

Abstract
Neurofibrillary tangles composed of hyperphosphorylated, aggregated tau are a common pathological feature of tauopathies, including Alzheimer's disease. Abnormal phosphorylation of tau by kinases or phosphatases has been proposed as a pathogenic mechanism in tangle formation. To investigate whether kinase inhibition can reduce tauopathy and the degeneration associated with it in vivo, transgenic mice overexpressing mutant human tau were treated with the glycogen synthase kinase-3 (GSK-3) inhibitor lithium chloride. Treatment resulted in significant inhibition of GSK-3 activity. Lithium administration also resulted in significantly lower levels of phosphorylation at several epitopes of tau known to be hyperphosphorylated in Alzheimer's disease and significantly reduced levels of aggregated, insoluble tau. Administration of a second GSK-3 inhibitor also correlated with reduced insoluble tau levels, supporting the idea that lithium exerts its effect through GSK-3 inhibition. Levels of aggregated tau correlated strongly with degree of axonal degeneration, and lithium-chloride-treated mice showed less degeneration if administration was started during early stages of tangle development. These results support the idea that kinases are involved in tauopathy progression and that kinase inhibitors may be effective therapeutically.
Keywords: phosphorylation, neurofibrillary tangles, Alzheimer's disease, tau protein
Neurofibrillary tangles (NFTs) primarily comprise aggregated, microtubule-associated protein tau (1). Tau in NFTs aggregates in the neuronal cell body, it is hyperphosphorylated at specific sites (2), and it takes on an abnormal conformation (ref. 3 and reviewed in ref. 4). Two proline-directed kinases, glycogen synthase kinase-3 (GSK-3) and cyclin-dependent kinase-5 are thought to be key factors in abnormal tau phosphorylation (reviewed in refs. 5 and 6). Mammalian GSK-3 exists as two isoforms, α and β, and, unlike most protein kinases, it is constitutively active in neurons (7, 8). In vitro work has shown that GSK-3β phosphorylates tau at multiple sites, and some of these sites are abnormally phosphorylated in neurodegenerative disease (7, 9). In mammalian cells, cotransfection of tau with GSK-3β leads to increased tau phosphorylation and microtubule rearrangement (9, 10). Transgenic mice overexpressing GSK-3β display tau hyperphosphorylation, disrupted microtubules, and apoptotic neurons (11). GSK-3β is involved in the formation of oligomeric tau fibrils in vitro (12), and it is associated with filamentous tau in transgenic models (13, 14). GSK-3β has been identified in association with NFTs in Alzheimer's disease (AD) brain (15, 16). Lithium, a medication for bipolar disorder, is a direct (17) and indirect (18, 19) inhibitor of GSK-3. In cultured neurons, lithium has been shown to suppress tau phosphorylation, enhance tau–microtubule binding, and promote microtubule assembly (20–22). In vivo, lithium has been shown to reduce insoluble tau (23) and ameliorate axonal transport deficiencies in transgenic Drosophila (24). Phiel et al. (25) have recently shown that lithium chloride (LiCl) significantly decreases β-amyloid (Aβ) production in vivo through inhibition of GSK-3 activity. These data suggest LiCl may have therapeutic relevance in the treatment of AD and related disorders. To test the effect of LiCl on pathogenic tau formation in vivo, we have treated JNPL3 transgenic mice overexpressing mutant human tau (26) with two GSK-3 inhibitors. We report significant reduction in aggregated tau and degeneration after treatment in these mice. These results suggest that kinase inhibitors may be therapeutically beneficial for the treatment of tauopathies, such as frontotemporal dementia and AD.
Materials and Methods
Animals. Transgenic mice [JNPL3 line (26)] overexpressing mutant human tau (P301L, 4R0N) were used in this study. Mice were heterozygous and on a mixed hybrid genetic background composed of C57BL/DBA2/SW, as published in ref. 26. These mice develop NFTs in the basal telencephalon, diencephalon, brainstem, and spinal cord, with severe pathology accompanied by degeneration in the spinal cord leading to dystonia, paralysis, and death in mice >12 months in age. Four individual, single-gender groups of JNPL3 mice between the ages of 8 and 11 months were administered LiCl (total, n = 22) or AR-A014418 (n = 10) and used for analyses with littermates split between treatment groups as much as possible. Mice did not display signs of dystonia when assessed for hindlimb clasping. One group of mice at a later stage (≈12 months of age, 11 were LiCl-treated and 12 were PBS-treated) was also tested. These mice had borderline-to-significant impairment of motor performance when assessed by rotarod (AccuRotor rotarod, 3-cm diameter, AccuScan Instruments, Columbus, OH) (four trials each at 10, 20, or 30 rpm, trials performed before treatment and then at 1-week intervals for 4 weeks) that worsened significantly during the 4-week treatment duration. All animals were maintained and killed according to National Institute of Health/Institutional Animal Care and Use Committee guidelines.
Kinase Inhibitor Treatment. Mice received i.p. injections of either 0.6 M LiCl (10 microliters per gram of body weight) or sterile 10 mM PBS (10 microliters per gram of body weight) daily for 30 days. Mice were killed 1 h after treatment by cervical dislocation. Brain regions were dissected and immediately snap-frozen on dry ice. Spinal cords were immersion-fixed in cold paraformaldehyde and paraffin-embedded. AR-A014418 (AstraZeneca, Sodertalje, Sweden) is a thiazole, N-(4-methoxybenzyl)-N′-(5-nitro-1,3-thiazol-2-yl) urea, which is a selective and potent inhibitor of GSK-3 among 26 other kinases tested (27). It has been shown to inhibit tau phosphorylation in cells expressing human tau, and it inhibits neurodegeneration mediated by Aβ peptide in hippocampal slices. AR-A014418 was reconstituted in 40% PEG400 and 40% dimethylamine/water and administered at a concentration of 30 μmol/kg. Drug or vehicle was administered twice daily for 1 month by oral gavage to female JNPL3 mice (n = 10 for each group).
Antibodies. The following monoclonal antibodies from Peter Davies (Albert Einstein University, New York) were used (specificity and isotype are given in parentheses): CP27 (human tau; mouse IgG2b), TG5 (mouse and human tau; mouse IgG2b), CP13 (phospho-Ser-202; mouse IgG1), PHF-1 (phospho-Ser-396/404; mouse IgG1), MC1 (abnormal tau conformation 5–15, 312–322; mouse IgG1). Also used were the following antibodies from Biosource International, Camarillo, CA: anti-tau pS262 (rabbit polyclonal), Anti-tau p422 (rabbit polyclonal), and GSK-3α/β (mouse IgG). Phospho-Akt (Ser-473, rabbit polyclonal, Cell Signaling Technology, Beverly, MA), phospho-GSK-3β pS9 (phospho-Ser-9 of GSK-3β, rabbit IgG polyclonal; Quality Controlled Biochemicals, Hopkinson, MA.), and GSK-3β (mouse IgG1, BD Transduction Laboratories, Lexington, KY) were also used. 3-repeat (RD3) and 4-repeat (RD4) tau-specific monoclonal antibodies (28) were a gift from R. de Silva (University College London, London).
Immunoprecipitation and Kinase Activity Assay. GSK-3β activity assay was performed on fresh mouse cortex by using a modification of the methods described in refs. 14 and 29. Briefly, mice were killed by cervical dislocation, and brains were dissected and homogenized in RIPA buffer (50 mM Tris, pH 8.0/150 mM NaCl/1% Nonidet P-40/0.5% sodium deoxycholate/0.1% SDS) containing protease and phosphatase inhibitors. After immunoprecipitation with GSK-3β antibody, aliquots of the immunocomplex were loaded on a 10% SDS/PAGE gel, and the activation state of GSK-3β was detected with GSK-3β phospho-Ser-9 antibody. The rest of the immunocomplex was subjected to kinase assay by using recombinant human tau as a substrate (Invitrogen).
Immunoblot Analyses of Heat-Stable Tau and Aggregated Tau. Frozen dissected tissues were homogenized in RIPA buffer without thawing by using a mechanical homogenizer (TH, Omni International, Marietta, GA). After being boiled for 5 min, protein aggregates were removed by centrifugation. Heat-stable samples containing 1–3 μg of protein (previously determined to fit in the linear range for quantification) were run on a 10% Tris-Tricine gel (Invitrogen) then electrophoretically transferred to a poly-(vinylidene difluoride) membrane. After blocking with 5% dry milk for 1 h, membranes were probed with primary antibodies, detected by using horseradish peroxidase-coupled secondary antibodies (Jackson ImmunoResearch), and visualized by enhanced chemiluminescence reagent (SuperSignal West Pico or Femto, Pierce) using a Fujifilm LAS3000 imaging system. For analysis of aggregated tau, Sarkosyl extractions were performed on mouse brainstem or cortex samples as described in ref. 30. Values presented are derived from densitometry arbitrary units.
Immunohistochemistry. Spinal cord tissues were drop-fixed without perfusion into 4% paraformaldehyde overnight, paraffin-embedded with an autoprocessor (Shandon, Pittsburgh, PA), and subjected to immunohistochemistry by using antibody MC1 as described in ref. 26. Luxol fast blue (LFB) staining was performed according to a standard protocol (31). Briefly, sections were deparaffinized and rehydrated to 95% alcohol and incubated overnight in 0.01% LFB solution. Sections were rinsed and differentiated in 0.05% LiCl solution. Sections were pre-treated in 5% periodic acid solution for 5 min, incubated in Schiff's reagent for 15 min, and developed by washing slides in tap water for 5 min. Slides were counterstained with 0.68% hematoxylin, dehydrated, and coverslipped.
Image Analysis. Images were taken from spinal cord sections at high power (×40). Images were captured by a Sony three-charge-coupled-device, color video camera and digitized with the intellicam 2.0 program (Matrox, Dorval, Canada). The images were imported into sigmascan pro 3.0 (SyStat Software, Richmond, CA), and the percentage area of immunostaining was determined.
Analysis of LFB-Stained Sections. The severity of white matter pathology was scored qualitatively from 0 to 3 by observing three abnormalities as described in ref. 32. Mice that were pathologically normal were given a score of 0. Mice that had myelin debris, increased vacuolization, and possibly a phagocyte were given a score of 1; mice that had myelin debris, vacuolization, and several phagocytes were scored as 2; and mice that had myelin debris, a large degree of vacuolization, and many phagocytes were given a score of 3.
Statistical Analysis. Statistical analysis was performed by using prism 3.0 (GraphPad, San Diego). Data were expressed as mean ± SD and analyzed by using a two-tailed unpaired t test, with results deemed significant at P < 0.05. Correlations were computed by using the Spearman test. Significance of correlations was computed by using Kendall's tau.
Results
LiCl is used therapeutically at plasma concentrations of between 0.2 and 1.5 milliequivalents (mEq) per liter for the treatment of bipolar disorders (33). The level of lithium and its effect on GSK-3 was determined in nontransgenic mice (Fig. 1 A–D, n = 5 for each time point). Our dosing paradigm resulted in plasma concentrations of ≈0.2 mEq/liter 24 h after injection, indicating that plasma lithium was maintained at physiologically relevant levels (Fig. 1 A). All mice used in this study were killed 1 h after final injection when the plasma lithium concentration was 3.2 ± 0.2 mEq/liter. Lithium has been reported to cause renal problems resulting in weight loss. We found no significant difference in weight after treatment with LiCl (mean weight ± SEM was 45.7 ± 4.5 g compared with 42.9 ± 2.9 g after 1 month of treatment). Western blot analysis showed no difference in the levels of GSK-3α or GSK-3β in homogenate from cortex after LiCl treatment when compared with vehicle-treated mice (Fig. 1B). Lithium is known to inhibit GSK-3β activity in two ways: by acting as a competitive inhibitor of Mg2+ (34) and by increasing inhibitory phosphorylation at Ser-9 through activation of the Akt/protein kinase B pathway (18). GSK-3β activity was measured by using two methods: (i) Immunoblots were probed with an antibody against the Ser-9 epitope of GSK-3β (Fig. 1C), and (ii) GSK-3β activity was measured directly by using an enzyme-based activity assay with recombinant tau (Fig. 1D). Quantification of Western blot analysis showed that phosphorylation at Ser-9 was increased by 64 ± 12% compared with control mice (P < 0.01) (Fig. 1C). Similarly, the activity assays showed a 57 ± 9% (P < 0.01) reduction in GSK-3β activity (Fig. 1D). Both methods of analysis indicated that GSK-3β activity was significantly reduced after administration of LiCl. We also confirmed that LiCl treatment correlated with activation of the Akt pathway (Fig. 1D) in addition to inhibitory phosphorylation of GSK-3β at Ser-9 (Fig. 1E) in JNPL3 mice (n = 6 for each treatment). PBS was used for control rather than NaCl because our published data have shown that NaCl has no significant effect on tau phosphorylation (29). Furthermore, the use of a second GSK-3 inhibitor strengthened our confidence that the effects seen with LiCl were not due to ionic imbalance. Inhibition of GSK-3β by lithium has been reported to increase tau mRNA containing exon 10 (35). By using 3-repeat and 4-repeat tau-specific antibodies (28), we assessed whether tau splice ratios were altered by lithium treatment. 4-Repeat tau did not change and 3-repeat tau was undetectable (data not shown), which is not surprising given that tau in adult mouse brain is exclusively 4-repeat (36) and that JNPL3 mice express only 4-repeat human tau with no regulatory sequence. Thus, our results are not due to a shift in tau isoform composition that might affect the pathology.
Fig. 1.

Plasma lithium concentration and GSK-3 expression and activity (act.). Measurement of plasma lithium concentration in treated nontransgenic animals over 24 h. Concentration was maintained at or above physiologically relevant levels (0.2 mEq/liter) throughout the treatment regime. (A) Plasma LiCl concentration in PBS-treated controls was minimal. (B) Western blot analysis shows no change in GSK-3α/β levels after LiCl injection. (C) Kinase activity assays followed by Western blot analyses show a 63.98 ± 12.43% increase in phosphorylation at the Ser-9 epitope (pS-9) of GSK-3β 1 h after LiCl administration, indicating a significant reduction in GSK-3 activity after LiCl treatment. (D) The inhibition of GSK-3β activity was confirmed by using an enzyme-based kinase activity assay with 32P. GSK-3β activity was reduced up to 24 h after injection when compared with controls (Veh) (n = 5 in all analyses). Three representative data are displayed. (E and F) In addition to enhanced phosphorylation at the inhibitory Ser-9 site of GSK-3 (E), lithium-treated JNPL3 mice showed enhanced phosphorylation at the Akt activating site (F)(n = 6 for each treatment). Two representative data are displayed.
Aggregate-free, heat-stable (soluble) tau was prepared from the brainstem of male mice (vehicle, n = 5; LiCl, n = 4) and assessed by immunoblotting (Fig. 2). Levels of human tau recognized by CP27 or total tau recognized by TG5 were unchanged after LiCl treatment (Fig. 2 A). After normalization to total levels of tau, LiCl treatment resulted in significantly decreased phosphorylation at putative GSK-3-directed sites, including Ser-202 (CP13) and Ser-396/404 (PHF-1). Phosphorylation at sites not recognized by GSK-3 (Ser-422 and Ser-262) appeared not to be affected in LiCl-treated mice. These data were confirmed in another set of male mice (vehicle, n = 5; LiCl, n = 6) that displayed significant reductions in tau phosphorylation at CP13 and PHF-1 epitopes (P < 0.05) but no change at the CP27 site (Fig. 2B). Similar results were obtained for CP13 and CP27 in a set of female mice (vehicle, n = 5; LiCl, n = 6) (data not shown).
Fig. 2.

Heat-stable tau fraction from the brainstem after lithium treatment. (A) Analysis of the heat-stable, soluble tau fraction from the brainstem showed no significant change in the amount of total tau (TG5, human and mouse tau) or human tau (CP27). After normalization to total levels of tau, lithium treatment resulted in decreased (P < 0.05) phosphorylation at GSK-3-directed sites, including Ser-202 (CP13) and Ser-396/404 (PHF-1). Sites not phosphorylated by GSK-3 (Ser-422 and Ser-262) appeared not to be affected in lithium-treated mice. Two representative data are displayed from five vehicle (Veh) and four LiCl-treated mice. *, P < 0.05. (B) These data were confirmed in another set of mice (vehicle, n = 5; LiCl, n = 6) showing reduction in tau phosphorylation at CP13 and PHF-1 epitopes (P < 0.05) but no change in total human tau levels (CP27). Two representative data are displayed.
We next examined whether LiCl treatment modulated tau aggregate formation in the cortex and the brainstem, a region prone to tangle formation in JNPL3 mice. Tau in the Sarkosyl pellet has been shown by immunoEM to be filamentous (14), and it is synonymous with that identified by immunohistochemistry in NFTs. There was a significant reduction of aggregated tau in the cortex of LiCl-treated male mice (n = 5) relative to controls (n = 5), as revealed by CP27 (Fig. 3A). Tau phosphorylated at Ser-202 and Ser-396/404 was also diminished by the treatment (Fig. 3A). In the brainstem, Sarkosyl-insoluble tau was similarly decreased by LiCl treatment (P < 0.01) in either male or female mice (vehicle, n = 5; LiCl, n = 6) (Fig. 3B). The insoluble tau fraction from LiCl-treated and vehicle-treated mice contained tau that migrated at ≈53 kDa. Tau from vehicle-treated female mice had an additional, hyperphosphorylated form that migrated at ≈64 kDa (Fig. 3B). Our observation that female JNPL3 mice have enhanced pathology compared with age-matched male littermates confirms previous reports (37). Although lithium is a relatively specific inhibitor of GSK-3, it has also been shown to inhibit the activity of some other kinases (38) or enhance kinases, such as phosphatidylinositol 3-kinase and Akt (18). To assess whether the effects on insoluble tau were due to direct inhibition of GSK-3, a second inhibitor, AR-A014418 was administered to JNPL3 mice. AR-A014418 has been shown to be a specific inhibitor of GSK-3 among 26 other kinases tested (27). Insoluble tau levels in the brainstem of mice treated with AR-A014418 were significantly reduced (P < 0.05; vehicle, n = 10; AR-A014418, n = 10), when compared with vehicle-treated animals (Fig. 3C).
Fig. 3.

Aggregated tau fraction from the cortex and the brainstem after lithium or AR-A014418 (AR) treatment. (A) There was a significant reduction of aggregated tau in the cortex of lithium-treated male mice relative to controls (n = 5 for each), as revealed by CP27 (P < 0.01). Two representative data are displayed. Tau phosphorylated at Ser-202 and Ser-396/404 was also diminished by the treatment (P < 0.05 and 0.01, respectively), but the significance was lost when normalized to CP27 (data not shown). Veh, vehicle. *, P < 0.05; **, P < 0.01. (B) In the brainstem, insoluble tau was similarly decreased by lithium treatment (P < 0.01; vehicle, n = 5; LiCl, n = 6) in either male or female mice. Four representative data are displayed. (C) Insoluble tau levels in the brainstem of mice treated with the GSK-3 inhibitor AR-A014418 were significantly reduced (P < 0.05, n = 10) when compared with vehicle-treated animals.
Pathology in the gray matter of spinal cord from mice was assessed by using antibody MC1 and Gallyas silver stain. A positive correlation was observed between insoluble tau levels and MC1-immunopositive cell staining (12-month-old mice, LiCl: R = 0.7, P = 0.005, n = 11; PBS: R = 0.5, P = 0.05, n = 12; 8-month-old mice, LiCl: R = 0.97, P = 0.003, n = 6; PBS, R = 0.88, P = 0.03, n = 6) (Fig. 4). No significant difference was found between the percentage immunostaining for MC1 and treatment in either group. The number of Gallyas-positive cell bodies was too low to be informative.
Fig. 4.
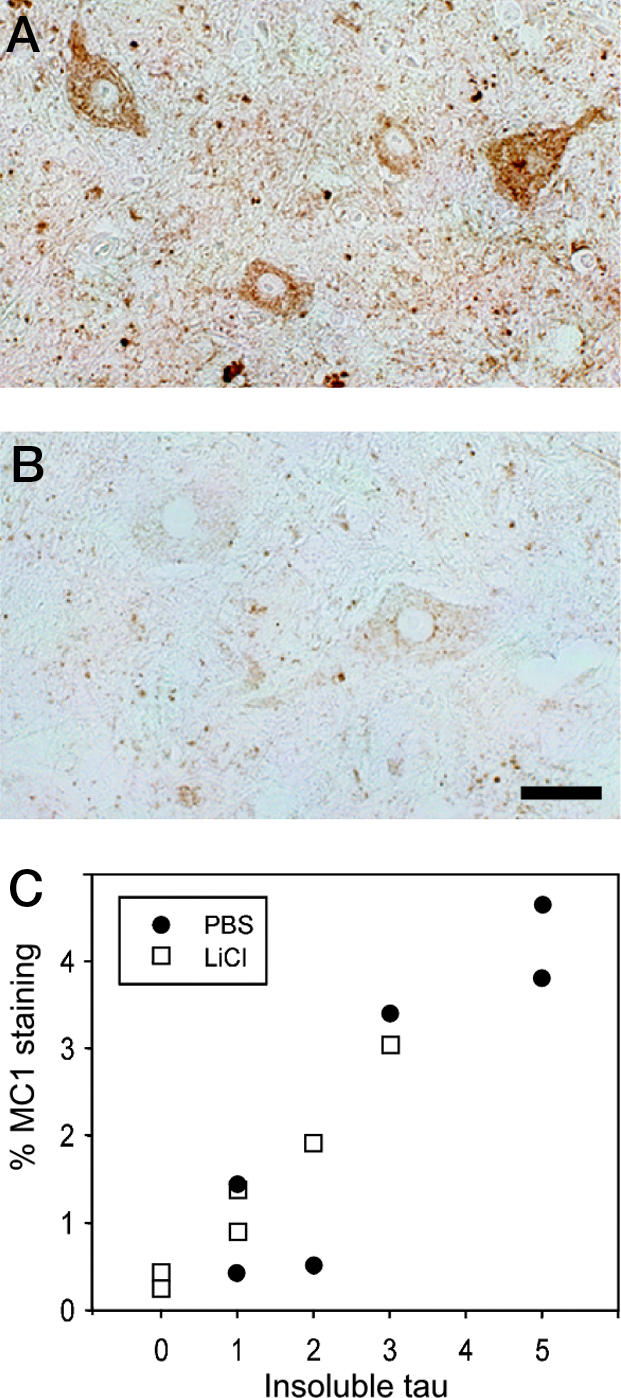
MC1 immunostaining. (A and B) Section from the spinal cord of a vehicle-treated mouse with high levels of insoluble tau (A) or a lithium-treated mouse with low levels of insoluble tau (B) stained with antibody MC1. (Scale bar, 90 μm.) (C) Insoluble tau levels correlated with MC1 staining in 12- and 8-month-old mice (8-month-old mice shown; vehicle, n = 6; LiCl, n = 6; R = 0.7, P = 0.005); however, LiCl treatment did not significantly reduce levels of MC1 immunostaining.
Most of the groups of mice were killed early during disease progression, before hindlimb paralysis developed. One older group (12 months old; LiCl, n = 11; vehicle, n = 12) did degenerate to the stage at which dystonia was significant, but we were unable to detect any retardation in LiCl-treated animals in the degree of motor impairment on the rotarod (data not shown). We were also unable to show a significant reduction in insoluble tau levels in the brainstem or reduced axonal degeneration with treatment in these mice, although variability at this stage was high. Administration starting at an early age for several months was not attempted because long-term administration of LiCl to mice appears to result in loss of GSK-3 inhibition (M. Hutton, personal communication).
To assess whether insoluble tau accumulation in the brainstem and spinal cord of the mice had a functional impact, the degree of axonal degeneration was assessed by staining spinal cord sections with LFB. Three measures of axonal degeneration were monitored: (i) degeneration of the myelin sheath indicated by accumulated debris, (ii) increased vacuolization, and (iii) influx of phagocytes as described by Zehr et al. (32). Fig. 5 shows the three indicators in a more severely affected JNPL3 mouse (Fig. 5A) compared with a mildly affected mouse (Fig. 5B). Each mouse was assessed for the three indicators and scored from 1 to 3, where 1 indicated the least degeneration and 3 indicated the most severe degeneration. Overall, correlation analysis with Kendall's tau indicated that the level of insoluble tau correlated significantly with the degree of axonal degeneration (P = 0.01, n = 9) (Fig. 5C). Furthermore, if treatment was started at an early stage in the degenerative process, the degree of axonal degeneration could be significantly reduced after LiCl treatment (Fig. 5D).
Fig. 5.

Axonal degeneration and insoluble tau levels. (A) Section from the spinal cord of a vehicle-treated (Veh) mouse with high levels of insoluble tau stained with LFB. The asterisk represents a vacuole, the arrowhead represents a phagocyte, and the arrows indicate degenerating myelin. This mouse scored a 3 on overall degeneration. (B) Section from a lithium-treated mouse with low levels of insoluble tau stained with LFB. The arrows indicate glial nuclei. This mouse scored a 0 on overall degeneration (Scale bar, 60 μm.) (C) Correlation between insoluble tau levels and degree of axonal degeneration for PBS-treated mice (n = 12). There was a significant correlation between the level of insoluble tau and degeneration (R = 0.6, P = 0.01). (D) In 8-month-old mice treated with LiCl, there was a significant reduction in the level of axonal degeneration compared with vehicle-treated mice (n = 6 for each group). *, P < 0.05.
Discussion
In tauopathy, tau is redistributed from the axons to the somatodendritic compartment of neurons, where it aggregates in a hyperphosphorylated, filamentous form as NFTs. Insoluble, filamentous, hyperphosphorylated tau isolated in Sarkosyl preparations is found in some transgenic mouse models of tauopathy, and it becomes more abundant and more hyperphosphorylated as pathology progresses to form mature, congophilic/argyrophilic tangles (26, 39, 40). Insoluble tau is therefore considered a highly relevant target for intervention.
To assess whether inhibition of GSK-3 is associated with reduced tauopathy in vivo, JNPL3 mice were administered LiCl. Because LiCl is not completely specific for GSK-3, a second GSK3 inhibitor, AR-A014418, was also tested. Treatment correlated with reduced phosphorylation of soluble tau at sites known to be phosphorylated by GSK-3β, suggesting that the inhibitors can reduce tau phosphorylation either directly or indirectly in these mice. LiCl treatment was associated with reduced levels of Sarkosyl-insoluble, filamentous tau in brainstem and cortex, which was also seen in brainstem after treatment with AR-A014418, confirming that the most likely mediator is GSK-3. Subsequent studies with AR-A014418 have confirmed these results in more JNPL3 mice and in a new model of tauopathy, the hTau line (39) (data not shown). Overall, our data showing a reduction in insoluble tau levels confirm and extend the findings of Perez et al. (23), strongly supporting LiCl impact on accumulation of this potentially pathogenic type of tau.
It is not known whether treatment reduced mature tangle load, because these studies were performed on mice at a relatively early stage of pathology progression and numbers of argyrophilic neurons were low or negligible, making it difficult to determine whether there was a statistically significant difference. Our data from older mice would suggest, however, that once pathology has progressed to a more advanced stage, with extensive argyrophilic tangles and degeneration in the brainstem and spinal cord, tauopathy is more refractive to intervention.
An antibody recognizing the abnormal conformation of tau in AD tangles, antibody MC1, showed a positive correlation with insoluble tau levels, confirming previous observations that this epitope recognizes a pathological form of tau that accumulates in cell bodies in older mice (26, 39). Levels of insoluble tau decreased with LiCl treatment, but MC1 immunostaining did not significantly decrease. This observation may be a technical issue in that the reduction in insoluble tau levels may be too subtle to be reflected by a significant change in cell body MC1, or it may be that there is a large excess of insoluble tau that is not in the MC1 conformation, which is supported by the observation that insoluble tau exists in younger mice before MC1-immunopositive cell bodies are observed (26). Alternatively, this finding may be more functionally relevant, suggesting that, although there is a general correlation between high levels of insoluble tau and formation of the MC1 epitope, there may be a disconnect with a pool of insoluble tau that is modulated by LiCl treatment. Interestingly, evidence is mounting that the degree of filamentous tau accumulating in tangles does not directly correlate with morphological cellular abnormalities (41), which suggests that immunohistochemically defined lesions are not the only pathway to cellular dysfunction.
Treatment with LiCl induced a reduction of tau phosphorylation at Ser-202 and Ser-396/404 but not at Ser-262 or Ser-422. Thus, a partial reduction of tau phosphorylation at specific GSK-3 epitopes seems to be enough to induce a reduction in tau aggregation levels. This observation could find an explanation in recent work by Necula and Kuret (42) that showed that Ser-202 and Ser-396/404 phosphorylation can enhance tau fibrillization in vitro, whereas Ser-262 and Ser-422 phosphorylation does not. Overall, these data suggest that inhibition of specific kinases phosphorylating epitopes known to promote tau fibrillization might be a valid approach to reducing aggregated tau levels in vivo.
Of note, in 8-month-old mice, the reduction in levels of insoluble tau correlated with a reduced degree of axonal degeneration, suggesting that the target of LiCl was functionally relevant. Because the levels of insoluble tau and phosphoepitopes of soluble and insoluble tau were reduced, it is not possible to identify which species may be responsible for the axonal degeneration seen in these mice. We have previously shown that modulating kinase activity can accelerate tauopathy in the JNPL3 mice (14), suggesting that abnormal phosphorylation is a pathogenic mechanism, and it would appear that reducing the activity of a kinase may be beneficial if treatment is started before irreversible damage is done.
AD is characterized by the accumulation of Aβ into amyloid plaques and by tauopathy. LiCl has also been shown to reduce Aβ levels in amyloid-forming transgenic mice by inhibiting GSK-3 activity (25). The effects we have observed after administration of LiCl to tangle-forming mice suggest that LiCl treatment of patients with AD could have a dual impact, not only reducing the accumulation of Aβ but also of pathogenic, insoluble tau. It will be interesting to see whether GSK-3 inhibitors other than LiCl also target Aβ and whether inhibitors targeting kinases other than GSK-3 also impact neurofibrillary pathogenesis, which would improve the range of inhibitors that could be developed for treating tauopathies. Furthermore, given that protein aggregation induced or enhanced by aberrant phosphorylation may be a common pathogenic mechanism in other neurodegenerative disorders, such as Parkinson's disease (43), the use of kinase inhibitors may have utility in treating diseases other than the tauopathies.
Acknowledgments
We thank Dr. Peter Davies and Dr. Rohan de Silva for supplying tau antibodies, Dr. Tom Cooper (Nathan S. Kline Institute) for plasma LiCl measurements, and Dr. Mike Hutton (Mayo Clinic College of Medicine, Jacksonville, FL) for helpful discussions. This work was supported by National Institute on Aging Grant AG172116, National Institute of Neurological Disorders and Stroke Grant NS40256 (to K.D. and D.D.), and an Alzheimer's Association grant (to K.D.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: Aβ, β-amyloid; AD, Alzheimer's disease; GSK-3, glycogen synthase kinase-3; LFB, Luxol fast blue; NFT, neurofibrillary tangle.
References
- 1.Friedhoff, P., von Bergen, M., Mandelkow, E. M. & Mandelkow, E. (2000) Biochim. Biophys. Acta 1502, 122–132. [DOI] [PubMed] [Google Scholar]
- 2.Grundke-Iqbal, I., Iqbal, K., Tung, Y. C., Quinlan, M., Wisniewski, H. M. & Binder, L. I. (1986) Proc. Natl. Acad. Sci. USA 83, 4913–4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weaver, C. L., Espinoza, M., Kress, Y. & Davies, P. (2000) Neurobiol. Aging 21, 719–727. [DOI] [PubMed] [Google Scholar]
- 4.Buee, L., Bussiere, T., Buee-Scherrer, V., Delacourte, A. & Hof, P. R. (2000) Brain Res. Brain. Res. Rev. 33, 95–130. [DOI] [PubMed] [Google Scholar]
- 5.Cruz, J. C. & Tsai, L. H. (2004) Curr. Opin. Neurobiol. 14, 390–394. [DOI] [PubMed] [Google Scholar]
- 6.Planel, E., Sun, X. & Takashima, A. (2002) Drug Dev. Res. 56, 491–510. [Google Scholar]
- 7.Hanger, D. P., Hughes, K., Woodgett, J. R., Brion, J. P. & Anderton, B. H. (1992) Neurosci. Lett. 147, 58–62. [DOI] [PubMed] [Google Scholar]
- 8.Woodgett, J. R. (1990) EMBO J. 9, 2431–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wagner, U., Utton, M., Gallo, J. M. & Miller, C. C. (1996) J. Cell Sci. 109, 1537–1543. [DOI] [PubMed] [Google Scholar]
- 10.Lovestone, S., Hartley, C. L., Pearce, J. & Anderton, B. H. (1996) Neuroscience 73, 1145–1157. [DOI] [PubMed] [Google Scholar]
- 11.Lucas, J. J., Hernandez, F., Gomez-Ramon, P., Moran, M. A., Hen, R. & Avila, J. (2001) EMBO J. 20, 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sato, S., Tatebayashi, Y., Akagi, T., Chui, D. H., Murayama, M., Miyasaka, T., Planel, E., Tanemura, K., Sun, X., Hashikawa, T., et al. (2002) J. Biol. Chem. 277, 42060–42065. [DOI] [PubMed] [Google Scholar]
- 13.Ishizawa, T., Sahara, N., Ishiguro, K., Kersh, J., McGowan, E., Lewis, J., Hutton, M., Dickson, D. W. & Yen, S. H. (2003) Am. J. Pathol. 163, 1057–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noble, W., Olm, V., Takata, K., Casey, E., Mary, O., Meyerson, J., Gaynor, K., LaFrancois, J., Wang, L., Kondo, T., et al. (2003) Neuron 38, 555–565. [DOI] [PubMed] [Google Scholar]
- 15.Pei, J. J., Tanaka, T., Tung, Y. C., Braak, E., Iqbal, K. & Grundke-Iqbal, I. (1997) J. Neuropathol. Exp. Neurol. 56, 70–78. [DOI] [PubMed] [Google Scholar]
- 16.Pei, J. J., Braak, E., Braak, H., Grundke-Iqbal, I., Iqbal, K., Winblad, B. & Cowburn, R. F. (1999) J. Neuropathol. Exp. Neurol. 58, 1010–1019. [DOI] [PubMed] [Google Scholar]
- 17.Phiel, C. J. & Klein, P. S. (2001) Annu. Rev. Pharmacol. Toxicol. 41, 789–813. [DOI] [PubMed] [Google Scholar]
- 18.Chalecka-Franaszek, E. & Chuang, D. M. (1999) Proc. Natl. Acad. Sci. USA 96, 8745–8750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang, F., Phiel, C. J., Spece, L., Gurvich, N. & Klein, P. S. (2003) J. Biol. Chem. 278, 33067–33077. [DOI] [PubMed] [Google Scholar]
- 20.Hong, M., Chen, D. C., Klein, P. S. & Lee, V. M. (1997) J. Biol. Chem. 272, 25326–25332. [DOI] [PubMed] [Google Scholar]
- 21.Munoz-Montano, J. R., Moreno, F. J., Avila, J. & Diaz-Nido, J. (1997) FEBS Lett. 411, 183–188. [DOI] [PubMed] [Google Scholar]
- 22.Takahashi, M., Yasutake, K. & Tomizawa, K. (1999) J. Neurochem. 73, 2073–2083. [PubMed] [Google Scholar]
- 23.Perez, M., Hernandez, F., Lim, F., Diaz-Nido, J. & Avila, J. (2003) J. Alzheimers Dis. 5, 301–308. [DOI] [PubMed] [Google Scholar]
- 24.Mudher, A., Shepherd, D., Newman, T. A., Mildren, P., Jukes, J. P., Squire, A., Mears, A., Berg, S., MacKay, D., Asuni, A. A., et al. (2004) Mol. Psychiatry 9, 522–530. [DOI] [PubMed] [Google Scholar]
- 25.Phiel, C. J., Wilson, C. A., Lee, V. M. & Klein, P. S. (2003) Nature 423, 435–439. [DOI] [PubMed] [Google Scholar]
- 26.Lewis, J., McGowan, E., Rockwood, J., Melrose, H., Nacharaju, P., Van Slegtenhorst, M., Gwinn-Hardy, K., Paul Murphy, M., Baker, M., Yu, X., et al. (2000) Nat. Genet. 25, 402–405. [DOI] [PubMed] [Google Scholar]
- 27.Bhat, R., Xue, Y., Berg, S., Hellberg, S., Ormo, M., Nilsson, Y., Radesater, A. C., Jerning, E., Markgren, P. O., Borgegard, T., et al. (2003) J. Biol. Chem. 278, 45937–45945. [DOI] [PubMed] [Google Scholar]
- 28.de Silva, R., Lashley, T., Gibb, G., Hanger, D., Hope, A., Reid, A., Bandopadhyay, R., Utton, M., Strand, C., Jowett, T., et al. (2003) Neuropathol. Appl. Neurobiol. 29, 288–302. [DOI] [PubMed] [Google Scholar]
- 29.Planel, E., Yasutake, K., Fujita, S. C. & Ishiguro, K. (2001) J. Biol. Chem. 276, 34298–34306. [DOI] [PubMed] [Google Scholar]
- 30.Greenberg, S. G. & Davies, P. (1990) Proc. Natl. Acad. Sci. USA 87, 5827–5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sheenan, D. C. & Hrapchak, B. B. (1980) Theory and Practice of Histotechnology (Battelle, Columbus, OH).
- 32.Zehr, C., Lewis, J., McGowan, E., Crook, J., Lin, W. L., Godwin, K., Knight, J., Dickson, D. W. & Hutton, M. (2004) Neurobiol. Dis. 15, 553–562. [DOI] [PubMed] [Google Scholar]
- 33.Marcus, W. L. (1994) J. Environ. Pathol. Toxicol. Oncol. 13, 73–79. [PubMed] [Google Scholar]
- 34.Ryves, W. J. & Harwood, A. J. (2001) Biochem. Biophys. Res. Commun. 280, 720–725. [DOI] [PubMed] [Google Scholar]
- 35.Hernandez, F., Perez, M., Lucas, J. J., Mata, A. M., Bhat, R. & Avila, J. (2004) J. Biol. Chem. 279, 3801–3806. [DOI] [PubMed] [Google Scholar]
- 36.Janke, C., Beck, M., Stahl, T., Holzer, M., Brauer, K., Bigl, V. & Arendt, T. (1999) Brain Res. Mol. Brain Res. 68, 119–128. [DOI] [PubMed] [Google Scholar]
- 37.Lewis, J., Dickson, D. W., Lin, W. L., Chisholm, L., Corral, A., Jones, G., Yen, S. H., Sahara, N., Skipper, L., Yager, D., et al. (2001) Science 293, 1487–1491. [DOI] [PubMed] [Google Scholar]
- 38.Davies, S. P., Reddy, H., Caivano, M. & Cohen, P. (2000) Biochem. J. 351, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andorfer, C., Kress, Y., Espinoza, M., de Silva, R., Tucker, K. L., Barde, Y. A., Duff, K. & Davies, P. (2003) J. Neurochem. 86, 582–590. [DOI] [PubMed] [Google Scholar]
- 40.Sahara, N., Lewis, J., DeTure, M., McGowan, E., Dickson, D. W., Hutton, M. & Yen, S. H. (2002) J. Neurochem. 83, 1498–1508. [DOI] [PubMed] [Google Scholar]
- 41.Andorfer, C., Acker, C., Kress, Y., Hof, P., Duff, K. & Davies, P. (2005) J. Neurosci., in press. [DOI] [PMC free article] [PubMed]
- 42.Necula, M. & Kuret, J. (2004) J. Biol. Chem. 279, 49694–49703. [DOI] [PubMed] [Google Scholar]
- 43.Frasier, M. & Wolozin, B. (2004) Exp. Neurol. 187, 235–239. [DOI] [PubMed] [Google Scholar]