

Abstract
Invasive fungal infections, with high morbidity and mortality, have become one of the most serious threats to human health. There are a few kinds of clinical antifungal drugs but large amounts of them are used, so there is an urgent need for a new structural type of antifungal drug. In this study, we carried out three rounds of structural optimisation and modification of the compound YW-01, which was obtained from the preliminary screening of the group, by using the strategy of scaffold hopping. A series of novel phenylpyrimidine CYP51 inhibitors were designed and synthesised. In vitro antifungal testing showed that target compound C6 exhibited good efficacy against seven common clinically susceptible strains, which was significantly superior to the clinical first-line drug fluconazole. Subsequently in vitro tests on metabolic stability and cytotoxicity revealed that C6 was safe and stable for hepatic microsomal function. Finally, C6 warranted further exploration as a possible novel structural type of CYP51 inhibitor.
We designed and synthesised a series of novel CYP51 inhibitors. Three rounds of structural optimisation and modification obtained compound C6 which showed superior antifungal activity.
1. Introduction
Human fungal infections could be divided into common and slight superficial fungal infections (SFIs) and life-threatening invasive fungal infections (IFIs).1–3 As modern medicine advances, many incurable SFIs in the past can be treated by drugs, but more and more people especially patients with weakened immune systems can become vulnerable to invasion by IFIs.4,5 In patients with COVID-19, fungal pathogens have been discovered to cause severe secondary infections, which has aroused our concern about IFIs.6 It is reported that about 300 fungal pathogens could cause human infections, among which Candida, Cryptococcus, and Aspergillus species are responsible for the main pathogenic factors of IFI cases and cause over 90% of life-threatening infections per year.7,8 The most significant contributor to IFIs, with a mortality rate of 20–40%, is the Candida species, which annually causes more than 4 million life-threatening infections worldwide.9–11 Traditional antifungal drugs including the four classes of polyenes, azoles, echinocandins and flucytosine have been widely used for the treatment of invasive candidiasis (Fig. 1).12,13
Fig. 1. Chemical structures of FDA-approved antifungal drugs and lead compound YW-01.

However, with the increasing number of IFI patients, antifungal drugs are prone to lead to fungal resistance. Fungal resistance leads to an increase in the scarcity of the limited number of antifungal drugs currently available, and thus the need for a new structural type of antifungal drug is urgent in the current situation.14,15 Since humans and fungi both belong to the same class of eukaryotic creatures and have a great deal of genetic similarity, there are very few antifungal medications that can effectively combat fungi.16,17 Fungal cells primarily depend on ergosterol for their membrane sterol, whereas mammalian cells rely on cholesterol for their functions.18–20 The enzyme lanosterol 14α-demethylase (CYP51) is a key part of ergosterol biosynthesis, making it a promising target for antifungal drugs.21–24
In our previous studies, we have discovered a class of structurally novel CYP51 inhibitors, among which the compound YW-01 exhibited moderate inhibitory activity against Candida albicans, Candida tropicalis and Cryptococcus neoformans (C. alb. MIC = 8 μg mL−1, C. tro. MIC = 4 μg mL−1, C. neo. MIC = 16 μg mL−1).25 In the present research, to further enhance the antifungal activity, we investigated the binding mode of compound YW-01 to the CYP51 protein by molecular docking. We speculated that the bulky tricyclic backbone forms collisions with the neighbouring amino acids Pro230 and Met508,19,26 which may be the main reason for the lower activity of the compound. To avoid the collision of small molecules with CYP51 amino acid residues, we used a scaffold hopping strategy to investigate the parent core part and designed and synthesized a series of compounds. By evaluating the in vitro antifungal activity, we found that 2-phenylpyrimidine compound A9 had great potential. Several studies showed that the addition of a pyrimidine portion to the hydrophobic side chain of CYP51 inhibitors may improve their antifungal activity.27 Thus, we designed and synthesized a series of 2-phenylpyrimidine derivatives (Fig. 2). Finally, we found that compound C6 exhibited good antifungal activities. We carried out a series of experiments on the selected compound C6, including in vitro antifungal activity, in vitro cytotoxicity and cytochrome P450 (CYP) inhibition experiments.
Fig. 2. Design strategies for novel CYP51 inhibitors.

2. Results and discussion
2.1. Chemistry
The synthetic route of the key intermediates 6a–g is illustrated in Scheme 1. Firstly, through the Delepine reaction, the bromoacetophenones 1a–g were reacted with hexamethylenetetramine and then refluxed to afford the amino derivatives 2a–g under acidic conditions.
Scheme 1. The synthetic route of intermediates 6a–g. Reagents and conditions: (a) hexamethylenetetramine, CHCl3, 50 °C; (b) 37% HCl, EtOH, reflux; (c) (Boc)2O, NaHCO3, MeOH, H2O, r.t.; (d) 37% CH2O (aq), NaHCO3, EtOH; (e) imidazole, CDI, CH3CN, 70 °C; (f) HCl, EA, r.t.

The intermediates 2a–g were protected with (Boc)2O and the compounds 3a–g were obtained. The compounds 4a–g were obtained through a Tollens condensation reaction of intermediates 3a–g and formaldehyde, and were converted to intermediates 5a–g through a substitution reaction with imidazole. The deprotection of intermediates 5a–g under acidic conditions produced the amino derivatives 6a–g in a remarkable yield.
Intermediate acids 9, 12 and 16a–q were prepared according to the procedures shown in Scheme 2. Cyclization of 1a with 7 provided thiazole ester 8. Intermediate 12 was synthesized via palladium-catalysed Suzuki coupling of commercially available 3-furan boronic acid 10 and 4-bromobenzoic acid 11. Intermediates 15a–q were synthesized from substituted phenylboronic acids 13a–q and 14 by a method similar to that for intermediate 12. These obtained esters 8 and 15a–q were subsequently converted to intermediate acids 9 and 16a–q in the presence of sodium hydroxide.
Scheme 2. The synthetic route of intermediates 9, 12 and 16a–q. Reagents and conditions: (f) EtOH, reflux; (g) NaOH, MeOH/H2O; (h) Pd(PPh3)4, K2CO3, dioxane/H2O, reflux.

The synthetic routes of designed compounds A1–A9 are summarized in Scheme 3. The target compounds A1–A9 were synthesized via an amide reaction of amino 6a and acids 9, 12, 16a and 17a–f in DMF at room temperature, which was optimized to employ PyBop as a coupling reagent for improved parallel processing.
Scheme 3. The synthetic route of the target compounds A1–A9. Reagents and conditions: (i) Py-BOP, DMAP, DIEA, DMF (anhydrous), r.t.

Finally, as depicted in Scheme 4, intermediates 6a–g and 16b–q undergo similar reactions to those described above to obtain compounds B1–B18 and C1–C6.
Scheme 4. The synthetic route of the target compounds B1–B17 and C1–C6. Reagents and conditions: (i) Py-BOP, DMAP, DIEA, DMF (anhydrous), r.t.

2.2. In vitro antifungal activity of synthesized compounds
The target compounds were evaluated for their in vitro antifungal activity against seven pathogenic fungi according to the CLSI M27-A3 guidelines by using FLC as the reference drug.28 The results of in vitro antifungal activities are summarized in Tables 1–3. As shown in the tables, compounds in the data were used as racemates for antifungal testing.
In vitro antifungal activities of compounds A1–A9 (MIC, μg mL−1)a.

| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd. | R1 | C. alb.(I) | C. alb.(II) | C. tro. | C. neo. | C. par. | C. gla. | C. kru. |
| A1 |

|
32 | 64 | >64 | >64 | >64 | >64 | >64 |
| A2 |
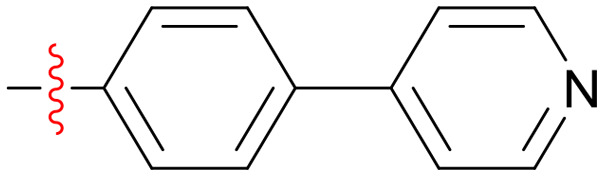
|
8 | 16 | 8 | >64 | >64 | >64 | 16 |
| A3 |

|
8 | 32 | >64 | >64 | >64 | >64 | >64 |
| A4 |

|
>64 | >64 | >64 | >64 | >64 | >64 | >64 |
| A5 |

|
>64 | >64 | >64 | >64 | >64 | >64 | >64 |
| A6 |

|
>64 | >64 | >64 | >64 | >64 | >64 | >64 |
| A7 |

|
16 | >64 | >64 | >64 | >64 | >64 | >64 |
| A8 |

|
>64 | >64 | >64 | >64 | >64 | >64 | >64 |
| A9 |

|
4 | 16 | 8 | >64 | >64 | 32 | 16 |
| FLC | 0.5 | 1 | 0.5 | 4 | 2 | 4 | 8 | |
Abbreviations: C. alb.(I), Candida albicans (ATCC SC5314); C. alb.(II), Candida albicans (CPCC400616); C. neo., Cryptococcus neoformans (CGMCC 2.3161); C. tro., Candida tropicalis (CGMCC 2.3739); C. kru., Candida krusei (GIM 2.1); C. par., Candida parapsilosis (GIM 2.190); C. gla., Candida glabrata (clinical isolation); FLC: fluconazole.
In vitro antifungal activities of compounds B1–B18 (MIC, μg mL−1)a.

| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd. | R2 | C. alb.(I) | C. alb.(II) | C. tro. | C. neo. | C. par. | C. gla. | C. kru. |
| B1 | 4-Me | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| B2 | 4-OMe | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| B3 | 4-Et | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| B4 | 4-CN | 4 | >64 | >64 | >64 | >64 | >64 | >64 |
| B5 | 3-Isopropyl | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| B6 | 4-Isopropyl | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| B7 | 3-tert-Butyl | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| B8 | 4-tert-Butyl | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| B9 | 3-CF3 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| B10 | 3-OCF3 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| B11 | 4-OCF3 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| B12 | 2-F | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| B13 | 3-F | 2 | 8 | 4 | 32 | 16 | 32 | 32 |
| B14 | 4-F | 16 | 32 | >64 | >64 | 64 | >64 | >64 |
| B15 | 2-Cl | 8 | 16 | 4 | >64 | 32 | >64 | 16 |
| B16 | 3-Cl | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| B17 | 4-Cl | 8 | 16 | 4 | >64 | >64 | >64 | >64 |
| B18 | 2-F, 4-Cl | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| FLC | 0.5 | 1 | 0.5 | 4 | 2 | 4 | 8 | |
Abbreviations: C. alb.(I), Candida albicans (ATCC SC5314); C. alb.(II), Candida albicans (CPCC400616); C. neo., Cryptococcus neoformans (CGMCC 2.3161); C. tro., Candida tropicalis (CGMCC 2.3739); C. kru., Candida krusei (GIM 2.1); C. par., Candida parapsilosis (GIM 2.190); C. gla., Candida glabrata (clinical isolation); FLC: fluconazole.
In vitro antifungal activities of compounds C1–C6 (MIC, μg mL−1)a.

| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd. | R3 | C. alb.(I) | C. alb.(II) | C. tro. | C. neo. | C. par. | C. gla. | C. kru. |
| C1 | 4-F | 16 | >64 | >64 | >64 | >64 | >64 | >64 |
| C2 | 4-Cl | 1 | 32 | >64 | >64 | >64 | >64 | >64 |
| C3 | 4-Br | 0.5 | 8 | 16 | >64 | >64 | >64 | >64 |
| C4 | 4-I | 1 | 32 | >64 | >64 | >64 | >64 | >64 |
| C5 | 4-Me | 1 | 4 | 1 | 64 | 4 | 8 | 4 |
| C6 | 4-OMe | 0.25 | 1 | 0.25 | 32 | 1 | 1 | 1 |
| FLC | 0.5 | 1 | 0.5 | 4 | 2 | 4 | 8 | |
Abbreviations: C. alb.(I), Candida albicans (ATCC SC5314); C. alb.(II), Candida albicans (CPCC400616); C. neo., Cryptococcus neoformans (CGMCC 2.3161); C. tro., Candida tropicalis (CGMCC 2.3739); C. kru., Candida krusei (GIM 2.1); C. par., Candida parapsilosis (GIM 2.190); C. gla., Candida glabrata (clinical isolation); FLC: fluconazole.
Firstly, we adopted the strategy of scaffold hopping in the design and synthesis of compound A1 to reduce the tricyclic fragment colliding with the narrow hydrophobic channel of the CYP51 protein. Unfortunately, compared with compound YW-01, the in vitro antifungal activity of the compound decreased significantly. We speculated that the rigid tricyclic fragment was opened, which increased the flexibility of the molecule, and collided with the narrow hydrophobic cavity of the CYP51 protein. To reduce the steric hindrance of the structure with the narrow hydrophobic cavity, we investigated other aryl heterocycles to replace the bulky tricyclic structure to improve the antifungal activity and designed and synthesised compounds A2–A9.
The results revealed that only compounds A2 and A9 displayed moderate antifungal activity, whereas the antifungal activities of the other compounds drastically diminished or vanished. Following careful deliberation, compound A9, which shared the same activity as the lead compound, was selected as the preferred compound and had further structure optimization.
Then, we verified the binding mode of compound A9 with CYP51 (PDB code 5TZ1) by molecular docking and found that it can effectively bind to CYP51 hydrophobic cavities, which proved the rationality of our design (Fig. 3A). Additionally, it was shown that when the hydrophobic side chain was phenylpyrimidine, the additional contact with Phe233 made it more stable in the small hydrophobic cavity, which may have been the primary driver of its increased activity.
Fig. 3. (A) Predicted binding mode of A9 in Candida albicans CYP51 (PDB: 5TZ1); (B) predicted binding mode of B13 in Candida albicans CYP51 (PDB: 5TZ1). The distances are shown as yellow dashed lines.

The 2-phenylpyrimidine fragment of compound A9 was located in the narrow hydrophobic cavity of CYP51, and by molecular docking, we found that there was still a part of the hydrophobic cavity near its benzene ring that was not occupied. Considering the para-position of the benzene ring as a potential metabolic site, its closure may be beneficial for the stability of the compound. Therefore, we carried out a second round of structural optimisation and modification. The examination of the A ring was mainly focused on the introduction of hydrophobic substituents in the para position with a view to forming hydrophobic interactions with the surrounding amino acids. The results of in vitro antifungal activities are summarized in Table 2. Regrettably, a range of compounds B1–B11 were synthesized, but only B4 demonstrated moderate antifungal properties at the para-cyano position substituted, with the others having no evident activity. This was proposed to be caused by the aliphatic hydrocarbons being too large, leading to them striking the narrow hydrophobic cavity. We then changed our strategy and synthesized compounds B12–B18 by introducing halogen atoms to the benzene ring to further explore their activity. Compounds B13 and B15 were found to have relatively good activities. Finally, we determined that compound B13 with a 3-position fluorine substitution, which had relatively higher antifungal activity against a variety of pathogenic fungi, was the preferred compound, and further molecular docking study was carried out. We found that B13 with its 3-position fluorine atom can form fluorine atom interaction with its surrounding amino acid residues Tyr64 and Ser378, which restricts its conformation and further reduces collisions with the narrow hydrophobic cavities, and may be the main reason for our activity enhancement. Under the guidance of the docking study (Fig. 3B), we found that the hydrophobic cavity adjacent to the B ring was not fully occupied by the naked benzene ring of compound B13. Therefore, we fixed ring A as a 3-position fluorine substitution and explored the structure–activity relationship of ring B. The substituents at the 4-position were examined, firstly by introducing halogen substituents, and compounds C1–C4 were designed and synthesized. Introducing halogen atoms led to a gradual increase in activity with increasing substituent size. However, the highest activity was observed with bromine, and it decreased with the introduction of iodine. Next, to further explore hydrophobic cavities, we considered the addition of hydrophobic aliphatic hydrocarbons. To our amazement, the antifungal activity of compounds C5 and C6 significantly increased. Of these, compound C6 showed excellent antifungal activities against C. albicans, C. tropicalis, C. glabrata, C. parapsilosis, and C. krusei with MIC values in the range of 0.25–1 μg mL−1, which were superior to those of the reference drug FLC (Fig. 4). This also indicated that compared to halogens, perhaps hydrophobic aliphatic hydrocarbons can better occupy this hydrophobic cavity.
Fig. 4. (A) Predicted binding mode of C6 in Candida albicans CYP51 (PDB: 5TZ1). The distances are shown as yellow dashed lines. (B) A schematic representation of the contacts between C6 and the binding site residues of Candida albicans CYP51 (PDB: 5TZ1) NBD.

2.3. In vitro cytotoxicity assay
Due to the high phylogenetic similarity between fungi and humans, the side effects of antifungal agents on mammalian cells should also be concerned. To further determine the selectivity of our compounds towards fungal cells, we tested our best compounds C5 and C6 by CCK-8 tests for their toxicity against three nucleated mammalian cell lines, MDA-MB-231, DU145 and L929. In parallel, we also used FDA-approved antifungal agent ketoconazole as a comparator. The results in Fig. 5 showed that C5 and C6 exhibited weak cytotoxicity against the above three cell lines, and when the concentration was increased to 32 μM, they still demonstrated quite good cell viability, significantly better than the control drug ketoconazole.
Fig. 5. Cell viability of C5, C6 and ketoconazole on mammalian cell lines. (A) Human breast cancer cells (MDA-MB-231); (B) human prostate cancer cells (DU145); (C) mouse lung fibroblast cells (L929). Bars represent the mean ± SD (n = 3).

2.4. Evaluating the metabolic stability of compound C6
Drug metabolic stability studies are an important part of innovative drug development. The metabolic stability of compounds is the basis for their efficacy and maintenance, and the liver is the organ where metabolism occurs most frequently. Therefore, we selected the preferred compound C6 for this investigation and in vitro assessed its stability on human liver microsomes. As shown in Table 4, compound C6 had good metabolic stability with a half-life of 14.8 minutes.
Stability assessment of compound C6 in human liver microsomes in vitro.
| Parameters | T 1/2 (min) | CLint(liver) (mL min−1 mg−1) | Remaining (%) (T = 60 min) |
|---|---|---|---|
| In human | 14.8 | 0.0936 | 6.33% |
2.5. Cytochrome P450 (CYP450) inhibition assay
The cytochrome P450 (CYP450) family of enzymes is crucial for the metabolism of many pharmaceuticals, and between 80% and 90% of routinely prescribed medications are broken down by these enzymes. CYP51 and CYP50 have structural and functional similarities, so an obvious side effect of most clinically used antifungal drugs (azole antifungals) is the inhibition of the CYP450 enzyme, resulting in drug–drug interactions. Therefore, in order to prove its safety, we evaluated the inhibitory activity of the target compound C6 on five major metabolic enzymes, which are CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4. The test results indicated that at a concentration of 10 μM, C6 had no significant inhibitory effect on CYP1A2, but the inhibition rate of the other CYP enzymes exceeded 80%, which may lead to the risk of drug–drug interactions (Table 5).
Evaluation of CYPa inhibition of compound C6in vitro.
| Isozyme | CYP1A2 | CYP2C9 | CYP2C19 | CYP2D6 | CYP3A4 |
|---|---|---|---|---|---|
| Inhibition at 10 μM (%) | 38.4 | 89.8 | 89.4 | 72.3 | 92.3 |
α-Naphthoflavone (CYP1A2), sulfaphenazole (CYP2C9), ticlopidine (CYP2C19), quinidine (CYP2D6), and ketoconazole (CYP3A4) were used as the positive controls, performed at 10 μM concentration.
3. Conclusions
Compound YW-01 was a structurally novel and very potential CYP51 inhibitor, but it showed only moderate antifungal activity against the clinically common Candida albicans. To improve its antifungal activities, we used the strategy of scaffold hopping to screen the hydrophobic side chains reaching the solvent region, which was guided by the results of molecular docking. We found that compound A9 with phenylpyrimidine as a side chain has relatively promising antifungal activity. We then carried out two rounds of structural optimisation and designed and synthesized a series of compounds based on compound A9, testing the substituents of the A and B rings. Inspiringly, we finally obtained compound C6 with strong antifungal activity, and its inhibitory effect on Candida albicans has reached four times that of the marketed drug FLC. We then performed further in vitro pharmacological evaluation. C6 exhibited strong fungal cell selectivity and a good safety profile, according to cytotoxicity tests. Moreover, the optimised compound C6 was evaluated for in vitro metabolic stability and showed good drug-like properties. In conclusion, C6 was a promising novel CYP51 inhibitor that warranted further structural modifications and studies.
4. Experiment
4.1. Chemistry
4.1.1. General
Unless otherwise specified, starting materials, reagents, and solvents were purchased from commercial sources and used without further purification. The reactions were monitored by TLC, which was performed on silica gel plates with fluorescence F-254 and visualized with UV light. Silica gel of 300–400 mesh was used for column chromatography. Fast column chromatography used Changzhou Santai Technology's rapid preparation liquid phase SepaBean™ MachineT. The melting points of the compounds were determined on a BüCHI melting point B-540 melting point apparatus and were uncorrected. Low-resolution electrospray ionization mass spectrometry was measured using an Agilent 6120 (quadrupole liquid chromatography) equipped Agilent 1200 LC/MS. Measurements of high-resolution mass spectra (HRMS) were made on Agilent Accurate-Mass Q-TOF 6530 (Santa Clara, CA, USA) equipment. Nuclear magnetic resonance (NMR) spectra were recorded on Bruker Avance III-600 or Bruker Avance III-400 instruments (600 or 400 MHz for 1H and 150 or 100 MHz for 13C) with TMS as an internal standard. The coupling constants (J) were reported in Hertz and the peak multiplicities were described as s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; and br, broad peak. Reverse phase HPLC was conducted on an Agilent 1260 Infinity chromatograph, which was equipped with a ZORBAX SB-C18 column (250 mm × 4.6 mm). The mobile phase A was methanol, and the mobile phase B was 30 mM NaH2PO4 in water (pH = 2.5). The purity of the final compounds was determined by HPLC and was 90% or higher unless specified otherwise.
4.1.2. General procedure for the synthesis of intermediates 2a–2g
Intermediates 2a–2g were synthesized according to a reported method.29
4.1.3. General procedure for the synthesis of intermediates 3a–3g
Intermediates 3a–3g were synthesized according to a reported method.29
4.1.4. General procedure for the synthesis of intermediates 4a–4g
Intermediates 4a–4g were synthesized according to a reported method. To a solution of the amino protected compounds 3a–3g (1.0 equiv.) in ethanol, NaHCO3 (0.5 M, 0.5 equiv.) and 37% CH2O (1.5 equiv.) were added. The mixture was stirred at 50 °C for 12 h and monitored by TLC. After completion, the organic solvent was removed under vacuum. The residue was dissolved in EA and washed with water and brine sequentially. The organic phase was dried over Na2SO4 and filtered. The filtrate was concentrated and purified on a silica gel column. White solid compounds 4a–4g were obtained.
4.1.5. General procedure for the synthesis of intermediates 5a–5g
Intermediates 4a–4g (1.0 equiv.) and CDI (1.2 equiv.) were dissolved in acetonitrile, and imidazole (1.0 equiv.) was added with stirring. After completion, the organic solvent was removed under vacuum. The residue was dissolved in EA and washed with water and brine sequentially. The organic phase was dried over Na2SO4 and filtered. The filtrate was concentrated and purified on a silica gel column.
4.1.6. General procedure for the synthesis of intermediates 6a–6g
Boc protected compounds 5a–5g (1.0 equiv.) were dissolved in EA, and HCl–EA (4 N, 6.0 equiv.) was added under stirring. After the reaction was monitored by TLC, the organic solvent was removed under vacuum. The mixture was then stirred at room temperature for 3 h. The residue solid was washed with ethyl acetate in ethyl acetate to give products 6a–6i as white solids.
4.1.7. General procedure for the synthesis of intermediate 8
To a stirring solution of intermediate 1a in ethanol was added ethyl thioglycinate (1.0 equiv.). The reaction mixture was heated to reflux carried out for 12 h and monitored by TLC. After completion, the solvent was evaporated using a rotovap and purified on a silica gel column.
4.1.8. General procedure for the synthesis of intermediate 12
Intermediate 12 was synthesized according to a reported method.29
4.1.9. General procedure for the synthesis of intermediates 15a–15s
K2CO3 (2.0 equiv.) was added to a solution of 14 (1.0 equiv.) and various substituted phenylboronic acids 13a–13s (1.0 equiv.) in dioxane/H2O (5 : 1). After degasification with Ar for 5 min, Pd(PPh3)4 (0.05 equiv.) was added, and the mixture was degassed again for 5 min. Under an Ar atmosphere, the resulting suspension was heated at 95 °C for 6 h and then cooled to room temperature. The organic solvent was removed under vacuum. The residue was dissolved in EA and successively washed with water and brine. The organic phase was filtered after being dried over Na2SO4. The filtrate was concentrated and purified on a silica column (petroleum ether/ethyl acetate = 30/1–20/1) to give the compounds 15a–15s as white solids.
4.1.10. General procedure for the synthesis of intermediates 9 and 16a–16s
Multiple esters (1.0 equiv.) were dissolved in a methanol/water 1 : 1 (30 mL) solution. Then NaOH (1.6 equiv.) was added slowly to the mixture under stirring. The reaction was stirred at room temperature for 4 h, and the solvent was concentrated under reduced pressure after the reaction was monitored by TLC. The reaction solution was poured into ice water and the pH was adjusted to 6–7 with 3 N HCl, and a large amount of white solid precipitated out. The resulting precipitate was collected by filtration as a white solid.
4.1.11. General procedure for the synthesis of compounds A1–A9 and B1–B18
Various carboxylic acids (1.0 equiv.), PyBop (2.0 equiv.) and DMAP (0.05 equiv.) were dissolved in DMF (10 mL) solution and stirred at room temperature for 0.5 h. Then DIEA (2.0 equiv.) and compound 6a (1.0 equiv.) were added. The reaction solution was then stirred at room temperature for about 2 h. After the reaction was monitored by TLC, the reaction solution was poured into water and extracted with EA, dried, evaporated to remove the solvent, and finally purified by silica chromatography to obtain the target compounds A1–A9 and B1–B18 as white solids.
4.1.12. General procedure for the synthesis of compounds C1–C6
The method used for synthesising compounds A1–A9 and B1–B18 was additionally applied to synthesise compounds C1–C6.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-5-phenylisoxazole-3-carboxamide (A1)
White solid, yield 73.5%. 1H NMR (600 MHz, DMSO-d6) δ 9.51 (d, J = 8.6 Hz, 1H), 8.01 (d, J = 7.0 Hz, 2H), 7.91–7.88 (m, 2H), 7.66 (d, J = 7.4 Hz, 1H), 7.63 (s, 1H), 7.54 (d, J = 7.5 Hz, 5H), 7.30 (s, 1H), 7.21 (s, 1H), 6.85 (s, 1H), 5.79 (dt, J = 9.0, 4.6 Hz, 1H), 4.60 (dd, J = 14.2, 4.4 Hz, 1H), 4.44 (dd, J = 14.2, 9.6 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 196.7, 171.2, 159.0 (d, J = 72.0 Hz), 138.4, 135.2, 134.2, 131.4, 129.8 (d, J = 7.4 Hz), 129.6, 129.2, 129.0, 128.8 (d, J = 6.5 Hz), 126.6, 126.3, 120.5, 100.4, 55.2, 46.2. ESI-MS (m/z): calcd for C22H18N4O3: [M + H]+, 387.14; found 387.5; [M + Na]+, 409.14; found 409.5. HRMS (ESI, m/z) calcd for C22H18N4O3, [M + H]+: 387.1412; found: 387.1437. HPLC purity: 95.42%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-4-(pyridin-4-yl)benzamide (A2)
White solid, yield 73.8%. 1H NMR (600 MHz, DMSO-d6) δ 9.27 (d, J = 8.4 Hz, 1H), 8.66 (d, J = 6.2 Hz, 2H), 8.03–8.00 (m, 2H), 7.92 (s, 1H), 7.86 (t, J = 8.1 Hz, 4H), 7.73 (d, J = 6.2 Hz, 2H), 7.63 (t, J = 7.4 Hz, 1H), 7.52 (t, J = 7.7 Hz, 2H), 7.33 (s, 1H), 6.97 (s, 1H), 5.79 (td, J = 9.0, 4.5 Hz, 1H), 4.65 (dd, J = 14.1, 4.4 Hz, 1H), 4.47 (dd, J = 14.1, 9.6 Hz, 1H). ESI-MS (m/z): calcd for C24H20N4O2: [M + H]+, 397.16; found 397.5. HRMS (ESI, m/z) calcd for C24H20N4O2, [M + H]+: 397.1620; found: 397.1651. HPLC purity: 93.67%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-4-(pyridin-3-yl)benzamide (A3)
White solid, yield 73.7%. 1H NMR (600 MHz, DMSO-d6) δ 9.20 (d, J = 8.4 Hz, 1H), 8.92 (s, 1H), 8.60 (d, J = 4.8 Hz, 1H), 8.11 (d, J = 8.0 Hz, 1H), 8.02 (d, J = 7.7 Hz, 2H), 7.82 (d, J = 10.0 Hz, 4H), 7.63 (s, 2H), 7.54–7.48 (m, 3H), 7.22 (s, 1H), 6.84 (s, 1H), 5.75 (d, J = 4.6 Hz, 1H), 4.59 (dd, J = 14.2, 4.5 Hz, 1H), 4.42 (dd, J = 14.2, 9.6 Hz, 1H). ESI-MS (m/z): calcd for C24H20N4O2: [M + H]+, 397.16; found 397.5; [M–H]−, 395.16, found 395.5. HRMS (ESI, m/z) calcd for C24H20N4O2, [M + H]+: 397.1620; found: 397.1643. HPLC purity: 98.48%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-5-phenylpicolinamide (A4)
White solid, yield 54.5%. 1H NMR (600 MHz, DMSO-d6) δ 9.2 (d, J = 8.4 Hz, 1H), 8.7 (dt, J = 4.8, 1.4 Hz, 1H), 8.1–8.1 (m, 1H), 8.0 (td, J = 8.2, 1.2 Hz, 2H), 7.9 (td, J = 7.7, 1.8 Hz, 1H), 7.8–7.8 (m, 1H), 7.7 (s, 1H), 7.7–7.6 (m, 1H), 7.5 (d, J = 7.8 Hz, 1H), 7.4 (ddd, J = 7.5, 4.8, 1.1 Hz, 1H), 7.3 (s, 1H), 6.9 (d, J = 15.4 Hz, 1H), 6.7 (s, 1H), 5.8 (td, J = 9.0, 4.5 Hz, 1H), 5.3 (dd, J = 5.5, 4.3 Hz, 1H), 4.6 (dd, J = 14.2, 4.5 Hz, 1H), 4.4 (dd, J = 14.2, 9.6 Hz, 1H). ESI-MS (m/z): calcd for C24H20N4O2: [M + H]+, 397.16; found 397.5; [M + Na]+, 419.16; found 419.5. HRMS (ESI, m/z) calcd for C24H20N4O2, [M + H]+: 397.1620; found: 397.1655. HPLC purity: 93.52%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-4-(pyrimidin-2-yl)benzamide (A5)
White solid, yield 74.3%. 1H NMR (600 MHz, DMSO-d6) δ 9.23 (d, J = 8.6 Hz, 1H), 9.21 (s, 1H), 9.17 (s, 2H), 8.04–8.01 (m, 2H), 7.91–7.85 (m, 4H), 7.65–7.61 (m, 2H), 7.52 (t, J = 7.7 Hz, 2H), 7.23 (s, 1H), 6.84 (s, 1H), 5.76 (dt, J = 8.4, 4.3 Hz, 1H), 4.60 (dd, J = 14.2, 4.5 Hz, 1H), 4.43 (dd, J = 14.2, 9.6 Hz, 1H). ESI-MS (m/z): calcd for C23H19N5O2: [M + H]+, 398.15; found398.5. HRMS (ESI, m/z) calcd for C23H19N5O2, [M + H]+: 398.1572; found: 398.1618. HPLC purity: 96.85%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-4-(pyrimidin-5-yl)benzamide (A6)
White solid, yield 73.5%. 1H NMR (600 MHz, DMSO-d6) δ 9.35 (d, J = 8.4 Hz, 1H), 8.94 (d, J = 2.3 Hz, 1H), 8.15 (dd, J = 8.3, 2.3 Hz, 1H), 8.12 (s, 1H), 8.11 (s, 1H), 8.07–8.02 (m, 3H), 7.67–7.63 (m, 2H), 7.53 (d, J = 12.7 Hz, 3H), 7.49 (d, J = 6.9 Hz, 1H), 7.25 (s, 1H), 6.85 (s, 1H), 5.80 (td, J = 9.0, 4.6 Hz, 1H), 4.61 (dd, J = 14.2, 4.6 Hz, 1H), 4.43 (dd, J = 14.2, 9.6 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 197.4, 166.1, 162.9, 158.4, 140.5, 138.4, 135.7, 135.4, 134.1, 129.2, 128.8 (d, J = 9.7 Hz), 128.2, 128.0, 120.9, 120.6, 55.4, 46.3. ESI-MS (m/z): calcd for C23H19N5O2: [M + H]+, 398.15; found 398.5; [M + Na]+, 420.15; found 420.5. HRMS (ESI, m/z) calcd for C23H19N5O2, [M + H]+: 398.1572; found: 398.1624. HPLC purity: 98.15%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-4-(furan-3-yl)benzamide (A7)
White solid, yield 63.5%. 1H NMR (600 MHz, DMSO-d6) δ 9.12 (d, J = 8.4 Hz, 1H), 8.28 (s, 1H), 8.03 (d, J = 6.8 Hz, 2H), 7.77 (t, J = 1.8 Hz, 1H), 7.74 (d, J = 8.5 Hz, 2H), 7.68 (d, J = 8.5 Hz, 3H), 7.63 (t, J = 7.3 Hz, 1H), 7.52 (t, J = 7.8 Hz, 2H), 7.24 (s, 1H), 7.01 (d, J = 1.9 Hz, 1H), 6.86 (s, 1H), 5.75 (td, J = 9.1, 4.7 Hz, 1H), 4.60 (dd, J = 14.1, 4.6 Hz, 1H), 4.43 (dd, J = 14.1, 9.5 Hz, 1H). ESI-MS (m/z): calcd for C23H19N3O3: [M + H]+, 386.14; found 386.5. HRMS (ESI, m/z) calcd for C23H19N3O3, [M + H]+: 386.1460; found: 386.1522. HPLC purity: 95.51%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-2-phenylthiazole-4-carboxamide (A8)
White solid, yield 63.5%. 1H NMR (600 MHz, DMSO-d6) δ 9.17 (d, J = 8.8 Hz, 1H), 8.29 (s, 1H), 8.06 (s, 1H), 8.05–8.03 (m, 3H), 7.64 (t, J = 7.4 Hz, 1H), 7.60 (s, 1H), 7.53 (dd, J = 5.4, 2.6 Hz, 5H), 7.18 (s, 1H), 6.82 (s, 1H), 5.83 (q, J = 4.4 Hz, 1H), 4.63 (dd, J = 14.2, 4.6 Hz, 1H), 4.51 (dd, J = 14.2, 9.1 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 197.1, 167.8, 160.6, 149.9, 135.3, 134.2, 132.8, 131.3, 129.9, 129.7 (d, J = 13.3 Hz), 129.2 (2C), 129.1–128.5 (m, 2C), 127.0, 125.6, 120.5, 113.0, 54.9, 46.5. ESI-MS (m/z): calcd for: C22H18N4O2S: [M + H]+, 403.12; found 403.5; [M + Na]+, 425.14; found 425.5; [M–H]−, 401.12; found 401.2. HRMS (ESI, m/z) calcd for C22H18N4O2S, [M + H]+: 403.1184; found: 403.1228. HPLC purity: 98.41%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-2-phenylpyrimidine-5-carboxamide (A9)
White solid, yield 61.3%. 1H NMR (600 MHz, DMSO-d6) δ 9.52 (d, J = 8.3 Hz, 1H), 9.13 (s, 2H), 8.42 (d, J = 6.9 Hz, 2H), 8.05 (d, J = 6.8 Hz, 2H), 7.72 (s, 1H), 7.66 (t, J = 7.4 Hz, 1H), 7.59–7.54 (m, 5H), 7.28 (s, 1H), 6.87 (s, 1H), 5.86 (td, J = 9.0, 4.6 Hz, 1H), 4.62 (dd, J = 14.2, 4.5 Hz, 1H), 4.43 (dd, J = 14.2, 9.4 Hz, 1H). ESI-MS (m/z): calcd for C23H19N5O2: [M + H]+, 398.15; found 398.5. HRMS (ESI, m/z) calcd for C23H19N5O2, [M + H]+: 398.1572.; found: 398.1611. HPLC purity: 94.86%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-2-(p-tolyl)pyrimidine-5-carboxamide (B1)
White solid, yield 73.5%. 1H NMR (600 MHz, DMSO-d6) δ 9.47 (d, J = 8.4 Hz, 1H), 9.09 (s, 2H), 8.31 (d, J = 8.3 Hz, 2H), 8.05–8.00 (m, 2H), 7.66 (t, J = 8.1 Hz, 2H), 7.55 (t, J = 7.8 Hz, 2H), 7.36 (d, J = 8.0 Hz, 2H), 7.26 (s, 1H), 6.85 (s, 1H), 5.84 (td, J = 9.0, 4.6 Hz, 1H), 4.61 (dd, J = 14.2, 4.6 Hz, 1H), 4.41 (dd, J = 14.2, 9.4 Hz, 1H), 2.39 (s, 3H). ESI-MS (m/z): calcd for C24H21N5O2: [M + H]+, 412.17; found 412.5. HRMS (ESI, m/z) calcd for C24H21N5O2, [M + H]+: 412.1729; found: 412.1768. HPLC purity: 95.28%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-2-(4-methoxyphenyl)pyrimidine-5-carboxamide (B2)
White solid, yield 74.2%. 1H NMR (600 MHz, DMSO-d6) δ 9.44 (d, J = 8.4 Hz, 1H), 9.06 (s, 2H), 8.37 (d, J = 9.0 Hz, 2H), 8.04 (d, J = 7.6 Hz, 2H), 7.69–7.63 (m, 2H), 7.55 (t, J = 7.7 Hz, 2H), 7.26 (s, 1H), 7.09 (d, J = 9.0 Hz, 2H), 6.85 (s, 1H), 5.83 (td, J = 8.8, 5.2 Hz, 1H), 4.61 (dd, J = 14.2, 4.6 Hz, 1H), 4.41 (dd, J = 14.2, 9.4 Hz, 1H), 3.85 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 196.6, 164.9, 163.1, 162.3, 156.6 (2C), 138.0, 134.8, 133.8, 130.1 (2C), 128.8 (2C), 128.4 (2C), 123.7, 120.1, 114.3, 55.4, 54.8. ESI-MS (m/z): calcd for C24H21N5O3: [M + H]+, 428.16; found 428.5; [M–H]−, 426.16; found 426.5. HRMS (ESI, m/z) calcd for C24H21N5O3, [M + H]+: 428.1678; found: 428.1712. HPLC purity: 99.96%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-2-(4-ethylphenyl)pyrimidine-5-carboxamide (B3)
White solid, yield 73.5%. 1H NMR (600 MHz, DMSO-d6) δ 9.6 (d, J = 8.3 Hz, 1H), 9.1 (s, 2H), 8.3 (d, J = 9.1 Hz, 2H), 8.2 (s, 1H), 8.1–8.0 (m, 2H), 7.6 (d, J = 7.4 Hz, 2H), 7.5 (t, J = 7.7 Hz, 3H), 7.3 (s, 1H), 6.9 (d, J = 6.8 Hz, 1H), 5.9 (d, J = 5.1 Hz, 1H), 4.6 (dd, J = 14.2, 4.6 Hz, 1H), 4.4 (dd, J = 14.2, 9.3 Hz, 1H), 3.0 (s, 2H), 1.7 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 196.9, 165.6, 163.5, 157.1 (2C), 148.4, 135.2, 134.3, 130.0–128.5 (m, 6C), 124.8, 55.2, 46.3 (d, J = 3.9 Hz), 26.4, 15.7. ESI-MS (m/z): calcd for C25H23N5O2: [M + H]+, 426.19; found 426.6. HRMS (ESI, m/z) calcd for C25H23N5O2, [M + H]+: 426.1885; found: 426.1922. HPLC purity: 89.28%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-2-(4-cyanophenyl)pyrimidine-5-carboxamide (B4)
White solid, yield 67.5%. 1H NMR (600 MHz, DMSO-d6) δ 9.57 (d, J = 8.3 Hz, 1H), 9.19 (s, 2H), 8.55 (d, J = 8.2 Hz, 2H), 8.04 (t, J = 8.7 Hz, 4H), 7.70–7.65 (m, 2H), 7.55 (t, J = 7.6 Hz, 2H), 7.27 (s, 1H), 6.85 (s, 1H), 5.86 (td, J = 8.6, 7.9, 3.5 Hz, 1H), 4.62 (dd, J = 14.2, 4.6 Hz, 1H), 4.42 (dd, J = 14.2, 9.4 Hz, 1H). ESI-MS (m/z): calcd for C24H18N6O2: [M + H]+, 423.15; found 423.2. HRMS (ESI, m/z) calcd for C24H18N6O2, [M + H]+: 423.1525; found: 423.1563. HPLC purity: 96.94%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-2-(3-isopropylphenyl)pyrimidine-5-carboxamide (B5)
White solid, yield 73.5%. 1H NMR (600 MHz, DMSO-d6) δ 9.50 (d, J = 8.4 Hz, 1H), 9.12 (s, 2H), 8.30 (s, 1H), 8.23 (t, J = 5.4 Hz, 1H), 8.05 (d, J = 7.1 Hz, 2H), 7.69–7.64 (m, 2H), 7.55 (t, J = 7.8 Hz, 2H), 7.49–7.45 (m, 2H), 7.27 (s, 1H), 6.85 (s, 1H), 5.85 (td, J = 8.9, 4.6 Hz, 1H), 4.62 (dd, J = 14.2, 4.6 Hz, 1H), 4.42 (dd, J = 14.2, 9.4 Hz, 1H), 3.01 (p, J = 6.9 Hz, 1H), 1.26 (d, J = 6.9 Hz, 6H). 13C NMR (151 MHz, DMSO-d6) δ 197.0, 165.7, 163.5, 157.1 (2C), 149.5, 138.5, 136.7, 135.2, 134.3, 130.4, 129.4 (2C), 129.3 (2C), 128.9 (2C), 126.5 (d, J = 14.2 Hz), 125.0, 120.6, 46.4, 33.9, 24.3, 24.3. ESI-MS (m/z): calcd for C26H25N5O2: [M + H]+, 440.20; found 440.6. HRMS (ESI, m/z) calcd for C26H25N5O2, [M + H]+: 440.2042; found: 440.2082. HPLC purity: 99.35%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-2-(4-isopropylphenyl)pyrimidine-5-carboxamide (B6)
White solid, yield 73.4%. 1H NMR (600 MHz, DMSO-d6) δ 9.47 (d, J = 8.4 Hz, 1H), 9.09 (s, 2H), 8.34 (d, J = 8.4 Hz, 2H), 8.04 (d, J = 7.1 Hz, 2H), 7.68–7.63 (m, 2H), 7.55 (t, J = 7.7 Hz, 2H), 7.42 (d, J = 8.2 Hz, 2H), 7.26 (s, 1H), 6.85 (d, J = 1.1 Hz, 1H), 5.84 (td, J = 9.0, 4.6 Hz, 1H), 4.61 (dd, J = 14.2, 4.5 Hz, 1H), 4.41 (dd, J = 14.2, 9.4 Hz, 1H), 2.97 (p, J = 6.9 Hz, 1H), 1.24 (d, J = 6.8 Hz, 6H). 13C NMR (151 MHz, DMSO-d6) δ 197.0, 165.6, 163.5, 157.1 (2C), 153.0, 138.5, 135.2, 134.5, 134.3, 129.3 (2C), 128.9 (4C), 127.4 (2C), 124.8, 120.6, 55.3, 46.4, 33.9, 24.1 (2C). ESI-MS (m/z): calcd for C26H25N5O2: [M + H]+, 440.20; found 440.6. HRMS (ESI, m/z) calcd for C26H25N5O2, [M + H]+: 440.2042; found: 440.2083. HPLC purity: 99.81%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-2-(3-(tert-butyl)phenyl)pyrimidine-5-carboxamide (B7)
White solid, yield 73.6%. 1H NMR (600 MHz, DMSO-d6) δ 9.49 (d, J = 8.4 Hz, 1H), 9.12 (s, 2H), 8.46 (t, J = 1.7 Hz, 1H), 8.23 (dt, J = 7.8, 1.2 Hz, 1H), 8.05 (d, J = 7.1 Hz, 2H), 7.70 (s, 1H), 7.68–7.62 (m, 2H), 7.57–7.54 (m, 2H), 7.48 (t, J = 7.8 Hz, 1H), 7.27 (s, 1H), 6.86 (s, 1H), 5.85 (td, J = 9.0, 4.6 Hz, 1H), 4.62 (dd, J = 14.2, 4.6 Hz, 1H), 4.42 (dd, J = 14.2, 9.4 Hz, 1H), 1.34 (s, 9H). 13C NMR (151 MHz, DMSO-d6) δ 197.0, 165.9, 163.5, 157.1 (2C), 151.7, 138.5, 136.5, 135.2, 134.3, 129.4–129.1 (m, 4C), 128.9 (d, J = 8.4 Hz), 126.2, 125.2, 125.0, 120.6, 55.3, 46.4, 35.0, 31.6. ESI-MS (m/z): calcd for C27H27N5O2: [M + H]+, 454.22; found 454.6. HRMS (ESI, m/z) calcd for C27H27N5O2, [M + H]+: 454.2198; found: 454.2244. HPLC purity: 97.67%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-2-(4-(tert-butyl)phenyl)pyrimidine-5-carboxamide (B8)
White solid, yield 72.4%. 1H NMR (600 MHz, DMSO-d6) δ 9.49 (d, J = 8.4 Hz, 1H), 9.12 (s, 2H), 8.46 (t, J = 1.7 Hz, 1H), 8.23 (dt, J = 7.8, 1.2 Hz, 1H), 8.05 (d, J = 7.1 Hz, 2H), 7.70 (s, 1H), 7.68–7.62 (m, 2H), 7.55 (t, J = 7.8 Hz, 2H), 7.48 (t, J = 7.8 Hz, 1H), 7.27 (s, 1H), 6.86 (s, 1H), 5.85 (td, J = 9.0, 4.6 Hz, 1H), 4.62 (dd, J = 14.2, 4.6 Hz, 1H), 4.42 (dd, J = 14.2, 9.4 Hz, 1H), 1.34 (s, 9H). ESI-MS (m/z): calcd for C27H27N5O2: [M + H]+, 454.22; found 454.6. HRMS (ESI, m/z) calcd for C27H27N5O2, [M + H]+: 454.2198; found: 454.2233. HPLC purity: 96.91%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-2-(3-(trifluoromethyl)phenyl)pyrimidine-5-carboxamide (B9)
White solid, yield 64.7%. 1H NMR (600 MHz, DMSO-d6) δ 9.55 (d, J = 8.3 Hz, 1H), 9.16 (s, 2H), 8.44 (d, J = 7.9 Hz, 1H), 8.33–8.26 (m, 1H), 8.05 (d, J = 7.0 Hz, 2H), 7.74–7.69 (m, 2H), 7.67 (t, J = 7.4 Hz, 1H), 7.61 (d, J = 8.0 Hz, 1H), 7.55 (t, J = 7.8 Hz, 2H), 7.28 (d, J = 1.3 Hz, 1H), 6.86 (s, 1H), 5.86 (td, J = 9.0, 4.6 Hz, 1H), 4.62 (dd, J = 14.2, 4.6 Hz, 1H), 4.42 (dd, J = 14.2, 9.4 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 196.9, 164.0, 163.3, 157.3 (2C), 149.4, 138.8 (d, J = 69.4 Hz), 135.2, 134.3, 131.7, 129.3 (2C), 128.9 (2C), 127.7, 125.7, 124.7, 121.4, 120.6 (d, J = 16.1 Hz), 119.7, 118.0, 55.3, 46.4. ESI-MS (m/z): calcd for C24H18F3N5O2: [M + H]+, 466.14; found 466.5. HRMS (ESI, m/z) calcd for C24H18F3N5O2, [M + H]+: 466.1446; found: 482.1444. HPLC purity: 98.47%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-2-(3-(trifluoromethoxy)phenyl)pyrimidine-5-carboxamide (B10)
White solid, yield 64.3%. 1H NMR (600 MHz, DMSO-d6) δ 9.58 (d, J = 8.4 Hz, 1H), 9.18 (s, 2H), 8.70–8.66 (m, 2H), 8.06 (d, J = 7.2 Hz, 2H), 7.96 (d, J = 7.7 Hz, 1H), 7.81 (t, J = 7.8 Hz, 1H), 7.70–7.65 (m, 2H), 7.56 (t, J = 7.9 Hz, 2H), 7.28 (s, 1H), 6.86 (s, 1H), 5.88 (td, J = 9.2, 4.6 Hz, 1H), 4.63 (dd, J = 14.3, 4.6 Hz, 1H), 4.44 (dd, J = 14.2, 9.4 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 197.0, 164.0, 163.3, 157.3 (2C), 138.5, 137.7, 135.2, 134.3, 132.5, 130.8, 129.3 (2C), 129.0–128.3 (m, 2C), 127.2, 125.7, 125.4, 124.9 (d, J = 3.8 Hz), 123.6, 121.8, 120.6, 55.3, 46.4. ESI-MS (m/z): calcd for C24H18F3N5O3: [M + H]+, 482.14; found 482.5. HRMS (ESI, m/z) calcd for C24H18F3N5O3, [M + H]+: 482.1395; found: 482.1428. HPLC purity: 96.68%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-2-(4-(trifluoromethoxy)phenyl)pyrimidine-5-carboxamide (B11)
White solid, yield 65.4%. 1H NMR (600 MHz, DMSO-d6) δ 9.53 (d, J = 8.7 Hz, 1H), 9.14 (s, 2H), 8.53 (d, J = 8.4 Hz, 2H), 8.05 (d, J = 7.7 Hz, 2H), 7.67 (dd, J = 17.5, 10.1 Hz, 2H), 7.55 (t, J = 7.8 Hz, 4H), 7.27 (s, 1H), 6.86 (s, 1H), 5.86 (td, J = 8.7, 3.6 Hz, 1H), 4.62 (dd, J = 14.2, 4.5 Hz, 1H), 4.42 (dd, J = 14.5, 9.6 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 197.0, 164.3, 163.3, 157.2, 151.3, 135.8, 135.2, 134.3, 130.9, 129.6, 129.3, 129.0, 128.9, 125.3, 123.0, 121.6, 121.3, 119.6, 117.9, 55.3, 46.5. ESI-MS (m/z): calcd for C24H18F3N5O3: [M + H]+, 482.14; found 482.5. HRMS (ESI, m/z) calcd for C24H18F3N5O3, [M + H]+: 482.1395; found: 482.1437. HPLC purity: 98.47%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-2-(2-fluorophenyl)pyrimidine-5-carboxamide (B12)
White solid, yield 70.5%. 1H NMR (600 MHz, DMSO-d6) δ 9.54 (d, J = 8.3 Hz, 1H), 9.15 (s, 2H), 8.04 (d, J = 7.8 Hz, 3H), 7.70–7.65 (m, 2H), 7.61 (dd, J = 5.7, 2.1 Hz, 1H), 7.55 (t, J = 7.7 Hz, 2H), 7.37 (t, J = 8.0 Hz, 2H), 7.27 (s, 1H), 6.85 (s, 1H), 5.85 (td, J = 8.8, 5.9 Hz, 1H), 4.61 (dd, J = 14.3, 4.5 Hz, 1H), 4.41 (dd, J = 14.2, 9.4 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 164.6, 163.3 (2C), 156.9 (2C), 138.5, 135.2, 134.3, 133.9–130.8 (m), 129.3 (2C), 128.9 (2C), 125.8 (d, J = 9.2 Hz), 125.1 (d, J = 3.6 Hz), 117.4 (d, J = 21.7 Hz), 55.3, 46.4. ESI-MS (m/z): calcd for C23H18FN5O2: [M + H]+, 416.14; found 416.5. HRMS (ESI, m/z) calcd for C23H18FN5O2, [M + H]+: 416.1478; found: 416.1533. HPLC purity: 92.74%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-2-(3-fluorophenyl)pyrimidine-5-carboxamide (B13)
White solid, yield 69.8%. 1H NMR (600 MHz, DMSO-d6) δ 9.53 (d, J = 8.3 Hz, 1H), 9.14 (s, 2H), 8.26 (d, J = 8.0 Hz, 1H), 8.11 (d, J = 10.0 Hz, 1H), 8.05 (d, J = 8.1 Hz, 2H), 7.71–7.65 (m, 2H), 7.63–7.59 (m, 1H), 7.55 (t, J = 7.7 Hz, 2H), 7.44 (t, J = 8.8 Hz, 1H), 7.27 (s, 1H), 6.85 (s, 1H), 5.86 (td, J = 8.9, 4.6 Hz, 1H), 4.62 (dd, J = 14.2, 4.6 Hz, 1H), 4.42 (dd, J = 14.2, 9.4 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 197.0, 164.3, 163.3, 162.2, 157.2, 139.2, 138.5, 135.2, 134.3, 131.5 (d, J = 8.3 Hz), 129.3 (2C), 128.9 (d, J = 3.6 Hz, 2C), 125.6, 124.8, 120.6, 119.1, 119.0, 115.1, 115.0, 55.3, 46.4. ESI-MS (m/z): calcd for C23H18FN5O2: [M + H]+, 416.14; found 416.5. HRMS (ESI, m/z) calcd for C23H18FN5O2, [M + H]+: 416.1478; found: 416.1540. HPLC purity: 99.01%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-2-(4-fluorophenyl)pyrimidine-5-carboxamide (B14)
White solid, yield 72.5%. 1H NMR (600 MHz, DMSO-d6) δ 9.50 (d, J = 8.3 Hz, 1H), 9.12 (s, 2H), 8.46 (dd, J = 8.9, 5.7 Hz, 2H), 8.04 (d, J = 7.0 Hz, 2H), 7.68–7.65 (m, 2H), 7.55 (t, J = 7.8 Hz, 2H), 7.37 (d, J = 8.9 Hz, 2H), 7.26 (s, 1H), 6.85 (d, J = 1.2 Hz, 1H), 5.84 (dt, J = 9.4, 4.5 Hz, 1H), 4.63–4.59 (dd, 1H), 4.44–4.39 (dd, 1H). 13C NMR (151 MHz, DMSO-d6) δ 197.0, 165.8, 164.6, 163.4, 157.2, 138.5, 135.2, 134.3, 133.3, 131.3 (d, J = 9.1 Hz), 129.3, 128.9, 125.0, 120.6, 116.5, 116.3, 55.3, 46.4. ESI-MS (m/z): calcd for C23H18FN5O2: [M + H]+, 416.14; found 416.5. HRMS (ESI, m/z) calcd for C23H18FN5O2, [M + H]+: 416.1478; found: 416.1550. HPLC purity: 97.95%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-2-(2-chlorophenyl)pyrimidine-5-carboxamide (B15)
Yellow solid, yield 64.3%. 1H NMR (600 MHz, DMSO-d6) δ 9.50 (d, J = 8.4 Hz, 1H), 9.12 (s, 2H), 8.41 (d, J = 6.7 Hz, 2H), 8.04 (d, J = 7.1 Hz, 3H), 7.65 (d, J = 7.4 Hz, 2H), 7.55 (s, 3H), 7.26 (s, 1H), 6.85 (s, 1H), 5.85 (td, J = 9.2, 4.6 Hz, 1H), 4.62 (dd, 1H), 4.41 (dd, J = 4.8 Hz, 1H). ESI-MS (m/z): calcd for C23H18ClN5O2: [M + H]+, 432.11; found 432.5. HRMS (ESI, m/z) calcd for C23H18ClN5O2, [M + H]+: 432.1154; found: 432.1228. HPLC purity: 96.37%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-2-(3-chlorophenyl)pyrimidine-5-carboxamide (B16)
White solid, yield 74.5%. 1H NMR (600 MHz, DMSO-d6) δ 9.51 (d, J = 8.6 Hz, 1H), 8.01 (d, J = 7.8 Hz, 2H), 7.92–7.87 (m, 2H), 7.67–7.62 (m, 2H), 7.56 (s, 1H), 7.54 (d, J = 6.8 Hz, 4H), 7.29 (s, 1H), 7.22 (s, 1H), 6.85 (s, 1H), 5.79 (td, J = 9.1, 4.4 Hz, 1H), 4.60 (dd, J = 14.2, 4.4 Hz, 1H), 4.44 (dd, J = 14.2, 9.6 Hz, 1H). ESI-MS (m/z): calcd for C23H18ClN5O2: [M + H]+, 432.11; found 432.5. HRMS (ESI, m/z) calcd for C23H18ClN5O2, [M + H]+: 432.1154; found: 432.1226. HPLC purity: 93.03%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-2-(4-chlorophenyl)pyrimidine-5-carboxamide (B17)
White solid, yield 75.3%. 1H NMR (600 MHz, DMSO-d6) δ 9.52 (d, J = 8.4 Hz, 1H), 9.13 (s, 2H), 8.41 (d, J = 8.6 Hz, 2H), 8.04 (d, J = 7.1 Hz, 2H), 7.69–7.64 (m, 2H), 7.62 (d, J = 8.7 Hz, 2H), 7.56–7.53 (m, 2H), 7.26 (s, 1H), 6.85 (s, 1H), 5.87–5.83 (m, 1H), 4.61 (dd, J = 14.3, 4.6 Hz, 1H), 4.42 (dd, J = 14.2, 9.4 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 197.0, 164.6, 163.3, 157.2 (2C), 138.5, 137.2, 135.6, 135.2, 134.3, 130.5 (2C), 129.5 (2C), 129.3 (2C), 128.9 (2C), 125.2, 55.3. ESI-MS (m/z): calcd for C23H18ClN5O2: [M + H]+, 432.11; found 432.5. HRMS (ESI, m/z) calcd for C23H18ClN5O2, [M + H]+: 393.1856; found: 432.1222. HPLC purity: 97.70%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-phenylpropan-2-yl)-2-(4-chloro-2-fluorophenyl)pyrimidine-5-carboxamide (B18)
White solid, yield 66.5%. 1H NMR (600 MHz, DMSO-d6) δ 9.56 (d, J = 8.3 Hz, 1H), 9.15 (s, 2H), 8.10 (t, J = 8.5 Hz, 1H), 8.03 (d, J = 7.3 Hz, 2H), 7.77 (s, 1H), 7.66 (t, J = 7.4 Hz, 1H), 7.62 (dd, J = 10.8, 2.1 Hz, 1H), 7.55 (t, J = 7.7 Hz, 2H), 7.47 (dd, J = 8.4, 2.1 Hz, 1H), 7.30 (s, 1H), 6.90 (s, 1H), 5.86 (td, J = 8.9, 4.5 Hz, 1H), 4.62 (dd, J = 14.3, 4.5 Hz, 1H), 4.42 (dd, J = 14.3, 9.4 Hz, 1H). ESI-MS (m/z): calcd for C23H17ClFN5O2: [M + H]+, 450.10; found 450.5. HRMS (ESI, m/z) calcd for C23H17ClFN5O2, [M + H]+: 450.1055; found: 450.1121. HPLC purity: 99.44%.
2-(3-Fluorophenyl)-N-(1-(4-fluorophenyl)-3-(1H-imidazol-1-yl)-1-oxopropan-2-yl)pyrimidine-5-carboxamide (C1)
Yellow solid, yield 63.5%. 1H NMR (600 MHz, DMSO-d6) δ 9.64 (d, J = 8.2 Hz, 1H), 9.14 (s, 2H), 8.25 (d, J = 7.8 Hz, 1H), 8.09 (d, J = 10.0 Hz, 1H), 7.87 (d, J = 7.8 Hz, 1H), 7.78 (d, J = 9.7 Hz, 1H), 7.73 (s, 1H), 7.60 (t, J = 8.0 Hz, 2H), 7.51 (t, J = 8.4 Hz, 1H), 7.43 (d, J = 9.2 Hz, 1H), 7.28 (s, 1H), 6.87 (s, 1H), 5.84–5.79 (m, 1H), 4.62 (dd, J = 14.1, 4.4 Hz, 1H), 4.44 (dd, J = 14.1, 9.4 Hz, 1H). ESI-MS (m/z): calcd for C23H17F2N5O2: [M–H]−, 432.14; found 431.9. HRMS (ESI, m/z) calcd for C23H17F2N5O2, [M + H]+: 434.1384; found: 434.1421. HPLC purity: 96.58%.
N-(1-(4-Chlorophenyl)-3-(1H-imidazol-1-yl)-1-oxopropan-2-yl)-2-(3-fluorophenyl)pyrimidine-5-carboxamide (C2)
Yellow solid, yield 65.5%. 1H NMR (600 MHz, DMSO-d6) δ 9.56 (d, J = 8.2 Hz, 1H), 9.13 (s, 2H), 8.25 (d, J = 7.8 Hz, 1H), 8.09 (d, J = 9.6 Hz, 1H), 8.01 (d, J = 8.5 Hz, 2H), 7.61 (d, J = 8.4 Hz, 4H), 7.46–7.36 (m, 2H), 6.95 (d, J = 7.1 Hz, 1H), 5.82 (s, 1H), 4.64 (d, J = 15.2 Hz, 1H), 4.45 (d, J = 14.5 Hz, 1H). ESI-MS (m/z): calcd for C23H17ClFN5O2: [M + H]+, 450.11; found 450.5. HRMS (ESI, m/z) calcd for C23H17ClFN5O2, [M + H]+: 450.1051; found: 450.1123. HPLC purity: 91.22%.
N-(1-(4-Bromophenyl)-3-(1H-imidazol-1-yl)-1-oxopropan-2-yl)-2-(3-fluorophenyl)pyrimidine-5-carboxamide (C3)
Yellow solid, yield 64.8%. 1H NMR (600 MHz, DMSO-d6) δ 9.56 (d, J = 8.3 Hz, 1H), 9.14 (s, 2H), 8.26 (d, J = 7.9 Hz, 1H), 8.10 (d, J = 10.3 Hz, 1H), 7.94 (d, J = 8.6 Hz, 2H), 7.76 (d, J = 8.6 Hz, 3H), 7.63–7.59 (m, 1H), 7.44 (td, J = 8.4, 2.7 Hz, 1H), 7.27 (s, 1H), 6.87 (s, 1H), 5.80 (td, J = 8.7, 4.6 Hz, 1H), 4.61 (dd, J = 14.2, 4.6 Hz, 1H), 4.42 (dd, J = 14.2, 9.3 Hz, 1H). ESI-MS (m/z): calcd for C23H17BrFN5O2: [M + H]+, 494.05; found 494.5. HRMS (ESI, m/z) calcd for C23H17BrFN5O2, [M + H]+: 494.0544; found: 494.0621. HPLC purity: 98.72%.
N-(3-(1H-Imidazol-1-yl)-1-(4-iodophenyl)-1-oxopropan-2-yl)-2-(3-fluorophenyl)pyrimidine-5-carboxamide (C4)
Yellow solid, yield 65.7%. 1H NMR (600 MHz, DMSO-d6) δ 9.58 (d, J = 8.9 Hz, 1H), 9.13 (s, 2H), 8.25 (d, J = 7.9 Hz, 1H), 8.10 (d, J = 10.3 Hz, 1H), 7.93 (d, J = 8.5 Hz, 2H), 7.76 (d, J = 8.5 Hz, 3H), 7.62–7.58 (m, 1H), 7.43 (t, J = 8.8 Hz, 1H), 7.28 (s, 1H), 6.88 (s, 1H), 5.79 (td, J = 8.6, 4.0 Hz, 1H), 4.61 (dd, J = 14.3, 4.5 Hz, 1H), 4.42 (dd, J = 14.2, 9.3 Hz, 1H). ESI-MS (m/z): calcd for C23H17FIN5O2: [M + H]+, 542.04; found 542.5. HRMS (ESI, m/z) calcd for C23H17FIN5O2, [M + H]+: 542.0445; found: 542.0483. HPLC purity: 92.91%.
N-(3-(1H-Imidazol-1-yl)-1-oxo-1-(p-tolyl)propan-2-yl)-2-(3-fluorophenyl)pyrimidine-5-carboxamide (C5)
White solid, yield 61.5%. 1H NMR (600 MHz, DMSO-d6) δ 9.59 (d, J = 8.4 Hz, 1H), 9.15 (s, 2H), 8.25 (d, J = 7.9 Hz, 1H), 8.09 (d, J = 9.1 Hz, 1H), 7.95 (d, J = 8.4 Hz, 2H), 7.82 (s, 1H), 7.61–7.58 (m, 1H), 7.42 (t, J = 8.5 Hz, 1H), 7.34 (d, J = 8.0 Hz, 2H), 7.32 (s, 1H), 6.91 (s, 1H), 5.85 (td, J = 9.0, 4.5 Hz, 1H), 4.62 (dd, J = 14.2, 4.5 Hz, 1H), 4.42 (dd, J = 14.2, 9.4 Hz, 1H), 2.36 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 195.8, 163.8 (d, J = 2.8 Hz), 163.3, 162.8, 161.7, 156.8 (2C), 144.4, 138.7 (d, J = 7.8 Hz), 138.0, 132.2, 131.1 (d, J = 8.8 Hz), 129.4 (2C), 128.6 (2C), 127.6, 125.1, 124.3, 120.5, 118.6, 118.5, 114.6, 114.5, 54.6, 46.3, 21.2. ESI-MS (m/z): calcd for C24H20FN5O2: [M + H]+, 430.16; found 430.5. HRMS (ESI, m/z) calcd for C24H20FN5O2, [M + H]+: 430.1635; found: 430.1670. HPLC purity: 97.07%.
N-(3-(1H-Imidazol-1-yl)-1-(4-methoxyphenyl)-1-oxopropan-2-yl)-2-(3-fluorophenyl)pyrimidine-5-carboxamide (C6)
White solid; yield: 68.2%; mp: 152.6–153.3 °C. 1H NMR (600 MHz, DMSO-d6) δ 9.50 (d, J = 8.5 Hz, 1H), 9.16 (s, 2H), 8.25 (d, J = 7.8 Hz, 1H), 8.10 (d, J = 8.6 Hz, 1H), 8.05 (d, J = 8.5 Hz, 2H), 7.71 (s, 1H), 7.60 (q, J = 7.7 Hz, 1H), 7.42 (t, J = 9.8 Hz, 1H), 7.27 (s, 1H), 7.07 (d, J = 8.6 Hz, 2H), 6.86 (s, 1H), 5.85 (td, J = 9.0, 4.7 Hz, 1H), 4.58 (dd, J = 14.2, 4.6 Hz, 1H), 4.40 (dd, J = 14.2, 9.4 Hz, 1H), 3.83 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 194.6, 163.7, 163.3, 162.8, 161.7, 156.8, 138.0, 131.2–130.8 (m), 128.2, 127.5, 125.1, 124.3, 120.2, 114.6, 114.5, 114.1, 55.6, 54.3, 46.2, 45.8. ESI-MS (m/z): calcd for C24H20FN5O3: [M + H]+, 446.06; found 446.5. HRMS (ESI, m/z) calcd for C24H20FN5O3, [M + H]+: 446.1584; found: 446.1643. HPLC purity: 98.98%.
4.2. Pharmacological assays
4.2.1. In vitro antifungal testing
In this paper, based on the M27-A3 bill proposed by the National Committee for Clinical Laboratory Standards (NCCLS), the antifungal activity of the compounds was tested. Seven strains of sensitive pathogenic fungus were selected for in vitro MIC determination and the positive control drug was fluconazole. The RPMI 1640 assay medium (Gibco) was buffered with morphinepropanesulfonic acid (Genview). The fungal cells were diluted with the RPMI 1640 medium to a concentration of 1–5 × 103 CFU mL−1 and 100 μL of cell suspension was seeded into a 96-well microplate. All of the compounds were dissolved in DMSO and serially diluted in the growth medium.
4.2.2. In vitro cytotoxicity assays
The MDA-MB-231, DU145 and L929 cells were obtained from ATCC and cultured using RPMI-1640 medium and Dulbecco's modified Eagle's medium (Gibco, Thermo Fisher Scientific, American). The medium was mixed with 10% fetal bovine serum (Gibco, Thermo Fisher Scientific, American) and 1% antibiotics (penicillin 10 000 U mL−1, streptomycin 100 mg mL−1) (Solarbio, Beijing, China), respectively.
The three cell lines (MDA-MB-231, DU145 and L929) with a suitable density (5 × 104 cells per well) were seeded in 96-well plates with 100 μL of cell suspension per well, and each group included at least 6 replicates for overnight adhesion culture at 37 °C and in 5% CO2. The control group was treated with complete medium, whereas the sample group was treated with the medium of the compounds at concentrations of 2, 4, 8, 16, 32 and 64 μM for 24 hours in 5% CO2 incubators at 37 °C. The cytotoxicity of molecular compounds was evaluated using the CCK-8 assay (ZOMANBIO). After removing the medium from each well and adding 100 μL fresh medium containing the CCK-8 reagent (10 microliters), absorbance detection was performed at a wavelength of 450 nm using a microplate reader (BioTek). The cell viability was calculated according to the formula provided in the CCK-8 manual.
4.2.3. Cytochrome P450 enzyme inhibition assay
Cytochrome P450 inhibition was evaluated in human liver microsomes (0.253 mg mL−1) using five probe substrates (CYP1A2, 90 μM phenacetin; CYP2C9; 8 μM diclofenac; CYP2C19, 20 μM, S-mephenytoin; CYP2D6, 4 μM dextromethorphan; CYP3A4, 2 μM midazolam and 50 μM testosterone) in the presence of compound C6 (10 μM). Adding mixed solution of liver microsome substrate into 2 μL series concentration-specific inhibitors and 1 μL compound incubated at 37 °C water bath for 5 min. Then, the NADPH regeneration system was added. After incubation at 37 °C for 10 min, the internal standard working solution was added to terminate the reaction. The incubation mixture was centrifuged and the supernatant was analyzed by LC-MS/MS.
4.2.4. Liver microsomal stability assay
After diluting liver microsomes to 0.59 mg mL−1 in 100 mM potassium phosphate buffer as a working solution, pipette 445 μL into pre-warmed “incubation” plates T60 and NCF60, followed by shaking at 37 °C for 10 min. Add 5 μL of complex working solution (100 μM) to both plates and mix well three times before transferring 54 μL of liver microsomes to another black plate. To the black plate was added 6 μL of NADPH solution followed by 180 μL of acetonitrile solution containing 200 ng mL−1 tolbutamide and 200 ng mL−1 labetalol. Add 50 μL of buffer to the NCF60 plate, mix well three times, and incubate at 37 °C for 60 min with shaking. Add 44 μL of NADPH cofactor to the T60 plate and incubate the plate at 37 °C for 60 min. At 5, 15, 30, 45, and 60 min, add 180 μL of quench solution to the quenching plate, mix once, and transfer 60 samples of the T60 plate to the quenching plate sequentially at each time point. The sample plate was shaken for 10 min, then centrifuged at 4000 rpm for 20 min at 4 °C, and 80 μL of supernatant was transferred to 240 μL of pure water and a vial of the mixed plate for 10 min. Each bioassay plate was capped and shaken for 10 min before liquid chromatography with tandem mass spectrometry (LC-MS/MS) analysis.
4.3. Molecular docking analysis
Molecular docking was performed using Schrödinger Maestro 12.8 software. The crystal structure of CYP51 (PDB code: 5TZ1) was taken from the protein database (PDB, https://www.rcsb.org/), and was processed through the Protein Preparation wizard tool by default, which includes adding hydrogen atoms, complementing the missing residue, deleting solvent water and irrelevant molecules and minimizing the energy of hydrogen atoms in the crystal structure with the OPLS_2005 force field. Then we use the Receptor Grid Generation module in the Schrödinger suite to prepare the docking box. We use LigPrep tool to handle ligands by default. Finally, the ligand docking is completed without other settings changes. The interaction of molecules docking is displayed by Discovery Studio2019 and visualized by PyMOL.
Abbreviations
- DMF
N,N-Dimethylformamide
- DMSO
Dimethyl sulfoxide
- EA
Ethyl acetate
- ACN
Acetonitrile
- CDI
Di(1H-imidazol-1-yl)methanone
Conflicts of interest
The authors declare that there are no conflicts of interests.
Supplementary Material
Acknowledgments
We gratefully acknowledge the Program for Innovative Research Team of the Ministry of Education and the Program for Liaoning Innovative Research Team in University.
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3md00589e
Notes and references
- Zhu P. Zhou T. Chen H. Chen X. Wang X. Kong L. Yang M. J. Med. Chem. 2023;66:7497–7515. doi: 10.1021/acs.jmedchem.3c00266. [DOI] [PubMed] [Google Scholar]
- Liu N. Tu J. Dong G. Wang Y. Sheng C. J. Med. Chem. 2018;61:5484–5511. doi: 10.1021/acs.jmedchem.7b01413. [DOI] [PubMed] [Google Scholar]
- Wu S. Wang Y. Liu N. Dong G. Sheng C. J. Med. Chem. 2017;60:2193–2211. doi: 10.1021/acs.jmedchem.6b01203. [DOI] [PubMed] [Google Scholar]
- Ivanov M. Ćirić A. Stojković D. Int. J. Mol. Sci. 2022;23(5):2756. doi: 10.3390/ijms23052756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfect J. R. Nat. Rev. Drug Discovery. 2017;16:603–616. doi: 10.1038/nrd.2017.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokas A. Nat. Microbiol. 2022;7:607–619. doi: 10.1038/s41564-022-01112-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy M. W. Walsh T. J. Expert Rev. Anti-infect. Ther. 2017;15:577–584. doi: 10.1080/14787210.2017.1328279. [DOI] [PubMed] [Google Scholar]
- Zhao L. Yin W. Sun Y. Sun N. Tian L. Zheng Y. Zhang C. Zhao S. Su X. Zhao D. Cheng M. Eur. J. Med. Chem. 2021;224:113715. doi: 10.1016/j.ejmech.2021.113715. [DOI] [PubMed] [Google Scholar]
- Brown G. D. Denning D. W. Levitz S. M. Science. 2012;336:647. doi: 10.1126/science.1222236. [DOI] [PubMed] [Google Scholar]
- Mehta D. Saini V. Bajaj A. RSC Med. Chem. 2023;14(9):1603–1628. doi: 10.1039/D3MD00151B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moudgal V. Sobel J. Expert Opin. Pharmacother. 2010;11:2037–2048. doi: 10.1517/14656566.2010.493875. [DOI] [PubMed] [Google Scholar]
- Wall G. Lopez-Ribot J. L. Antibiotics. 2020;9(8):445. doi: 10.3390/antibiotics9080445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angiolella L. Microorganisms. 2022;10(2):409. doi: 10.3390/microorganisms10020409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahiya S. Sharma N. Punia A. Choudhary P. Gulia P. Parmar V. S. Chhillar A. K. Curr. Drug Targets. 2022;23:116–125. doi: 10.2174/1389450122666210719124143. [DOI] [PubMed] [Google Scholar]
- Almeida F. Rodrigues M. L. Coelho C. Front. Microbiol. 2019;10:214. doi: 10.3389/fmicb.2019.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederhold N. Infect. Drug Resist. 2017;10:249–259. doi: 10.2147/IDR.S124918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campoy S. Adrio J. L. Biochem. Pharmacol. 2017;133:86–96. doi: 10.1016/j.bcp.2016.11.019. [DOI] [PubMed] [Google Scholar]
- Yan Z. Huang Y. Zhao D. Li Z. Wang X. Guo M. Wei Y. Wang Y. Mou Y. Hou Z. Guo C. J. Med. Chem. 2023;66:13247–13265. doi: 10.1021/acs.jmedchem.3c01254. [DOI] [PubMed] [Google Scholar]
- Hargrove T. Y. Friggeri L. Wawrzak Z. Qi A. Hoekstra W. J. Schotzinger R. J. York J. D. Guengerich F. P. Lepesheva G. I. J. Biol. Chem. 2017;292:6728–6743. doi: 10.1074/jbc.M117.778308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H. Zhang W. Zhang M. Kudo M. Aoyama Y. Yoshida Y. Sheng C. Song Y. Yang S. Zhou Y. Lü J. Zhu J. J. Med. Chem. 2003;46:474–485. doi: 10.1021/jm020362c. [DOI] [PubMed] [Google Scholar]
- Lass-Flörl C. Drugs. 2011;71:2405–2419. doi: 10.2165/11596540-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Aoyama Y. Yoshida Y. Sato R. J. Biol. Chem. 1984;259:1661–1666. doi: 10.1016/S0021-9258(17)43459-4. [DOI] [PubMed] [Google Scholar]
- Masiá Canuto M. Gutiérrez Rodero F. Lancet Infect. Dis. 2002;2:550–563. doi: 10.1016/S1473-3099(02)00371-7. [DOI] [PubMed] [Google Scholar]
- Mani Chandrika K. V. S. Sharma S. Bioorg. Med. Chem. 2020;28:115398. doi: 10.1016/j.bmc.2020.115398. [DOI] [PubMed] [Google Scholar]
- Yin W. Zhang Y. Cui H. Jiang H. Liu L. Zheng Y. Wu T. Zhao L. Sun Y. Su X. Li S. Zhao D. Cheng M. Eur. J. Med. Chem. 2021;225:113740. doi: 10.1016/j.ejmech.2021.113740. [DOI] [PubMed] [Google Scholar]
- Yin W. Liu L. Jiang H. Wu T. Cui H. Zhang Y. Gao Z. Sun Y. Qin Q. Zhao L. Su X. Zhao D. Cheng M. Eur. J. Med. Chem. 2022;233:114195. doi: 10.1016/j.ejmech.2022.114195. [DOI] [PubMed] [Google Scholar]
- Heeres J. Meerpoel L. Lewi P. Molecules. 2010;15:4129–4188. doi: 10.3390/molecules15064129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne P., CLSI 2008A, 2008, vol. 28, pp. 6–12 [Google Scholar]
- Zhao L. Sun N. Tian L. Sun Y. Chen Y. Wang X. Zhao S. Su X. Zhao D. Cheng M. Eur. J. Med. Chem. 2019;183:111689. doi: 10.1016/j.ejmech.2019.111689. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.