

Abstract
Age-related macular degeneration (AMD) is the most frequent cause of irreversible blindness in the elderly in developed countries. Our previous studies implicated activation of complement in the formation of drusen, the hallmark lesion of AMD. Here, we show that factor H (HF1), the major inhibitor of the alternative complement pathway, accumulates within drusen and is synthesized by the retinal pigmented epithelium. Because previous linkage analyses identified chromosome 1q25-32, which harbors the factor H gene (HF1/CFH), as an AMD susceptibility locus, we analyzed HF1 for genetic variation in two independent cohorts comprised of ≈900 AMD cases and 400 matched controls. We found association of eight common HF1 SNPs with AMD; two common missense variants exhibit highly significant associations (I62V, χ2 = 26.1 and P = 3.2 × 10-7 and Y402H, χ2 = 54.4 and P = 1.6 × 10-13). Haplotype analysis reveals that multiple HF1 variants confer elevated or reduced risk of AMD. One common at-risk haplotype is present at a frequency of 50% in AMD cases and 29% in controls [odds ratio (OR) = 2.46, 95% confidence interval (1.95-3.11)]. Homozygotes for this haplotype account for 24% of cases and 8% of controls [OR = 3.51, 95% confidence interval (2.13-5.78)]. Several protective haplotypes are also identified (OR = 0.44-0.55), further implicating HF1 function in the pathogenetic mechanisms underlying AMD. We propose that genetic variation in a regulator of the alternative complement pathway, when combined with a triggering event, such as infection, underlie a major proportion of AMD in the human population.
Age-related macular degeneration (AMD) is the leading cause of irreversible vision loss (1, 2), affecting ≈50 million individuals worldwide. AMD is characterized by a progressive loss of central vision attributable to degenerative and neovascular changes that occur at the interface between the neural retina and the underlying choroid. At this location are the retinal photoreceptors, the retinal pigmented epithelium (RPE), a basement membrane complex known as Bruch's membrane (BM) and a network of choroidal capillaries.
The prevailing view is that AMD is a complex disorder stemming from the interaction of multiple genetic and environmental risk factors (3, 4). Familial aggregation studies indicate that a genetic contribution can be identified in up to 25% of the cases (5). Thus, AMD appears to be a product of the interaction between multiple susceptibility loci rather than a collection of single-gene disorders. The number of loci involved, the attributable risk conferred, and the interactions between various loci remain obscure.
Linkage analyses and candidate gene screening have provided limited insight into the genetics of AMD. Reliable associations of ABCA4 (6, 7) and ApoE (8, 9) have been reported. A recent study suggests a minor association with Fibl5 (10), although this has yet to be confirmed. Genome-wide linkage analyses (4, 11) have linked one AMD phenotype (ARMD1; MIM 603075) to chromosomal region 1q25-q31 (12). Fibl6 has been tentatively identified as the causal gene (13), although it does not account for a significant disease load (14, 15). The identification of overlapping loci on chromosome 1q by several groups (11, 16) indicates that this locus likely harbors a major AMD-associated gene.
In AMD and diseases such as Alzheimer's (17), atherosclerosis (18), and glomerulonephritis (19), characteristic lesions and deposits contribute to disease pathogenesis and progression. Although the molecular bases of these diseases may be diverse, the deposits contain many shared molecular constituents that are attributable, in part, to local inflammation and activation of the complement cascade, a key element of the innate immune system in host defense. Drusen are the hallmark deposits associated with early AMD (eAMD), and recent studies have implicated local inflammation and activation of the complement cascade in their formation (20-30). Drusen contain complement activators, inhibitors, activation-specific complement fragments, and terminal pathway components, including the membrane attack complex (MAC). The MAC is a lytic complex that is lethal to foreign pathogens but also to local host cells and tissues in various disease processes.
Individuals with membranoproliferative glomerulonephritis (MPGN) type II (MPGNII), a rare (≈1:1,000,000) kidney disease characterized by uncontrolled activation of the alternative complement pathway, often develop ocular drusen in the macula. These are indistinguishable in composition and appearance from those in AMD (23, 31-33). Furthermore, one patient diagnosed with MPGNII harbors a mutation in the factor H gene (HF1), a major inhibitor of the alternative complement pathway (P. Zipfel, personal communication). Additionally, individuals in a few extended families with MPGN type III, a related disorder, show linkage to a region of chromosome mapped in 1q31-1q32 (34) that overlaps the locus identified in linkage studies for AMD. These collective findings provided the impetus for examining whether HF1 is involved in the development of AMD and MPGN type II.
In this investigation, we determined the frequencies of HF1 sequence variants in AMD and MPGN type II patients and matched controls and analyzed their association with disease phenotype. We also examined HF1 transcription and the distribution of HF1 protein in the macular RPE/choroid complex from normal and AMD donors.
Methods
Patients. Two independent groups of AMD cases and age-matched controls were used for this study. Individuals were of European-American descent, over the age of 60, and enrolled under Institutional Review Board (Columbia University and University of Iowa) approved protocols. These groups consisted of 404 unrelated patients with clinically documented AMD (mean age 79.5 ± 7.8) and 131 unrelated control individuals (mean age 78.4 ± 7.4; matched by age and ethnicity) from the University of Iowa and 550 unrelated patients with clinically documented AMD (mean age, 71.32 ± 8.9 years) and 275 unrelated controls matched by age and ethnicity (mean age, 68.84 ± 8.6 years) from Columbia University. Patients were examined by trained ophthalmologists.
All stereo fundus photographs were graded by C.C.W.K. and individuals trained by her according to standardized classification systems (35, 36). Controls did not exhibit any distinguishing signs of macular disease or have a known family history of AMD (stage 0). AMD patients were subdivided into phenotypic categories [eAMD (stages 1a, 1b, 2a, 2b, and 3), geographic atrophy (GA; stage 4) and exudative (choroidal neovascularization; stage 4)] AMD based on the classification of their most severe eye at the time of their recruitment. Genomic DNA was generated from peripheral blood leukocytes by using QIAamp DNA Blood Maxi kits (Qiagen, Valencia, CA).
Human Donor Eyes. Human donor eyes were obtained from the Iowa Lions, Oregon Lions, and Central Florida Lions Eye Banks within 5 h of death. Fundi and/or posterior poles were graded by using the same grading criteria described above (35, 36). DNA derived from 38 unrelated donors with clinically documented AMD (mean age, 81.5 ± 8.6) and 19 unrelated control donors (mean age, 80.5 ± 8.8; matched by age and ethnicity) were used for single-strand conformation polymorphism (SSCP) analyses to examine genotype-phenotype relationships. Total RNA was prepared from donor retina, RPE/choroid, and RPE cells and from cultures of donor-derived RPE cells (37) and used for RT-PCR and real-time quantitative RT-PCR analyses.
Immunohistochemistry. Posterior poles were processed as described in ref. 20; some were embedded directly in OCT compound without prior fixation. Tissues were sectioned to a thickness of 6-8 μm, and immunolabeling was performed as described in ref. 20. Adjacent sections were incubated with secondary antibody alone to serve as controls. Some immunolabeled specimens were prepared and viewed by confocal laser scanning microscopy, as described in ref. 24. The antibodies used are listed in the legend to Fig. 1.
Fig. 1.

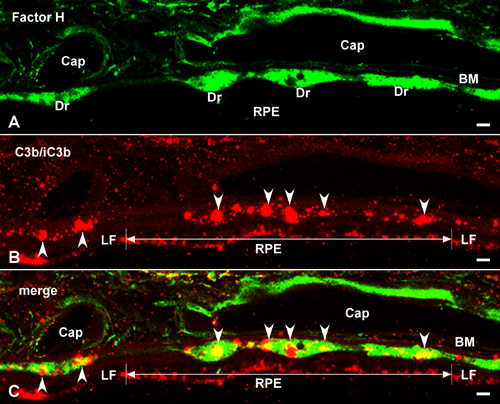
Immunolocalization of HF1 (A-H) and MAC (C5b-9) (I-L) in the RPE/choroid (Chor) complex. Ret, retina; Dr, drusen. (A-D) Autofluorescent RPE lipofuscin granules are red (Cy3 channel). (A and B) Confocal immunofluorescence images from an 84-year-old male with atrophic AMD. Anti-HF1 antibody (Advanced Research Technologies, San Diego) labels substructural elements (arrows) in drusen and the subRPE space (green; Cy2 channel). These elements also display IR by using antibodies raised against C3 fragments (iC3b, C3d, and C3dg). The anti-HF1 labeling in the lumens of choroidal capillaries (asterisks) reflects circulating HF1. (Scale bars: A, 5 μm; B, 3 μm.) (C and D) Confocal localization of HF1 in drusen and the subRPE space in an 83-year-old male with AMD (anti-HF1; Quidel, San Diego) (green). (C) Drusen IR is homogeneous. (D) HF1 IR is present throughout the choroid and in the subRPE space (arrowheads). (Scale bars: C, 10 μm; D, 20 μm.) (E-L) Brown pigment in the RPE cytoplasm and choroid is melanin. (E and F) Localization of HF1 in drusen in a 79-year-old donor eye. (E) Anti-HF1 monoclonal antibody-labeling (Quidel) (purple reaction product) is apparent in drusen, along BM, and on the choroidal capillary walls (arrows). (F) Control section of the same eye with no primary antibody. Labeling is absent. (Scale bars: E, 10 μm; F, 10 μm.) (G and H) Localization of HF1 in the macula. (G) Extensive labeling is present along BM, the choroidal capillary walls, and intercapillary pillars (arrows) in a 78-year-old with AMD. (H) Control section from the macula of a donor without AMD; much less labeling is apparent. (Scale bars: G, 20 μm; H, 20 μm.) (I and J) Localization of C5b-9 in the RPE-choroid underlying the macula (I) and extramacula (J) in the same eye of an 81-year-old AMD donor. (I) Intense anti-C5b-9 IR is associated with drusen, BM, and the choroidal capillary endothelium. (J) Outside the macula, there is only sporadic labeling in the vicinity of BM. (Scale bars: I, 20 μm; J, 20 μm.) (K and L) Localization of C5b-9 in the macula from a donor with AMD (K) and from a second donor without AMD (L). (K) Anti-C5b-9 labeling is associated primarily with the choroidal capillary walls (black arrows) and intercapillary pillars (white arrows). Labeling is much more intense in the AMD eye. Note the strong similarity to the anti-HF1 labeling pattern in the macula from the same donor (G). (Scale bars: K, 15 μm; L, 20 μm.)
Mutation Screening and Analysis. Coding and adjacent intronic regions of HF1 were examined for variants by using SSCP analyses, denaturing high performance liquid chromatography (DHPLC) and direct sequencing. Primers for SSCP, DHPLC, and DNA sequencing analyses (Table 2, which is published as supporting information on the PNAS web site) were designed to amplify each exon and its adjacent intronic regions with macvector software (Accelrys, San Diego). PCR-derived amplicons were screened for sequence variation as described in refs. 6 and 15. All changes detected by SSCP and DHPLC were confirmed by bidirectional sequencing according to standard protocols. Statistical analyses, including χ2 and Fisher's exact tests were performed as described in ref. 38. Detailed protocols are provided in Supporting Text: Statistical Analyses, which is published as supporting information on the PNAS web site.
Genotyping. SNPs were discovered through data mining (Ensembl database, dbSNP; Celera Discovery System, Applid Biosystems) and through sequencing. Assays for variants with >10% frequency in test populations were purchased from Applied Biosystems as Validated, Inventoried SNP Assays-On-Demand or submitted to an Applied Biosystems Assays-By-Design pipeline. The technique used was identical to that described in ref. 37. Briefly, 5 ng of DNA were subjected to 50 cycles on an Applied Biosystems 9700 384-well thermocycler, and plates were read in an Applied Biosystems 7900 HT Sequence Detection System.
Results
Factor H at the RPE-Choroid Interface. The distribution of HF1 within the macular and extramacular RPE/choroid complex was assessed in the eyes of six donors with early AMD and three donors of similar age without AMD or drusen (Fig. 1). In donors with AMD, intense and specific HF1 immunoreactivity (IR) was present in drusen, the subRPE space, and around the choroidal capillaries (Fig. 1 A-E and G). HF1 antibodies generally labeled drusen homogeneously (Fig. 1 C and E). In some cases, substructural elements within drusen (Fig. 1 A and B) react with antibodies against C3 fragments (e.g., iC3b) that are known HF1 ligands (25, 27). HF1 IR is more robust in donors with AMD compared with age-matched controls and most pronounced within the macula (Fig. 1 G and H). The distribution of HF1 in the macula (Fig. 1G) was highly similar to that of C5b-9 (Fig. 1 I-K); in both cases, labeling included the choroidal capillaries. Extramacular locations showed less HF1 and C5b-9 IR (Fig. 1J). Little or no C5b-9 IR in the RPE/choroid was observed in donors under the age of 50 and without AMD (Fig. 1L).
The RPE Is a Local Source of HF1. Appropriately sized amplicons for HF1 and FHL1 (truncated isoform) gene products were generated from cultured human RPE and from freshly isolated RPE and the RPE/choroid complex, but not neural retina, derived from donor eyes with and without AMD (Fig. 4, which is published as supporting information on the PNAS web site). Real-time quantitative RT-PCR assays confirmed that transcripts for HF1 and FHL1 are abundant in the RPE and choroid, approaching levels observed in the liver (Fig. 5, which is published as supporting information on the PNAS web site).
HF1 Variants Are Associated with AMD. To determine whether variants of HF1 are associated with AMD, all 22 coding exons and their 50- to 100-bp flanking intronic sequences were screened by SSCP in the University of Iowa cohort (some exons were screened in a 50% subset of the cohort; see Table 1 and Table 3, which is published as supporting information on the PNAS web site). A total of 26 sequence variants were detected: 17 SNPs in the coding region, including 5 synonymous and 12 nonsynonymous substitutions, and 9 intronic SNPs (Fig. 2). Coding-region SNPs included previously described common nonsynonymous variants, such as I62V in exon 2, Y402H in exon 9, and D936E in exon 18 (Figs. 2 and 3 and Table 1). A common variant in the intron 2 splice acceptor site, IVS2-18insTT, was also detected. Five rare (<0.5%) variants were also detected (data not shown) in AMD patients and controls, suggesting no significant role for rare HF1 alleles in the disorder. Detailed genotyping data were generated for seven of these SNPs in the Iowa cohort (Fig. 2 and Tables 1 and 3; see also Table 4, which is published as supporting information on the PNAS web site), and association analyses were performed by using a case-control study design. Highly significant associations with AMD were found with several variants, including the nonsynonymous I62V (χ2 = 15.0, P = 1.1 × 10-4) and Y402H (χ2 = 49.4, P = 2.1 × 10-12) variants and the IVS2 variant (χ2 = 22.2, P = 2.4 × 10-6) (Table 1). The strongest association with AMD in this cohort was observed with the synonymous A473A variant in exon 10 [odds ratio (OR) = 3.42, 95% confidence interval (CI) (2.27-5.15)].
Table 1. HF1 SNP association with AMD.
| lowa cohort
|
Columbia cohort
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Location | Designation | dbSNP ID | Cont, n | Case, n | χ2 | P | OR | 95% Cl | Cont, n | Case, n | χ2 | P | OR | 95% Cl |
| Promoter | Promoter | rs3753394 | — | — | — | — | — | — | 264 | 549 | 2.84 | 0.089 | 1.23 | 0.98-1.52 |
| IVS1 | IVS1 | rs529825 | — | — | — | — | — | — | 266 | 547 | 25.4 | 4.66×10−7 | 1.92 | 1.49-2.48 |
| Exon 2 | I62V | rs800292 | 68 | 228 | 15 | 1.1 × 10−4 | 2.79 | 1.67-4.65 | 261 | 546 | 26.1 | 3.21 × 10−7 | 1.95 | 1.51-2.52 |
| IVS2 | insTT | — | 126 | 390 | 22.21 | 2.44 × 10−6 | 2.38 | 1.65-3.44 | 273 | 549 | 28.4 | 9.87 × 10−8 | 2.04 | 1.57-2.63 |
| IVS6 | IVS6 | rs3766404 | — | — | — | — | — | — | 271 | 546 | 23.0 | 1.59 × 10−6 | 2.11 | 1.56-2.85 |
| Exon 7 | A307A | rs1061147 | 131 | 404 | 49.4 | 2.09 × 10−12 | 2.82 | 2.11-3.78 | 262 | 547 | 59.6 | 1.16 × 10−14 | 2.34 | 1.89-2.91 |
| Exon 9 | Y402H | rs1061170 | 131 | 403 | 49.4 | 2.09 × 10−12 | 2.82 | 2.11-3.78 | 272 | 549 | 54.4 | 1.64 × 10−13 | 2.25 | 1.79-2.75 |
| Exon 10 | A473A | rs2274700 | 68 | 221 | 35.14 | 3.07 × 10−9 | 3.42 | 2.27-5.15 | 264 | 542 | 45.4 | 1.61 × 10−11 | 2.10 | 1.69-2.61 |
| IVS10 | IVS10 | rs203674 | — | — | — | — | — | — | 264 | 545 | 66.1 | 4.29 × 10−16 | 2.44 | 1.97-3.03 |
| Exon 13 | Q672Q | rs3753396 | 129 | 404 | 0.21 | 0.65 | 1.12 | 0.76-1.64 | 265 | 545 | 2.05 | 0.15 | 1.24 | 0.94-1.65 |
| Exon 18 | D936E | rs1065489 | 67 | 223 | 0.64 | 0.8 | 0.809 | 0.51-1.56 | 264 | 536 | 0.53 | 0.46 | 1.12 | 0.85-1.49 |
The frequency of allele 1 and allele 2 from each SNP was compared between cases and controls (Cont) and the Yates χ2 and P values were calculated along with the OR and 95% confidence interval (95% CI). The actual counts of each genotype are provided in Table 3. —, no data available.
Fig. 2.

Schematic of the HF1/CFH gene. The approximate locations of the 11 SNPs used in the analyses are shown on top of the diagram. The encoded HF1 protein contains 20 SCRs, a RGD domain, and N-linked glycosylation sites (potential sites “P”). Approximate binding sites for pathogens and other substrates are depicted below the diagram based on data published in refs. 40 and 41. The map is not drawn to scale.
Fig. 3.

Association analysis of HF1 haplotypes. A set of the eight most informative SNPs in the HF1 gene were selected and analyzed for pairwise linkage disequilibrium in the Columbia cases and controls. R2 and D′ values are shown. All of the haplotypes with a frequency of >3% are displayed. The ORs for the comparison of cases and controls were calculated, and 95% CIs are shown in brackets. The estimated frequencies of the haplotype in cases (CAS) and controls (CON) are also shown. The major risk haplotype (H1) is shown in dark shading, and the protective haplotypes are shown in light shading. SNPs exclusively found in these risk and protective haplotypes are boxed. Analysis of diplotypes was performed to assess dominant and recessive effects. Homozygotes for the H1 haplotype (H1/H1) are significantly at risk, and H2/H2 individuals are significantly protected. Detailed information pertaining to analyses and access to additional data is provided in Supporting Text: Statistical Analyses.
The same two nonsynonymous SNPs were highly associated with AMD in an independent cohort from Columbia University (I62V: χ2 = 26.1, P = 3.2 × 10-7 and Y402H: χ2 = 54.4, P = 1.6 × 10-13) (Table 1). Several additional intronic SNPs were selected (based on their frequency and the availability of commercial assays) in addition to those examined in the Iowa cohort and screened in the Columbia cohort (for a total of 11 SNPs). The strongest association in this cohort was observed with SNP rs203674 in IVS10 (χ2 = 66.1, P = 4.29 × 10-16) [OR = 2.44, 95% CI (1.97-3.03)]. Although the OR is modest, the variant was very common; 30.5% of the cases were homozygous for allele B, compared with 12.9% of the controls. The Q672Q and D936E variants located in exons 13 and 18 in the C-terminal region of HF1 showed no statistically significant associations. Thus, the N-terminal region of HF1, which includes domains involved in pathogen and substrate molecule recognition (Fig. 2), is associated with AMD. Data derived from the two cohorts, including the significant associations with genotyped SNPs with AMD, and the frequencies and extents of associations were strikingly similar (Tables 1 and 4).
The observed associations were highly significant when the entire AMD patient cohorts were compared with controls (Table 1). When major subphenotypes of AMD, including eAMD, choroidal neovascularization, and GA were analyzed separately, the association was prominent in cases with eAMD and choroidal neovascularization. The GA group deviated from the general trend in some cases, especially with the haplotype defined by exon 13 (Q672Q) and 18 (D936E) alleles (data not shown). Although this deviation may be significant in terms of varying etiology, it did not reach statistical significance, most likely because of the relatively small numbers of GA cases examined.
Linkage disequilibrium (LD) analysis performed on both cohorts showed extensive LD across an extended region of HF1. Results of the Columbia cohort are shown in Fig. 3; complete genotyping data for all samples are available upon request (Supporting Text: Statistical Analyses). Three SNPs in the exon 2-3 region were in virtually complete LD, as were the A307A and Y402H variants in exons 7 and 9 and the Q672Q and D936E variants in exons 13 and 18. Haplotype estimation in cases and controls identified the most frequent at-risk haplotype in 50% of cases versus only 29% of controls [OR = 2.46, 95% CI (1.95-3.11)]. Homozygotes for this haplotype were present in 24.2% of cases and 8.3% of the controls [OR = 3.51, 95% CI (2.13-5.78)]. Two common protective haplotypes were found in 34% of controls and 18% of cases [OR = 0.48, 95% CI (0.33-0.69) and OR = 0.54, 95% CI (0.33-0.69)]. The SNPs that distinguish the risk and protective haplotypes are mainly contained in a region between exons 2 and 11; SNPs outside this area, e.g., the promoter SNP and the SNPs in exons 13 and 18, add little effect.
Discussion
HF1 Polymorphisms in AMD and MPGNII. The data presented here link a major proportion of AMD cases in two independent cohorts to specific polymorphisms in the complement regulatory gene HF1/CFH (39-41). Haplotype analysis shows the most frequent at-risk haplotype is present in half of individuals with AMD, compared with 29% of controls. The magnitude of the observed association is striking when compared to those of genetic abnormalities previously linked to AMD (6, 7, 12-14). The frequencies and extent of SNP associations are similar in the two cohorts; several SNPs show highly significant association with AMD in each. The associations are particularly strong in individuals with eAMD and choroidal neovascularization and less so for individuals with GA. No other associations with specific ocular phenotypes were discovered. Several protective haplotypes were also identified, further implicating HF1 function in AMD pathogenesis.
Individuals with MPGNII, a rare renal disease associated with HF1 deficiency, also develop early onset macular drusen, the hallmark lesion of AMD. The same 11 SNPs shown in Table 1 were also genotyped in 20 unrelated MPGNII patients. Approximately 70% of the MPGNII cases harbored the HF1 at-risk haplotype, providing further support for the concept that specific HF1 haplotypes are a key factor in drusen formation and confer increased susceptibility to macular pathology associated with AMD.
Functional Implications of HF1 Polymorphisms. Most of the AMD-associated HF1 SNPs lie within important functional domains of the encoded protein (Fig. 2), consisting of 20 short consensus repeats (SCR). The SCRs contain binding sites for C3b, heparin, sialic acid, and C-reactive protein (Fig. 2). Thus, these SNPs might affect HF1 function through variability in expression levels, binding efficiencies, and/or other properties. For example, the exon 2 I62V variant is located in SCR2, which includes a C3b binding site, and the exon 9 Y402H variant lies within SCR7 domain, which binds heparin and C-reactive protein. Interestingly, analysis of the effect of the TT insertion in the IVS2 splice site variant (https://splice.cmh.edu) suggests creation of a new cryptic splice acceptor 6 bp upstream of the natural acceptor site. Some of the studied SNPs might also affect the expression of HF1 isoforms. For example, I62V is present in a predicted exon splice enhancer (42). The functional consequences of other common SNPs might be modest because they are involved in late-onset phenotypes and not subjected to rigorous evolutionary constraint (43).
HF1 variants with more substantial effects are implicated in earlier-onset, severe diseases, such as atypical hemolytic uremic syndrome (aHUS) (40, 41, 44-46). HF1 mutations that lead to aHUS are typically missense mutations that limit the inhibitory functions of FH1. Although putative disease-causing mutations have been identified in only ≈25-35% of aHUS patients after complete screening of HF1 (44), a disease-associated haplotype defined by variants -257C>T (promoter), A473A (exon 13), and D936E (exon 18) predominates in aHUS patients without identifiable disease-causing mutations (44). The at-risk haplotype in aHUS does not overlap with that of AMD and/or MPGN. It is noteworthy that aHUS mutations cluster in the 3′ end of the HF1 gene, which produces only full-length HF1. In contrast, the AMD at-risk haplotype is located in regions that produce the full-length HF1 and truncated (FHL1) proteins. Thus, it may be important to determine the role of these two HF1-derived proteins in AMD.
The presence of the at-risk HF1 haplotype, in combination with an infectious agent or atypical activator of the alternative pathway, might substantially increase one's susceptibility to disease. It is plausible that different forms of the HF1 gene emerged in response to pathogens that activate the alternative complement pathway. Weakly acting HF1 haplotypes could provide reduced complement inhibition and stronger protection against bacterial infection. Weak alleles could also predispose individuals to the host cell/tissue damage that can be a consequence of complement activation. The combined effect of these factors most likely determines the severity of the resulting disease phenotype, which ranges from AMD to MPGN.
Biological Model of HF1 Dysfunction in AMD. A primary function of the complement system is to provide defense against infectious agents (47-49). Activation of complement triggers an amplifying, proteolytic cascade that leads to modifications of activating surfaces, the release of soluble proinflammatory anaphylatoxins and to the formation of the MAC, a macromolecular complex that promotes cell lysis through the formation of transmembrane pores. Uncontrolled activation of complement can lead to bystander damage in host cells and tissues. As a result, HF1 and other circulating and membrane-associated proteins have evolved to modulate the system (48).
A spectrum of complement components, including terminal pathway complement components, activation-specific fragments, and modulators, has been identified either within drusen (and/or adjacent RPE cells), along BM, and/or associated with the choriocapillaris in AMD (21-24, 26-28, 50, 51). There is evidence that cell-mediated events may also contribute to this process (29, 52-54).
Here, we show that HF1, a modulator of the C3 component of complement, is also a constituent of drusen in human donors with a history of AMD. HF1 colocalizes with its ligand C3b/iC3b in amyloid-containing substructural elements within drusen (Fig. 6, which is published as supporting information on the PNAS web site), implicating these structures as candidate complement activators within the subRPE space (25, 50). HF1 and MAC, as shown by C5b-9 IR, codistribute at the RPE-choroid interface and are most robust in the macular regions of eyes from donors with prior histories of AMD.
The strikingly similar distributions of HF1 and C5b-9 imply that significant amounts of MAC are generated and deposited at the RPE-choroid interface. This finding suggests that the HF1 protein encoded by the at-risk HF1 haplotype(s) may have attenuated complement inhibitory function. HF1 variants associated with AMD may put RPE and choroidal cells at sustained risk for alternative pathway-mediated complement attack. These findings are consistent with the fact that AMD pathology is manifested primarily in the macula and that complement activation at the level of BM is a key element in the process of drusen formation and the disruption of BM integrity (55) that is associated with late-stage neovascular AMD.
The data obtained in this study may also provide insights regarding the roles of established risk factors for AMD, such as smoking history, the most consistently documented AMD risk factor (1, 2). Cigarette smoke has been shown to activate the alternative complement pathway through the modification of C3 in vitro (56). Similar processes acting in vivo could promote inflammatory events at the RPE-choroid interface in the eye that hasten drusen formation and exacerbate the genetic susceptibility to AMD that is conferred by the at-risk HF1 haplotype.
The results of this investigation provide strong evidence that a specific common haplotype of the complement regulator HF1 predispose individuals to AMD. The results also implicate abnormalities in HF1-mediated regulation of alternative pathway complement activation and pathogenic agents that activate the system in a substantial proportion of AMD cases. Thus, molecules involved in complement activation and its regulation become prime targets for therapeutic intervention in AMD.
Supplementary Material
Acknowledgments
We thank Drs. D. Bok, P. Dudley, M. Niesman, L. Donoso, P. Luthert, B. Martin, R. Mullins, N. Aptsiauri, Y. Wang, Q. Lu, V. Chong, A. Hughes, V. Sheffield, M. Radeke, and P. Johnson for insights and helpful discussions; Drs. J. Hoballah, J. Sharp, T. Krezowik, T. Weingeist, C. Boldt, J. Folk, T. Johnson, M. Wilkinson, A. Lotery, D. Zumbro, E. Stone, J. Sanders, P. Gouras, C. McAvoy, S. Thompson, L. Arbisser, and A. Arbisser for unending commitment and assistance in recruiting patients; L. Buckta, K. Betts, K. Peterson, C. Sweet, R. Lee, S. Baruah, J. Roos, R. Wolfe, S. McCormick, J. Donahue, J. Smith, S. Imhoff, K. Kelly and staff, T. Ali and staff, B. Murphy and staff, and T. Fisher for technical support; J. Requard and the staffs of the Iowa Lions, Oregon Lions, and Central Florida Lions Eye Banks; and participating retirement and senior centers in Iowa for dedication to this research endeavor. Most of all, we thank the individuals and families who unselfishly donated their time and/or the eyes of their loved ones to this research program; science would not move forward without their participation. This work was supported by National Institutes of Health Grants EY11515 (to G.S.H.), EY13435 (to R.A.), EY11527 (to L.V.J.), and EY11521 (to D.H.A.); Department of Health and Human Services Contract NO1-CO-12400 (to M.D.); the American Macular Degeneration Foundation (G.S.H.); the International Retina Research Foundation (G.S.H.); the Eye Research Institute (G.S.H.); the Foundation Fighting Blindness (R.A.); the Ruth and Milton Steinbach Fund (G.S.H. and R.A.); gifts to the Center for the Study of Macular Degeneration; and unrestricted grants to the University of Iowa and Columbia University from Research to Prevent Blindness, Inc. G.S.H. holds an Honorary Professorship in the School of Medicine, Queen's University, Belfast.
Author contributions: G.S.H., M.D., and R.A. designed research; G.S.H., D.H.A., L.V.J., L.S.H., A.J.T., L.I.H., J.L.H., H.A.S., J.D.B., R.J.H.S., J.S., C.C.W.K., I.B., A.K.O., J.B., J.Z., J.E.M., B.G., M.D., and R.A. performed research; G.S.H. contributed new reagents/analytic tools; G.S.H., D.H.A., L.V.J., L.S.H., A.J.T., L.I.H., J.L.H., H.A.S., J.D.B., R.J.H.S., C.C.W.K., I.B., A.K.O., J.B., J.Z., J.E.M., B.G., M.D., and R.A. analyzed data; G.S.H., D.H.A., L.V.J., M.D., and R.A. wrote the paper; and G.S.H., J.L.H., H.A.S., K.M.G., G.S., S.R.R., S.C., L.A.Y., G.R.B., J.C.M., and R.T.S. contributed patient DNA samples and clinical history information.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: AMD, age-related macular degeneration; eAMD, early AMD; RPE, retinal pigmented epithelium; MAC, membrane attack complex; BM, Bruch's membrane; MPGN, membranoproliferative glomerulonephritis; SSCP, single-strand conformation polymorphism; GA, geographic atrophy; IR, immunoreactivity; OR, odds ratio; CI, confidence interval.
See Commentary on page 7053.
References
- 1.Klein, R., Peto, T., Bird, A. & Vannewkirk, M. R. (2004) Am. J. Ophthalmol. 137, 486-495. [DOI] [PubMed] [Google Scholar]
- 2.van Leeuwen, R., Klaver, C. C., Vingerling, J. R., Hofman, A. & de Jong, P. T. (2003) Eur. J. Epidemiol. 18, 845-854. [DOI] [PubMed] [Google Scholar]
- 3.Klein, M. L. & Francis, P. J. (2003) Ophthalmol. Clin. N. Am. 16, 567-574. [DOI] [PubMed] [Google Scholar]
- 4.Tuo, J., Bojanowski, C. M. & Chan, C. C. (2004) Prog. Retinal Eye Res. 23, 229-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klaver, C. C., Wolfs, R. C., Assink, J. J., van Duijn, C. M., Hofman, A. & de Jong, P. T. (1998) Arch. Ophthalmol. 116, 1646-1651. [DOI] [PubMed] [Google Scholar]
- 6.Allikmets, R., Shroyer, N. F., Singh, N., Seddon, J. M., Lewis, R. A., Bernstein, P. S., Peiffer, A., Zabriskie, N. A., Li, Y., Hutchinson, A., et al. (1997) Science 277, 1805-1807. [DOI] [PubMed] [Google Scholar]
- 7.Allikmets, R. (2000) Am. J. Hum. Genet. 67, 487-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klaver, C. C., Kliffen, M., van Duijn, C. M., Hofman, A., Cruts, M., Grobbee, D. E., van Broeckhoven, C. & de Jong, P. T. (1998) Am. J. Hum. Genet. 63, 200-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Souied, E. H., Benlian, P., Amouyel, P., Feingold, J., Lagarde, J. P., Munnich, A., Kaplan, J., Coscas, G. & Soubrane, G. (1998) Am. J. Ophthalmol. 125, 353-359. [DOI] [PubMed] [Google Scholar]
- 10.Stone, E. M., Braun, T. A., Russell, S. R., Kuehn, M. H., Lotery, A. J., Moore, P. A., Eastman, C. G., Casavant, T. L. & Sheffield, V. C. (2004) N. Engl. J. Med. 351, 346-353. [DOI] [PubMed] [Google Scholar]
- 11.Weeks, D. E., Conley, Y. P., Tsai, H. J., Mah, T. S., Rosenfeld, P. J., Paul, T. O., Eller, A. W., Morse, L. S., Dailey, J. P., Ferrell, R. E., et al. (2001) Am. J. Ophthalmol. 132, 682-692. [DOI] [PubMed] [Google Scholar]
- 12.Klein, M. L., Schultz, D. W., Edwards, A., Matise, T. C., Rust, K., Berselli, C. B., Trzupek, K., Weleber, R. G., Ott, J., Wirtz, M. K., et al. (1998) Arch. Ophthalmol. 116, 1082-1088. [DOI] [PubMed] [Google Scholar]
- 13.Schultz, D. W., Klein, M. L., Humpert, A. J., Luzier, C. W., Persun, V., Schain, M., Mahan, A., Runckel, C., Cassera, M., Vittal, V., et al. (2003) Hum. Mol. Genet. 12, 3315-3323. [DOI] [PubMed] [Google Scholar]
- 14.Abecasis, G. R., Yashar, B. M., Zhao, Y., Ghiasvand, N. M., Zareparsi, S., Branham, K. E., Reddick, A. C., Trager, E. H., Yoshida, S., Bahling, et al. (2004) Am. J. Hum. Genet. 74, 482-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayashi, M., Merriam, J. E., Klaver, C. C., Zernant, J., Bergen, A. A., Smith, R. T., Chang, S., Merriam, J. C. & Allikmets, R. (2004) Ophthalmic Genet. 25, 111-119. [DOI] [PubMed] [Google Scholar]
- 16.Iyengar, S. K., Song, D., Klein, B. E., Klein, R., Schick, J. H., Humphrey, J., Millard, C., Liptak, R., Russo, K., Jun, G., et al. (2004) Am. J. Hum. Genet. 74, 20-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Akiyama, H., Barger, S., Barnum, S., Bradt, B., Bauer, J., Cole, G. M., Cooper, N. R., Eikelenboom, P., Emmerling, M., Fiebich, B. L., et al. (2000) Neurobiol. Aging 21, 383-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Torzewski, J., Bowyer, D. E., Waltenberger, J. & Fitzsimmons, C. (1997) Atherosclerosis 132, 131-138. [DOI] [PubMed] [Google Scholar]
- 19.Schwertz, R., Rother, U., Anders, D., Gretz, N., Scharer, K. & Kirschfink, M. (2001) Pediatr. Allergy Immunol. 12, 166-172. [DOI] [PubMed] [Google Scholar]
- 20.Hageman, G. S., Mullins, R. F., Russell, S. R., Johnson, L. V. & Anderson, D. H. (1999) FASEB J. 13, 477-484. [DOI] [PubMed] [Google Scholar]
- 21.Hageman, G. S., Luthert, P. J., Victor Chong, N. H., Johnson, L. V., Anderson, D. H. & Mullins, R. F. (2001) Prog. Retinal Eye Res. 20, 705-732. [DOI] [PubMed] [Google Scholar]
- 22.Mullins, R. F., Russell, S. R., Anderson, D. H. & Hageman, G. S. (2000) FASEB J. 14, 835-846. [PubMed] [Google Scholar]
- 23.Mullins, R. F., Aptsiauri, N. & Hageman, G. S. (2001) Eye 15, 390-395. [DOI] [PubMed] [Google Scholar]
- 24.Anderson, D. H., Mullins, R. F., Hageman, G. S. & Johnson, L. V. (2002) Am. J. Ophthalmol. 134, 411-431. [DOI] [PubMed] [Google Scholar]
- 25.Anderson, D. H., Talaga, K. C., Rivest, A. J., Barron, E., Hageman, G. S. & Johnson, L. V. (2004) Exp. Eye Res. 78, 243-256. [DOI] [PubMed] [Google Scholar]
- 26.Johnson, L. V., Ozaki, S., Staples, M. K., Erickson, P. A. & Anderson, D. H. (2000) Exp. Eye Res. 70, 441-449. [DOI] [PubMed] [Google Scholar]
- 27.Johnson, L. V., Leitner, W. P., Staples, M. K. & Anderson, D. H. (2001) Exp. Eye Res. 73, 887-896. [DOI] [PubMed] [Google Scholar]
- 28.Crabb, J. W., Miyagi, M., Gu, X., Shadrach, K., West, K. A., Sakaguchi, H., Kamei, M., Hasan, A., Yan, L., Rayborn, M. E., et al. (2002) Proc. Natl. Acad. Sci. USA 99, 14682-14687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Penfold, P. L., Madigan, M. C., Gillies, M. C. & Provis, J. M. (2001) Prog. Retin. Eye Res. 20, 385-414. [DOI] [PubMed] [Google Scholar]
- 30.Espinosa-Heidmann, D. G., Suner, I. J., Hernandez, E. P., Monroy, D., Csaky, K. G. & Cousins, S. W. (2003) Invest. Ophthalmol. Visual Sci. 44, 3586-3592. [DOI] [PubMed] [Google Scholar]
- 31.O'Brien, C., Duvall-Young, J., Brown, M., Short, C. & Bone, M. (1993) Br. J. Ophthalmol. 77, 778-780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Colville, D., Guymer, R., Sinclair, R. A. & Savige, J. (2003) Am. J. Kidney Dis. 42, E2-5. [DOI] [PubMed] [Google Scholar]
- 33.McAvoy, C. E. & Silvestri, G. (September 17, 2004) Eye, 10.1038/sj.eye.6701697. [DOI]
- 34.Neary, J. J., Conlon, P. J., Croke, D., Dorman, A., Keogan, M., Zhang, F. Y., Vance, J. M., Pericak-Vance, M. A., Scott, W. K. & Winn, M. P. (2002) J. Am. Soc. Nephrol. 13, 2052-2057. [DOI] [PubMed] [Google Scholar]
- 35.Bird, A. C., Bressler, N. M., Bressler, S. B., Chisholm, I. H., Coscas, G., Davis, M. D., de Jong, P. T., Klaver, C. C., Klein, B. E., Klein, R., et al. (1995) Surv. Ophthalmol. 39, 367-374. [DOI] [PubMed] [Google Scholar]
- 36.Klaver, C. C., Assink, J. J., van Leeuwen, R., Wolfs, R. C., Vingerling, J. R., Stijnen, T., Hofman, A. & de Jong, P. T. (2001) Invest. Ophthalmol. Visual Sci. 42, 2237-2241. [PubMed] [Google Scholar]
- 37.Hu, J. & Bok, D. (2001) Mol. Vision 7, 14-19. [PubMed] [Google Scholar]
- 38.Gold, B., Kalush, F., Bergeron, J., Scott, K., Mitra, N., Wilson, K., Ellis, N., Huang, H., Chen, M., Lippert, R., et al. (2004) Cancer Res. 64, 8891-8900. [DOI] [PubMed] [Google Scholar]
- 39.Zipfel, P. F. (2001) Semin. Thromb. Hemostasis 27, 191-199. [DOI] [PubMed] [Google Scholar]
- 40.Zipfel, P. F., Skerka, C., Hellwage, J., Jokiranta, S. T., Meri, S., Brade, V., Kraiczy, P., Noris, M. & Remuzzi, G. (2002) Biochem. Soc. Trans. 30, 971-978. [DOI] [PubMed] [Google Scholar]
- 41.Rodriguez de Cordoba, S., Esparza-Gordillo, J., Goicoechea de Jorge, E., Lopez-Trascasa, M. & Sanchez-Corral, P. (2004) Mol. Immunol. 41, 355-367. [DOI] [PubMed] [Google Scholar]
- 42.Wang, Z., Rolish, M. E., Yeo, G., Tung, V., Mawson, M. & Burge, C. B. (2004) Cell 119, 831-845. [DOI] [PubMed] [Google Scholar]
- 43.Thomas, P. D. & Kejariwal, A. (2004) Proc. Natl. Acad. Sci. USA 101, 15398-15403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caprioli, J., Castelletti, F., Bucchioni, S., Bettinaglio, P., Bresin, E., Pianetti, G., Gamba, S., Brioschi, S., Daina, E., Remuzzi, G., et al. (2003) Hum. Mol. Genet. 12, 3385-3395. [DOI] [PubMed] [Google Scholar]
- 45.Zipfel, P. F. (2001) Trends Immunol. 22, 345-348. [DOI] [PubMed] [Google Scholar]
- 46.Zipfel, P. F., Skerka, C., Caprioli, J., Manuelian, T., Neumann, H. H., Noris, M. & Remuzzi, G. (2001) Int. Immunopharmacol. 1, 461-468. [DOI] [PubMed] [Google Scholar]
- 47.Morgan, B. P. & Walport, M. J. (1991) Immunol. Today 12, 301-306. [DOI] [PubMed] [Google Scholar]
- 48.Morgan, B. P. (1999) Crit. Rev. Immunol. 19, 173-198. [PubMed] [Google Scholar]
- 49.Kinoshita, T. (1991) Immunol. Today 12, 291-295. [DOI] [PubMed] [Google Scholar]
- 50.Johnson, L. V., Leitner, W. P., Rivest, A. J., Staples, M. K., Radeke, M. J. & Anderson, D. H. (2002) Proc. Natl. Acad. Sci. USA 99, 11830-11835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mullins, R. F., Johnson, L. V., Anderson, D. H. & Hageman, G. S. (1997) Ophthalmology 104, 288-294. [DOI] [PubMed] [Google Scholar]
- 52.Seddon, J. M., Gensler, G., Milton, R. C., Klein, M. L. & Rifai, N. (2004) J. Am. Med. Assoc. 291, 704-710. [DOI] [PubMed] [Google Scholar]
- 53.Cousins, S. W., Espinosa-Heidmann, D. G. & Csaky, K. G. (2004) Arch. Ophthalmol. 122, 1013-1018. [DOI] [PubMed] [Google Scholar]
- 54.Miller, D. M., Espinosa-Heidmann, D. G., Legra, J., Dubovy, S. R., Suner, I. J., Sedmak, D. D., Dix, R. D. & Cousins, S. W. (2004) Am. J. Ophthalmol. 138, 323-328. [DOI] [PubMed] [Google Scholar]
- 55.Chong, N. H., Keonin, J., Luthert, P. J., Frennesson, C. I., Weingeist, D. M., Wolf, R. L., Mullins, R. F. & Hageman, G. S. (2005) Am. J. Pathol. 166, 241-251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kew, R. R., Ghebrehiwet, B. & Janoff, A. (1985) J. Clin. Invest. 75, 1000-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}