

Abstract
A multicomponent-derived synthesis of arylidene isoquinolinones decorated with phenolic moieties is described. The series demonstrated good DPPH trapping and, in the case of sinapic acid-containing analogs, excellent activity against lipoperoxidation; EPR also demonstrated that one derivative scavenged hydroxyl radicals. In addition, some compounds showed excellent inhibition of α-glucosidase activity and, according to both Lineweaver–Burk plots and molecular docking, they act as non-competitive or mixed inhibitors. In vitro assay also demonstrated that two compounds significantly reduced the plasma glucose levels after sucrose administration. In summary, the studied isoquinolinones become novel compounds with dual action (antioxidant and α-glucosidase inhibition) against diabetes and related metabolic diseases, whose optimization would lead to more potent candidates.
A series of phenolic isoquinolinones with dual action (antioxidant and α-glucosidase inhibition) is shown for the first time. In vivo assays also demonstrate the reduction of blood glucose after administration of sucrose.
Introduction
Chronic diseases reduce the quality and life expectancy of patients and require an enormous economic burden for their treatment. Among them, type II diabetes mellitus (DMTII) is one of the most preoccupant metabolic disorders with high rates of mortality and morbidity. In 2019, around 4.2 million adults died from diabetes (11.3% of total deaths worldwide),1 which highlights the urgency for new treatments.
Diabetes progression and development is highly related to inflammation, which is an essential response mediated by the immune system against harmful stimuli such as xenobiotics, pathogens, radiation or injuries.2 Being equally important in repairing and regenerating the tissue, inflammation plays a vital role in homeostasis and health.3 However, when uncontrolled, inflammation may become an enemy instead of an ally, contributing to the pathogenesis of a myriad of chronic disorders such as diabetes, metabolic syndrome, arthritis and even cancer.4 Among the gamut of factors related to chronic inflammation, undoubtedly, oxidative stress is one of the most intrinsically related. This imbalance between free radicals (both endogenous – generated by cell metabolism – and exogenous – from external sources) and antioxidants5 is also responsible for diabetes progression. During hyperglycaemia, enhanced pathways, such as glucose oxidation and enzyme-independent glycation, lead to the exacerbated production of ROS, which in turn results in glucose oxidation, thus starting a vicious cycle.6 Meanwhile, ROS may either destroy pancreatic beta cells or impair insulin signalling (mainly activating NF-κB – nuclear factor κB – and PKC – protein kinase C), therefore conducing to both types of diabetes.7
Hence, the rational control of oxidative stress may improve metabolic and inflammation-related diseases, being the systematic search of new antioxidants one of the most studied approaches. Among them, phenols are ubiquitous molecules whose free radical scavenging is attributed to their capability for hydrogen atom transfer (HAT),8 but the sequential proton-loss electron-transfer (SPLET) is also involved.9 On the other hand, multicomponent reactions (MCRs) are considered masterpieces in drug research programs owing to their feasibility for the expedite construction of complex pharmacologically relevant scaffolds. Besides, MCRs offer several advantages over traditional synthesis, highlighting the good yields with simple purifications steps, the atom economy, and the possibility of creating diversity-oriented synthesis (DOS) programs.10 Specifically, when the MCR incorporates a phenolic feature, potential antioxidant properties are expected.11 For example, the Groebke–Blackburn–Bienaymé multicomponent reaction allowed the synthesis of imidazo[1,2-a]pyridines (1) with remarkable DPPH (2,2-diphenyl-1-picryl-hydrazyl) scavenging activity (Fig. 1).12 Besides, the Ugi four component reaction (Ugi 4-CR) is considered as a crucial transformation for constructing phenol-containing antioxidants. Ismaili and coworkers reported a series of peptides containing ferulic and caffeic acid moieties, where compound 2 excelled in both DPPH and ORAC (oxygen radical capacity) assays.13 In this sense, our group has been focused on the diversity-oriented synthesis of potentially bioactive heterocycles, especially acting as antioxidants, through Ugi 4-CR post-modification protocols. Accordingly, we prepared a series of benzopyrrolizidines (like compound 3) which showed excellent DPPH scavenging and reduced lipoperoxidation.14 More recently, a cascade deprotection/cyclization/oxidation process allowed the synthesis of pyrazinones (such as 4) with significant antioxidant and anti-inflammatory properties according to an in vivo murine model.15
Fig. 1. Some aza-heterocycles with antioxidant properties obtained through multicomponent processes.

Although a variety of phenolic compounds has been prepared to date, the antioxidant properties of phenol-containing 3-isoquinolinones have not yet been achieved. Hence, encouraged by our previous works and inspired by the significant antioxidant properties of ferulic and sinapic acids, in this work, we describe a convenient strategy for the synthesis of novel isoquinolinones featuring phenolic arylidene moieties. Moreover, in an attempt to construct molecules with dual activity related to diabetes control, we report their antioxidant capacity as well as their α-glucosidase inhibition – a validated biological target for DMTII treatment.16 The design of the series (Fig. 2) maintained a phenolic group attached to the cinnamic moiety (as the feature with antioxidant properties seen in compound 3) and allowed the incorporation of different substitution patterns (in terms of electronic and steric properties) at the 2-position of the isoquinolinone to examine the effect on the pharmacological activity.
Fig. 2. Design of isoquinolinones as potential antioxidant and α-glucosidase inhibitors.

Results and discussion
Chemistry
The design of the compounds considered a dihydro-3-isoquinolinone ring bearing an arylidene at the 4-position as well as an aromatic ring at the 2-position (10, Scheme 1). We soon discovered that Ugi 4-CR assembled the core necessary for the isoquinoline construction through a palladium-catalyzed process. Therefore, 2-bromobenzaldehydes (5), ferulic/sinapic acid (8) – key intermediates for the later intramolecular Mizoroki–Heck reaction – several isocyanides (6) and amines (7) reacted to afford the corresponding α-acylamino carboxamides 9a–r under reflux conditions. The lack of complications at this stage of the synthesis (yields ranging from 26 to 84% isolated yield) allowed us to proceed to the sp2–sp2 hybridized carbon–carbon bond formation; only anilines bearing a trifluoromethyl substituent (9o and 9p) gave poor yields, but stirring at room temperature afforded better results.
Scheme 1. Two-step synthesis of the arylidene isoquinolinones 10a–r. Reagents and conditions: i) InCl3, methanol, sealed vial, reflux, 8 h (or r.t. for three days when 4-(trifluromethyl)aniline was used); ii) Pd(AcO)2, PPh3, K2CO3, sealed vial, reflux, 5–9 h.

To this end, the carboxamides 9a–r contained an alkene and an aryl bromide that served as pivotal groups for performing the conventional intramolecular Mizoroki–Heck transformation. Fortunately, a typical system consisting in palladium acetate(ii)/triphenylphosphine (acting as a catalyst and ligand, respectively) gave a satisfactory 6-exo-trig cyclization (85% of isolated yield) for 10a when using potassium carbonate as a base (Table 1). Given that result, we employed the palladium-catalyzed reaction for converting the remaining carboxamides to the corresponding isoquinolinones 10b–10r in good yields, the analogue 10h (a sinapic acid derivative) being the exception with a scarce result (39%). Notably, the X-ray structure of 10e (Fig. 3; CCDC number 1995525) evidenced that the palladium-mediated process favored the six-membered ring over the seven-membered ring (via 7-endo-trig pathway); additionally, the alkene kept the most stable “E” geometry, and the lactam ring assumes a “quasi-boat” conformation in accordance with some related heterocycles.17 We want to highlight that no other significant by-products nor the Z isomer were detected in TLC.
Structures and isolated yields of arylidene isoquinolones 10a–10r. The yields of the corresponding Ugi adducts 9a–9r are shown in parentheses.
 10a; 75% (84%) 10a; 75% (84%) |
 10b; 76% (51%) 10b; 76% (51%) |
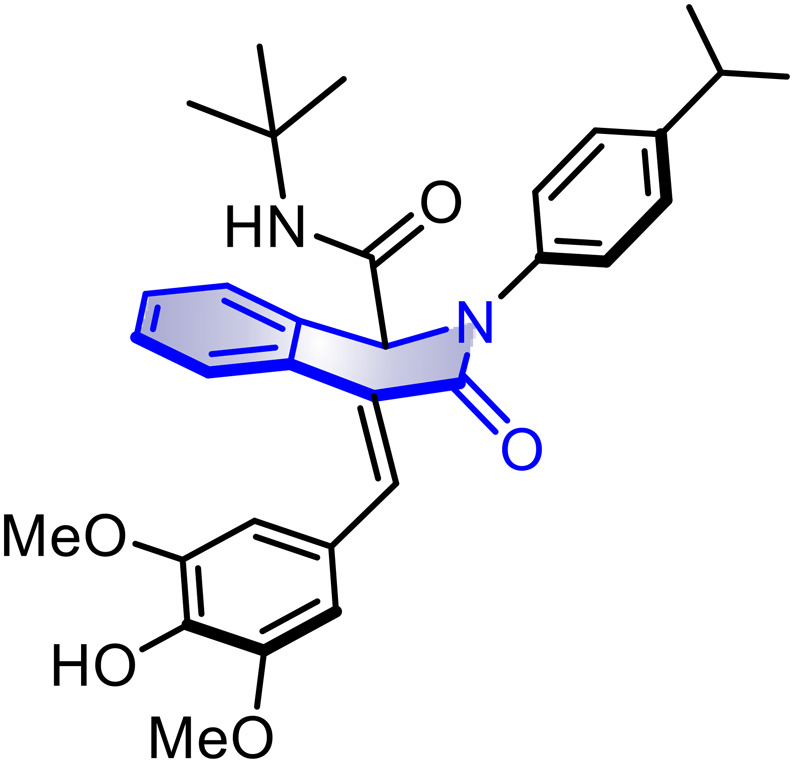 10c; 50% (78%) 10c; 50% (78%) |
 10d; 68% (48%) 10d; 68% (48%) |
 10e; 85% (57%) 10e; 85% (57%) |
 10f; 68% (60%) 10f; 68% (60%) |
 10g; 72% (52%) 10g; 72% (52%) |
 10h; 39% (62%) 10h; 39% (62%) |
 10i; 47% (60%) 10i; 47% (60%) |
 10j; 80% (53%) 10j; 80% (53%) |
 10k; 68% (61%) 10k; 68% (61%) |
 10l; 64% (51%) 10l; 64% (51%) |
 10m; 77% (83%) 10m; 77% (83%) |
 10n; 79% (73%) 10n; 79% (73%) |
 10o; 73% (31%) 10o; 73% (31%) |
 10p; 71% (26%) 10p; 71% (26%) |
 10q; 65% (55%) 10q; 65% (55%) |
 10r; 63% (72%) 10r; 63% (72%) |
||
Fig. 3. X-ray structure of isoquinolinone 10e (thermal ellipsoids are drawn at 50% probability for all atoms except for hydrogen, CCDC number 1995525).

According to the X-ray data, the isoquinolinone retained the E geometry from the alkene, though the Mizoroki–Heck process of related cinnamates proceeded preferably with an inversion of the aryl in the alkene.18,19 Therefore, a plausible mechanism for the transformation could involve a palladium(ii)-mediated geometry isomerization of I to II (as previously reported)20 prior the cross-coupling reaction (Scheme 2). Once the Z-isomer is obtained, oxidative addition and subsequent steps gave the isolated isoquinolinones 10.
Scheme 2. Proposed mechanism for the synthesis of isoquinolinones 10.

The protocol allowed the synthesis of isoquinolinones bearing t-butyl and cyclohexyl groups attached at the amide moiety. Moreover, the cross-coupling reaction was compatible with a methoxy group attached to the aryl ring containing the bromine atom (example 10d). Since we decided to explore the preliminary antioxidant effect of this nucleus, we maintained the ferulic and sinapic portions (well-established natural phenolic antioxidants)21,22 as the carboxylic acid components. Anilines with different properties in terms of steric and electrostatic nature were added to analyze their global effect in biological activity. Thus, phenyl (10i and 10j), benzyl (10k and 10l), and phenyl bearing electron-donating (methyl and hydroxy) and electron-withdrawing groups (fluorine and trifluoromethyl) decorated the isoquinolinone ring, while isopropyl was used to evaluate the effect of steric hindrance.
Antioxidant evaluation
Ferulic/sinapic acid-containing isoquinolinones 10a–10r were tested for their antioxidant capacity by using DPPH assay (2,2-diphenyl-1-picrylhidrazyl radical) as a HAT-mechanism test and thiobarbituric acid reactive substances (TBARS) as a lipoperoxidation model. The lack of DPPH scavenging at 100 μM for 10a points out the requirement of a phenol group at the isoquinolinone core (Table 2), since the hydroxy group is a structural feature required to neutralize the DPPH radical through a hydrogen-atom transfer mechanism. Although none of the compounds demonstrated a relevant capacity to inhibit the DPPH radical, ferulic acid-containing isoquinolinones (percentage of inhibition ranging from 65–77%) surpassed the scavenging of sinapic acid derivatives (less than 38% of scavenging). The only exception was 10p, which showed a significant DPPH scavenging of 90% – the best activity in all the series! These outcomes contrast with the previously observed elsewhere, in which the incorporation of methoxy groups reduces the bond dissociation enthalpy and thus enhances the antioxidant activity through the HAT mechanism.23 Apparently, neither the isocyanide nor the electrostatic properties of the amine moiety influenced the biological effect.
In vitro antioxidant capacity of isoquinolinones 10a–r.
| Compounda | % DPPH scavenging | % TBARS scavenging |
|---|---|---|
| 10a | 00.0 | 6.1 |
| 10b | 67.3 | 92.4 |
| 10c | 37.6 | 96.8 |
| 10d | 69.2 | 95.4 |
| 10e | 68.0 | 95.7 |
| 10f | 35.4 | 97.7 |
| 10g | 72.7 | 70.7 |
| 10h | 37.5 | 96.5 |
| 10i | 66.8 | 95.5 |
| 10j | 40.6 | 97.8 |
| 10k | 77.1 | 93.1 |
| 10l | 36.2 | 96.8 |
| 10m | 67.3 | 90.5 |
| 10n | 36.1 | 96.8 |
| 10o | 65.9 | 64.3 |
| 10p | 89.9 | 99.4 |
| 10q | 72.2 | 96.4 |
| 10r | 33.1 | 98 |
| α-Tocopherol | 87.4 | 79.1 |
| Quercetin | 97.9 | 95.7 |
Compounds were tested at a concentration of 100 μM.
DPPH is the most used radical to assess the antioxidant capability of substances,24 but it has a limited scope of biological relevance. To circumvent this restriction, other models that mimic endogenous radicals can be performed. The TBARS simulates lipid peroxidation through the quantification of end products generated from membrane decomposition,25 so, in our opinion, it is a more significant model than the DPPH-scavenging test. Regarding the isoquinolinones, TBARS assay also corroborated the vital role of the OH group in the arylidene ring: 10a did not show any inhibition of the MDA–TBA (malondialdehyde–thiobarbituric acid) adduct even at 100 μM (Table 2). Contrary to the DPPH test, all sinapic acid-containing derivatives inhibit more efficiently the lipoperoxidation than ferulic isoquinolinones, with an inhibition near 100%. 10r becomes an exceptional case: despite having two phenol groups, TBARS reduction dropped to 57%. It is well known that high conjugation stabilizes radicals.26 However, if we analyze the X-ray conformation (Scheme 1), there is a lack of coplanarity at the enamide system, which reduces the number of resonance structures; thus, the antioxidant effect of the isoquinolinones may be attributed to other mechanisms (such as SPLET) instead of simply HAT.
We determined the IC50 for the compounds with an inhibition higher than 95% (Fig. 4). None of the ferulic acid-containing molecules displayed an IC50 lower than 10 μM, being less active than the reference antioxidants (in this case, we used known antioxidants such as α-tocopherol and quercetin, which have a phenolic portion like isoquinolinones 10a–r). To our pleasant surprise, the presence of a sinapic acid moiety (except the dihydroxylated 10r) induced an excellent antioxidant effect, with values lower than 5 μM and surpassing the activity of the potent vitamin E (α-tocopherol). In general, those compounds having bulky groups with an sp3 carbon (isopropyl – 10c and 10f – and benzyl – 10l) or trifluoromethyl (10p) showed excellent antioxidant effects. Surprisingly, isoquinolinone 10f matched the activity of the flavonoid quercetin, a potent antioxidant which scavenges toxic radicals and inhibits enzymes with oxidizing properties.27
Fig. 4. IC50 values of the most active isoquinolinones. The SEM values were calculated with three different experiments.

Due to the significant results achieved, we wanted to explore if compound 10f could scavenge other biological relevant radicals besides those leading to lipoperoxidation. Specifically, the hydroxyl radical is the most damaging free radical against proteins and DNA, thus leading to cancer development.28 Since the hydroxyl radical has an ephemeral existence, Electron Paramagnetic Resonance (EPR) becomes one of the most advantageous techniques to quantify it. As shown in Fig. 5, the isoquinolinone 10f scavenged HO· (generated by the Fenton reaction and using DMPO as a spin trap) in a concentration-dependent manner. At 10 μM, 10f reduced the hydroxyl concentration to almost 25% and reached 76% inhibition at 106 μM. The outcomes described before point out the antioxidant activity of 10f: although it possesses a moderate DPPH scavenging (36% at 100 μM), it showed excellent properties for reducing lipoperoxidation and hydroxyl radicals, two-well known contributors related to chronic diseases.
Fig. 5. Hydroxyl radical scavenging of 10f measured by EPR. a.u. symbolizes arbitrary units.

Inhibition of α-glucosidase
Besides antioxidant properties of isoquinolinones 10a–r, we evaluated if compounds could have a secondary activity against DMTII and thus become molecules with dual action. The inhibition of α-glucosidase delays carbohydrate absorption from diet and prevents the elevation of glucose levels during the postprandial period, thus regulating DMTII, therefore being an attractive target for investigation.29 Notwithstanding, only three polyol α-glucosidase inhibitors are available for DMTII treatment (acarbose, voglibose and miglitol),30 but their attributable negative side-effects lead to the discovery of new drugs. Accordingly, the search for novel α-glucosidase inhibitors is a common strategy for diabetes control, but previous works have demonstrated the benefits of combining them with an antioxidant portion due to the close relationship between diabetes and oxidative stress.16,31,32
Therefore, inspired by some reports concerning the α-glucosidase inhibition of some natural products containing isoquinolines (such as schulzeines33 and magnoflorine34), we tested the series against S. cerevisiae α-glucosidase, a model commonly used for rapid screening of new antidiabetic candidates.35–37 Only 4 analogs lacked inhibition even at 100 μM (10b, 10p–r, Fig. 6); quercetin served as the positive control because it has a phenolic moiety and has been reported as a glucosidase inhibitor.38 Again, a hydroxy group at the aniline moiety had a negative impact on the activity. The IC50 of most of the compounds falls into the range of 30–68 mM, and four of them showed an IC50 below 32 mM (10d–e, 10g and 10o). Structurally, those compounds share some patterns: ferulic acid served as the acid in the multicomponent set and they bore aliphatic chains or a CF3 (in the case 10o). We can deduce that a ferulic acid moiety is crucial for the inhibition since their sinapic acid counterparts had less activity (see Fig. 4). The most active compounds 10e and 10o (IC50 of 14.8 ± 0.3 μM and 13.7 ± 0.6 μM, respectively), besides the aforementioned characteristics, had a cyclohexyl moiety and displayed a comparable inhibition to the detected for quercetin (15.6 ± 1.7 μM; acarbose had an IC50 value of 4.7 ± 0.2 mM!).
Fig. 6. IC50 values of isoquinolinones 10a–r and quercetin against α-glucosidase.

Isoquinolinones showed herein had a distinct structure compared to the substrate and inhibitors used in therapeutics; therefore, a non-competitive inhibition could be expected. To further confirm this hypothesis, we performed a kinetic study by Lineweaver–Burk plot analysis using two concentrations: the IC50 and 2 × IC50 (in the case of 10g, we tested the IC50 and 0.5 × IC50 due to its insolubility, Fig. 7). The double reciprocal plots (1/V vs. 1/[substrate]) of the most active α-glucosidase inhibitors (10e, 10g and 10o) clearly show that they do not act as competitive inhibitors: 10e and 10g displayed a mixed inhibition while 10o behaved as a non-competitive inhibitor (straight lines intersect at the x axis with the same Michaelis–Menten constant). Other inhibitors of α-glucosidase are proposed as mixed inhibitors like the flavonoid luteolin, which binds both the apoenzyme and the catalytic domain.39 Contrarily, quercetin binds into the catalytic cavity, acting as a competitive inhibitor as previously reported.40
Fig. 7. Lineweaver–Burk plots of α-glucosidase inhibition for isoquinolinones 10e, 10g and 10o and quercetin (Q). IC50 and 2 × IC50 were tested for 10e and 10o and IC50 and 0.5 × IC50 for 10g.

Molecular docking helped us to identify a plausible cavity for the isoquinolinone–receptor interaction. We modeled S. cerevisiae α-glucosidase with Alphafold41,42 and considered the most active compounds (10e and 10o) as well as the low-active analog 10b (both enantiomers were docked). None of the compounds bound into the catalytic pocket formed by the Asp214, Glu276 and Asp349 triad but they interacted within a surrounding pocket previously reported (Fig. 8A).43 Curiously, we did not find a clear differentiation in the (R)- and (S)-enantiomer binding pose.
Fig. 8. A) Binding pose of the tested isoquinolinones inside an allosteric pocket of α-glucosidase. B) Alternative site of interaction. None of the compounds were docked inside the catalytic pocket. Color code: (R)-10b = orange; (S)-10b = green; (R)-10e = pink; (S)-10e = yellow; (R)-10o = purple; (S)-10o = black.

The complex is stabilized by hydrophobic interactions and, in the case of (R)-10o and (R)-10e, a hydrogen bond with Ala358 is detected (2.5 and 2.2 Å, respectively). Notwithstanding, autodock vina identified an alternative pocket far away from the catalytic site in which each enantiomer significantly differs in orientation and conformation: (R)-enantiomers bind to a more superficial region while (S)-enantiomers are located in a deeper cavity (Fig. 8B). Into this pocket, the principal interactions of isoquinolinones with the yeast α-glucosidase correspond to hydrogen bonds (see Table 3).
Hydrogen bond distance of isoquinolinones into the alternative site of yeast α-glucosidase.
| Compound | Hydrogen bond distance (Å) | |
|---|---|---|
| Glu304 | Other amino acid | |
| (R)-10b | — | 2.6 (His63) |
| (S)-10b | 3.2 | — |
| (R)-10e | 2.2 | 2.1 (Asn241) |
| 2.7 (Arg312) | ||
| (S)-10e | — | 2.3 (Arg222) |
| 2.7 (Trp242) | ||
| (R)-10o | — | 2.5 (His239) |
| (S)-10o | 3.3 | 3.1 (Thr307) |
The two-dimensional interaction plot of the most active compound ((R)-10o) inside the allosteric pocket revealed that a hydrogen bond is formed between NH of the amide group and Ala358 (Fig. 9). In addition, the complex is stabilized by several alkyl and π–π interactions with surrounding amino acids and even fluorine atoms interact with Phe318 and Thr319. It is possible that compound 10e (a mixed inhibitor) binds to both the region near the catalytic domain and the allosteric site shown in Fig. 8B, while 10o, on the opposite, binds exclusively to the allosteric site. It is interesting to note that the only structural difference between 10e and 10o is the replacement of an isopropyl chain by CF3, and probably the halogen interaction may play a key role during inhibition.
Fig. 9. Two-dimensional plot and interactions of the best-scored pose of (R)-10o inside an allosteric pocket of α-glucosidase.

Once we identified that isoquinolinones may act as inhibitors of S. cerevisiae α-glucosidase, we moved to predict if compounds are able to bind to the human homolog (lysosomal acid-α-glucosidase). The PBD code 5NN8 serves as a template for the docking study. Acarbose was docked into the catalytic domain of the enzyme (Glide docking score of −13.07), achieving a similar pose to the co-crystalized inhibitor (Fig. 10), thus validating the employed methodology. The polysaccharide is stabilized by five hydrogen-bonds with His674 and aspartic acids 282, 404, 518 and 616.
Fig. 10. Docking of acarbose into the catalytic domain of α-glucosidase: blue: docked compound; orange: co-crystalized acarbose.

We also employed the above methodology for the in silico evaluation of isoquinolinones inside the catalytic pocket of human α-glucosidase. Excepting (S)-10b, all the tested compounds were satisfactorily docked and conserved some electrostatic interactions as acarbose did (Fig. 11A). (R)-10o (the most active derivative) had the best Glide score (Table 4) and was stabilized by a hydrogen bond with Asp518. The remaining compounds displayed a similar pose which was assumed by hydrogen bond formation between the hydroxy and alkoxy substituents with His674 and Asp404 (Table 4). It is worth noting that the (R)-enantiomers had better affinity than the (S)-enantiomers in all the cases, attributed to a steric hindrance between the arylamine region of (S)-isomers and Asp282, an interaction not detected with the (R)-isomers.
Fig. 11. A) Binding pose of the tested isoquinolinones inside human α-glucosidase. Colour code: (R)-10b = orange; (R)-10e = pink; (S)-10e = yellow; (R)-10o = purple; (S)-10o = black. B) Two-dimensional plot of 10o inside human α-glucosidase.

Relevant docking data from selected isoquinolinones inside human α-glucosidase.
| Compd | Glide score | Hydrogen bond distance (Å) | ||
|---|---|---|---|---|
| His674 | Asp404 | Asp518 | ||
| (R)-10b | −10.5 | 1.9 | 1.6 | — |
| (R)-10e | −10.0 | 2.1 | 2.1 | — |
| (S)-10e | −9.7 | 1.6 | 1.9 | — |
| (R)-10o | −10.7 | — | — | 1.6 |
| (S)-10o | −6.2 | 2.4 | 1.6 | — |
| 1.9 | ||||
The two-dimensional diagram of the most active analog (R)-10o (Fig. 11B) indicates the principal interactions with amino acids surrounding the catalytic domain: the hydroxyl group forms hydrogen bonds with Asp518 and Trp481 while the phenyl ring of the arylidene moiety shows a π–π stacking with Trp376; other amino acids stabilize the complex through hydrophobic interactions.
In vivo sucrose tolerance test
Motivated by the above results, we questioned if compounds could reduce blood glucose levels after the administration of sucrose (a disaccharide containing an α-glycosidic bond). We tested the same compounds used in the molecular docking study (10e, 10o and 10p), and two compounds with good activity according to TBARS assay (10f and 10g). The drug acarbose (a potent α-glucosidase inhibitor) was used as the positive control. For our delight, all compounds (at a dose of 50 mg kg−1) reduced the glucose levels released from sucrose hydrolysis at 0.5 h (maximum peak, Fig. 12). 10p had low glucose reduction followed by 10e and 10o (17 and 26%, respectively). Additionally, the glucose levels at 3 h after administration were higher than the control group (Tween 80). On the other hand, the most active isoquinolinones significantly deplete the glucose concentration to 20%, for 10g, and 29% for 10f. Due to these encouraging outcomes, we decided to evaluate those analogs at a higher concentration (i.e. 100 mg kg−1); in this case, the activity resulted in an amazing surprise: 10g reduced blood glucose in 41% and 10f in 54%, surpassing the activity of acarbose. After analyzing the AUC of the sucrose tolerance test (Fig. S77 in the ESI†), only 10o, 10g and 10f showed a statistically significant difference with respect to the control; in addition, the glucose reduction of 10g and 10f had a dose-dependent relationship.
Fig. 12. Sucrose tolerance assay of the selected isoquinolinones. A) Compounds with moderate and low activity (10e, 10o and 10p); B) plot for 10g at 50 and 100 mg kg−1; C) plot for 10f at 50 and 100 mg kg−1. *p < 0.05; **p < 0.01; ***p < 0.001.

An oral glucose tolerance test for 10g and 10o demonstrated that the reduction is not attributable to glucose absorption itself since we did not observe any change even at 50 mg kg−1 (Fig. S78 in the ESI†); the same result was found after checking the AUC: no glucose reduction occurred after oral administration of the isoquinolinones (S79 in the ESI†). It is worth noting that none of the tested isoquinolinones showed an appreciable cytotoxicity against the healthy cell line COS-7 (see Table S3 in the ESI†) nor any toxicity when administered in vivo. We have discussed the relationship between diabetes and inflammation, so biochemical related parameters (like TNF-α and IL-6)44 after administration of 10f will be quantified in future works.
Conclusions
We discovered a series of phenolic isoquinolinones with dual activity obtained through a two-step multicomponent-derived protocol. Accordingly, compounds showed moderate DPPH scavenging, particularly the ferulic acid derivatives, while sinapic acid-containing analogs lessened lipoperoxidation in TBARS assay. In addition, 10f was also capable of scavenging hydroxyl radicals according to the EPR technique. Regarding the α-glucosidase activity, several compounds inhibited the enzyme and two of them (10e and 10o) overcame the activity of quercetin; the kinetic and molecular docking studies indicated that isoquinolinones act as non-competitive or as mixed inhibitors. Finally, five isoquinolinones depleted the blood glucose levels after sucrose administration at a dose of 50 mg kg−1, 10f being the most remarkable compound. This sinapic acid-containing derivative, which also became the most potent in TBARS, can be considered as a novel lead for the design of antidiabetic drugs with an additional antioxidant effect.
Experimental section
General methods
Solvents and reagents were purchased from Merck-Aldrich and were employed without further purification. UV and sulfuric vanillin were used as revealing agents. 1H-NMR and 13C-NMR spectra were recorded with a JEOL Eclipse-300 spectrometer and a Bruker Avance III-400 spectrometer; CDCl3 and DMSO-d6 were employed as solvents. NMR coupling constants are reported in Hertz (Hz), while chemical shifts (δ) are reported in ppm. Signal splitting patterns are described as: singlet (s), doublet (d), triplet (t), quartet (q), quintet (quint), septet (sept), broad signal (br s), doublet of doublets (dd) or multiplet (m). Mass spectra were recorded with a JMST100LC AccuTOF LC with an ionSense DART SVP100 controller ionization source.
General methodology for the synthesis of α-acylaminocarboxamides 9a–r
Method A
The corresponding benzaldehyde 5 (0.891 mmol), aniline derivative (7) (0.965 mmol) and InCl3 were dissolved in 9 mL of MeOH in a vial. The mixture was stirred for 1 h at r.t. Then, the carboxylic acid 8 (0.805 mmol) was added and, after 15 min, the corresponding isocyanide 6 (0.891 mmol) was dropped. The multicomponent reaction was heated at 85 °C for 12 h. The methanol was eliminated through vacuum distillation and finally purified by column chromatography.
Method B
The corresponding benzaldehyde 5 (0.891 mmol), aniline derivative (7) (0.965 mmol) and InCl3 were dissolved in 9 mL of MeOH in a round-bottom flask and the mixture was stirred for 1 h at rt. Then, the carboxylic acid 8 (0.805 mmol) was added and, after 15 min, the corresponding isocyanide 6 (0.891 mmol) was dropped. The multicomponent reaction was stirred at r.t. for 24 h. After the completion of the reaction, the solvent was eliminated through vacuum distillation and finally purified by column chromatography.
N-(1-(2-Bromophenyl)-2-(tert-butylamino)-2-oxoethyl)-N-(4-isopropylphenyl)cinnamamide (9a)
Prepared according to method A. Purified by flash chromatography Hex–AcOEt (80 : 20). Yellowish foam in 84% yield. 1H-NMR (CDCl3/TMS, 300 MHz) δ: 7.72 (d, J = 15.6 Hz, 1H), 7.51–7.47 (m, 1H), 7.26 (s, 5H), 7.18–7.14 (m, 2H), 7.11–7.06 (m, 1H), 7.03–6.99 (m, 4H), 6.42 (s, 1H), 6.26 (d, J = 15.3 Hz, 1H), 5.67 (s, 1H), 2.81 (sept, J = 6.9 Hz, 1H), 1.37 (s, 9H), 1.16 (d, J = 6.9 Hz, 6H); 13C-NMR (CDCl3/TMS, 75.5 MHz) δ: 168.6, 166.7, 149.0, 142.2, 136.9, 135.4, 134.7, 132.8, 132.2, 130.2, 129.9, 129.6, 128.7, 128.1, 127.2, 126.8, 126.4, 119.0, 64.7, 51.9, 33.8, 28.8, 24.0; IR (KBr, cm−1): 3309, 3064, 3031, 2964, 2933, 1672, 1648, 1551, 1365, 1234; MS (DART+) m/z: 533 [M + H]+; HRMS m/z calcd for C30H3479Br1N2O2 [M + H]+, 533.18037; found 533.18090.
(E)-N-(1-(2-Bromophenyl)-2-(tert-butylamino)-2-oxoethyl)-3-(4-hydroxy-3-methoxyphenyl)-N-(4-isopropylphenyl)acrylamide (9b)
Prepared according to method A. Purified by flash chromatography Hex–AcOEt (75 : 25). Yellowish foam in 51% yield. 1H-NMR (CDCl3/TMS, 300 MHz) δ: 7.62 (d, J = 15.3 Hz, 1H), 7.51–7.48 (m 1H), 7.20–7.13 (m, 2H), 7.11–7.05 (m, 1H), 7.03–6.97 (m, 4H), 6.85–6.78 (m, 2H), 6.70 (s, 1H), 6.43 (s, 1H), 6.06 (d, J = 15.3 Hz, 1H), 5.73 (s, 1H), 3.76 (s, 3H), 2.81 (sept, J = 6.9 Hz, 1H), 1.37 (s, 9H), 1.14 (d, J = 6.9 Hz, 6H); 13C-NMR (CDCl3/TMS, 75.5 MHz) δ: 168.7, 167.0, 148.9, 147.4, 146.6, 142.4, 137.0, 134.8, 132.8, 132.2, 130.3, 129.8, 127.9, 127.2, 126.6, 126.4, 122.2, 116.7, 114.7, 110.0, 64.6, 55.8, 51.8, 33.8, 28.8, 24.0; IR (KBr, cm−1): 3550, 3352, 3066, 2963, 2931, 2872, 1687, 1647, 1594, 1513, 1271; MS (DART+) m/z: 579 [M + H]+; HRMS m/z calcd for C31H3679Br1N2O4 [M + H]+, 579.18584; found 579.18534.
(E)-N-(1-(2-Bromophenyl)-2-(tert-butylamino)-2-oxoethyl)-3-(4-hydroxy-3,5-dimethoxyphenyl)-N-(4-isopropylphenyl)acrylamide (9c)
Prepared according to method A. Purified by flash chromatography Hex–AcOEt (70 : 30). Off white foam in 78% yield. 1H-NMR (CDCl3/TMS, 300 MHz) δ: 7.58 (d, J = 15.3 Hz, 1H), 7.52–7.49 (m, 1H), 7.20–7.17 (m, 2H), 7.04–6.99 (m, 5H), 6.47 (s, 2H), 6.43 (s, 1H), 6.03 (d, J = 15.6 Hz, 1H), 5.71 (s, 1H), 5.67 (s, 1H), 3.76 (s, 6H), 2.81 (sept, J = 6.6 Hz, 1H), 1.37 (s, 9H), 1.13 (d, J = 6.9 Hz, 6H); 13C-NMR (CDCl3/TMS, 75.5 MHz) δ: 168.7, 166.8, 149.0, 147.1, 142.4, 137.0, 136.5, 134.8, 132.8, 132.2, 130.4, 129.9, 127.2, 126.9, 126.6, 126.4, 117.3, 104.9, 64.5, 56.2, 51.8, 33.8, 28.8, 24.1; IR (KBr, cm−1): 3491, 3429, 3337, 3068, 2962, 2932, 2873, 1683, 1649, 1613, 1513, 1327, 1220, 1117; MS (DART+) m/z: 609 [M + H]+; HRMS m/z calcd for C32H3879Br1N2O5 [M + H]+, 609.19641; found 609.19640.
(E)-N-(1-(2-Bromo-4-methoxyphenyl)-2-(tert-butylamino)-2-oxoethyl)-3-(4-hydroxy-3-methoxyphenyl)-N-(4-isopropylphenyl)acrylamide (9d)
Prepared according to method A. Purified by flash chromatography Hex–AcOEt (70 : 30). White foam in 48% yield. 1H-NMR (CDCl3/TMS, 300 MHz) δ: 7.62 (d, J = 15.6 Hz, 1H), 7.36 (d, J = 9.0 Hz, 1H), 7.20–7.07 (m, 2H), 7.03–7.00 (m, 2H), 6.86–6.78 (m, 2H), 6.71–6.70 (m, 2H), 6.61 (dd, J = 8.7 and 3.0 Hz, 1H), 6.40 (s, 1H), 6.07 (d, J = 15.4 Hz, 1H), 5.91 (br s, 1H), 5.75 (s, 1H), 3.76 (s, 3H), 3.48 (s, 3H), 2.82 (sept, J = 6.9 Hz, 1H), 1.37 (s, 9H), 1.16 (d, J = 6.9 Hz, 6H); 13C-NMR (CDCl3/TMS, 75.5 MHz) δ: 168.8, 166.9, 158.6, 149.0, 147.4, 146.6, 142.4, 136.9, 135.6, 133.2, 130.5, 128.0, 126.6, 122.1, 116.9, 116.8, 116.7, 116.6, 114.7, 110.1, 64.2, 55.8, 55.4, 51.8, 33.8, 28.8, 24.1, 24.0; IR (KBr, cm−1): 3495, 3430, 3366, 3066, 2963, 2935, 2873, 1693, 1664, 1594, 1512, 1249; MS (DART+) m/z: 609 [M + H]+; HRMS m/z calcd for C32H3779Br1N2O5 [M + H]+, 609.19641; found 609.19619.
(E)-N-(1-(2-Bromophenyl)-2-(cyclohexylamino)-2-oxoethyl)-3-(4-hydroxy-3-methoxyphenyl)-N-(4-isopropylphenyl)acrylamide (9e)
Prepared according to method A. Purified by flash chromatography Hex–AcOEt (70 : 30). White foam in 57% yield. 1H-NMR (CDCl3/TMS, 300 MHz) δ: 7.63 (d, J = 15.3 Hz, 1H), 7.50 (dd, J = 7.65 and 1.5 Hz, 1H), 7.16–7.09 (m, 2H), 7.06–6.96 (m, 5H), 6.86–6.78 (m, 2H), 6.71 (d, J = 1.8 Hz, 1H), 6.50 (s, 1H), 6.06 (d, J = 15.6 Hz,1H), 5.94 (s, 1H), 5.73 (d, J = 8.1 Hz, 1H), 3.93–3.80 (m, 1H), 3.76 (s, 3H), 2.81 (sept, J = 6.9 Hz, 1H), 2.01–1.86 (m, 2H), 1.73–1.55 (m, 3H), 1.42–1.29 (m, 2H), 1.20–1.03 (m, 3H), 1.15 (d, J = 6.9 Hz, 6H); 13C-NMR (CDCl3/TMS, 75.5 MHz) δ: 168.6, 167.1, 149.0, 147.4, 146.6, 142.5, 137.0, 134.7, 132.8, 132.3, 130.3, 129.9, 127.9, 127.2, 126.7, 126.4, 122.2, 116.6, 114.7, 110.0, 64.3, 55.8, 49.0, 33.8, 33.0, 32.9, 25.6, 25.0, 24.9, 24.0; IR (KBr, cm−1): 3517, 3497, 3409, 3296, 3068, 2957, 2931, 2855, 1648, 1593, 1513, 1271; MS (DART+) m/z: 605 [M + H]+; HRMS m/z calcd for C33H3879Br1N2O4 [M + H]+, 605.20149; found 605.20156.
(E)-N-(1-(2-Bromophenyl)-2-(cyclohexylamino)-2-oxoethyl)-3-(4-hydroxy-3,5-dimethoxyphenyl)-N-(4-isopropylphenyl)acrylamide (9f)
Prepared according to method A. Purified by flash chromatography Hex–AcOEt (70 : 30). Off white foam in 60% yield. 1H-NMR (CDCl3/TMS, 300 MHz) δ: 7.59 (d, J = 15.6 Hz, 1H), 7.50 (dd, J = 7.35 and 1.8 Hz, 1H), 7.21–7.14 (m, 2H), 7.12–7.08 (m, 1H), 7.07–7.03 (m, 1H), 7.02–6.96 (m, 3H), 6.51 (s, 1H), 6.48 (s, 2H), 6.04 (d, J = 15.3 Hz, 1H), 5.7 (d, J = 8.1 Hz, 1H), 5.68 (s, 1H), 3.88–3.80 (m, 1H), 3.77 (s, 6H), 2.81 (sept, J = 6.9 Hz, 1H), 2.03–1.83 (m, 2H), 1.74–1.55 (m, 3H), 1.47–1.32 (m, 2H), 1.25–0.99 (m, 3H), 1.14 (d, J = 6.9 Hz, 6H); 13C-NMR (CDCl3/TMS, 75.5 MHz) δ: 168.6, 166.9, 149.0, 147.1, 142.5, 137.0, 136.6, 134.7, 132.8, 132.3, 130.4, 129.9, 127.2, 126.9, 126.6, 126.4, 117.1, 104.9, 64.2, 56.2, 49.0, 33.8, 33.0, 32.9, 25.6, 25.0, 24.9, 24.0 (two signals, CH3); IR (KBr, cm−1): 3514, 3495, 3430, 3317, 3068, 3002, 2931, 2854, 1670, 1649, 1615, 1512, 1327, 1219, 1116; MS (DART+) m/z: 635 [M + H]+; HRMS m/z calcd for C34H4079Br1N2O5 [M + H]+, 635.21206; found 635.21192.
(E)-N-(1-(2-Bromophenyl)-2-(cyclohexylamino)-2-oxoethyl)-3-(4-hydroxy-3-methoxyphenyl)-N-(p-tolyl)acrylamide (9g)
Prepared according to method A. Purified by flash chromatography Hex–AcOEt (65 : 35). White foam in 52% yield. 1H-NMR (CDCl3/TMS, 300 MHz) δ: 7.63 (d, J = 15.6 Hz, 1H), 7.52–7.49 (m, 1H), 7.19–7.13 (m, 2H), 7.12–7.06 (m, 1H), 7.05–7.00 (m, 2H), 6.96–6.93 (AA′BB′, 2H), 6.83 (dd, J = 8.25 and 1.5 Hz, 1H), 6.79 (d, J = 8.1 Hz, 1H), 6.73 (d, J = 1.5 Hz, 1H), 6.50 (s, 1H), 6.07 (d, J = 15.3 Hz, 1H), 6.00 (br s, 1H), 5.73 (d, J = 8.1 Hz, 1H), 3.93–3.82 (m, 1H), 3.79 (s, 3H), 2.25 (s, 3H), 2.02–1.85 (m, 2H), 1.74–1.55 (m, 3H), 1.42–1.26 (m, 2H), 1.22–0.99 (m, 3H); 13C-NMR (CDCl3/TMS, 75.5 MHz) δ: 168.7, 167.1, 147.4, 146.6, 142.6, 137.9, 136.7, 134.7, 132.8, 132.3, 130.2, 130.0, 129.4, 127.9, 127.3, 126.4, 121.7, 116.5, 114.8, 110.7, 62.2, 56.0, 49.0, 33.0, 32.9, 25.6, 25.0, 24.9, 21.2; IR (KBr, cm−1): 3520, 3500, 3290, 3065, 3010, 2930, 2854, 1648, 1592, 1513, 1269; MS (DART+) m/z: 577 [M + H]+; HRMS m/z calcd for C31H3479Br1N2O4 [M + H]+, 577.17019; found 577.17004.
(E)-N-(1-(2-Bromophenyl)-2-(cyclohexylamino)-2-oxoethyl)-3-(4-hydroxy-3,5-dimethoxyphenyl)-N-(p-tolyl)acrylamide (9h)
Prepared according to method A. Purified by flash chromatography Hex–AcOEt (65 : 35). White foam in 62% yield. 1H-NMR (CDCl3/TMS, 300 MHz) δ: 7.61 (d, J = 15.3 Hz, 1H), 7.52–7.49 (m, 1H), 7.22–7.13 (m, 2H), 7.12–7.06 (m, 1H), 7.05–7.00 (m, 2H), 6.96–6.93 (AA′BB′, 2H), 6.50 (s, 1H), 6.49 (s, 2H), 6.05 (d, J = 15.6 Hz, 1H), 5.74–5.70 (m, 2H), 3.91–3.82 (m, 1H), 3.79 (s, 6H), 2.24 (s, 3H), 2.04–1.86 (m, 2H), 1.73–1.58 (m, 3H), 1.40–1.30 (m, 2H), 1.22–1.03 (m, 3H); 13C-NMR (CDCl3/TMS, 75.5 MHz) δ: 168.6, 166.9, 147.1, 142.7, 137.9, 136.6, 134.6, 132.8, 132.3, 130.3 (br s), 130.0, 129.3, 127.3, 126.9, 126.4, 117.0, 105.2, 64.2, 56.4, 49.0, 33.0, 32.9, 25.6, 25.0, 24.9, 21.2; IR (KBr, cm−1): 3503, 3415, 3347, 3308, 3066, 3002, 2931, 2853, 1651, 1606, 1512, 1215, 1113; MS (DART+) m/z: 607 [M + H]+; HRMS m/z calcd for C32H3679Br1N2O5 [M + H]+, 607.18076; found 608.18056.
(E)-N-(1-(2-Bromophenyl)-2-(cyclohexylamino)-2-oxoethyl)-3-(4-hydroxy-3-methoxyphenyl)-N-phenylacrylamide (9i)
Prepared according to method A. Purified by flash chromatography Hex–AcOEt (65 : 35). Yellowish foam in 60% yield. 1H-NMR (CDCl3/TMS, 400 MHz) δ: 7.64 (d, J = 15.6 Hz, 1H), 7.50 (dd, J = 5.7 and 1.6 Hz, 1H), 7.38–7.29 (m, 1H), 7.17–7.15 (m, 4H), 7. 07–6.97 (m, 3H), 6.86–6.78 (m, 2H), 6.73 (d, J = 1.6 Hz, 1H), 6.53 (s, 1H), 6.05 (d, J = 15.2 Hz, 1H), 5.92 (br s, 1H), 5.72 (d, J = 8.0 Hz, 1H), 3.91–3.82 (m, 1H), 3.78 (s, 3H), 2.03–1.87 (m, 2H), 1.74–1.59 (m, 3H), 1.38–1.27 (m, 2H), 1.21–1.04 (m, 3H); 13C-NMR (CDCl3/TMS, 100 MHz) δ: 168.6, 166.9, 147.5, 146.7, 142.8, 139.4, 135.6, 132.9, 132.2, 130.6, 130.0, 128.8, 128.2, 127.9, 127.3, 126.4, 122.0, 116.4, 114.8, 110.4, 64.2, 56.0, 49.0, 33.0, 32.9, 25.6, 25.0, 24.9; IR (KBr, cm−1): 3526, 3504, 3399, 3337, 3304, 3065, 2931, 2854, 1647, 1591, 1514, 1270; MS (DART+) m/z: 563 [M + H]+; HRMS m/z calcd for C30H3279Br1N2O4 [M + H]+, 563.15454; found 563.15465.
(E)-N-(1-(2-Bromophenyl)-2-(cyclohexylamino)-2-oxoethyl)-3-(4-hydroxy-3,5-dimethoxyphenyl)-N-phenylacrylamide (9j)
Prepared according to method A. Purified by flash chromatography Hex–AcOEt (65 : 35). Yellowish foam in 53% yield. 1H-NMR (CDCl3/TMS, 300 MHz) δ: d (7.62, J = 15.3 Hz, 1H), 7.49 (dd, J = 7.35 and 1.8 Hz, 1H), 7.29 (br s, 1H), 7.16–7.13 (m, 5H), 7.05–6.96 (m, 2H), 6.53 (s, 1H), 6.48 (s, 2H), 6.03 (d, J = 1.3 Hz, 1H), 5.72–5.68 (m, 2H), 3.94–3.81 (m, 1H), 3.77 (s, 6H), 2.04–1.87 (m, 2H), 1.76–1.56 (m, 3H), 1.41–1.29 (m, 2H), 1.24–1.03 (m, 3H); 13C-NMR (CDCl3/TMS, 75.5 MHz) δ: 168.6, 166.7, 147.1, 142.9, 139.3, 136.6, 134.5, 132.9, 132.2, 130.6, 130.0, 128.7, 128.1, 127.3, 126.8, 126.4, 116.8, 105.0, 64.4, 56.3, 49.0, 33.0, 32.9, 25.6, 25.0, 24.9; IR (KBr, cm−1): 3505, 3411, 3346, 3064, 3003, 2931, 2852, 1651, 1592, 1513, 1326, 1293, 1216, 1114; MS (DART+) m/z: 593 [M + H]+; HRMS m/z calcd for C31H3479Br1N2O5 [M + H]+, 593.16511; found 539.16546.
(E)-N-Benzyl-N-(1-(2-bromophenyl)-2-(cyclohexylamino)-2-oxoethyl)-3-(4-hydroxy-3-methoxyphenyl)acrylamide (9k)
Prepared according to method A. Purified by flash chromatography Hex–AcOEt (65 : 35). White foam in 61% yield. 1H-NMR (CDCl3/TMS, 300 MHz) δ: 7.73 (d, J = 15.0 Hz, 1H), 7.61 (d, J = 6.3 Hz, 1H), 7.40 (d, J = 6.9 Hz, 1H), 7.28–7.23 (m, 2H), 7.15 (br s, 5H), 7.00–6.93 (m, 1H), 6.85–6.78 (m, 2H), 6.60 (d, J = 15.0 Hz, 1H), 6.33 (s, 1H), 5.99 (s, 1H), 5.67 (d, J = 7.5 Hz, 1H), 4.86 (d, J = 18.0 Hz, 1H), 4.66 (d, J = 1.8.0 Hz, 1H), 3. 81 (br s, 4H), 1.99–1.82 (m, 2H), 1.73–1.56 (m, 3H), 1.43–1.23 (m, 2H), 1.16–0.97 (m, 3H); 13C-NMR (CDCl3/TMS, 75.5 MHz) δ: 168.5 (2C), 147.7, 146.8, 144.4, 137.9, 134.8, 133.3, 131.2, 130.2, 128.4, 128.2, 127.7, 127.0, 126.7, 126.5, 122.6, 115.4, 114.8, 109.8, 62.5, 56.0, 50.0, 48.8, 32.8, 25.6, 24.8; IR (KBr, cm−1): 3624, 3509, 3290, 3064, 3031, 2931, 2853, 1645, 1592, 1514, 1272, 1030; MS (DART+) m/z: 577 [M + H]+; HRMS m/z calcd for C31H3479Br1N2O4 [M + H]+, 577.17019; found 577.17017.
(E)-N-Benzyl-N-(1-(2-bromophenyl)-2-(cyclohexylamino)-2-oxoethyl)-3-(4-hydroxy-3,5-dimethoxyphenyl)acrylamide (9l)
Prepared according to method A. Purified by flash chromatography Hex–AcOEt (65 : 35). Yellowish foam in 51% yield. 1H-NMR (CDCl3/TMS, 300 MHz) δ: 7.68 (d, J = 14.7 Hz, 1H), 7.62 (d, J = 6.9 Hz, 1H), 7.42 (d, J = 8.1 Hz, 1H), 7.14 (br s, 7H), 6.60–6.55 (m, 3H), 6.35 (s, 1H), 5.77–5.70 (m, 2H), 4.86 (d, J = 17.4 Hz, 1H), 4.66 (d, J = 16.8 Hz, 1H), 3.80 (br s, 7H), 1.96–1.82 (m, 2H), 1.70–1.54 (m, 3H), 1.37–1.27 (m, 2H), 1.17–0.99 (m 3H); 13C-NMR (CDCl3/TMS, 75.5 MHz) δ: 168.5, 168.3, 147.2, 144.3, 138.2, 136.8, 134.9, 133.3, 131.3, 130.3, 128.4, 127.8, 127.0, 126.7, 126.6, 126.5, 116.1, 105.0, 62.5, 56.4, 53.6, 50.0, 48.8, 32.8, 25.6, 24.8; IR (KBr, cm−1): 3520, 3427, 3253, 3064, 3011, 2933, 2853, 1643, 1601, 1514, 1462, 1338, 1215, 1119; MS (DART+) m/z: 607 [M + H]+; HRMS m/z calcd for C32H3679Br1N2O5 [M + H]+, 607.18076; found 607.18095.
(E)-N-(1-(2-Bromophenyl)-2-(cyclohexylamino)-2-oxoethyl)-N-(4-fluorophenyl)-3-(4-hydroxy-3-methoxyphenyl)acrylamide (9m)
Prepared according to method A. Purified by flash chromatography Hex–AcOEt (75 : 25). Yellowish foam in 83% yield. 1H-NMR (CDCl3/TMS, 300 MHz) δ: 7.65 (d, J = 15.3 Hz, 1H), 7.53–7.50 (m, 1H), 7.33 (br s, 1H), 7.143–7.08 (m, 2H), 7.06–6.99 (m, 2H), 6.87–6.79 (m, 5H), 6.74 (d, J = 1.5 Hz, 1H), 6.52 (s, 1H), 6.02 (d, J = 15.6 Hz, 1H), 5.68 (d, J = 8.1 Hz, 1H), 3.90–3.82 (m, 1H), 3.80 (s, 3H), 2.03–1.86 (m, 2H), 1.75–154 (m, 3H), 1.42–1.29 (m, 2H), 1.21–0.99 (m, 3H); 13C-NMR (CDCl3/TMS, 75.5 MHz) δ: 168.7, 166.9, 162.1 (d, J = 247.6 Hz), 147.6, 146.7, 143.2, 135.3 (d, J = 3.0 Hz), 134.5, 133.0, 132.6 (br s), 132.1, 130.2, 127.7, 127.5, 126.4, 122.0, 116.0, 115.6 (d, J = 22.7 Hz), 114.8, 110.4, 64.0, 56.0, 49.1, 33.0, 32.9, 25.6, 25.0, 24.9; IR (KBr, cm−1): 3520, 3489, 3301, 3073, 2932, 2854, 1649, 1594, 1509, 1271, 1215; MS (DART+) m/z: 581 [M + H]+; HRMS m/z calcd for C30H3179Br119F1N2O4 [M + H]+, 581.14512; found 581.14531.
(E)-N-(1-(2-Bromophenyl)-2-(cyclohexylamino)-2-oxoethyl)-N-(4-fluorophenyl)-3-(4-hydroxy-3,5-dimethoxyphenyl)acrylamide (9n)
Prepared according to method A. Purified by flash chromatography Hex–AcOEt (65 : 35). Yellowish foam in 73% yield. 1H-NMR (CDCl3/TMS, 400 MHz) δ: 7.63 (d, J = 15.2 Hz, 1H), 7.53 (dd, J = 5.85 and 1.6 Hz, 1H), 7.10 (td, J = 5.4 and 2.4 Hz, 2H), 7.04 (td, J = 5.7 and 2.0 Hz, 2H), 6.84 (br s, 3H), 6.51–6.50 (m, 3H), 6.01 (d, J = 15.6 Hz, 1H), 5.69 (s, 1H), 5.65 (d, J = 8.0 Hz, 1H), 3.89–3.83 (m, 1H), 3.80 (s, 6H), 2.02–1.87 (m, 2H), 1.74–1.58 (m, 3H), 1.38–1.29 (m, 2H), 1.20–1.03 (m, 3H); 13C-NMR (CDCl3/TMS, 100 MHz) δ: 168.7, 166.8, 162.1 (d, J = 247 Hz), 147.2, 143.4, 136.9, 135.2, 134.5, 133.0, 132.5 (br s), 132.1, 130.2, 127.5, 126.7, 126.4, 116.4, 115.5 (d, J = 22 Hz), 105.2, 64.0, 56.4, 49.1, 33.0, 32.9, 25.6, 25.0, 24.9; IR (KBr, cm−1): 3509, 3451, 3422, 3350, 3071, 3006, 2931, 2854, 1652, 1611, 1510, 1296, 1216; MS (DART+) m/z: 611 [M + H]+; HRMS m/z calcd for C37H4478Br119F1N5O5 [M + H]+, 611.15569; found 611.15576.
(E)-N-(1-(2-Bromophenyl)-2-(cyclohexylamino)-2-oxoethyl)-3-(4-hydroxy-3-methoxyphenyl)-N-(4-(trifluoromethyl)phenyl)acrylamide (9o)
Prepared according to method B. Purified by flash chromatography Hex–AcOEt (70 : 30). White foam in 31% yield. 1H-NMR (CDCl3/TMS, 300 MHz) δ: 7.66 (d, J = 15.3 Hz, 1H), 7.55–7.49 (m, 2H), 7.46–7.41 (m, 3H), 7.15–7.10 (m, 1H), 7.06–7.01 (m, 2H), 6.86–6.79 (m, 2H), 6.69 (d, J = 1.2 Hz, 1H), 6.54 (s, 1H), 6.20 (br s, 1H), 5.94 (d, J = 15.3 Hz, 1H), 5.63 (d, J = 8.1 Hz, 1H), 3.94–3.81 (m, 1H), 3.77 (s, 3H), 2.03–1.86 (m, 2H), 1.73–1.56 (m, 3H), 1.41–1.30 (m, 2H), 1.21–1.02 (m, 3H); 13C-NMR (CDCl3/TMS, 75.5 MHz) δ: 168.5, 166.5, 147.7, 146.7, 143.6, 142.7 (q, J = 1.51 Hz), 134.1, 133.1, 131.9, 131.3, 130.4, 130.1 (q, J = 32.47 Hz), 127.6, 127.5, 126.3, 125.73 (q, J = 3.78 Hz), 122.2, 115.7, 114.8, 110.2, 64.1, 55.9, 49.2, 32.94, 32.9, 25.6, 25.0, 24.9; 19F-NMR (CDCl3/TMS, 376.5 MHz) δ: −62.62; IR (KBr, cm−1): 3533, 3431, 3266, 3074, 3014, 2934, 2856, 1672, 1649, 1604, 1516, 1323, 1282, 1167, 1124; MS (DART+) m/z: 631 [M + H]+; HRMS m/z calcd for C31H31BrF3N2O4 [M + H]+, 631.14193; found 631.14190.
(E)-N-(1-(2-Bromophenyl)-2-(cyclohexylamino)-2-oxoethyl)-3-(4-hydroxy-3,5-dimethoxyphenyl)-N-(4-(trifluoromethyl)phenyl)acrylamide (9p)
Prepared according to method B. Purified by flash chromatography Hex–AcOEt (70 : 30). Yellowish foam in 26% yield. 1H-NMR (CDCl3/TMS, 400 MHz) δ: 7.63 (d, J = 15.3 Hz, 1H), 7.53–7.52 (m, 1H), 7.51–7.50 (m, 1H), 7.48 (br s, 1H), 7.44–7.42 (m, 2H), 7.13 (dd, J = 7.0 and 2.4 Hz, 1H), 7.08–7.01 (m, 2H), 6.54 (s, 1H), 6.47 (s, 2H), 5.91 (dd, J = 15.6 Hz, 1H), 5.77–5.70 (m, 2H), 3.89–3.80 (m, 1H), 3.77 (s, 6H), 2.03–1.87 (m, 2H), 1.75–1.57 (m, 3H), 1.40–1.29 (m, 3H), 1.22–1.04 (m, 3H); 13C-NMR (CDCl3/TMS, 100 MHz) δ: 168.5, 166.4, 147.2, 143.6, 142.8, 137.0, 134.1, 133.2, 131.9, 131.4 (br s), 130.4, 130.1 (q, J = 32.0 Hz), 127.6, 126.5, 126.3, 125.7, (q, J = 4.0 Hz), 123.8 (q, J = 270 Hz), 116.3, 105.1, 64.1, 58.3, 49.2, 33.0, 32.9, 25.6, 25.0, 24.9; 19F-NMR (CDCl3/TMS, 376.5 MHz) δ: −62.68; IR (KBr, cm−1): 3535, 3354, 3304, 3072, 3004, 2933, 2854, 1657, 1609, 1514, 1325, 1216, 1116; MS (DART+) m/z: 661 [M + H]+; HRMS m/z calcd for C32H3379Br119F3N2O5 [M + H]+, 661.15249; found 661.15237.
(E)-N-(1-(2-Bromophenyl)-2-(cyclohexylamino)-2-oxoethyl)-3-(4-hydroxy-3-methoxyphenyl)-N-(4-hydroxyphenyl)acrylamide (9q)
Prepared according to method A. Purified by flash chromatography Hex–AcOEt (65 : 35). White solid in 55% yield. 1H-NMR (CDCl3-DMSOd6/TMS, 300 MHz) δ: 8.78 (br s, 1H), 7.45 (d, J = 15.3 Hz, 1H), 7.37–7.33 (m, 1H), 6.97–6.94 (m, 1H), 6.91–6.84 (m, 2H), 6.80–6.69 (m, 1H), 6.66–6.62 (m, 3H), 6.47 (br s, 2H), 6.37 (s, 1H), 6.30 (d, J = 8.1 Hz, 1H), 5.97 (d, J = 15.6 Hz, 1H), 3.75–3.68 (m, 1H), 3.65 (s, 3H), 1.86–1.69 (m, 2H), 1.60–1.44 (m, 3H), 1.26–0.96 (m, 5H); 13C-NMR (CDCl3-DMSOd6/TMS, 75.5 MHz) δ: 168.6, 167.1, 156.6, 147.9, 147.1, 142.1, 134.7, 132.4, 131.9, 131.2 (br s), 130.4, 129.5, 127.1, 127.0, 126.1, 121.5, 116.0, 115.1, 110.8, 63.8, 55.7, 48.6, 32.6, 32.5, 25.3, 24.8, 24.6; IR (KBr, cm−1): 3397, 3253, 3068, 2932, 2855, 1647, 1593, 1514, 1275, 1027, 1004; MS (DART+) m/z: 579 [M + H]+; HRMS m/z calcd for C30H3279Br1N2O5 [M + H]+, 579.14946; found 576.14981.
(E)-N-(1-(2-Bromophenyl)-2-(cyclohexylamino)-2-oxoethyl)-3-(4-hydroxy-3,5-dimethoxyphenyl)-N-(4-hydroxyphenyl)acrylamide (9r)
Prepared according to method A. Purified by flash chromatography Hex–AcOEt (60 : 40). Yellow foam in 72% yield. 1H-NMR (CDCl3-DMSOd6/TMS, 300 MHz) δ: 8.64 (br s, 1H), 8.03 (s, 1H), 7.54 (d, J = 15.6 Hz, 1H), 7.46–7.43 (m, 1H), 7.08–7.05 (m, 1H), 6.98–6.92 (m, 2H), 6.57 (br s, 2H), 6.47 (s, 2H), 6.45 (s,1H), 6.21 (br s, 1H), 6.07 (d, J = 15.6 Hz, 1H), 5.98 (d, J = 8.1 Hz, 1H), 3.84–3.76 (m, 1H), 3.74 (s, 6H), 1.98–1.80 (m, 2H), 1.69–1.52 (m, 3H), 1.39–1.24 (m, 2H), 1.20–1.02 (m, 3H); 13C-NMR (CDCl3-DMSOd6/TMS, 75.5 MHz) δ: 168.7, 167.2, 160.5, 156.9, 147.2, 142.6, 136.8, 134.7, 132.7, 132.2, 130.6, 129.8, 127.2, 126.7, 126.3, 116.8, 115.4, 105.1, 64.1, 56.4, 48.8, 32.9, 32.7, 24.9, 24.8, 24.76; IR (KBr, cm−1): 3429, 3325, 3265, 3007, 2932, 2854, 1649, 1604, 1513, 1301, 1214, 1116; MS (DART+) m/z: 609 [M + H]+; HRMS m/z calcd for C31H3479Br1N2O6 [M + H]+, 609.16002; found 609.15988.
General procedure for the synthesis of isoquinolinones 10a–r
In a vial, the corresponding α-acilaminocarboxamide 9 (0.094 mmol), Pd(AcO)2 (0.0094 mmol), triphenylphosphine (0.0188 mmol) and K2CO3 (0.188 mmol) were suspended in PhMe (5 mL, 0.02 M) and degassed by bubbling nitrogen. The vial was sealed and submitted to reflux for 5–9 h. After completing the reaction, the toluene was removed under reduced pressure and the crude product was purified by column chromatography.
(E)-4-Benzylidene-N-(tert-butyl)-2-(4-isopropylphenyl)-3-oxo-1,2,3,4-tetrahydroisoquinoline-1-carboxamide (10a)
Purified by flash chromatography Hex–AcOEt (85 : 15). Yellow solid in 75% yield. 1H-NMR (CDCl3/TMS, 300 MHz) δ: 7.78–7.74 (AA′BB′, 2H), 7.67–7.64 (m, 1H), 7.47–7.42 (m, 1H), 7.38–7.34 (m, 2H), 7.32–7.28 (m, 2H), 7.26 (s, 5H), 7.18 (s, 1H), 5.92 (s, 1H), 5.11 (s, 1H), 2.90 (sept, J = 6.9 Hz, 1H), 1.24 (d, J = 6.9 Hz, 6H), 1.19 (s, 9H); 13C-NMR (CDCl3/TMS, 75.5 MHz) δ: 168.11, 164.9, 147.9, 139.5, 137.9, 135.3, 134.9, 131.8, 130.3, 130.0, 129.1, 128.9, 128.3, 128.0, 127.5, 126.5, 125.7, 124.3, 69.7, 52.0, 33.9, 28.5, 24.1; IR (KBr, cm−1): 3417, 3328, 3059, 3028, 296, 2928, 1691, 1650, 1512, 1224; MS (DART+) m/z: 453 [M + H]+; HRMS m/z calcd for C30H33N2O2 [M + H]+, 453.25420; found 453.25404.
(E)-N-(tert-Butyl)-4-(4-hydroxy-3-methoxybenzylidene)-2-(4-isopropylphenyl)-3-oxo-1,2,3,4-tetrahydroisoquinoline-1-carboxamide (10b)
Purified by flash chromatography Hex–AcOEt (75 : 25). Yellow solid in 76% yield. 1H-NMR (CDCl3/TMS, 400 MHz) δ: 7.74 (s, 1H), 7.51 (d, J = 7.6 Hz, 1H), 7.46 (d, J = 7.6 Hz, 1H), 7.29 (s, 5H), 7.21–7.17 (m, 1H), 7.03–7.01 (m, 1H), 6.96 (d, J = 1.6 Hz, 1H), 6.85 (d, J = 8.0 Hz, 1H), 5.13 (s, 1H), 6.02 (s, 1H), 5.90 (s, 1H), 3.69 (s, 3H), 2.95 (sept, J = 6.8 Hz, 1H), 1.28 (s, 9H), 1.28 (d, J = 6.8 Hz, 6H); 13C-NMR (CDCl3/TMS, 100 MHz) δ: 168.5, 166.7, 147.9, 146.9, 146.5, 139.6, 136.2, 133.1, 130.7, 128.4, 128.2, 128.1, 127.7, 127.5, 127.4, 127.1, 125.4, 123.9, 114.8, 111.7, 69.8, 55.8, 52.0, 33.9, 28.7, 24.1; IR (KBr, cm−1): 3363, 3059, 3037, 2963, 2931, 2871, 1696, 1644, 1599, 1512, 1258; MS (DART+) m/z: 499 [M + H]+; HRMS m/z calcd for C31H35N2O4 [M + H]+, 499.25968; found 499.25949.
(E)-N-(tert-Butyl)-4-(4-hydroxy-3,5-dimethoxybenzylidene)-2-(4-isopropylphenyl)-3-oxo-1,2,3,4-tetrahydroisoquinoline-1-carboxamide (10c)
Purified by flash chromatography Hex–AcOEt (70 : 30). Yellow solid in 50% yield. 1H-NMR (CDCl3/TMS, 400 MHz) δ: 7.69 (s, 1H), 7.47 (d, J = 7.5 Hz, 1H), 7.42 (d, J = 7.5 Hz, 1H), 7.24 (br s, 5H), 7.18–7.13 (m, 1H), 6.67(s, 2H), 6.04 (s, 1H), 5.75 (s, 1H), 5.09 (s, 1H), 3.67 (s, 6H), 2.90 (sept, J = 6.9 Hz, 1H), 1.24 (s, 9H), 1.23 (d, J = 6.3 Hz, 6H); 13C-NMR (CDCl3/TMS, 100 MHz) δ: 168.5, 166.6, 147.9, 147.3, 147.0, 139.5, 136.1, 135.9, 133.1, 130.6, 128.4, 128.3, 127.5, 127.2, 126.0, 125.4, 106.5, 69.6, 56.2, 52.0, 33.9, 28.7, 24.0; IR (KBr, cm−1): 3529, 3502, 3430, 3331, 3054, 3003, 2963, 2932, 2870, 1686, 1646, 1609, 1511, 1217, 1116; MS (DART+) m/z: 529 [M + H]+; HRMS m/z calcd for C32H37N2O5 [M + H]+, 529.27025; found 529.27027.
(E)-N-(tert-Butyl)-4-(4-hydroxy-3-methoxybenzylidene)-2-(4-isopropylphenyl)-6-methoxy-3-oxo-1,2,3,4-tetrahydroisoquinoline-1-carboxamide (10d)
Purified by flash chromatography Hex–AcOEt (65 : 35). Yellow solid in 68% yield. 1H-NMR (CDCl3/TMS, 300 MHz) δ: 7.65 (s, 1H), 7.46–7.37 (m, 1H), 7.27 (br s, 4H), 6.97 (br s, 3H), 6.84–5.72 (m, 2H), 6.08 (br s, 2H), 5.07 (s, 1H), 3.83 (s, 3H), 3.70 (s, 3H), 2.93 (br s, 1H), 1.27 (br, 15 H); 13C-NMR (CDCl3/TMS, 75.5 MHz) δ: 168.5, 166.8, 159.5, 147.9, 146.6, 146.5, 139.6, 134.4, 134.3, 129.5, 127.5, 127.4, 127.38, 127.3, 125.4, 123.6, 122.9, 114.8, 114.6, 111.7, 70.0, 55.9, 55.5, 52.1, 33.9, 28.7, 24.0; IR (KBr, cm−1): 3497, 3367, 3252, 3066, 2964, 2933, 2872, 1692, 1645, 1595, 1512, 1277, 1250; MS (DART+) m/z: 529 [M + H]+; HRMS m/z calcd for C32H37N2O5 [M + H]+, 529.27025; found 529.27010.
(E)-N-Cyclohexyl-4-(4-hydroxy-3-methoxybenzylidene)-2-(4-isopropylphenyl)-3-oxo-1,2,3,4-tetrahydroisoquinoline-1-carboxamide (10e)
Purified by flash chromatography Hex–AcOEt (65 : 35). Yellow solid in 85% yield. 1H-NMR (CDCl3/TMS, 400 MHz) δ: 7.67 (s, 1H), 7.42 (d, J = 7.2 Hz, 1H), 7.37 (d, J = 7.6 Hz, 1H), 7.20–7.16 (m, 5H), 7.07 (td, J = 5.7 and 1.2 Hz, 1H), 6.91 (dd, J = 6.2 and 1.6 Hz, 1H), 6.86 (d, J = 2.0 Hz, 1H), 6.73 (d, J = 8.0 Hz, 1H), 5.99 (d, J = 8.4 Hz, 1H), 5.96 (s, 1H), 5.11 (s, 1H), 3.73–3.64 (m, 1H), 3.61 (s, 3H), 2.83 (sept, J = 6.8 Hz, 1H), 1.80–1.60 (m, 2H), 1.60–1.43 (m, 3H), 1.25–1.13 (m, 2H), 1.17 (d, J = 6.8 Hz, 6H), 1.04–0.88 (m, 3H); 13C-NMR (CDCl3/TMS, 100 MHz) δ: 168.4, 166.7, 148.0, 146.9, 146.5, 139.5, 136.5, 132.6, 130.7, 128.3, 128.1, 127.7, 127.5, 127.44, 127.4, 127.0, 125.6, 123.9, 114.8, 111.9, 69.2, 55.9, 48.8, 33.9, 32.9, 25.3, 24.7, 24.6, 24.0; IR (ATR, cm−1); 3536, 3475, 3415, 3370, 3315, 3065, 2956, 2930, 2854, 1691, 1648, 1512, 1252; MS (DART+) m/z: 525 [M + H]+; HRMS m/z calcd for C33H37N2O4 [M + H]+, 525.27533; found 525.27562.
(E)-N-Cyclohexyl-4-(4-hydroxy-3,5-dimethoxybenzylidene)-2-(4-isopropylphenyl)-3-oxo-1,2,3,4-tetrahydroisoquinoline-1-carboxamide (10f)
Purified by flash chromatography Hex–AcOEt (65 : 35). Yellow solid in 68% yield. 1H-NMR (CDCl3/TMS, 400 MHz) δ: 7.75 (s, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.48 (d, J = 7.8 Hz, 1H), 7.28 (br s, 5H), 7.20–7.16 (m, 1H), 6.71 (s, 2H), 6.06 (d, J = 8.0 Hz, 1H), 5.77 (s, 1H), 5.21 (s, 1H), 3.82–3.79 (m, 1H), 3.72 (s, 6H), 2.93 (sept, J = 6.8 Hz, 1H), 1.95–1.74 (m, 2H), 1.68–1.54 (m, 3H), 1.38–1.32 (m, 2H), 1.27 (d, J = 7.2 Hz, 6H), 1.21–1.00 (m, 3H), 13C-NMR (CDCl3/TMS, 100 MHz) δ: 168.4, 166.6, 148.0, 147.1, 139.5, 136.5, 136.0, 132.7, 130.5, 128.4, 128.3, 127.9, 127.5, 12.46, 127.39, 126.0, 125.5, 106.7, 69.3, 56.3, 48.8, 33.9, 33.0, 32.9, 25.3, 24.7, 24.6, 24.0; IR (KBr, cm−1): 3518, 3503, 3427, 3363, 3329, 3061, 3035, 2932, 2855, 1678, 1654, 1607, 1512, 1217, 1114; MS (DART+) m/z: 555 [M + H]+; HRMS m/z calcd for C34H39N2O5 [M + H]+, 555.28590; found 555.28605.
(E)-N-Cyclohexyl-4-(4-hydroxy-3-methoxybenzylidene)-3-oxo-2-(p-tolyl)-1,2,3,4-tetrahydroisoquinoline-1-carboxamide (10g)
Purified by flash chromatography Hex–AcOEt (65 : 35). Yellow solid in 72% yield. 1H-NMR (CDCl3/TMS, 400 MHz) δ: 7.74 (s, 1H), 7.49 (dd, J = 8.0 and 0.8 Hz, 1H), 7.45 (dt, J = 7.6 and 0.8 Hz, 1H), 7.26 (td, J = 7.6 and 1.2 Hz, 1H), 7.20 (b s, 4H), 7.15 (td, J = 7.6 and 1.2 Hz, 1H), 6.99–6.96 (m, 1H), 6.93 (d, J = 2.0 Hz, 1H), 6.80 (d, J = 8.0 Hz, 1H), 6.07 (d, J = 8.4 Hz, 1H), 6.02 (br s, 1H), 5.18 (s, 1H), 3.81–3.72 (m, 1H), 3.68 (s, 3H), 2.34 (s, 3H), 1.86–1.65 (m, 2H), 1.61–1.49 (m, 3H), 1.34–1.22 (m, 2H), 1.13–0.83 (m, 3H); 13C-NMR (CDCl3/TMS, 100 MHz) δ: 168.4, 166.7, 146.9, 146.5, 139.3, 137.2, 136.5, 132.6, 130.7, 130.0, 128.3, 128.1, 127.7, 127.5, 127.4, 127.0, 125.7, 123.9, 114.8, 111.9, 69.3, 55.9, 48.8, 32.9, 32.88, 25.3, 24.7, 24.6, 21.1; IR (KBr, cm−1): 3453, 3422, 3368, 3326, 3064, 3036, 2930, 2855, 1668, 1649, 1597, 1513, 1284, 1249; MS (DART+) m/z: 497 [M + H]+; HRMS m/z calcd for C31H33N2O4 [M + H]+, 497.24403; found 497.24409.
(E)-N-Cyclohexyl-4-(4-hydroxy-3,5-dimethoxybenzylidene)-3-oxo-2-(p-tolyl)-1,2,3,4-tetrahydroisoquinoline-1-carboxamide (10h)
Purified by flash chromatography Hex–AcOEt (65 : 35). Yellow solid in 39% yield. 1H-NMR (CDCl3/TMS, 400 MHz) δ: 7.72 (s, 1H), 7.51 (dd, J = 8.0 and 1.2 Hz, 1H) 7.47–7.45 (m, 1H), 7.27 (td, J = 7.6 and 1.2 Hz, 1H), 7.22 (br s, 4H), 7.16 (td, J = 7.6 and 1.2 Hz, 1H), 6.69 (s, 2H), 6.02 (d, J = 8.4 Hz, 1H), 5.70 (br s, 1H), 5.18 (s, 1H), 3.81–3.75 (m, 1H), 3.71 (s, 6H), 2.36 (s, 3H), 1.88–1.66 (m, 2H), 1.57–1.43 (m, 3H), 1.33–1.24 (m, 2H), 1.07–0.93 (m, 3H); 13C-NMR (CDCl3/TMS, 100 MHz) δ: 168.4, 166.6, 147.1, 139.3, 137.3, 136.6, 136.0, 132.7, 130.5, 130.1, 128.5, 128.3, 127.9, 127.6, 127.4, 126.0, 125.6, 106.7, 69.3, 56.3, 48.8, 33.1, 32.9, 25.3, 24.7, 24.65, 21.2; IR (KBr, cm−1): 35013457, 3415, 3062, 3008, 2928, 2855, 1712, 1663, 1606, 1512, 1249, 1218, 1114; MS (DART+) m/z: 527 [M + H]+; HRMS m/z calcd for C32H35N2O5 [M + H]+, 527.25460; found 527.25473.
(E)-N-Cyclohexyl-4-(4-hydroxy-3-methoxybenzylidene)-3-oxo-2-phenyl-1,2,3,4-tetrahydroisoquinoline-1-carboxamide (10i)
Purified by flash chromatography Hex–AcOEt (65 : 35). Yellow solid in 47% yield. 1H-NMR (CDCl3/TMS, 400 MHz) δ: 7.68 (s, 1H), 7.44 (dd, J = 7.8 and 1.2 Hz, 1H), 7.40–7.38 (m, 1H), 7.35–7.31 (m, 2H), 7.29–7.26 (m, 2H), 7.24–7.22 (m, 1H), 7.21–7.19 (m, 1H), 7.09 (td, J = 7.6 and 1.2 Hz, 1H), 6.93–6.91 (m, 1H), 6.87 (d, J = 1.6 Hz, 1H), 6.74 (d, J = 8.0 Hz, 1H), 5.95 (d, J = 8.0 Hz, 1H), 5.86 (br s, 1H), 5.13 (s, 1H), 3.73–3.66 (m, 1H), 3.62 (s, 3H), 1.81–1.63 (m, 2H), 1.54–1.44 (m, 3H), 1.26–1.15 (m, 2H), 1.07–0.84 (m, 3H); 13C-NMR (CDCl3/TMS, 100 MHz) δ: 168.3, 166.7, 146.9, 146.5, 142.0, 136.7, 132.6, 130.7, 129.5, 128.4, 128.2, 127.8, 127.5, 127.4, 127.3, 127.0, 125.8, 123.9, 114.8, 111.9, 69.2, 55.9, 48.8, 33.0, 32.9, 25.4, 24.7, 24.7; IR (KBr, cm−1): 3512, 3438, 3398, 3070, 3056, 2929, 2854, 1667, 1647, 1595, 1514, 1284, 1248, 1032; MS (DART+) m/z: 483 [M + H]+; HRMS m/z calcd for C30H31N2O4 [M + H]+, 483.22838; found 483.22812.
(E)-N-Cyclohexyl-4-(4-hydroxy-3,5-dimethoxybenzylidene)-3-oxo-2-phenyl-1,2,3,4-tetrahydroisoquinoline-1-carboxamide (10j)
Purified by flash chromatography Hex–AcOEt (86 : 13). Yellow solid in 80% yield. 1H-NMR (CDCl3/TMS, 400 MHz) δ: 7.67 (s, 1H), 7.45 (dd, J = 6 and 0.8 Hz, 1H), 7.41–7.39 (m, 1H), 7.36–7.34 (m, 2H), 7.30–7.27 (m, 2H), 7.25–7.22 (m, 1H), 7.19 (s, 1H), 7.11 (td, J = 5.7 and 1.2 Hz, 1H), 6.29 (s, 2H), 5.91 (d, J = 8.4 Hz, 1H), 5.62 (s, 1H), 5.14 (s, 1H), 3.76–3.69 (m, 1H), 3.65 (s, 6H), 1.84–1.64 (m, 2H), 1.58–1.44 (m, 3H), 1.26–1.14 (m, 2H), 1.07–0.86 (m, 3H); 13C-NMR (CDCl3/TMS, 100 MHz) δ: 168.4, 166.6, 147.1, 142.0, 136.8, 136.1, 132.7, 130.5, 129.5, 128.5, 128.3, 127.9, 127.6, 127.5, 127.3, 126.0, 125.8, 106.7, 69.2, 56.3, 48.8, 33.1, 33.0, 25.4, 24.7, 24.66; IR (KBr, cm−1): 3546, 3410, 3363, 3318, 3068, 3003, 2931, 2853, 1677, 1653, 1606, 1512, 1320, 1217, 1138; MS (DART+) m/z: 513 [M + H]+; HRMS m/z calcd for C31H33N2O5 [M + H]+, 513.23895; found 513.23917.
(E)-2-Benzyl-N-cyclohexyl-4-(4-hydroxy-3-methoxybenzylidene)-3-oxo-1,2,3,4-tetrahydroisoquinoline-1-carboxamide (10k)
Purified by flash chromatography Hex–AcOEt (65 : 35). Yellow solid in 68% yield. 1H-NMR (CDCl3/TMS, 300 MHz) δ: 7.73 (s, 1H), 7.47 (d, J = 7.8 Hz, 1H), 7.35 (d, J = 7.2 Hz, 1H), 7.28 (br s, 5H), 7.23–7.18 (m, 1H), 7.13–7.08 (m, 1H), 6.96–6.94 (m, 1H), 6.91 (br s, 1H), 6.81 (d, J = 8.1 Hz, 1H), 5.92 (s, 1H), 5.63 (d, J = 8.4 Hz, 1H), 5.26 (d, J =14.7 Hz, 1H), 4.81 (s, 1H), 4.43 (d, J = 14.7 Hz, 1H), 3.67 (s, 3H), 3.62–3.55 (m, 1H), 1.60–1.45 (m, 5H), 1.22–1.15 (m, 2H), 0.98–0.83 (m, 3H); 13C-NMR (CDCl3/TMS, 75.5 MHz) δ: 168.1, 166.9, 146.8, 146.5, 136.2, 136.1, 132.0, 130.5, 129.0, 128.6, 128.4, 128.0, 128.0, 127.6, 127.4, 127.1, 126.7, 123.8, 114.7, 111.7, 65.3, 55.9, 50.6, 48.5, 32.9, 32.7, 25.3, 24.6, 22.55; IR (KBr, cm−1): 3546, 3307, 3065, 3033, 2931, 2854, 1665, 1644, 1596, 1514, 1448, 1250; MS (DART+) m/z: 497 [M + H]+; HRMS m/z calcd for C31H33N2O4 [M + H]+, 497.24403; found 497.24424.
(E)-2-Benzyl-N-cyclohexyl-4-(4-hydroxy-3,5-dimethoxybenzylidene)-3-oxo-1,2,3,4-tetrahydroisoquinoline-1-carboxamide (10l)
Purified by flash chromatography Hex–AcOEt (65 : 35). Yellow solid in 64% yield. 1H-NMR (CDCl3/TMS, 400 MHz) δ: 7.65 (s, 1H), 7.42 (d, J = 7.6 Hz, 1H), 7.29 (d, J = 7.2 Hz, 1H), 7.23–7.18 (m, 5H), 7.11 (td, J = 5.7 and 1.2 Hz, 1H), 7.04 (td, J = 5.85 and 1.2 Hz, 1H), 6.59 (s, 2H), 5.74 (s, 1H), 5.60 (d, J = 8.0 Hz, 1H), 5.17 (d, J = 14.8 Hz, 1H), 4.81 (s, 1H), 4.38 (d, J = 14.8 Hz, 1H), 3.61 (s, 6H), 3.57–3.47 (m, 1H), 1.58–1.50 (m, 2H), 1.45–1.39 (m, 3H), 1.20–1.07 (m, 2H), 0.95–0.78 (m, 3H), 13C-NMR (CDCl3/TMS, 100 MHz) δ: 168.0, 166.8, 147.1, 136.2, 136.0, 132.1, 130.3, 129.0, 128.5, 128.4, 128.1, 128.0, 127.4, 127.0, 126.0, 106.6, 65.3, 56.2, 50.6, 48.4, 32.9, 32.6, 32.4, 25.3, 24.5, 24.49; IR (KBr, cm−1): 3525, 3499, 3420, 3294, 3034, 2999, 2928, 2852, 1644, 1604, 1514, 1450, 1257, 1117; MS (DART+) m/z: 527 [M + H]+; HRMS m/z calcd for C32H35N2O5 [M + H]+, 527.25460; found 527.25409.
(E)-N-Cyclohexyl-2-(4-fluorophenyl)-4-(4-hydroxy-3-methoxybenzylidene)-3-oxo-1,2,3,4-tetrahydroisoquinoline-1-carboxamide (10m)
Purified by flash chromatography Hex–AcOEt (65 : 35). Yellow solid in 77% yield. 1H-NMR (CDCl3/TMS, 400 MHz) δ: 7.68 (s, 1H), 7.46 (d, J = 7.6 Hz, 1H), 7.37 (d, J = 7.6 Hz, 1H), 7.27–7.19 (m, 3H), 7.12–7.09 (m, 1H), 7.04–7.0 (m, 2H), 6.92 (d, J = 8.4 Hz, 1H), 6.87 (s, 1H), 6.74 (d, J = 8.0 Hz, 1H), 5.84 (d, J = 8.4 Hz, 1H), 5.81 (s, 1H), 5.07 (s, 1H), 3.71–3.68 (m, 1H), 3.63 (s, 3H), 1.82–1.62 (m, 2H), 1.55–1.45 (m, 3H), 1.28–1.16 (m, 2H), 1.03–0.89 (m, 3H); 13C-NMR (CDCl3/TMS, 100 MHz) δ: 168.2, 166.8, 161.4 (d, J = 245 Hz), 147.0, 146.5, 138.0, 137.9, 136.9, 132.4, 130.8, 128.5, 128.3, 128.0, 127.9, 127.23. 127.2, 127.0, 123.9, 116.3 (d, J = 22 Hz), 111.9, 69.3, 55.9, 48.9, 33.0, 25.4, 24.7; IR (KBr, cm−1): 3525, 3498, 3301, 3073, 3008, 2933, 2855, 1670, 1649, 1594, 1510, 1217; MS (DART+) m/z: 501 [M + H]+; HRMS m/z calcd for C30H3019F1N2O4 [M + H]+, 501.21896; found 501.21889.
(E)-N-Cyclohexyl-2-(4-fluorophenyl)-4-(4-hydroxy-3,5-dimethoxybenzylidene)-3-oxo-1,2,3,4-tetrahydroisoquinoline-1-carboxamide (10n)
Purified by flash chromatography Hex–AcOEt (65 : 35). Yellow solid in 79% yield. 1H-NMR (CDCl3/TMS, 400 MHz) δ: 7.66 (s, 1H), 7.46 (d, J = 7.2 Hz, 1H), 7.36 (d, J = 7.2 Hz, 1H), 7.25–7.22 (m, 2H), 7.21–7.18 (m, 1H), 7.10 (td, J = 7.6 and 1.2 Hz, 1H), 7.02–6.98 (m, 2H), 6.61 (s, 1H), 6.60 (s, 1H), 5.94 (d, J = 8.0 Hz, 1H), 5.72 (s, 1H), 5.07 (s, 1H), 3.75–3.66 (m, 1H), 3.63 (s, 6H), 1.75–1.63 (m, 2H), 1.54–1.43 (m, 3H), 1.23–1.14 (m, 2H), 1.03–0.83 (m, 3H); 13C-NMR (CDCl3/TMS, 100 MHz) δ: 168.1, 166.8, 161.3 (d, J = 245.0 Hz), 147.1, 144.9, 136.8, 136.1, 132.4, 130.7, 129.0 (d, J = 4.0 Hz), 128.4, 128.0, 127.9, 127.6, 127.0, 125.9, 11.24 (d, J = 22.0 Hz), 106.7, 69.1, 56.3, 48.8, 32.9, 32.88, 25.3, 24.6; IR (KBr, cm−1): 3508, 3406, 3342, 3316, 3071, 2931, 2853, 1675, 1658, 1509, 1320, 1217, 1114; MS (DART+) m/z: 531 [M + H]+; HRMS m/z calcd for C31H3219F1N2O5 [M + H]+, 531.22952; found 531.22928.
(E)-N-Cyclohexyl-4-(4-hydroxy-3-methoxybenzylidene)-3-oxo-2-(4-(trifluoromethyl)phenyl)-1,2,3,4-tetrahydroisoquinoline-1-carboxamide (10o)
Purified by flash chromatography Hex–AcOEt (70 : 30). Yellow solid in 73% yield. 1H-NMR (CDCl3/TMS, 400 MHz) δ: 7.76 (s, 1H), 7.65–7.63 (AA′BB′, 2H), 7.53 (dd, J = 7.6 and 0.8 Hz, 1H), 7.50–7.48 (AA′BB′, 2H), 7.44 (dd, J = 7.6 and 1.2 Hz, 1H), 7.28 (td, J = 7.6 and 1.2 Hz, 1H), 7.18 (td, J = 7.6 and 1.2 Hz, 1H), 6.98–6.95 (m, 1H), 6.92 (d, J = 2.0 Hz, 1H), 6.79 (d, J = 8.4 Hz, 1H), 6.07 (br s, 1H), 5.98 (d, J = 8.4 Hz, 1H), 5.21 (s, 1H), 3.79–3.73 (m, 1H), 3.69 (s, 3H), 1.82–1.70 (m, 2H), 1.61–1.50 (m, 3H), 1.31–1.23 (m, 2H), 1.08–0.96 (m, 3H); 13C-NMR (CDCl3/TMS, 100 MHz) δ: 167.9, 166.9, 147.1, 146.6, 145.1, 137.4, 132.3, 130.8, 128.9 (q, J = 33.0 Hz), 128.5, 128.3, 128.0, 127.2, 126.9, 126.8, 126.41–126.37 (m), 126.0, 123.9, 114.8, 111.9, 68.4, 55.9, 49.0, 32.9, 25.3, 24.6; 19F-NMR (CDCl3/TMS, 376.5 MHz) δ: −62.49; IR (KBr, cm−1): 3527, 3496, 3407, 3314, 3067, 3007, 2934, 2856, 1684, 1657, 1613, 1514, 1324, 1284, 1124, 1067; MS (DART+) m/z: 551 [M + H]+; HRMS m/z calcd for C31H3019F3N2O4 [M + H]+, 551.21577; found 551.21554.
(E)-N-Cyclohexyl-4-(4-hydroxy-3,5-dimethoxybenzylidene)-3-oxo-2-(4-(trifluoromethyl)phenyl)-1,2,3,4-tetrahydroisoquinoline-1-carboxamide (10p)
Purified by flash chromatography Hex–AcOEt (70 : 30). Yellow solid in 71% yield. 1H-NMR (CDCl3/TMS, 400 MHz) δ: 7.74 (s, 1H), 7.65–7.63 (AA′BB′, 2H), 7.54 (d, J = 7.6 Hz, 1H), 7.50–7.48 (AA′BB′, 2H), 7.44 (d, J = 7.2 Hz, 1H), 7.28 (td, J = 7.6 and 1.2 Hz, 1H), 7.19 (td, J = 7.6 and 1.2 Hz, 1H), 6.68 (s, 2H), 5.94 (d, J = 8.4 Hz, 1H), 5.79 (br s, 1H), 5.20 (s, 1H), 3.80–3.73 (m, 1H), 3.69 (s, 6H), 1.82–1.70 (m, 2H), 1.60–1.49 (m, 3H), 1.49–1.30 (m, 2H), 1.07–0.95 (m, 3H); 13C-NMR (CDCl3/TMS, 100 MHz) δ: 167.8, 166.7, 147.0, 144.9, 137.3, 136.2, 132.3, 130.5, 128.8 (d, J = 33.0 Hz), 128.5, 128.4, 127.7, 127.2, 127.0, 126.3 (d, J = 3.0 H), 125.9, 125.6, 125.2, 106.7, 68.31, 56.2, 48.9, 32.8, 24.2, 24.5; 19F-NMR (CDCl3/TMS, 376.5 MHz) δ: −62.51; IR (KBr, cm−1): 3493, 3411, 3316, 3081, 3009, 2932, 2855, 1657, 1615, 1513, 1325, 1248, 1116; MS (DART+) m/z: 581 [M + H]+; HRMS m/z calcd for C32H3219F3N2O5 [M + H]+, 581.22633; found 581.22636.
(E)-N-Cyclohexyl-4-(4-hydroxy-3-methoxybenzylidene)-2-(4-hydroxyphenyl)-3-oxo-1,2,3,4-tetrahydroisoquinoline-1-carboxamide (10q)
Purified by flash chromatography Hex–AcOEt (50 : 50). Yellow solid in 65% yield. 1H-NMR (CDCl3-DMSOd6/TMS, 400 MHz) δ: 8.56 (s, 1H), 7.72 (s, 1H), 7.65–7.60 (m, 1H), 1.55–7.51 (m,1H), 7.46–7.41 (m, 1H), 7.22–7.19 (m, 1H), 7.12–7.08 (m, 1H), 6.97–6.95 (AA′BB′, 2H), 6.90 (br s, 1H), 6.76 (d, J = 8.0 Hz, 1H), 6.70–6.67 (AA′BB′, 2H), 6.63 (s, 1H), 6.48 (d, J = 8.0 Hz, 1H), 5.12 (s, 1H); 3.73–3.66 (m, 1H); 3.64 (s, 3H), 1.78–1.67 (m, 2H), 1.60–1.47 (m, 3H), 1.29–1.15 (m, 2H), 1.10–0.90 (m, 3H); 13C-NMR (CDCl3-DMSOd6/TMS, 100 MHz) δ:168.6, 167.4, 156.4, 146.9, 146.7, 136.2, 133.4, 132.2, 132.1, 130.9, 128.7, 128.6, 128.1, 127.4, 127.1, 127.0, 123.7, 116.5, 114.9, 112.1, 69.3, 55.8, 48.9, 32.8, 32.7, 25.3, 24.7, 24.6; IR (KBr, cm−1): 3650, 3401, 3265, 3063, 2930, 2855, 1659, 1592, 1517, 1424, 1244, 1166, 1030; MS (DART+) m/z: 499 [M + H]+; HRMS m/z calcd for C30H31N2O5 [M + H]+, 499.22330; found 499.22311.
(E)-N-Cyclohexyl-4-(4-hydroxy-3,5-dimethoxybenzylidene)-2-(4-hydroxyphenyl)-3-oxo-1,2,3,4-tetrahydroisoquinoline-1-carboxamide (10r)
Purified by flash chromatography Hex–AcOEt (40 : 60). Dark yellow solid in 63% yield. 1H-NMR (CDCl3-DMSOd6/TMS, 400 MHz) δ: 8.42 (s, 1H), 7.74 (m, 1H), 7.66–7.63 (m, 1H), 7.57–7.53 (m, 1H), 7.25 (td, J = 7.6 and 1.2 Hz, 1H), 7.14 (td, J = 7.8 and 1.2 Hz, 1H), 7.00–6.98 (AA′BB′, 2H), 6.72–6.70 (AA′BB′, 2H), 6.69 (s, 2H), 6.26 (d, J = 8.0 Hz, 1H), 5.75 (s, 1H), 5.15 (s, 1H), 3.84–3.74 (m, 1H), 3.70 (s, 6H), 1.84–1.68 (m, 2H), 1.64–1.51 (m, 3H), 1.34–1.19 (m, 2H), 1.10–0.93 (m, 3H); 13C-NMR (CDCl3-DMSOd6/TMS, 100 MHz) δ: 168.6, 167.3, 156.5, 147.1, 136.4, 135.9, 132.8, 132.3, 132.2, 131.8, 128.8, 128.7, 128.3, 127.5, 127.3, 127.1, 116.7, 106.8, 69.6, 56.3, 48.9, 32.9, 32.8, 25.4, 24.7, 24.67; IR (KBr cm−1): 3414, 3385, 3364, 3298, 3064, 2931, 2854, 1669, 1601, 1512, 1320, 1220, 1172, 1118; MS (DART+) m/z: 529 [M + H]+; HRMS m/z calcd for C31H33N2O6 [M + H]+, 529.23386; found 529.23376.
In vitro antioxidant assays. DPPH-scavenging test
On a 96-well plate, 50 μL of isoquinolinones 10a–r were placed at the corresponding concentration in DMSO. Then, 150 μL of DPPH in EtOH (133.33 μM) was added (final concentration of 100 μM). The plate was incubated for 30 min with stirring at 37 °C in the dark. Finally, the absorbance at 515 nm was measured. The activity was expressed as reduction percentage and was calculated using the following formula: % scavenging = (C − E) × 100/C, where C is the absorbance of the control (DPPH at 10 μM) and E is the absorbance of the test sample.
Materials and methods for TBARS
Animals
Adult male Wistar rats (200–250 g) were provided by the Instituto de Fisiología Celular, UNAM. Procedures and care of animals were conducted in conformity with the Mexican Official Norm for Animal Care and Handling (NOM-062-ZOO-1999). The rats were maintained at 23 ± 2 °C in a 12/12 h light–dark cycle with free access to food and water.
Rat brain homogenate preparation
Animal sacrifice was carried out with CO2 to avoid unnecessary pain. The cerebral tissue (whole brain) was rapidly dissected and homogenized in phosphate-buffered saline (PBS) solution (0.2 g of KCl, 0.2 g of KH2PO4, 8 g of NaCl, and 2.16 g of NaHPO4·7H2O/L, pH 7.4) to produce a 1/10 (w/v) homogenate. Then, the homogenate was centrifuged for 10 min at 800 rcf (relative centrifugal field) to yield a pellet, which was discarded. The supernatant protein content was measured using Folin and Ciocalteu's phenol reagent and adjusted with PBS at 2.666 mg of protein per mL.
Induction of lipid peroxidation and thiobarbituric acid reactive substance (TBARS) quantification
50 μL of 20 μM EDTA and 50 μL of each isoquinolinone concentration dissolved in DMSO (50 μL of DMSO for control group) were added to the supernatant (375 μL) and the mixture was incubated at 37 °C for 30 min. Lipid peroxidation started by adding 50 μL of freshly prepared FeSO4 solution (100 μM, final concentration 10 μM) incubated at 37 °C for 1 h. The TBARS content was determined as follows: a 500 μL portion of TBA reagent (1% 2-thiobarbituric acid in 0.05 N NaOH and 30% trichloroacetic acid, in 1 : 1 proportion) was added to each tube, and the final suspension was cooled on ice for 10 min, centrifuged at 13 400 rcf for 5 min, and heated at 80 °C in a water bath for 30 min. After cooling at room temperature, the absorbance of 200 μL of supernatant was measured at λ = 540 nm in a microplate reader. The concentration of TBARS was calculated by interpolation with a standard curve of tetramethoxypropane (TMP) as a precursor of MDA. Results were expressed as nmol of TBARS per mg of protein. The inhibition ratio [IR (%)] was calculated using the following formula: IR = (C − E) × 100/C, where C is the absorbance of the control and E is the absorbance of the test sample. α-Tocopherol was used as the positive standard. All data are represented as mean ± standard error (SEM). The 50% inhibitory concentration (IC50) was estimated by means of linear regression.
Hydroxyl radical measurement
EPR measurements were performed at room temperature using a JEOL JES-TE300 spectrometer operated at 100 kHz modulation frequency and a cylindrical cavity in the TE 011 mode. The external calibration of the magnetic field was conducted with a JEOL ES-FC5 precision gaussmeter. The spectral acquisition and manipulations were conducted using the ES-IPRITS/TE program. EPR spectra were recorded as the first derivative. All experiments were conducted using a quartz flat cell.
Hydroxyl radicals were generated by means of the Fenton reaction, using FeSO4·7H2O 20 μM prepared in acid water at pH = 2.5; H2O2 (30%) 200 μM in the presence of 6000 μM of 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) in a phosphate buffered solution (PBS) at pH = 7.4. DMPO was used as a spin trap. Compound 10f was tested at final concentrations of 106, 60, 10.66 and 3.3 μM in acetone. Reaction mixtures of FeSO4·7H2O, DMPO and H2O2 showed a quartet signal with intensity 1 : 2 : 2 : 1 and hyperfine coupling constants (hfcc) aH = aN = 1.49 mT, g = 2.0057 assigned to the DMPO–OH adduct. The intensity was determined by the peak-height of the second signal at different concentrations of 10f. For all spectra collection, the spectrometer settings were: center field = 334.5 ± 4 mT; microwave power = 8 mW; microwave frequency = 9.44 GHz; modulation width = 0.1 mT; time constant = 2 min; amplitude = 250; sweep time = 120 s; and accumulation = 1 scan. The signal intensity was determined as a double integral.45
α-Glucosidase inhibition assay
Inhibition was evaluated using a previously reported method,46 with few modifications. A solution (25 μL) of tested samples in DMSO–H2O (1 : 1) was added to 150 μL of phosphate buffer solution (PBS, 67 mM, pH 6.8) and incubated at 37 °C for 10 min with 25 μL of reduced glutathione (3 mM in PBS) and 25 μL of 0.2 U mL−1 in PBS solution of α-glucosidase type I (Sigma cat. G5003-100UN). The substrate solution (25 μL, 23.2 mM p-nitrophenyl-α-d-glucopyranoside, Sigma N1377-1G, in PBS) was added and incubated at 37 °C for an additional 15 min with shaking. The reaction mixture was stopped with CaCO3 1 M (50 μL) and after 5 min of agitation, the optical density was determined at 405 nm. Quercetin was used as the positive standard. The inhibition percentage was calculated by the equation:Inhibition (%) = [(A control − A sample)/A control] × 100where A = the absorbance at 405 nm of the sample and control.
α-Glucosidase kinetic studies
To determine the kinetics of enzyme inhibitors, IC50 and 2 × IC50 concentrations were selected. With each inhibitor concentration, the α-glucosidase activity was assayed by varying p-nitrophenyl-α-d-glucopyranoside (PNPG) concentration from 1 to 23.1 mM. The kinetic inhibition was carried out in a similar manner to the inhibition of α-glucosidase protocol. The enzyme and inhibitor were preincubated for 10 min, the reaction was started with different concentrations of PNPG at 37 °C and quenched with CaCO3 and the absorbance at 405 nm was determined. The Line-weaver Burk plot was constructed to determine the kinetics of enzyme inhibition.
α-Glucosidase modeling and molecular docking
The α-glucosidase model was obtained from the AlphaFold Protein Structure Database using the Uniprot identification P38158. The model was subjected to energy minimization using the UCSF Chimera 1.16.47 The generation of pdbqt files for protein and compounds was completed using AutoDock Tools (ADT).48 The grid dimension used for the receptor is 80 × 74 × 85 Å (grid box). Molecular docking between the receptor and compounds was performed using AutoDock Vina49 and the results were visualized and represented using PyMol (Molecular Graphics System, version 2.5.0 Schrödinger, LLC).
Human α-glucosidase was retrieved from PFB (5NN8) and prepared using the Protein Preparation Wizard from Schrödinger. Compounds were prepared by using the module Ligprep from Schrödinger and were docked into the catalytic site (10 × 10 × 10 Å grid box) using the extra precision (XP) algorithm in GLIDE. Acarbose was docked using the same protocol. Binding poses, distances and edition of figures were obtained from the same docking suite.
Animals
Mice were used and kept under standard animal facility conditions (12 h/12 h light/dark, 18–22 °C, 45–60% humidity), and free access to food (Purina chow™, Mexico) and water. All animal procedures complied with our Federal Regulations for Animal Experimentation and Care (SAGARPA, NOM-062-ZOO.1999, Mexico), and were approved for the Institutional Animal Care and Use Committee (Protocol 1368, F.E.S. Iztacala, UNAM, issued on September 28, 2020).
Oral sucrose tolerance tests.
Male CDI mice (6–7 weeks) were used for this study. The normoglycemic mice were randomly divided into three groups (n = 6): group 1: vehicle (10% Tween 80), group 2: test compounds (50 and 100 mg kg−1), and group 3: positive control (acarbose, 3 mg kg−1). 30 min after administration of test samples, a dose of 3 g kg−1 of sucrose solution was administered to each mouse. Then, blood samples were collected from the caudal vein at time 0 (before oral administration), 0.5, 1, 1.5, 2 and 3 h after vehicle, positive control and isoquinolinone administration. The blood glucose concentration was estimated using a commercial glucometer (Accu-Chek, Performa; Roche). The percentage variation of glycemia for each group was calculated in relation to the initial (0 h) level, according to the formula: % variation of glycemia = [(Gx − G0)/G0] × 100, where G0 was the initial glycemia value and Gx were the glycemia values at each time, respectively.50
Author contributions
Eduardo HV participated in the synthesis and characterization of organic compounds as well as the manuscript preparation and correction. Siseth MC performed the docking studies and revised the manuscript. Samuel ES and Diana AT evaluated the derivatives in the in vivo model. Antonio NC performed the in vitro assays.
Conflicts of interest
The authors declare no conflict of interest.
Supplementary Material
Acknowledgments
Financial support from PAPIIT, UNAM (IA205123) is gratefully acknowledged. The authors are grateful for the analytical support from the “Instituto de Química, UNAM”: Virginia Gómez Vidales (EPR), Elizabeth Huerta-Salazar (NMR support); María del Carmen González García and Javier Flores (MS) and Adriana Romo-Pérez (IR).
Electronic supplementary information (ESI) available. CCDC 1995525. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3md00585b
Notes and references
- Saeedi P. Salpea P. Karuranga S. Petersohn I. Malanda B. Gregg E. W. Unwin N. Wild S. H. Williams R. Diabetes Res. Clin. Pract. 2020;162:108086. doi: 10.1016/j.diabres.2020.108086. [DOI] [PubMed] [Google Scholar]
- Chen L. Deng H. Cui H. Fang J. Zuo Z. Deng J. Li Y. Wang X. Zhao L. Oncotarget. 2018;9:7204–7218. doi: 10.18632/oncotarget.23208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greten F. R. Grivennikov S. I. Immunity. 2019;51:27–41. doi: 10.1016/j.immuni.2019.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen E. J., in Bioactive Food as Dietary Interventions for Diabetes, ed. R. R. Watson and V. R. Preedy, Academic Press, 2nd edn, 2019, pp. 3–17 [Google Scholar]
- Ma X. Chen Z. Wang L. Wang G. Wang Z. Dong X. Wen B. Zhang Z. Front. Pharmacol. 2018;9:782. doi: 10.3389/fphar.2018.00782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengstie M. A. Chekol Abebe E. Behaile Teklemariam A. Tilahun Mulu A. Agidew M. M. Teshome Azezew M. Zewde E. A. Agegnehu Teshome A. Front. Mol. Biosci. 2022;9:1002710. doi: 10.3389/fmolb.2022.1002710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri B. Dey S. Das T. Sarkar M. Banerjee J. Dash S. K. Biomed. Pharmacother. 2018;107:306–328. doi: 10.1016/j.biopha.2018.07.157. [DOI] [PubMed] [Google Scholar]
- Hernández-Vázquez E. Castañeda-Arriaga R. Ramírez-Espinosa J. J. Medina-Campos O. N. Hernández-Luis F. Chaverri J. P. Estrada-Soto S. Eur. J. Med. Chem. 2015;100:106–118. doi: 10.1016/j.ejmech.2015.06.010. [DOI] [PubMed] [Google Scholar]
- Xue Y. Zheng Y. Zhang L. Wu W. Yu D. Liu Y. J. Mol. Model. 2013;19:3851–3862. doi: 10.1007/s00894-013-1921-x. [DOI] [PubMed] [Google Scholar]
- Nasiriani T. Javanbakht S. Nazeri M. T. Farhid H. Khodkari V. Shaabani A. Top. Curr. Chem. 2022;380:50. doi: 10.1007/s41061-022-00403-8. [DOI] [PubMed] [Google Scholar]
- Buskes M. J. Coffin A. Troast D. M. Stein R. Blanco M.-J. ACS Med. Chem. Lett. 2023;14:376–385. doi: 10.1021/acsmedchemlett.3c00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X. Shang Q. Bo C. Hu L. Zhou Y. ARKIVOC. 2018;2018:184–193. [Google Scholar]
- Benchekroun M. Pachón-Angona I. Luzet V. Martin H. Oset-Gasque M.-J. Marco-Contelles J. Ismaili L. Bioorg. Chem. 2019;85:221–228. doi: 10.1016/j.bioorg.2018.12.029. [DOI] [PubMed] [Google Scholar]
- Miranda L. D. Hernández-Vázquez E. J. Org. Chem. 2015;80:10611–10623. doi: 10.1021/acs.joc.5b01742. [DOI] [PubMed] [Google Scholar]
- Hernández-Vázquez E. Chávez-Riveros A. Nieto-Camacho A. Miranda L. D. ChemMedChem. 2019;14:132–146. doi: 10.1002/cmdc.201800634. [DOI] [PubMed] [Google Scholar]
- Barmak A. Niknam K. Mohebbi G. ACS Omega. 2019;4:18087–18099. doi: 10.1021/acsomega.9b01906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolak-Wojciechowska J. Czylkowski R. Karczmarzyk Z. Paluchowska M. H. Rys B. Szneler E. Mokrosz M. J. J. Mol. Struct. 2002;612:39–47. doi: 10.1016/S0022-2860(02)00070-4. [DOI] [Google Scholar]
- Botella L. Nájera C. J. Org. Chem. 2005;70:4360–4369. doi: 10.1021/jo0502551. [DOI] [PubMed] [Google Scholar]
- Taylor J. G. Correia C. R. D. J. Org. Chem. 2011;76:857–869. doi: 10.1021/jo102134v. [DOI] [PubMed] [Google Scholar]
- Matsuura R. Karunananda M. K. Liu M. Nguyen N. Blackmond D. G. ACS Catal. 2021;11:4239–4246. doi: 10.1021/acscatal.1c00783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tańska M. Mikołajczak N. Konopka I. Food Chem. 2018;240:679–685. doi: 10.1016/j.foodchem.2017.08.007. [DOI] [PubMed] [Google Scholar]
- Chen C. Oxid. Med. Cell. Longevity. 2016;2016:3571614. doi: 10.1155/2016/3571614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. Yang J. Ma L. Li J. Shahzad N. Kim C. K. Sci. Rep. 2020;10:2611. doi: 10.1038/s41598-020-59451-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akar Z. Küçük M. Doğan H. J. Enzyme Inhib. Med. Chem. 2017;32:640–647. doi: 10.1080/14756366.2017.1284068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar Diaz De Leon J. Borges C. R. J. Visualized Exp. 2020;(159) doi: 10.3791/61122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.-Z. Zhou X.-L. Huo B.-Q. Chen D.-Z. Liu Z.-H. Sheng X.-H. Front. Chem. 2019;7:850. doi: 10.3389/fchem.2019.00850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D. Hu M.-J. Wang Y.-Q. Cui Y.-L. Molecules. 2019;24:1123. doi: 10.3390/molecules24061123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayouf N. Charef N. Saoudi S. Baghiani A. Khennouf S. Arrar L. J. Ethnopharmacol. 2019;239:111914. doi: 10.1016/j.jep.2019.111914. [DOI] [PubMed] [Google Scholar]
- Dirir A. M. Daou M. Yousef A. F. Yousef L. F. Phytochem. Rev. 2022;21:1049–1079. doi: 10.1007/s11101-021-09773-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrington M. S. Davis S. N. Expert Opin. Pharmacother. 2019;20:2229–2235. doi: 10.1080/14656566.2019.1672660. [DOI] [PubMed] [Google Scholar]
- Mphahlele M. J. Magwaza N. M. Gildenhuys S. Setshedi I. B. Bioorg. Chem. 2020;97:103702. doi: 10.1016/j.bioorg.2020.103702. [DOI] [PubMed] [Google Scholar]
- Hussain F. Khan Z. Jan M. S. Ahmad S. Ahmad A. Rashid U. Ullah F. Ayaz M. Sadiq A. Bioorg. Chem. 2019;91:103128. doi: 10.1016/j.bioorg.2019.103128. [DOI] [PubMed] [Google Scholar]
- Bowen E. G. Wardrop D. J. J. Am. Chem. Soc. 2009;131:6062–6063. doi: 10.1021/ja9005755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M. B. Mishra S. M. J. Funct. Foods. 2012;4:79–86. doi: 10.1016/j.jff.2011.08.002. [DOI] [Google Scholar]
- Wang G. Peng Y. Xie Z. Wang J. Chen M. MedChemComm. 2017;8:1477–1484. doi: 10.1039/C7MD00173H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghadam Farid S. Noori M. Nazari Montazer M. Khalili Ghomi M. Mollazadeh M. Dastyafteh N. Irajie C. Zomorodian K. Mirfazli S. S. Mojtabavi S. Faramarzi M. A. Larijani B. Iraji A. Mahdavi M. Sci. Rep. 2023;13:4392. doi: 10.1038/s41598-023-31080-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D. Wang Q. Zhang W. Hu B. Zhou L. Zeng X. Sun. Y. J. Agric. Food Chem. 2015;63:3694–3703. doi: 10.1021/acs.jafc.5b00420. [DOI] [PubMed] [Google Scholar]
- Barber E. Houghton M. J. Williamson G. Foods. 2021;10:1939. doi: 10.3390/foods10081939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son H.-U. Yoon E.-K. Yoo C.-Y. Park C.-H. Bae M.-A. Kim T.-H. Lee C. H. Lee K. W. Seo H. Kim K.-J. Lee S.-H. Biomolecules. 2019;9:828. doi: 10.3390/biom9120828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proença C. Freitas M. Ribeiro D. Oliveira E. F. T. Sousa J. L. C. Tomé S. M. Ramos M. J. Silva A. M. S. Fernandes P. A. Fernandes E. J. Enzyme Inhib. Med. Chem. 2017;32:1216–1228. doi: 10.1080/14756366.2017.1368503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi M. Anyango S. Deshpande M. Nair S. Natassia C. Yordanova G. Yuan D. Stroe O. Wood G. Laydon A. Žídek A. Green T. Tunyasuvunakool K. Petersen S. Jumper J. Clancy E. Green R. Vora A. Lutfi M. Figurnov M. Cowie A. Hobbs N. Kohli P. Kleywegt G. Birney E. Hassabis D. Velankar S. Nucleic Acids Res. 2022;50:D439–D444. doi: 10.1093/nar/gkab1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J. Evans R. Pritzel A. Green T. Figurnov M. Ronneberger O. Tunyasuvunakool K. Bates R. Žídek A. Potapenko A. Bridgland A. Meyer C. Kohl S. A. A. Ballard A. J. Cowie A. Romera-Paredes B. Nikolov S. Jain R. Adler J. Back T. Petersen S. Reiman D. Clancy E. Zielinski M. Steinegger M. Pacholska M. Berghammer T. Bodenstein S. Silver D. Vinyals O. Senior A. W. Kavukcuoglu K. Kohli P. Hassabis D. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye G.-J. Lan T. Huang Z.-X. Cheng X.-N. Cai C.-Y. Ding S.-M. Xie M.-L. Wang B. Eur. J. Med. Chem. 2019;177:362–373. doi: 10.1016/j.ejmech.2019.05.045. [DOI] [PubMed] [Google Scholar]
- Sidhom P. A. El-Bastawissy E. Ibrahim M. A. A. Shawky A. M. Salama A. El-Moselhy T. J. Med. Chem. 2023;66:991–1010. doi: 10.1021/acs.jmedchem.2c01818. [DOI] [PubMed] [Google Scholar]
- Granados-Oliveros G. Torres E. Zambrano M. Nieto-Camacho A. Gómez-Vidales V. Res. Chem. Intermed. 2018;44:3407–3424. doi: 10.1007/s11164-018-3315-2. [DOI] [Google Scholar]
- Ye X.-P. Song C.-Q. Yuan P. Mao R.-G. Chin. J. Nat. Med. 2010;8:349–352. doi: 10.3724/SP.J.1009.2010.00349. [DOI] [Google Scholar]
- Pettersen E. F. Goddard T. D. Huang C. C. Couch G. S. Greenblatt D. M. Meng E. C. Ferrin T. E. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Morris G. M. Huey R. Lindstrom W. Sanner M. F. Belew R. K. Goodsell D. S. Olson A. J. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt J. Santos-Martins D. Tillack A. F. Forli S. J. Chem. Inf. Model. 2021;61:3891–3898. doi: 10.1021/acs.jcim.1c00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chávez-Silva F. Cerón-Romero L. Arias-Durán L. Navarrete-Vázquez G. Almanza-Pérez J. Román-Ramos R. Ramírez-Ávila G. Perea-Arango I. Villalobos-Molina R. Estrada-Soto S. J. Ethnopharmacol. 2018;212:1–7. doi: 10.1016/j.jep.2017.10.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.