

Abstract
Determining 3D intermediate structures during the biological action of proteins in real time under ambient conditions is essential for understanding how proteins function. Here we use time-resolved Laue crystallography to extract short-lived intermediate structures and thereby unveil signal transduction in the blue light photoreceptor photoactive yellow protein (PYP) from Halorhodospira halophila. By analyzing a comprehensive set of Laue data during the PYP photocycle (forty-seven time points from one nanosecond to one second), we track all atoms in PYP during its photocycle and directly observe how absorption of a blue light photon by its p-coumaric acid chromophore triggers a reversible photocycle. We identify a complex chemical mechanism characterized by five distinct structural intermediates. Structural changes at the chromophore in the early, red-shifted intermediates are transduced to the exterior of the protein in the late, blue-shifted intermediates through an initial “volume-conserving” isomerization of the chromophore and the progressive disruption of hydrogen bonds between the chromophore and its surrounding binding pocket. These results yield a comprehensive view of the PYP photocycle when seen in the light of previous biophysical studies on the system.
Keywords: intermediates, mechanism, signal transduction, time-resolved crystallography, singular value decomposition
In the photoreceptor photoactive yellow protein (PYP) from Halorhodospira halophila, the absorption of a blue light photon by its p-coumaric acid (pCA) chromophore results in a fast trans-cis isomerization that generates a structural signal, ultimately resulting in a negative phototactic response of the bacterium (1). PYP has been studied extensively by visible absorption spectroscopy, Fourier transform infrared, resonance Raman, NMR, and crystallography (reviewed in refs. 2 and 3), and was the first member of the Per-Arnt-Sim (PAS) super-family (4) to have its structure determined (5). Upon exposure to blue light, PYP undergoes a fully reversible photocycle from the dark state, through two early spectroscopic intermediates (I0 and 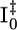 ) (6) that decay quickly to a red-shifted intermediate denoted pR (or I1). I1 decays in turn to a blue-shifted state denoted pB (or I2), which then reverts to the dark state (7-9) (Fig. 1A).
) (6) that decay quickly to a red-shifted intermediate denoted pR (or I1). I1 decays in turn to a blue-shifted state denoted pB (or I2), which then reverts to the dark state (7-9) (Fig. 1A).
Fig. 1.

Overview of PYP (46). (A) The room-temperature PYP photocycle (6, 9). (B) Structure of PYP. The pCA chromophore is shown in yellow; secondary structure is labeled using the notation of Rubinstenn et al. (47). (C) Structure of the chromophore binding pocket of PYP. Hydrogen bonds are shown as green dotted lines and chromophore atoms are labeled according to Borgstahl et al. (5).
The structure of PYP in the dark state has the canonical α/β Per-Arnt-Sim (PAS) fold supplemented by two N-terminal helices (αA and αA′) packed on the back of its β-sheet (Fig. 1B). The chromophore is covalently linked to the protein through a thioester bond to Cys69 and is stabilized in the dark state as a phenolate anion (10, 11) by a hydrogen bonding network consisting of residues Tyr42, Glu46, and Thr50 (Fig. 1C). The pCA carbonyl oxygen (O1′) has a partial negative charge due to resonance stabilization of the chromophore between phenolic and quinonic forms (12) and makes a hydrogen bond with the backbone amide of Cys69 (Fig. 1C). Access to the completely buried chromophore-binding site might be gated by Arg52, whose closed state is stabilized by hydrogen bonds between its guanidino group and the backbone carbonyl oxygens of Tyr98 and Thr50.
The PYP photocycle (Fig. 1 A), deduced from transient visible absorption spectroscopy, is usually described as a simple linear, irreversible progression between intermediate spectroscopic states. However, the existence of biphasic transitions between pR and pB, and pB and the ground state, shows that this mechanism must be an oversimplification (2). Different methodologies (13-17) identify multiple pR- and pB-like species and complex pathways for their interconversions. In the pR-like species, the chromophore hydrogen bonding network is either fully or partially conserved (13, 14), and in the pB-like species, the pCA chromophore is protonated after breakage of the hydrogen bonding network (18). After protonation, structural changes occur in the surrounding protein (19, 20) and involve unfolding of the two N-terminal helices (21, 22). Although direct evidence is lacking (1), it is thought that pB is the signaling state of the protein (2, 3) and that structural changes in the N-terminal helices mediate its signaling activity (3).
Although spectroscopic data have provided useful information on the optical characteristics of PYP intermediates and their kinetics, these data generally provide limited information about their 3D structures. Previous attempts to characterize the high-resolution structures of these intermediates with time-resolved crystallography have either suffered from lower signal-to-noise levels that made data interpretation difficult (23, 24) or did not cover the entire time range of the photocycle (25). In this study, we have collected and analyzed time-resolved crystallographic data from 47 time points during the photocycle of wild-type PYP, spanning the time range from 1 ns to >1 s. We obtain a complete view of the PYP photocycle in the crystal over this time range and reconstruct its pathway for chromophore isomerization via a series of intermediates.
Materials and Methods
Experimental Data Collection. Protein was expressed, purified, and crystallized as described (26, 27). Time-resolved crystallographic data were collected at BioCARS 14-ID-B beamline at the Advanced Photon Source (APS) and at beamline ID09B at the European Synchrotron Radiation Facility (ESRF). All experiments were performed in a pump-probe mode with the crystal illuminated by a pump laser pulse followed by a controlled time delay and exposure to the x-ray probe pulse. Data collected at the ESRF covered the early to middle time range of 1 ns to 10 μs, and data collected at the APS covered the middle to late time range of 6 μs to 1.33 s. In total, 47 time points from 1 ns to 1.33 s, spanning the complete photocycle, were analyzed by singular value decomposition (SVD) (see supporting information, which is published on the PNAS web site). APS data used laser illumination from both sides of the crystal to significantly increase the extent of photoactivation (28).
For all data sets, time was the fast variable. That is, we collected data at all desired time points at one angular setting; advanced the angular setting to survey a different region of reciprocal space and repeated all time points; and continued in this way until the crystal no longer provided usable data. This method of data collection greatly minimizes systematic errors between time points and facilitates SVD analysis. To facilitate the comparison of the ESRF and APS data sets, the 10-μs time point was taken in all data sets.
Data Processing. All APS data were processed with laueview (29), with the exception of data from two crystals. These two data sets and all of the ESRF data were processed with precognition (Renz Research, Westmont, IL), a recently developed program for Laue data analysis. Time-resolved experiments yield structure factor amplitudes of the initial, dark state |FD(hkl)| and time-dependent structure factor amplitudes |F(hkl, t)|. From these amplitudes, time-dependent difference structure factor amplitudes ΔF(hkl, t) = |F(hkl, t)| - |FD(hkl)| are obtained for each time point t. To increase the signal-to-noise ratio and the completeness, data from several crystals were merged via weighted averaging of ΔF(hkl, t). In this way, we obtained data sets of high completeness and redundancy to 1.6-Å resolution (supporting information). Experimental, weighted difference electron density maps Δρ(t) were then generated by Fourier transformation of wΔF(hkl, t) where w is a weighting factor to weigh down observations with high experimental errors (30): w = 1/(1 + σ2/〈σ2〉).
Kinetic Analysis of Time-Resolved Crystallographic Data. In general, two or more intermediates coexist at each time point in the PYP photocycle. Consequently, each time-dependent difference electron density map Δρ(t) consists of a mixture of difference density features. Separation of this mixture into the time-independent difference electron densities of the intermediates is essential to determine the reaction mechanism and the structure of each intermediate. It has been demonstrated that this deconvolution can be efficiently achieved by SVD analysis (25, 31, 32). In addition, SVD serves as a convenient noise filter. Because of a large difference of the signal-to-noise ratio, we analyzed the ESRF and APS data separately at the SVD stage, but recombined these data when performing posterior analysis, a process in which the experimental difference maps are compared quantitatively with corresponding calculated difference maps generated from the structures of the intermediates and various candidate mechanisms (31). SVD yielded four distinct difference density maps of intermediates. After calculation of difference structure factors (amplitudes and phases) from these intermediate difference densities, difference refinement (33) was performed as described (32, 34) to generate five distinct intermediate structures.
Refinement was performed in shelx-97 (35), and model building and viewing of electron density maps was performed in xtalview (36). The difference electron density calculated from these structures was then used to elucidate the detailed chemical mechanism in posterior analysis. A number of candidate mechanisms based on the general scheme involving five intermediates identified by SVD were used to fit the data and were assessed by the magnitude of the total squared deviation between experimental and calculated maps over all 47 time points of features above +3σ or below -3σ present in either the experimental or calculated maps.
Results and Discussion
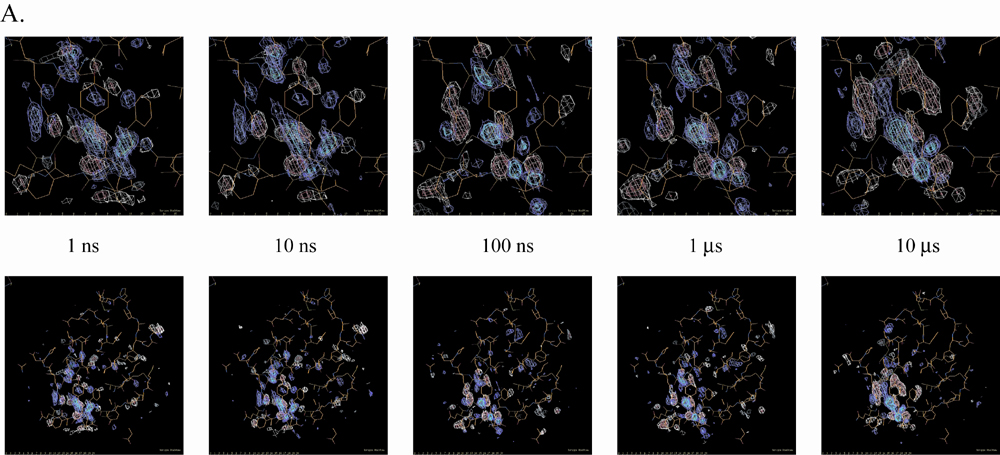
Structural Progression Through the Photocycle. From an initial SVD kinetic analysis of 47 time points from one nanosecond to one second, four relaxation times (τ1 = 20 ns, τ2 = 180 μs, τ3 = 5 ms, and τ4 = 52 ms) were observed. We generated four intermediate difference density maps based on simple irreversible candidate mechanisms for the ESRF data (α → β) and APS data (β → γ → δ → dark) (see supporting information), where α, β, γ, and δ denote proposed intermediate states. Both the APS and ESRF data contain the β state, and although the APS data has a higher signal-to-noise ratio (see supporting information), close inspection of the ESRF and APS maps shows similar strong difference density features in the chromophore binding pocket (compare Fig. 2 B and G with C and H).
Fig. 2.

Difference electron density maps for distinct chemical states (α, β, γ, and δ) from an initial kinetic analysis of 47 time points from 1 ns to 1 s. Chromophore-binding pocket (A-E) and whole protein views (F-J) of the difference maps associated with the “α” (A and F) and “β” (B and G) states derived from the ESRF data and from the “β” (C and H), “γ” (D and I), and “δ” (E and J) states from the APS data. Difference maps are contoured at -4σ (red), -3σ (pink), +3σ (cyan), and +4σ (blue). These chemical states are in the order of occurrence in time through the photocycle of PYP. The population of the α, β, γ, and δ states are peaked around at nanoseconds, microseconds, milliseconds, and subseconds time range, respectively (see text and Figs. 3 and 4B).
The α and β states, predicted to occur early in the photocycle (from nanoseconds to hundreds of microseconds), show little signal outside the chromophore binding pocket (Fig. 2 F-H). The γ and δ states have difference features in the chromophore-binding pocket similar to those found in pB (18). In the surrounding protein, the γ state (populated in the early millisecond regime) has difference features on helix αB, proximal to the chromophore, whereas the δ state also has signal on the more distal N-terminal helices αA and αA′, >20 Å distant from the chromophore (32). Thus, we observe a progression of structural signal from the chromophore to the putative signaling region of the protein at the N terminus.
Intermediate Structures. The next step in the analysis is to determine whether each of the proposed intermediate maps indeed correspond to a single structure. The α, γ, and δ states were modeled satisfactorily with single intermediate structures (Fig. 3), whereas the β state required two intermediate structures to account for the observed difference electron density (Fig. 3) (see supporting information).
Fig. 3.

Chromophore-binding pocket views of refined intermediate structures and mechanism for the isomerization and rotation of the pCA chromophore upon absorption of blue light. Five distinct structural intermediates (ICP, pRCW, pRE46Q, pB1, and pB2) were identified from four chemical states (α, β, γ, and δ) shown in Fig. 2. ICP is shown twice to demonstrate the biphasic pathways to pRCW and pRE46Q. Isomerization and rotation about single bonds are shown by arrows; hydrogen bonds are dotted. A bicycle pedal mechanism (44), which couples trans-cis isomerization of the C2─C3 double bond with rotation about a nonadjacent single bond, is used for the dark state to ICP transition. Further rotations about single bonds result in the pB1 conformation. pB2 reverts thermally to the dark state with no further detectable intermediates.
Two of these five room temperature intermediate structures are analogous to those observed in cryotrapping experiments (37), and our results and analysis establish the direct correspondence between the low-temperature intermediates and those in ambient conditions. The first structure identifies the α state (Fig. 3). We refer to it as ICP for intermediate cis planar-like, because of the planarity between the aromatic ring and the carbonyl group of the chromophore and because of its close similarity to a cis-planar structure from a cryotrapped intermediate (37). Similarly, we refer to one of the two structures used to fit the β state (Fig. 3) as pRCW for pR cis wobble-like, because of the loss of planarity between the aromatic ring and carbonyl group of the chromophore and because of its close similarity to a structure from the same cryotrapping experiment (37). In both ICP and pRCW, the chromophore has isomerized about the C2─C3 double bond to a cis conformation while maintaining the strained distal hydrogen-bonding network. To maintain the distal hydrogen bonding network between the phenolate oxygen and the side chains of Tyr42 and Glu46, a concomitant rotation about the S─C1 single bond has occurred. The major structural differences between the two intermediates are the planarity of the chromophore (there is a much more significant deviation from planarity in pRCW) and the position of the cysteine sulfur of residue 69 (caused by a difference in torsional angles, see Table 1 and Fig. 3). These differences are related to the temporal progression of the chromophore conformation during the PYP photocycle (see The Mechanism of Chromophore Isomerization).
Table 1. Relevant atom-to-atom distances (in Å), torsional angles (in degrees) and deviation from planarity (DP) of the chromophore (using atoms from the cysteine sulfur of Cys69 to the chromophore phenolate oxygen) of the five structural intermediates during the wild-type PYP photocycle.
| Dark | ICP | pRCW | pRE46Q | pB1 | pB2 | |
|---|---|---|---|---|---|---|
| O4′-Tyr42, Å | 2.51 | 2.6 | 2.8 | 2.7 | 5.2 | 5.0 |
| O4′-Glu46, Å | 2.58 | 2.7 | 2.5 | 5.5 | 8.0 | 7.8 |
| O1-Cys69, Å | 2.82 | 4.9 | 3.7 | 2.8 | 2.9 | 2.9 |
| Tyr42-Thr50, Å | 2.89 | 2.8 | 2.8 | 3.0 | 2.9 | 2.8 |
| N-Cα-Cβ-S, ° | 83 | 109 | −35 | 95 | 89 | 90 |
| Cα-Cβ-S-C1, ° | −84 | −97 | 94 | −77 | −72 | −79 |
| Cβ-S-C1-C2, ° | 175 | −35 | −170 | 179 | 180 | −180 |
| Cβ-S-C1-O1, ° | 5 | 147 | 9 | −1 | −3 | 0 |
| S-C1-C2-C3, ° | 174 | 179 | −175 | −179 | 167 | 172 |
| C1-C2-C3-C1′, ° | 169 | −2 | 21 | −23 | −27 | −24 |
| DP | 0.17 | 0.06 | 0.27 | 0.30 | 0.43 | 0.44 |
O1, the chromophore carbonyl oxygen; O4′, the chromophore phenol(ate) oxygen; Tyr42, the hydroxyl oxygen of Tyr42; Glu46, the Oε2 of Glu46; Cys69, the backbone amide nitrogen of Cys69; Thr50, the Oγ of Thr50; DP, rms. deviation from planarity of the chromophore from the sulfur of Cys 69 to the phenolate oxygen. DPs were calculated by using CCP4 program geomcalc (48). The dark state structure is from Anderson et al. (46).
The second of the two structures used to fit the β state has a chromophore conformation similar to the pR intermediates from time-resolved Laue studies of E46Q PYP (32, 34) (Fig. 3) and, thus, we refer to it as pRE46Q. Unlike ICP and pRCW, where a strained distal hydrogen bonding network is preserved, the chromophore phenolate oxygen in pRE46Q has completely broken its hydrogen bond to Glu46 while retaining its hydrogen bond to Tyr42 (although its unusually short length of 2.51 Å in the dark state has inflated to 2.7 Å; see Table 1). We did not determine the spectra of these early intermediates, but based on the time ranges when these intermediates are populated, we propose that ICP is analogous to 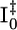 and the pRE46Q and pRCW structures correspond to two pR species observed by resonance Raman spectroscopy (14) (see below).
and the pRE46Q and pRCW structures correspond to two pR species observed by resonance Raman spectroscopy (14) (see below).
We refer to the later intermediates (from the γ and δ states) as pB1 and pB2 (Fig. 3), and predict that they have pB-like absorption spectra. This expectation is based on the fact that their chromophore conformations are similar to those found in previous pB structures (18, 25, 32, 34); whereas the cis-conformation of chromophore has fully relaxed, the hydrogen bonding network to Tyr42 and Glu46 is completely broken (Table 1), and Arg52 has been ejected into the solvent (Fig. 3). Small structural shifts in the N-terminal helices αA and αA′ are present in the later pB structure, pB2, but are absent in the early structure, pB1.
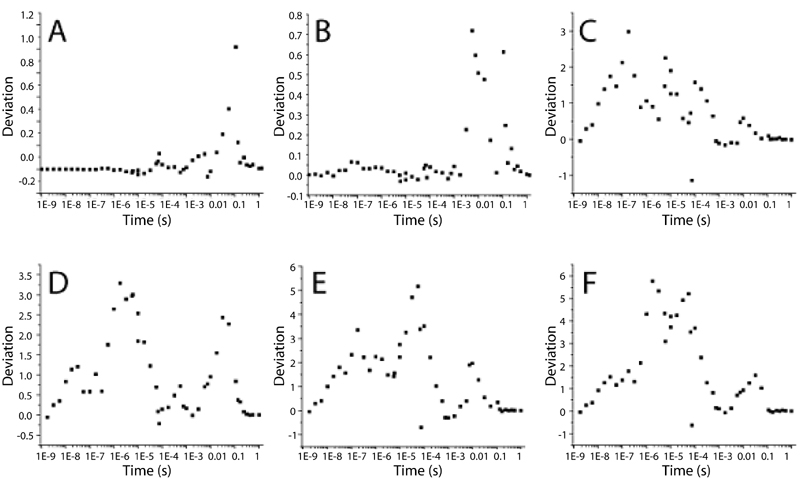
The Chemical Kinetic Mechanism of Wild-Type PYP. Generally speaking, our nanosecond to millisecond relaxation times from SVD are similar to those observed in solution (6, 9, 13, 17, 19, 38, 39). Our shortest relaxation time of τ1 = 20 ns is well correlated with a 3-ns decay (6). Time constants measured at later times using various spectroscopic methods (9, 13, 17, 19, 38, 39) are rather scattered, but most experiments independently showed two common time constants: one in the range of ≈113-370 μs and another one in the range of ≈1.0-10 ms. Our observed relaxation times of τ2 = 180 μs and τ3 = 5 ms fall well within these two ranges, so it appears that our results are in good agreement with other spectroscopic measurements, at least up to the millisecond time range. Relaxation times in the final decay to the ground state show the largest difference compared to those in the solution. Here, a process at τ4 = 52 ms is observed in the crystal, whereas in solution relaxation times are in the range of ≈350-583 ms at about pH 7 (13, 17, 19, 38) and up to 2 s and longer at lower pH values (9, 16). It is plausible that the reversal to the ground state may be faster in the crystal than in solution because of the crystal context. We conclude that the time scales of the photocycle in the crystal and solution broadly match up except the final decay to the ground state.
Most previous attempts to understand the PYP photocycle are based on the apparent relaxation times only. In our approach, we performed a final kinetic analysis (posterior analysis) using the calculated difference density associated with each of the intermediate structures shown in Fig. 3 and the candidate chemical kinetic mechanism shown in Fig. 4A. Our posterior analysis introduces a six-state model with parallel kinetic pathways and transforms four apparent relaxation times from SVD into seven model-specific rate coefficients. We chose this overall mechanism because it is relatively simple, consistent with strong constraints placed by the SVD analysis (see supporting information), and broadly consistent with previously proposed photocycle mechanisms (2, 3). In the proposed mechanism, the first intermediate, ICP (corresponding to the α state), decays in parallel to pRCW and pRE46Q (the β state). These two intermediates both convert to the pB1 intermediate, which then can decay either to pB2 (and subsequently to the dark state) or directly to the dark state. A very good fit in the posterior analysis was obtained by using seven rate coefficients shown in Fig. 4 (see supporting information), which results in the time-dependent relative concentrations for each intermediate shown in Fig. 4B.
Fig. 4.

Properties of the chemical kinetic mechanism of the wild-type PYP photocycle. (A) General chemical kinetic mechanism used to fit the data. The dashed arrow indicates the light-driven reaction from pG to the first intermediate observed here, ICP. (B) Predicted concentrations of intermediates [ICP (red), pRCW (magenta), pRE46Q (purple), pB1 (blue), pB2 (cyan), and the dark state (black)] after posterior analysis with rate coefficients (s-1): k1 = 4.8 × 107; k2 = 3.7 × 107; k3 = 3.0 × 103; k4 = 3.3 × 104; k5 = 55; k6 = 100; and k7 = 7.1. The seven rate coefficients correspond to time constants of 21 ns, 27 ns, 333 μs, 30 μs, 18 ms, 10 ms, and 141 ms, respectively.
The direct visualizing power of time-resolved crystallography, combined with the extended time range and improved signal-to-noise ratio represented in this work, revealed the coexistence of two distinct pR-like intermediate structures (pRCW and pRE46Q), which allow us to go one step further in building a more comprehensive mechanism rather than relying on the relaxation times only. The coexistence of two intermediate structures for the pR-like species requires at least two more time constants, as shown in Fig. 4A. In our mechanism, these pRCW and pRE46Q build up with similar time constants (21 ns and 27 ns, respectively), but the decay of pRE46Q (30 μs) is 10 times faster than that of pRCW (333 μs). We note that the presence of two conformations in pR was also implicated in earlier resonance Raman studies (14), and the two predicted structures are markedly similar to our pRCW and pRE46Q structures, although the time constants related to these structures could not be obtained from resonance Raman studies.
Our mechanism adds one more time constant for the direct decay of the pB1 to the ground state to account for the fast decay and the low occupancy of pB2. Therefore, there are two pathways for the return to the ground state: one directly from pB1 and another through pB2. The observed biphasic decay to the dark state with rate coefficients of 55 s-1 and 7 s-1 is notably close to the values of 20 s-1 and 6 s-1 observed during decay from a pB-like photostationary state by single crystal microspectrophotometry (40).
Another key finding relates to the latest blue shifted intermediate, pB2: a clear signal at the N terminus (Fig. 2 J) is observed with wild-type PYP. A previous time-resolved crystallographic study (25) did not capture this signal, probably because of a poorer signal-to-noise ratio in their diffraction data. Time-resolved Fourier transform infrared studies (38) on wild-type PYP in both solution and crushed crystals suggested that the structure of the putative signaling state is not developed in crystals, but only in solution. Our results provide direct evidence that a signaling state is also formed in the single crystal, although the extent of this signal may be suppressed by the crystal context.
In wild-type PYP we have identified two structures for pR (pRCW and pRE46Q) but only one (pRE46Q) in E46Q PYP (34). In E46Q PYP, early intermediates corresponding to ICP and pRCW are predicted to be less stable because of the weaker hydrogen bond between the chromophore phenolate oxygen and Gln46. These early intermediates might be expected to have shorter lifetimes, perhaps in the hundreds of picoseconds time range (41), which is beyond the time resolution of 10 ns provided by the previous time-resolved crystallographic study on the E46Q mutant (34). This hypothesis is supported by the refinement of cryotrapped intermediates, which show ICP- and pRCW-like species in both wild-type and E46Q PYP (37, 42).
The Mechanism for Chromophore Isomerization. The high-resolution structures of the intermediates identified in this study make it possible to visualize the complex mechanism for chromophore isomerization in wild-type PYP in unprecedented detail (Fig. 3). In a photoreceptor, the initial photochemistry of the chromophore must be coupled to the protein in a manner that ultimately generates a biological signal. In the case of PYP, this photochemistry is trans-cis isomerization about the C2─C3 bond of the pCA chromophore. Isomerization about the double bond, although embedded in a densely packed medium such as a protein, should be “volume-conserving” (43). That is, the isomerization should be accompanied by rotation about one or more nearby bonds to minimize the volume swept out by the chromophore atoms, and thus minimize displacement of adjacent atoms on the ultrafast time scale.
To convert from the trans dark state to the earliest intermediate we observe, ICP, a bicycle pedal mechanism is involved (Fig. 3) in which trans-to-cis isomerization is accompanied by rotation about a single bond, positioned one bond away from the double bond (44). The hydrogen-bonding network is maintained during this correlated rotation (45). Retaining the network generates strain in the chromophore, which now contains the energy required to drive successive steps in the photocycle. The first step is conversion to either pRCW (Fig. 3 Upper) or pRE46Q (Fig. 3 Lower). Conversion from ICP to pRCW requires rotation about three single bonds, namely the Cα─Cβ, Cβ─S and S─C1 single bonds. This pathway partially relieves the strain in the chromophore yet maintains the hydrogen-bonding network.
Conversion from ICP to pRE46Q requires significant rotation only about one bond, namely Cβ─S1. This conversion breaks the hydrogen bond between the chromophore and residue 46, but reestablishes the hydrogen bond between the chromophore carbonyl oxygen and the backbone amide of Cys69. Both pRCW and pRE46Q convert to the pB1 chromophore conformation by completely breaking the phenolate oxygen hydrogen-bonding network, with the transition from pRCW requiring additional rotations about the Cα─Cβ and Cβ─S bonds. Complete disruption of the hydrogen-bonding network upon formation of pB1 results in structural shifts throughout αB, which are then transduced throughout the protein.
PYP harnesses the energy of the blue light photon by adopting a strained chromophore conformation that maintains its hydrogen bond network and contains the energy required for further steps in the photocycle. The subsequent breakage of hydrogen bonds results in the fully relaxed cis conformation associated with pB. These structural changes of the chromophore are coupled to the protein to generate a structural signal at its N terminus, which is thought to mediate the biological signaling activity of PYP (3). Structural changes originating at the chromophore are transduced to the N terminus from the chromophore hydrogen bonding network, to helix αB that contains both Tyr42 and Glu46, through a conserved helix capping motif involving the backbone of Asn43, to the N-terminal αA and αA′ helices via the side chain of Asn43. In this manner, changes in chromophore conformation are associated with the differential signaling activity of PYP.
Conclusions
Forty-seven time-resolved Laue time points were collected during the wild-type PYP photocycle from 1 ns to 1 s. A kinetic analysis of the data allowed the observation of five distinct structural intermediates during the photocycle. The three early intermediates, which either partially or completely preserve the distal hydrogen bonding network, are identified as red-shifted species, whereas the two late intermediates, in which the hydrogen bonding is completely broken, are identified as blue-shifted species. The extended time range and improved signal-to-noise ratio represented in this work now allows us to identify the mixture of pRE46Q and pRCW that was not possible in an earlier time-resolved Laue study, enhancing the fitting procedure in the kinetic analysis and defining rate coefficients more accurately. The chromophore isomerization pathway involves “volume-conserving” rotations. The ensuing disruption of the hydrogen bonds to the chromophore phenolate oxygen allows energy absorbed by the chromophore to be released in a controlled fashion to the surrounding protein, resulting in structural changes in the distal two N-terminal helices. These results allow for a comprehensive view of the PYP photocycle when seen in the light of previous biophysical studies on the system.
Supplementary Material
Acknowledgments
We have benefited from helpful discussions with Sean Crosson, Jason Key, Zhong Ren, and Xiaojing Yang. This work was supported by National Institutes of Health Grant GM36452 (to K.M.), the Deutsche Forschungsgemeinschaft, Sonderforschungsbereich 533 (to M.S.), the European Union Grants HPRI-CT-1999-50004 and HPRN-CT-00160 (to M.W.), and Korea Research Foundation Grant KRF-2004-003-C00100 (to H.I.). The Bio-CARS facility is supported by National Institutes of Health Grant RR07707 (to K.M.), and the Advanced Photon Source is supported by the Department of Energy. H.I. acknowledges the financial support from the Damon Runyon Cancer Research Foundation.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: PYP, photoactive yellow protein; APS, Advanced Photon Source; ESRF, European Synchrotron Radiation Facility; SVD, singular value decomposition.
Data deposition: The structures reported in this paper have been deposited in the Protein Data Bank, www.pdb.org [PDB ID codes 1TS8 (ICP), 1TS7 (pRCW + pRE46Q), 1TS6 (pB1), and 1TS0 (pB2)].
References
- 1.Sprenger, W. W., Hoff, W. D., Armitage, J. P. & Hellingwerf, K. J. (1993) J. Bacteriol. 175, 3096-3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hellingwerf, K. J., Hendriks, J. & Gensch, T. (2003) J. Phys. Chem. A 107, 1082-1094. [Google Scholar]
- 3.Cusanovich, M. A. & Meyer, T. E. (2003) Biochemistry 42, 4759-4770. [DOI] [PubMed] [Google Scholar]
- 4.Taylor, B. L. & Zhulin, I. B. (1999) Microbiol. Mol. Biol. Rev. 63, 479-506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borgstahl, G. E., Williams, D. R. & Getzoff, E. D. (1995) Biochemistry 34, 6278-6287. [DOI] [PubMed] [Google Scholar]
- 6.Ujj, L., Devanathan, S., Meyer, T. E., Cusanovich, M. A., Tollin, G. & Atkinson, G. H. (1998) Biophys. J. 75, 406-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meyer, T. E., Yakali, E., Cusanovich, M. A. & Tollin, G. (1987) Biochemistry 26, 418-423. [DOI] [PubMed] [Google Scholar]
- 8.Meyer, T. E., Tollin, G., Hazzard, J. H. & Cusanovich, M. A. (1989) Biophys. J. 56, 559-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoff, W. D., van Stokkum, I. H., van Ramesdonk, H. J., van Brederode, M. E., Brouwer, A. M., Fitch, J. C., Meyer, T. E., van Grondelle, R. & Hellingwerf, K. J. (1994) Biophys. J. 67, 1691-1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoff, W. D., Dux, P., Hard, K., Devreese, B., Nugteren-Roodzant, I. M., Crielaard, W., Boelens, R., Kaptein, R., van Beeumen, J. & Hellingwerf, K. J. (1994) Biochemistry 33, 13959-13962. [DOI] [PubMed] [Google Scholar]
- 11.Baca, M., Borgstahl, G. E., Boissinot, M., Burke, P. M., Williams, D. R., Slater, K. A. & Getzoff, E. D. (1994) Biochemistry 33, 14369-14377. [DOI] [PubMed] [Google Scholar]
- 12.Genick, U. K., Soltis, S. M., Kuhn, P., Canestrelli, I. L. & Getzoff, E. D. (1998) Nature 392, 206-209. [DOI] [PubMed] [Google Scholar]
- 13.Brudler, R., Rammelsberg, R., Woo, T. T., Getzoff, E. D. & Gerwert, K. (2001) Nat. Struct. Biol. 8, 265-270. [DOI] [PubMed] [Google Scholar]
- 14.Unno, M., Kumauchi, M., Hamada, N., Tokunaga, F. & Yamauchi, S. (2004) J. Biol. Chem. 279, 23855-23858. [DOI] [PubMed] [Google Scholar]
- 15.Van der Horst, M. A., Van Stokkum, I. H., Crielaard, W. & Hellingwerf, K. J. (2001) FEBS Lett. 497, 26-30. [DOI] [PubMed] [Google Scholar]
- 16.Borucki, B., Otto, H., Joshi, C. P., Gasperi, C., Cusanovich, M. A., Devanathan, S., Tollin, G. & Heyn, M. P. (2003) Biochemistry 42, 8780-8790. [DOI] [PubMed] [Google Scholar]
- 17.Joshi, C. P., Borucki, B., Otto, H., Meyer, T. E., Cusanovich, M. A. & Heyn, M. (2005) Biochemistry 44, 656-665. [DOI] [PubMed] [Google Scholar]
- 18.Genick, U. K., Borgstahl, G. E., Ng, K., Ren, Z., Pradervand, C., Burke, P. M., Srajer, V., Teng, T. Y., Schildkamp, W., McRee, D. E., et al. (1997) Science 275, 1471-1475. [DOI] [PubMed] [Google Scholar]
- 19.Hendriks, J., Gensch, T., Hviid, L., van Der Horst, M. A., Hellingwerf, K. J. & van Thor, J. J. (2002) Biophys. J. 82, 1632-1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Borucki, B., Devanathan, S., Otto, H., Cusanovich, M. A., Tollin, G. & Heyn, M. P. (2002) Biochemistry 41, 10026-10037. [DOI] [PubMed] [Google Scholar]
- 21.Van der Horst, M. A., Van Stokkum, I. H., Crielaard, W. & Hellingwerf, K. J. (2001) FEBS Lett. 497, 26-30. [DOI] [PubMed] [Google Scholar]
- 22.Craven, C. J., Derix, N. M., Hendriks, J., Boelens, R., Hellingwerf, K. J. & Kaptein, R. (2000) Biochemistry 39, 14392-14399. [DOI] [PubMed] [Google Scholar]
- 23.Perman, B., Srajer, V., Ren, Z., Teng, T., Pradervand, C., Ursby, T., Bourgeois, D., Schotte, F., Wulff, M., Kort, R., et al. (1998) Science 279, 1946-1950. [DOI] [PubMed] [Google Scholar]
- 24.Ren, Z., Perman, B., Srajer, V., Teng, T. Y., Pradervand, C., Bourgeois, D., Schotte, F., Ursby, T., Kort, R., Wulff, M. & Moffat, K. (2001) Biochemistry 40, 13788-13801. [DOI] [PubMed] [Google Scholar]
- 25.Schmidt, M., Pahl, R., Srajer, V., Anderson, S., Ren, Z., Ihee, H., Rajagopal, S. & Moffat, K. (2004) Proc. Natl. Acad. Sci. USA 101, 4799-4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McRee, D. E., Tainer, J. A., Meyer, T. E., Van Beeumen, J., Cusanovich, M. A. & Getzoff, E. D. (1989) Proc. Natl. Acad. Sci. USA 86, 6533-6537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Imamoto, Y., Ito, T., Kataoka, M. & Tokunaga, F. (1995) FEBS Lett. 374, 157-160. [DOI] [PubMed] [Google Scholar]
- 28.Baxter, R. H., Ponomarenko, N., Srajer, V., Pahl, R., Moffat, K. & Norris, J. R. (2004) Proc. Natl. Acad. Sci. USA 101, 5982-5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ren, Z. & Moffat, K. (1995) J. Appl. Cryst. 28, 461-481. [Google Scholar]
- 30.Ursby, T. & Bourgeois, D. (1997) Acta Crystallogr. A 53, 564-575. [Google Scholar]
- 31.Schmidt, M., Rajagopal, S., Ren, Z. & Moffat, K. (2003) Biophys. J. 84, 2112-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rajagopal, S., Anderson, S., Srajer, V., Schmidt, M., Pahl, R. & Moffat, K. (2005) Structure (London) 13, 55-63. [DOI] [PubMed] [Google Scholar]
- 33.Terwilliger, T. C. & Berendzen, J. (1995) Acta Crystallogr. D 51, 609-618. [DOI] [PubMed] [Google Scholar]
- 34.Anderson, S., Srajer, V., Pahl, R., Rajagopal, S., Schotte, F., Anfinrud, P., Wulff, M. & Moffat, K. (2004) Structure (London) 12, 1039-1045. [DOI] [PubMed] [Google Scholar]
- 35.Sheldrick, G. M. & Schneider, T. R. (1997) Methods Enzymol. 277, 319-343. [PubMed] [Google Scholar]
- 36.McRee, D. E. (1999) J. Struct. Biol. 125, 156-165. [DOI] [PubMed] [Google Scholar]
- 37.Anderson, S. (2003) Ph.D. thesis (University of Chicago, Chicago).
- 38.Xie, A., Kelemen, L., Hendriks, J., White, B. J., Hellingwerf, K. J. & Hoff, W. D. (2001) Biochemistry 40, 1510-1517. [DOI] [PubMed] [Google Scholar]
- 39.Takeshita, K., Imamoto, Y., Kataoka, M., Tokunaga, F. & Terazima, M. (2002) Biochemistry 41, 3037-3048. [DOI] [PubMed] [Google Scholar]
- 40.Ng, K., Getzoff, E. D. & Moffat, K. (1995) Biochemistry 34, 879-890. [DOI] [PubMed] [Google Scholar]
- 41.Devanathan, S., Lin, S., Cusanovich, M. A., Woodbury, N. & Tollin, G. (2000) Biophys. J. 79, 2132-2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anderson, S., Srajer, V. & Moffat, K. (2004) Photochem. Photobiol. 80, 7-14. [DOI] [PubMed] [Google Scholar]
- 43.Liu, R. S. & Hammond, G. S. (2001) Chemistry 7, 4537-4544. [DOI] [PubMed] [Google Scholar]
- 44.Warshel, A. (1976) Nature 260, 679-683. [DOI] [PubMed] [Google Scholar]
- 45.Imamoto, Y., Kataoka, M. & Liu, R. S. (2002) Photochem. Photobiol. 76, 584-589. [DOI] [PubMed] [Google Scholar]
- 46.Anderson, S., Crosson, S. & Moffat, K. (2004) Acta Crystallogr. D 60, 1008-1016. [DOI] [PubMed] [Google Scholar]
- 47.Rubinstenn, G., Vuister, G. W., Mulder, F. A., Dux, P. E., Boelens, R., Hellingwerf, K. J. & Kaptein, R. (1998) Nat. Struct. Biol. 5, 568-570. [DOI] [PubMed] [Google Scholar]
- 48.Bailey, S. (1994) Acta Crystallogr. D 50, 760-763.15299374 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}