

Abstract
Annotation of immunologic gene function in vivo typically requires the generation of knockout mice, which is time-consuming and low-throughput. We previously developed CHimeric IMmune Editing (CHIME), a CRISPR-Cas9 bone marrow delivery system for constitutive, ubiquitous deletion of single genes. Here we describe X-CHIME, four new CHIME-based systems for modular and rapid interrogation of gene function combinatorially (C-CHIME), inducibly (I-CHIME), lineage-specifically (L-CHIME), or sequentially (S-CHIME). We use C-CHIME and S-CHIME to assess the consequences of combined deletion of Ptpn1 and Ptpn2, an embryonic lethal gene pair, in adult mice. We find that constitutive deletion of both PTPN1 and PTPN2 leads to bone marrow hypoplasia and lethality, while inducible deletion after immune development leads to enteritis and lethality. These findings demonstrate that X-CHIME can be used for rapid mechanistic evaluation of genes in distinct in vivo contexts and that PTPN1 and PTPN2 have some functional redundancy important for viability in adult mice.
Introduction
The in vivo annotation of gene function in hematopoietic cells typically requires the generation of constitutive knockout (KO) or conditional KO (cKO) mouse strains1–3. Combinations of single gene KO strains or cKO strains with Cre-expressing or Tamoxifen-responsive Cre-ERT2-expressing strains enable assessment of gene interactions4, cell-lineage-specific gene functions, or the temporal roles of genes5. However, such studies often require time-consuming crosses of mice to make KO and cKO strains6,7. To avoid these crosses, several strategies have been employed to enable disruption of genes in intact mice using RNA interference or CRISPR-Cas98,9, but the broad applicability of these approaches is limited by low editing penetrance in immune cells and immunogenicity9–11. We previously created the CHIME (CHimeric IMmune Editing) system to circumvent some of these limitations by knocking out genes in primary hematopoietic stem cells and reintroducing them into an irradiated host, where they differentiate into mature immune cells that are KO for a given gene12.
Beyond KO of single genes, more advanced CRISPR-based approaches have been used in cancer cells in vitro to efficiently KO two genes13 as well as to inducibly or sequentially KO genes14. However, the deployment of these powerful techniques to more complex contexts, such as primary cells in vivo, is not straightforward. Here we describe four new systems optimized and benchmarked for perturbation of genes in hematopoietic cells in vivo. We demonstrate efficient combinatorial deletion of two genes in T cells and myeloid cells with C-CHIME, tightly controlled inducible deletion of CD2 in T cells using I-CHIME, lineage-specific deletion of CD2 in CD8+ T cells with L-CHIME, and sequential deletion of CD28 and CD2 in T cells using S-CHIME.
We previously used CHIME to investigate the non-receptor tyrosine phosphatase PTPN2 (also known as TCPTP) in CD8+ T cells in viral and tumor contexts12,15 and found that PTPN2 restrains antitumor immunity by negatively regulating cytokine signaling in T cells as well as in tumor cells15–17. These findings have motivated development of PTPN2-deficient CAR T cells18 and small-molecule inhibitors of PTPN219,20 as cancer immunotherapies. PTPN2 shares strong homology with PTPN121,22, which also restrains T cell responses to tumors23. Therefore, we asked if KO of both PTPN1 and PTPN2 in immune cells would further improve tumor immunity and tumor growth control. Given PTPN1 and PTPN2 double KO (DKO) is embryonic lethal in mice24, we used the X-CHIME system to study the consequences of combined or sequential deletion of PTPN1 and PTPN2 in hematopoietic cells in adult mice.
Results
C-CHIME enables KO of two genes in diverse immune lineages
DKO studies enable assessment of epistasis, gene synergies, and gene buffering. We previously created CHIME for constitutive deletion of genes in hematopoietic cells in vivo12,15. To extend this system to enable knockout of two genes for evaluating gene interactions in vivo, we developed C-CHIME, which utilizes a lentiviral vector containing two distinct gRNA cloning sites and tracrRNAs driven by the human U6 and mouse U6 promoters13, as well as the fluorophore violet-excited GFP (Vex)25 for tracking transduced populations (Figure 1a). To generate C-CHIME bone marrow chimeras (BMCs), we transduced lineage− Sca-1+ c-Kit+ (LSK) cells (Extended Data Figure 1a) from Rosa26-Cas9-expressing donors (Cas9ON) with the C-CHIME lentiviral vector containing gRNAs targeting Cd2 and Cd28 (Figure 1b). We found an ~90% reduction in the expression of either CD2 or CD28 on CD8+ and CD4+ T cells (Figures 1c–1e and Extended Data Figures 1b–1e) following targeting with the dual gRNA expression vector, indicating efficient knockout of CD2 and CD28. To assess the potential for promoter or gRNA competition, we also generated BMCs with a single gRNA expression vector targeting Cd2 or Cd28 and found that the single KO efficiency of CD2 or CD28 on CD8+ and CD4+ T cells was comparable to the KO efficiency achieved with the dual gRNA expression vector (Figures 1d, 1e and Extended Data Figures 1d, 1e). Moreover, C-CHIME led to ~90% reduction in both CD2 and CD28 expression on CD8+ and CD4+ T cells (Figure 1f and Extended Data Figure 1f). Importantly, 93% and 97% of all KO CD8+ and CD4+ T cells, respectively, were DKO for CD2 and CD28 (Figure 1f and Extended Data Figure 1f), indicating the majority of KO cells were DKO cells. We also assessed KO efficiency by examining insertion-deletion (indel) formation in the DNA of these BMCs. We observed ~95% Cd2 indel frequency and ~85% Cd28 indel frequency in cells receiving both Cd2 and Cd28 targeting gRNAs (Figures 1g and 1h). To assess the capability of this system to KO genes in other hematopoietic lineages, we targeted Fcgr1 and Mertk, which led to 58% KO of each gene and 50% DKO of both genes on red-pulp macrophages (Figures 1i–1l and Extended Data Figure 1g). This indicates that of the KO cells, 79% are DKO for both MERTK and FcγRI. Thus, our C-CHIME system allows for efficient editing of two genes in both adaptive and innate immune lineages.
Figure 1: C-CHIME enables knockout of two genes in adaptive and innate immune lineages.
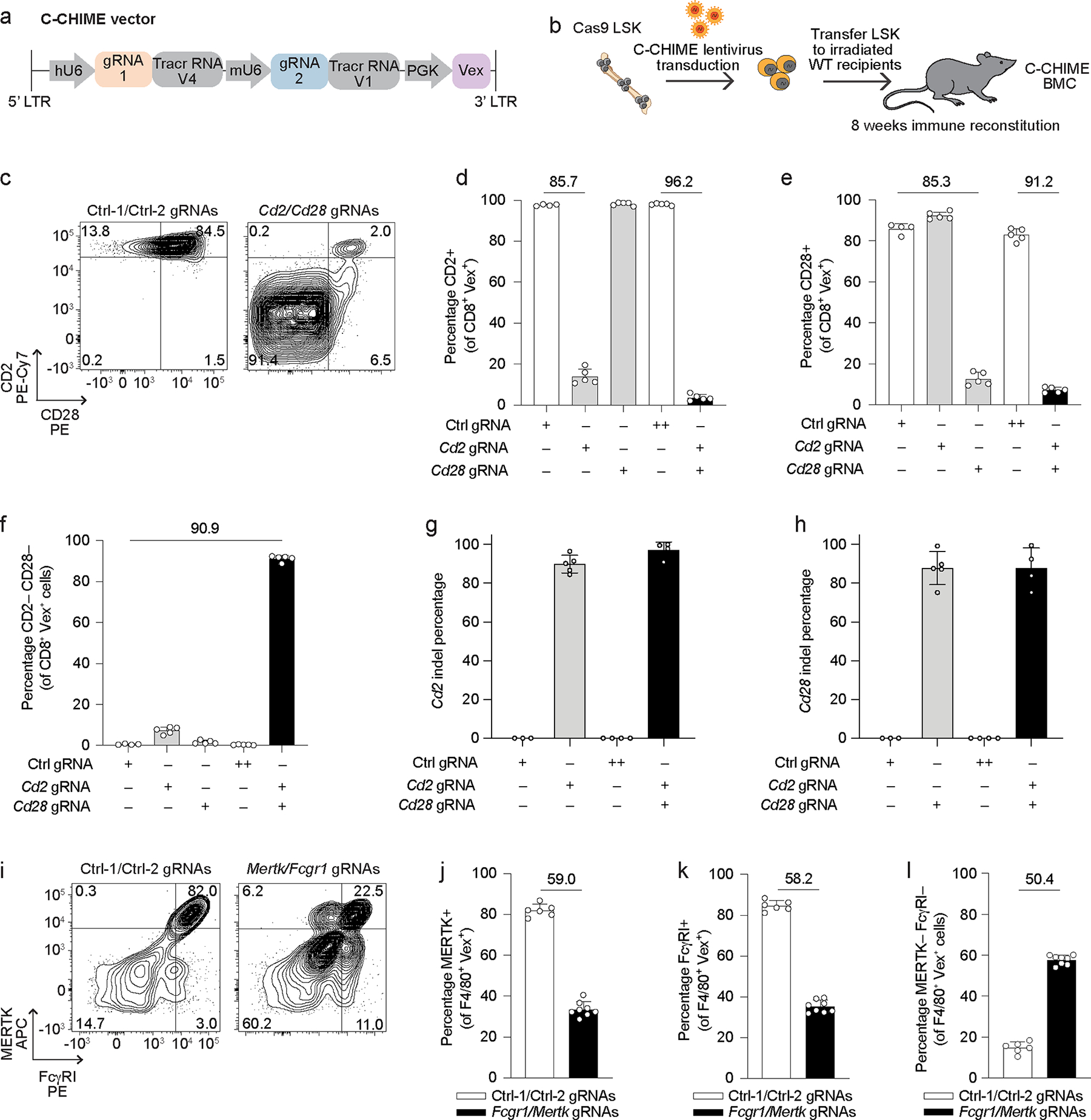
(a) Schematic of the C-CHIME vector containing two sgRNA expression cassettes. (b) Schematic for creation of C-CHIME BMCs. (c) Representative flow cytometry plots of CD2 and CD28 expression on Vex+ CD8+ splenocytes from BMCs containing two non-targeting control gRNAs (left) or Cd2 and Cd28 gRNAs (right). (d, e) Quantification of CD2 (d) or CD28 (e) expression on Vex+ CD8+ splenocytes from (c) as well as relevant single gRNA controls. (f) Quantification of the percentage of cells lacking expression of both CD2 and CD28 on Vex+ CD8+ splenocytes from (c) as well as relevant single gRNA controls. (g, h) Quantification of indels in Cd2 (g) or Cd28 (h) following next-generation sequencing of Vex+ CD8+ splenocytes from (c). Note for (d-h) ++ indicates two control gRNAs. (i) Representative flow cytometry plots of MERTK and FcγRI expression on Vex+ F4/80+ splenic red-pulp macrophages from BMCs containing two non-targeting control gRNAs (left) or Mertk and Fcgr1 gRNAs (right). (j) Quantification of MERTK expression on Vex+ F4/80+ splenic macrophages from (i). (k) Quantification of FcγRI expression on Vex+ F4/80+ splenic macrophages from (i). (l) Quantification of the percentage of cells lacking expression of both MERTK and FcγRI on Vex+ F4/80+ splenic macrophages from (i). Data are representative of two experiments with at least n = 4 mice per group. Bar graphs represent the mean and error bars represent s.d. Calculated knockout efficiency is denoted above bars for indicated comparisons.
I-CHIME enables inducible deletion of genes in T cells
Many genes have different functions at distinct stages of an immune response, making constitutive KO studies insufficient to characterize the functions of these genes. To study such genes, we developed an inducible CHIME system (I-CHIME), which enables temporal control of gene deletion. Although the original Rosa26-Cas9 mouse has a transcriptional stop flanked by loxP sites (LSL), enabling inducibility, we reasoned that induction of the gRNA as opposed to Cas9 would be preferable due to the immunogenicity of Cas926,27. In addition, induction of Cas9 might result in limiting Cas9 expression, which could reduce KO efficiency28. Thus, we crossed the Rosa26-Cas9ON strain to UBC-Cre-ERT2 mice. We also modified our CHIME lentiviral gRNA expression vector to incorporate a tracrRNA disrupted by a loxP-flanked transcriptional stop, modeled after the CRISPR-Switch-On system14. Administration of Tamoxifen to Rosa26-Cas9ON UBC-Cre-ERT2 mice would induce Cre-ERT2 localization to the nucleus and thereby enable excision of the transcriptional stop in the lentiviral vector and expression of the full gRNA-tracrRNA (Extended Data Figure 2a). Following Cre-mediated recombination, a residual loxP site would remain in the tracrRNA, which would not impact gRNA activity14. We created BMCs containing a control or Cd2-targeting gRNA to benchmark this system. Deletion of CD2 on CD4+ and CD8+ T cells was less than 10% at both Tamoxifen dosages assessed at the protein and DNA levels (Extended Data Figures 2b–2d). Low formation of indels could be due to inefficient excision of the transcriptional stop and thus low expression of functional Cd2 gRNA. To assess this, we performed a targeted PCR to determine the efficiency of excision of the transcriptional stop and found that the transcriptional stop was excised in ~80% of cells (Extended Data Figure 2e). Therefore, indel formation is the rate-limiting step in this system, which may be due to steric hindrance of gRNA expression, through binding of Cre to the residual loxP site14.
Given the Cre-ERT2 I-CHIME system had low KO efficiency, we created a second system where the transcriptional stop was flanked by FRT sites and could be excised by the recombinase FlpO. The published FlpO-ERT2 mouse was made by transgenic expression of FlpO-ERT2 from the Rosa26 locus29 and thus would only permit one copy of Rosa26-Cas9 following crossing of the Rosa26-FlpO-ERT2 strain with Rosa26-Cas9ON mice. Therefore, we used the H11-Cas9 mouse that has a Cas9 expression cassette in the H11 locus30, instead of the Rosa26 locus, so that the donor mice could contain two copies of Cas928. To benchmark our constitutive CHIME system using H11-Cas9 donor cells, we created BMCs to examine deletion efficiency of CD2 in CD4+ and CD8+ T cells and found that H11-Cas9 BMCs had comparable KO efficiency to the Rosa26-Cas9ON BMCs (Extended Data Figures 2f, 2g). We thus crossed H11-Cas9 mice with Rosa26-FlpO-ERT2 mice to create a Rosa26-FlpO-ERT2 H11-Cas9 mouse strain. Administration of Tamoxifen to Rosa26-FlpO-ERT2 H11-Cas9 mice enables excision of the transcriptional stop in the I-CHIME vector by FlpO-ERT2 and productive gRNA-tracrRNA transcription (Figure 2a). Following FlpO activity, a single FRT site remains in the tracrRNA stem loop that does not impair gRNA activity14.
Figure 2: I-CHIME enables inducible deletion of genes in T cells.
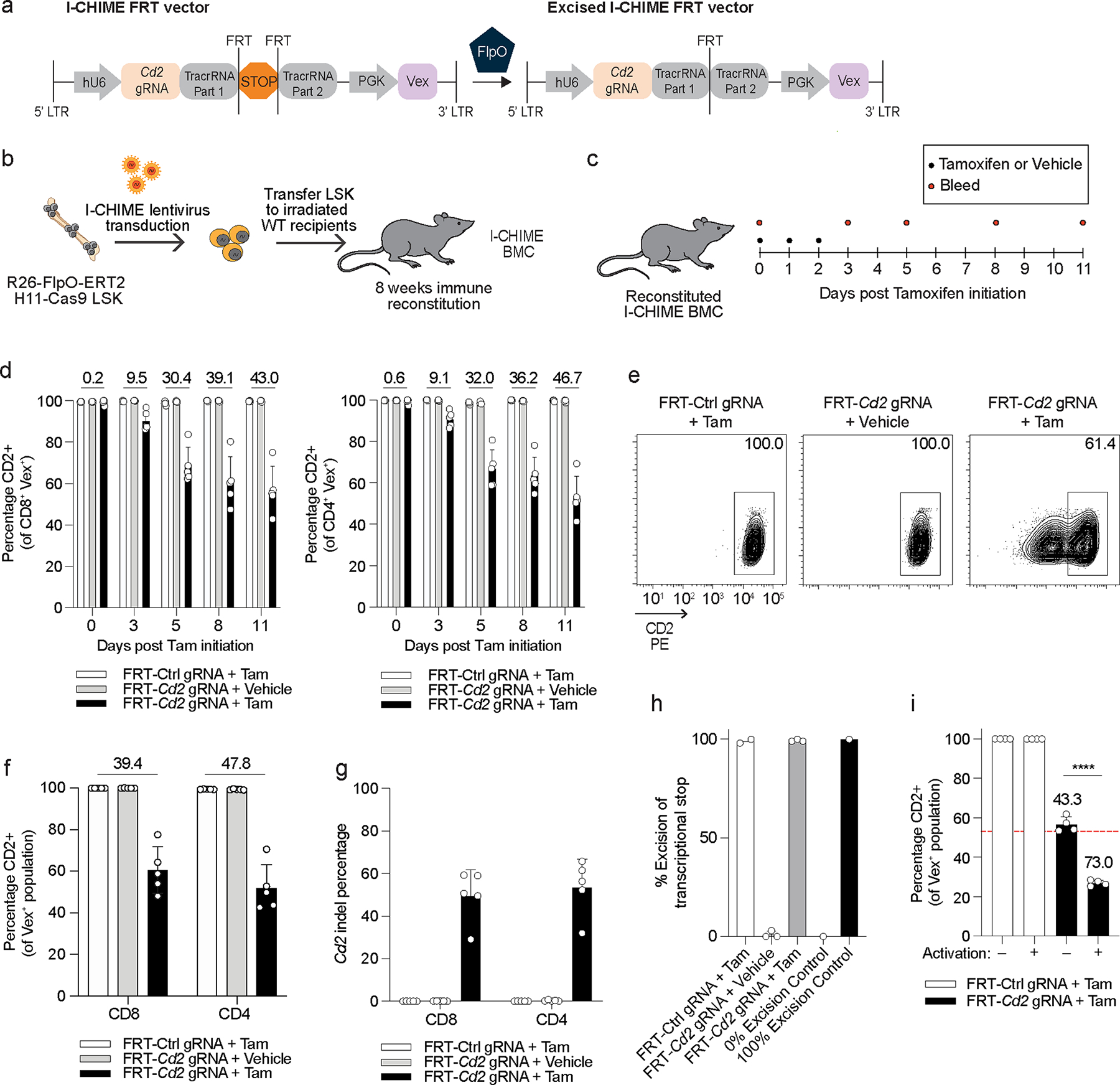
(a) Schematic of I-CHIME FRT vector before and after FlpO-mediated excision. (b) Schematic for creation of I-CHIME BMCs. (c) Timeline of Tamoxifen treatment and serial bleeds of I-CHIME BMCs. (d) Quantification of CD2 expression on CD8+ (left) and CD4+ (right) Vex+ peripheral blood cells as in (c). Serial measurements were taken from the same mice. (e) Representative flow cytometry plots of CD2 expression on CD8+ Vex+ splenocytes from I-CHIME BMCs with non-targeting control or Cd2 gRNAs, 11 days after initiation of Tamoxifen or vehicle control treatment. (f) Quantification of CD2 expression on CD8+ and CD4+ Vex+ splenocytes on day 11 after initiation of Tamoxifen or vehicle treatment as in (e). (g) Quantification of indels in Cd2 following next-generation sequencing of Vex+ CD8+ and Vex+ CD4+ splenocytes as in (e). (h) Quantification of % excision as in (e). (i) CD8+ Vex+ splenocytes were isolated from I-CHIME BMCs containing control or Cd2 gRNAs, 11 days after initiation of Tamoxifen as in (e), and were either activated with CD3 and CD28 cross-linking antibodies and IL-2 or maintained in a naive state with IL-7. Quantification of CD2 expression on day 6 post culture. The red dotted line represents average CD2 expression on the Cd2-targeted CD8+ Vex+ splenocytes prior to culture (p < 0.0001). Data (a-g) are representative of two experiments with at least n = 5 mice per group. Data (h) are representative of two experiments with at least n = 2 mice per group. Data (i) are representative of one experiment with n = 4 mice per group. Bar graphs represent the mean and error bars represent s.d. Calculated knockout efficiency is denoted above bars for indicated comparisons and in (i) each Cd2 gRNA is compared to the control gRNA for the corresponding activation condition. Statistical significance was assessed by a two-sided Student’s unpaired t-test (i).
To determine the optimal dosage and treatment window of Tamoxifen, we created BMCs containing control or Cd2-targeting gRNAs and, following hematopoietic reconstitution, treated these mice with Tamoxifen or a vehicle (Figure 2b). All dosages and treatment windows led to similar KO efficiency but the 2 mg dosage had the most consistent KO efficiency (Extended Data Figure 2h) and therefore was used for all subsequent experiments. To assess the deletion efficiency and inducibility of the FlpO-based system in CD8+ and CD4+ T cells, we treated BMCs containing a control or Cd2-targeted gRNA with Tamoxifen or vehicle and serially sampled blood from these BMCs to determine the kinetics of CD2 deletion (Figure 2c). CD2 expression on peripheral blood CD8+ and CD4+ T cells gradually decreased following Tamoxifen treatment (Figure 2d). CD2 surface expression was not reduced in Cd2-targeted BMCs that received the vehicle treatment, demonstrating that the I-CHIME vector is tightly controlled, a hallmark of a favorable inducible system. At 11 days post Tamoxifen initiation, surface CD2 expression was reduced by 45% on splenic CD4+ and CD8+ T cells in mice with the Cd2 gRNA compared with the control gRNA (Figures 2e, 2f). Furthermore, the Cd2 indel percentage was ~50% in T cells and ~62% in LSKs (Figure 2g and Extended Data Figure 2i), suggesting a potential inefficiency in T cells, although this would need to be confirmed by targeting additional genes. We next determined if the transcriptional stop cassette was efficiently excised following Tamoxifen treatment and found ~100% excision of the transcriptional stop in CD8+ T cells (Figure 2h and Extended Data Figure 2j), whereas vehicle treatment led to minimal excision (<2%). These results indicate that the transcriptional stop is efficiently excised following Tamoxifen treatment and that the rate-limiting step is either gRNA expression post excision of the transcriptional stop or the formation of indels. Given T cell activation can improve KO efficiency31, we asked if activation would improve the KO efficiency achieved via I-CHIME. To do this, we isolated naive Vex+ CD8+ T cells from Tamoxifen-treated I-CHIME BMCs containing control or Cd2-targeting gRNAs and either activated them in vitro with CD3/CD28 cross-linking antibodies and IL-2 or kept them naive. Activated CD8+ T cells had significantly reduced expression (~30%) of CD2, compared with naive CD8+ T cells (Figure 2i). These findings demonstrate that I-CHIME enables ~45% deletion of CD2 in naive T cells with tight inducible control and that subsequent T cell activation can significantly increase the KO efficiency of I-CHIME to ~75%.
Specific deletion of genes in CD8+ T cells using L-CHIME
To rapidly assess gene function in individual immune cell lineages without the need for floxed mice, we developed a lineage-specific KO system (L-CHIME: lineage-specific CHIME). To create the L-CHIME system we crossed Cas9ON mice to E8I-Cre mice (CD8-specific Cre)32. We paired these mice with our loxP-STOP-loxP-gRNA lentiviral vector (Extended Data Figure 3a) to enable deletion of genes specifically in CD8+ T cells (Extended Data Figure 3b). To test the stringency of this system we created BMCs using our loxP-gRNA vector and Cas9ON bone marrow donors that did not express Cre. In the absence of Cre, a functional gRNA should not be expressed and thus this approach allows evaluation of the rate of spontaneous expression of functional gRNA. CD2 was not spontaneously deleted on CD4+ T cells, CD8+ T cells, or Ly6c+ monocytes (Extended Data Figures 3c–3e). Thus, the L-CHIME vector is tightly controlled.
We next evaluated the KO efficiency of CD2 by creating BMCs using E8I-Cre Cas9ON donor mice and the loxP-gRNA vector containing a gRNA targeting Cd2. There was ~40% reduction in CD2 protein expression on CD8+ T cells and 2% reduction in CD4+ T cells (Extended Data Figures 3f, 3g). To determine the efficiency of Cre excision of the loxP-flanked transcriptional stop we performed a targeted PCR and found high excision efficiency (>95%) (Extended Data Figures 3h, 3i), suggesting the lower KO efficiency was not due to inefficiency at the excision step but instead occurred in KO formation after excision. Since we had shown that T cell activation improved KO efficiency in the I-CHIME system (Figure 2i), we asked if T cell activation would improve KO efficiency in the L-CHIME system in vivo and in vitro. First, we assessed the L-CHIME system during a tumor challenge, a context where CD8+ T cells will become activated. Analysis of tumor-infiltrating T cells revealed a reduction in CD2 expression of ~82% in CD8+ T cells and 3% in CD4+ T cells (Extended Data Figure 3j), suggesting activation significantly increased KO efficiency while retaining lineage specificity. In addition, we isolated naive Vex+ CD8+ T cells from E8I-Cre L-CHIME BMCs containing control or Cd2 gRNAs and either activated them or kept them naive in vitro. Consistent with our in vivo findings, activation of CD8+ T cells in vitro significantly improved the KO efficiency of L-CHIME (by ~20%) with ~60% reduction of CD2 expression in activated CD8+ T cells compared to ~40% reduction in CD2 expression in naive CD8+ T cells (Extended Data Figure 3k). Overall, L-CHIME led to efficient KO of CD2 with high specificity in CD8+ T cells, and for both the L-CHIME and I-CHIME systems T cell activation improves KO efficiency.
S-CHIME enables sequential KO of paired genes
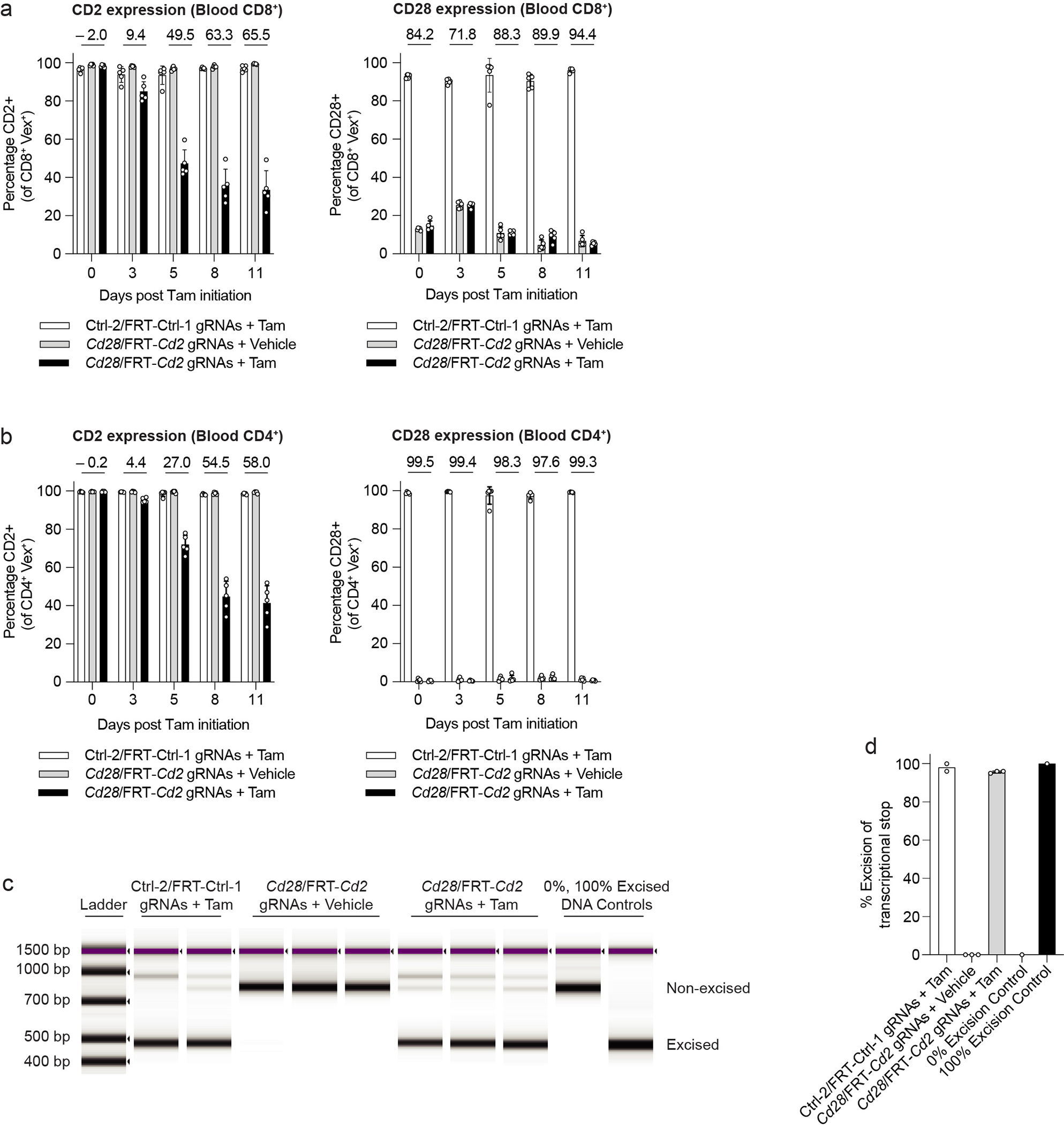
While C-CHIME allows for epistasis studies of paired genes in vivo, there are instances where double knockout of two genes can lead to synthetic lethality or different outcomes depending on the sequence or timing of deletion. To overcome this problem, we developed a sequential knockout system (S-CHIME), in which one gene is constitutively deleted while the other gene can be temporally deleted with the use of Tamoxifen, as in the I-CHIME system. The S-CHIME system uses a vector containing two gRNAs, where only the first tracrRNA contains an FRT-stop-FRT sequence (Figures 3a, 3b), and Rosa26-FlpO-ERT2 H11-Cas9 donor LSK cells (Figure 3b), so expression of the first gRNA can be induced upon Tamoxifen administration. We targeted Cd2 and Cd28 on T cells, assessing inducible KO of CD2 and constitutive KO of CD28. To examine the deletion kinetics of CD2 using S-CHIME, we treated BMCs with Tamoxifen or vehicle control and analyzed CD2 expression over time with serial bleeds (Figure 3c). The Tamoxifen treated group containing the Cd2 gRNA had a 65% decrease in CD2 expression at day 11 after Tamoxifen initiation in blood CD8+ and CD4+ T cells (Extended Data Figures 4a, 4b). CD2 was not reduced in the presence of the vehicle, indicating the system is tightly controlled (Extended Data Figures 4a, 4b). There was ~90% reduction in expression of CD28 (Extended Data Figures 4a, 4b) and 60% DKO of CD2 and CD28 on CD8+ and CD4+ T cells (Figure 3d). Comparable KO efficiencies were observed on splenic (Figures 3e–3j) and blood (Figure 3d and Extended Data Figures 4a, 4b) CD4+ and CD8+ T cells. We also assessed excision of the transcriptional stop in the S-CHIME vector and found ~100% excision (Extended Data Figures 4c, 4d), consistent with the results observed with the I-CHIME and L-CHIME vectors (Figure 2h and Extended Data Figures 2j, 3h, 3i). Thus, S-CHIME enables efficient, constitutive KO of one gene and inducible KO of a second gene in vivo.
Figure 3: S-CHIME enables sequential constitutive and inducible knockout of paired genes.
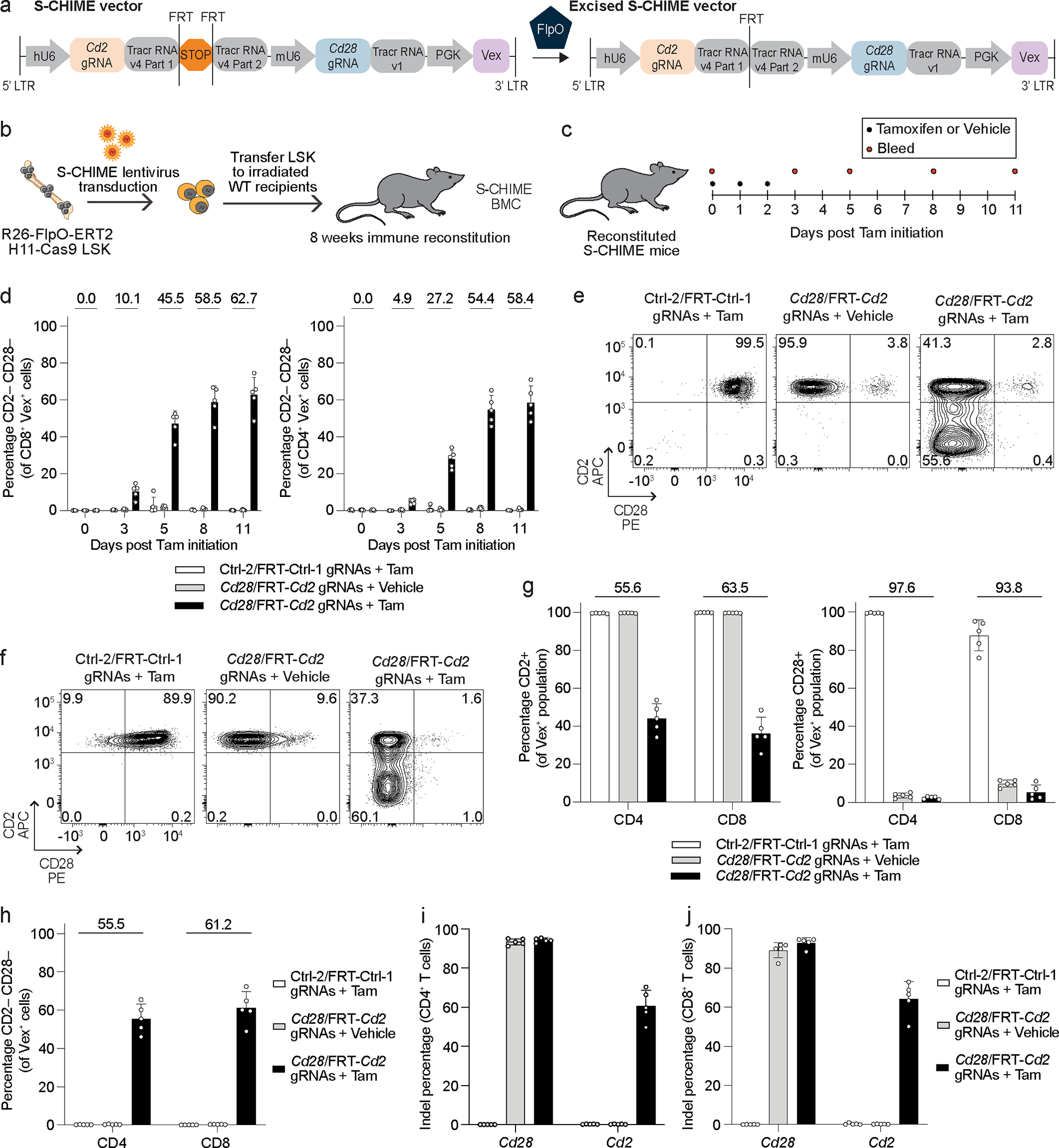
(a) Schematic of S-CHIME vector before and after FlpO-mediated excision. (b) Schematic for creation of S-CHIME BMCs. (c) Timeline of Tamoxifen treatment and serial bleeds of S-CHIME BMCs. (d) Quantification of the percentage of cells lacking expression of CD2 and CD28 on CD8+ (left) and CD4+ (right) Vex+ peripheral blood cells following the experimental treatment timeline (c). Repeat measurements were taken from the same mice. (e, f) Representative flow cytometry plots of CD2 and CD28 expression on (e) CD4+ Vex+ and (f) CD8+ Vex+ splenocytes from S-CHIME BMCs with non-targeting control gRNAs (one constitutive and one inducible) or Cd2 (inducible) and Cd28 (constitutive) gRNAs, 11 days after initiation of Tamoxifen or vehicle control treatment. (g) Quantification of CD2 expression (left) or CD28 expression (right) on CD4+ Vex+ and CD8+ Vex+ splenocytes as in (e, f). (h) Quantification of the percentage of cells lacking expression of both CD2 and CD28 on CD4+ Vex+ and CD8+ Vex+ splenocytes as in (e, f). (i, j) Quantification of indels in Cd2 following next-generation sequencing of (i) Vex+ CD4+ and (j) Vex+ CD8+ splenocytes as in (e, f). Data are representative of two experiments with at least n = 5 mice per group. Bar graphs represent the mean and error bars represent s.d. Calculated knockout efficiency is denoted above bars for indicated comparisons.
DKO of PTPN1 and PTPN2 leads to bone marrow hypoplasia
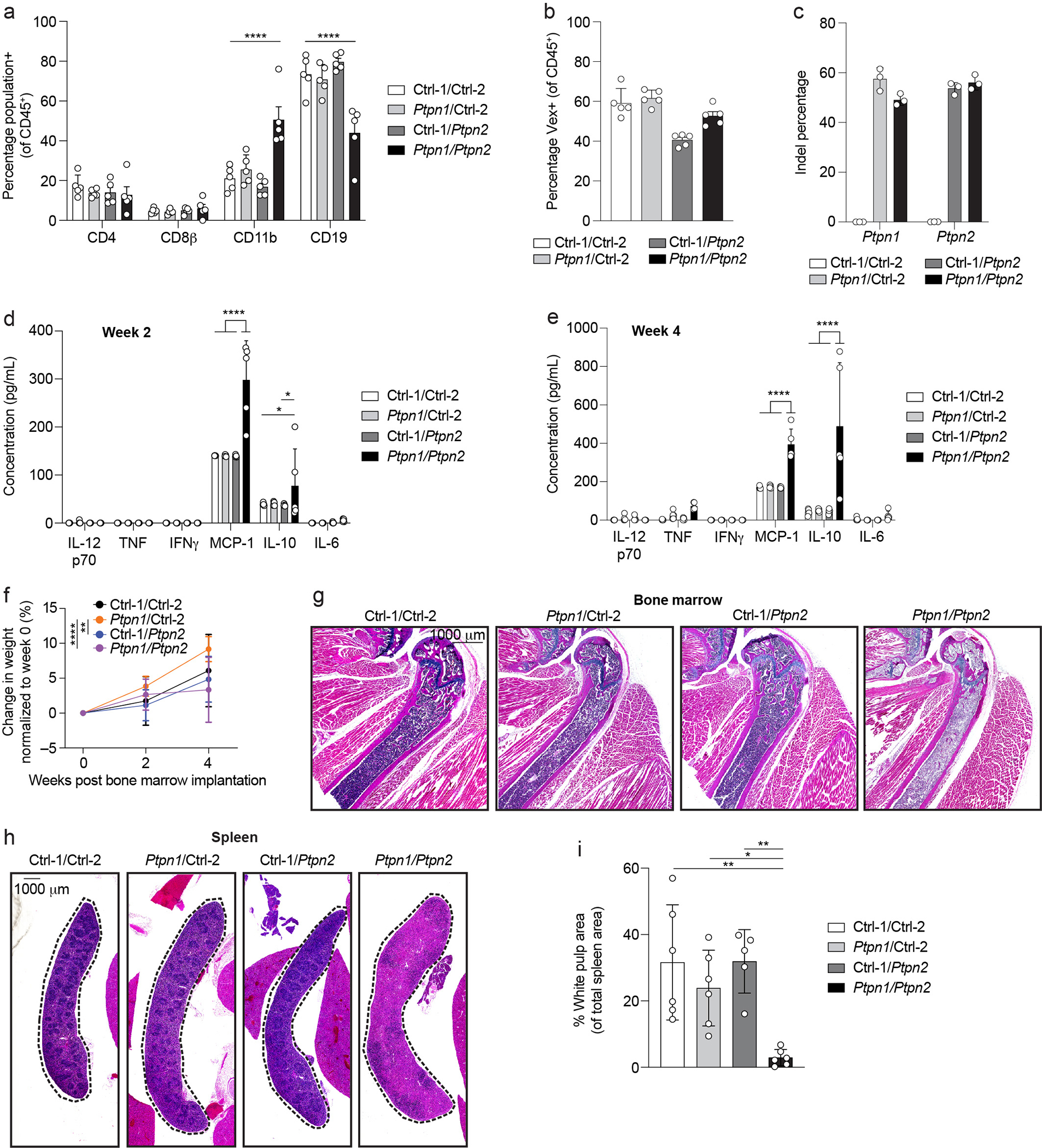
Embryonic lethal single genes or synthetic lethal pairs of genes are difficult to study in mice because they preclude analysis post embryogenesis. We hypothesized that X-CHIME could be a useful platform to bypass this issue because X-CHIME (1) only deletes genes in the hematopoietic compartment and (2) is performed in adult mice, enabling studies post embryogenesis. DKO of PTPN1 and PTPN2 is embryonic lethal24, but whether this phenotype is hematopoietic cell-intrinsic33 is not clear. We used C-CHIME to create BMCs with Ptpn1 and Ptpn2 gRNAs (Figure 4a). Between four and five weeks after bone marrow implantation, the PTPN1/PTPN2 DKO BMCs spontaneously died (Figure 4b). This lethality was associated with a significant increase in the percentage of blood CD11b+ cells and a significant decrease in the percentage of blood CD19+ cells (Extended Data Figure 5a). Vex expression and KO analyses revealed ~50% transduction across the groups and ~50% KO of PTPN1 and PTPN2, indicating that KO of PTPN1 and PTPN2 did not selectively disadvantage cells in the blood (Extended Data Figures 5b, 5c). PTPN1/PTPN2 DKO BMCs had elevated levels of the proinflammatory marker MCP-1 and the anti-inflammatory marker IL-10, at two- and four-weeks post bone marrow implantation (Extended Data Figures 5d, 5e), suggesting systemic inflammation. The weights of PTPN1/PTPN2 DKO BMCs did not significantly differ from Control-1/Control-2 BMCs, indicating weight loss is not associated with lethality of the PTPN1/PTPN2 DKO BMCs (Extended Data Figure 5f). Necropsies at approximately four weeks post bone marrow implantation identified atrophic thymi and anemic livers in PTPN1/PTPN2 DKO BMCs (Supplementary Table 1) as well as significant decreases in leukocytes and hematopoiesis in the bone marrow of PTPN1/PTPN2 DKO BMCs (Figures 4c–4e, and Extended Data Figure 5g), consistent with PTPN2 germline KO mice33,34. Moreover, the PTPN1/PTPN2 DKO BMC spleens were devoid of white-pulp regions compared to controls (Extended Data Figures 5h, 5i), consistent with expanded red-pulp regions in PTPN2 KO animals33.
Figure 4: Constitutive deletion of PTPN1 and PTPN2 leads to lethality associated with bone marrow hypoplasia.
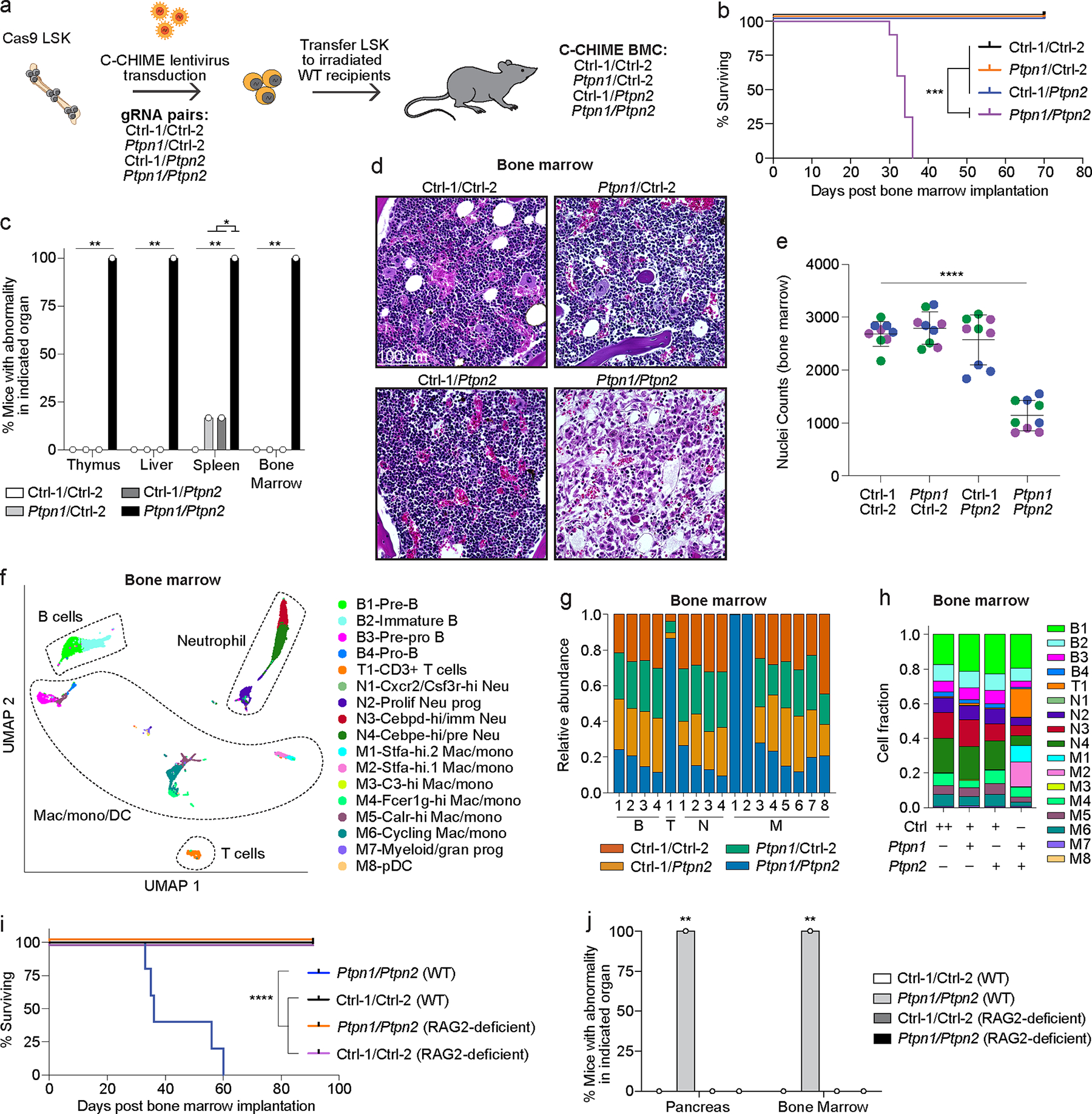
(a) Schematic for creation of control/control, control/Ptpn1, control/Ptpn2, and Ptpn1/Ptpn2 gRNA-containing BMCs. (b) Survival curve of BMCs as in (a) (p = 0.0001). (c) Quantification of pathologic findings pooled from two independent experiments for organs in which there was a significant increase in pathology in the BMCs. (d) Representative hematoxylin and eosin (40x magnification) images of BMC bone marrow sections four weeks after bone marrow implantation. Scale bar indicates 100 μm. (e) Quantification of cell nuclei in (d) per image. Dots of the same color reflect quantification of 3 representative images per mouse and the distinct colors represent replicate animals (p < 0.0001). (f) UMAP clustering of 10x scRNAseq from bone marrow samples of BMCs as in (d). (g, h) Quantification of cell cluster proportions within each cluster (g) or within each BMC group (h). The x-axis in (g) corresponds to the clusters B1–4, T1, N1–4, and M1–8 described in (f). Note ++ indicates two control gRNAs (h). (i) Survival curve of BMCs created by nucleofecting either WT or RAG2-deficient bone marrow stem cells with two control gRNAs or Ptpn1/Ptpn2 gRNAs, followed by implantation of WT bone marrow stem cells into WT recipients and RAG2-deficient bone marrow stem cells into RAG2-deficient recipients (p < 0.0001). (j) Quantification of pathologic findings for the pancreas and bone marrow from BMCs in (i) (p = 0.0079). Data are representative of two experiments with at least n = 3 mice per group except for the 10x scRNAseq data, which was one experiment with n = 3 mice per group. Bar graphs represent the mean and error bars represent s.d. Statistical significance was assessed by a two-sided Mantel-Cox test (b, i), two-sided Fisher’s exact test (c, j), or one-sided one-way ANOVA (e).
To further characterize the cellular changes associated with PTPN1/PTPN2 DKO BMCs, we performed 10x scRNAseq on Vex+ CD45+ cells, which are enriched for KO and DKO cells, in the bone marrow and spleen, four weeks after bone marrow implantation. UMAP clustering (Supplementary Table 2) identified clusters from major immune lineages, including B cells, T cells, macrophages, monocytes, and neutrophils (Figure 4f and Extended Data Figures 6a–6c). In addition, there were three new clusters in the bone marrow (two Stfa-expressing myeloid clusters and a CD3+ cluster) specific to the PTPN1/PTPN2 DKO BMC samples (Figures 4g, 4h). The spleens of PTPN1/PTPN2 DKO BMCs had a cluster of Stfa-expressing neutrophils (Extended Data Figures 6b, 6d, 6e) as well as a Granzyme-expressing CD8+ T cell effector cluster (Extended Data Figures 6b–6e).
To examine the contribution of T cells to the lethality of PTPN1/PTPN2 DKO BMCs, we nucleofected wild-type (WT) or RAG2-deficient bone marrow stem cells with Cas9-gRNA ribonucleoprotein complexes targeting Ptpn1 and Ptpn2 (or controls). Here we used a nucleofection-based approach as we do not have Cas9-expressing RAG2-deficient mice for standard lentiviral-based approaches. While PTPN1/PTPN2 DKO BMCs on the WT background died (Figure 4i), albeit with slightly delayed kinetics compared to the lentiviral system (Figure 4b), PTPN1/PTPN2 DKO BMCs on the RAG2-deficient background were viable, suggesting a requirement for B and T cells for the lethality observed. Necropsies of these BMCs revealed that PTPN1/PTPN2 DKO BMCs on the WT background had atrophic pancreases and bone marrow, whereas the other groups did not (Figure 4j). Notably, pancreatic atrophy was not observed with the lentiviral C-CHIME system, perhaps due to the differences in the kinetics of the lethality. In summary, C-CHIME enabled assessment of PTPN1/PTPN2 DKO in the bone marrow post embryogenesis and demonstrated that there is a hematopoietic cell-intrinsic component to the lethality observed following DKO of PTPN1 and PTPN2, likely mediated by B and T cells,.
Sequential DKO of PTPN1 and PTPN2 leads to enteritis
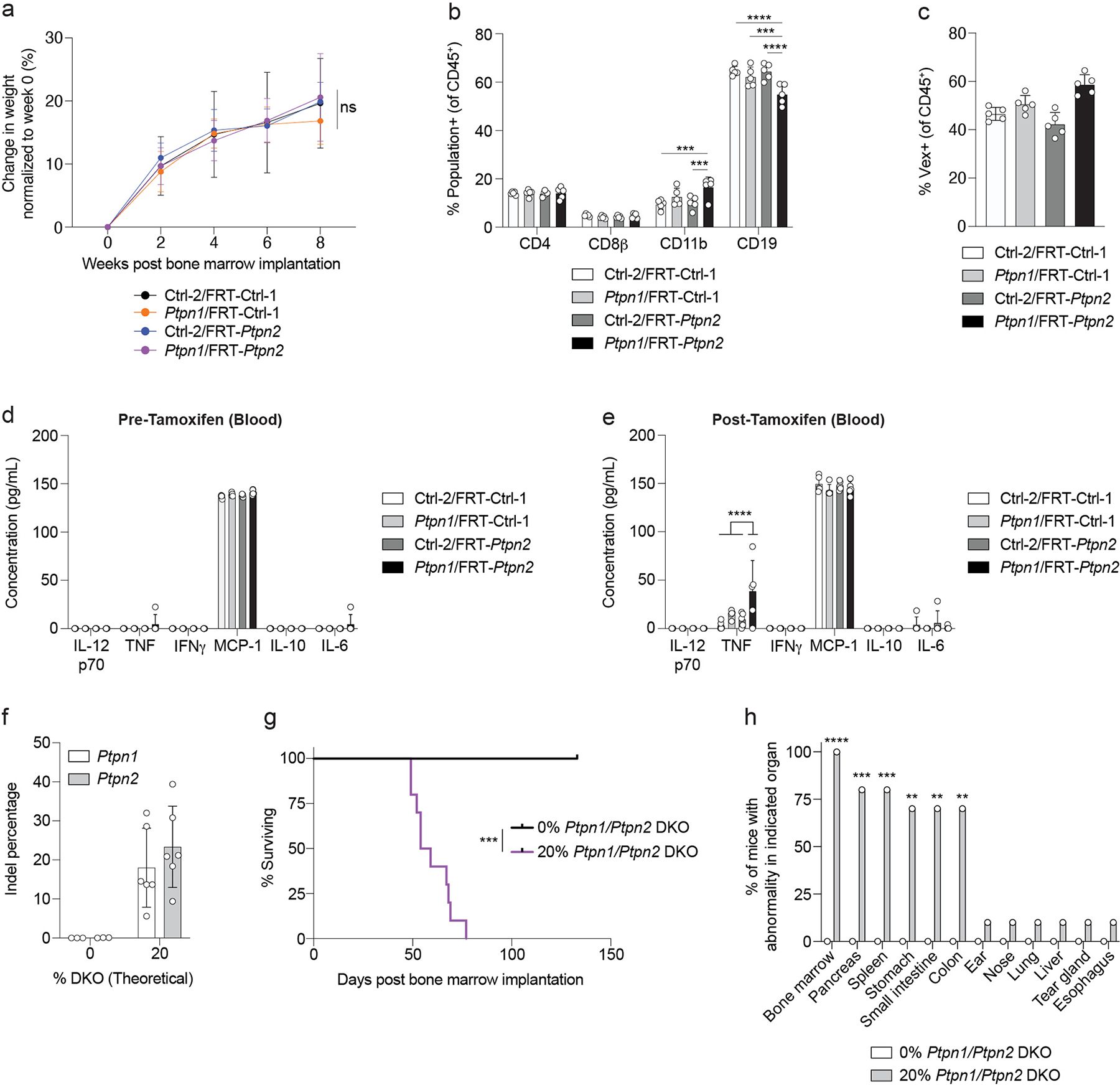
Constitutive PTPN1 and PTPN2 DKO during immune development leads to bone marrow hypoplasia and lethality, but it is unclear if PTPN1 and PTPN2 are required for maintenance of a mature immune system. Thus, we asked if sequential DKO of PTPN1 and PTPN2 after immune development would avert the lethality observed following constitutive DKO of PTPN1 and PTPN2. We created BMCs using our S-CHIME system with constitutive expression of control or Ptpn1-targeting gRNAs paired with inducible expression of control or Ptpn2-targeting gRNAs (Figure 5a). We monitored the weight of the BMCs during hematopoietic reconstitution and did not observe significant weight loss (Extended Data Figure 7a). We treated reconstituted BMCs with Tamoxifen to induce expression of the control or Ptpn2 gRNAs and monitored their weights. PTPN1/PTPN2 DKO BMCs exhibited a significant decrease in weight compared to controls after Tamoxifen treatment (Figure 5b). A subset of the PTPN1/PTPN2 DKO BMCs died within a week of Tamoxifen administration without reaching a weight loss endpoint (Figure 5c). Given the rapid deterioration of these mice, we monitored KO efficiency in peripheral blood cells 7 days after Tamoxifen initiation, instead of at the peak of KO efficiency, typically observed 11 days post Tamoxifen initiation. PTPN2 KO efficiency (inducible KO) was ~20% and PTPN1 KO efficiency was ~70% (Figure 5d). Thus, the PTPN1/PTPN2 DKO BMCs rapidly lost weight and died despite a low overall DKO efficiency (maximum DKO% of 20%) at this time point. Importantly, we did not observe significant morbidity or mortality in control, PTPN1 KO, or PTPN2 KO BMCs, indicating the weight loss and lethality in the PTPN1/PTPN2 DKO BMCs were not due to Tamoxifen administration or FlpO-ERT2 activation35,36.
Figure 5: Constitutive deletion of PTPN1 and inducible deletion of PTPN2 in hematopoietic cell reconstituted mice leads to enteritis and lethality.
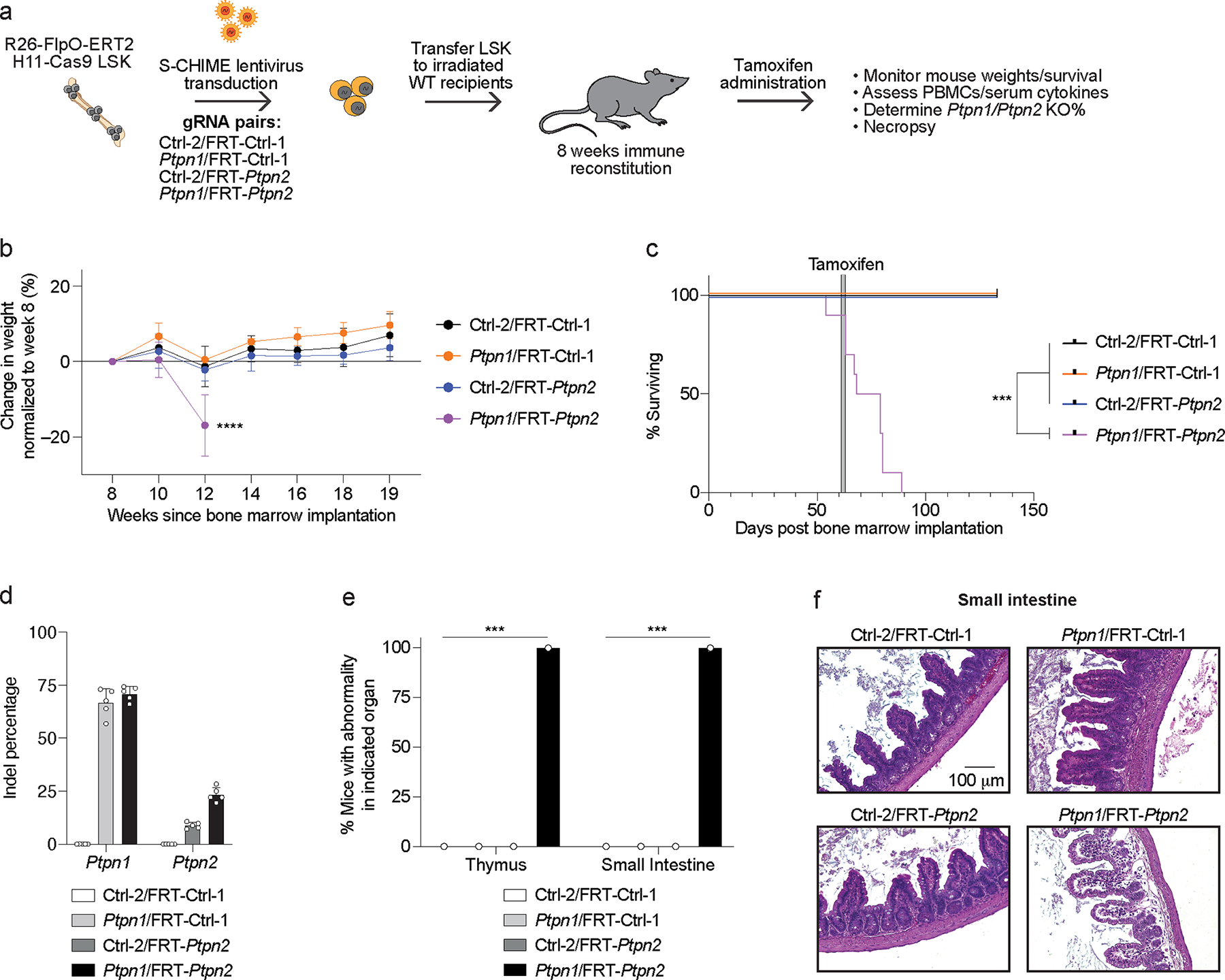
(a) Schematic for creation of Ctrl-2/FRT-Ctrl-1, Ptpn1/FRT-Ctrl-1, Ctrl-2/FRT-Ptpn2, and Ptpn1/FRT-Ptpn2 gRNA-containing BMCs. (b) Weight loss curves normalized to week 8 weight after bone marrow implantation for BMCs as in (a). Serial measurements were taken from the same mice (p < 0.0001). (c) Survival curve of BMCs as in (a). Time of Tamoxifen administration is indicated by the gray bar (p = 0.0006). (d) Quantification of indels in Ptpn1 and Ptpn2 following next-generation sequencing of peripheral blood cells from BMCs 7 days after Tamoxifen initiation. (e) Quantification of pathologic findings pooled from two independent experiments for organs in which there was a significant increase in pathology in the BMCs. (f) Representative hematoxylin and eosin (20x magnification) images of the small intestine 2 weeks after Tamoxifen treatment of BMCs. Scale bar indicates 100 μm. Data are representative of two experiments with at least n = 3 mice per group. Bar graphs represent the mean and error bars represent s.d. Statistical significance was assessed by one-sided one-way ANOVA (b), two-sided Mantel-Cox test (c), or two-sided Fisher’s exact test (e).
To determine the mechanism of the weight loss and lethality, we monitored the peripheral blood for population changes and found an increase in CD11b+ cells and a decrease in CD19+ cells (Extended Data Figure 7b), similar to constitutive deletion of PTPN1/PTPN2 (Extended Data Figure 5a). Peripheral blood leukocytes in the different BMC groups had similar Vex expression, suggesting DKO of PTPN1 and PTPN2 did not selectively disadvantage immune populations in the blood (Extended Data Figure 7c). We also assessed serum cytokines before and after Tamoxifen administration and found a subtle but significant increase in TNF post treatment (Extended Data Figures 7d, 7e). Necropsies revealed that PTPN1 and PTPN2 DKO S-CHIME BMCs had significant inflammation and edema of the small intestine (Figures 5e, 5f), which may explain the rapid weight loss observed (Figure 5b). Notably, the lack of bone marrow pathology contrasted with the constitutive DKO of PTPN1/PTPN2 (Figures 4c–4e).
Given the different phenotypes observed between constitutive vs. sequential PTPN1/PTPN2 DKO, we sought to rule out a technical difference due to the predicted maximal percentage of DKO (~50% with the constitutive DKO system compared with ~20% with the sequential DKO system). We created BMCs with PTPN1/PTPN2 DKO bone marrow stem cells mixed with unedited bone marrow stem cells, such that the resulting BMCs were ~20% constitutively PTPN1/PTPN2 DKO (Extended Data Figure 7f). 20% constitutive DKO of PTPN1/PTPN2 was lethal, but with delayed kinetics (Extended Data Figure 7g) compared to 50% constitutive DKO (Figure 4b). Consistent with the 50% constitutive DKO BMCs, the 20% DKO BMCs had bone marrow atrophy (Extended Data Figure 7h). Pancreatic atrophy and inflammation were found in several additional organs, including the stomach, small intestine, and colon (Extended Data Figure 7h). Collectively, these data suggest that the BMCs with lower penetrance of constitutive DKO of PTPN1/PTPN2 show features characteristic of both the constitutive (bone marrow atrophy) and sequential (enteritis) DKO BMCs.
Discussion
Genetic perturbation of immune cells has revealed many mechanisms of immunity1–3. However, the use of genetic perturbation is limited by the temporal and lineage-specific functions of genes, as well as gene buffering and redundancy. To understand gene functions in vivo, a suite of tools that enable inducible, lineage-specific, combinatorial, and sequential gene KOs is needed. Traditional germline KO/cKO approaches require time-consuming crosses and are non-modular. Moreover, CRISPR-based tools for inducible, combinatorial, and sequential gene KO have been benchmarked in cancer cells in vitro but remain untested in more complex contexts, such as primary immune cells in vivo. To address these issues, we developed X-CHIME: four modifications of CHIME, that enable rapid combinatorial, inducible, lineage-specific, and sequential KO of genes in hematopoietic cells. Deletion of PTPN1 and PTPN2 using X-CHIME served as a proof-of-concept to illustrate how distinct mechanistic insights can be obtained with single vs. double KO and constitutive vs. inducible KO. Collectively, our findings demonstrate the power of the X-CHIME platform for combinatorial and temporal perturbation of genes in hematopoietic cells and suggest that PTPN1 and PTPN2 have functional redundancy in adult mice that is required for viability.
Lentivirus-based combinatorial KO approaches have not been optimized for use in diverse immune lineages in vivo. The C-CHIME system fills this gap by enabling efficient DKO of two genes, in diverse immune lineages: 91% in CD8+ and CD4+ T cells and 50% in macrophages. Future work is needed with a larger set of targeted genes to determine whether these differences in KO efficiency are gene/gRNA-intrinsic or cell-intrinsic due to potentially different levels of Cas9/gRNA expression or fidelity of DNA repair. Moreover, by creating KOs in an intact immune system, X-CHIME enables in vivo assessment of genes in disease models such as cancer or viral infection12,15. In addition to lentiviral approaches to KO genes, nucleofection of Cas9-gRNA ribonucleoprotein complexes into immune cells is an orthogonal approach to rapidly KO genes in hematopoietic cells37,38. Nucleofection and lentiviral approaches are complementary: here we used nucleofection to KO genes in a preexisting KO mouse strain, bypassing time-consuming crosses. However, because nucleofection transiently delivers the gRNA without integration, it cannot be readily used for sequential KO or pooled screening approaches39. C-CHIME and S-CHIME solve this problem through lentiviral integration of the gRNAs, thus enabling both sequential KO and screening studies. C-CHIME and S-CHIME are dynamic new tools that complement existing approaches used to modify hematopoietic cells in vivo.
Temporal control of gene knockout enables analysis of the function of genes at distinct times, evaluation of genes essential for mouse development, and modeling therapeutic inhibition of proteins. Inducible approaches utilizing baseline repression of a gRNA or Cas9 expression that can be relieved with an inducing agent are often leaky, leading to CRISPR-mediated indel formation in the absence of the inducing agent40–43. CRISPR-Switch-On utilizes a gRNA-expression vector that is “Off” prior to induction and can be induced via Cre or Flp-mediated excision of a transcriptional stop in the tracrRNA14, thus circumventing leakiness and immunogenicity concerns. Adaptation of the Cre-based CRISPR-Switch-On system to immune cells revealed low editing efficiency, highlighting the importance of benchmarking and optimizing CRISPR-based systems in immune cells. Thus, we optimized a Flp-based system to study inducible deletion of genes in hematopoietic cells in vivo, which led to ~50% indel formation, despite ~100% excision of the transcriptional stop. We suspect that in our L-CHIME and I-CHIME systems Cre and Flp bind to residual loxP and FRT sites following excision of the transcriptional stop, hindering transcription of the gRNA and subsequent indel formation. In vitro and in vivo stimulation significantly improved KO efficiency, which may be the result of Cre/Flp dissociating from the loxP/FRT sites during proliferation. We further modified the I-CHIME system to enable sequential deletion of genes in hematopoietic cells without leakiness using S-CHIME. To facilitate future vector designs where both gRNAs can be induced, the S-CHIME vector contains two gRNA-expression cassettes which may enable studies to model therapeutic inhibition of two proteins in vivo.
DKO of PTPN1 and PTPN2 is lethal in mice24,33, but the role of the immune system in this phenotype is unclear. Our work shows that the lethality observed in these mice has an immune-intrinsic component, characterized by changes in bone marrow cellularity, thymic atrophy, and decreased splenic white pulp areas, consistent with phenotypes seen in Ptpn2 KO mice and Ptpn2+/− Ptpn1−/− KO mice24,33,34. Furthermore, the lethality observed following DKO of PTPN1 and PTPN2 requires B and T cells, suggesting that the novel lymphoid populations seen in PTPN1/PTPN2 DKO BMCs may be directly causing the lethality. In contrast, sequential DKO of PTPN1/PTPN2 in adult mice led to enteritis, weight loss, and death, but was not associated with bone marrow defects, consistent with the association of single-nucleotide polymorphisms (SNPs) in PTPN2 with Crohn’s disease and ulcerative colitis44,45. DKO of PTPN1 and PTPN2 in ~20% of the immune system prolonged the lifespan of the BMCs but ultimately led to lethality associated with bone marrow atrophy as well as enteritis, suggesting PTPN1 and PTPN2 may be required for defined windows in specific cell types during immune development. Despite the lethality we observed following DKO of PTPN1 and PTPN2, two recent studies demonstrated that therapeutic inhibition of PTPN1 and PTPN2 leads to immune-mediated tumor control in mice in the absence of toxicity19,20, suggesting that therapeutic inhibition of PTPN1 and PTPN2 may avoid the significant toxicities associated with DKO. Collectively, the distinct phenotypes observed with single KO vs. DKO, 50% DKO vs. 20% DKO, and constitutive DKO vs. inducible DKO demonstrate the complex functions of PTPN1 and PTPN2 in immunity. Together, these findings further illuminate PTPN1 and PTPN2 biology in vivo and demonstrate the power of X-CHIME as a modular platform to rapidly assess genes both combinatorially and temporally for biological and therapeutic studies in hematopoietic cells.
Methods:
Mouse strains
Seven- to ten-week-old male and female mice were used as bone marrow recipients for all experiments, and seven- to fourteen-week-old male and female mice were used as donors for BMC experiments. Wild-type C57BL/6J mice (Jax# 000664) were purchased from the Jackson Laboratory. Rosa26-Cas9-expressing (Cas9ON) mice12 were used for all C-CHIME experiments. For the I-CHIME and S-CHIME studies we crossed H11-Cas9 mice (B6.129(Cg)-Igs2tm1.1(CAG-Cas9*)Mmw/J Jax# 028239)30 to Rosa26-FlpO-ERT2 mice (B6N.129S6(Cg)-Gt(ROSA)26Sortm3(CAG-flpo/ERT2)Alj/J Jax# 019016)29. We then crossed to homozygosity for H11-Cas9. In the Cre-based version of the I-CHIME system, we crossed the Cas9ON strain to UBC-Cre-ERT2 mice (B6.Cg-Ndor1Tg(UBC-cre/ERT2)1Ejb/1J Jax# 007001)46. We then crossed to homozygosity for Rosa26-Cas9. For the L-CHIME studies, we crossed the Cas9ON strain to E8I-Cre mice (C57BL/6-Tg(Cd8a-cre)1Itan/J Jax# 008766)32, and then crossed to homozygosity at both alleles. RAG2-deficient mice (B6.Cg-Rag2tm1.1Cgn/J Jax# 008449) were purchased from the Jackson Laboratory. Mice were genotyped using PCR or Transnetyx probes. No statistical methods were used to pre-determine sample sizes but our sample sizes are similar to those reported in previous publications12,15. Data exclusion was not used. Data collection and analysis were not performed blind to the conditions of the experiments. Age and sex-matched animals were used for each experiment. For BMC experiments, LSK donor and LSK recipients were sex-matched. All attempts to reproduce our findings were successful. Randomization was not used as groups were predetermined when BMCs were created. All experimental mice were housed in specific pathogen-free conditions with a 12-hour light cycle and a 12-hour dark cycle, 30–70% relative humidity, and 21.1°C ambient temperature. Mice were used in accordance with animal care guidelines from the Harvard Medical School Standing Committee on Animals and the National Institutes of Health (Protocol number: IS00000041-6).
Guide RNA design and cloning
gRNAs were designed using the Broad CRISPR algorithm47. gRNAs for single knockout studies were cloned into our lentiviral vectors using BsmBI (NEB Cat# R0580L) and BfuAI (NEB Cat# R0701L) restriction digests. Plasmids were amplified in Stbl3 bacteria (Invitrogen Cat# C737303) and extracted using a HiSpeed Plasmid Maxi kit (Qiagen Cat# 12662). The following gRNAs were used:
Control-1 gRNA: 5’-GCGAGGTATTCGGCTCCGCG-3’
Control-2 gRNA: 5’-GCTTTCACGGAGGTTCGACG-3’
Control-3 gRNA: 5’-ATTGTTCGACCGTCTACGGG-3’
Cd2 gRNA: 5’-CCGATGATGAGAAACGACAG-3’
Cd28 gRNA: 5’-GCTTGTGGTAGATAGCAACG-3’
Fcgr1 gRNA: 5’-AGAGTACCATATAGCAAGGG-3’
Mertk gRNA: 5’-AGGTACGGTTAGGACAGACG-3’
Ptpn1 gRNA: 5’-GGTGCAATTTAATCCGACTG-3’
Ptpn2 gRNA: 5’-CTCACTTCCATTATACCACC-3’
Plasmids
For single gene knockout studies, we utilized pXPR_053 (Addgene ID: 113591). For C-CHIME studies we utilized pXPR_219 (Addgene ID: 164559). For I-CHIME studies we utilized pXPR_070 (Addgene ID: 164265) and pXPR_068 (Addgene ID: 164555). For L-CHIME studies we also utilized pXPR_070. For S-CHIME studies we utilized pXPR_220 (Addgene ID: 164560). See Supplementary Note 1 for a detailed description of plasmids.
Cell lines
MB49 cells (Sigma-Aldrich Cat# SCC148), MC38 (gift from Dario Vignali, University of Pittsburgh), and 293x cells (gift from Cigall Kadoch, Dana Farber Cancer Institute) were cultured in DMEM+/+/+ (Gibco Cat# 11995073) supplemented with 10% FBS (Sigma Cat# F2442) and 1% penicillin/streptomycin (VWR Cat# 97063-708). 293x cells (HEK variant, which are a commonly misidentified cell line) were authenticated by producing high-titer lentivirus. All cell lines were confirmed to be mycoplasma negative and free of standard pathogens.
Lentivirus production and titer
293x cells were transfected using polyethylenimine (Polysciences Cat# 24765-2) with the packaging plasmids pMD2.G (Addgene ID: 12259) and psPAX2 (Addgene ID: 12260), and gRNA-containing plasmids. Lentivirus-containing supernatant was collected 72 hours post-transfection and spun at 854g for 5 minutes to remove cells. The supernatant was filtered through a 45 μm low-protein binding filter (Whatman Cat# EW-29705-54), placed in ultracentrifuge tubes (Beckman Coulter Cat #344058), and spun at 20,000 RPM (71,934.8g) at 4°C for 2 hours in a Beckman Coulter ultracentrifuge with an SW28 rotor. Concentrated lentivirus was then resuspended in SFEM media (STEMCELL Technologies Cat# 9600) overnight before freezing at −80°C. Lentivirus was titered on 293x cells to determine viral particles per mL.
Bone marrow isolation and bone marrow chimera setup
Femurs, tibias, pelvises, and vertebral columns were isolated from Cas9-expressing (Cas9ON) donor mice and crushed with a mortar and pestle to liberate bone marrow. Bone marrow was filtered twice through a 70 μm filter and then enriched using CD117 microbeads (Miltenyi Biotec Cat# 130-091-224). CD117-enriched bone marrow was stained with CD117-APC (different clone than microbeads), lineage-PE (Gr1, CD11b, CD5, CD3ε, Ter119, B220), and Sca1-BV421 antibodies. Cells were sorted on a BD Aria IIu. Sorted cells were then placed in a 37°C incubator overnight in SFEM media containing TPO (100 ng/mL) (VWR Cat# 10770-954), SCF (100 ng/mL) (VWR Cat# 10773-132), Flt3 ligand (50 ng/mL) (VWR Cat# 10780-340), IL-7 (50 ng/mL) (VWR Cat# 10780-184), and 1% penicillin/streptomycin (VWR Cat# 97063-708) at a concentration of 150,000–250,000 cells per mL. The next day, LSK cells were spin transduced (692g, 27°C, 1.5 hours) on Retronectin (Takara Bio Cat# T100B) coated plates with lentivirus at a multiplicity of infection (MOI) of 30. This MOI was chosen such that LSK transduction would be approximately 30–40%. Following the spin, cells were supplemented with additional SFEM media containing cytokines (TPO, SCF, Flt3 ligand, and IL-7) and penicillin/streptomycin and placed back into a 37°C incubator overnight. The next morning cells were checked for viability and absence of contamination. CD45.2+ wild type recipients were then irradiated two times spaced 3–6 hours apart with 600 rad per time (1200 rad total) using a Cesium-137 irradiator, and ~50,000 LSK cells were transferred intravenously via the tail vein into irradiated recipients. Mice were administered Sulfatrim antibiotic (0.8 mg/mL Sulfamethoxazole and 0.16 mg/mL Trimethoprim) (Pharmaceutical Associates, Inc. Cat# NDC 00121-0854-16) in their drinking water for one week. Mice were then allowed to reconstitute a minimum of 8 weeks prior to use in experiments.
Extended LSK cell culture
For some experiments the LSK were cultured in vitro for up to one week to facilitate sorting on the Vex transduction marker. This was done by following the bone marrow isolation and bone marrow chimera setup protocol above with the following change: Polyvinyl alcohol-containing (Sigma-Aldrich Cat# P8136-250G) media was used as previously described48. Following up to one week of culture, cells were sorted for Vex expression on a BD Aria IIu and injected into irradiated recipients.
Flow cytometry and cell sorting
Samples were stained for 30 minutes on ice in the dark with antibodies in FACS buffer, containing PBS–/– (VWR Cat# 82020-066) with 2 mM EDTA (VWR Cat# 45001-122) and 1% FBS. Following staining, cells were washed twice with FACS buffer. Cells were then resuspended in FACS buffer and either (1) analyzed on the following instruments: BD LSR II, BD Symphony A5, BD Celesta, or Beckman Coulter CytoFLEX or (2) sorted on a BD Aria IIu. CytExpert Version 2.3.1.22 and FACSDIVA Versions 9.0.1 and 9.4 software were used for data collection. Compensation was performed with single color controls. Gating was performed using fluorescence-minus-one (FMO) controls where necessary. Samples were analyzed using Flowjo Version 10.8.1 software.
Antibodies and dyes used for flow cytometry
Antibodies from Biolegend: B220, CD11b, CD19, CD3ε, CD2, CD4, CD45, CD5, CD8α, CD8β, c-Kit, FcγRI, F4/80, Gr-1, Isotype IgG2b κ, Ly6c, MERTK, Sca-1, Ter-119 (1:100 dilution); CD28 (1:200 dilution); Streptavidin and TruStain fcX (1:50 dilution). Near-IR Fixable Live/Dead (1:500 dilution) is from Thermo Fisher Scientific. Please see Reporting Summary for additional details on antibody clones and catalog numbers.
Spleen and lymph node processing
Spleens were smashed through a 70 μm filter to create single cell suspensions. T cells were enriched from spleens using CD90.2 microbeads (Miltenyi Biotec Cat# 130-121-278). Alternatively, spleens were lysed with ACK lysing buffer (Quality Biological Cat# 118-156-721) for 1 minute at 22°C. Lymph nodes were isolated and smashed through a 70 μm filter to create single cell suspensions.
Calculation of KO%
Single gene:
100*(Average % of cells that are Gene− in the target gRNA sample – Average % of cells that are Gene− in the control gRNA sample) / (100 – Average % of cells that are Gene− in the control gRNA sample)
Two genes:
100*(Average % of cells that are Gene1− Gene2− in the target gRNA sample – Average % of cells that are Gene1− Gene2− in the control gRNA sample) / (100 – Average % of cells that are Gene1− Gene2− in the control gRNA sample)
Calculation of DKO% of all KO cells
100*(Average % of cells that are Gene1− Gene2− in the target gRNA sample) / [(Average % of cells that are Gene1− Gene2− in the target gRNA sample) + (Average % of cells that are Gene1− Gene2+ in the target gRNA sample) + (Average % of cells that are Gene1+ Gene2− in the target gRNA sample)]
Indel analysis
DNA was isolated from a minimum of 15,000 cells using a DNeasy Blood & Tissue kit (Qiagen Cat# 69504), and a PCR flanking the region of the gRNA edit was performed. The overall PCR product was ~220 bp and the potential edit was within 100 bp of one end of the PCR product. This product was purified using a QIAquick PCR purification kit (Qiagen Cat# 28106). Samples were then submitted to the MGH CCIB DNA Core Facility at Massachusetts General Hospital (Cambridge, MA) for the addition of sequencing adapters and sequencing on a MiSeq. Following sequencing, indel formation was analyzed using the CRISPR Ebert Pipeline on basepairtech.com with gRNA-specific bed files. To perform indel analysis from blood, ~250 μL of blood was collected via retro-orbital bleed and lymphocytes isolated using a Histopaque 1083 (Sigma-Aldrich Cat# 10831-6X100ML) gradient spin (854g, 23°C, 20 minutes). Lymphocytes were then processed as above starting at the DNA isolation step.
Excision PCR
To assess excision of the transcriptional stop from samples transduced with the lentiviral vectors pXPR_068, pXPR_070, and pXPR_220, a PCR was performed using primers flanking this region followed by a PCR cleanup using a QIAquick PCR purification kit. Products were analyzed on an Agilent TapeStation 2200 or 4150 with TapeStation Analysis Software 4.1.1. Using this software, the intensities of the two products were analyzed and normalized by the size of the products. We then calculated the ratio of the excised product intensity over the total product intensity to calculate the % excision. For the PCR of the I-CHIME pXPR_070 samples, we analyzed products on a 1% agarose gel, quantified band intensity using ImageJ, and calculated % excision.
Tamoxifen treatment
Tamoxifen (Sigma-Aldrich Cat# T5648-5G) was dissolved in molecular grade ethanol (Koptec Cat# 64-1-5) to a concentration of 200 mg/mL, then was diluted to a concentration of 10 mg/mL in sunflower seed oil (Sigma-Aldrich Cat# S5007-250ML), and then was sonicated for 60 minutes at 37°C with vortexing every 10 minutes. This solution was then frozen at −20°C. To make the vehicle control, the same steps were performed in the absence of Tamoxifen. For the I-CHIME experiment with the Cre-based system, BMCs were injected intraperitoneally with 1 mg or 2 mg of Tamoxifen or vehicle control daily for up to 5 days. For the I-CHIME Tamoxifen titration experiment with the FlpO-based system, BMCs were injected daily with 1 mg of Tamoxifen for 3 days or 5 days or 2 mg of Tamoxifen for 3 days. For all other I-CHIME and S-CHIME experiments, BMCs were injected daily with 2 mg Tamoxifen or equivalent volume of vehicle intraperitoneally for 3 consecutive days.
Serial bleeding
Chimeric mice to be given Tamoxifen or vehicle were bled via the tail vein prior to the first treatment (day 0), and serially bled on days 3, 5, 8, 11 following the first bleed. ACK lysis was performed, and peripheral blood lymphocytes were stained with antibodies and analyzed on a BD Celesta.
CD8+ T cell culture
Spleens and lymph nodes were isolated from BMCs, naive CD8+ T cells were enriched using Miltenyi’s Naive CD8a+ T cell Isolation Kit (Miltenyi Cat# 130-096-543), and Vex+ CD8+ T cells were sorted.
To keep CD8+ T cells naive, Vex+ CD8+ T cells were cultured for 3–6 days in complete RPMI media [RPMI 1640 with L-glutamine and no Sodium Pyruvate pH 7.0–7.4 (Fisher Scientific Cat# 11-875-093), 10% Fetal Bovine Serum, 1% Penicillin-Streptomycin, 10 mM HEPES (VWR Cat# 97064-360), 55 μM BME (VWR Cat# 97064-588), 1% Sodium Pyruvate (Thermo Fisher Scientific Cat# 11360070), and 1% Non-essential amino acids (Thermo Fisher Scientific Cat# 11-140-050)], containing 1 ng/mL recombinant murine IL-7 (VWR Cat# 10780-184).
To activate CD8+ T cells, 100,000 naive Vex+ CD8+ T cells were stimulated on plate-bound anti-CD3 (2–4 μg/mL, Clone 145–2C11) (VWR Cat# 103252-148) and anti-CD28 (2–4 μg/mL, Clone 37.51) (VWR Cat# 103252-290) and supplemented with 100–200 U/mL IL-2 (VWR Cat# 103717-092) in complete RPMI media for 3–6 days.
Tumor injection
Mice were anesthetized with 2.5% 2,2,2-Tribromoethanol (VWR Cat# AAA18706-22), shaved, and injected midway between the stomach and the flank intradermally with 2e6 MC38 tumor cells. Tumors were measured every 2–3 days once palpable using a caliper. Tumor volume was calculated as: 1/2 × D × d2 where D is the longer diameter, and d is the shorter diameter. Mice were sacrificed when the tumor volume reached 2 cm3 or upon ulceration, as per our animal protocol guidelines. The maximal tumor volume of 2 cm3 was not exceeded in these studies.
Tumor-infiltrating lymphocyte isolation
Tumors were excised, mechanically minced, and incubated in Collagenase Type 1 (Worthington Biochemical Cat# LS004194) for 20 minutes at 37°C with gentle agitation. Collagenase was then neutralized with FBS-containing RPMI media. Tumors were mechanically dissociated through a 70 μm filter. Lymphocytes were enriched using an Optiprep gradient (Sigma-Aldrich Cat# D1556).
Nucleofection of bone marrow stem cells
CD117-enriched fractions were obtained from bones (as above). S. pyogenes Cas9 protein (IDT Cat# 1081059) was mixed with synthetic gRNAs (Synthego) at a molar ratio of 8.1:1 (gRNA: Cas9) in nuclease-free water for 10 minutes to make Cas9-gRNA RNPs. Cas9-gRNA RNPs (25 μL) were then mixed with CD117-enriched cells in 100 μL of P3 buffer (Lonza Cat# V4XP-3024), loaded into nucleofector cuvettes (Lonza Cat# V4XP-3024), and nucleofected using a Lonza 4D nucleofector with the DL100 program. Immediately following nucleofection, RPMI media with 1% FBS was added to the cells which were placed in a 37°C incubator for 1 hour to recover. Cells were then counted and ~500,000 cells were intravenously transferred into irradiated mice (as above).
To create BMCs with 20% DKO of PTPN1/PTPN2, bone marrow stem cells were isolated and nucleofected with Ptpn1 and Ptpn2 gRNAs. Nucleofected cells were then mixed with WT non-nucleofected cells at ratios that would result in ~20% DKO in the recipient animal (estimating transferred bone marrow would constitute ~85% of the subsequent immune system and ~70% of transferred cells would be DKO). The %DKO in BMCs was validated using next-generation sequencing from the blood.
Necropsy
Mice were euthanized, dissected to expose inner organs, and fixed at 22°C for four days in 250 mL Bouin’s solution (Sigma-Aldrich Cat# HT10132-1L). Fixed mice were submitted to the Dana Farber/Harvard Cancer Center Rodent Histopathology Core for tissue sectioning (5 μm thick) and hematoxylin and eosin (H&E) staining. All organs were analyzed for abnormalities by a rodent histopathologist blinded to group identities.
Imaging
H&E stained sections were imaged with a Hamamatsu ORCA-Flash4.0 V3 Digital CMOS camera attached to a Nikon Ti inverted microscope used for widefield imaging. The microscope had a Prior ProScanIII motorized stage, a Nikon Z drive focus accessory, and a Lumencor SOLA LED light engine. All images were acquired with the Nikon Elements Acquisition Software Version 5.21.03 build 1489. Fiji was used to quantify the number of cells in the 40x bone marrow images. Fiji v1.53q and QuPath v0.3.0 were used to quantify white pulp regions in the spleen. See Supplementary Note 2 for a detailed description of imaging acquisition parameters and analyses.
Assessment of mouse weights
BMCs were weighed the day they received LSK as an initial starting weight. They were then weighed every two weeks for the duration of the experiment. Weight loss or gain was normalized to initial starting weight and plotted as a percentage deviation from initial weight. For S-CHIME studies, weight loss or gain was also normalized to the weight of the mouse prior to Tamoxifen administration (week 8). In addition, mice with decreasing body condition (lethargy, poor grooming, and hunched appearance) were weighed more frequently to ensure they did not exceed 20% weight loss.
Survival analysis
BMCs were monitored twice per week to assess survival after receiving new bone marrow. Mice with decreasing body condition (lethargy, poor grooming, and hunched appearance) were monitored daily. Mice were euthanized if moribund or had weight loss exceeding 20%, as per our animal protocol guidelines.
Cytokine analysis
Blood was collected from mice via tail vein or retro-orbital bleed. Blood was incubated for 30 minutes at 4°C in the absence of anticoagulants to enable coagulation. Blood was then spun at 20,000g at 4°C for 15 minutes to separate the serum. Serum was frozen at −80°C until use. The BD CBA Mouse Inflammation Kit (BD Cat# 552364) was used to assess cytokine concentrations in the serum.
Single-cell RNAseq and analyses
Sample prep
Chimeric mice were created following the bone marrow isolation and BMC setup methods listed above. Mice were euthanized four weeks post-setup, and the femurs, tibias, pelvises, vertebral columns, and spleens were isolated and pooled together by group. Bone marrow was extracted from bones with a mortar and pestle. The bone marrow and spleens were smashed through a 70 μm filter followed by red blood cell lysis with ACK lysis buffer. Cells were counted, split evenly between 4 tubes, and stained with Biolegend anti-mouse TotalSeq™-B antibodies (Hashtag B1–4) using 1 μg of antibody for 2 million cells. The samples were pooled back together, keeping distinct organ and BMC groups separate, and were then sorted on live, CD45+ Vex+ cells. 40,000 sorted cells were loaded onto the Chromium Controller (10x Genomics), and gene and cell surface expression libraries were created according to the Chromium Next GEM Single Cell 3’ Reagent Kits v3.1 (Dual Index) user guide. Sequencing was performed on an Illumina NovaSeq 6000 (S2, 100 cycle setting, paired-end reads: 28 bp read 1, 90 bp read 2). Associated sequencing data has been deposited on the Broad Institute’s Single Cell Portal: https://singlecell.broadinstitute.org/single_cell (Accession: SCP1945).
See Supplementary Note 3 for a detailed description of single-cell RNAseq analyses.
Statistical analyses
Statistical analyses were performed using GraphPad Prism 9 software. The data were considered statistically significant with p values <0.05 and for significant values: *p < .05, **p < .01, ***p < .001, ****p < .0001. We used a Student’s t-test for single comparisons of two groups, one-way ANOVA for single comparisons with groups greater than two, two-way ANOVA for multiple comparisons within groups, the Mantel-Cox test for survival curve analyses, and Fisher’s exact test for contingency analyses. For t-tests and ANOVA tests, data distribution was assumed to be normal, but this was not formally tested.
Extended Data
Extended Data Fig. 1. Additional data related to Figure 1.
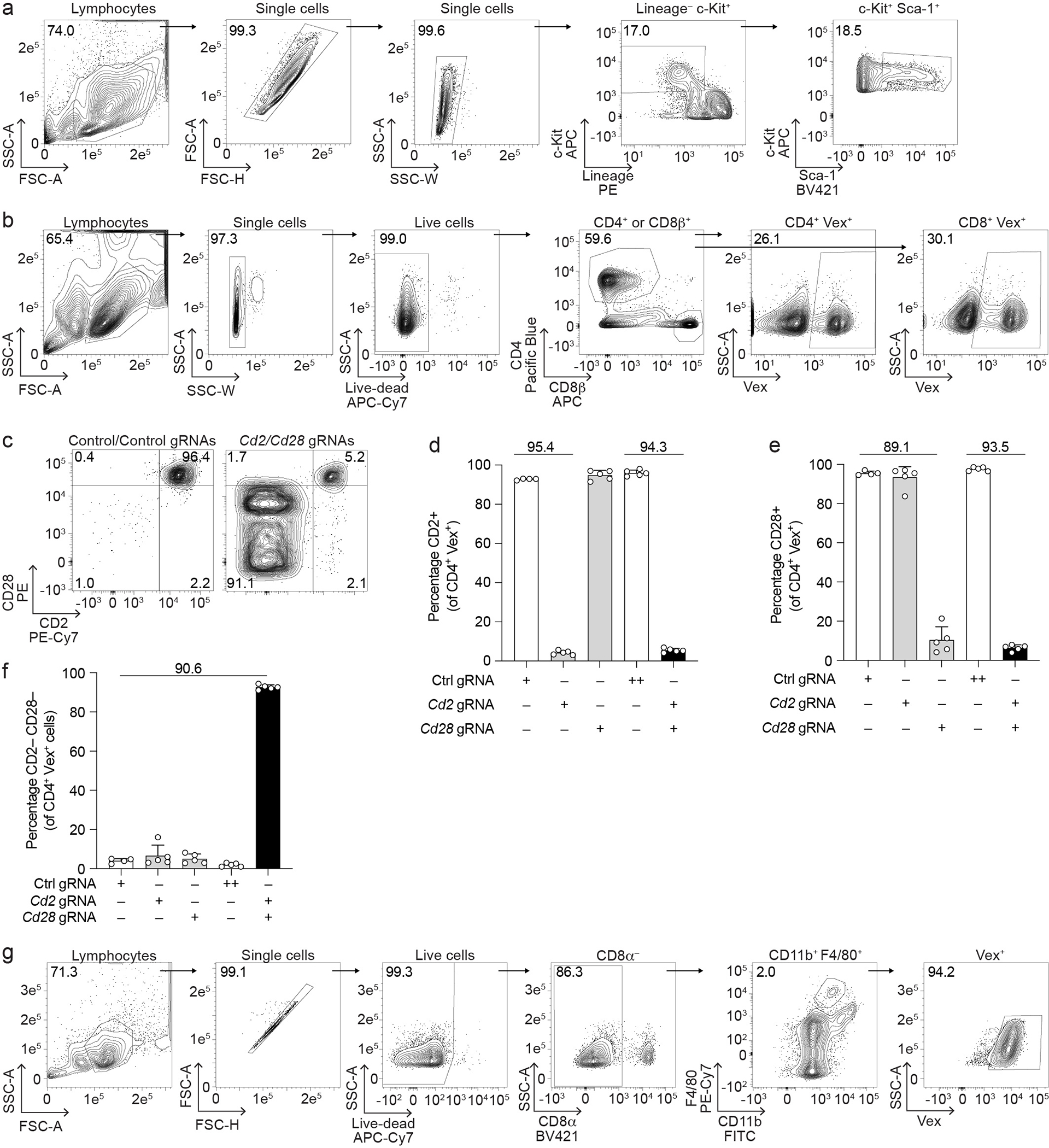
(a) Gating strategy for isolation of LSK from bone marrow for all BMC studies. (b) Gating strategy for isolation of Vex+ CD4+ or Vex+ CD8+ T cells from mouse peripheral blood or spleens. (c) Representative flow cytometry plots of CD2 and CD28 expression on Vex+ CD4+ splenocytes from BMCs containing two non-targeting control gRNAs (left) or Cd2 and Cd28 gRNAs (right). (d, e) Quantification of CD2 (d) or CD28 (e) expression on Vex+ CD4+ splenocytes from (c) as well as relevant single gRNA controls. (f) Quantification of the percentage of cells lacking expression of both CD2 and CD28 on Vex+ CD4+ splenocytes from (c) as well as relevant single gRNA controls. Note ++ indicates two control gRNAs in (d-f). (g) Gating strategy for splenic red-pulp macrophages from mouse spleens. Data are representative of two experiments with at least n = 4 mice per group. Bar graphs represent the mean and error bars represent s.d. Calculated knockout efficiency is denoted above bars for indicated comparisons.
Extended Data Fig. 2. Additional data related to Figure 2.
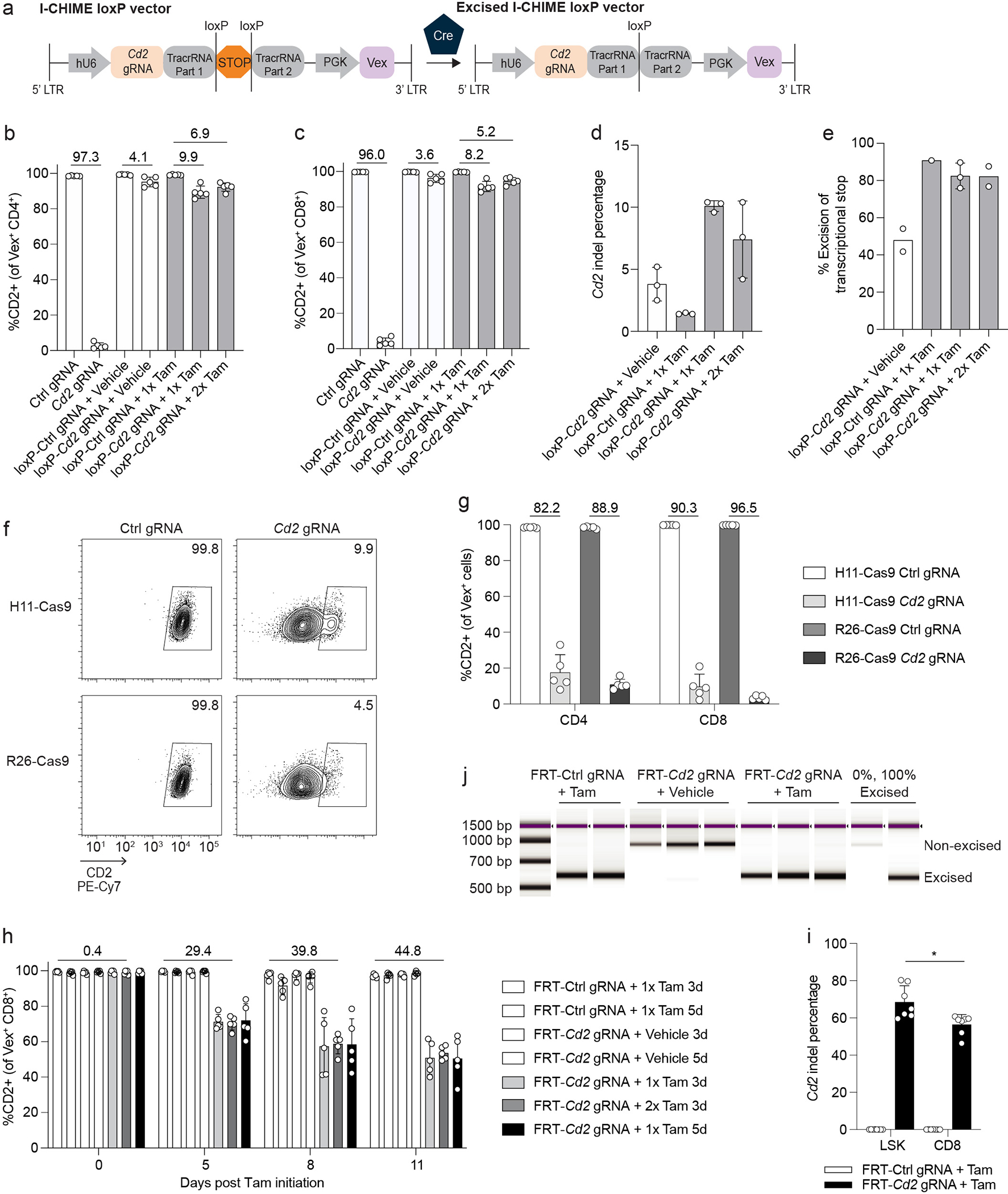
(a) Schematic of I-CHIME loxP vector before and after Cre-mediated excision. (b-c) Quantification of CD2 expression on (b) Vex+ CD4+ and (c) Vex+ CD8+ splenocytes from Cre-ERT2 I-CHIME BMCs containing a constitutive control or Cd2-targeted gRNA, or inducible control or Cd2-targeted gRNA treated with vehicle, 1 mg Tamoxifen (1x in panel), or 2 mg Tamoxifen (2x in panel). (d) Quantification of indels in Cd2 following next-generation sequencing of Vex+ CD8+ splenocytes as in (c). (e) Quantification of % excision of PCR products following an I-CHIME vector-specific PCR of Vex+ CD8+ splenocytes isolated from BMCs containing a non-targeting control or Cd2 gRNA and treated with vehicle control, 1 mg Tamoxifen (1x in panel), or 2 mg Tamoxifen (2x in panel). (f) Representative flow cytometry plots of CD2 expression on Vex+ CD8+ splenocytes from BMCs containing a non-targeting control or Cd2 gRNA. BMCs were created using H11-Cas9 donors or R26-Cas9 donors. (g) Quantification of CD2 expression on Vex+ CD4+ and Vex+ CD8+ splenocytes from (f). (h) Quantification of CD2 expression on Vex+ CD8+ peripheral blood T cells from FlpO-ERT2 I-CHIME BMCs containing an inducible control or Cd2-targeted gRNA treated with vehicle (3 days or 5 days), 1 mg Tamoxifen (3 days or 5 days) (1x Tamoxifen in panel), or 2 mg Tamoxifen (3 days) (2x Tamoxifen in panel). Serial measurements were taken from the same mice. (i) Quantification of indels in Cd2 following next-generation sequencing of Vex+ CD8+ T cells and Vex+ LSKs (as in Figure 2g). The bar graph represents pooled data from two independent experiments (p = 0.0146). (j) TapeStation gel of PCR products following an I-CHIME vector-specific PCR of Vex+ CD8+ splenocytes isolated from BMCs containing a non-targeting control or Cd2 gRNA and treated with Tamoxifen or vehicle control. Genomic DNA that is 0% excised and 100% excised were included as controls. Data are representative of one experiment with at least n = 2 mice per group except (i), which is representative of two experiments with at least n = 3 mice per group. Bar graphs represent the mean and error bars represent s.d. Calculated knockout efficiency is denoted above bars for indicated comparisons. Statistical significance was assessed by a two-sided Student’s paired t-test (i).
Extended Data Fig. 3. Specific deletion of genes in CD8+ T cells using L-CHIME.
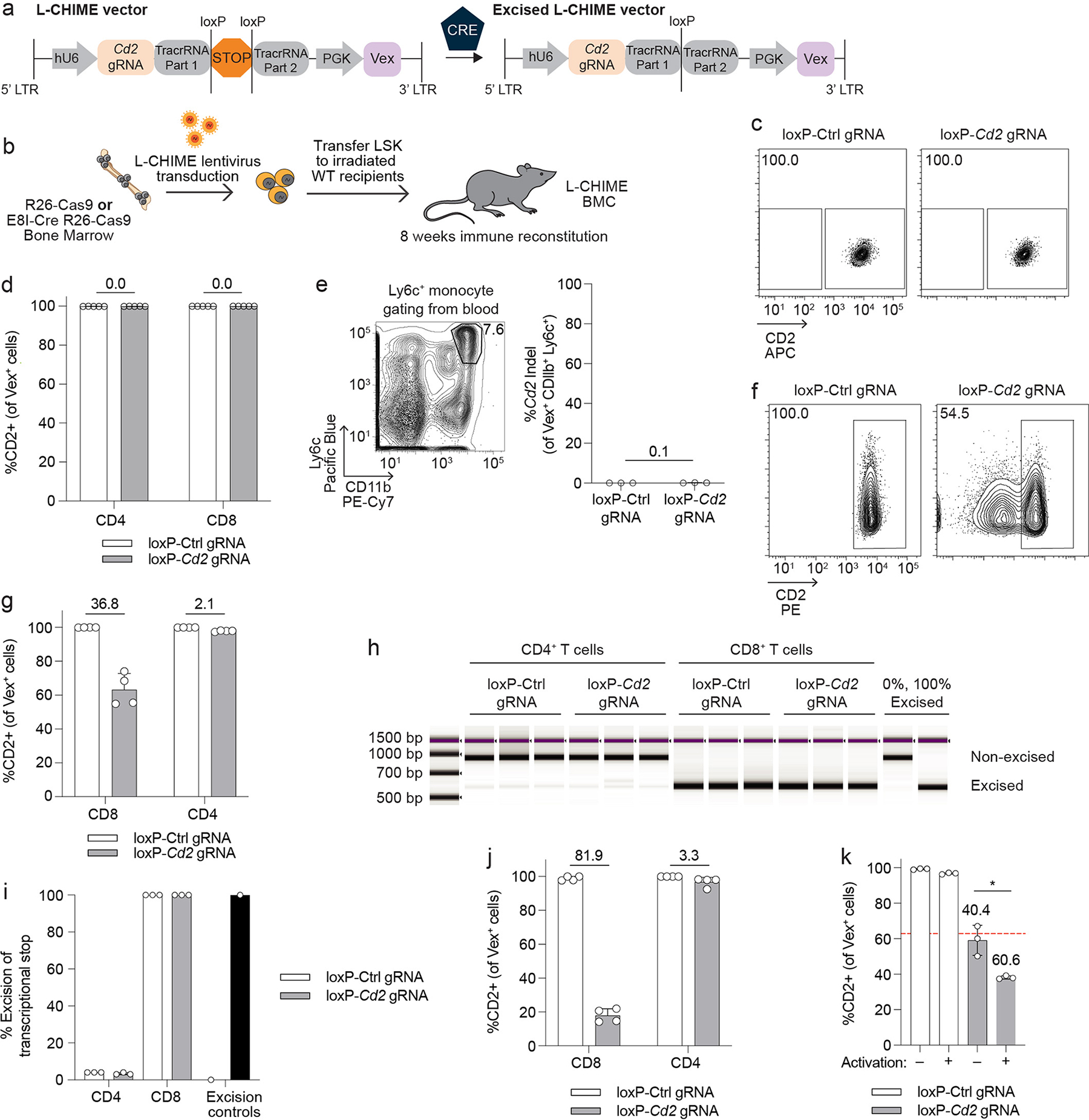
(a) Schematic of L-CHIME vector before and after Cre-mediated excision. (b) Schematic for creation of L-CHIME BMCs. (c) Representative flow cytometry plots of CD2 expression on splenic CD8+ T cells from BMCs created using Cas9ON bone marrow (no Cre present) and the L-CHIME vector containing control or Cd2-targeting gRNAs. (d) Quantification of CD2 expression on Vex+ CD4+ and Vex+ CD8+ splenocytes from BMCs in (c). (e) (Left) Representative gating of Ly6c+ monocytes from the blood. (Right) Quantification of indels in Cd2 following next-generation sequencing of Vex+ Ly6c+ monocytes from BMCs in (c). (f) Representative flow cytometry plots of CD2 expression on Vex+ CD4+ and Vex+ CD8+ T cells isolated from BMCs created using E8I-Cre Cas9ON bone marrow (Cre present only in CD8+ T cells) and the L-CHIME vector containing control or Cd2-targeting gRNAs. (g) Quantification of CD2 expression on Vex+ CD4+ and Vex+ CD8+ splenocytes from BMCs in (f). (h) TapeStation gel of PCR products following an L-CHIME vector-specific PCR of Vex+ CD4+ and Vex+ CD8+ splenocytes containing a non-targeting control or Cd2 gRNA. Genomic DNA that is 0% excised and 100% excised were included as controls. (i) Quantification of % excision as in (h). (j) Quantification of CD2 expression on Vex+ CD4+ and Vex+ CD8+ tumor-infiltrating lymphocytes from BMCs as in (f) challenged with MC38 tumors. (k) CD8+ Vex+ splenocytes were isolated from L-CHIME BMCs containing control or Cd2 gRNAs and were either activated with CD3 and CD28 cross-linking antibodies and IL-2 or maintained in a naive state with IL-7. Quantification of CD2 expression on day 3 post culture. The red dotted line represents the average CD2 expression on the Cd2-targeted CD8+ Vex+ splenocytes prior to culture (p = 0.0131). Data are representative of two experiments with at least n = 3 mice per group. Bar graphs represent the mean and error bars represent s.d. Calculated knockout efficiency is denoted above bars for indicated comparisons and in (k) each Cd2 gRNA is compared to the control gRNA for the corresponding activation condition. Statistical significance was assessed by a two-sided Student’s unpaired t-test (k).
Extended Data Fig. 4. Additional data related to Figure 3.
(a) Quantification of CD2 (left) or CD28 (right) expression on Vex+ CD8+ peripheral blood T cells from (Figure 3d). Serial measurements were taken from the same mice. (b) Quantification of CD2 (left) or CD28 (right) expression on Vex+ CD4+ peripheral blood T cells from (Figure 3d). Serial measurements were taken from the same mice. (c) TapeStation gel of PCR products following an S-CHIME vector-specific PCR of Vex+ CD8+ splenocytes containing (in the inducible gRNA cassette) a non-targeting control or Cd2 gRNA and treated with Tamoxifen or vehicle control. Genomic DNA that is 0% excised and 100% excised were included as controls. (d) Quantification of % excision in (c). Knockout data are representative of two experiments with at least n = 5 mice per group. Excision data are representative of one experiment with at least n = 2 mice per group. Bar graphs represent the mean and error bars represent s.d. Calculated knockout efficiency is denoted above bars for indicated comparisons.
Extended Data Fig. 5. Additional data related to Figure 4.
(a) Quantification of peripheral blood leukocytes assessed by flow cytometry from BMCs (as in Figure 4a) 4 weeks after bone marrow implantation (p < 0.0001). (b) Quantification of Vex expression on CD45+ leukocytes from peripheral blood 4 weeks after bone marrow implantation. (c) Quantification of indels in Ptpn1 (left) and Ptpn2 (right) following next-generation sequencing of peripheral blood cells from BMCs (as in Figure 4a) 4 weeks after bone marrow implantation. (d, e) CBA analysis of serum cytokines from BMCs (as in Figure 4a) 2 weeks (d) or 4 weeks (e) after bone marrow implantation (*Ctrl-1/Ctrl-2 vs. Ptpn1/Ptpn2 p = 0.0480, *Ctrl-1/Ptpn2 vs. Ptpn1/Ptpn2 p = 0.0342, ****p < 0.0001). (f) Weight loss curves normalized to initial starting weight after bone marrow implantation for BMCs (as in Figure 4a). Repeat measurements were taken from the same mice (**p = 0.0027, ****p < 0.0001). (g) Hematoxylin and eosin (10x magnification) scanned images of representative BMC bone marrow sections 4 weeks after bone marrow implantation. Scale bar indicates 1000 μm. (h) Hematoxylin and eosin (10x magnification) scanned images of BMC spleens 4 weeks post bone marrow implantation. Scale bar indicates 1000 μm. (i) Quantification of white pulp area in spleens in (h) (**Ctrl-1/Ctrl-2 vs. Ptpn1/Ptpn2 p = 0.0021, *Ptpn1/Ctrl-2 vs. Ptpn1/Ptpn2 p = 0.0259, **Ctrl-1/Ptpn2 vs. Ptpn1/Ptpn2 p = 0.0029). The bar graph represents pooled data from two independent experiments. Data are representative of two experiments with at least n = 3 mice per group. Bar graphs represent the mean and error bars represent s.d. Statistical significance was assessed by one-sided two-way ANOVA (a, d, e, f) or one-sided one-way ANOVA (i).
Extended Data Fig. 6. Additional data related to Figure 4.
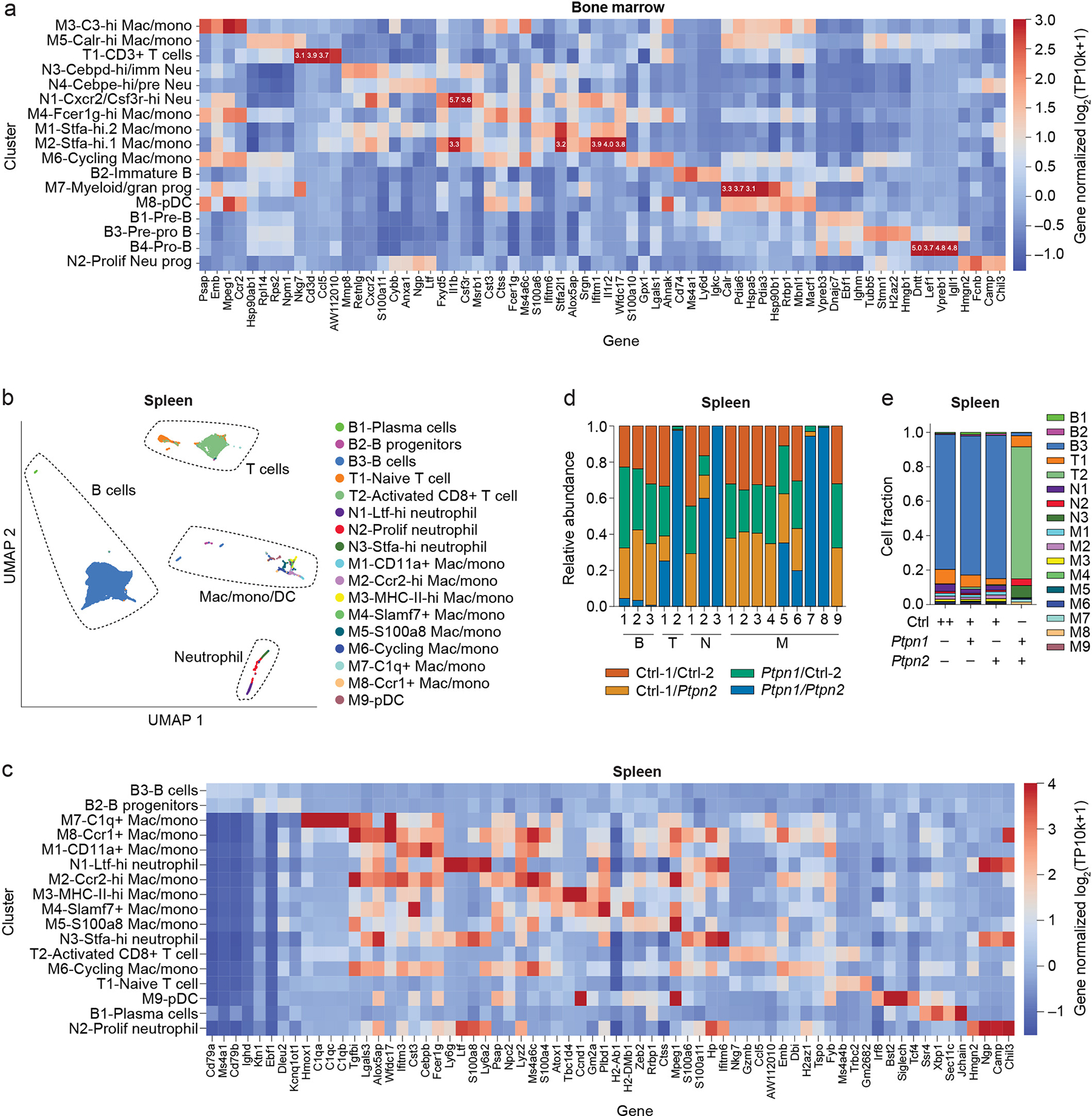
(a) Heat map of genes used to annotate UMAP clusters of 10x scRNAseq from bone marrow samples of BMCs (as in Figure 4f). Note values added to the heat map represent the intensity of saturated values that exceed the intensity scale. (b) UMAP clustering of 10x scRNAseq from spleen samples of BMCs (as in Figure 4a). (c) Heat map of genes used to annotate UMAP clusters of 10x scRNAseq from spleen samples of BMCs (as in Extended Data Figure 6b). (d, e) Quantification of cell cluster proportions from the spleen within each cluster (d) or within each BMC group (e). Data are representative of one experiment with n = 3 mice per group.
Extended Data Fig. 7. Additional data related to Figure 5.
(a) Weight loss curves normalized to initial starting weight after bone marrow implantation for BMCs (as in Figure 5a). Serial measurements were taken from the same mice. (b) Quantification of peripheral blood leukocytes assessed by flow cytometry from BMCs (as in Figure 5a) 4 weeks after bone marrow implantation (CD11b: ***Ctrl-1/Ctrl-2 vs. Ptpn1/FRT-Ptpn2 p = 0.0002, ***Ctrl-2/FRT-Ptpn2 vs. Ptpn1/FRT-Ptpn2 p = 0.0008; CD19: ***p = 0.0002, ****p < 0.0001). (c) Quantification of Vex expression on CD45+ leukocytes from peripheral blood 4 weeks after bone marrow implantation. (d, e) CBA analysis of serum cytokines from BMCs one week prior to (d) or one week after (e) Tamoxifen administration (p < 0.0001). (f) Ptpn1 and Ptpn2 indel percentage in the blood from BMCs created by nucleofecting bone marrow stem cells with Ptpn1/Ptpn2 gRNAs, mixing with non-nucleofected bone marrow stem cells, and implanting into irradiated recipients. (g) Survival curve of BMCs as in (f) (p = 0.0005). (h) Quantification of pathologic findings for indicated organs from BMCs in (g). Data are representative of two experiments with at least n = 5 mice per group, except (f) which is representative of two experiments with at least n = 3 mice per group. The graph in (h) represents pooled data from two independent experiments. Bar graphs represent the mean and error bars represent s.d. Statistical significance was assessed by one-sided two-way ANOVA (a, b, d, e), two-sided Mantel-Cox test (g), or two-sided Fisher’s exact test (h).
Supplementary Material
Acknowledgements
This work was supported by U19AI133524 from the National Institute of Allergy and Infectious Diseases to A.H.S and J.G.D., U54 CA225088 from the National Cancer Institute to A.H.S., and P01 AI108545 from the National Institute of Allergy and Infectious Diseases to A.H.S. A.H.S. also received funding for this work from Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA. We thank the Dana-Farber/Harvard Cancer Center in Boston, MA, for use of the Rodent Histopathology Core, which performed necropsies. Dana-Farber/Harvard Cancer Center is supported in part by NCI Cancer Center Support Grant #NIH 5 P30 CA06516. We thank the Center for Computational and Integrative Biology (CCIB) at Massachusetts General Hospital for the use of the CCIB DNA Core Facility (Cambridge, MA), which performed CRISPR sequencing for indel assessment. We thank the Microscopy Resources on the North Quad (MicRoN) core, RRID:SCR_019195 at Harvard Medical School for microscopy and analysis services.
Footnotes
Competing Interests Statement
A.H.S. has patents/pending royalties on the PD-1 pathway from Roche and Novartis. A.H.S. is on advisory boards for Elpiscience, Selecta, Bicara, Monopteros, Fibrogen, Alixia, IOME, Corner Therapeutics, GlaxoSmithKline, Amgen, and Janssen. A.H.S. has received research funding from Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA related to this project and Vertex, Moderna, Quark/Iome, AbbVie, and Erasca unrelated to this project. J.G.D. consults for Microsoft Research, Abata Therapeutics, Servier, Maze Therapeutics, BioNTech, Sangamo, and Pfizer. J.G.D. consults for and has equity in Tango Therapeutics. J.G.D. serves as a paid scientific advisor to the Laboratory for Genomics Research, funded in part by GlaxoSmithKline. J.G.D. receives funding support from the Functional Genomics Consortium: AbbVie, Bristol Myers Squibb, Janssen, Vir Biotechnology, and Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA. J.G.D.’s interests were reviewed and are managed by the Broad Institute in accordance with its conflict-of-interest policies. W.N.H. is an employee of and holds equity in Arsenal Biosciences. The remaining authors declare no competing interests.
Code availability
The custom code used for single-cell RNAseq analyses has been deposited on GitHub.
Data availability
The data and materials that support the findings of this study are available from the corresponding author upon reasonable request. Source data for all graphs in the Figures and Extended Data Figures are listed within the Supplementary Information. All 10x scRNA sequencing data from this study has been deposited in the Broad Institute’s Single Cell Portal and are accessible through the accession code SCP1945. M. musculus genome GRCm39 is accessible here: https://ftp.ensembl.org/pub/release-106/fasta/mus_musculus/dna. Gene annotations from Ensembl release 106 are accessible here: https://ftp.ensembl.org/pub/release-106/gtf/mus_musculus/.
References
- 1.Gu H, Marth JD, Orban PC, Mossmann H & Rajewsky K Deletion of a DNA Polymerase β Gene Segment in T Cells Using Cell Type-Specific Gene Targeting. Science 265, 103–106 (1994). [DOI] [PubMed] [Google Scholar]
- 2.Thomas KR & Capecchi MR Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 51, 503–512 (1987). [DOI] [PubMed] [Google Scholar]
- 3.Jaenisch R Transgenic Animals. Science 240, 1468–1474 (1988). [DOI] [PubMed] [Google Scholar]
- 4.Woo S-R et al. Immune Inhibitory Molecules LAG-3 and PD-1 Synergistically Regulate T-cell Function to Promote Tumoral Immune Escape. Cancer Res 72, 917–927 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paterson AM et al. Deletion of CTLA-4 on regulatory T cells during adulthood leads to resistance to autoimmunity. J Exp Medicine 212, 1603–1621 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang H et al. One-Step Generation of Mice Carrying Mutations in Multiple Genes by CRISPR/Cas-Mediated Genome Engineering. Cell 153, 910–918 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang H et al. One-Step Generation of Mice Carrying Reporter and Conditional Alleles by CRISPR/Cas-Mediated Genome Engineering. Cell 154, 1370–1379 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yin H, Kauffman KJ & Anderson DG Delivery technologies for genome editing. Nat Rev Drug Discov 16, 387–399 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Wei T et al. Delivery of Tissue-Targeted Scalpels: Opportunities and Challenges for In Vivo CRISPR/Cas-Based Genome Editing. Acs Nano 14, 9243–9262 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldstein JM et al. In Situ Modification of Tissue Stem and Progenitor Cell Genomes. Cell Reports 27, 1254–1264.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng Q et al. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat Nanotechnol 15, 313–320 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.LaFleur MW et al. A CRISPR-Cas9 delivery system for in vivo screening of genes in the immune system. Nat Commun 10, 1668 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen JP et al. Combinatorial CRISPR–Cas9 screens for de novo mapping of genetic interactions. Nat Methods 14, 573–576 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chylinski K et al. CRISPR-Switch regulates sgRNA activity by Cre recombination for sequential editing of two loci. Nat Commun 10, 5454 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.LaFleur MW et al. PTPN2 regulates the generation of exhausted CD8+ T cell subpopulations and restrains tumor immunity. Nat Immunol 20, 1335–1347 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manguso RT et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547, 413–418 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goh PK et al. PTPN2 elicits cell autonomous and non–cell autonomous effects on antitumor immunity in triple-negative breast cancer. Sci Adv 8, eabk3338 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wiede F et al. PTPN2 phosphatase deletion in T cells promotes anti-tumour immunity and CAR T-cell efficacy in solid tumours. Embo J 39, e103637 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang S et al. A small molecule inhibitor of PTP1B and PTPN2 enhances T cell anti-tumor immunity. Nat. Commun. 14, 4524 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baumgartner CK et al. The PTPN2/PTPN1 inhibitor ABBV-CLS-484 unleashes potent anti-tumour immunity. Nature 1–13 (2023) doi: 10.1038/s41586-023-06575-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asante-Appiah E et al. The YRD Motif Is a Major Determinant of Substrate and Inhibitor Specificity in T-cell Protein-tyrosine Phosphatase*. J Biol Chem 276, 26036–26043 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Jia Z, Barford D, Flint AJ & Tonks NK Structural Basis for Phosphotyrosine Peptide Recognition by Protein Tyrosine Phosphatase 1B. Science 268, 1754–1758 (1995). [DOI] [PubMed] [Google Scholar]
- 23.Wiede F et al. PTP1B Is an Intracellular Checkpoint that Limits T-cell and CAR T-cell Antitumor Immunity. Cancer Discov 12, 752–773 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heinonen KM, Bourdeau A, Doody KM & Tremblay ML Protein tyrosine phosphatases PTP-1B and TC-PTP play nonredundant roles in macrophage development and IFN-γ signaling. Proc National Acad Sci 106, 9368–9372 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson MT et al. Simultaneous fluorescence-activated cell sorter analysis of two distinct transcriptional elements within a single cell using engineered green fluorescent proteins. Proc National Acad Sci 93, 8508–8511 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Charlesworth CT et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat Med 25, 249–254 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dubrot J et al. In vivo screens using a selective CRISPR antigen removal lentiviral vector system reveal immune dependencies in renal cell carcinoma. Immunity 54, 571–585.e6 (2021). [DOI] [PubMed] [Google Scholar]
- 28.Chu VT et al. Efficient CRISPR-mediated mutagenesis in primary immune cells using CrispRGold and a C57BL/6 Cas9 transgenic mouse line. Proc National Acad Sci 113, 12514–12519 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lao Z, Raju GP, Bai CB & Joyner AL MASTR: A Technique for Mosaic Mutant Analysis with Spatial and Temporal Control of Recombination Using Conditional Floxed Alleles in Mice. Cell Reports 2, 386–396 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chiou S-H et al. Pancreatic cancer modeling using retrograde viral vector delivery and in vivo CRISPR/Cas9-mediated somatic genome editing. Gene Dev 29, 1576–1585 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seki A & Rutz S Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J Exp Medicine 215, 985–997 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maekawa Y et al. Notch2 integrates signaling by the transcription factors RBP-J and CREB1 to promote T cell cytotoxicity. Nat Immunol 9, 1140–1147 (2008). [DOI] [PubMed] [Google Scholar]
- 33.You-Ten KE et al. Impaired Bone Marrow Microenvironment and Immune Function in T Cell Protein Tyrosine Phosphatase–deficient Mice. J Exp Medicine 186, 683–693 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wiede F et al. Strain-Dependent Differences in Bone Development, Myeloid Hyperplasia, Morbidity and Mortality in Ptpn2-Deficient Mice. Plos One 7, e36703 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huh WJ et al. Tamoxifen Induces Rapid, Reversible Atrophy, and Metaplasia in Mouse Stomach. Gastroenterology 142, 21–24.e7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bohin N, Carlson EA & Samuelson LC Genome Toxicity and Impaired Stem Cell Function after Conditional Activation of CreERT2 in the Intestine. Stem Cell Rep 11, 1337–1346 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schumann K et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc National Acad Sci 112, 10437–10442 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang R et al. CRISPR/Cas9-targeting of CD40 in hematopoietic stem cells limits immune activation mediated by anti-CD40. Plos One 15, e0228221 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Doench JG Am I ready for CRISPR? A user’s guide to genetic screens. Nat Rev Genet 19, 67–80 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Dow LE et al. Inducible in vivo genome editing with CRISPR/Cas9. Nat Biotechnol 33, 390–394 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aubrey BJ et al. An Inducible Lentiviral Guide RNA Platform Enables the Identification of Tumor-Essential Genes and Tumor-Promoting Mutations In Vivo. Cell Reports 10, 1422–1432 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Sun N et al. Development of drug-inducible CRISPR-Cas9 systems for large-scale functional screening. Bmc Genomics 20, 225 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lundin A et al. Development of an ObLiGaRe Doxycycline Inducible Cas9 system for pre-clinical cancer drug discovery. Nat Commun 11, 4903 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Glas J et al. PTPN2 Gene Variants Are Associated with Susceptibility to Both Crohn’s Disease and Ulcerative Colitis Supporting a Common Genetic Disease Background. Plos One 7, e33682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spalinger MR, McCole DF, Rogler G & Scharl M Role of Protein Tyrosine Phosphatases in Regulating the Immune System. Inflamm Bowel Dis 21, 645–655 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only references
- 46.Ruzankina Y et al. Deletion of the Developmentally Essential Gene ATR in Adult Mice Leads to Age-Related Phenotypes and Stem Cell Loss. Cell Stem Cell 1, 113–126 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doench JG et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilkinson AC et al. Long-term ex vivo haematopoietic-stem-cell expansion allows nonconditioned transplantation. Nature 571, 117–121 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data and materials that support the findings of this study are available from the corresponding author upon reasonable request. Source data for all graphs in the Figures and Extended Data Figures are listed within the Supplementary Information. All 10x scRNA sequencing data from this study has been deposited in the Broad Institute’s Single Cell Portal and are accessible through the accession code SCP1945. M. musculus genome GRCm39 is accessible here: https://ftp.ensembl.org/pub/release-106/fasta/mus_musculus/dna. Gene annotations from Ensembl release 106 are accessible here: https://ftp.ensembl.org/pub/release-106/gtf/mus_musculus/.