

Abstract
We describe here the tyrosine kinase activity of human biliverdin reductase (BVR) and its potential role in the insulin-signaling pathway. BVR is both a substrate for insulin receptor (IR) tyrosine kinase (IRK) activity and a kinase for serine phosphorylation of IR substrate 1 (IRS-1). Our previous studies have revealed serine/threonine kinase activity of BVR. Y198, in the YMKM motif found in the C-terminal domain of BVR, is shown to be a substrate for insulin-activated IRK. This motif in IRS proteins provides a docking site for proteins that contain a Src homology 2 domain. Additionally, Y228 in the YLSF sequence and Y291 are IRK substrates; the former sequence provides optimum recognition motif in the tyrosine phosphatase, SHP-1, and for SHC (Src homology 2 domain containing transfroming protein 1). BVR autophosphorylates N-terminal tyrosines Y72 and Y83. Serine residues in IRS-1 are targets for BVR phosphorylation, and point mutation of serine residues in the kinase domain of the reductase inhibits phosphotransferase activity. Because tyrosine phosphorylation of IRS-1 activates the insulin signaling pathway and serine phosphorylation of IRS-1 blocks insulin action, our findings that insulin increases BVR tyrosine phosphorylation and that there is an increase in glucose uptake in response to insulin when expression of BVR is “knocked down” by small interfering RNA suggest a potential role for BVR in the insulin signaling pathway.
Keywords: dual-specificity kinases, insulin receptor substrates, insulin signaling pathway, heme metabolic enzymes
Biliverdin reductase (BVR) is an evolutionarily conserved soluble enzyme found primarily in mammalian species. The human reductase was recently identified as a serine/threonine kinase (1) sharing conserved catalytic domains that define such kinases (2). Before this, the enzyme was solely considered in the context of its reductase activity and conversion of the open tetrapyrrole biliverdin to bilirubin (3-5). Biliverdin is the product of the isomer-specific cleavage of heme (Fe-protoporphyrin IX) by the heme oxygenase isozymes, HO-1 and HO-2, reviewed in refs. 6 and 7. BVR was also found to translocate to the nucleus in cells treated with cGMP (8) and function as a transcription factor for activator protein 1-regulated genes and activation of c-jun and CREB/ATF-2 (9-11).
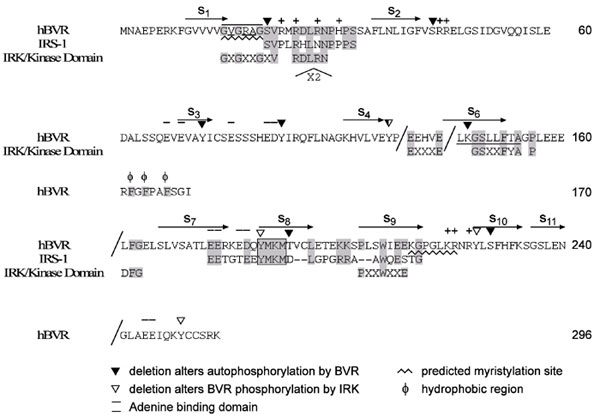
Analysis of the primary and the secondary structures of BVR (5, 12, 13) identify features that signify its relevance to protein tyrosine kinase (PTK)-regulated functions and its contribution to cell signaling as an adaptor/scaffold protein (see Fig. 7, which is published as supporting information on the PNAS web site). This includes the Y198MKM sequence that in insulin receptor (IR) substrate (IRS) proteins functions as the binding site for effector proteins with a Src homology 2 (SH-2) domain, such as phosphatidylinositol 3-kinase (PI3-kinase) (14, 15). In both BVR and IRS-1, four acidic residues are located N-terminal to the YMXM sequence, a feature frequently associated with tyrosine residues that are substrates for PTKs (2). PTKs are a multigenic family exclusive to higher organisms (2). In BVR, a threonine, and in three of four copies of YMXM in IRS-1, a serine residue flanks the C-terminal of the sequence. In addition, sequence alignment of IR tyrosine kinase (IRK) and BVR shows a similarity of key functional residues in corresponding regions of the tyrosine kinase and the reductase (Fig. 7). Whereas the N-terminal segment of BVR is the nucleotide-binding domain, the C terminus consists of a six-stranded β-sheet that would provide an ideal docking/protein:protein interaction site.
The action of insulin as a metabolic regulator and a growth factor is PTK-dependent; PTKs function in cell signaling pathways involved in growth, differentiation, and mobility, and in the development of diseases such as diabetes and cancer. The coupling of the intracellular kinase domain of IR with its substrate is an essential step in the initiation of the signaling cascade (16, 17). Autophosphorylation on tyrosine residues and activation of IRK that results from conformational change in the kinase, after insulin binding to the extracellular domain of the receptor, serves as a recognition signal for IRS proteins (IRS-1-IRS-7) (14, 15, 18).
Signaling complexes around tyrosyl phosphate are also assembled with the adaptor/scaffold proteins, including the small family of SH-2-containing tyrosine phosphatases, SHP-1 and SHP-2, that regulate signaling events for growth factors and cytokines (19, 20). Not all tyrosine kinases are membrane-bound, e.g., CLK1 and STY0 are primarily present in the cytoplasm (21). Insulin signaling is inhibited by IRS-1 serine phosphorylation, which has been considered as a mechanism for insulin resistance (22). In human IRS-1, a number of serines have been identified as important residues, including S307, S312, and S616. Several serine/threonine kinases including JNK and PKC are known to phosphorylate IRS-1 (23-25).
The above combination of BVR features inspired the present investigation, which has identified BVR as a new IRK substrate family member. The investigation has characterized the motifs Y198MKM and Y228LSF, together with Y291, as IRK tyrosine phosphorylation sites, whereas tyrosines at BVR positions 72 and 83 are identified as targets of autophosphorylation. This finding defines human BVR as a member of the rare family of dual-specificity (serine/threonine/tyrosine) kinases. Data suggest that both the tyrosine and the serine/threonine kinase activities of BVR contribute to insulin action and glucose uptake.
Materials and Methods
Materials. Human insulin, poly-Glu/Tyr (4:1), and DL-DTT were purchased from Sigma-Aldrich. IR beta (IRK) and IRS-1 peptide Y608 were purchased from Biomol. Monoclonal antiphosphotyrosine antibodies were obtained from Zymed, and [γ-32P]ATP was purchased from PerkinElmer. 2-Deoxy-[1-3H]glucose was purchased from Amersham Pharmacia Biotech. Raytide was purchased from Oncogene Science. Genistein was from Calbiochem. Myelin basic protein (MBP) and protein A/G plus-agarose were obtained from Santa Cruz Biotechnology. IRS-1 peptides KKHADDGAMPMS616PGVA (peptide 1) and RTES307ITATS312PAS315MVGGKP (peptide 2) were generated by Synpep (Dublin, CA).
Expression Vector Construction. A full-length BVR clone cDNA (5) was used as a PCR template to amplify the coding sequence, and the resulting fragment was gel-purified and ligated into expression vector pcDNA3 (Invitrogen). Transformants in Escherichia coli DH5α were verified by PCR, restriction analysis, and DNA sequencing. The pGEX 4T-2/human BVR vector was prepared as described in ref. 1. Mutant variants of both pGEX 4T-2 and pcDNA3 constructs were obtained by using a QuikChange XL site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions, using PAGE purified primers, and extensively characterized by restriction analysis, PCR, and DNA sequencing. For details, see Supporting Methods and Table 1, which are published as supporting information on the PNAS web site.
GST-Fusion Protein Expression in E. coli and Purification. Transformation of INV chemically competent cells (Amersham Pharmacia) with the pGEX 4T-2/hBVR construct or the various mutants, followed by expression of the plasmid genes, yielded human BVR fused to GST. GST-tagged proteins were purified by using glutathione-Sepharose 4B as described in ref. 1.
Cell Culture Transfection and Infection of 293A. 293A cells were grown in 10-cm plates with DMEM containing 10% FBS and 1% penicillin-G/streptomycin for 24 h or until a confluency of 70% was reached. Cells were subsequently transfected with 4 μg of pcDNA3 mutant BVR Y198 by using Transfectin lipid reagent (Bio-Rad) and incubated for 48 h at 37°C. In addition, non-transfected cells were infected with pSuper-Retro-siBVR (11) for 24 h in the presence of 4 μg/ml polybrene. Cells were starved in serum-free DMEM 16 h before insulin treatment (50 nM).
Measurement of BVR Reductase Activity. 293A cells treated with insulin were lysed in buffer containing a protease inhibitor mixture and phosphatase inhibitors (10 mM NaF/1 mM NaVO4). BVR conversion of biliverdin to bilirubin was measured as before at pH 6.7 by using NADH as the cofactor (26). Specific activity is expressed as μmol of bilirubin per min per mg of protein and is shown as activity relative to the control.
BVR Autophosphorylation and Kinase Activity. To detect autophosphorylation, wild-type and mutant GST-hBVR were incubated in 50 mM Hepes (pH 8.4) in the presence of 30 mM MnCl2 and 1 mM DTT for 2 h at 30°C. The reaction was started by the addition of 10 μM ATP labeled with 10 μCi of [γ-32P]ATP (1 Ci = 37 GBq) and was stopped by adding Laemmli sample buffer. Samples were boiled for 3 min and applied to 8% SDS/PAGE gels, transferred to PVDF membrane, and autoradiographed. To measure kinase activity, GST-BVR (5 μg) was incubated in 50 μl of 50 mM Hepes (pH 8.4), 20 mM MgCl2, 30 mM MnCl2, 1 mM DTT, and 10 μM ATP labeled with 10 μCi of [γ-32P]ATP with 5 μg of substrate [IRS-1, Raytide, or poly-Glu/Tyr (4:1)] for 2 h at 30°C. The reaction was terminated by adding 120 μl of 10% H3PO4, and a 100-μl aliquot was directly transferred to P81 Whatman filters, which were washed in 0.75% phosphoric acid at room temperature, then washed in acetone, dried, and counted in a liquid scintillation counter.
BVR Phosphorylation by the IRK. Phosphorylation of BVR by IRK was examined by using GST-hBVR fusion protein and the 48-kDa cytoplasmatic domain of the β-subunit of IRK. For routine assay, 5 μg of BVR was used, and the reductase was incubated in 50 μl of IRK kinase buffer (50 mM Hepes, pH 8.0/20 mM MgCl2/1 mM DTT) in the presence of 0.05 μg of IRK and 10 μM ATP labeled with 10 μCi of [γ-32P]ATP for 2 h at 30°C. For time course studies, the volume of the assay system was doubled. Reactions were processed as described above for the autophosphorylation samples.
Glucose Uptake. Glucose uptake was assessed by using 2-deoxy-[1-3H]glucose as described by Braiman et al. (27) with minor modifications. Briefly, after insulin treatment, cells in six-well plates were incubated (15 min) in 1 ml of PBS containing 1 μCi/ml 2-deoxy-[1-3H]glucose and either 5 mM or 200 mM glucose. The solution was aspirated rapidly and wells were washed three times with cold PBS. Cells were solubilized by addition of 200 μl of 1% (wt/vol) SDS. Radioactivity was measured and normalized to protein concentration. Specific uptake was determined by subtracting nonspecific binding in the presence of 200 mM glucose from the total uptake (in the presence of 5 mM glucose). Assessments were made in triplicate, and experiments were repeated three times.
Immunoprecipitation and Immunoblotting. 293A Cells in 10-cm plates were transfected with appropriate plasmids. Cells were washed twice with cold PBS, harvested, lysed in 300 μl of cold modified RIPA buffer (11), sonicated (twice for 3 sec), and centrifuged at 10,000 × g for 15 min. Protein concentration was determined by Bradford assay. Polyclonal hBVR antibody (28) was added to the lysate at a 1:50 dilution. After 3 h at 4°C, protein A/G agarose beads were added, incubated for 1 h, and centrifuged. The beads were washed twice with RIPA buffer containing protease inhibitors followed by three washes with cold PBS. Samples were subjected to 8% SDS/PAGE and transferred to PVDF, and membranes were probed with antiphosphotyrosine or anti-hBVR antibodies and visualized by using the ECL system (PerkinElmer).
Identification of Y198 Residue by Mass Spectrometry. Autophosphorylated BVR, and BVR either untreated or treated with IRK in the presence of 1 mM ATP were subjected to SDS/PAGE and stained with Coomassie blue (Bio-Rad). The stained protein was excised, subjected to tryptic hydrolysis, and analyzed with a MALDI-TOF mass spectrometer at either WEM Biochem (Toronto) or the University of Rochester.
Results
BVR Is a Substrate for the IRK and Is Phosphorylated in Vitro on Tyrosine. Based on the protein structure of BVR, and its resemblance to certain features of IRK and IRS-1, it was hypothesized that the reductase might be a substrate for IRK and a member of the PTK family. We find that BVR is a phosphorylation target of IRK; in the presence of IRK, BVR phosphorylation was detectable at 1 h and gradually increased up to 4 h (Fig. 1a). The lag period for BVR phosphorylation reflects a requirement for IRK activation and autophosphorylation. Reaction with antiphosphotyrosine antibodies (Fig. 1b) indicates that BVR is phosphorylated on tyrosine residues by IRK and/or is autophosphorylated. IRK treatment increases antityrosine antibody binding to BVR, and this is abolished by the inclusion of EDTA, a kinase inhibitor, or inhibited by the general PTK inhibitor genistein (data not shown). Autophosphorylation was implied by the basal phosphotyrosine immunoreactivity of BVR expressed in E. coli, which has no endogenous PTKs.
Fig. 1.

BVR is a substrate for IRK. (a) Time course of BVR phosphorylation by IRK. Ten micrograms of purified wtBVR was incubated in 100 μl of kinase buffer (pH 8.0) containing 0.1 μg of IRK and [γ-32P]ATP up to 4 h. At the indicated times, samples were subjected to 8% SDS/PAGE and transferred to PVDF, and phosphorylated proteins were visualized by autoradiography. (b) IRK phosphorylates BVR on tyrosine residues. Five micrograms of purified wtBVR was incubated in IRK phosphorylation buffer for 2 h. The reaction mixtures, with the indicated additions, were resolved as described above, blotted onto PVDF, and analyzed by using antiphosphotyrosine antibodies with the ECL detection system.
Tyrosines at Positions Y198, Y228, and Y291 Are Targets for IRK-Mediated Phosphorylation. Five of the six tyrosines in BVR meet the general criterion ascribed to targets of viral PTKs, i.e., the presence of at least one acidic amino acids N-terminal to the tyrosine (2). Point mutation analysis has identified Y198MXM as the main target of IRK; the Y198 mutation reduced the extent of phosphorylation by nearly 70% compared with wtBVR (Fig. 2a). The time course of phosphorylation of the Y198 mutant also shows it to be a less effective substrate for IRK (cf. Fig. 2b vs. Fig. 1b). The finding that mutation of Y198 did not completely abolish phosphorylation of BVR suggested that it is not the only IRK target. To identify other targets of IRK, BVRs with single point mutations at each tyrosine were examined for phosphorylation by IRK (Fig. 2c). Mutations at Y228 and Y291 also significantly decreased BVR phosphorylation by IRK (40% and 49%, respectively), whereas those at Y72, Y83, and Y98 residues did not. The assignment of Y228 and Y291 as targets of IRK was supported by the finding that little or no phosphorylation was detected with either of the double mutant Y198+Y228 or Y198+Y291 (Fig. 2d). Further support for phosphorylation of Y198 by IRK was sought by mass spectrometry. Purified GST-BVR grown in E. coli was subjected to IRK phosphorylation (see Materials and Methods). While both IRK-treated and nontreated BVR had a peak for peptide EDQY198MK at 813 Da, only the IRK-treated sample had a peak at 895 Da for the phosphorylated peptide.
Fig. 2.

BVR Y198 residue is a target site for IRK phosphorylation. (a) Y198 residue of BVR is phosphorylated by IRK. Phosphorylation of purified wtBVR or Y198 mutant BVR by IRK was assessed by autoradiography as described in Fig. 1a. (b) Time course of Y198 mutant BVR phosphorylation by IRK. Purified human BVR carrying the Y198 mutation was incubated with IRK in kinase assay. Samples were analyzed as in Fig. 1a. (c) Effect of mutation of tyrosine residues on BVR phosphorylation by IRK. Purified wtBVR or the indicated BVR tyrosine mutants were incubated with IRK for 2 h at 30°C in the presence of [γ-32P]ATP as described in the text. Subsequently, samples were subjected to SDS/PAGE and stained with Coomassie blue, and the bands were excised and their radioactivity measured. The experiment was repeated three times. (d) Further identification of BVR tyrosine residues phosphorylated by IRK. Double mutants of BVR were used as substrates for IRK, and analysis was carried out as described in Fig. 1a.
Autophosphorylation Sites of BVR Differ from Those of IRK-Mediated Phosphorylation. We identified BVR tyrosine autophosphorylation sites using the same tyrosine point mutants. Elimination of the IRK phosphorylation targets Y198, Y228, and Y291 did not result in significant changes in autophosphorylation of BVR, whereas mutations at Y72 and Y83 caused a marked decrease (Fig. 3a). The phosphotransferase activity of BVR was further explored by using the tyrosine kinase substrates Raytide and poly-Glu/Tyr (4:1). Raytide was effectively phosphorylated by BVR (Fig. 3b), as was poly-Glu/Tyr (data not shown). Y98 is neither a site of autophosphorylation nor a target for IRK.
Fig. 3.

Human BVR autophosphorylates on tyrosine residues and is a tyrosine kinase. (a) Effect of tyrosine mutations of BVR on autophosphorylation. wtBVR and BVR with the indicated tyrosine mutations were used in the autophosphorylation assay described in Materials and Methods. (b) BVR is a tyrosine kinase. Five micrograms of purified BVR in the presence or absence of 5 μg of Raytide was incubated in 50 μl of kinase buffer with [γ-32P]ATP for 2 h at 30°C. Reaction mixtures were transferred onto a P81 Whatman filters, and bound radioactivity was measured by liquid scintillation counting.
BVR Is Autophosphorylated in the Presence of Mn2+ but Not Mg2+. Autophosphorylation of BVR increased with increasing pH in the range 6.7-8.7 (data not shown). BVR tyrosine kinase activity is seen only in the presence of Mn2+ (Fig. 4a). When Mn2+ was replaced with Mg2+, Ca2+, or Zn2+, autophosphorylation of BVR was almost abolished. Zn2+, but not Ca2+ or Mg2+, proved to be inhibitory to BVR autophosphorylation, and this was not overcome by Mn2+. The effect of Zn2+ is consistent with the previous identification of human BVR as a Zn2+-binding protein with a likely binding site in the C-terminal segment of the protein (5). Because of the extensive interaction between the N-terminal ATP-binding domain of the protein and the carboxy terminal α-helix structure (12, 13), the inhibitory effect of Zn2+ could reflect disruption of proper folding of the protein.
Fig. 4.

Autophosphorylation of BVR is an Mn2+-dependant reaction. (a) Metal dependency of BVR kinase activity. The effect of divalent metal ions (30 mM MnCl2/20 mM MgCl2/20 mM CaCl2/20 mM Zn acetate) singly and in combination on BVR autophosphorylation was analyzed as described in Materials and Methods. (b) Inhibition of BVR autophosphorylation by a PTK inhibitor. Phosphorylation of wtBVR by IRK was examined in the presence of 200 μM genistein. The reactions were performed in the presence of 20 mM MgCl2.
Autophosphorylation of BVR does not occur in the presence of Mg2+ (Fig. 4a); however, IRK effectively used this metal ion to phosphorylate BVR. By using different conditions of pH and Mn2+ and Mg2+, we are able to differentiate BVR autophosphorylation and IRK-specific phosphorylation signals.
IRS-1 Is a Substrate for BVR Kinase Activity. Previous studies had shown that MBP and caseines are substrates for BVR serine/threonine kinase activity. Key serine residues of BVR that are contained in known kinase motifs were mutated to alanine, resulting in marked decreases in the ability of BVR to autophosphorylate (Fig. 5a). In addition, T202 flanking the YMKM motif was also found to be an autophosphorylation target. In IRS-1, a serine residue flanks this motif.
Fig. 5.

Human BVR is a kinase for IRS. (a) Identification of serine residue targets of BVR autophosphorylation. Recombinant human BVR with mutations on the indicated residues were used in the autophosphorylation assay. (b) IRS-1 is a substrate for BVR. Five micrograms of IRS-1 was incubated with 5 μg of BVR, HO-1, HO-2, or the known substrate, MBP. Bound radioactivity to P81 paper was normalized to that of BVR alone. (c) The presence of IRS increases phosphorylation of BVR by IRK. Phosphorylation of BVR by IRK was examined as in Fig. 1a, in the presence of increasing molar ratio of IRS:BVR. (d) ATP binding to BVR is necessary for it to phosphorylate IRS-1. The G17 to alanine mutant BVR construct was expressed, purified, and examined for kinase activity by using IRS-1 as the substrate as in b.
Several proteins were examined as potential substrates for BVR kinase activity, including IRS-1, HO-1, HO-2, and MBP. Both Mn2+ and Mg2+ were present in the assay as autophosphorylation of BVR is permitted. IRS-1 and MBP are clearly substrates for kinase activity (Fig. 5b), whereas HO-1 and HO-2 are not. Further confirmation of kinase activity of BVR was provided by increased MBP phosphorylation in the presence of BVR. In the presence of increasing amounts of IRS, there was an increase in phosphorylation of BVR by IRK up to an equimolar ratio of IRS:BVR (Fig. 5c). This concentration-dependent enhancement of BVR phosphorylation indicates an interaction of the IRK substrates. Further confirmation of BVR kinase activity was provided by a mutation at G17 in the ATP-binding site. The G17 mutation reduces IRS phosphorylation by BVR (Fig. 5d).
We examined whether IRS is a substrate for the serine or tyrosine kinase activity of BVR. The IRS-1 wild-type sequence of amino acids 605-620 contains three potential phosphorylation sites: T608, Y612, and S616. The mutant IRS-1 peptide (peptide 1) contained neither Y612 nor T608 (both mutated to alanine). Both the mutated and unmutated peptides were phosphorylated to the same extent (1,049 ± 78 cpm and 1,081 ± 183 cpm, respectively), indicating that S616 is the site of BVR phosphorylation. The IRS-1 peptide 2 contained S307, S312, and S315, which are the residues that have also been implicated as sites of serine phosphorylation and insulin resistance, and this peptide is also a suitable substrate for BVR (1,198 ± 230 cpm).
BVR Is an Antagonist to Insulin-Mediated Glucose Uptake by the Cell. Because the above data revealed that BVR is a substrate of IRK and that it phosphorylates IRS, it was hypothesized that BVR may participate in the insulin signaling pathway. To examine this, the effects of insulin treatment on BVR activity, tyrosine phosphorylation, and glucose uptake in cells were examined. The results are shown in Fig. 6. As noted in Fig. 6a, treatment of cells with insulin led to a 2-fold increase in BVR reductase activity after 1 h, which returned to basal level by 6 h. BVR activity increased as the result of in vitro phosphorylation by IRK, with the highest activity at 40 min (Fig. 6b). This finding suggests that phosphorylation at Y198, Y228, and/or Y291 are responsible for the increased BVR activity. BVR tyrosine phosphorylation was increased in cells within 8 min of insulin treatment (Fig. 6c). The relevance of Y198 phosphorylation to insulin-mediated glucose uptake was examined by using cells infected with small interfering RNA for human BVR and in cells transfected with pcDNA3-Y198 mutant BVR. The cells were then treated with insulin, and the rate of uptake of labeled glucose was assessed. “Knock-down” of BVR by small interfering RNA for human BVR significantly increased insulin-mediated glucose uptake (Fig. 6d) compared with controls infected with vector alone (P ≤ 0.05). Uptake by the cells transfected with Y198 mutant BVR was also increased significantly (P ≤ 0.05) but to a lesser extent than that of small interfering RNA. The findings are consistent with BVR being antagonistic to insulin effect on glucose uptake and its action being regulated via tyrosine Y198 phosphorylation and insulin-mediated activation of BVR.
Fig. 6.

“Knock-down” BVR and Y198 deletion increase glucose uptake into 293A cells upon insulin treatments. (a) Insulin treatment increases BVR activity. 293A cells were treated with insulin (50 nM). At the indicated times, cell lysates were prepared and the reduction of biliverdin to bilirubin was measured, expressed as μmol bilirubin per min per mg, and normalized to the control. (b) BVR phosphorylation by IRK in vitro increases BVR reductase activity. Purified BVR was phosphorylated by IRK for the indicated periods. Reactions were terminated by diluting with PBS and freezing at -20°C. BVR activity was determined as in a and normalized to that of the control (43.8 μmol per min per mg). (c) Insulin treatment increases BVR tyrosine phosphorylation. Cell lysates were obtained from cells treated with insulin for 8 or 30 min and immunoprecipitated with anti-human BVR antibodies. The phosphorylated BVR was subjected to Western blot and visualized by using antityrosine antibodies and the ECL system. (d) Effect of insulin treatment on glucose uptake by cells infected with small interfering RNA for human BVR or transfected with Y198 mutant BVR. Cells were infected/transfected with the constructs shown, treated with 50 nM insulin for 15 min, and subsequently incubated in 1 ml of PBS containing 5 mM glucose and 1 μCi/ml 2-deoxy-[1-3H]glucose for 15 min. Experimental details are provided in the text. Data are presented as mean ± SD of three experiments with triplicate samples in each.
Discussion
Primary and secondary structural features of BVR (5, 12, 13), and experiments that suggested posttranslational modification of BVR (26), made consideration that the protein is a component of the PTK-regulated signaling cascade and a substrate for the IRK plausible. The tyrosine-phosphorylated IRK substrates dock with molecules containing SH-2 phosphotyrosine-binding sites, such as PI3-kinase; the primary sequences adjacent to phosphotyrosine confer SH-2 domain recognition specificity. This study tested, and validated, two hypotheses: that BVR is a substrate for IRK and that the reductase is a PTK. The investigation also has identified tyrosine residues that are phosphorylation sites for IRK and those that are autophosphorylated. The N-terminal domain tyrosines Y72 and Y83 are autophosphorylation targets, and the C-terminal domain tyrosines (Y198, Y228, and Y291) are substrates for IRK. Because mutation of the sixth tyrosine, Y98, affected neither phosphorylation of BVR by IRK nor autophosphorylation of BVR, no function can yet be assigned to this residue. The study has also identified IRS-1, which is the primary target of phosphorylation by IRK for insulin-mediated glucose uptake, as a substrate for BVR serine/threonine kinase activity.
Collectively, the findings define BVR as a component of insulin signaling pathway. Furthermore, the identification of the human BVR as a tyrosine kinase characterizes the protein as one of a rare group of dual-specificity kinases, which have the ability to autophosphorylate on all three hydroxy amino acids (21, 29-32).
Although domains of the dual-specificity kinases are indistinguishable from those of serine/threonine kinases, autophosphorylation of tyrosine residues seems to obey certain criteria (2). They generally show a preference for Mn2+ over Mg2+, and much reduced activity with Ca2+; these are seen with BVR (Fig. 4a). The BVR kinase activity described here, which was measured under conditions specific to tyrosine kinases by using recombinant human BVR expressed in E. coli, doubtlessly reflects that of the enzyme itself, because the E. coli genome does not encode PTKs.
In context of kinase activity, there is certain resemblance between BVR and IRK, and, in the context of being substrate for IRK, there are similarities between IRS-1 and BVR in residues that flank the key tyrosine phosphorylation sites (Fig. 7). The secondary structure of BVR, unlike IRS proteins, does not have a defined and conserved pleckstrin homology domain. This domain is the phosphotyrosine-binding region of receptor substrates and consists of a sheet of seven β-strands at one end that interact with a C-terminal helix (33, 34). The phosphotyrosine-binding motif of IRS proteins interact with the NPXY motif of IR (35, 36). The primary structures of pleckstrin homology domains are divergent, but the secondary structure is highly conserved. A parallel structure that is present in BVR consists of a six-strand β-sheet and extensive interaction between the N-terminal domain and the C-terminal helix; this would provide an ideal structure for protein:protein interaction. A point mutation introduced in the adenine-binding domain (37) of BVR (GXGXXG) significantly decreased phosphorylation of IRS by BVR (Fig. 5). Evidently, the N-terminal domain of BVR has a key role in the transfer of phosphate to IRS-1, whereas its C-terminal domain functions as a binding site and acceptor of phosphates from IRK.
We have identified two potential SH-2 protein-docking sites in BVR; one is the Y198MKM motif. Many insulin responses associated with cell growth and glucose metabolism are mediated through IRS-1 and IRS-2 complexes (18). Interaction of IRS with IRK causes tyrosine phosphorylation of YMXM motifs of IRS proteins (IRS-1-IRS-7) that, in turn, serve as docking sites for SH-2-containing proteins and activation of insulin signaling. The human IRS protein has four copies of the YMXM motif. BVR has one such motif, followed by a threonine, which is a target of BVR autophosphorylation. In the case of insulin modulation of glucose uptake, docking involves binding of PI3-kinase to phosphorylated IRS and activation of PKB/Akt (38). The PI3-kinase pathway is a major arm of insulin signaling; PI3-kinase binding to the IRS and its interaction with downstream substrates leads to modulation of a variety of effector functions in the cell, including glucose transport. The phosphorylated YMXM motif is the preferred binding site for the P85 and P55 regulatory subunits of PI3-kinase. Based on the defined specificity of Src family for binding site (14, 15, 39), the BVR Y198MKM motif is predicted to be an ideal site for PI3-kinase.
The Y228LSF motif meets criteria for an optimum site for tyrosine phosphorylation of proteins that assemble into a multiprotein complex that recruits and/or facilitates relocation by SH-2 domain-containing polypeptides (14), including the SH-2 domain of Src family members and tyrosine phosphatases (19, 20). The presence of more than one SH-2-binding motif in a docking/scaffolding protein is not unusual; the IRK substrate Gab1 is phosphorylated on multiple tyrosine residues, not all of which are associated with a YMXM motif (40).
Serine phosphorylation of the IRS proteins reduces the ability to interact with the receptor and to function as a molecular docking site. Serine phosphorylation sites have been mapped to several residues including S307, S312, and S616 in human IRS-1 (23, 41-43); insulin resistance has been linked to serine phosphorylation of IRS-1. Our finding that BVR phosphorylates synthetic IRS-1 peptides specifically lacking tyrosine, but otherwise identical to IRS-1 peptides used as IRK substrates, indicates that IRS-1 is a likely substrate for BVR serine/threonine kinase activity.
Although most PTKs are associated with cell membranes, there are some that are not receptor-associated. BVR is a nonreceptor tyrosine kinase found mainly in the cytoplasm, although activation/hyperphosphorylation of the reductase, for instance by cGMP, leads to nuclear translocation (8). This relocalization is relevant to BVR's gene regulatory activity as a member of the leucine zipper family of transcription factors (9) and supports a role for the protein in context of its potential function as an anchoring/docking protein. In the C-terminal domain of the protein, downstream from the Y198MKM and immediately preceding Y228LSF, there is a sequence that contains a number of positively charged residues, K219GPGLKRNR, which is a potential myristoylation site. The sequence is similar to the Src myristoylation signal KDPSQRRN (44); the positively charged residues function in binding to membrane phospholipids. The GPG sequence preceding the charged residues permits maximum flexibility of folding of the BVR polypeptide. Previous studies have shown that replacement of positively charged residues in this sequence abrogates nuclear localization of the protein (8).
Three observations suggest a role for BVR in the mechanism of insulin resistance. The presence of IRS increases phosphorylation of BVR by IRK, BVR directly phosphorylates IRS (Fig. 5), and insulin-mediated glucose uptake is increased when BVR expression is “knocked down” (Fig. 6). The phosphorylation, by BVR, of IRS-1 peptides containing serine phosphorylation sites known to negatively affect glucose uptake is consistent with this concept. It is reinforced by the observation that under assay conditions that favor IRK activity, but not BVR autophosphorylation, phosphorylation of BVR is increased when both BVR and IRS-1 are available to IRK. This increased phosphorylation likely reflects a direct interaction of BVR and IRS, because there is precedence for this. A change in conformation of a kinase initiated by ligand binding can function both in directing proteins to subcellular targets and in modulating their activity. For instance, ligand binding to PKB/Akt can activate or inhibit its kinase activity by inducing conformational changes in the kinase that allow its activation/phosphorylation by PDK (33, 45). Similarly, changes in the conformation of PKC isoforms upon substrate binding or protein:protein interaction unmasks the catalytic domain/phosphorylation site (46). In the case of BVR, because four tyrosines in the protein potentially can be phosphorylated by IRK, a change in conformation of the protein caused by IRS binding may position a larger number of tyrosine residues for phosphorylation by IRK.
Insulin activation of the mitogen-activated protein kinase (MAPK) pathway, the second major arm for insulin signaling, primarily leads to activation of downstream substrates that function in gene expression/mutagenesis; therefore, activation of BVR by IRK would likely effect a wide spectrum of functions in the cell. This would include the previously reported activation and induction of a number of genes in cell signaling pathways, including MAPK and cAMP regulated genes by BVR (10). The principle of BVR silencing could be used to overcome insulin resistance, and increased expression of BVR could be of value in increasing expression of the genes that function in cell growth and differentiation.
Supplementary Material
Acknowledgments
We thank Ms. J. Boyce for preparing the manuscript. This work was supported by National Institutes of Health Grant ES04066.
Author contributions: N.L.-M. and M.D.M. designed research; N.L.-M., J.S., M.D.T., A.K., and Z.H. performed research; M.D.M. analyzed data; M.D.M. wrote the paper; and M.D.M. directed and funded the research.
Abbreviations: BVR, biliverdin reductase; IR, insulin receptor; IRK, IR tyrosine kinase; IRS, IR substrate; MBP, myelin basic protein; PI3-kinase, phosphatidylinositol 3-kinase; PTK, protein tyrosine kinase; SH-2, Src homology 2; wtBVR, wild-type BVR.
References
- 1.Salim, M., Brown-Kipphut, B. A. & Maines, M. D. (2001) J. Biol. Chem. 276, 10929-10934. [DOI] [PubMed] [Google Scholar]
- 2.Hunter, T. & Cooper, J. A. (1985) Annu. Rev. Biochem. 54, 897-930. [DOI] [PubMed] [Google Scholar]
- 3.Kutty, R. K. & Maines, M. D. (1981) J. Biol. Chem. 256, 3956-3962. [PubMed] [Google Scholar]
- 4.Fakhrai, H. & Maines, M. D. (1992) J. Biol. Chem. 267, 4023-4029. [PubMed] [Google Scholar]
- 5.Maines, M. D., Polevoda, B. V., Huang, T. J. & McCoubrey, W. K., Jr. (1996) Eur. J. Biochem. 235, 372-381. [DOI] [PubMed] [Google Scholar]
- 6.Maines, M. D. (1992) Heme Oxygenase: Clinical Applications and Functions (CRC Press, Boca Raton, FL).
- 7.Maines, M. D. (1997) Annu. Rev. Pharmacol. Toxicol. 37, 517-554. [DOI] [PubMed] [Google Scholar]
- 8.Maines, M. D., Ewing, J. F., Huang, T. J. & Panahian, N. (2001) J. Pharmacol. Exp. Ther. 296, 1091-1097. [PubMed] [Google Scholar]
- 9.Ahmad, Z., Salim, M. & Maines, M. D. (2002) J. Biol. Chem. 277, 9226-9232. [DOI] [PubMed] [Google Scholar]
- 10.Kravets, A., Hu, Z., Miralem, T., Torno, M. D. & Maines, M. D. (2004) J. Biol. Chem. 279, 19916-19923. [DOI] [PubMed] [Google Scholar]
- 11.Miralem, T., Hu, Z. B., Torno, M. D., Lelli, K. M. & Maines, M. D. (2005) J. Biol. Chem., in press. [DOI] [PubMed]
- 12.Kikuchi, A., Park, S. Y., Miyatake, H., Sun, D., Sato, M., Yoshida, T. & Shiro, Y. (2001) Nat. Struct. Biol. 8, 221-225. [DOI] [PubMed] [Google Scholar]
- 13.Whitby, F. G., Phillips, J. D., Hill, C. P., McCoubrey, W. & Maines, M. D. (2002) J. Mol. Biol. 319, 1199-1210. [DOI] [PubMed] [Google Scholar]
- 14.Songyang, Z., Shoelson, S. E., McGlade, J., Olivier, P., Pawson, T., Bustelo, X. R., Barbacid, M., Sabe, H., Hanafusa, H., Yi, T., et al. (1994) Mol. Cell. Biol. 14, 2777-2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Myers, M. G., Jr., Zhang, Y., Aldaz, G. A., Grammer, T., Glasheen, E. M., Yenush, L., Wang, L. M., Sun, X. J., Blenis, J., Pierce, J. H. & White, M. F. (1996) Mol. Cell. Biol. 16, 4147-4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lavan, B. E., Fantin, V. R., Chang, E. T., Lane, W. S., Keller, S. R. & Lienhard, G. E. (1997) J. Biol. Chem. 272, 21403-21407. [DOI] [PubMed] [Google Scholar]
- 17.White, M. F. & Yenush, L. (1998) Curr. Top Microbiol. Immunol. 228, 179-208. [DOI] [PubMed] [Google Scholar]
- 18.White, M. F. (2002) Am. J. Physiol. Endocrinol. Metab. 283, E413-E422. [DOI] [PubMed] [Google Scholar]
- 19.Saxton, T. M., Henkemeyer, M., Gasca, S., Shen, R., Rossi, D. J., Shalaby, F., Feng, G. S. & Pawson, T. (1997) EMBO J. 16, 2352-2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pawson, T. & Scott, J. D. (1997) Science 278, 2075-2080. [DOI] [PubMed] [Google Scholar]
- 21.Menegay, H. J., Myers, M. P., Moeslein, F. M. & Landreth, G. E. (2000) J. Cell Sci. 113, 3241-3253. [DOI] [PubMed] [Google Scholar]
- 22.Tanti, J. F., Gremeaux, T., van Obberghen, E. & Le Marchand-Brustel, Y. (1994) J. Biol. Chem. 269, 6051-6057. [PubMed] [Google Scholar]
- 23.De Fea, K. & Roth, R. A. (1997) J. Biol. Chem. 272, 31400-31406. [DOI] [PubMed] [Google Scholar]
- 24.Lee, Y. H., Giraud, J., Davis, R. J. & White, M. F. (2003) J. Biol. Chem. 278, 2896-2902. [DOI] [PubMed] [Google Scholar]
- 25.Liu, Y. F., Paz, K., Herschkovitz, A., Alt, A., Tennenbaum, T., Sampson, S. R., Ohba, M., Kuroki, T., LeRoith, D. & Zick, Y. (2001) J. Biol. Chem. 276, 14459-14465. [DOI] [PubMed] [Google Scholar]
- 26.Huang, T. J., Trakshel, G. M. & Maines, M. D. (1989) J. Biol. Chem. 264, 7844-7849. [PubMed] [Google Scholar]
- 27.Braiman, L., Alt, A., Kuroki, T., Ohba, M., Bak, A., Tennenbaum, T. & Sampson, S. R. (1999) Mol. Endocrinol. 13, 2002-2012. [DOI] [PubMed] [Google Scholar]
- 28.Maines, M. D. & Trakshel, G. M. (1993) Arch. Biochem. Biophys. 300, 320-326. [DOI] [PubMed] [Google Scholar]
- 29.Ben-David, Y., Letwin, K., Tannock, L., Bernstein, A. & Pawson, T. (1991) EMBO J. 10, 317-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duncan, P. I., Howell, B. W., Marius, R. M., Drmanic, S., Douville, E. M. & Bell, J. C. (1995) J. Biol. Chem. 270, 21524-21531. [DOI] [PubMed] [Google Scholar]
- 31.Johnson, K. W. & Smith, K. A. (1991) J. Biol. Chem. 266, 3402-3407. [PubMed] [Google Scholar]
- 32.Lindberg, R. A., Quinn, A. M. & Hunter, T. (1992) Trends Biochem. Sci. 17, 114-119. [DOI] [PubMed] [Google Scholar]
- 33.Blomberg, N., Baraldi, E., Nilges, M. & Saraste, M. (1999) Trends Biochem. Sci. 24, 441-445. [DOI] [PubMed] [Google Scholar]
- 34.Dhe-Paganon, S., Ottinger, E. A., Nolte, R. T., Eck, M. J. & Shoelson, S. E. (1999) Proc. Natl. Acad. Sci. USA 96, 8378-8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haslam, R. J., Koide, H. B. & Hemmings, B. A. (1993) Nature 363, 309-310. [DOI] [PubMed] [Google Scholar]
- 36.Pawson, T. (1995) Nature 373, 573-580. [DOI] [PubMed] [Google Scholar]
- 37.Hanks, S. K., Quinn, A. M. & Hunter, T. (1988) Science 241, 42-52. [DOI] [PubMed] [Google Scholar]
- 38.Backer, J. M., Myers, M. G., Jr., Shoelson, S. E., Chin, D. J., Sun, X. J., Miralpeix, M., Hu, P., Margolis, B., Skolnik, E. Y., Schlessinger, J., et al. (1992) EMBO J. 11, 3469-3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Myers, M. G., Jr., Sun, X. J. & White, M. F. (1994) Trends Biochem. Sci. 19, 289-293. [DOI] [PubMed] [Google Scholar]
- 40.Rocchi, S., Tartare-Deckert, S., Murdaca, J., Holgado-Madruga, M., Wong, A. J. & Van Obberghen, E. (1998) Mol. Endocrinol. 12, 914-923. [DOI] [PubMed] [Google Scholar]
- 41.Feinstein, R., Kanety, H., Papa, M. Z., Lunenfeld, B. & Karasik, A. (1993) J. Biol. Chem. 268, 26055-26058. [PubMed] [Google Scholar]
- 42.Strack, V., Hennige, A. M., Krutzfeldt, J., Bossenmaier, B., Klein, H. H., Kellerer, M., Lammers, R. & Haring, H. U. (2000) Diabetologia 43, 443-449. [DOI] [PubMed] [Google Scholar]
- 43.Rui, L., Yuan, M., Frantz, D., Shoelson, S. & White, M. F. (2002) J. Biol. Chem. 277, 42394-42398. [DOI] [PubMed] [Google Scholar]
- 44.DeClue, J. E., Vass, W. C., Papageorge, A. G., Lowy, D. R. & Willumsen, B. M. (1991) Cancer Res. 51, 712-717. [PubMed] [Google Scholar]
- 45.Leevers, S. J., Vanhaesebroeck, B. & Waterfield, M. D. (1999) Curr. Opin. Cell Biol. 11, 219-225. [DOI] [PubMed] [Google Scholar]
- 46.Newton, A. C. (2003) Biochem. J. 370, 361-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}