for regression models. A non-parametric bootstrap procedure was used to estimate confidence intervals for AUC measurements. Other multivariate linear regressions with elastic net regularization were performed using a similar procedure.
We compared the predictive performance of the CSF peptides to existing biomarker or demographic data. A non-parametric permutation procedure was used to compare performance for logistic regression models or linear models trained using CSF peptides and existing biomarker or demographic data. Our null hypothesis was that across participants the CSF peptides showed no difference in performance to existing biomarker or demographic data. We computed the true difference in performance for the CSF peptides and existing biomarker data. We then randomly permuted the estimation generated CSF peptides and existing biomarkers for each participant and recomputed the difference in performance. Significance was established using 1000 permutations.
Supplementary Material
Acknowledgments:
We are indebted to all individuals who have selflessly volunteered their time to participate in this study, and to the ADNI teams responsible for recruitment, retention, and study participation. Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf
Funding:
This work was supported by the National Institute of Aging (AG066511) to AIL, (AG061357) to AIL and NTS, (AG056533, AG072120 and AG075827) to APW and TSW, (AG070937) to JJL, (RAG079170) to TSW and by the Foundation of NIH (AMP-AD 2.0 Proteomics) to AIL and NTS. ADNI is supported by the National Institute on Aging (AG024904), the National Institute of Biomedical Imaging and Bioengineering, the Department of Defense (W81XWH-12-2-0012 and through generous contributions from the following: AbbVie, Alzheimer’s Association, Alzheimer’s Drug Discovery Foundation, Araclon Biotech, BioClinica Inc., Biogen, Bristol-Myers Squibb Company, CereSpir Inc., Cogstate, Eisai Inc., Elan Pharmaceuticals Inc., Eli Lilly and Company, EuroImmun, F. Hoffmann-La Roche Ltd and its affiliated company Genentech Inc., Fujirebio, GE Healthcare, IXICO Ltd., Janssen Alzheimer Immunotherapy Research & Development LLC., Johnson & Johnson Pharmaceutical Research & Development LLC., Lumosity, Lundbeck, Merck & Co. Inc., Meso Scale Diagnostics LLC., NeuroRx Research, Neurotrack Technologies, Novartis Pharmaceuticals Corporation, Pfizer Inc., Piramal Imaging, Servier, Takeda Pharmaceutical Company, Transition Therapeutics, The Canadian Institutes of Health Research
Footnotes
Competing interests: DND, NTS, and AIL are founders of EmTheraPro. RH is a consultant for EmTheraPro. TSW is a co-founder of revXon. JJL is a consultant for Roche Diagnostics. A.J.S. is on the Scientific Advisory Board for Bayer Oncology, Eisai; on the Dementia Advisory Board for Siemens Medical Solutions USA, Inc.; on the MESA Observational Study Monitoring Board for NIH NHLBI; on Multiple ADRC and consortium External Advisory Board and the Editorial Office Support as Editor-in-Chief, Brain Imaging and Behavior for Springer-Nature Publishing. All other authors declare no competing financial interests.
Data and materials availability:
The results published here are in whole or in part based on data obtained from the AMP-AD Knowledge Portal (https://adknowledgeportal.synapse.org). The AMP-AD Knowledge Portal is a platform for accessing data, analyses, and tools generated by the AMP-AD Target Discovery Program and other programs supported by the National Institute on Aging to enable open-science practices and accelerate translational learning. The data, analyses, and tools are shared early in the research cycle without a publication embargo on secondary use. Data are available for general research use according to the following requirements for data access and data attribution (https://adknowledgeportal.synapse.org/#/DataAccess/Instructions). Raw MS files and the SRM peptide data that were generated and analyzed as part of this study are available on Synapse (Project SynID: 52288633). For up-to-date information on ADNI, see https://www.adni-info.org. All ADNI data are available the ADNI website (https://adni.loni.usc.edu/). These SRM data are also available at https://adni.loni.usc.edu/ as the “Emory University CSF Targeted MS” version 2022-10-19. Scripts for data analysis are also available at: DOI 10.5281/zenodo.8188292.
References and Notes
- 1.Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, Liu E, Molinuevo JL, Montine T, Phelps C, Rankin KP, Rowe CC, Scheltens P, Siemers E, Snyder HM, Sperling R, Contributors, NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement 14, 535–562 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT, Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 1, a006189 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K, Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112, 389–404 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Braak H, Thal DR, Ghebremedhin E, Del Tredici K, Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 70, 960–969 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Hampel H, Cummings J, Blennow K, Gao P, Jack CR Jr., Vergallo A, Developing the ATX(N) classification for use across the Alzheimer disease continuum. Nat Rev Neurol 17, 580–589 (2021). [DOI] [PubMed] [Google Scholar]
- 6.Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah RC, Wilson RS, Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 66, 1837–1844 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Knopman DS, Parisi JE, Salviati A, Floriach-Robert M, Boeve BF, Ivnik RJ, Smith GE, Dickson DW, Johnson KA, Petersen LE, McDonald WC, Braak H, Petersen RC, Neuropathology of cognitively normal elderly. J Neuropathol Exp Neurol 62, 1087–1095 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Price JL, McKeel DW Jr., Buckles VD, Roe CM, Xiong C, Grundman M, Hansen LA, Petersen RC, Parisi JE, Dickson DW, Smith CD, Davis DG, Schmitt FA, Markesbery WR, Kaye J, Kurlan R, Hulette C, Kurland BF, Higdon R, Kukull W, Morris JC, Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging 30, 1026–1036 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson ECB, Carter EK, Dammer EB, Duong DM, Gerasimov ES, Liu Y, Liu J, Betarbet R, Ping L, Yin L, Serrano GE, Beach TG, Peng J, De Jager PL, Haroutunian V, Zhang B, Gaiteri C, Bennett DA, Gearing M, Wingo TS, Wingo AP, Lah JJ, Levey AI, Seyfried NT, Large-scale deep multi-layer analysis of Alzheimer’s disease brain reveals strong proteomic disease-related changes not observed at the RNA level. Nat Neurosci 25, 213–225 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson ECB, Dammer EB, Duong DM, Ping L, Zhou M, Yin L, Higginbotham LA, Guajardo A, White B, Troncoso JC, Thambisetty M, Montine TJ, Lee EB, Trojanowski JQ, Beach TG, Reiman EM, Haroutunian V, Wang M, Schadt E, Zhang B, Dickson DW, Ertekin-Taner N, Golde TE, Petyuk VA, De Jager PL, Bennett DA, Wingo TS, Rangaraju S, Hajjar I, Shulman JM, Lah JJ, Levey AI, Seyfried NT, Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat Med 26, 769–780 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mostafavi S, Gaiteri C, Sullivan SE, White CC, Tasaki S, Xu J, Taga M, Klein HU, Patrick E, Komashko V, McCabe C, Smith R, Bradshaw EM, Root DE, Regev A, Yu L, Chibnik LB, Schneider JA, Young-Pearse TL, Bennett DA, De Jager PL, A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat Neurosci 21, 811–819 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neff RA, Wang M, Vatansever S, Guo L, Ming C, Wang Q, Wang E, Horgusluoglu-Moloch E, Song WM, Li A, Castranio EL, Tcw J, Ho L, Goate A, Fossati V, Noggle S, Gandy S, Ehrlich ME, Katsel P, Schadt E, Cai D, Brennand KJ, Haroutunian V, Zhang B, Molecular subtyping of Alzheimer’s disease using RNA sequencing data reveals novel mechanisms and targets. Sci Adv 7, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seyfried NT, Dammer EB, Swarup V, Nandakumar D, Duong DM, Yin L, Deng Q, Nguyen T, Hales CM, Wingo T, Glass J, Gearing M, Thambisetty M, Troncoso JC, Geschwind DH, Lah JJ, Levey AI, A Multi-network Approach Identifies Protein-Specific Co-expression in Asymptomatic and Symptomatic Alzheimer’s Disease. Cell Syst 4, 60–72 e64 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wingo AP, Dammer EB, Breen MS, Logsdon BA, Duong DM, Troncosco JC, Thambisetty M, Beach TG, Serrano GE, Reiman EM, Caselli RJ, Lah JJ, Seyfried NT, Levey AI, Wingo TS, Large-scale proteomic analysis of human brain identifies proteins associated with cognitive trajectory in advanced age. Nat Commun 10, 1619 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wingo AP, Fan W, Duong DM, Gerasimov ES, Dammer EB, Liu Y, Harerimana NV, White B, Thambisetty M, Troncoso JC, Kim N, Schneider JA, Hajjar IM, Lah JJ, Bennett DA, Seyfried NT, Levey AI, Wingo TS, Shared proteomic effects of cerebral atherosclerosis and Alzheimer’s disease on the human brain. Nat Neurosci 23, 696–700 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Higginbotham L, Ping L, Dammer EB, Duong DM, Zhou M, Gearing M, Hurst C, Glass JD, Factor SA, Johnson ECB, Hajjar I, Lah JJ, Levey AI, Seyfried NT, Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer’s disease. Sci Adv 6, eaaz9360 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watson CM, Dammer EB, Ping L, Duong DM, Modeste E, Carter EK, Johnson ECB, Levey AI, Lah JJ, Roberts BR, Seyfried NT, Quantitative Mass Spectrometry Analysis of Cerebrospinal Fluid Biomarker Proteins Reveals Stage-Specific Changes in Alzheimer’s Disease. medRxiv, 2022.2008.2030.22279370 (2022). [Google Scholar]
- 18.Foote M, Zhou Y, 14-3-3 proteins in neurological disorders. Int J Biochem Mol Biol 3, 152–164 (2012). [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou M, Haque RU, Dammer EB, Duong DM, Ping L, Johnson ECB, Lah JJ, Levey AI, Seyfried NT, Targeted mass spectrometry to quantify brain-derived cerebrospinal fluid biomarkers in Alzheimer’s disease. Clin Proteomics 17, 19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cornell B, Toyo-Oka K, 14-3-3 Proteins in Brain Development: Neurogenesis, Neuronal Migration and Neuromorphogenesis. Front Mol Neurosci 10, 318 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Villemagne VL, Dore V, Burnham SC, Masters CL, Rowe CC, Imaging tau and amyloid-beta proteinopathies in Alzheimer disease and other conditions. Nat Rev Neurol 14, 225–236 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Blennow K, Mattsson N, Scholl M, Hansson O, Zetterberg H, Amyloid biomarkers in Alzheimer’s disease. Trends Pharmacol Sci 36, 297–309 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Zetterberg H, Bendlin BB, Biomarkers for Alzheimer’s disease-preparing for a new era of disease-modifying therapies. Mol Psychiatry 26, 296–308 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olsson B, Lautner R, Andreasson U, Ohrfelt A, Portelius E, Bjerke M, Holtta M, Rosen C, Olsson C, Strobel G, Wu E, Dakin K, Petzold M, Blennow K, Zetterberg H, CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol 15, 673–684 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Jack CR Jr., Wiste HJ, Therneau TM, Weigand SD, Knopman DS, Mielke MM, Lowe VJ, Vemuri P, Machulda MM, Schwarz CG, Gunter JL, Senjem ML, Graff-Radford J, Jones DT, Roberts RO, Rocca WA, Petersen RC, Associations of Amyloid, Tau, and Neurodegeneration Biomarker Profiles With Rates of Memory Decline Among Individuals Without Dementia. JAMA 321, 2316–2325 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou M, Haque RU, Dammer EB, Duong DM, Ping L, Johnson ECB, Lah JJ, Levey AI, Seyfried NT, Targeted mass spectrometry to quantify brain-derived cerebrospinal fluid biomarkers in Alzheimer’s disease. Clin Proteomics 17, 19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bader JM, Geyer PE, Muller JB, Strauss MT, Koch M, Leypoldt F, Koertvelyessy P, Bittner D, Schipke CG, Incesoy EI, Peters O, Deigendesch N, Simons M, Jensen MK, Zetterberg H, Mann M, Proteome profiling in cerebrospinal fluid reveals novel biomarkers of Alzheimer’s disease. Mol Syst Biol 16, e9356 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Visser PJ, Reus LM, Gobom J, Jansen I, Dicks E, van der Lee SJ, Tsolaki M, Verhey FRJ, Popp J, Martinez-Lage P, Vandenberghe R, Lleo A, Molinuevo JL, Engelborghs S, Freund-Levi Y, Froelich L, Sleegers K, Dobricic V, Lovestone S, Streffer J, Vos SJB, Bos I, Adni A. B. Smit, Blennow K, Scheltens P, Teunissen CE, Bertram L, Zetterberg H, Tijms BM, Cerebrospinal fluid tau levels are associated with abnormal neuronal plasticity markers in Alzheimer’s disease. Mol Neurodegener 17, 27 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bai B, Wang X, Li Y, Chen PC, Yu K, Dey KK, Yarbro JM, Han X, Lutz BM, Rao S, Jiao Y, Sifford JM, Han J, Wang M, Tan H, Shaw TI, Cho JH, Zhou S, Wang H, Niu M, Mancieri A, Messler KA, Sun X, Wu Z, Pagala V, High AA, Bi W, Zhang H, Chi H, Haroutunian V, Zhang B, Beach TG, Yu G, Peng J, Deep Multilayer Brain Proteomics Identifies Molecular Networks in Alzheimer’s Disease Progression. Neuron 106, 700 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.ADNI Procedures Manual(Accessed: URL: https://adni.loni.usc.edu/wp-content/uploads/2008/07/adni2-procedures-manual.pdf
- 31.Shaw LM, Trojanowski JQ, for the Alzheimer’s Disease Neuroimaging Initiative, Methods Report, ADNI3: Batch analyses of Ab42, t-tau, and p-tau181 in ADNI1, GO, 2 CSF samples using the fully automated Roche Elecsys and cobas e immunoassay analyzer system”(Accessed: 12/13/2022) URL: https://adni.loni.usc.edu/wp-content/uploads/2018/04/PPT-set-102-PPSB-FTF-Boston-10-31-2017.pptx
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The results published here are in whole or in part based on data obtained from the AMP-AD Knowledge Portal (https://adknowledgeportal.synapse.org). The AMP-AD Knowledge Portal is a platform for accessing data, analyses, and tools generated by the AMP-AD Target Discovery Program and other programs supported by the National Institute on Aging to enable open-science practices and accelerate translational learning. The data, analyses, and tools are shared early in the research cycle without a publication embargo on secondary use. Data are available for general research use according to the following requirements for data access and data attribution (https://adknowledgeportal.synapse.org/#/DataAccess/Instructions). Raw MS files and the SRM peptide data that were generated and analyzed as part of this study are available on Synapse (Project SynID: 52288633). For up-to-date information on ADNI, see https://www.adni-info.org. All ADNI data are available the ADNI website (https://adni.loni.usc.edu/). These SRM data are also available at https://adni.loni.usc.edu/ as the “Emory University CSF Targeted MS” version 2022-10-19. Scripts for data analysis are also available at: DOI 10.5281/zenodo.8188292.