

Abstract
Urocortin 3 (UCN3) is a peptide hormone expressed in pancreatic islets of Langerhans of both human alpha and human beta cells and solely in murine beta cells. UCN3 signaling acts locally within the islet to activate its cognate receptor, corticotropin releasing hormone receptor 2 (CRHR2), which is expressed by delta cells, to potentiate somatostatin (SST) negative feedback to reduce islet cell hormone output. The functional importance of UCN3 signaling in the islet is to modulate the amount of SST tone allowing for finely tuned regulation of insulin and glucagon secretion. UCN3 signaling is a hallmark of functional beta cell maturation, increasing the beta cell glucose threshold for insulin secretion. In doing so, UCN3 plays a relevant functional role in accurately maintaining blood glucose homeostasis. Additionally, UCN3 acts as an indicator of beta cell maturation and health, as UCN3 is not expressed in immature beta cells and is downregulated in dedifferentiated and dysfunctional beta cell states. Here, we review the mechanistic underpinnings of UCN3 signaling, its net effect on islet cell hormone output, as well as its value as a marker for beta cell maturation and functional status.
Keywords: pancreatic islet, UCN3, CRH, glucagon, insulin, SST, beta cell maturity
Introduction
Pancreatic islets are mini organs that reside in the pancreas that are the source of two of the most important glucoregulatory hormones our bodies produce. Best-known of course is insulin, which is produced and secreted solely by pancreatic beta cells, which are the most abundant islet endocrine cell in both mouse and human pancreas. The other major endocrine hormone that originates from the pancreas is glucagon, which is processed from pre-pro-glucagon precursor by pancreatic alpha cells. Fine-tuning and modulating the activity of beta and alpha cells are a host of paracrine factors. Some of these are byproducts of cellular metabolism, such as ATP and Ca2+, which are released by beta cells activated by nutrient stimulation. And Zn2+ ions are co-crystalized with insulin and co-released upon stimulation of beta cell exocytosis. However, other paracrine factors are dedicated signaling molecules that serve no other role than to relay a signal from one cell to the next. There are easily several dozen paracrine signals that contribute to the regulation of beta cell fate and function, which have been reviewed in detail elsewhere [1, 2]. One of the most abundant of these signals is the peptide hormone Urocortin 3 (UCN3), which is the specific focus of this review. We review the basic properties of UCN3, the role of UCN3 as a paracrine signal within the islet, and the utility of Ucn3 as a beta cell maturation marker as UCN3 1) appears late during development 2) is downregulated early in diabetes, and 3) is readily detectable.
Urocortin3 is a peptide hormone of to the CRH family of neuropeptides
UCN3 was discovered simultaneously by two groups. Hsu and Hsueh described their discovery of UCN3 alongside of the discovery of a second putative peptide hormone and named these stresscopin and stresscopin-related peptide (now known as UCN3) [3]. Contemporaneously, Wylie Vale’s group discovered these same two peptide hormones and named them Urocortin2 (UCN2) [4] and UCN3 [5] for their sequence similarity to the then known peptide hormones Urocortin (UCN) and Corticotropin Releasing Hormone (CRH). The latter is of course known as the principal hypothalamic releasing hormone that activates the endocrine stress response by stimulating the release of Adrenocorticotropic Hormone (ACTH), culminating with glucocorticoid release from the adrenal cortex. CRH and the Urocortins are pre-pro-proteins that share an unmistakable, yet relatively modest amino acid similarity concentrated towards the N-terminus that encodes the mature peptide hormones [6]. UCN and CRH both have consensus dibasic cleavage sites that lead to the processing of 40 and 41 amino acid mature peptides, respectively. In contrast, the 161 amino acid (163 aa in mouse) UCN3 precursor is also a pre-pro-hormone, but without a consensus dibasic cleavage site. Instead, UCN3 is most-commonly considered to be cleaved at a single lysine residue (K119 in human UCN3), resulting in a putative 38 amino acid mature peptide that is amidated at its N-terminus [5]. Most studies to date that use synthetic UCN3 have used this 38-mer, which is bioactive and has high affinity (low nM) for its receptors [5, 7].
UCN3 is the most abundant beta cell hormone after insulin and IAPP
UCN3 is among the most abundant of all transcripts expressed by beta cells. Among signaling molecules encoded in our DNA, it is the third-most abundant beta cell signal encoded in our DNA after insulin and islet amyloid polypeptide (IAPP) [8]. Ucn3 mRNA levels are approximately 1000-fold lower than those of Ins2 in mouse beta cells based on bulk RNA-Seq of FACS-purified alpha, beta, and delta cells [9]. Comparison of total islet peptide content in mouse confirms that UCN3 content is approximately 3 orders of magnitude below that of insulin [8]. This probably reflects the exceptional abundance of insulin more than it should lead to the interpretation of UCN3 as a rare signal. Nevertheless, detecting the release of UCN3 from islet is challenging simply because the levels released are relatively low compared to insulin. UCN3 release from clonal MIN6 increased dose-dependently with glucose stimulation and is robustly stimulated by depolarization with KCl [10]. UCN3 secretion from primary mouse islets also is stimulated by glucose, but this could only be detected by accumulating UCN3 release from a pool of 1000 islets over the course of 4 hours [8]. This observation provides important context for the physiological role that UCN3 plays: at levels of secretion several orders of magnitude below those of insulin, local concentrations of UCN3 within the islet are high enough to activate its cognate receptors, but UCN3 would dilute out quickly in the portal and general circulation. Indeed, circulating UCN3 levels are relatively low at a reported 50 pg/g in mouse [11] and slightly higher in human plasma [12]. While these levels would correspond to low nM concentrations of UCN3, it is worth noting that plasma UCN3 levels are not measured routinely and validating such assays is not trivial. It is likely that UCN3 released from beta cells plays a predominantly paracrine role via the local activation of target cells within the islet and makes little contribution to any circulating levels of UCN3.
Species differences in UCN3 and CRH expression within the islet
Within the islet field, there is ample debate about the species differences between the human pancreas and the rodent models that are so prevalent as model systems of pancreas physiology and disease. Similarities between the rodent and primate pancreas abound: islets in each contain a mixed population of beta, alpha, and delta cells that generally speaking are similar in the expression and regulation of the secretion of the major hormones that they each release. However, the overlap in islet physiology between species notwithstanding, there are multiple aspects where the rodent and primate pancreas differ. These differences are well-documented [13, 14] and emphasized as they represent interesting experiments of nature that stand to teach us something about islet physiology. One of these notable species differences is with regards to the expression of UCN3 and CRH within the islet. In the mouse pancreas, UCN3 is expressed highly selectively in the beta cell and is not detected at the protein level in either alpha or delta cells [10]. Comprehensive bulk transcriptome expression patterns of FACS-purified alpha, beta, and delta cells collected from transgenic reporter lines generated for this purpose reveal specific Ucn3 mRNA expression in mouse beta cells [9]. Single cell RNA-seq data from dissociated mouse islet cells confirm these findings [15].
In contrast to rodent islets, UCN3 expression in human and non-human primate islets is notably not restricted to beta cells. Instead, in pancreases from human donors without diabetes, Ucn3 mRNA abundance and staining intensity are approximately equally abundant in beta and alpha cells (Figure 1) [16]. UCN3 expression is also detected in human pancreatic polypeptide cells (Figure 2) and is absent from delta cells in both species. This latter observation rules out an autocrine role for UCN3 as delta cells are the major target for the signal that is UCN3 as will be described in detail below. A related species difference between rodents and primates is that CRH, the original hypothalamic releasing factor that triggers the endocrine stress response, is expressed by human alpha cells at levels comparable to the expression of UCN3 [16]. CRH is known to trigger a Ca2+ response in beta cells [17, 18] and stimulates cAMP, phosphorylates Erk1/2, protects against beta cell apoptosis and potentiates glucose-stimulated insulin secretion [19]. These actions are mediated by the CRHR1 receptor, which is a G protein-coupled receptor related to the incretin receptors with many of the same downstream effects. This places CRH on a short list with the proglucagon-derived peptides (glucagon and glucagon-like peptide 1) as alpha cell-derived peptides capable of potentiating glucose-stimulated insulin secretion in a paracrine manner [20–22]. It is notable that the local actions of alpha cell-derived CRH are directly insulinotropic and would result in the lowering of plasma glucose, while the actions of CRH releasing from hypothalamic paraventricular nucleus projecting onto the median eminence culminate in the release of glucocorticoids which result in an increase in plasma glucose. Therefore, the hypothalamic and intra-islet CRH systems likely reflect two functionally independent systems that each happen to use the same ligand-receptor pair but operate in response to different physiological cues and likely at non-overlapping anatomical compartments.
Figure 1: Species difference between rodent and primate islet expression of Ucn3 and Crh.
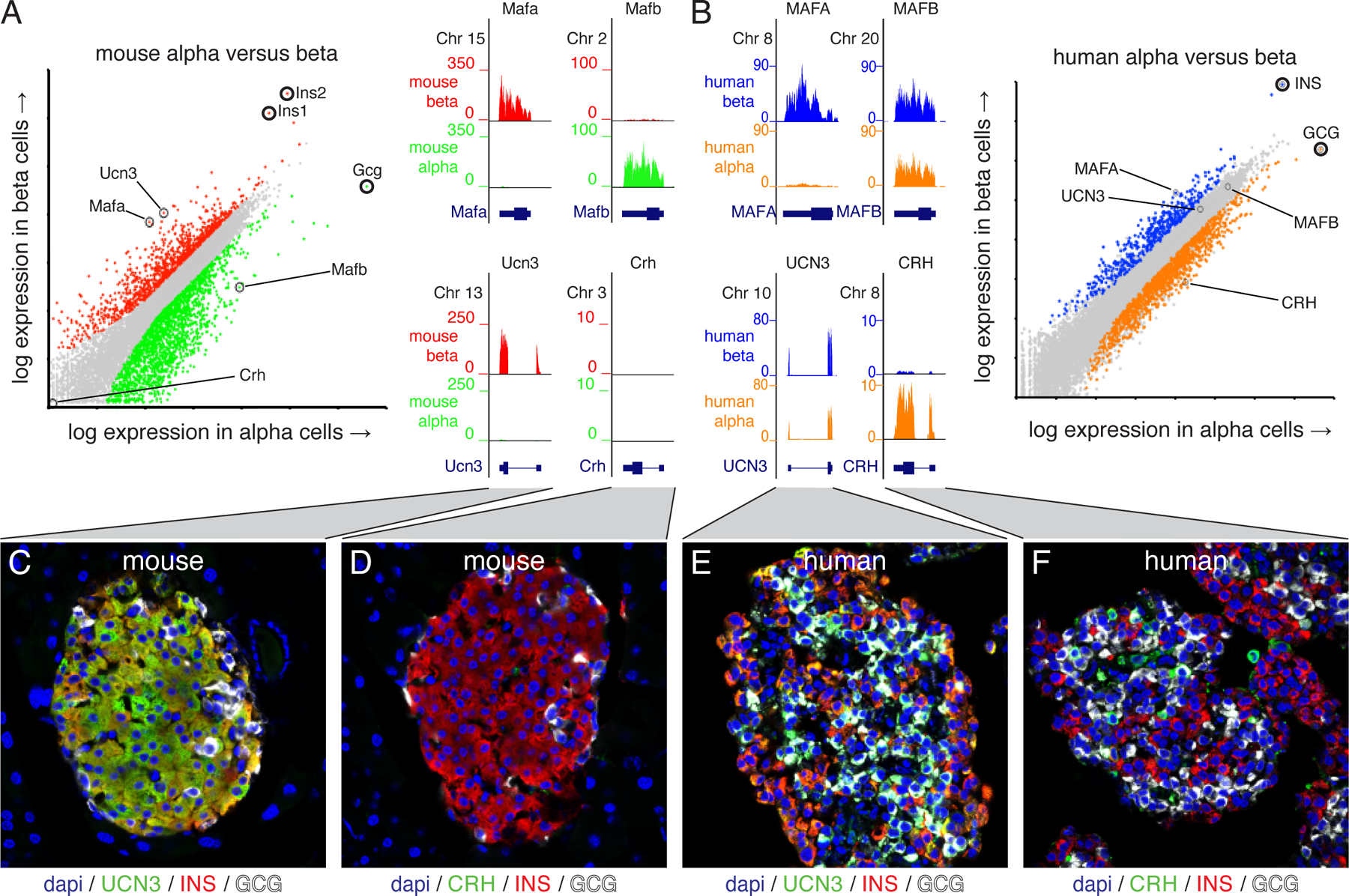
The expression of two sets of genes, Mafa/Mafb and Ucn3/Crh, that markedly differ in their expression between alpha and beta cells in mouse (A) and human (B) islets. Mafa is restricted to beta cells of both species, while Mafb is selectively expressed in mouse alpha cells (A) but expressed in human beta cells as well (B). Ucn3 is highly selective for mouse beta cells while Crh is detected in neither mouse islet population (A, C, D). By contrast, UCN3 expression is abundant in human alpha and beta cells, while CRH is enriched in human alpha cells (B, E, F). Human RNA-seq data from [78]. Figure originally published in [16] under a Creative Commons Attribution 4.0 International License and reused without alteration.
Figure 2: Expression of Ucn3 in PP cells.
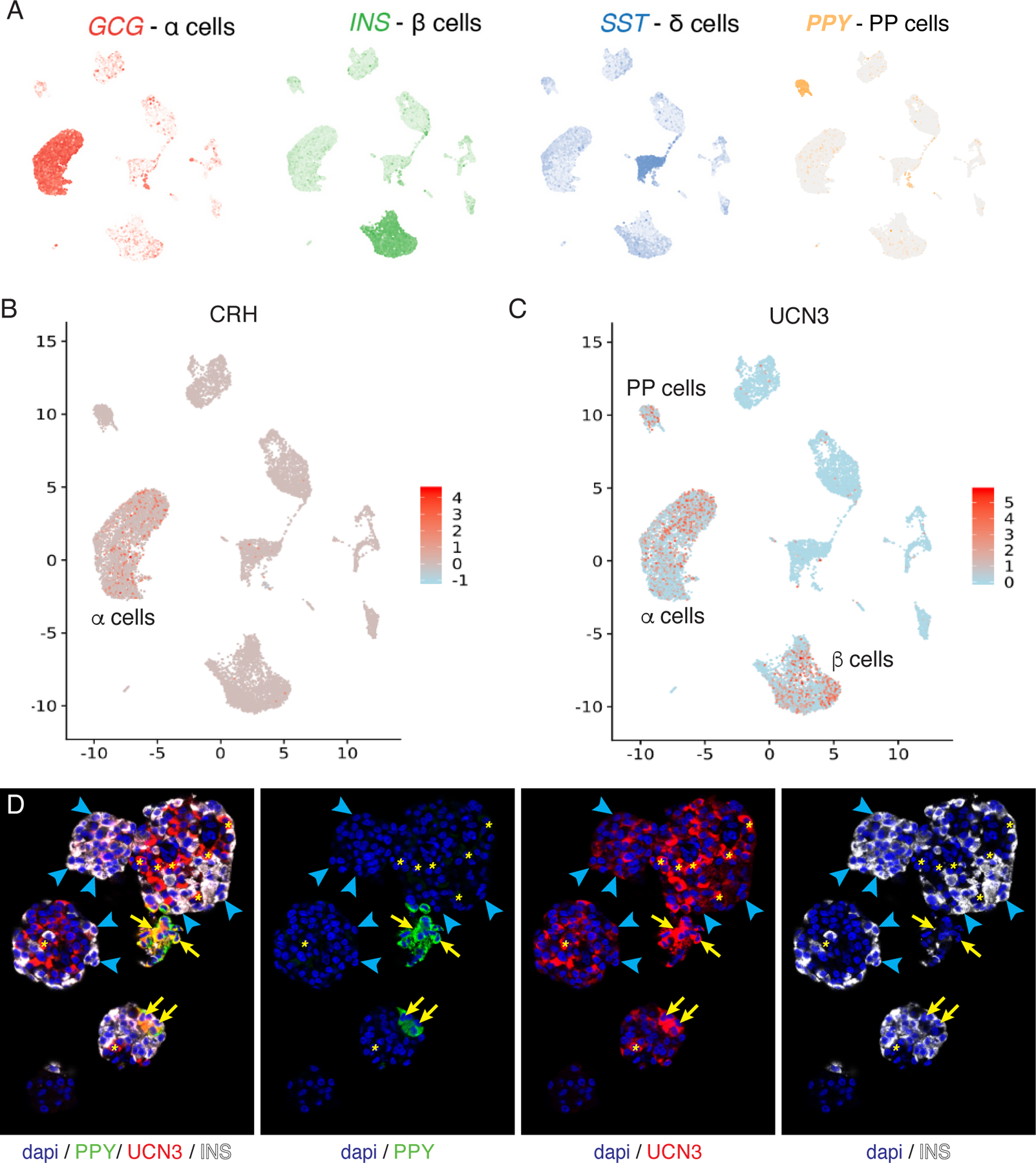
The expression of UCN3 is absent from pancreatic delta cells, but present in human and mouse PP cells, characterized by the expression of pancreatic polypeptide (Ppy/PPY). Human single cell RNA-seq data from [79] with the main endocrine cell clusters as defined by the expression of GCG, INS, SST and PPY (A). Expression of Crh is restricted to human alpha cells (B), while Ucn3 expression is detected in PP cells as well as in alpha and beta cells (C). Note how the mosaic detection of Crh and Ucn3 reflects dropout rather that biologically meaningful heterogeneous expression. The expression of UCN3 in PP cells is confirmed at the protein level by the observation that UCN3 and PPY overlap within the same cells of human pancreatic islets, as indicated by yellow areas. Blue arrow heads indicate overlap between UCN3 and insulin.
UCN3 expression and function outside of the pancreatic islet
The central nervous system (CNS) is a major hub for CRF-urocortin derived signaling, where CRF and Urocortins direct the behavioral and physiological stress response and regulate metabolic homeostasis via CRFR1 and CRFR2 that are reviewed in detail elsewhere [23–25]. UCN3 is expressed in neurons located in the median preoptic nucleus (MPO), the perifornical area of the hypothalamus, the medial amygdala, the bed nucleus of the stria terminalis, and superior para-olivary nucleus [5, 26, 27]. UCN3 immunoreactive fibers are also located in the ventral premammillary nucleus (PMV) where they are hypothesized to play a role in conveying sensory information to modulate behavioral and neuroendocrine responses [28]. Indeed, CRFR2 mutant mice are hypersensitive to stress and display increased anxiety-like behavior and display increased UCN3 expression in the PVN [29]. Additionally, UCN3 expression was shown in nuclei connected to the accessory olfactory system using a tau-lacZ reporter gene in replace of the endogenous Ucn3 open reading frame, where they found evidence in support of a role for UCN3 in establishing social memories [26].
UCN3 is expressed in tissues of the gastrointestinal tract besides pancreatic islet endocrine cells. UCN3 expression has been reported in the stomach, small intestine, colon, and rectum [3, 5, 30–32]. Saruta et al., showed via both immunohistochemistry and mRNA in situ hybridization the presence of UCN3 expression in myenteric and submucosal nervous plexus, vascular endothelial and smooth muscle cells of blood vessels in subserosa, in smooth muscle layers of the large intestine, and in enterochromaffin cells [33]. More recently, a single cell mRNA sequencing study was able to identify rare UCN3 positive cells in the small intestine [34]. Systemically administered UCN3 delays gastric emptying, which can be prevented by co-administration of the CRHR2-specific antagonist Astressin2b [35]. However, it is undetermined whether endogenous UCN3 helps control the rate of gastric emptying under physiological conditions and the mechanism by which UCN3 would inhibit gastric emptying has not been established [30].
Other peripheral sites of UCN3 expression include the adrenal cortex, gestational tissues, heart, kidney, skin, and adipose tissue [5, 36–38]. Fukuda et al., reported UCN3 expression in the adrenal cortex using immunohistochemistry and mRNA in situ hybridization techniques and found the peptide colocalized with its cognate receptor CRFR2 in more than 85% of adrenocortical cells [39]. However, they did not map a physiological function to the presence of UCN3 ligand and receptor co-expression in adrenocortical cells. UCN3 was also shown to localize in gestational tissues, including the placenta, decidua, and fetal membranes [36] where they propose UCN3 to be a regulator of placental vascular endothelial tone, but did not present evidence for this function. The heart and kidney also contain UCN3 expressing cells [37], where UCN3 acts as a vasodilator of the cardiovascular system through CRFR2 [40]. UCN3 expression was also reported in adipose tissue using RT qPCR and exogenous administration of the ligand was shown to inhibit adipocyte inflammatory responses [38].
UCN3 is co-released with insulin
Glucose-stimulated insulin secretion is a well characterized pathway, where glucose is metabolized via glycolysis, and subsequent oxidation of acetyl CoA in the tricarboxylic acid cycle leads to efficient production of ATP through oxidative phosphorylation. When the extracellular glucose concentration is above the glucose threshold for insulin secretion, the relative rate of ATP production compared to its breakdown into ADP is high enough to close ATP sensitive potassium channels, which results in a depolarizing effect on the beta cell membrane potential. Depolarization of the beta cell membrane causes the opening of L-type voltage gated calcium channels (and P/Q type voltage-gated calcium channels in human beta cells), which leads to calcium influx and facilitates the exocytosis of insulin granules [41, 42]. UCN3 and insulin are not only expressed by the beta cell, super-resolution structured illumination microscopy demonstrates that UCN3 is packaged alongside insulin into secretory granules [8] (Figure 3). This would imply that UCN3 and insulin are co-secreted under conditions that stimulate beta cell secretion. Indeed, UCN3 and insulin are released dose-dependently from MIN6 insulinoma cells stimulated with increasing concentrations of glucose and with depolarizing (30 mM) concentrations of KCl [43]. Stimulation of primary islets over the course of 4 hours with 16.8 mM glucose also leads to a detectable rise in UCN3 [8]. Therefore, without ruling out a scenario of differential release of UCN3 and insulin via ‘kiss-and-run’ exocytosis [44], the fact that UCN3 and insulin are both peptide hormones of approximately similar size suggests that the simplest model of the regulation of UCN3 release is one where UCN3 is co-released with insulin under conditions that stimulate beta cells.
Figure 3: Colocalization of UCN3 and insulin in beta cell secretory granules.
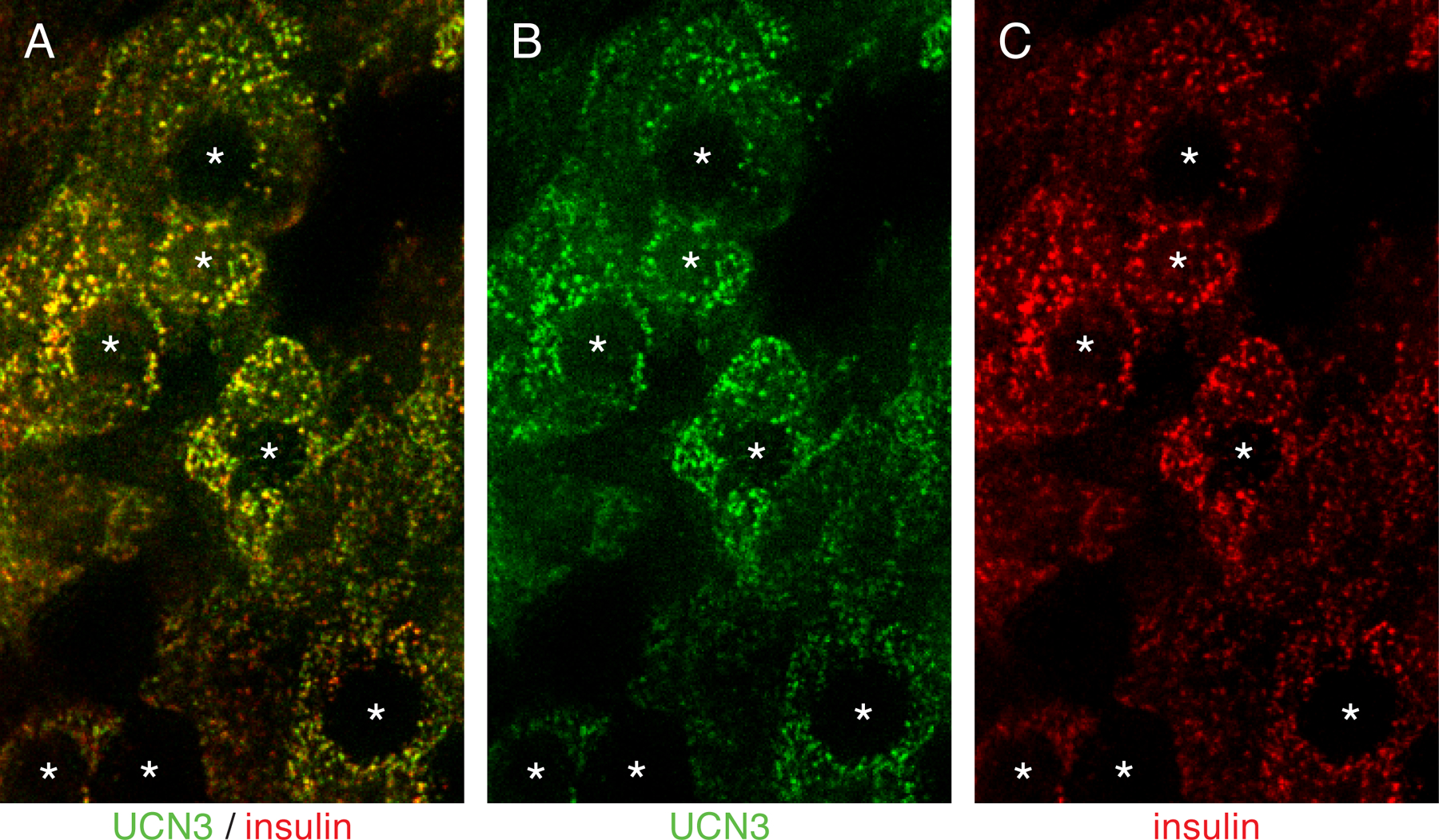
Structured illumination microscopy demonstrates that UCN3 and insulin overlap with high fidelity in the secretory vesicles of mouse beta cells. Nuclear shades are indicated by asterisks.
The receptor for UCN3 in the pancreas is CRHR2α expressed by delta cells
CRH family peptides exclusively signal through the corticotropin-releasing hormone receptors 1 and 2. These two G protein-coupled receptors are members of the class B family of GPCRs that includes the receptors for glucagon, glucose-stimulated insulinotropic peptide (GIP) and glucagon-like peptides-1 and −2 (GLP-1 and GLP-2). Members of the class B GPCR family canonically couple to Gas, leading to the activation of adenylate cyclase and the generation of cyclic AMP (cAMP), and CRHR1 and CRHR2 are no exception [45]. CRH has high affinity for CRHR1 and slightly lower affinity for CRHR2, while UCN is a high affinity agonist to both CRHRs [6]. In contrast, UCN2 and UCN3 are rather selective CRHR2 agonists, which effectively means that UCN3 under physiological circumstances will act exclusively via CRHR2 [6, 45].
CRHR2 can be transcribed as an alpha and a beta isoform that arise from the use of alternative starting exons from a single Crhr2 gene. In primates, a gamma isoform is reported as well. To generate the CRHR2 beta isoform exons 1, 2, are spliced to exon 4–14 and further downstream. In contrast, Crhr2α is generated from an alternative starting exon 3 that is spliced to exons 4 and downstream. Since exons 1–2 (beta) and 3 (alpha) are preceded by their own proximal promotor and encode largely the signal peptide, both CRHR2 isoforms share similar pharmacology, but different regulation of their expression [7]. CRHR2α is the predominant isoform expressed by the CNS, with CRHR2β expressed at select sites including the choroid plexus. Early reviews on the distribution of the alpha and beta CRHR2 isoform tend to attribute all peripherally expressed CRHR2 to the beta isoform. While skeletal and cardiac muscle, each major sites of CRHR2 expression, indeed express the beta isoform [7], pancreatic delta cells and any islet-derived endocrine cell lines that express CRHR2 express the alpha isoform [8, 46].
UCN3 stimulates SST secretion
The evidence of UCN3 stimulating delta cells directly to inhibit insulin secretion is strong. The alpha isoform of Crhr2 – the only known UCN3 receptor – is expressed by delta cells. UCN3 stimulation of islets increases SST secretion. Importantly, the Crhr2-specific peptide antagonist Astressin2b [35] reduces glucose-stimulated SST secretion in dynamic perfusion assays, which allows for a rise in glucose-stimulated insulin secretion [8]. These observations not only demonstrate that delta cells are the direct target of UCN3 but that endogenous UCN3 acting in a paracrine fashion is responsible for a substantial portion of the glucose-stimulated insulin secretion. Another line of evidence in support of UCN3 activating delta cells is provided by the observations that Ucn3 null mice [43], have reduced delta cell numbers, islet SST content and islet Sst mRNA expression [8]. Alpha and beta cell numbers and glucagon and insulin expression were not affected confirming the specificity of the delta cell defect in Ucn3 null mice.
Mechanistically, delta cells just like beta cells require a combination of a nutrient-stimulus to trigger secretion, and cAMP from the activation of a Gas-coupled GPCR to maximize secretion. Stimulation of SST release by glucose from intact islets can be blocked in full by either the KATP channel agonist diazoxide, or the L-type voltage-gated calcium channel antagonist isradipine, demonstrating that both channels are required for the islet to respond to UCN3. Importantly, this blockade cannot be rescued by exogenous UCN3, which demonstrates that KATP channels and voltage-gated calcium channels are required in a delta cell-autonomous fashion to enable the potentiation of SST secretion by UCN3. Activation of CRHR2α by UCN3 then is similar to the role played by activation of incretin and glucagon receptors on beta cells: all these receptors are related members of the class B family of GPCRs that canonically lead to the activation of Gαs, including the dissociation of the Gas subunit and activation of adenylyl cyclase, which converts ATP into cAMP. Indeed, UCN3 stimulation of delta cells leads to a robust induction of cAMP levels [Noguchi et al., submitted]. This rise in cAMP then promotes SST secretion from the delta cell, presumably the activation of EPAC and PKA-dependent downstream pathways mediate the potentiation of insulin secretion by incretin signaling in beta cells. The important contribution of cAMP to glucose-stimulated SST was confirmed by a recent paper that demonstrates a glucose-dependent source of cAMP [47] that is fully consistent with the role of UCN3 that we described a few years earlier [8]. In addition to the rapid effects of UCN3 on the secretion of SST, UCN3 stimulation also leads to the activation of a transcriptional complex that includes the cullin 4B-RING E3 ligase and polycomb repressive complex 2 that reduces the expression of Cav1.2 and adenylate cyclase 6 [48]. This presumably represents an epigenetic negative feedback mechanism that de-sensitizes delta cells from continuous UCN3 stimulation and prevent sustained SST release.
UCN3 inhibits -not stimulates - insulin secretion
Despite the unambiguous evidence regarding the role of UCN3 in stimulating delta cells, there remains quite a bit of confusion and more than a few casual references to outdated literature that suggests that UCN3 is an insulinotropic agent. This is not helpful to our field moving forward, which is why it is worthwhile to dedicate a paragraph on how views on the role of UCN3 within the islet have shifted, based on current insights and observations. Shortly after UCN3 was discovered, the original role attributed to UCN3 was that it stimulates insulin secretion [10, 43], implying that beta cells express CRHR2 receptors. Clonal MIN6 beta cells indeed do express low levels of CRHR2α, which are insufficient to elicit a cAMP response [46]. However, Crhr2α levels can be upregulated by dexamethasone pre-treatment to render MIN6 cells directly sensitive to UCN3 stimulation [46]. In vivo a scenario where UCN3 would activate beta cells would effectively constitute and autocrine positive feedback loop of insulin (and UCN3) secretion. Such feedback loops are exceptions in physiology and the model of UCN3 acting directly on the beta cell to stimulate insulin secretion has subsequently been disproven by the discovery that CRHR2α is selectively expressed by SST-secreting delta cells in the pancreatic islets, as discussed earlier [8]. Indeed, UCN3 stimulates SST secretion and simultaneously reduces static and dynamic insulin release from intact islets. This finding established the currently accepted role for UCN3 signaling to amplify SST secretion thereby inhibiting, rather than activating insulin secretion [8]. Even though this paper revised the earlier model with the regard to the islet cell type that expresses CRHR2 that is therefore capable of responding to locally released UCN3, the primary data in the more recent 2015 paper and the earlier papers that concluded UCN3 is insulinotropic is at times remarkably similar. For example, both papers show that the acute administration of UCN3 peptide causes a significant reduction in glucose tolerance in rats [10] and mice [8]. Conversely, the genetic deletion of UCN3 improves glucose tolerance [43]. These observations are hard to reconcile with an insulinotropic role of UCN3 but are perfectly consistent with a model where UCN3 stimulates SST to inhibit insulin release, thereby aggravating glucose tolerance. Furthermore, co-administration of SST antagonists with UCN3 prevents the reduction of glucose tolerance that is induced by UCN3 alone [8], demonstrating that intact SST signaling is required for UCN3 to inhibit insulin release and aggravate glucose tolerance. A possible caveat to these glucose tolerance experiments is that systemic administration of CRHR2 agonists stimulates gastric emptying, which could lead to faster glucose uptake to explain the peak in glucose intolerance. However, the glucose tolerance tests in question were intraperitoneal glucose tolerance tests on fasted animals; conditions where the rate of gastric emptying does not contribute to glucose (in)tolerance.
UCN3 as a beta cell maturation marker
As one of the principal regulators of plasma glucose levels, insulin expression is a hallmark feature of the β-cell. As such, labeling insulin-positive cells is a reliable way to identify beta cells among other endocrine cell types in cultured islets and pancreatic tissues. However, this distinguishing feature alone does not convey the functional maturity status of a β-cell, since insulin is expressed relatively early in β-cell development prior to the induction of key genes encoding proteins that confer the necessary traits for glucose responsiveness such as the Glut2 glucose transporter. Among these maturity markers, UCN3 stands out as perhaps the most suitable single-marker that can be used to identify functionally mature beta cells as it 1) appears late in development, 2) disappears early in disease and 3) can be easily detected by immunofluorescence using antibodies that are highly specific [49–51].
During mouse development, insulin-positive cells are detectable as early as E9.5. By E13.5 insulin-expressing cells are widespread throughout pancreatic islets [52, 53]. In contrast, the onset of UCN3 expression occurs at E17.5 and the UCN3 peptide is not homogeneously expressed across most β cells until postnatal day 14 [49]. This late onset places it well after the expression of other known maturity markers such as neuroD [54] and MAFA [53], or the downregulation of MAFB, which in mice is initially expressed by immature beta cells before they down regulate it during maturation [55].
While UCN3 is expressed in both beta cells and alpha cells of human islets, it remains a reliable marker of mature beta cells in humans. Similar to the observed timeline in rodents, UCN3 expression is nearly undetectable in immature beta cells at the onset of insulin expression occurring at the beginning of the fetal stage between 8 to 9 weeks post-conception. In the ensuing weeks, insulin/UCN3 co-positive cells increase in frequency as more beta cells differentiate and acquire glucose sensing capacity. It is not until the end of the first trimester that UCN3 expression is evident in a majority of insulin-positive cells, placing its onset well-after other markers of β-cell maturity [56]. Further supporting this notion that UCN3 is tightly correlated with β-cell maturity is the delayed induction of UCN3 in beta cells derived from human embryonic stem cell (hESC)-derived pancreatic endoderm following their engraftment into mice [49]. In culture, UCN3 expression is nearly undetectable in the hESC-derived pancreatic endoderm at any stage, with the exception being a small subpopulation of polyhormonal cells. But after 128 days in recipient mice, strong UCN3 immunoreactivity is observed in 79% of insulin-positive cells that arise from the engrafted hESC-derived pancreatic endoderm. Notably, these insulin/UCN3 co-positive cells were shown to express other established maturity markers such as Pdx1 and Nkx6.1, further validating its sensitivity as a maturity marker even in the context of stem cell derived beta cells. As such, it is becoming increasingly common for UCN3 to be included in marker panels used to evaluate the degree of maturity achieved in β-like cells differentiated from human stem cell-derived progenitors, along with insulin secretory profiles and other measures of functional maturity [57–62].
The physiological role of UCN3 signaling
The physiological setpoint for the plasma glucose concentration in both mouse and human is in large part determined by the net balance between insulin and glucagon. Changes in the release of either of these hormones alters the uptake of plasma glucose or production of hepatic glucose via gluconeogenesis and glycogenolysis, respectively. SST secretion in turn provides important negative paracrine feedback on both alpha and beta cells [63, 64] (Figure 4) as well as autocrine feedback inhibition to limit SST secretion [65]. Since UCN3 amplifies SST secretion from delta cells and is co-secreted with insulin in response to hyperglycemia, it acts as a modulator to constrain insulin output under these conditions [66]. The contribution of this feedback inhibition by the delta onto the beta cell is manifested as exaggerated glucose-stimulated insulin secretion from Ucn3 null islets [8] leading to improved glucose tolerance that we discussed earlier [43]. The glucose set point in Ucn3 null mice at the start of the glucose tolerance test (i.e. after an overnight fast) is relatively normal, and the glucose-stimulated calcium response of Ucn3 null beta cells proceeds in the slowly oscillating calcium synchronized waves of calcium that are indistinguishable from the responses of healthy beta cells [67]. This means that UCN3 has little effect over the calcium response elicited by glucose in beta cells, which is in line with UCN3 activating the release of SST which in turn activate Gαi proteins and limit cAMP – less so calcium.
Figure 4: Overview of paracrine signaling in the islet involving UCN3.
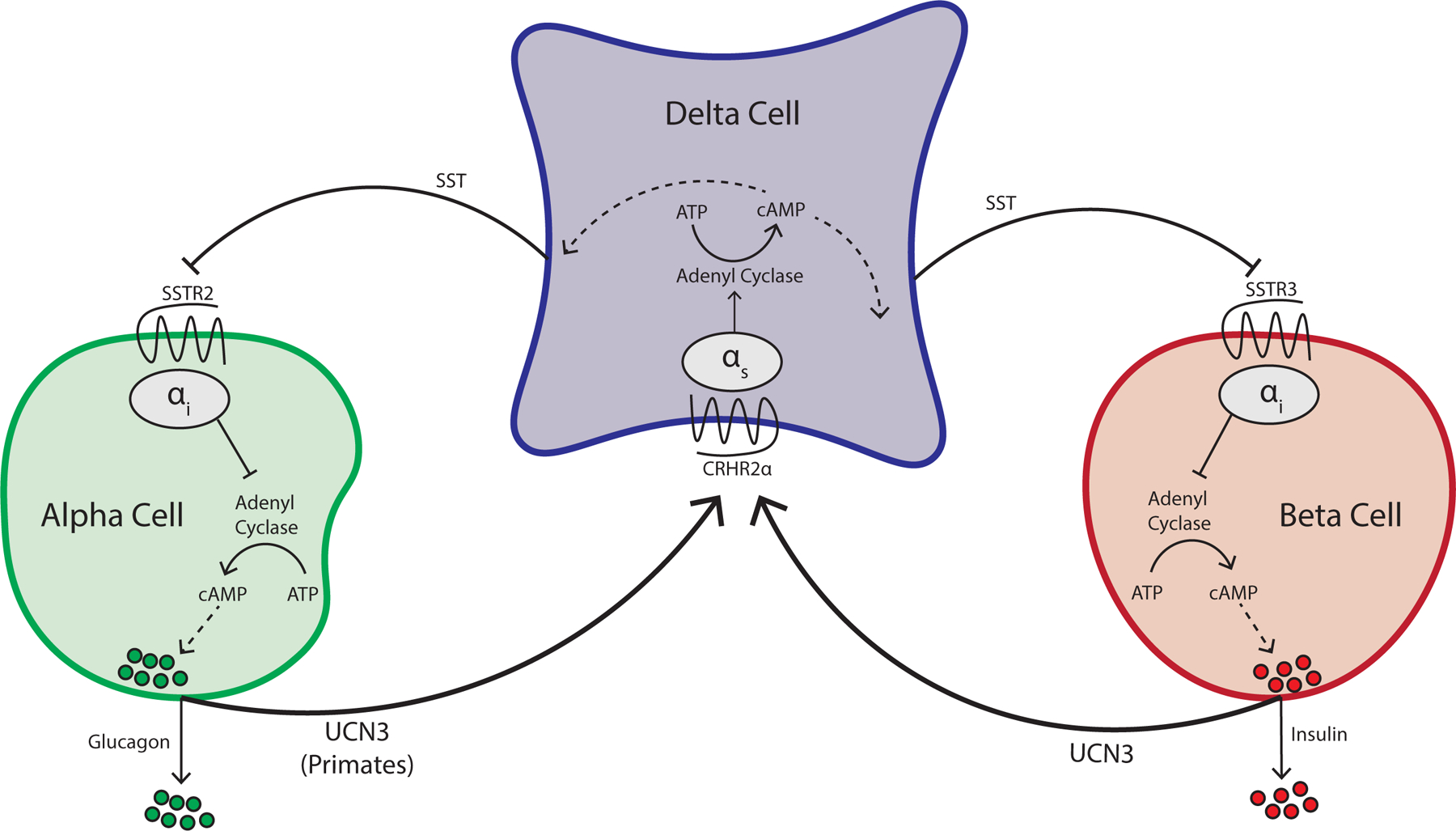
Beta cells co-secrete UCN3 along with insulin to activate its cognate receptor CRHR2α, which is selectively expressed on delta cells and amplifies SST negative feedback on alpha and beta cells. CRHR2 is a Gαs-coupled G-protein coupled receptor, which raises cAMP in delta cells to potentiate SST secretion. UCN3 is also expressed by alpha cells in primate, but not rodent islets.
To understand the physiological contribution of beta cell UCN3 to glucose control, it is helpful to return to the perinatal period in mice where the timing of UCN3 expression in beta cells aligns closely with an increase in the glucose threshold for insulin secretion. This transition is correlated with a marked suppression of plasma insulin levels that is associated with an uptick in non-fasting blood glucose setpoint that persists through adulthood [68]. Premature induction of UCN3 in mouse beta cells at the same level as endogenous UCN3 expression during the early postnatal maturation period is sufficient to prematurely raise the blood glucose setpoint by suppressing insulin secretion. This observation establishes causality for the role of UCN3 as an activator of somatostatin-mediated feedback inhibition on β-cell insulin secretion in mice [8]. In essence, UCN3 signaling acts to stimulate SST negative feedback on the beta cell, thereby attenuating insulin secretion. This UCN3-driven reduction in insulin secretion requires greater plasma glucose to stimulate the same level of insulin secretion that occurs in the absence of UCN3 [8].
UCN3 as a dedifferentiation marker in Type 2 Diabetes
Numerous studies in both rodent and human islets have demonstrated that beta cells gradually reduce the expression of key transcription factors along with other maturity markers, while also upregulating specific endocrine progenitor genes during the progression of type 2 diabetes (T2D) [69]. This change is thought to specify a population of beta cells that have undergone dedifferentiation in the face of metabolically stressful conditions associated with constant hyperglycemia. Dedifferentiation in beta cells loosely refers to a loss of cell identity resulting in an altered phenotype, rather than a strict reversal towards an early, progenitor-like state. In this context, the utility of UCN3 as a biomarker of functional, mature beta cells makes it well suited for identifying dedifferentiated beta cells under diabetic conditions as well.
Downregulation of UCN3 in beta cells has been shown to occur relatively early in various mouse models of T2D. For example, beta cells from obese leptin deficient (C57BL/6-LepOb/Ob) and leptin receptor deficient (C57BL/6-Leprdb/db) diabetic mice both show significantly lower UCN3 transcript and peptide content when compared to lean age matched controls [8, 51]. Notably, this downregulation occurs early in disease, prior to the well-characterized reduction of insulin expression in the beta cells of overtly diabetic mice. These findings were recently corroborated by whole-genome RNA-sequencing of lineage traced beta cells that were isolated in a different strain of obese leptin deficient mice (BTBR-LepOb/Ob), where UCN3 was the only marker that was consistently upregulated during maturation and downregulated under diabetic conditions [70]. Downregulation of UCN3 in beta cells is also observed in non-obese, insulin-dependent diabetic (Ins2Akita) mice suggesting that these changes are not directly mediated by deficiencies in leptin signaling or congenital obesity [51] In fact, this same study demonstrated that transient induction of hyperglycemia (≥500 mg/dl) in wild-type mice with the insulin-receptor antagonist S961 over a period 7 days was sufficient to drastically reduce UCN3 expression. Perhaps the more striking observation was that UCN3 expression was restored in mice that recovered normoglycemia after withdrawal of the S961 treatment, once again highlighting the close correlation between β-cell status and UCN3 expression. In addition, the conditional islet cell-specific Nkx6.1 knockout mice is another model of the loss of beta cell identity where UCN3 is downregulated [71]. Since Nkx6.1 plays an essential role in regulating genes involved in maintaining the molecular and functional traits of beta cells, Nkx6.1 ablation results in rapid-onset diabetes due to destabilized β-cell identity and accompanying defects in insulin secretion. Here, although the expression of markers such as Pdx1 and NeuroD were unaffected by Nkx6.1 deletion, Ucn3 transcript and peptide levels were markedly reduced in the beta cells of these mice.
Similar observations can be made in islets of humans and non-human primate models with regards to UCN3 expression under diabetic conditions. In islets of human T2D donors, UCN3 is selectively downregulated in beta cells, but not alpha cells [8]. In line with these observations in human T2D donors, pre-diabetic macaques that had developed insulin resistance as a consequence of chronic high-fat diet already displayed severe reduction of UCN3 immunoreactivity in their beta cells, but not alpha cells within the same islets. The early loss of UCN3 from beta cells would be reflected in the loss of delta cell activity and the removal of somatostatin-mediated beta cell inhibition that would maximize insulin secretion in the face of rising peripheral insulin resistance. Indeed, reversal of the loss of UNC3 in beta cells of ob/ob mice via the transgenic re-expression of Ucn3 at levels comparable to normal endogenous Ucn3 mRNA led to the immediate aggravation of diabetes via the renewed partial suppression of insulin release [8].
The loss in UCN3 expression by beta cells is not reflected in the rising plasma UCN3 levels reported in newly diagnosed T2D human subjects with metabolic syndrome compared to their lean counterparts [12]. Since UCN3 is hardly detectable in beta cells of T2D donors, elevated circulating UCN3 would have explained by the close association with metabolic syndrome and increased UCN3 expression in other tissues, such as subcutaneous adipose tissue [72]. While the expression and the role of UCN3 outside the context of pancreatic islets is not well understood, its role in the islet and utility as a marker of β-cell maturity and de-differentation is clear.
Connections of UCN3 to Type 1 Diabetes
In line with the observation that UCN3 is downregulated in mouse models of T2D and in non-human primates and human patients with (pre)diabetes, UCN3 expression in residual beta cells from donors with Type 1 Diabetes is lower compared to donors without diabetes. There are two likely scenarios to account for this selective reduction in Type 1 Diabetes and they are not mutually exclusive. The first, and perhaps most likely scenario is that UCN3 is downregulated in residual beta cells as a consequence of the cellular stresses of the pro-inflammatory and/or autoimmune-mediated insults that are prevalent locally within the islet environment. Indeed, single cell RNA-Seq data of beta cells in the NOD model reflect a loss of UCN3 expression that correlates with increased markers of cell cycle arrest and cellular senescence [15]. Also in the context of T1D, it has been documented that UCN3 expression in intraportal islet allotransplant recipients is lost in all beta cells, but retained in the large majority of alpha cells within the same graft [73]. It is notable that this sustained loss in UCN3 expression was accompanied by overall graft function as measured by sustained insulin independence and HbA1c values in the normal range [73]. This indicates that beta cell function can be dissociated from UCN3 expression, in line with the recent demonstration that direct genetic deletion of UCN3 by itself does not affect expression of other beta cell maturity markers such as MafA, Slc2a2, and Pdx1 and also does not interfere with normal pulsatile beta cell calcium responses to hyperglycemia [67].
An alternative explanation for the observation of UCN3 negative beta cells in donors with T1D may reflect the possibility that such UCN3-negative beta cells represent an immature-like subset of beta cells resembling UCN3-negative virgin beta [74] cells that are long-lived and turn over slowly [75] and are selectively spared from cytotoxic T cell-mediated death. Indeed, a peptide derived from the UCN3 signal peptide (UCN31–9) is a beta cell epitope present in the HLA-I peptidome [76], which means that it is presented naturally in the context of MHC class I (HLA-A2) by beta cells. This combination of HLA-A2/UCN31–9 was detected by naïve CD8+ T cells that circulate in donors with and without T1D, but were enriched among pancreas-infiltrating cytotoxic T cells [76]. Whether these UCN31–9 -selective cytotoxic T cells play a direct role in cytotoxic T cell-mediated beta cell destruction has not been definitively shown. Whether human alpha present the same UCN31–9 peptide in the context of HLA-A2 is not known. Since UCN31–9 is not unique among the HLA-I peptidome, which also contains conventional epitopes derived from ChromograninA, ChromograninB, and Pcsk2 that are not unique to human beta cells [76], the question of the susceptibility of beta cells to subsequent autoimmune attach may relate to the upregulation of HLA-I by beta cells [77].
Outstanding questions
In conclusion, UCN3 is a peptide hormone that is a relative newcomer to the islet field, despite being expressed rather abundantly. Its physiological role is as the main intra-islet activator of somatostatin secretion from neighboring delta cells to activate islet-intrinsic feedback inhibition to restrain insulin secretion. This feedback inhibition is established relatively late in development by the relatively late onset of UCN3 expression in comparison to essentially all other genes that establish beta cell identity. However, there are also several areas of UCN3 biology in general and its intersection with islet physiology that remain unknown. One of these is the contribution of UCN3 in human and non-human primate alpha cells, where we lack a direct demonstration of the contributions of alpha cell-derived UCN3 to the activation of somatostatin feedback and whether this feedback is aimed primarily at alpha or beta cells. Related to this, the transcriptional and epigenetic mechanisms that regulate the expression of UCN3 during developmental time, in diabetes and differentially in primate and non-primate alpha cells are not fully understood. And another outstanding question that is broader than just the pancreatic islet regards the processing of endogenous UCN3 as there has to date not been a direct demonstration of the exact processing site(s) of mature, bioactive UCN3 peptide isolated from an endogenous site. While answers to these outstanding questions are hopefully forthcoming, the reader is hopefully left with the impression of UCN3 is a novel peptide hormone that plays an important role in the modulation of plasma glucose levels as the principal intra-islet activator of delta cell somatostatin release. Related to this role, and perhaps reflecting an adaptive response by the islets to maximize insulin output in the face of rising insulin demand in Type 2 Diabetes, the loss of UCN3 is an early indicator of islet dysfunction that is furthermore readily detectable by standard immunohistochemical techniques. These properties warrant the inclusion of UCN3 in any panel of beta cell markers that are used to either assess the maturation of stem cell-derived beta-like cells or their dedifferentiation in the context of diabetes.
Funding.
Research in the Huising lab was supported by grants from the JDRF (CDA-2-2013-54 & 2-SRA-2021-1054), ADA (1-19-IBS-078) and NIH/NIDDK (R01 DK110276).
Duality of Interest.
MOH has received funding and honoraria from Crinetics Inc. to investigate somatostatin analogs that are not discussed in this review.
References
- [1].Noguchi GM, Huising MO, Integrating the inputs that shape pancreatic islet hormone release, Nature Metabolism 1(12) (2020) 1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hartig SM, Cox AR, Paracrine signaling in islet function and survival, J Mol Med (Berl) 98(4) (2020) 451–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hsu SY, Hsueh AJ, Human stresscopin and stresscopin-related peptide are selective ligands for the type 2 corticotropin-releasing hormone receptor, Nature medicine 7(5) (2001) 605–11. [DOI] [PubMed] [Google Scholar]
- [4].Reyes TM, Lewis K, Perrin MH, Kunitake KS, Vaughan J, Arias CA, Hogenesch JB, Gulyas J, Rivier J, Vale WW, Sawchenko PE, Urocortin II: a member of the corticotropin-releasing factor (CRF) neuropeptide family that is selectively bound by type 2 CRF receptors, Proc Natl Acad Sci U S A 98(5) (2001) 2843–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lewis K, Li C, Perrin MH, Blount A, Kunitake K, Donaldson C, Vaughan J, Reyes TM, Gulyas J, Fischer W, Bilezikjian L, Rivier J, Sawchenko PE, Vale WW, Identification of urocortin III, an additional member of the corticotropin-releasing factor (CRF) family with high affinity for the CRF2 receptor, Proc. Natl. Acad. Sci. USA 98(13) (2001) 7570–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lovejoy DA, Chang BS, Lovejoy NR, del Castillo J, Molecular evolution of GPCRs: CRH/CRH receptors, J Mol Endocrinol 52(3) (2014) T43–60. [DOI] [PubMed] [Google Scholar]
- [7].Hillhouse EW, Grammatopoulos DK, The molecular mechanisms underlying the regulation of the biological activity of corticotropin-releasing hormone receptors: implications for physiology and pathophysiology, Endocr Rev 27(3) (2006) 260–86. [DOI] [PubMed] [Google Scholar]
- [8].van der Meulen T, Donaldson CJ, Caceres E, Hunter AE, Cowing-Zitron C, Pound LD, Adams MW, Zembrzycki A, Grove KL, Huising MO, Urocortin3 mediates somatostatin-dependent negative feedback control of insulin secretion, Nat Med 21(7) (2015) 769–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].DiGruccio MR, Mawla AM, Donaldson CJ, Noguchi GM, Vaughan J, Cowing-Zitron C, van der Meulen T, Huising MO, Comprehensive alpha, beta and delta cell transcriptomes reveal that ghrelin selectively activates delta cells and promotes somatostatin release from pancreatic islets, Mol Metab 5(7) (2016) 449–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Li C, Chen P, Vaughan J, Blount A, Chen A, Jamieson PM, Rivier J, Smith MS, Vale W, Urocortin III is expressed in pancreatic beta-cells and stimulates insulin and glucagon secretion, Endocrinology 144(7) (2003) 3216–24. [DOI] [PubMed] [Google Scholar]
- [11].Jamieson PM, Cleasby ME, Kuperman Y, Morton NM, Kelly PA, Brownstein DG, Mustard KJ, Vaughan JM, Carter RN, Hahn CN, Hardie DG, Seckl JR, Chen A, Vale WW, Urocortin 3 transgenic mice exhibit a metabolically favourable phenotype resisting obesity and hyperglycaemia on a high-fat diet, Diabetologia 54(9) (2011) 2392–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Alarslan P, Unal Kocabas G, Demir I, Guler A, Bozkaya G, Aslanipour B, Calan M, Increased urocortin 3 levels are associated with the risk of having type 2 diabetes mellitus, Journal of diabetes 12(6) (2020) 474–482. [DOI] [PubMed] [Google Scholar]
- [13].Kim A, Miller K, Jo J, Kilimnik G, Wojcik P, Hara M, Islet architecture: A comparative study, Islets 1(2) (2009) 129–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dolensek J, Rupnik MS, Stozer A, Structural similarities and differences between the human and the mouse pancreas, Islets 7(1) (2015) e1024405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Thompson PJ, Shah A, Ntranos V, Van Gool F, Atkinson M, Bhushan A, Targeted Elimination of Senescent Beta Cells Prevents Type 1 Diabetes, Cell Metab 29(5) (2019) 1045–1060 e10. [DOI] [PubMed] [Google Scholar]
- [16].Benner C, van der Meulen T, Caceres E, Tigyi K, Donaldson CJ, Huising MO, The transcriptional landscape of mouse beta cells compared to human beta cells reveals notable species differences in long non-coding RNA and protein-coding gene expression, BMC Genomics 15(1) (2014) 620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kanno T, Suga S, Nakano K, Kamimura N, Wakui M, Corticotropin-releasing factor modulation of Ca2+ influx in rat pancreatic beta-cells, Diabetes 48(9) (1999) 1741–6. [DOI] [PubMed] [Google Scholar]
- [18].Kageyama K, Kimura R, Suga S, Ogawa Y, Suda T, Wakui M, Modulation of Ca2+ influx by corticotropin-releasing factor (CRF) family of peptides via CRF receptors in rat pancreatic beta-cells, Peptides 27(7) (2006) 1814–9. [DOI] [PubMed] [Google Scholar]
- [19].Huising MO, van der Meulen T, Vaughan JM, Matsumoto M, Donaldson CJ, Park H, Billestrup N, Vale WW, CRFR1 is expressed on pancreatic beta cells, promotes beta cell proliferation, and potentiates insulin secretion in a glucose-dependent manner, Proc. Natl. Acad. Sci. USA 107(2) (2010) 912–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cappozi ME, Svendsen B, Encisco SE, Lewandowski SL, Martin MD, Lin H, Jaffe JL, Coch RW, Haldeman JM, MacDonald PE, Merrins MJ, D’Alessio DA, Campbell JE, Beta-cell tone is defined by proglucagon peptides through cyclic AMP signalling, JCI Insight 4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Svendsen B, Larsen O, Gabe MBN, Christiansen CB, Rosenkilde MM, Drucker DJ, Holst JJ, Insulin Secretion Depends on Intra-islet Glucagon Signaling., Cell reports 25(5) (2018). [DOI] [PubMed] [Google Scholar]
- [22].Zhu L, Dattaroy D, Pham J, Wang L, Barella LF, Cui Y, Wilkins KJ, Roth BL, Hochgeschwender U, Matschinsky FM, Kaestner KH, Doliba NM, Wess J, Intra-islet glucagon signaling is critical for maintaining glucose homeostasis., JCI Insight 23(5) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kuperman Y, Chen A, Urocortins: emerging metabolic and energy homeostasis perspectives, Trends Endocrinol Metab 19(4) (2008) 122–9. [DOI] [PubMed] [Google Scholar]
- [24].Fekete EM, Zorrilla EP, Physiology, pharmacology, and therapeutic relevance of urocortins in mammals: ancient CRF paralogs, Front Neuroendocrinol 28(1) (2007) 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Deussing JM, Chen A, The Corticotropin-Releasing Factor Family: Physiology of the Stress Response, Physiol Rev 98(4) (2018) 2225–2286. [DOI] [PubMed] [Google Scholar]
- [26].Deussing JM, Breu J, Kuhne C, Kallnik M, Bunck M, Glasl L, Yen YC, Schmidt MV, Zurmuhlen R, Vogl AM, Gailus-Durner V, Fuchs H, Holter SM, Wotjak CT, Landgraf R, de Angelis MH, Holsboer F, Wurst W, Urocortin 3 modulates social discrimination abilities via corticotropin-releasing hormone receptor type 2, J Neurosci 30(27) (2010) 9103–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Li C, Vaughan J, Sawchenko PE, Vale WW, Urocortin III-immunoreactive projections in rat brain: partial overlap with sites of type 2 corticotrophin-releasing factor receptor expression, J Neurosci 22(3) (2002) 991–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cavalcante JC, Sita LV, Mascaro MB, Bittencourt JC, Elias CF, Distribution of urocortin 3 neurons innervating the ventral premammillary nucleus in the rat brain, Brain Res 1089(1) (2006) 116–25. [DOI] [PubMed] [Google Scholar]
- [29].Bale TL, Picetti R, Contarino A, Koob GF, Vale WW, Lee KF, Mice deficient for both corticotropin-releasing factor receptor 1 (CRFR1) and CRFR2 have an impaired stress response and display sexually dichotomous anxiety-like behavior, J Neurosci 22(1) (2002) 193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhang X, Liu Y, Qi J, Tian Z, Tang N, Chen D, Li Z, Progress in understanding the roles of Urocortin3 (UCN3) in the control of appetite from studies using animal models, Peptides 121 (2019) 170124. [DOI] [PubMed] [Google Scholar]
- [31].Ushikai M, Asakawa A, Sakoguchi T, Tanaka C, Inui A, Centrally administered urocortin 3 inhibits food intake and gastric emptying in mice, Endocrine 39(2) (2011) 113–7. [DOI] [PubMed] [Google Scholar]
- [32].Yuan PQ, Wu SV, Tache Y, Urocortins and CRF type 2 receptor isoforms expression in the rat stomach are regulated by endotoxin: role in the modulation of delayed gastric emptying, Am J Physiol Gastrointest Liver Physiol 303(1) (2012) G20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Saruta M, Takahashi K, Suzuki T, Fukuda T, Torii A, Sasano H, Urocortin 3/stresscopin in human colon: possible modulators of gastrointestinal function during stressful conditions, Peptides 26(7) (2005) 1196–206. [DOI] [PubMed] [Google Scholar]
- [34].Grun D, Lyubimova A, Kester L, Wiebrands K, Basak O, Sasaki N, Clevers H, van Oudenaarden A, Single-cell messenger RNA sequencing reveals rare intestinal cell types, Nature 525(7568) (2015) 251–5. [DOI] [PubMed] [Google Scholar]
- [35].Rivier J, Gulyas J, Kirby D, Low W, Perrin MH, Kunitake K, DiGruccio M, Vaughan J, Reubi JC, Waser B, Koerber SC, Martinez V, Wang L, Tache Y, Vale W, Potent and long-acting corticotropin releasing factor (CRF) receptor 2 selective peptide competitive antagonists, Journal of medicinal chemistry 45(21) (2002) 4737–47. [DOI] [PubMed] [Google Scholar]
- [36].Imperatore A, Florio P, Torres PB, Torricelli M, Galleri L, Toti P, Occhini R, Picciolini E, Vale W, Petraglia F, Urocortin 2 and urocortin 3 are expressed by the human placenta, deciduas, and fetal membranes, Am J Obstet Gynecol 195(1) (2006) 288–95. [DOI] [PubMed] [Google Scholar]
- [37].Takahashi K, Totsune K, Murakami O, Shibahara S, Urocortins as cardiovascular peptides, Peptides 25(10) (2004) 1723–31. [DOI] [PubMed] [Google Scholar]
- [38].Dermitzaki E, Liapakis G, Androulidaki A, Venihaki M, Melissas J, Tsatsanis C, Margioris AN, Corticotrophin-Releasing Factor (CRF) and the urocortins are potent regulators of the inflammatory phenotype of human and mouse white adipocytes and the differentiation of mouse 3T3L1 pre-adipocytes, PLoS One 9(5) (2014) e97060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Fukuda T, Takahashi K, Suzuki T, Saruta M, Watanabe M, Nakata T, Sasano H, Urocortin 1, urocortin 3/stresscopin, and corticotropin-releasing factor receptors in human adrenal and its disorders, J Clin Endocrinol Metab 90(8) (2005) 4671–8. [DOI] [PubMed] [Google Scholar]
- [40].Wiley KE, Davenport AP, CRF2 receptors are highly expressed in the human cardiovascular system and their cognate ligands urocortins 2 and 3 are potent vasodilators, Br J Pharmacol 143(4) (2004) 508–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Rorsman P, Braun M, Regulation of insulin secretion in human pancreatic islets, Annu. Rev. Physiol 75 (2013) 155–79. [DOI] [PubMed] [Google Scholar]
- [42].Rorsman P, Ashcroft FM, Pancreatic beta-Cell Electrical Activity and Insulin Secretion: Of Mice and Men, Physiol Rev 98(1) (2018) 117–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Li C, Chen P, Vaughan J, Lee KF, Vale W, Urocortin 3 regulates glucose-stimulated insulin secretion and energy homeostasis, Proc. Natl. Acad. Sci. USA 104(10) (2007) 4206–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].MacDonald PE, Braun M, Galvanovskis J, Rorsman P, Release of small transmitters through kiss-and-run fusion pores in rat pancreatic beta cells, Cell Metab 4(4) (2006) 283–90. [DOI] [PubMed] [Google Scholar]
- [45].Grammatopoulos DK, Insights into mechanisms of corticotropin-releasing hormone receptor signal transduction, Br J Pharmacol 166(1) (2012) 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Huising MO, Pilbrow AP, Matsumoto M, van der Meulen T, Park H, Vaughan JM, Lee S, Vale WW, Glucocorticoids differentially regulate the expression of CRFR1 and CRFR2alpha in MIN6 insulinoma cells and rodent islets, Endocrinology 152(1) (2011) 138–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Denwood G, Tarasov A, Salehi A, Vergari E, Ramracheya R, Takahashi H, Nikolaev VO, Seino S, Gribble F, Reimann F, Rorsman P, Zhang Q, Glucose stimulates somatostatin secretion in pancreatic delta-cells by cAMP-dependent intracellular Ca(2+) release, J Gen Physiol 151(9) (2019) 1094–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Li Q, Cui M, Yang F, Li N, Jiang B, Yu Z, Zhang D, Wang Y, Zhu X, Hu H, Li PS, Ning SL, Wang S, Qi H, Song H, He D, Lin A, Zhang J, Liu F, Zhao J, Gao L, Yi F, Xue T, Sun JP, Gong Y, Yu X, A cullin 4B-RING E3 ligase complex fine-tunes pancreatic delta cell paracrine interactions, J Clin Invest 127(7) (2017) 2631–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].van der Meulen T, Xie R, Kelly OG, Vale WW, Sander M, Huising MO, Urocortin 3 marks mature human primary and embryonic stem cell-derived pancreatic alpha and beta cells, PLoS One 7(12) (2012) e52181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].van der Meulen T, Huising MO, Role of transcription factors in the transdifferentiation of pancreatic islet cells, J Mol Endocrinol 54(2) (2015) R103–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Blum B, Roose AN, Barrandon O, Maehr R, Arvanites AC, Davidow LS, Davis JC, Peterson QP, Rubin LL, Melton DA, Reversal of beta cell de-differentiation by a small molecule inhibitor of the TGFbeta pathway, eLife 3 (2014) e02809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Teitelman G, Alpert S, Polak JM, Martinez A, Hanahan D, Precursor cells of mouse endocrine pancreas coexpress insulin, glucagon and the neuronal proteins tyrosine hydroxylase and neuropeptide Y, but not pancreatic polypeptide, Development 118(4) (1993) 1031–9. [DOI] [PubMed] [Google Scholar]
- [53].Matsuoka TA, Artner I, Henderson E, Means A, Sander M, Stein R, The MafA transcription factor appears to be responsible for tissue-specific expression of insulin, Proc Natl Acad Sci U S A 101(9) (2004) 2930–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Naya FJ, Huang HP, Qiu Y, Mutoh H, DeMayo FJ, Leiter AB, Tsai MJ, Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice, Genes Dev 11(18) (1997) 2323–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Artner I, Hang Y, Mazur M, Yamamoto T, Guo M, Lindner J, Magnuson MA, Stein R, MafA and MafB regulate genes critical to beta-cells in a unique temporal manner, Diabetes 59(10) (2010) 2530–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].van der Meulen T, Huising MO, Maturation of Stem Cell-Derived Beta cells Guided by the Expression of Urocortin 3, Rev. Diabet. Stud 11(1) (2014) 115–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, O’Dwyer S, Quiskamp N, Mojibian M, Albrecht T, Yang YH, Johnson JD, Kieffer TJ, Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells, Nat Biotechnol (2014). [DOI] [PubMed] [Google Scholar]
- [58].Singh R, Cottle L, Loudovaris T, Xiao D, Yang P, Thomas HE, Kebede MA, Thorn P, Enhanced structure and function of human pluripotent stem cell-derived beta-cells cultured on extracellular matrix, Stem Cells Transl Med 10(3) (2021) 492–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Nair GG, Liu JS, Russ HA, Tran S, Saxton MS, Chen R, Juang C, Li ML, Nguyen VQ, Giacometti S, Puri S, Xing Y, Wang Y, Szot GL, Oberholzer J, Bhushan A, Hebrok M, Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived beta cells, Nature cell biology 21(2) (2019) 263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Ghazizadeh Z, Kao DI, Amin S, Cook B, Rao S, Zhou T, Zhang T, Xiang Z, Kenyon R, Kaymakcalan O, Liu C, Evans T, Chen S, ROCKII inhibition promotes the maturation of human pancreatic beta-like cells, Nat Commun 8(1) (2017) 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Veres A, Faust AL, Bushnell HL, Engquist EN, Kenty JH, Harb G, Poh YC, Sintov E, Gurtler M, Pagliuca FW, Peterson QP, Melton DA, Charting cellular identity during human in vitro beta-cell differentiation, Nature 569(7756) (2019) 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Velazco-Cruz L, Song J, Maxwell KG, Goedegebuure MM, Augsornworawat P, Hogrebe NJ, Millman JR, Acquisition of Dynamic Function in Human Stem Cell-Derived beta Cells, Stem Cell Reports 12(2) (2019) 351–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Xu SFS, Andersen DB, Izarzugaza JMG, Kuhre RE, Holst JJ, In the rat pancreas, somatostatin tonically inhibits glucagon secretion and is required for glucose-induced inhibition of glucagon secretion, Acta Physiol (Oxf) (2020) e13464. [DOI] [PubMed] [Google Scholar]
- [64].Lai BK, Chae H, Gomez-Ruiz A, Cheng P, Gallo P, Antoine N, Beauloye C, Jonas JC, Seghers V, Seino S, Gilon P, Somatostatin Is Only Partly Required for the Glucagonostatic Effect of Glucose but Is Necessary for the Glucagonostatic Effect of KATP Channel Blockers, Diabetes 67(11) (2018) 2239–2253. [DOI] [PubMed] [Google Scholar]
- [65].Ipp E, Rivier J, Dobbs RE, Brown M, Vale W, Unger RH, Somatostatin analogs inhibit somatostatin release, Endocrinology 104(5) (1979) 1270–3. [DOI] [PubMed] [Google Scholar]
- [66].Huising MO, van der Meulen T, Huang JL, Pourhosseinzadeh MS, Noguchi GM, The Difference delta-Cells Make in Glucose Control, Physiology (Bethesda) 33(6) (2018) 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Huang JL, Lee S, Hoek P, van der Meulen T, Van R, Huising MO, Genetic deletion of Urocortin 3 does not prevent functional maturation of beta cells, J Endocrinol 246(1) (2020) 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Blum B, Hrvatin SS, Schuetz C, Bonal C, Rezania A, Melton DA, Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3, Nat. Biotechnol 30(3) (2012) 261–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Talchai C, Xuan S, Lin HV, Sussel L, Accili D, Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure, Cell 150(6) (2012) 1223–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Nimkulrat SD, Bernstein MN, Ni Z, Brown J, Kendziorski C, Blum B, The Anna Karenina Model of beta-Cell Maturation in Development and Their Dedifferentiation in Type 1 and Type 2 Diabetes, Diabetes 70(9) (2021) 2058–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Taylor BL, Liu FF, Sander M, Nkx6.1 is essential for maintaining the functional state of pancreatic beta cells, Cell reports 4(6) (2013) 1262–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Kavalakatt S, Khadir A, Madhu D, Hammad M, Devarajan S, Abubaker J, Al-Mulla F, Tuomilehto J, Tiss A, Urocortin 3 Levels Are Impaired in Overweight Humans With and Without Type 2 Diabetes and Modulated by Exercise, Front Endocrinol (Lausanne) 10 (2019) 762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Anderson SJ, White MG, Armour SL, Maheshwari R, Tiniakos D, Muller YD, Berishvili E, Berney T, Shaw JAM, Loss of end-differentiated beta-cell phenotype following pancreatic islet transplantation, Am J Transplant 18(3) (2018) 750–755. [DOI] [PubMed] [Google Scholar]
- [74].van der Meulen T, Mawla AM, DiGruccio MR, Adams MW, Nies V, Dolleman S, Liu S, Ackermann AM, Caceres E, Hunter AE, Kaestner KH, Donaldson CJ, Huising MO, Virgin Beta Cells Persist throughout Life at a Neogenic Niche within Pancreatic Islets, Cell Metab 25(4) (2017) 911–926 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Lee S, Zhang J, Saravanakumar S, Flisher MF, Grimm DR, van der Meulen T, Huising MO, Virgin beta-Cells at the Neogenic Niche Proliferate Normally and Mature Slowly, Diabetes 70(5) (2021) 1070–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Gonzalez-Duque S, Azoury ME, Colli ML, Afonso G, Turatsinze JV, Nigi L, Lalanne AI, Sebastiani G, Carre A, Pinto S, Culina S, Corcos N, Bugliani M, Marchetti P, Armanet M, Diedisheim M, Kyewski B, Steinmetz LM, Buus S, You S, Dubois-Laforgue D, Larger E, Beressi JP, Bruno G, Dotta F, Scharfmann R, Eizirik DL, Verdier Y, Vinh J, Mallone R, Conventional and Neo-antigenic Peptides Presented by beta Cells Are Targeted by Circulating Naive CD8+ T Cells in Type 1 Diabetic and Healthy Donors, Cell Metab 28(6) (2018) 946–960 e6. [DOI] [PubMed] [Google Scholar]
- [77].Lombardi A, Tsomos E, Hammerstad SS, Tomer Y, Interferon alpha: The key trigger of type 1 diabetes, J Autoimmun 94 (2018) 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Nica AC, Ongen H, Irminger JC, Bosco D, Berney T, Antonarakis SE, Halban PA, Dermitzakis ET, Cell-type, allelic, and genetic signatures in the human pancreatic beta cell transcriptome, Genome Res (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Mawla AM, Huising MO, Navigating the Depths and Avoiding the Shallows of Pancreatic Islet Cell Transcriptomes, Diabetes 68(7) (2019) 1380–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]